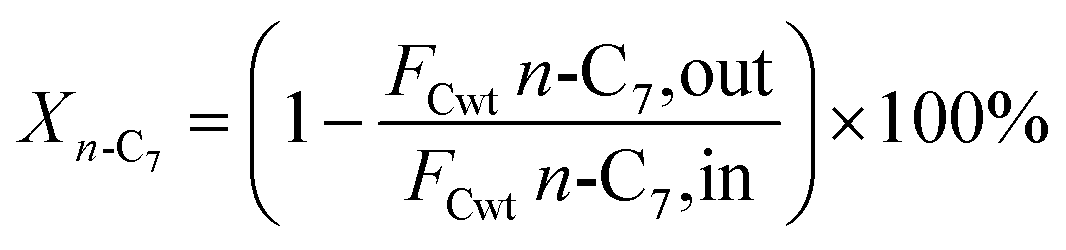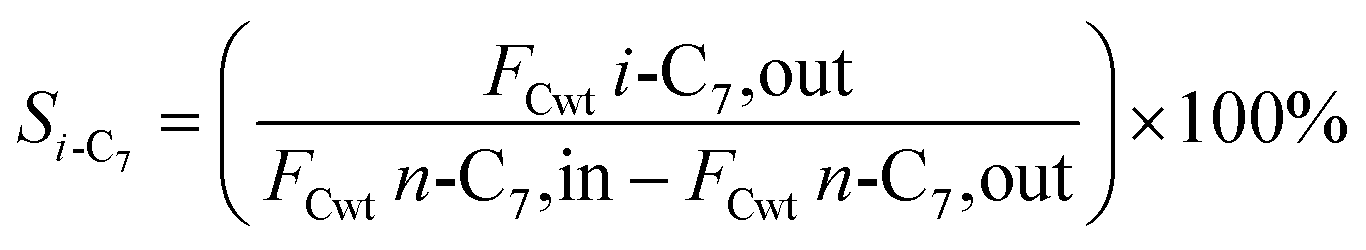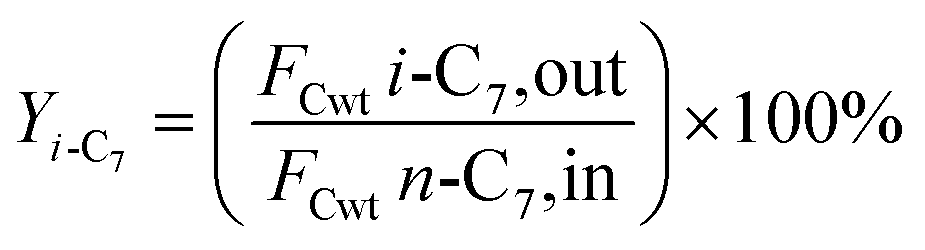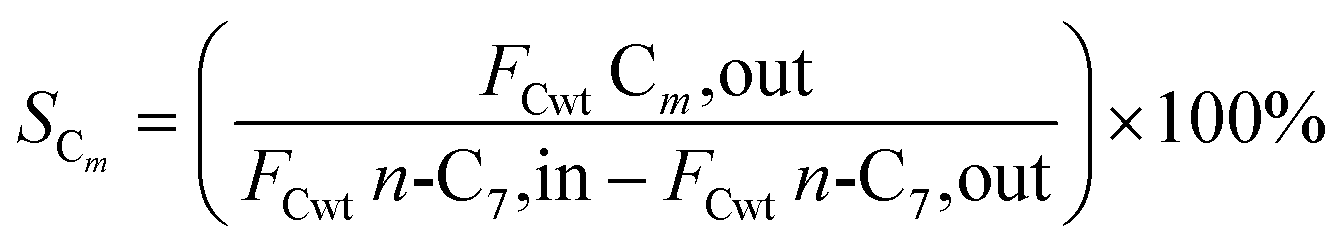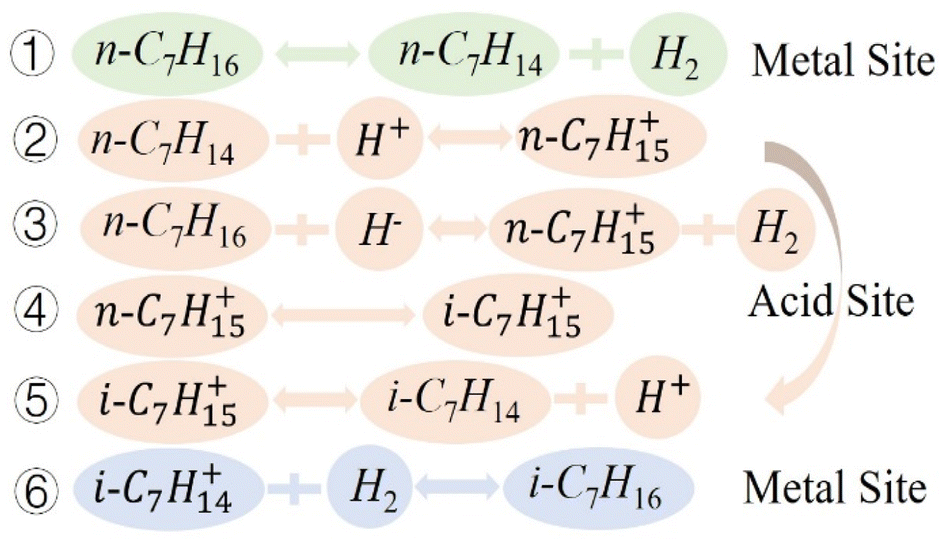DOI:
10.1039/D3RA08454J
(Paper)
RSC Adv., 2024,
14, 4105-4115
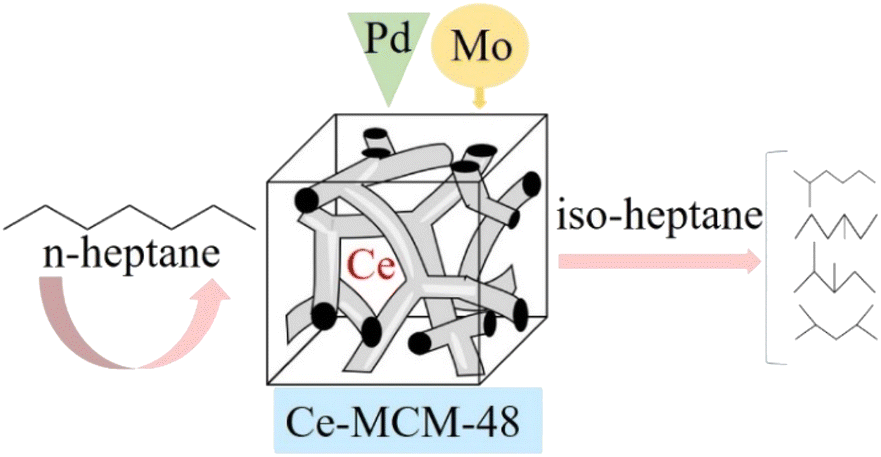
Study on the preparation and n-heptane isomerization performance of MoOx-Pd/Ce-MCM-48 catalyst†
Received
11th December 2023
, Accepted 22nd January 2024
First published on 30th January 2024
Abstract
A Ce-MCM-48 molecular sieve was prepared by the hydrothermal synthesis method. Using Ce-MCM-48 as the support, a series of MoOx Pd/Ce-M catalysts were prepared by the impregnation method by introducing MoOx and Pd. XRD, N2 adsorption desorption, SEM, TEM, NH3-TPD, Py-IR, FT-IR, and ICP-MS were used to characterize the physicochemical properties. The performance of n-heptane isomerization in a micro fixed bed device was evaluated. The results showed that the synthesized Ce-MCM-48 was mesoporous, with a spherical particle morphology, a long-range ordered pore structure, weakly acidic sites on the surface, and an increase of B and L acids. The 2% MoOx-Pd/Ce-M catalyst was used for the probe reaction of n-heptane hydroisomerization; when the reduction temperature was 400 °C, the reduction time was 2 hours, the reaction temperature was 300 °C, the WHSV was 7.6 h−1, the conversion rate was 58.7%, the selectivity was 91.2%, and the maximum yield was 53.5%. The product distribution of multiple C7 isomers increased the selectivity of multi branched isoheptane. The addition of an appropriate amount of MoOx would improve the performance of n-heptane isomerization.
