
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular aggregation by hydrogen bonding in cold-crystallization behavior of mixed nucleobases analyzed by temperature-controlled infrared spectroscopy†
Akinori Honda‡
 *,
Ryo Nozawa and
Kazuo Miyamura*
*,
Ryo Nozawa and
Kazuo Miyamura*
Department of Chemistry, Faculty of Science, Tokyo University of Science, 1-3 Kagurazaka, Shinjuku-ku, Tokyo 162-8601, Japan. E-mail: honda-akinori@rs.tus.ac.jp; miyamura@rs.tus.ac.jp
First published on 25th January 2024
Abstract
The cold-crystallization behaviors of dodecyl-substituted nucleobases (adenine, uracil, and thymine) were analyzed. The dodecyl derivative from uracil alone did not exhibit cold crystallization; however, a mixture of adenine and uracil derivatives at a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 exhibited cold crystallization. These results are similar to the thermal behavior of dodecyl derivatives of adenine and thymine alone and in mixtures reported in a previous study. Temperature-controlled infrared spectroscopy was used to observe the molecular assembly states of the liquid, supercooled state, and cold-crystallized compounds. Hydrogen-bonded molecular pairs in the high-temperature liquid state, multiple hydrogen-bonded networks in the supercooled state, and reverse Hoogsteen-type complementary hydrogen bonds in cold-crystallized compounds were observed using infrared spectroscopy. The heterogeneity of the system, due to multiple types of hydrogen bonding, retarded the crystallization rate, resulting in supercooling and cold crystallization. Infrared spectroscopy, which can be used to measure the aggregation state of molecules, including the liquid and supercooled states, is an effective analytical method for clarifying the process of cold crystallization.
:1 exhibited cold crystallization. These results are similar to the thermal behavior of dodecyl derivatives of adenine and thymine alone and in mixtures reported in a previous study. Temperature-controlled infrared spectroscopy was used to observe the molecular assembly states of the liquid, supercooled state, and cold-crystallized compounds. Hydrogen-bonded molecular pairs in the high-temperature liquid state, multiple hydrogen-bonded networks in the supercooled state, and reverse Hoogsteen-type complementary hydrogen bonds in cold-crystallized compounds were observed using infrared spectroscopy. The heterogeneity of the system, due to multiple types of hydrogen bonding, retarded the crystallization rate, resulting in supercooling and cold crystallization. Infrared spectroscopy, which can be used to measure the aggregation state of molecules, including the liquid and supercooled states, is an effective analytical method for clarifying the process of cold crystallization.
Introduction
The thermal analysis of thermal-storage materials is becoming increasingly important for solving energy problems. Cold crystallization is a latent-heat-storage phenomenon involving supercooling.1–5 Molecules that exhibit cold crystallization are expected to retain their heat-storage state (the supercooled state) for long periods.6,7 Cold crystallization of small molecules is typically observed at low temperatures, at approximately room temperature,8,9 and is expected to be applied to heat-storage materials for low-temperature waste heat.Recently, small molecules exhibiting cold crystallization via supercooled liquids, glass,10–19 liquid crystals,20–24 and ionic liquid crystals25,26 have been reported. To develop better thermal-storage materials, the mechanism of the thermal behavior during cold crystallization must be analyzed in detail. Another important factor in cold crystallization is the supercooling phenomenon that occurs during cooling; therefore, the molecular assembly in the supercooled liquid or glass state must be investigated. Previously, the cold crystallization behavior of supercooled liquids was difficult to analyze because of the effect of averaging the molecular structures. In liquids, observing the aggregation state of molecules is difficult because the conformations of the molecules are averaged. As a result, the analysis of liquids to elucidate the mechanism of the cold crystallization of small molecules has remained stagnant. In a previous study,27 the cold crystallization of mixed nucleobases (alkyl derivatives of adenine and thymine) was reported. Complementary hydrogen bonding was observed to have an important effect on thermal behavior. The mode of hydrogen bonding was analyzed using Fourier-transform infrared (FTIR) spectroscopy. Therefore, molecular aggregation, including that in liquids, can be analyzed by investigating hydrogen bonds. Although the information is still averaged, FTIR analysis provides more detailed data on molecular aggregation in liquids.
In this study, the thermal behavior of the dodecyl derivative of uracil (U-C12), in addition to the derivatives of adenine (A-C12) and thymine (T-C12) used in a previous study (Fig. 1), was investigated at a temperature-controlled stage using FTIR spectroscopy. The hydrogen bonding in the molecular assemblies formed in the liquid, supercooled-liquid, and cold-crystallized states was analyzed. Complementary hydrogen bonds are formed in the AT (adenine–thymine) and AU (adenine–uracil) pairs. The thermal behavior and temperature-dependent changes in hydrogen bonding in a mixed system of dodecyl derivatives were investigated. Molecular aggregation in the liquid and supercooled states was successfully achieved. The results will be useful for understanding the cold-crystallization behaviors.
