
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNovel spectrofluorometric approach for assessing vilazodone by blocking photoinduced electron transfer: analytical performance, and greenness–blueness evaluation†
Sayed M. Derayeaa,
Hadeer A. Elhamdy *b,
Khalid M. Badr El-Dina and
Mohamed Orabyb
*b,
Khalid M. Badr El-Dina and
Mohamed Orabyb
aDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Minia University, Minia 61519, Egypt. E-mail: adelhadeer33@gmail.com
bDepartment of Pharmaceutical Analytical Chemistry, Faculty of Pharmacy, Sohag University, Sohag 82524, Egypt
First published on 29th January 2024
Abstract
In this paper, vilazodone (VLD), a serotonin modulator prescribed for major depressive disorder, was investigated using a rapid, highly sensitive, and eco-friendly spectrofluorometric approach. The native fluorescence of VLD, originating from its indole moiety, exhibited an emission peak at 486 nm upon excitation at 241 nm. However, the presence of a piperazinyl nitrogen atom in the VLD structure, acting as an electron donor, significantly diminished the fluorescence intensity through photoinduced electron transfer (PET) to the indole ring. However, by protonating this nitrogen atom using 0.02 M Teorell–Stenhagen buffer (pH 3.5), inhibition of the PET process effectively blocked electron transfer, restoring the fluorescent properties of the drug. Further, an enhancement in the fluorescence was achieved by employing methanol as the solvent, resulting in a 1.5-fold increase. The combined use of PET blockage and methanol enabled the detection of VLD at levels as low as 0.78 ng mL−1. Calibration analysis demonstrated linearity within the range 5–400 ng mL−1, exhibiting a correlation coefficient of 0.9998 and a limit of quantification of 2.37 ng mL−1. The method obeyed the requirements of International Council on Harmonization (ICH). The proposed approach was applied for the accurate measurement of VLD in pharmaceutical tablets, content uniformity testing based on USP requirements, and determining VLD concentration in spiked human plasma. Moreover, the environmental impact, in addition to practical effectiveness, of the proposed approach was evaluated using different metrics.
1. Introduction
Vilazodone (VLD) is a potent medication that is widely used in the treatment of major depressive disorder. As a selective serotonin reuptake inhibitor (SSRI) and a partial agonist of the serotonin 5-HT1A receptor, VLD offers a unique pharmacological profile for managing depressive symptoms.1 The chemical structure of VLD is shown in Fig. 1, and its formula can be given as 5-[4-[4-(5-cyano-1H-indol-3-yl)butyl]piperazin-1-yl]-1benzofuran-2 carboxamide. VLD is a piperazine-indole derivative that has properties as an agonist with partial activity at the 5-HT1a receptor and as a selective serotonin reuptake inhibitor (SSRI),2 which have seen its use in therapy for major depression.3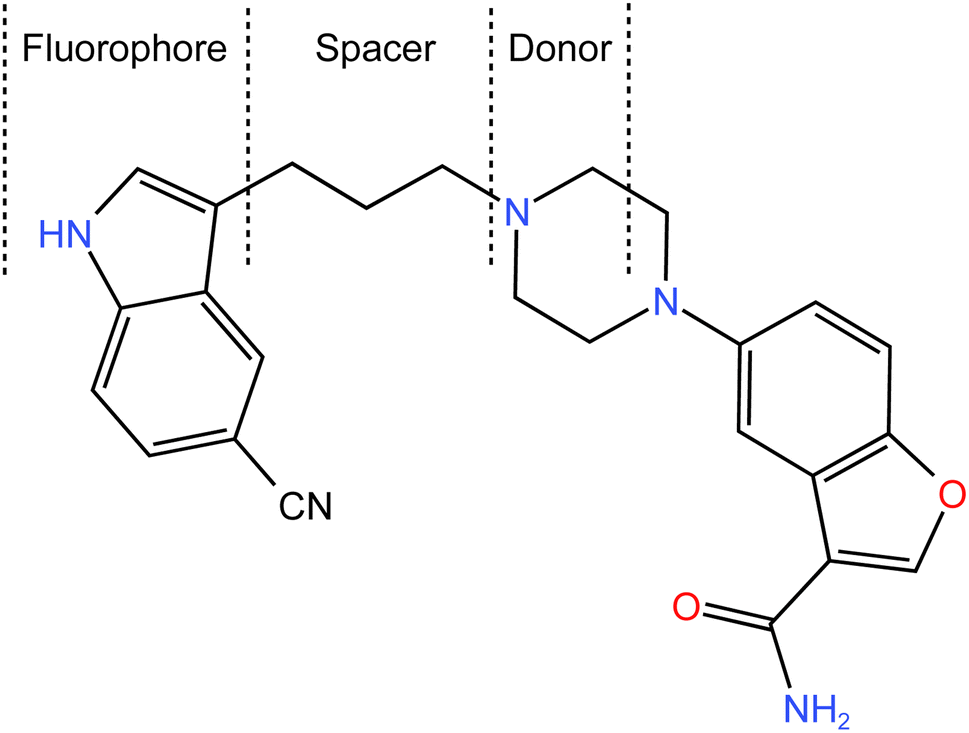 | ||
| Fig. 1 Chemical structure of VLD, showing the components involved in the photoinduced electron-transfer process (fluorophore, spacer, and donor (receptor)). | ||
Accurate and sensitive analytical methods are essential for the characterization and quantification of VLD in pharmaceutical formulations and biological samples. Few articles have been published reporting methods for testing VLD, which included spectrophotometry,4–7 spectrofluorimetry,8 HPTLC,7,9 HPLC,8,10–13 LC/MS,14,15 and UPLC.16,17 Although chromatographic methods8,10–13 have the merits of sensitivity and can be applied to separate different compounds in combinations and in the pharmacokinetic study of drugs and studies of degradation products, they utilize expensive equipment, high-purity reagents, and well-trained personnel.14,16,17 Alternatively, spectrofluorimetry, a powerful analytical technique, has gained considerable attention in pharmaceutical analysis owing to its high sensitivity and selectivity. In addition, it is simple, rapid, and utilizes less expensive solvents.