1. Introduction
In recent years, with the implementation of environmental regulations, automobile standards have simultaneously tightened regarding pollutant limits.1 Octane number is the main indicator for measuring the combustion performance of gasoline. The octane number of n-heptane is set to zero, and isomerization of alkanes is a convenient and environmentally friendly way to increase the octane number of gasoline. The conversion of n-heptane to multi substituted isomers through isomerization reaction can significantly increase the octane value of the component,2 which is an important research direction for improving gasoline quality. The problem of a large amount of n-heptane in straight run gasoline and reformed gasoline light naphtha urgently needs to be solved.3,4 The ideal mature technology for isomerization of n-C7+ (n-alkanes with C7 and above) has not yet been achieved.5,6 Therefore, the isomerization of long-chain alkanes, represented by n-heptane, has attracted great attention.
The core of n-heptane isomerization technology is the catalyst. The commonly used n-heptane isomerization catalyst is a metal acid support bifunctional catalyst supported by molecular sieves.7 MCM-48 is a mesoporous molecular sieve with a three-dimensional cubic pore structure, large specific surface area, stable skeleton, and long-range order. Due to the unique pore structure of MCM-48, good dispersion and transport performance, MCM-48 has a strong shape selective effect on some large molecules. However, due to the lack of catalytic active sites in pure silicon MCM-48 molecular sieve, some functionalization modifications of mesoporous molecular sieve are needed, such as adding heteroatoms to improve the activity of the catalyst.8 Ce, as an additive, can easily disperse well in MCM-48 under appropriate Si/Ce molar ratio conditions, causing the increase of specific surface area.9 Ce doping can also increase the surface acidity of the catalyst,10 enhance the dispersion of the precious metal active component Pt, and inhibit adverse reactions such as hydrogenation and cracking.11 Liang et al.12 prepared activated carbon supported Pd–Ce bimetallic catalysts and found that Ce doping can reduce the size of Pd particles and increase the content of Pd2+ species, indicating good catalytic performance of the catalyst. Palladium is a metal with hydrogenation function and exhibits high hydrogenation activity in different catalytic reactions. During the activation process of the catalyst, palladium can promote the reduction of surface atoms and also play a certain role in the formation of acidic properties in alkane isomerization catalysts.13
Introducing other heterogeneous components to improve the microstructure and electronic properties of catalysts is an important means to enhance the performance of the catalysts. In recent years, research results on the functional compounds of molybdenum in petrochemical applications have showed that molybdenum has strong hydrogen activation ability, molybdenum oxide has hydrogenation dehydrogenation activity, and MoO3 contains both Brønsted acid and Lewis acid.14 Teh L. P. et al. prepared a series of Pt and molybdenum trioxide modified HBEA zeolite catalysts for n-heptane isomerization, and found that the Pt/MoO3-HBEA catalyst significantly improved the n-heptane conversion rate and isoheptane rate.2 MoOx and Moδ+ species are responsible for isomerization and dehydrogenation functions, respectively. Sotelo-Boyás et al. modeled and simulated the bifunctional mechanism of n-heptane isomerization on molybdenum carbide catalysts, taking into account acid and metal sites. They believed that the low cracking selectivity of molybdenum carbide makes it a potential catalyst for n-heptane and heavy paraffin isomerization.6 Molybdenum is prone to form a layered structure and also an electronic additive that can synergistically catalyze with composite catalysts prepared by metal doping.15 Harmel et al.16 reported the possibility of MoOx catalyst combined with mesoporous solid acid to replace Pt for n-heptane hydroisomerization, which has been proven that an optimized non precious metal MoOx catalyst, when used in combination with mesoporous solid acids, can be similar to the corresponding platinum catalyst in the n-heptane conversion process. Therefore, molybdenum compounds exhibit excellent catalytic effects in heptane isomerization reactions. Although some progress has been made in previous studies. How to prepare Mo modified mesoporous Ce-MCM-48 molecular sieve loaded with Pd to catalyze n-heptane isomerization is not reported.
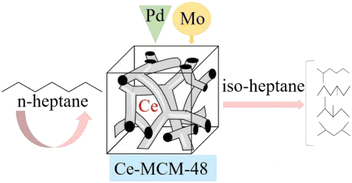
Based on the excellent properties of molybdenum compounds, in order to investigate the effect of Mo introduction on MoOx Pd/Ce-M catalyst for n-heptane isomerization, Ce-MCM-48 molecular sieve was prepared by hydrothermal synthesis method, and a series of Mo modified MoOx Pd/Ce-MCM-48 metal composite catalysts were prepared by distributed impregnation method. The synthesized catalyst was characterized through a series of analyses such as XRD, SEM, EDS, and FTIR, and the influence of MoOx on the catalyst structure, functional groups, and acid distribution was studied. Comparing with unmodified Pd/Ce-MCM-48 catalysts, Pd NPs as catalytic active sites and MoOx as metal active components, the effect of Mo introduction the catalytic performance of n-heptane isomerization reaction was investigated so as to develop a new and excellent catalyst for n-heptane isomerization, and provide a basic theoretical basis for the production of clean energy.
2. Experimental section
2.1 Materials
Tetraethoxysilane (TEOS, Tianjin Damao Chemical Reagent Factory, AR), palladium nitrate (Pd(NO3)2·2H2O, Sinopharm Chemical Reagent Co. Ltd., AR), Ammonium molybdate tetrahydrate ((NH4)6Mo7O24·4H2O, AR), cetyltrimethylammonium bromide (CTAB, Sinopharm Chemical Reagent Co. Ltd., AR), sodium hydroxide (NaOH, Tianjin Kemiou Chemical Reagent Co. Ltd., GR), absolute ethyl alcohol (Liaoning Quanrui Reagent Co. Ltd., AR), n-heptane (Shenyang East China Reagent Factory, AR), deionized water (Heilongjiang Bayi Agricultural University), and hydrogen (Daqing Xuelong Gas Station, volume fraction 99.99%).
2.2 Preparation of Ce-MCM-48
Ce-MCM-48 was prepared by hydrothermal synthesis method. The method was as follows. Under the condition of magnetic stirring at 35 °C, 0.8960 g NaOH was dissolved in 50 mL deionized water, Ce(NO3)·6H2O was added and stirred for 30 minutes. Then 5.30 g CTAB was added to the above solution gradually and stirred for 90 minutes. Then TEOS was added to the above solution slowly. The molar ratio of raw materials was n(TEOS)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n(CTAB):n(H2O):n(NaOH):n(Ce) = 1.0:0.65:62:0.5:0.02. The gel was transferred into a stainless steel autoclave with polytetrafluoroethylene lining, and crystallized at 110 °C for 76 h. The product was filtered and separated through a Buchner funnel, washed with deionized water until the pH value was neutral, and dried at 105 °C for 12 hours. Ce-MCM-48 was prepared by calcining in air at 550 °C for 6 hours to remove the template agent.