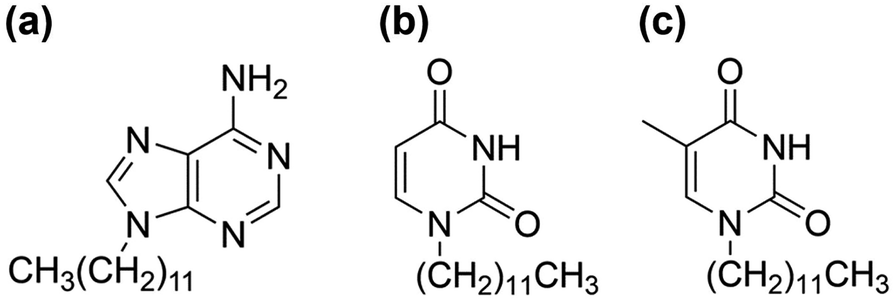 | ||
| Fig. 1 Chemical structures of (a) A-C12, (b) U-C12, and (c) T-C12. | ||
Experimental
Synthesis of U-C12
A-C12 and T-C12 were synthesized as previously described.27 U-C12 was synthesized using reagents obtained from Tokyo Chemical Industry and Kanto Chemical. Uracil powder (0.34 g, 3.0 mmol) and potassium carbonate (0.47 g, 3.4 mmol) were added to 10 mL of N,N-dimethylformamide. The mixture was then stirred at room temperature for 10 min. Liquid-state 1-bromododecane (0.85 g, 3.4 mmol) was added to the mixture, which was heated at 60 °C for 48 h. The product was purified by solvent extraction using CHCl3 and saturated aqueous NaCl solutions. The CHCl3 solution was collected and concentrated using a rotary evaporator. The product was purified via recrystallization from a 1:3 v/v ethyl acetate/hexane solution. The white crystalline powder was collected using suction filtration and dried under reduced pressure (U-C12, 0.14 g, 16% yield). Elemental analysis was performed using an analyzer (2400II, PerkinElmer, USA). The elemental analysis of C16H28N2O2 is as follows—calculated: C, 68.53; H, 10.07; N, 9.99%, observed: C, 68.48; H, 10.29; N, 10.00%. 1H nuclear magnetic resonance (NMR) spectra were recorded using a spectrometer (JNM-ECZ500R, JEOL, Japan). 1H-NMR (acetone-d6, 500 MHz): δ 9.87 (s, 1H, N–H), 7.55 (d, J = 8.0 Hz, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–N), 5.51 (d, J = 8.0 Hz, 1H, CHC–N), 3.73 (t, J = 7.4 Hz, 2H, N–CH2–C), 1.66 (quin, J = 6.9 Hz, 2H, N–C–CH2–C), 1.36–1.22 (m, 18H, C–CH2–C), 0.86 (t, J = 7.2 Hz, 3H, C–CH3).
CH–N), 5.51 (d, J = 8.0 Hz, 1H, CHC–N), 3.73 (t, J = 7.4 Hz, 2H, N–CH2–C), 1.66 (quin, J = 6.9 Hz, 2H, N–C–CH2–C), 1.36–1.22 (m, 18H, C–CH2–C), 0.86 (t, J = 7.2 Hz, 3H, C–CH3).
Thermal analysis
Differential scanning calorimetry (DSC) was conducted using a calorimeter (DSC 3500 Sirius, NETZSCH, Germany). The scan rate was 10 °C min−1, and the temperature range was −50 to +160 °C. The mixed system was prepared at a molar ratio of 1:1, and the molecules were mixed by convection by maintaining them at 160 °C for 1 h after the first heating process.
Powder X-ray diffraction (PXRD) analysis
PXRD patterns were measured using a diffractometer (MiniFlex, Rigaku, Japan) with a heat stage and CuKα (λ = 1.54059 Å). Cooling of the samples was performed outside the diffractometer using a cool plate (SCP-85, AS ONE, Japan). Preparation of the mixed system was conducted in the same method as described above.FTIR spectroscopy