VLD has native fluorescence derived from its indole moiety. However, the presence of a piperazinyl nitrogen atom attached to the aliphatic bridge in the VLD structure introduces a significant challenge. This nitrogen atom acts as an electron donor and initiates photoinduced electron-transfer (PET), leading to a substantial reduction in the fluorescent signal.
To overcome this limitation, innovative strategies are required to effectively block the photoinduced electron transfer and restore the fluorescence of VLD. In this study, a highly sensitive spectrofluorimetric approach was proposed that employs PET blocking to enhance the detection and quantification of VLD. By protonating the piperazinyl nitrogen atom using the Teorell–Stenhagen buffer system (pH 3.5), the PET process could be efficiently inhibited, allowing the re-emergence of the drug's fluorescence.
Further, the choice of solvent plays a crucial role in enhancing the fluorescence signal. By carefully selecting the appropriate solvent, further fluorescence enhancement could be achieved, thus further improving the sensitivity and accuracy of VLD quantification.
In this research article, a comprehensive investigation of the spectrofluorimetric analysis of VLD is presented utilizing PET blocking and using methanol as a solvent. The developed approach allows the rapid, highly sensitive, and eco-friendly detection of VLD in pure form, and in pharmaceutical tablets. The great sensitivity of the suggested method could expand its application to examine VLD in biological samples. Furthermore, the ultimate simplicity of the current approach enabled its use in the content uniformity testing of commercial VLD tablets. The method's performance characteristics, including its limit of detection, linearity, and precision, were thoroughly evaluated, and validated according to regulatory guidelines. Moreover, the environmental impact of the proposed method was assessed using established indices, including the National Environmental Method Index (NEMI), eco-scale, GAPI, and AGREE, all highlighting its greenness, applicability, and sustainable attributes. In addition, the practical usefulness of the proposed method was evaluated using the Blue Applicability Grade Index (BAGI).
Overall, this study aimed to provide an innovative and reliable spectrofluorimetric approach for the assessment of VLD, contributing to the advancement of analytical methods in pharmaceutical analysis and facilitating the quality control of VLD-containing formulations.
2. Experimental
2.1. Instrumentation
Spectrofluorometric measurements were conducted using a Jasco FP-8350 spectrofluorometer (Hachioji, Tokyo, Japan). The instrument features a 150 W Xe-arc lamp and a PMT tuned to 400 V. The emission and excitation monochromator slits were both 5 nm wide, and the scanning speed was 1000 nm min−1. Aquatron Water Still A4000D system producing double-distilled water (Cole-Parmer, Staffordshire, UK), Jenway 3510 pH meter (Staffordshire, UK), and Mettler Toledo 5-digit balance (Greifensee, Switzerland) were also used in the study.For measurement of the absorbance for the reported method, a T80 double beam UV-VIS spectrophotometer (PG Instruments, Leicestershire, UK) was used. Two matched 1 cm quartz cells were utilized in all the measurements.