:n(CTAB):n(H2O):n(NaOH):n(Ce) = 1.0:0.65:62:0.5:0.02. The gel was transferred into a stainless steel autoclave with polytetrafluoroethylene lining, and crystallized at 110 °C for 76 h. The product was filtered and separated through a Buchner funnel, washed with deionized water until the pH value was neutral, and dried at 105 °C for 12 hours. Ce-MCM-48 was prepared by calcining in air at 550 °C for 6 hours to remove the template agent.
2.3 Preparation of catalyst
The catalyst was prepared using a two-step impregnation method. (NH4)6Mo7O24·4H2O was dissolved in deionized water, then Ce-MCM-48 was immersed in the (NH4)6Mo7O24·4H2O solution. The above solution was sonicated at 50 °C for 1 hour, standed still for 12 hours, then dried at a Teflon coated electric heating plate at 90 °C for 6 hours, dried at 105 °C for 5 hours in an electric hot air drying oven, and calcined at 550 °C for 6 hours. Pd(NO3)2·2H2O was dissolved in ethanol, and the above calcined sample powder was immersed into the Pd(NO3)2·2H2O ethanol solution. The mass percentage of Pd loaded was 0.4%. The above solution was sonicated at 50 °C for 1 hour, standed still for 12 hours, then dried at a Teflon coated electric heating plate at 90 °C for 8 hours, dried at 105 °C for 5 hours in an electric hot air drying oven, and calcined at 400 °C for 4 hours. A series of catalysts were obtained with different MoOx loadings, labeled as yMoOx-Pd/Ce-M (y = 1%, 2%, and 3%). The contrast sample without loaded MoOx was labeled as Pd/Ce-MCM-48.
2.4 Characterization of catalysts
X-ray diffraction (XRD) was performed using a copper radiation source X-ray diffractometer (SmartLab SE type, Japan Institute of Science) for crystal structure analysis. Structural parameters of samples was determined by Micromeritics TriStar II specific surface area and pore size analyzer from the United States. Actual metal content in catalysts was characterized by Agilent7800 inductively coupled plasma mass spectrometry (ICP-MS) from the United States. The SEM micro surface morphology was recorded by Hitachi High Technologies SU5000 scanning electron microscopy from Japan for analysis. Energy dispersive spectroscopy (EDS) uses Oxford Ultimate MAX40 for analysis. The TEM images were recorded by a JEM-2100Plus transmission electron microscope produced in Japan, with an acceleration voltage of 100–120 kV. Acid strength distribution of the sample was tested on the Micromeritics Autochem II 2920 temperature programmed desorption instrument manufactured in the United States. Dry samples are pretreated at 450 °C for 2 hours. Fourier-transform infrared spectroscopy spectra of pyridine (Py-IR) were recorded on a Germany Brook Tensor 27 infrared spectrometer. The sample was subjected to vacuum treatment at 400 °C for 4 hours before analysis. The chemical state of surface elements of the sample was characterized using Thermo Scientific ESCALAB 250Xi. The chemical state of surface elements in the sample was characterized using X-ray photoelectron spectroscopy (XPS, Thermo Scientific ESCALAB 250Xi). The FT-IR spectrum was analyzed using a TENSOR II infrared spectrometer manufactured by Brooke Technology (Hong Kong) Co., Ltd. in Germany to determine the chemical structure of the sample. KBr plates were pressed and the wavenumber range was 400–4000 cm−1. The isomeric products of heptane were analyzed by a GC-7980A gas chromatograph in Zhengzhou, China.
2.5 Catalytic performance evaluation