A thermally controlled FTIR spectroscopic system was constructed, as shown in Fig. S1.† A spectrometer (FT/IR-4200, JASCO, Japan) was used to obtain the FTIR spectra. A stage (FTIR600, Linkam, UK) was used to control the sample temperature. The sample was inserted between CaF2 plates, and the plates were mounted on a stage with heat-resistant tape. Dry nitrogen gas was passed through the window of the stage to prevent condensation.Results and discussion
DSC analysis of U-C12, and mixture of A-C12 and U-C12 (AU-C12)
A DSC diagram of U-C12 is shown in Fig. 2a. Powder crystals of U-C12 were enclosed in an aluminum pan and used for the DSC analysis. In the first heating process, U-C12 melted with two endothermic peaks (peak a: 174 J g−1, 75 °C; peak b: 2 J g−1, 86 °C). In the first cooling process, crystallization peak c with an enthalpy of 85 J g−1 was observed at 54 °C, followed by a solid–solid transition (peak d: 10 J g−1, 31 °C). The corresponding transition peak e (7 J g−1, 38 °C) was observed in the second heating process. The U-C12 compounds melted, corresponding to peak f (108 J g−1, 86 °C).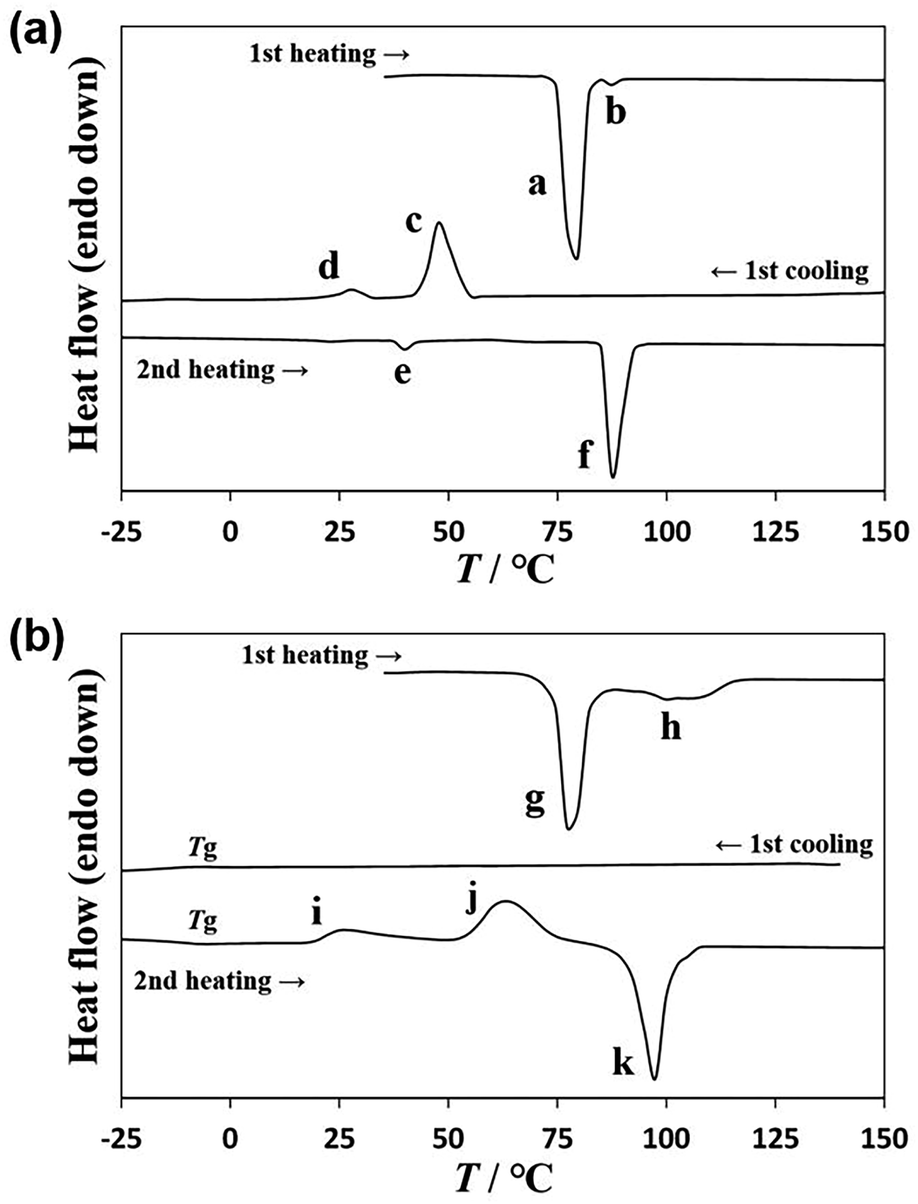 | ||
| Fig. 2 DSC diagram of (a) U-C12 and (b) AU-C12. | ||
The DSC diagram of AU-C12 is shown in Fig. 2b. During the first heating process, two melting peaks (peak g: 125 J g−1, 75 °C; peak h: 46 J g−1, around 100 °C) were observed. To ensure uniform mixing, the mixture was maintained at 160 °C for 1 h. Thereafter, the AU-C12 compounds were cooled, and no crystallization peaks appeared in the first cooling process. A glass transition Tg was observed at −8 °C. AU-C12 adopted supercooled liquid and glass states. The corresponding Tg was observed at −4 °C in the second heating process. In addition, cold crystallization (peak i: 28 J g−1, 19 °C; peak j: 77 J g−1, around 63 °C) was observed. Finally, the cold-crystallized sample melted at 92 °C, with an endothermic peak corresponding to an enthalpy of 104 J g−1 (peak k). In a previous study,27 A-C12 and T-C12 exhibited crystallization during cooling and melting during heating, whereas AT-C12 (a mixture of A-C12 and T-C12) exhibited supercooling and cold crystallization. Similarly, AU-C12 exhibited supercooling during cooling and cold crystallization during heating.