2.2. Materials and reagents
VLD was kindly obtained from Rameda Co. for Pharmaceutical Industries & Diagnostic Reagents (6th of October City, Egypt). Vilaphoria ® tablets (20 mg VLD) were bought via the local market. Analytical grade acetonitrile, methanol, ethanol, β-cyclodextrin (β-CD), Tween 80, and spectroscopic-grade perchloric acid were obtained from Merck (Darmstadt, Germany). Analytical grade sodium hydroxide was supplied by Fischer Scientific (Loughborough, United Kingdom). Sodium dodecyl sulfate (SDS) of analytical grade, cetyltrimethylammonium bromide (CTAB), acetone, sulfuric, phosphoric, acetic, and hydrochloric acid were purchased from El Nasr Pharmaceutical& Chemical Co. (Cairo, Egypt).2.3. Stock solutions
To make the solution of VLD (1000 μg mL−1), 25 mg of VLD was dispersed in methanol in an amount equal 25 mL. Next, 10 mL of that solution (1000 μg mL−1) was placed in a volumetric flask measuring 100 mL and methanol was properly added to achieve a 100 μg mL−1 solution of VLD. Various concentrations of standard VLD were made utilizing a similar solvent. These solutions were preserved in the fridge for use in the subsequent work.2.4. General assay procedures
Standard VLD solutions at concentrations from 5–400 ng mL−1 were put in 5 mL volumetric flasks, and 0.5 mL of Teorell–Stenhagen buffer (0.02 M, pH 3.5) was incorporated and then the flask was filled to the mark using methanol. The contents were then thoroughly mixed. The fluorescence intensity of the solutions was determined at 486 nm after excitation at 241 nm. The blank experiment was prepared in a similar way but without the addition of VLD solution. The resulting fluorescence vs. VLD concentrations were plotted to obtain a graph and the linear regression equation was obtained.2.5. Procedures for method validation
In the evaluation of the accuracy, precision, and robustness of the proposed method, different standard solutions of the studied drug under the specified conditions were analyzed using the general procedure. The values of the measured fluorescence intensities were utilized to calculate the drug concentrations by applying a linear regression equation for the calibration curve. The % recovery was calculated by dividing the found drug concentration by the claimed concentration in each standard solution, and then multiplying this by 100. The analysis was carried out in triplicate and the standard deviation was estimated.2.6. Procedures for assessing VLD tablets
Ten Vilaphoria® tablets (20 mg per tablet) were weighed and then pulverized using a mortar and pestle. A weight of the finely powdered tablets corresponding to 20 mg VLD was added in to a 50 mL volumetric flask, and then 30 mL of methanol was introduced, and the solution was sonicated for 30 min and filled to the mark with the same the solvent. The contents of the flask were subjected to filtering, and the initial portion of the filtrate was discarded. Aliquots of this stock solution were diluted to provide concentrations that fell inside the linear range. The general assay procedure was performed as mentioned previously. The nominal concentration in the test sample solution was computed using the previously estimated regression equation.For analyzing the commercial tablets with the reported method,5 different standard solutions of the drug (1.0–6.0 μg mL−1) were prepared in methanol. The absorbance of each solution was measured at 241 nm against blank methanol. The calibration curve for the reported method was constructed by plotting the drug concentration versus the absorbance. After that, the powdered commercial tablet was extracted with methanol as described above. A portion of the filtrate was diluted with methanol to give 3.0 μg mL−1 as a final concentration and its absorbance was measured at 241 nm. The obtained absorbance was utilized to determine the drug concentration in the analyzed sample solution. The obtained concentration was divided by the claimed concentration (3.0 μg mL−1) in the sample solution and then multiplied by 100 to get the recovery %. All measurements were carried out in triplicate.
2.7. Procedure for assessing the content uniformity
The content uniformity testing of VLD in tablet formulation was performed following US guidelines (Chap. 905).18 Here, 10 Vilaphoria® 20 mg tablets were individually examined to affirm that their contents were homogeneous using the formerly mentioned procedures under the analysis of VLD tablets.2.8. Procedures for evaluating VLD in human plasma spiked with VLD
Blood was collected from the forearm vein of a healthy volunteer and placed in a series of EDTA tubes. The procedures for collecting plasma from healthy volunteers were carried out according to the Recommendations of the Declaration of Helsinki.19 All experiments that included the use of human plasma were performed in accordance with the Guidelines Applied to Research on Human Subjects of Minia University and approved by the “Commission on the Ethics of Scientific Research”, Faculty of Pharmacy, Minia University. In all cases, informed written consent was obtained from each participant before donating the blood samples. A volume of 5 mL of blood was separated into plasma by 20 min centrifuging at 4000 rpm. The obtained plasma was stored at −20 °C. Next, 1.0 mL of plasma was transferred into a clean tube and mixed with 1.0 mL of VLD standard solution (5–400 μg mL−1) and 2.0 mL acetonitrile was added to precipitate plasma protein. The tube was vortex-mixed for 1 min before centrifuging for 10 min at 4000 rpm. After transferring the transparent supernatant onto a 5 mL volumetric flask, the general assay method was carried out. Experiments were also performed for the blank prepared using drug-free plasma.3. Results and discussion
The molecular structure of VLD contains two moieties that may be responsible for its native fluorescence: indole and benzofuran (Fig. 1). Actually, the indole moiety is the fluorophore since it possesses a conjugated system of double bonds with a rigid aromatic structure. This conjugation allows for the absorption of ultraviolet (UV) or visible light, leading to the excitation of electrons to higher energy levels. Upon relaxation, these excited electrons emit light, resulting in fluorescence. On the other hand, the benzofuran moiety does not possess the same features as the indole ring. Consequently, the benzofuran moiety is not typically responsible for fluorescence in this compound. The piperazinyl moiety in VLD contains two nitrogen atoms. The distal nitrogen atom from the benzofuran, which is attached to the aliphatic chain, can act as a potential electron donor. In its protonated form (when the pH is slightly acidic, between 2.0 and 6.0), this piperazinyl nitrogen atom becomes positively charged. This protonation alters the electronic properties of the molecule and disrupts the electron-transfer process. However, in its neutral unprotonated form (at pH higher than 6.0), the piperazinyl nitrogen is not protonated and can readily donate electrons to the indole moiety. In this state, the excited state of the indole can undergo the PET process, which involves the transfer of electrons from the donor (piperazinyl nitrogen) to the electron acceptor (indole). The structural properties of the drug make it susceptible to the PET process as in the case of amino(ethanesulfonate) anthracene indicator20 and others.21,22 VLD contains the indole ring as a fluorophore, butyl bridge as a spacer, and the proximal piperazine nitrogen as an electron donor (Fig. 1). The PET process effectively quenches the fluorescence emission of the drug. When the pH of the medium is 6.0 or lower, the piperazinyl moiety becomes protonated, which hinders its ability to donate electrons. As a result, the inhibition of PET takes place, allowing the excited state of the molecule to persist for a longer duration and emit fluorescence. Therefore, the high fluorescence exhibited by VLD at pH between 2.0 and 6.0 can be attributed to the protonation of the piperazinyl moiety, which inhibits the photoinduced electron-transfer process and allows for the augmentation of the fluorescence emission.VLD has an emission wavelength of 486 nm after an excitation wavelength of 241 nm (Fig. 2) and displayed an enhancement in VLD fluorescence upon the addition of Teorell–Stenhagen buffer (pH 3.5). In addition, an additional enhancement of the fluorescence intensity of VLD was observed in the presence of methanol as a diluting solvent.