The hydrogenation isomerization reaction of n-heptane was performed on a self-assembled fixed bed microreactor. The reaction temperature was 280–380 °C, pressure was 10 bar, H2/n-C7H16 = 12/1 mol mol−13 and WHSV was 7.6 h−1. The catalyst loading amount was 0.40 g. The inner diameter of the reactor tube was 6 mm. Before catalytic performance evaluation, the catalyst was activated in an H2 flow at 400 °C for 4 hours and cooled to the reaction temperature range. Start sampling after the reaction conditions were stable, the composition was analyzed by a gas chromatograph equipped with a FID detector. The catalytic performance was evaluated by the conversion rate of n-heptane (x), product selectivity (s), yield (y), and product distribution. The conversion rate, isomer selectivity and yield, and cracking product selectivity (Scm) were calculated from eqn (1)–(4). FCwt n, C7,out and FCwt n, C7,in was the amount of n-heptane flowing out and into the reactor. FCwt i, C7,out was the amount of isoheptane generated, FCwt Cm,out was the amount of cracking product Cm (m = 1–6).17| |
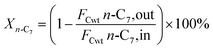 | (1) |
| |
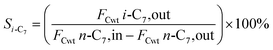 | (2) |
| |
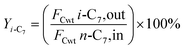 | (3) |
| |
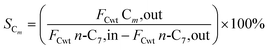 | (4) |
3. Results and discussion
3.1 The effect of MoOx on the structure of catalysts
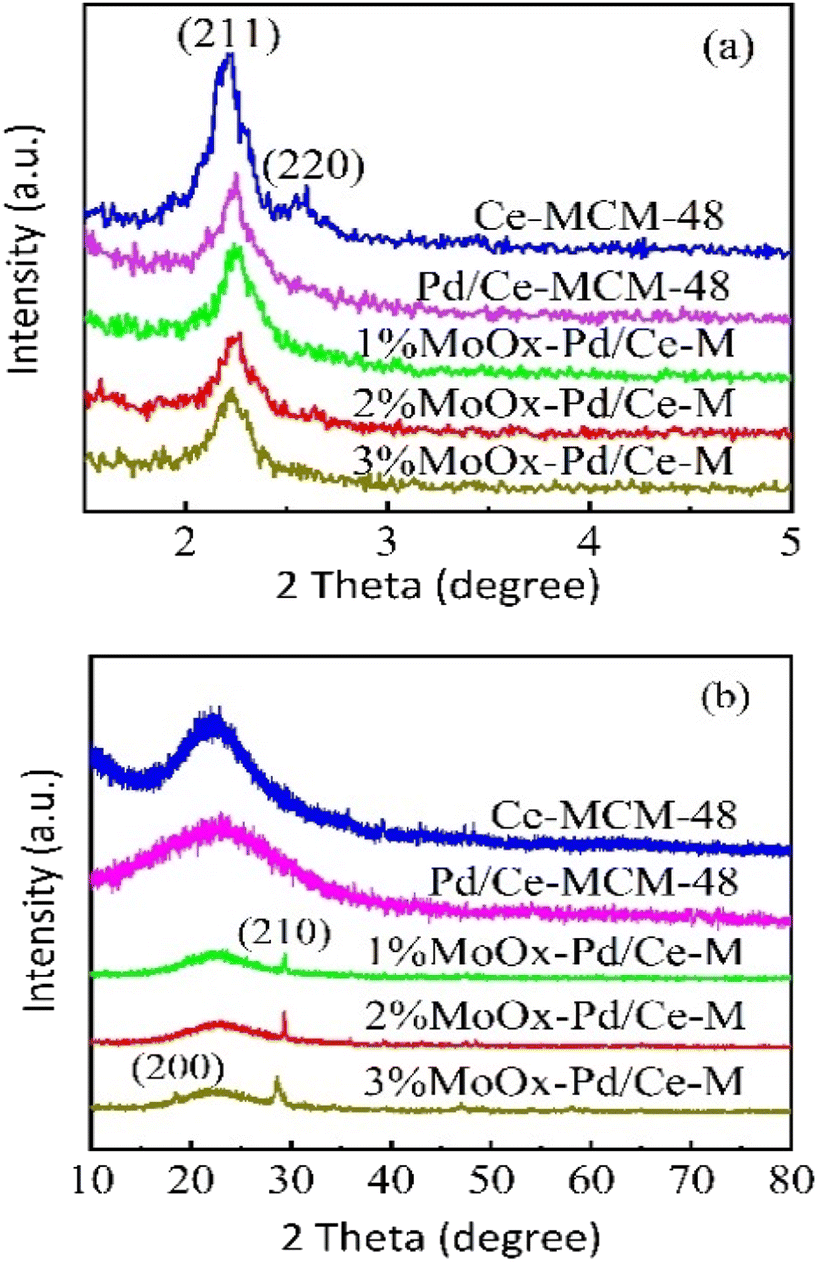
3.1.1 XRD characterization analysis. XRD pattern of Ce-MCM-48 and yMoOx Pd/Ce-M was shown in Fig. 1. As shown in Fig. 1(a), strong diffraction characteristics of MCM-48 mesoporous material crystal system (211) and (220) were observed 2θ = 2–3° in Ce-MCM-48 (ref. 18) and an ordered Ia 3d three-dimensional spatial group were formed.19 Characteristic peaks at (420) and (332) were not shown in Ce-MCM-48, possibly due to the substitution of some Si atoms by Ce after doping with rare earth elements, which disrupted some silicon oxygen tetrahedral symmetry.20 However, the Ce-MCM-48 also exhibited long-range ordering and no impurity crystal phase formation, indicating the good purity and crystallinity.20 Catalyst samples prepared after loading Mo and Pd, the peak positions of the characteristic diffraction peaks (211) and (220) had hardly changed, but the peak intensity had decreased, indicating that the structure of Ce-MCM-48 had been retained and all samples were still mesoporous materials. From the wide-angle XRD spectrum in Fig. 1(b), an amorphous silica broad peak appeared at 2θ = 20–30° in all samples.21 Compared with Ce-MCM-48, the characteristic diffraction peak intensity decreased after loading Mo. It was speculated that the molybdenum oxide species of the catalyst had deposited on the surface or outer pores of Ce-MCM-48, causing some changes in the structure of the support.22 And a weak MoO3 crystal phase diffraction peak appeared.16 When the loading amount of MoOx was 1% and 2%, the characteristic diffraction peaks corresponded to (210) crystal planes. When the loading amount increased to 3%, the characteristic diffraction peaks corresponded to (200) and (210) crystal planes, which was consistent with the hexagonal crystal structure (JCPDS 21-0509), indicating the presence of polymerized hexagonal MoO3 species on the catalyst surface, which corresponding to the typical XRD diffraction peaks of (NH4)6Mo7O24·4H2O and MoO3 reported in literature.7 In addition, no peak of palladium oxide was observed, indicating that Pd oxide particles were too small or highly dispersed in the amorphous walls of MCM-48,23 which could not be detected by XRD.
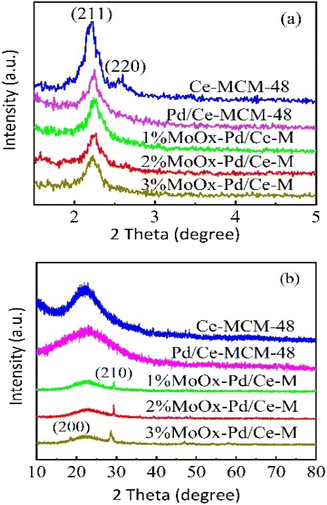 |
| | Fig. 1 XRD patterns of Ce-MCM-48 and yMoOx-Pd/Ce-M. (a) Small-angle XRD and (b) wide-angle XRD. | |
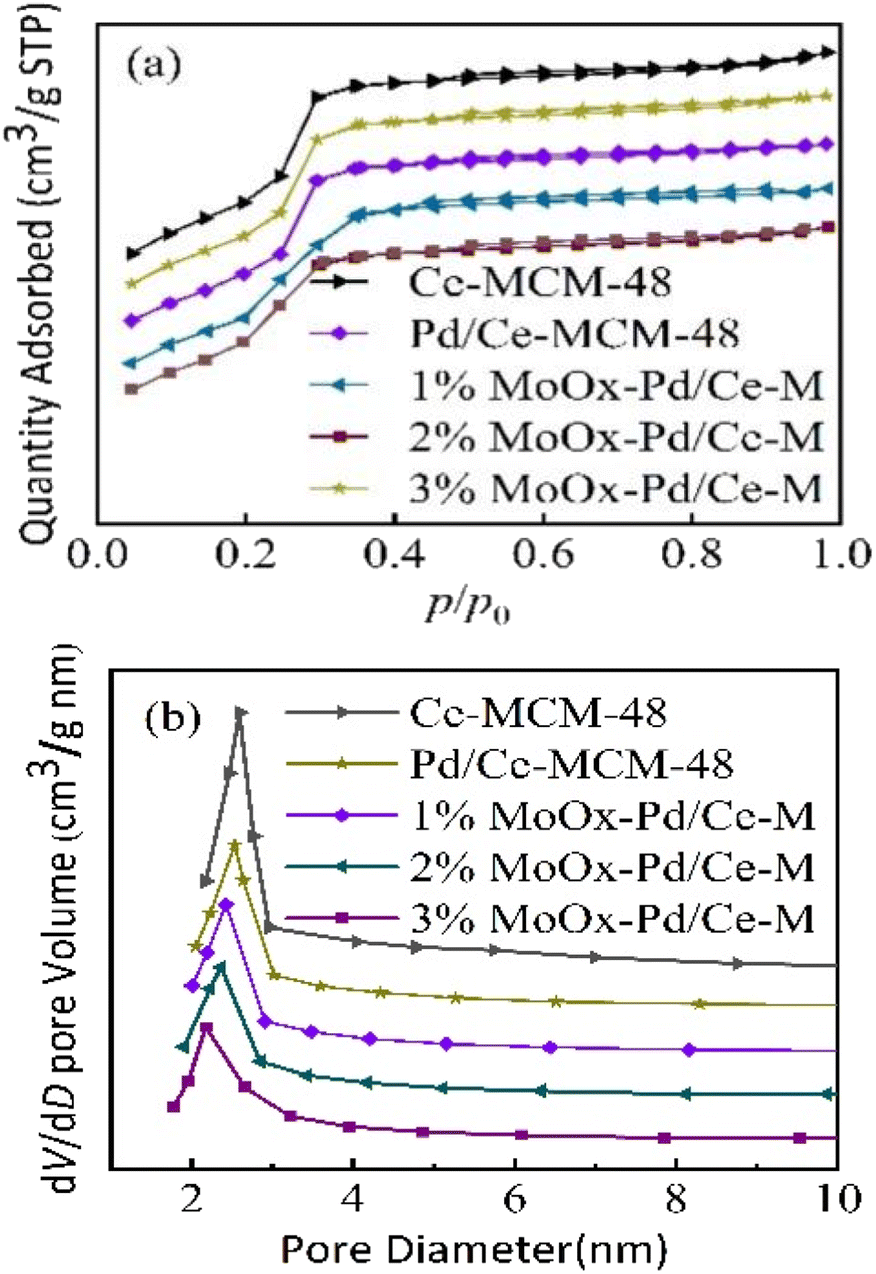
3.1.2 N2 adsorption desorption characterization and acidic characteristics. Fig. 2 showed the N2 adsorption desorption isotherms and pore size distribution curves of Ce-MCM-48 and yMoOx Pd/Ce-M. As shown in Fig. 2(a), to all samples, there was a significant leap between 0.2 and 0.4, and the curve conformed to the Langmuir IV adsorption isotherm, which was a characteristic of the cubic mesoporous material MCM-48.18 As shown in Table 1, the physical structural parameters of the samples, and the Mo content in the ICP-MS characterization results basically conformed to the set ratio, indicating the