Powder X-ray diffraction analysis
The thermal processes observed in the DSC scans were analyzed using powder X-ray diffraction (PXRD). The PXRD pattern of U-C12 is shown in Fig. 3a. The blue line indicates the pattern of crystals formed between peaks c and d in Fig. 2a. The red line shows the pattern of crystals formed below the temperature of peak d. The black line shows the pattern of crystals formed between peaks e and f. The PXRD patterns indicated by the blue and black lines were identical, whereas those indicated by the red line were different. In addition, the patterns were different from those of the sample measured at room temperature before thermal treatment (Fig. S2†). This suggests that U-C12 exhibits crystal polymorphisms.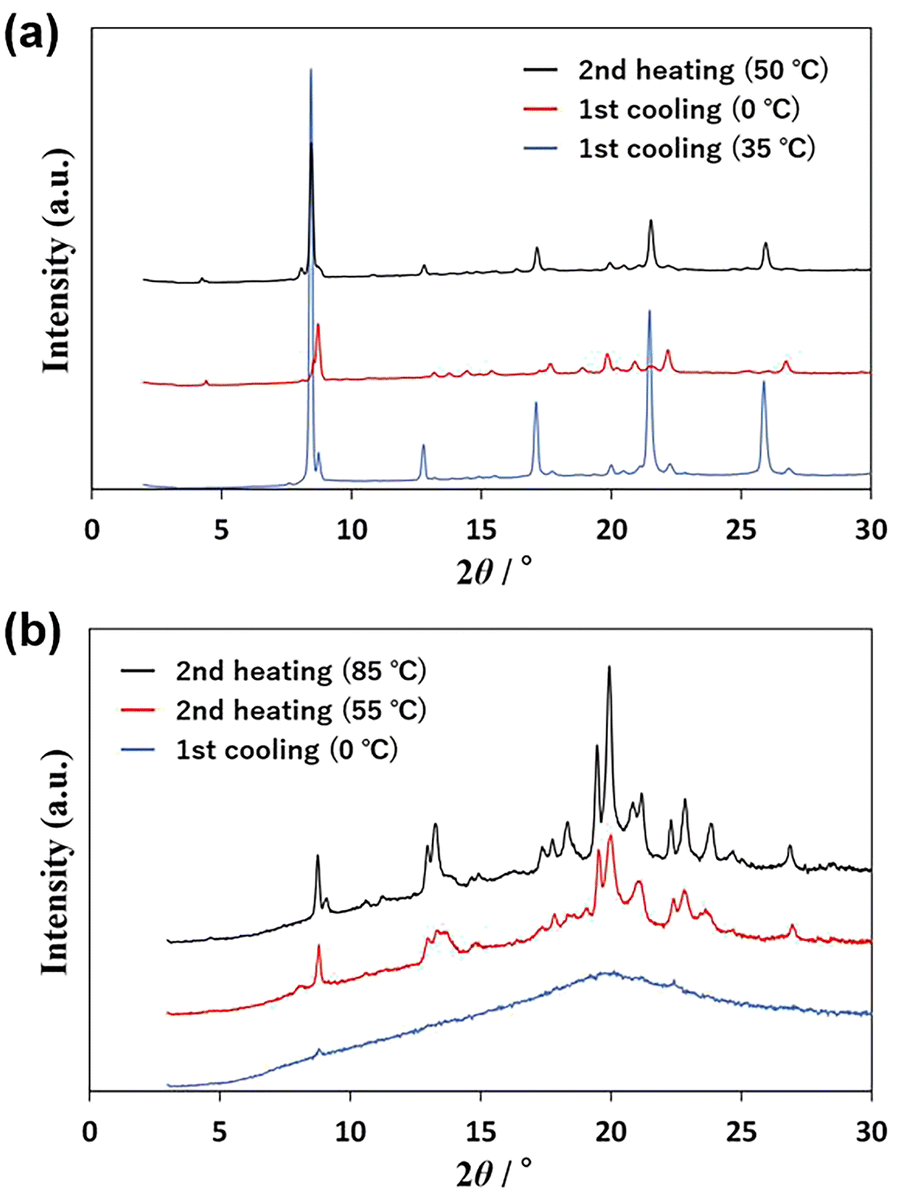 | ||
| Fig. 3 PXRD patterns of (a) U-C12 and (b) AU-C12. | ||
Fig. 3b shows the PXRD patterns of AU-C12. The blue line shows the pattern at 0 °C in the first cooling process. The pattern exhibits a broad peak (halo) at 2θ = 20°, suggesting that AU-C12 adopts an amorphous state and isotropic liquid state. The red line indicates the pattern of crystals formed at 55 °C (above the temperature of peak i in Fig. 2b) during the second heating process. The red line shows a crystalline pattern with many peaks, suggesting that AU-C12 underwent cold crystallization during heating from the supercooled state. The black line shows the pattern of the crystals formed between peaks j and k. The intensities of the peaks in the pattern corresponding to the red line increased, whereas new diffraction peaks (e.g., peaks at 9.2°, 11.2°, 14.8°, 17.4°, and 18.3°) appeared. Peak j corresponds to the growth of crystals formed at peak i and the cold crystallization of other crystalline states. Fig. S2† shows a comparison of the PXRD patterns of A-C12, U-C12, and AU-C12. At room temperature, the pattern of AU-C12 was a superposition of those of A-C12 and U-C12. On the other hand, after cold crystallization, the pattern of AU-C12 was different from those of A-C12 and U-C12. The results suggest that AU-C12 exhibits the new crystalline phases by cold crystallization.