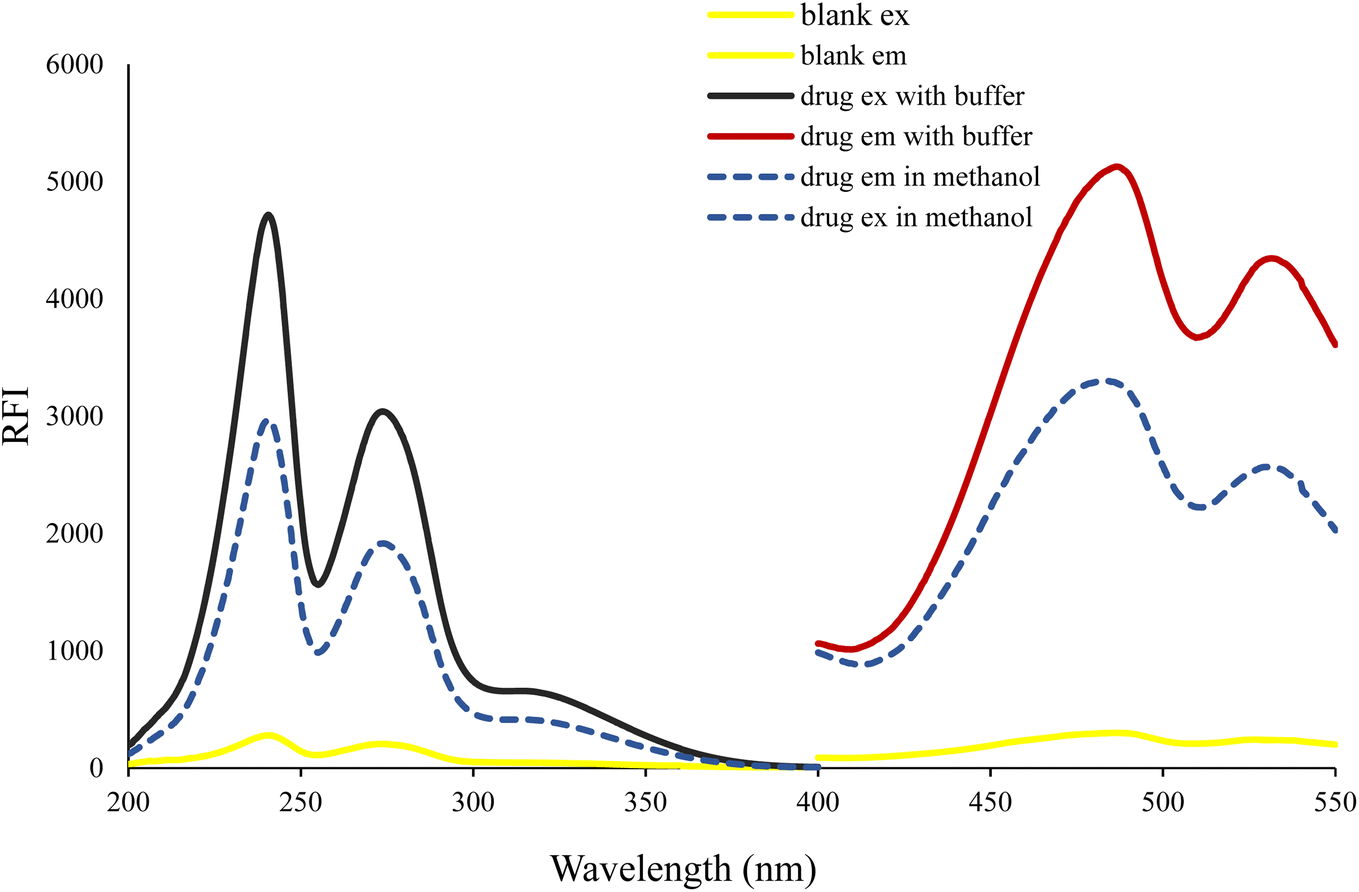 | ||
| Fig. 2 Excitation and emission spectra of 200 ng mL−1 VLD and the excitation and emission spectra of the blank in methanol. | ||
3.1 Improvement of the experimental parameters
The influence of various experimental parameters on the native fluorescence intensity of VLD were examined to get the optimal parameters to maximize the fluorescence intensity.To further confirm the protonation of the studied drug at pH below 6.0, the effect of the pH on the absorption spectrum of the drug was studied. The results are presented in the ESI (S1†) Although the change was not high, it was clear that increasing the pH led to a slight but distinct decrease in absorbance at 241 nm. Furthermore, a very slight red shift in the λmax at 241 nm was also observed upon increasing the pH above 6.0. Both changes in the position and absorbance confirmed the deprotonation of the drug by increasing the pH beyond 6.0.