formation of Mo containing heterostructures in the catalyst complex,24 which was consistent with the XRD results in Fig. 1(b). As shown in Fig. 2(b) and Table 1, the specific surface area and pore volume of Ce-MCM-48-0.02 decreased, and there was a certain amount of Ce present in the pores and skeleton. The increase in pore size could be explained by the radius Ce4+ > Si4+, and the bond length Ce–O > Si–O,20 which caused the skeleton to stretch and the pore wall to thicken. As shown in Fig. 2(b), the pore size distribution of the prepared samples was 2.17–2.59 nm. After impregnating Mo and Pd on the Ce-MCM-48 support, the specific surface area and pore volume of the catalyst yMoOx Pd/Ce-M decreased, indicating that the metal had been loaded onto the support. With the increase of Mo loading, the specific surface area and pore volume further decreased, which was due to the decomposition of (NH4)6Mo7O24·4H2O to generate α-MoO3. MoOx atomic clusters had Mo–O bonds (0.1690–0.2350 nm) smaller than the mesoporous pore size, and some MoOx atomic clusters were located in the phase cage of the molecular sieve, causing the decrease of the molecular sieve cell, cell parameters, specific surface area, and pore volume.25 And it also indicated that some metals were distributed on the inner surface of the support. The average pore size decreased gradually may be the insertion of some metals into the MCM-48 lattice, which blocked part of the pores.24 This was different from the conclusion drawn by Yang et al. that the zinc modified Pt/SAPO-11 catalyst metal is located on the outer surface of the support, and the pore volume of the sample is almost constant.26
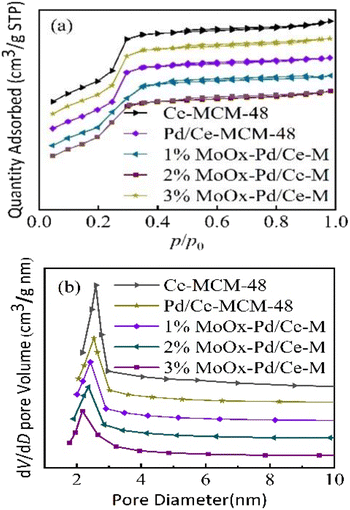 |
| | Fig. 2 (a) N2 adsorption–desorption isotherms and (b) pore size distributions of Ce-MCM-48 and yMoOx-Pd/Ce-M. | |
Table 1 Physical properties of the support
| Sample |
Mo loading percentage (wt%) |
Surface area (m2 g−1) |
Micropore volume (cm3 g−1) |
Mesopore volume (cm3 g−1) |
Pore volume (cm3 g−1) |
Average pore diameter (nm) |
| Ce-MCM-48 |
— |
973 |
0.02 |
0.95 |
0.97 |
2.59 |
| Pd/Ce-MCM-48 |
— |
954 |
0.03 |
0.92 |
0.95 |
2.53 |
| 1%MoOx-Pd/Ce-M |
98.4 |
875 |
0.04 |
0.87 |
0.91 |
2.42 |
| 2%MoOx-Pd/Ce-M |
99.2 |
860 |
0.06 |
0.81 |
0.87 |
2.36 |
| 3%MoOx-Pd/Ce-M |
99.1 |
822 |
0.09 |
0.73 |
0.82 |
2.17 |
3.1.3 TEM and SEM characterization. As shown in Fig. 3, the TEM image showed that Ce-MCM-48 had a regularly arranged long-range cubic pore structure.8 The change of Pd/Ce-MCM-48 and 2% MoOx-Pd/Ce-M was not significant, indicating that the skeleton structure was not damaged. Oxides were precipitated and enriched on the surface of Pd/Ce-MCM-48 catalyst.10 The aggregation degree of 2% MoOx Pd/Ce-M catalyst particles became loose. After doped with Mo, the surface metal obtained good dispersion, which was consistent with the SEM characterization results. The difference in catalytic performance of bifunctional catalysts mainly came from the properties of metals, including the dispersion and distribution of metals, which enhanced the balance and intimated relationship between the metal and acid sites of the catalyst.27
 |
| | Fig. 3 TEM images of (a) Ce-MCM-48; (b) Pd/Ce-MCM-48; (c) 2% MoOx-Pd/Ce-M. | |
As shown in Fig. 4, the SEM and microstructure analysis of the catalyst sample, and the results showed that Pd/Ce-M exhibited a spherical particle morphology, with tight spherical aggregation and a smooth surface. After Mo doping, the change of the particle size of the 2% MoOx Pd/Ce-M catalyst was not significant, with a small amount of hemispherical particles and uniformly dispersed coking on the surface, which may be nanosheets15 or oxides5 produced by Mo doping. The EDS spectral confirmed that Pd, Ce, and Mo metals were successfully uniformly distributed on the outer surface, which was consistent with the characterization results of N2 adsorption and desorption.