Temperature-controlled IR spectroscopy
A previous study revealed that complementary hydrogen bonding between A-C12 and T-C12 is a factor in cold crystallization. In this study, the temperature-controlled IR spectra of AT-C12 and AU-C12 were acquired using the equipment shown in Fig. S1.† The changes in hydrogen bonding with temperature were investigated in detail.Fig. 4 shows the IR spectra of liquid-state A-C12, T-C12, and AT-C12 at 160 °C. The spectrum of AT-C12 (red line) was the same as that of A-C12. The spectrum of A-C12 exhibited peaks at 3478 cm−1 (asymmetric stretching vibration of free NH2 with one hydrogen-bonded H atom), approximately 3320 cm−1 (NH2 bend overtone), and 3170 cm−1 (hydrogen-bonded NH stretch).28 The spectrum of T-C12 showed peaks at 3418 cm−1 (NH stretch of thymine monomer), approximately 3200 cm−1 (NH stretch of stacked thymine dimer), and 3050 cm−1 (NH stretch of hydrogen-bonded thymine dimer).29,30 A peak was also observed at 3478 cm−1, arising from the free NH2 stretching of A-C12 hydrogen-bonded to T-C12.31 The peaks of T-C12 did not appear in the spectrum of AT-C12, suggesting that the hydrogen-bonded pairs of A-C12 and T-C12 were predominant in the high-temperature liquid state of AT-C12.
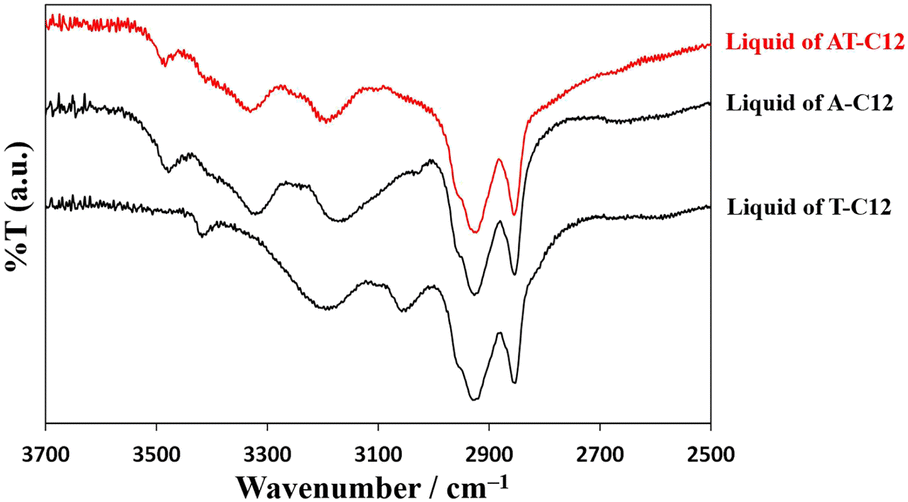 | ||
| Fig. 4 Comparison of infrared spectra of A-C12, T-C12, and AT-C12. | ||
Fig. 5a shows the variation in the IR spectra with the temperature of AT-C12. The spectrum of the high-temperature liquid state at 160 °C was the same as that in Fig. 4. The sample was cooled to −20 °C, and the spectrum of the supercooled state was measured. The peak at 3478 cm−1 disappeared, suggesting that both H atoms of the NH2 group of A-C12 formed hydrogen bonds, thereby forming a hydrogen-bonding network. The other peaks remained broad and are considered to be the averaged peaks corresponding to multiple types of complementary hydrogen bonds. The sample was re-heated, and the spectrum of the cold-crystallized solid was recorded at 60 °C. The spectrum was the same as that of the co-crystal formed by AT-C12, as shown at the bottom of Fig. 5a. The cold-crystallized sample formed complementary hydrogen bonds in a reverse Hoogsteen pattern.27 A stable crystal state with a single hydrogen-bonding pattern was formed; therefore, the peaks related to the hydrogen bonds (approximately 3100–3500 cm−1) became sharper. The hydrogen-bonded NH2 peaks of the reverse Hoogsteen structure appeared at higher wavenumbers than those of the Hoogsteen and Watson–Crick structures.32,33 Therefore, the peak of the cold-crystallized sample forming the reverse Hoogsteen structure showed a higher wavenumber shift relative to that of the averaged peak in the spectrum of the supercooled state. In the co-crystal, one H atom of NH2 formed a complementary hydrogen bond, whereas another H atom formed a hydrogen bond with an adjacent AT-C12 pair.27 This supports the conclusion that the disappearance of the peak at 3478 cm−1 was caused by the formation of a hydrogen-bonding network via NH2. In summary, a schematic diagram of the hydrogen-bonded pair with free NH2 in the high-temperature liquid state is shown in Fig. 6a, and multiple types of hydrogen bonds are shown in Fig. S3.† A diagram of the hydrogen-bonding network in supercooled liquid and cold-crystallized states is shown in Fig. 6b.