At low pH values, below the pKa of the relevant ionizable group, the nitrogen of the indole ring is likely to be protonated (>NH2+). This protonation can disrupt the electronic structure and conjugation within the indole ring, leading to a decrease in fluorescence efficiency. Protonation of the indole nitrogen can also increase the internal conversion or nonradiative decay processes, reducing the population of the excited state and hence decreasing the fluorescence intensity. Furthermore, in highly acidic medium (below pH 2.0), the cyano group attached to the indole moiety can be converted into the carboxylic functional group. The conversion of the cyano group (–CN) into carboxylic group (–COOH) can have a significant impact on the fluorescence properties of a molecule, often resulting in fluorescence quenching. This quenching effect is due to the higher electron-withdrawing nature of the carboxylic group compared to the cyano group, which can decrease the electron density within the indole fluorophore. The reduced electron density can lead to a less favorable excited state and an increased likelihood of nonradiative decay pathways, resulting in a decreased fluorescence intensity or shortened fluorescence lifetime. Furthermore, the fluorescence strength of VLD at pH 2.5–4.5 was increased by 1.5-fold compared with methanol alone. Hence, 0.02 M Teorell–Stenhagen solution of buffer at pH 3.5 was chosen for the following work (Fig. S1†).
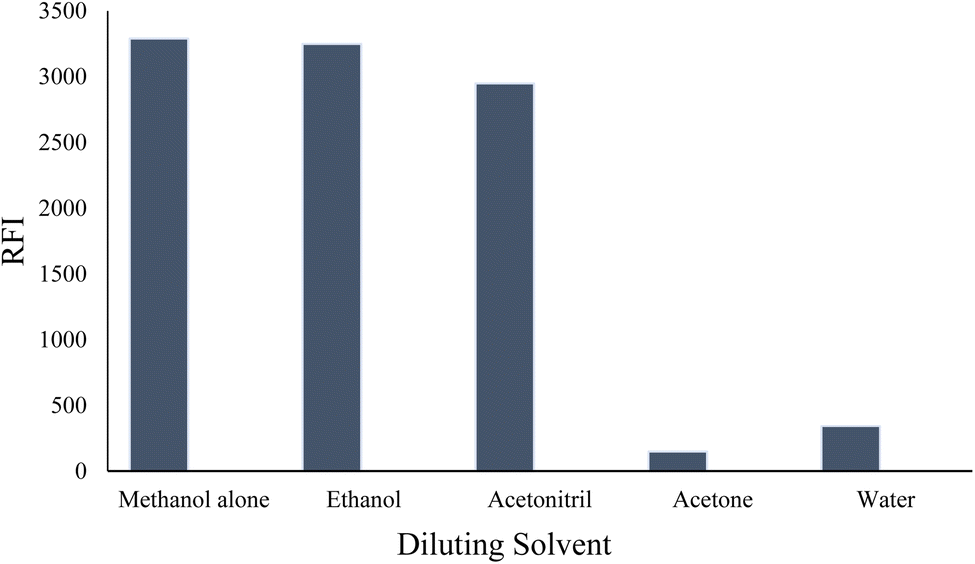 | ||
| Fig. 3 Effect of different diluting solvents on the relative fluorescence intensity (RFI) of 200 ng per mL VLD. | ||
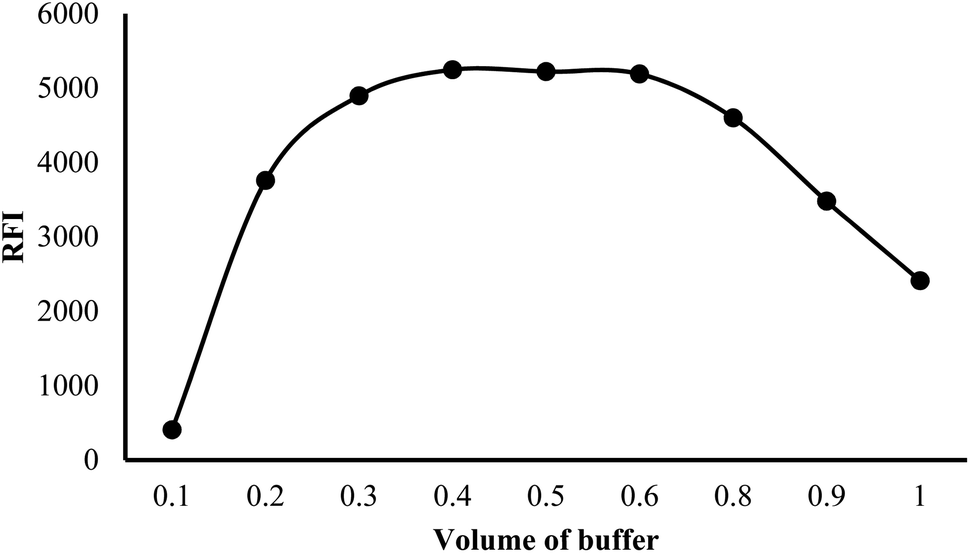 | ||
| Fig. 4 Effect of different volumes of buffer on the relative fluorescence intensity (RFI) of 200 ng per mL VLD. | ||
The presence of an electron-transfer state in tryptophan to a nearby amide in the protein backbone, which is available in polar environments, may result in a quenching of the fluorescence.23 Thus, the amide group attached to the benzofuran ring in VLD promoted fluorescence quenching, arising from a loss of energy owing to an electron transfer from the aromatic indole ring to this amide. Since the electron-transfer state is expected to be high in energy, normally this would not be a possible outcome, but because of its large dipole, such a state should be more accessible in polar solvents, such as water, which is a polar protic solvent with a relatively high dielectric constant.