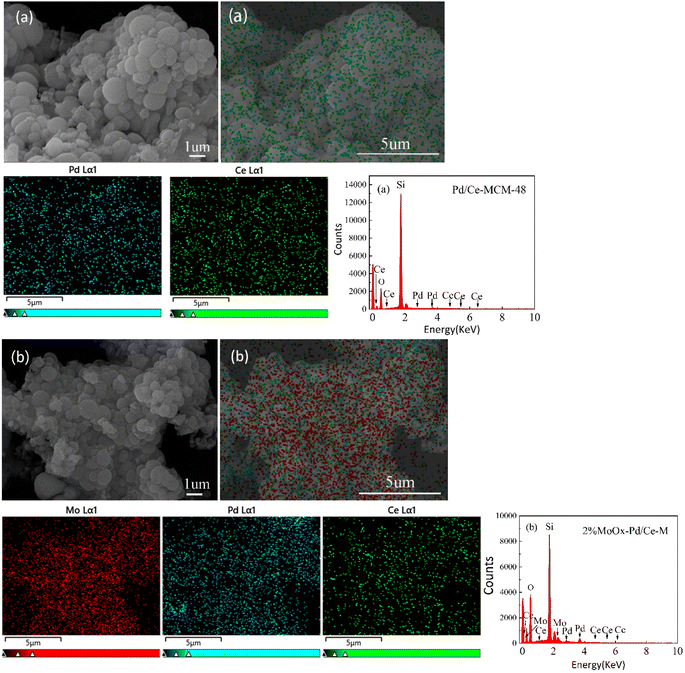 |
| | Fig. 4 SEM images, EDX spectrum, and SEM mapping of (a) Pd/Ce-MCM-48 and (b) 2%MoOx-Pd/Ce-M. | |
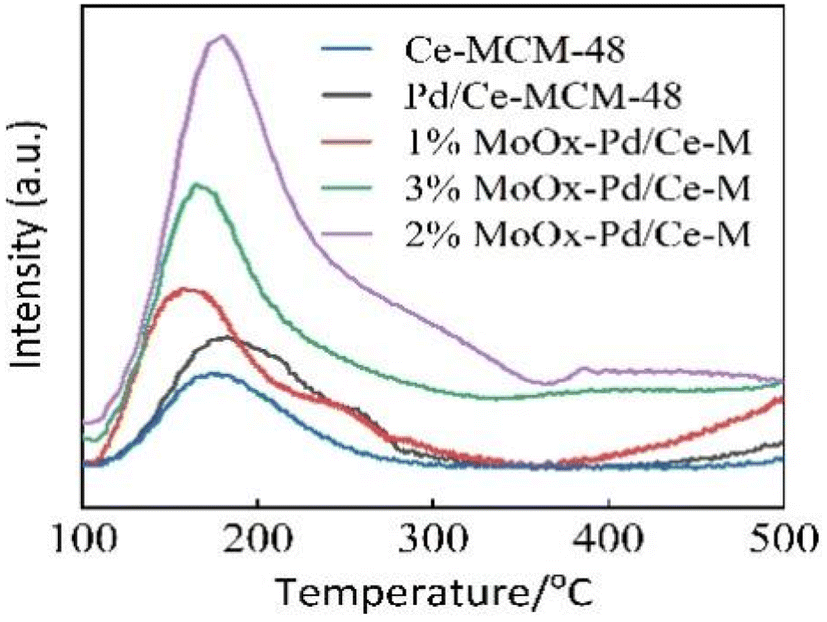
3.1.4 Acidic properties. Fig. 5 showed the NH3-TPD spectra, and the acid distribution of all samples was similar in the temperature range of 100–500 °C, indicating that metals had a certain influence on the acid distribution of the support.26 Ce-MCM-48 exhibited a desorption peak, indicating the presence of acid sites on the surface of Ce-MCM-48.5 Ce-MCM-48 had certain acidic sites due to the doping of Ce on the skeleton,10 which was not sufficient for heptane isomerization. After doping with Mo, the desorption peaks of 1% MoOx-Pd/Ce-M and 3% MoOx-Pd/Ce-M slightly shifted towards lower temperatures within the range of 100–250 °C, with an increase in weak acids. This may be due to the dope of Mo species on the surface of Ce-MCM-48.2 Due to the strong electron donating ability of MoOx atomic clusters, the composite catalyst 2% MoOx-Pd/Ce-M further generated more intermediate acid sites at the desorption peak at 300 °C.28 Isomerization reactions mainly occurred on intermediate acid sites, which would be beneficial for the reaction.29 The decrease in acidity of 3% MoOx-Pd/Ce-M may be due to MoOx being a positively charged atomic cluster, which could mask some of the acidic sites of the catalyst and reduce the number of acid centers in the molecular sieve.13 Fig. 1 in the supporting information† showed the Py-IR spectra of Pd/Ce-MCM-48 and 2% MoOx-Pd/Ce-M catalysts. The peak observed at 1540 cm−1 in the Pd/Ce-MCM-48 catalyst sample was attributed to the presence of Brønsted acid centers. The peak at 1450 cm−1 corresponded to the adsorption of pyridine on Lewis acid sites, while the peak at 1490 cm−1 was a characteristic peak generated by the adsorption of pyridine on B and L acid sites.30,31 Table 2 shows the type and acid content of the acid center on the catalysts. The dope of Mo can increase the acidity of B acid and L acid in the prepared 2% MoOx-Pd/Ce-M catalyst. Mo![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond was formed due to the interaction between MoO3 and Ce-MCM-48. MoOx/ZrO2 mixed oxides to promote Pt HMS catalyst for n-heptane isomerization was studied by N. Parsafard et al. The addition of 12 wt% molybdenum oxide to ZrO2/HMS also obtained the result of an increase in acidity.32 In theory, the number of Brønsted acid sites would affect the performance of isomerization, generating more carbocations in alkane isomerization reactions and increasing the conversion rate of the reaction.2
O bond was formed due to the interaction between MoO3 and Ce-MCM-48. MoOx/ZrO2 mixed oxides to promote Pt HMS catalyst for n-heptane isomerization was studied by N. Parsafard et al. The addition of 12 wt% molybdenum oxide to ZrO2/HMS also obtained the result of an increase in acidity.32 In theory, the number of Brønsted acid sites would affect the performance of isomerization, generating more carbocations in alkane isomerization reactions and increasing the conversion rate of the reaction.2
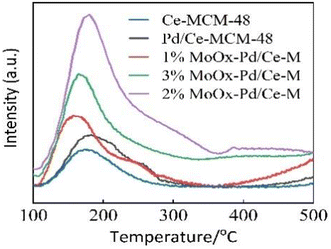 |
| | Fig. 5 NH3-TPD profiles of samples. | |
Table 2 Acidity of Pd/Ce-MCM-48 and 2%MoOx-Pd/Ce-M
| Sample |
Brønsted acid (mmol g−1) |
Lewis acid (mmol g−1) |
Amount of acid site (mmol g−1) |
Amount of acid sitea (mmol g−1) |
| The acid content is calculated from NH3-TPD. |
| Pd/Ce-MCM-48 |
1.39 |
2.82 |
4.21 |
2.76 |
| 2%MoOx-Pd/Ce-M |
1.76 |
4.11 |
5.87 |
3.15 |
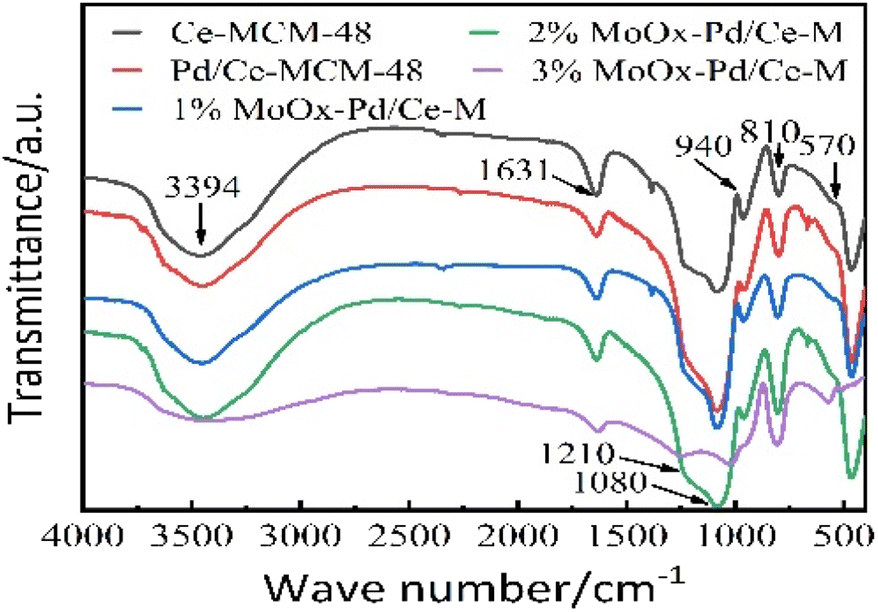