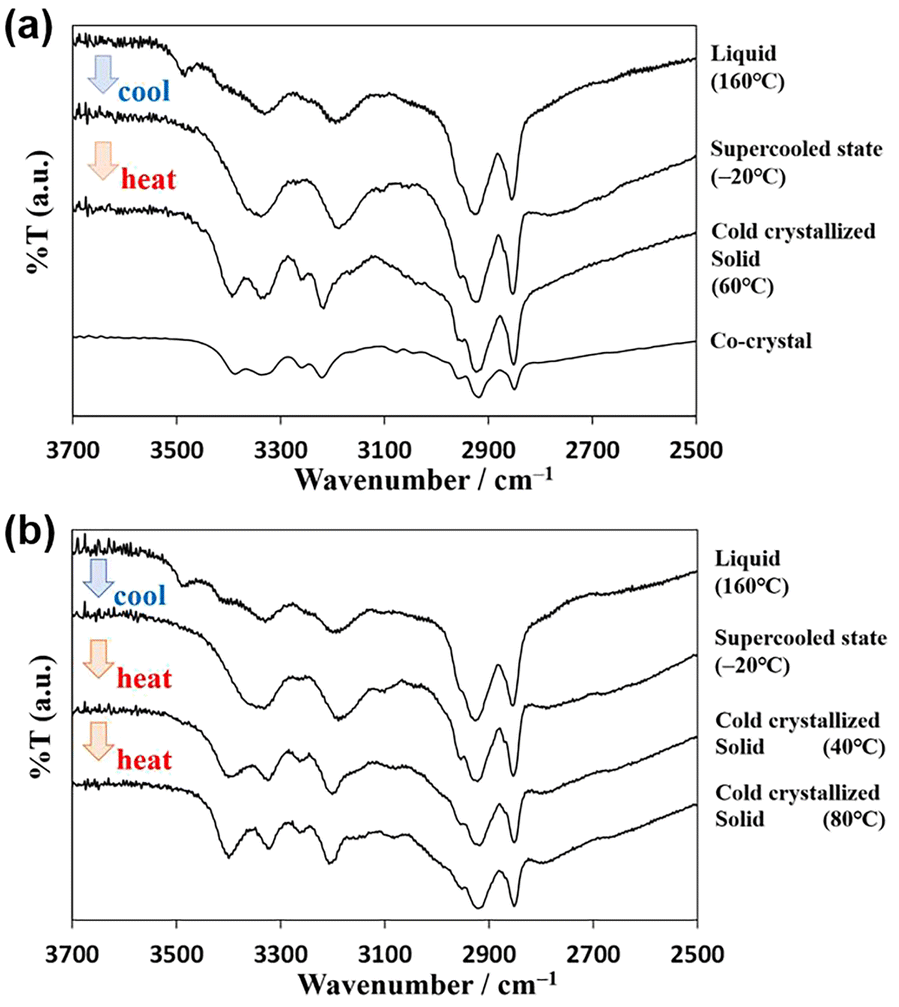 | ||
| Fig. 5 Infrared spectra of (a) AT-C12 and (b) AU-C12 in cold crystallization behavior. | ||
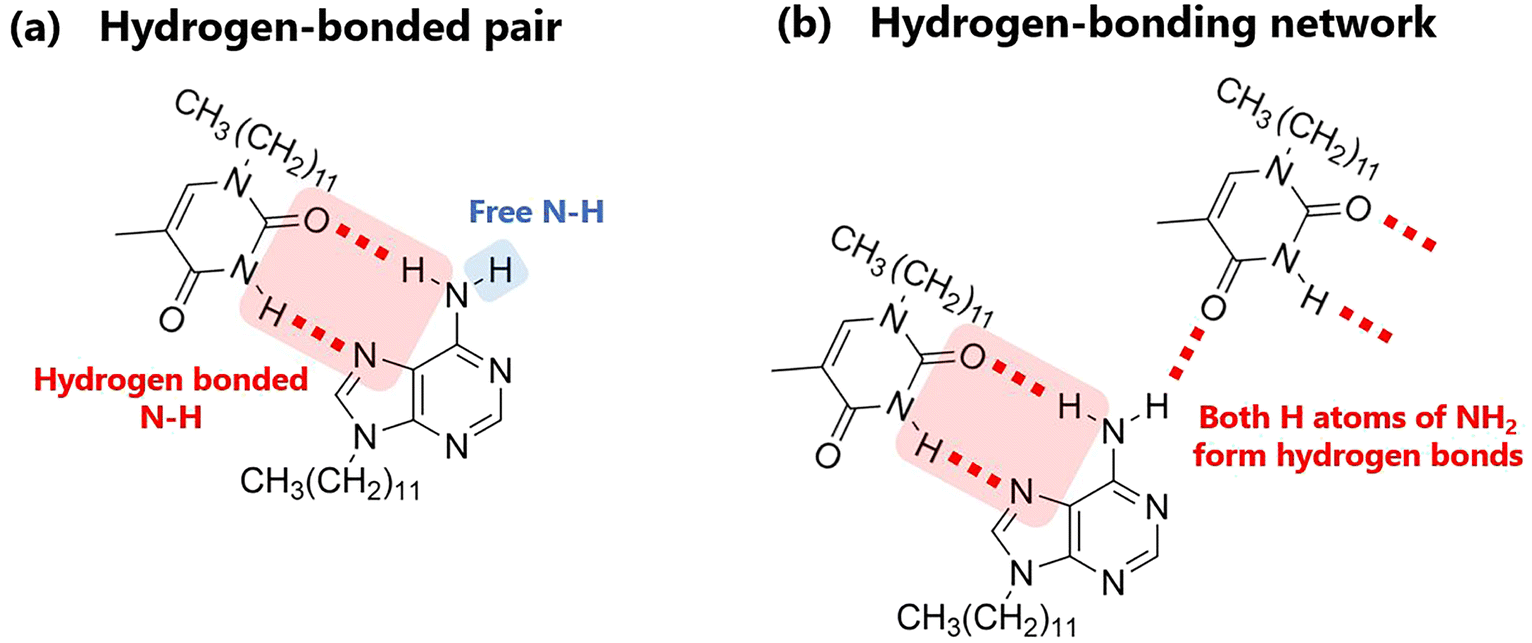 | ||
| Fig. 6 Schematics of (a) hydrogen-bonded pair and (b) hydrogen-bonding network. | ||
Fig. 5b shows the temperature-controlled IR spectrum of AU-C12, which changed with sample temperature. The spectrum of AU-C12 was almost identical to that of AT-C12. The FT-IR spectrum of the high-temperature liquid at 160 °C showed the asymmetric stretching vibration (3490 cm−1) of free NH2 with one hydrogen-bonded H atom. Similar to AT-C12, a hydrogen-bonded dimer of AU-C12 was formed, and no hydrogen-bonding network was formed in the high-temperature liquid. In the spectrum of the supercooled state at −20 °C, the peak of free NH2 disappeared, and averaged broad peaks of the hydrogen-bonding network were observed. Sharp peaks owing to the complementary hydrogen bonds of the reverse Hoogsteen pattern were observed in the spectrum of the cold-crystallized sample at 40 °C. These results suggest that the changes in the cold-crystallization behavior of the molecular assembly were the same for AT-C12 and AU-C12. A gradual increase in the absorption intensity at 3399 cm−1 in the temperature range of 40–80 °C was observed for AU-C12. This peak change was attributed to crystal polymorphism. As neither the IR spectra nor the PXRD patterns showed significant changes, the cold-crystallized compounds corresponding to peaks i and j in the DSC had almost the same structure. Both crystals exhibited a reverse Hoogsteen structure, and only the micro-orientations of the molecules were considered different. Fig. S4† shows the IR spectra measured at a step of 10 °C. The spectra of AT-C12 and AU-C12 change in response to cold crystallization (around 40 °C) and melting (around 100 °C). Therefore, the IR measurement proved to be a useful method to reveal the process of cold crystallization.