3.2 Validation of the method
The validation of the analytical process was carried out in accordance with the International Council on Harmonization (ICH) guidelines.24 The study included assessing the linearity, range, limit of detection (LOD), limit of quantification (LOQ), accuracy, precision, and robustness.| Parameters | Value |
|---|---|
| Linear range (ng mL−1) | 5–400 |
| Slope (b) | 23.72 |
| Standard deviation of slope (Sb) | 0.20 |
| Intercept (a) | 131.54 |
| Standard deviation of intercept (Sa) | 5.63 |
| Correlation coefficient (r) | 0.9998 |
| Determination coefficient (r2) | 0.9996 |
| Number of determinations | 5 |
| Limit of detection (LOD ng mL−1) | 0.78 |
| Limit of quantification (LOQ ng mL−1) | 2.37 |
3.3 Applications
AV; AV = KS +|M − ![[X with combining macron]](https://www.rsc.org/images/entities/i_char_0058_0304.gif) | | |
is the mean of the % recovery of the individual content. The AV must be smaller than the maximum permitted AV (L1 = 15). The aforementioned equation changes depending on the value of .| — If 98.5% ≤ ≤ 101.5%, then M = (AV = KS). |
| — If < 98.5%, then M = 98.5% (AV = 98.5 − + KS). |
| — If > 101.5%, then M = 101.5% (AV = − 101.5 + KS). |
The obtained AV value using the suggested spectrofluorometric method for the analysis of the 10 Vilaphoria 20 mg tablets was lower than the L1 value, thus affirming the uniformity of the studied tablet formulations (Table 6).
| Tablet number | Proposed method |
|---|---|
| 1 | 102.08 |
| 2 | 98.35 |
| 3 | 98.10 |
| 4 | 99.49 |
| 5 | 97.23 |
| 6 | 98.12 |
| 7 | 101.85 |
| 8 | 101.62 |
| 9 | 98.33 |
| 10 | 97.07 |
| Mean (X) | 99.22 |
| Standard deviation (S) | 1.93 |
| Acceptance value (AV) | 5.12 |
| Maximum allowed acceptance value (L1) | 15 |
3.4 Method greenness evaluation
Green processes are those having no or minimal usage of dangerous chemicals, the elimination of waste, and a decrease in energy consumption.The method was evaluated in accordance with the National Environmental Method Index (NEMI),25 which aims to avoid the use of permanent, bio-accumulative, and toxic solvents (PBTs). Methanol used in the developed spectrofluorometric method is not a PBT solvent. Also, the Teorell–Stenhagen buffer pH of the proposed method was 3.5, which is not judged as corrosive. Furthermore, the volume of the waste was not greater than 50 mg or 50 mL. According to these results, the proposed fluorometric method conserved solvents while producing low amounts of waste. As a result of these factors, it achieved each of the four quadrants of the greenness profile and is regarded as an eco-friendly green approach, as shown in Table 7.
| Parameters | Penalty points | NEMI pictogram | |
|---|---|---|---|
| a If the score is greater than 75, it represents excellent green analysis. If the score is greater than 50, it represents acceptable green analysis. If the score is less than 50, it represents inadequate green analysis.b NEMI = National Environmental Method Index; PBT = persistent, bio-accumulative, and toxic solvents. | |||
| Reagents | None | 0 |  |
| Instrument | Spectrofluorometer | 0 | |
| Solvent | Methanol | 6 | |
| Teorell–Stenhagen buffer (0.02 M) | 0 | ||
| Energy consumption | (Less than 0.1 kW h per sample) | 0 | |
| Occupational hazard | (Analytical process hermitization) | 0 | |
| Waste | (10 mL) | 3 | |
| Total penalty points | 9 | ||
| Analytical eco-scale total scorea | 91 | ||
The eco-scale is a straightforward method that can be used in the practice of quality control laboratories. The subsequent equation (analytical eco-scale score = 100 − total penalty)26 is employed to determine the penalty point value for each of the procedure's defined parameters, such as the quantity of chemicals used, dangers to employees, waste production, and energy consumption. If the score is greater than 75, the analytical method is considered green. The eco-scale score of the developed fluorometric method was found to be 91 (Table 7), which is regarded as environmentally friendly.