3.1.5 FT-IR spectroscopic analysis. Fig. 6 showed the FT-IR spectra of Ce-MCM-48 and yMoOx Pd/Ce-M. All catalysts exhibited spectral vibration peaks at 3394, 1631, 1210, 1080, 940, and 810 cm−1. According to references,2,5 the hydroxyl vibration peaks of water molecules were located at 3394 and 1631 cm−1. The external and internal asymmetric stretching of the Si–O–Si framework was attributed to 1210 and 1080 cm−1. The vibration peaks that appear at 940 cm−1 and 810 cm−1 were attributed to the symmetric stretching of external silanol groups (Si–OH) and Si–O–Si. 940 cm−1 for Si–O-R+ groups were increased by the silicon tensile vibration of Ce-MCM-48, which was commonly considered as evidence of the addition of metal cations in the framework of mesoporous silicates.5 After the introduction of MoOx, the infrared band of silanol groups appeared and the asymmetric tensile strength of Si–O–Si increased, indicating that MoOx groups participated in the molecular sieve framework through interactions with silanol groups.2 The changes in absorption peaks at 628, 872, and 994 cm−1 were attributed to Mo–O–Mo stretching vibration, Mo–O asymmetric stretching vibration, and MoO double bond oxygen stretching vibration.25
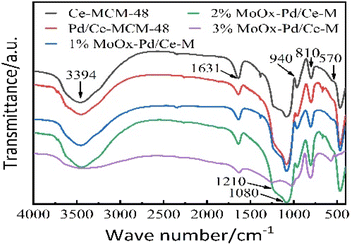 |
| | Fig. 6 FT-IR spectra of Ce-MCM-48 and yMoOx-Pd/Ce-M. | |
3.2 Catalytic performance
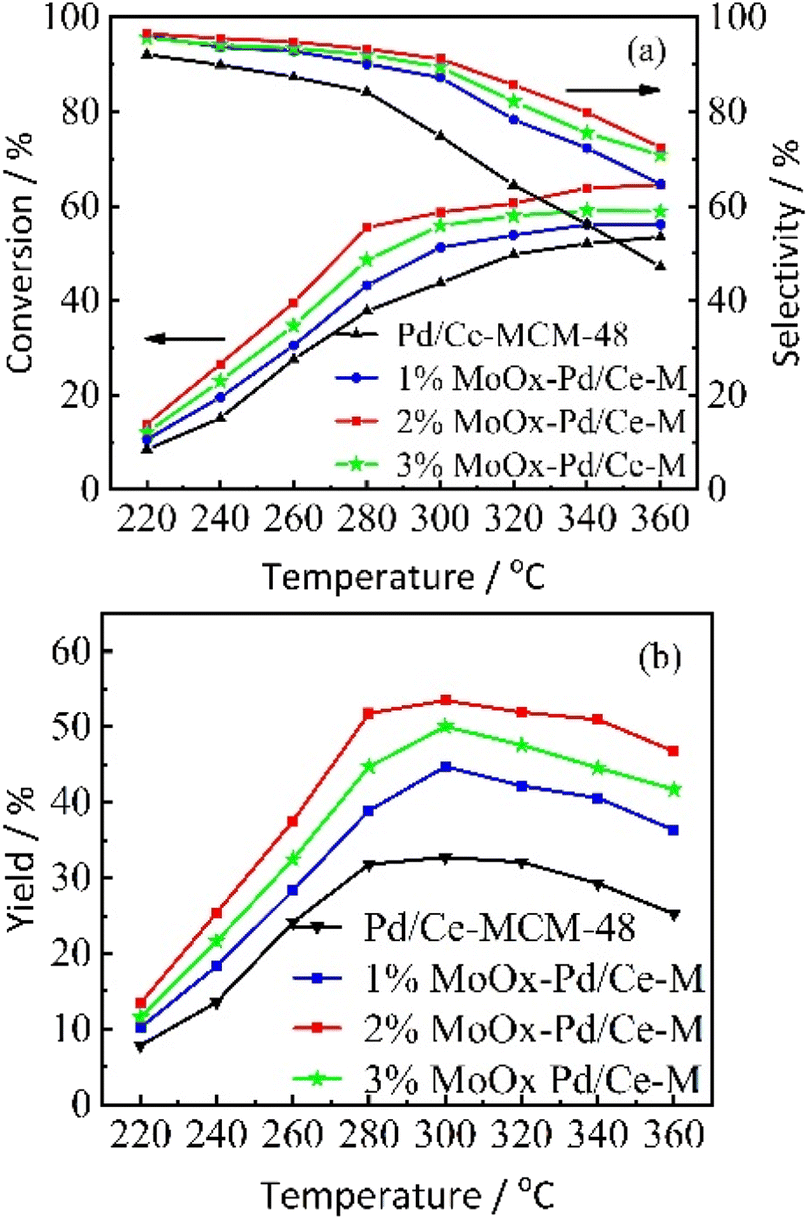
3.2.1 Effect of reaction temperature and different Mo contents on the isomerization performance of n-heptane. n-Heptane hydroisomerization was used as a model reaction, Pd/Ce-MCM-48 and yMoOx-Pd/Ce-M catalysts with different Mo contents were tested. Fig. 7 showed the variation of the conversion rate of n-heptane, selectivity of isoheptane (Fig. 7(a)), and yield (Fig. 7(b)) with the reaction temperature. With the increase of reaction temperature, the conversion rate of n-heptane increased, with the highest conversion rate of 2% MoOx-Pd/Ce-M. The selectivity for isoheptane decreased with increasing reaction temperature, and high temperatures irreversibly damaged the structure of some catalysts.33 The rate of hydrocracking reaction increased at higher temperatures.4 The selectivity order of catalysts was 2%MoOx-Pd/Ce-M > 1%MoOx-Pd/Ce-M > 3%MoOx-Pd/Ce-M > Pd/Ce-MCM-48. When the reaction temperature was 300 °C, the selectivity of 2% MoOx-Pd/Ce-M was 91.2%, and the conversion rate was 58.7%. The maximum yield of isoheptane was 53.5%. Due to the limitations of spatial structure and geometric configuration, some molecular sieves generally have difficulty diffusing multi branched isomeric products out of the pores of the catalyst, resulting in the occurrence of cracking side reactions. Catalysts prepared using mesoporous molecular sieves as supports could effectively avoid this problem. As shown in Fig. 7(a), the conversion rate and selectivity of yMoOx-Pd/Ce-M catalyst were higher than Pd/Ce-MCM-48. The Mo content increased from 1% to 2%, the isomer selectivity and catalytic activity increased, and the yield of the 2% MoOx-Pd/MCM-48 catalyst was the highest. The factors that determined catalytic performance were complex, and metal functions or geometric structural patterns should also be considered.31
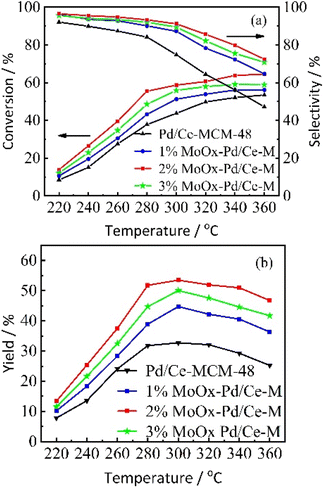 |
| | Fig. 7 The variation of catalytic performance of Pd/Ce-MCM-48 and yMoOx-Pd/Ce-M catalysts with reaction temperature. (a) The conversion rate of n-heptane, selectivity of isoheptane, and (b) yield. | |
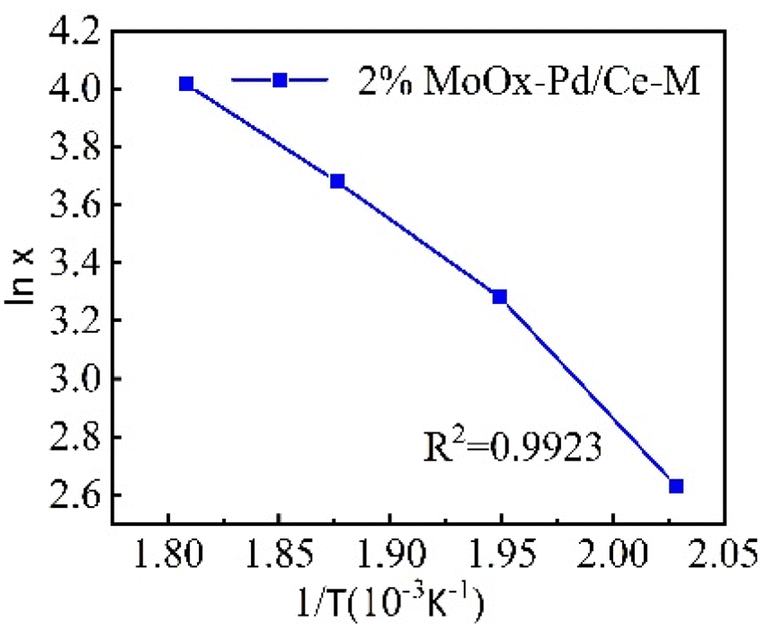