Temperature-controlled IR spectral measurements revealed that hydrogen-bonded pairs were formed in the high-temperature liquid state, but no hydrogen-bonding network was formed. In the supercooled state, multiple types of hydrogen bonds were formed, and one type of hydrogen-bonded structure (the reverse Hoogsteen structure) was formed during cold crystallization. Hence, the heterogeneity of the system, owing to multiple types of hydrogen bonds, reduced the crystallization rate during cooling, and cold crystallization occurred.
Conclusions
The molecular assemblies formed in each state (liquid, supercooled liquid, and cold-crystallized) were successfully determined using IR spectroscopy. The spectral changes were combined with the DSC and PXRD data to investigate the thermal behavior of the dodecyl-substituted nucleobases (adenine, uracil, and thymine). Various hydrogen bonds delayed crystallization during cooling, leading to cold crystallization. Temperature-controlled IR spectroscopy is a powerful technique for analyzing the cold-crystallization behaviors and is expected to contribute to future research on cold crystallization.Author contributions
A. Honda: conceptualization, investigation, validation, visualization, writing – original draft, writing – review & editing; R. Nozawa: investigation, validation, visualization; K. Miyamura: supervision, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by JSPS KAKENHI (Grant Number JP21K14725).Notes and references
- S. Puupponen, V. Mikkola, T. Ala-Nissila and A. Seppälä, Appl. Energy, 2016, 172, 96 CrossRef CAS.
- G. Englmair, Y. Jiang, M. Dannemand, C. Moser, H. Schranzhofer, S. Furbo and J. Fan, Energy Build., 2018, 180, 159 CrossRef.
- C. Rathgeber, S. Hiebler, R. Bayón, L. F. Cabeza, G. Zsembinszki, G. Englmair, M. Dannemand, G. Diarce, O. Fellmann, R. Ravotti, D. Groulx, A. C. Kheirabadi, S. Gschwander, S. Höhlein, A. König-Haagen, N. Beaupere and L. Zalewski, Appl. Sci., 2020, 10, 7968 CrossRef CAS.
- H. Zhou, L. Lv, Y. Zhang, M. Ji and K. Cen, Sol. Energy Mater. Sol. Cells, 2021, 230, 111244 CrossRef CAS.
- G. Wang, C. Xu, W. Kong, G. Englmair, J. Fan, G. Wei and S. Furbo, J. Energy Storage, 2021, 40, 102780 CrossRef.
- M. R. Yazdani, J. Etula, J. B. Zimmerman and A. Seppälä, Green Chem., 2020, 22, 5447 RSC.
- K. Turunen, M. R. Yazdani, A. Santasalo-Aarnio and A. Seppälä, Sol. Energy Mater. Sol. Cells, 2021, 230, 111273 CrossRef CAS.