Another trend, called GAPI,27 can be used to assess how environmentally friendly an analytical process is, from sample collection to final analysis. The greenness of each stage in the process of analysis is evaluated using a pictogram, which has three color levels: green, yellow, and red, in the GAPI tool. The suggested technique revealed 5 yellow, 7 green, and 3 red areas when evaluated using the GAPI metric. These regions are related to the toxicity and solvent volume used. As a result, the suggested approach had minimal environmental impact, see Fig. 5a.
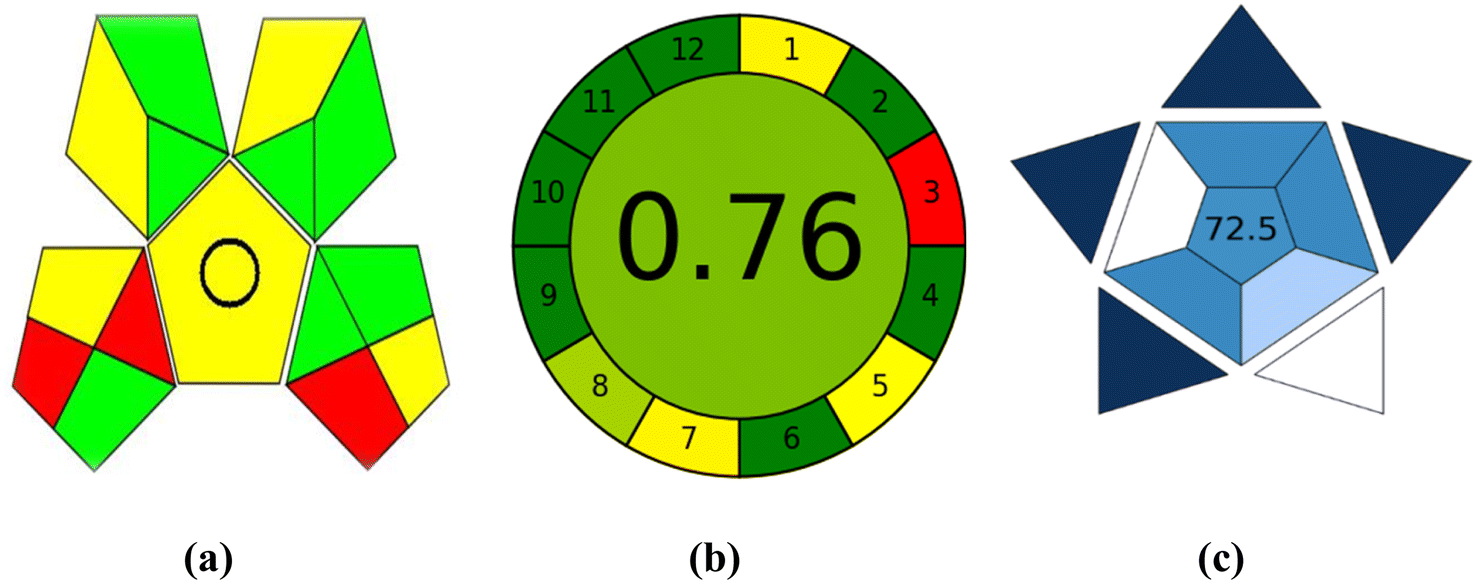 | ||
| Fig. 5 Evaluation of the greenness using GAPI (a) and AGREE (b) metrics and the blueness using BAGI (c) of the proposed spectrofluorometric method. | ||
The most recent metric is AGREE.28 The submitted criteria for the AGREE metric are flexible and can be weighted a different way, drawing design ideas from the 12 significance principles. Twelve input variables are graded from 0 to 1. The total of each principle's assessment results is the final evaluation result. The result is presented as a graph that resembles a clock, with the overall score and a color representation in the middle (Fig. 5b). The evaluation can be carried out with special software, which also generates a report in addition to an auto-generated graph. The AGREE evaluation depends on numerous variables, including the solvent type, volume used, quantity of chemicals throughout each run, and solvent toxicity to individuals and the surrounding environment. According to the AGREE evaluation metric, the proposed method received an overall score of 0.76.
3.5 Blueness evaluation
The Blue Applicability Grade Index (BAGI)29 complements the established green analytical chemistry metrics. It is a new tool that can be used to assess an analytical method's practical considerations. In order to create a pictogram and a score that illustrate the usefulness and effectiveness of an analytical approach, BAGI takes into account 10 factors (Table S2†). The BAGI metric tool yields two different sets of results: an asteroid-shaped pictogram as a graphic illustration and a score in number at the center of the pictogram. It is advised that the final score should be greater than 60 in order for the analytical procedure to be deemed “practical”. The asteroid-shaped pictogram, which is made up of several shades of blue to represent varying degrees of compliance (dark blue for high, blue for moderate, light blue for low, and white for non-compliance), serves as a visual representation of the assessment result. The proposed approach achieved an overall score of 72.5 in the final rating, as shown in the pictogram's center in Fig. 5c.4. Conclusion
A highly sensitive, simple, accurate, and green spectrofluorimetric was generated to measure VLD in its pure form tablet formulation and in spiked human plasma. The technique depends on boosting VLD's intrinsic fluorescence by the addition of Teorell–Stenhagen buffer (0.02 M, pH 3.5). The method was validated according to the ICH guideline and was found to be linear in the range of 5–400 ng mL−1. The method was also shown to be accurate, precise, and robust. The method's greenness was assessed following the eco-scale, the National Environmental Method Index (NEMI), GABI, and AGREE metrics. In addition, the practical effectiveness of the analytical procedure was assessed using the BAGI metric. Furthermore, the suggested spectrofluorometric method was used to test the content uniformity of VLD in pharmaceutical tablets. The proposed more-sensitive approach made it possible to analyze VLD in spiked human plasma. Since the proposed method requires low time and effort, and utilizes environmentally friendly solvents, it can be useful in quality control laboratories.Conflicts of interest
There are no conflicts to declare.References
- D. Guay, Consult. Pharm., 2012, 27, 857–867 CrossRef.
- T. de Paulis, Invest. DrugsJ., 2007, 10, 193–201 CAS.
- M. P. Cruz, Pharmacol. Ther., 2012, 37, 28 Search PubMed.
- S. R. Chaudhari, V. K. Salunkhe, H. S. Deore and A. A. Shirkhedkar, Sustainable Chem. Pharm., 2021, 24, 100534 CrossRef.
- K. R. Vivekkumar, F. C. Chetan, D. P. Pritam and J. S. Sanjay, J. Pharm. Res., 2014, 13, 20–22 CrossRef.
- C. Mazi, S. Karaderi and F. Arioz, Int. J. Pharm. Res. Allied Sci., 2018, 7, 146–152 Search PubMed.
- S. S. Panda and R. K. V. Bera, Indian Drugs, 2023, 60, 61–66 CrossRef.
- R. I. El-Bagary, H. Hashem, M. A. Fouad and S. T. Mahmoud, Int. J. Pharm. Res. Scholars, 2014, 3, 113–119 Search PubMed.
- K. R. Vivekkumar, F. C. Chetan and J. S. Sanjay, J. Pharm. Res., 2014, 13, 71–73 CrossRef.
- A. M. El-Kosasy, M. H. Rahman and S. H. Abdelaal, Int. J. Pharm., Chem. Biol. Sci., 2018, 8, 319–329 Search PubMed.
- S. Ghosh, S. Venkatesh and B. V. V. Ravikumar, Int. J. PharmTech Res., 2015, 7, 204–211 Search PubMed.
- A. Petruczynik, K. Wroblewski, M. Szultka-Mlynska, B. Buszewsk, H. Karakula-Juchnowicz, J. Gajewski, J. Morylowska-Topolska and M. Waksmundzka-Hajnosi, Acta Pol. Pharm., 2017, 74, 765–775 Search PubMed.
- M. M. Annapurna, K. Anusha, A. S. Sharief and V. Sindhuri, Res. J. Pharm. Technol., 2017, 10, 1077–1080 CrossRef.
- W. Sui, X. Yang, W. Yu, Y. Jin, X. Luan, X. Wang and H. Xu, J. Pharm. Biomed. Anal., 2014, 98, 228–234 CrossRef PubMed.
- B. Ghosh, P. Mandal, S. Chakraborty, R. Bera, C. Saha, S. Poddar, S. Barma, J. Khanam, S. Karmakar and T. Pal, J. Anal. Pharm. Res., 2021, 10, 1–12 CrossRef.
- M. Iqbal, E. Ezzeldin, K. A. Al-Rashood and A. A. Bajrai, J. Anal. Toxicol., 2015, 39, 106–112 CrossRef PubMed.
- R. El-Bagary, H. Hashem, M. Fouad and S. Tarek, J. Chromatogr. Sci., 2016, 54, 1365–1372 Search PubMed.
- M. Rockville, The United States Pharmacopoeia 30, the National Formulary 25 US Pharmacopeial Convention, Electronic version, 2007 Search PubMed.
- P. Rickham, Br. Med. J., 1964, 2, 177 CrossRef.
- M. A. Cardona, C. J. Mallia, U. Baisch and D. C. Magri, RSC Adv., 2016, 6, 3783–3791 RSC.
- S. K. Dwivedi, R. C. Gupta, P. Srivastava, P. Singh, B. Koch, B. Maiti and A. Misra, Anal. Chem., 2018, 90, 10974–10981 CrossRef.
- H. Niu, J. Liu, H. M. O’Connor, T. Gunnlaugsson, T. D. James and H. Zhang, Chem. Soc. Rev., 2023, 52, 2322–2357 RSC.
- A. P. Osysko and P. L. Muíño, Biophys. J., 2013, 104, 346a CrossRef.
- ICH Harmonised Tripartite Guideline, 2005 Search PubMed.
- L. H. Keith, L. U. Gron and J. L. Young, Chem. Rev., 2007, 107, 2695–2708 CrossRef.
- K. Aken, L. L. Strekowski and L. Patiny, Beilstein J. Org. Chem., 2006, 2, 3 Search PubMed.
- J. Płotka-Wasylka, Talanta, 2018, 181, 204–209 CrossRef.
- F. Pena-Pereira, W. Wojnowski and M. Tobiszewski, Anal. Chem., 2020, 92, 10076–10082 CrossRef.
- N. Manousi, W. Wojnowski, J. Płotka-Wasylka and V. Samanidou, Green Chem., 2023, 25, 7598–7604 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3ra08034j |
| This journal is © The Royal Society of Chemistry 2024 |