- A. Honda, Y. Takahashi, Y. Tamaki and K. Miyamura, Chem. Lett., 2016, 45, 211 CrossRef CAS.
- A. Honda, S. Kakihara, M. Kawai, T. Takahashi and K. Miyamura, Cryst. Growth Des., 2021, 21, 6223 CrossRef CAS.
- G. Szklarz, K. Adrjanowicz, J. Knapik-Kowalczuk, K. Jurkiewicz and M. Paluch, Phys. Chem. Chem. Phys., 2017, 19, 9879 RSC.
- R. Chang, Q. Fu, Y. Li, M. Wang, W. Du, C. Chang and A. Zeng, CrystEngComm, 2017, 19, 335 RSC.
- K. Iwase, Y. Toyama, I. Yoshikawa, Y. Yamamura, K. Saito and H. Houjou, Bull. Chem. Soc. Jpn., 2018, 91, 669 CrossRef CAS.
- Y. Tsujimoto, T. Sakurai, Y. Ono, S. Nagano and S. Seki, J. Phys. Chem. B, 2019, 123, 8325 CrossRef CAS.
- A. A. Boopathi, S. Sampath and T. Narasimhaswamy, New J. Chem., 2019, 43, 9500 RSC.
- M. T. Viciosa, J. J. M. Ramos and H. P. Diogo, Int. J. Pharm., 2020, 584, 119410 CrossRef CAS PubMed.
- S. E. Lapuk, T. A. Mukhametzyanov, C. Schick and A. V. Gerasimov, Int. J. Pharm., 2021, 599, 120427 CrossRef CAS PubMed.
- A. Dołęga and P. M. Zieliński, J. Non-Cryst. Solids, 2022, 575, 121198 CrossRef.
- A. Dołęga, E. Juszyńska-Gałązka, A. Deptuch, S. Baran and P. M. Zieliński, Thermochim. Acta, 2022, 707, 179100 CrossRef.
- T. Rozwadowski, H. Noda, Ł. Kolek, M. Ito, Y. Yamamura, H. Saitoh and K. Saito, Phys. Chem. Chem. Phys., 2023, 25, 724 RSC.
- Ł. Kolek, M. Massalska-Arodź, K. Adrjanowicz, T. Rozwadowski, K. Dychtoń, M. Drajewicz and P. Kula, J. Mol. Liq., 2020, 297, 111913 CrossRef.
- Ł. Kolek, M. Jasiurkowska-Delaporte, E. Juszyńska-Gałązka and T. Rozwadowski, J. Mol. Liq., 2021, 339, 117076 CrossRef.
- A. Deptuch, M. Jasiurkowska-Delaporte, M. Urbańska and S. Baran, J. Mol. Liq., 2022, 368, 120612 CrossRef CAS.
- A. Drzewicz, E. Juszyńska-Gałązka, M. Jasiurkowska-Delaporte and P. Kula, CrystEngComm, 2022, 24, 3074 RSC.
- A. Deptuch, A. Lelito, E. Juszyńska-Gałązka, M. Jasiurkowska-Delaporte and M. Urbańska, Phys. Chem. Chem. Phys., 2023, 25, 12379 RSC.
- K. Ishino, H. Shingai, Y. Hikita, I. Yoshikawa, H. Houjou and K. Iwase, ACS Omega, 2021, 6, 32869 CrossRef CAS PubMed.
- K. Iwase, Y. Hikita, I. Yoshikawa, H. Houjou and K. Ishino, J. Phys. Chem. C, 2022, 126, 10668 CrossRef CAS.
- A. Kimijima, A. Honda, K. Nomoto and K. Miyamura, CrystEngComm, 2019, 21, 3142 RSC.
- P. Ottiger, J. A. Frey, H.-M. Frey and S. Leutwyler, J. Phys. Chem. A, 2009, 113, 5280 CrossRef CAS PubMed.
- Y. Nosenko, M. Kunitski and B. Brutschy, J. Phys. Chem. A, 2011, 115, 9429 CrossRef CAS PubMed.
- T. Fornaro, M. Biczysko, S. Monti and V. Barone, Phys. Chem. Chem. Phys., 2014, 16, 10112 RSC.
- J. A. Frey, A. Müller, H.-M. Frey and S. Leutwyler, J. Chem. Phys., 2004, 121, 8237 CrossRef CAS PubMed.
- Y. Nosenko, M. Kunitski, T. Stark, M. Göbel, P. Tarakeshwar and B. Brutschy, J. Phys. Chem. A, 2011, 115, 11403 CrossRef CAS PubMed.
- Y. Nosenko, M. Kunitski, T. Stark, M. Göbel, P. Tarakeshwar and B. Brutschy, Phys. Chem. Chem. Phys., 2013, 15, 11520 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Photograph of the FTIR system; PXRD of A-C12, U-C12, and AU-C12; image of multiple types of hydrogen bonds; FTIR spectra measured at a step of 10 °C. See DOI: https://doi.org/10.1039/d3ra08293h |
| ‡ Present address: Department of Applied Chemistry, Faculty of Science and Engineering, Chuo University, 1-13-27 Kasuga, Bunkyo-ku, Tokyo 112-8551, Japan. |
| This journal is © The Royal Society of Chemistry 2024 |