From the above results, within the range of reaction temperature of 220–360 °C, there was a linear relationship between the catalytic activity of 2% MoOx Pd/MCM-48 catalyst for n-heptane isomerization reaction and the reaction temperature. We plotted with conversion rate lnx as the vertical axis and T−1 as the horizontal axis (Figure.8). According to the Arrhenius equation, based on the slope, the apparent activation energy Ea of the 2% MoOx Pd/Ce-M catalyst was 52 kJ mol−1. A lower activation energy indicated a faster adsorption rate of the catalyst, Langmuir–Hinshelwood seemed to be a suitable model for this reaction.34
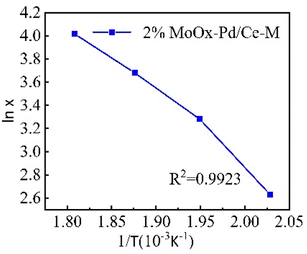 |
| | Fig. 8 Arrhenius plot. | |
MoO3 usually formed a structure similar to ReO3, which was connected by MoO6 octahedral edges and angles to form a one-dimensional chain like structure, a two-dimensional layered structure, or a three-dimensional network structure. This was conducive to the high dispersion of Mo on the surface of, support and the presence of MoO and Mo–O–Mo functional groups could both improve catalyst activity,35,36 which was consistent with the FT-IR results. MoOx loaded catalyst could provide both metal and acidic sites, Ce-MCM-48 had a large mesoporous structure, which increased the diffusion rate and created a good balance and intimate relationship between the metal and acid sites of the catalyst.27 When the Mo content increased from 2% to 3%, the isomerization efficiency of the catalyst did not continue to increase, instead showed a downward trend. It was speculated that excessive Mo caused certain agglomeration sintering, which had a certain destructive effect on the distribution of active components on the catalyst surface, masking some active substances and leading to a decrease in catalyst activity,27 which was consistent with the acidic properties of Section 3.1.4. The comparison of the catalytic performance for n-heptane hydroisomerization with those reported in literature was shown in Table 3.
Table 3 The effect of metal modified catalyst on the hydroisomerization properties of n-heptane
| Catalyst |
Reaction temperature/K |
Reduction temperature/K |
Conversion/% |
Selectivity/% |
Reference |
| 2%MoOx-Pd/Ce-M |
573 |
673 |
58.7 |
91.2 |
This paper |
| Pt/MCM48-HZSM5 |
573 |
673 |
87.7 |
61.7 |
41 |
| 0.5%Pt/5%MoO3-HBEA |
523 |
673 |
81.6 |
43.5 |
2 |
| 1% Pt/SBA-15 |
673 |
723 |
85 |
— |
4 |
| Pt/MIL-53 (Al) |
553 |
673 |
68.3 |
65.4 |
29 |
| Pt/SBA-16-HZSM-5 |
473 |
673 |
71.8 |
70.4 |
42 |
| PZH-0.3 |
533 |
723 |
81.1 |
94.2 |
43 |
| Pt/Al-MCM-48 |
473 |
673 |
78.8 |
81.9 |
3 |
| Pt/FSMO |
523 |
673 |
70.9 |
63.6 |
7 |
| 0.05%Pt1@CeOx/SAPO-11 |
653 |
— |
45.4 |
74.2 |
33 |
| 0.5%Pd/WO3–ZrO2 |
523 |
— |
84.9 |
79.5 |
13 |
| MOO3/FST |
573 |
673 |
84.0 |
60.9 |
14 |
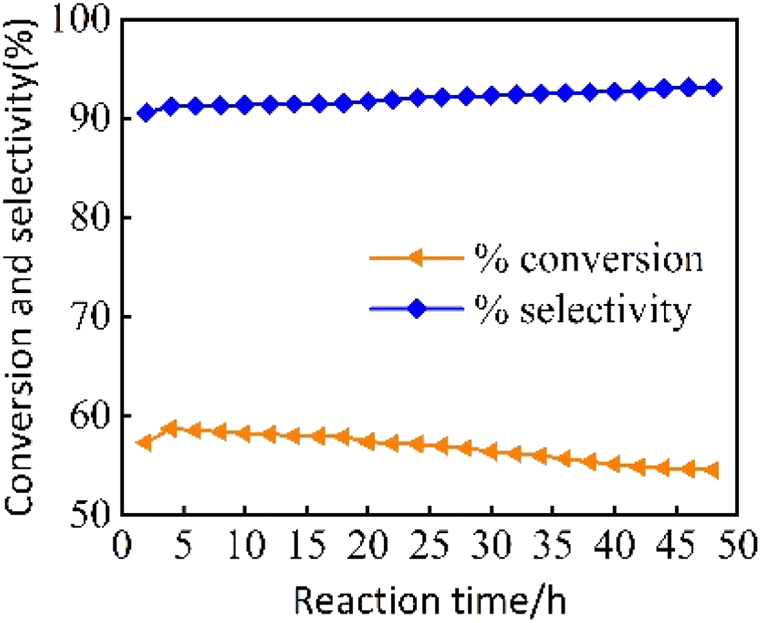
3.2.2 The effect of reaction time on the catalytic performance of 2% MoOx Pd/Ce-M. As shown in Fig. 9, the stability of the 2% MoOx Pd/Ce-M n-heptane isomerization catalyst was investigated at a reaction temperature of 300 °C, a reduction temperature of 400 °C, a reduction time of 2 hours, a hydrogen flow rate of 30 mL min−1, and WHSV = 7.6 h−1 for 48 hours. When the reaction time was 4 hours, the conversion rate of n-heptane was 58.7%, and the isomerization selectivity was 91.2%. When the reaction time was 48 hours, with extension of reaction time, the conversion rate of n-heptane decreased to 54.5%, the selectivity of isoheptane increased by 93.1%. The possible reason was that a small amount of carbon deposits had masked some of the active substances.
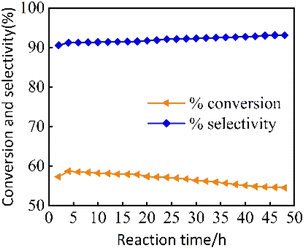 |
| | Fig. 9 The variation of catalytic performance of Pd/Ce-MCM-48 and yMoOx-Pd/Ce-M catalysts with reaction temperature. | |
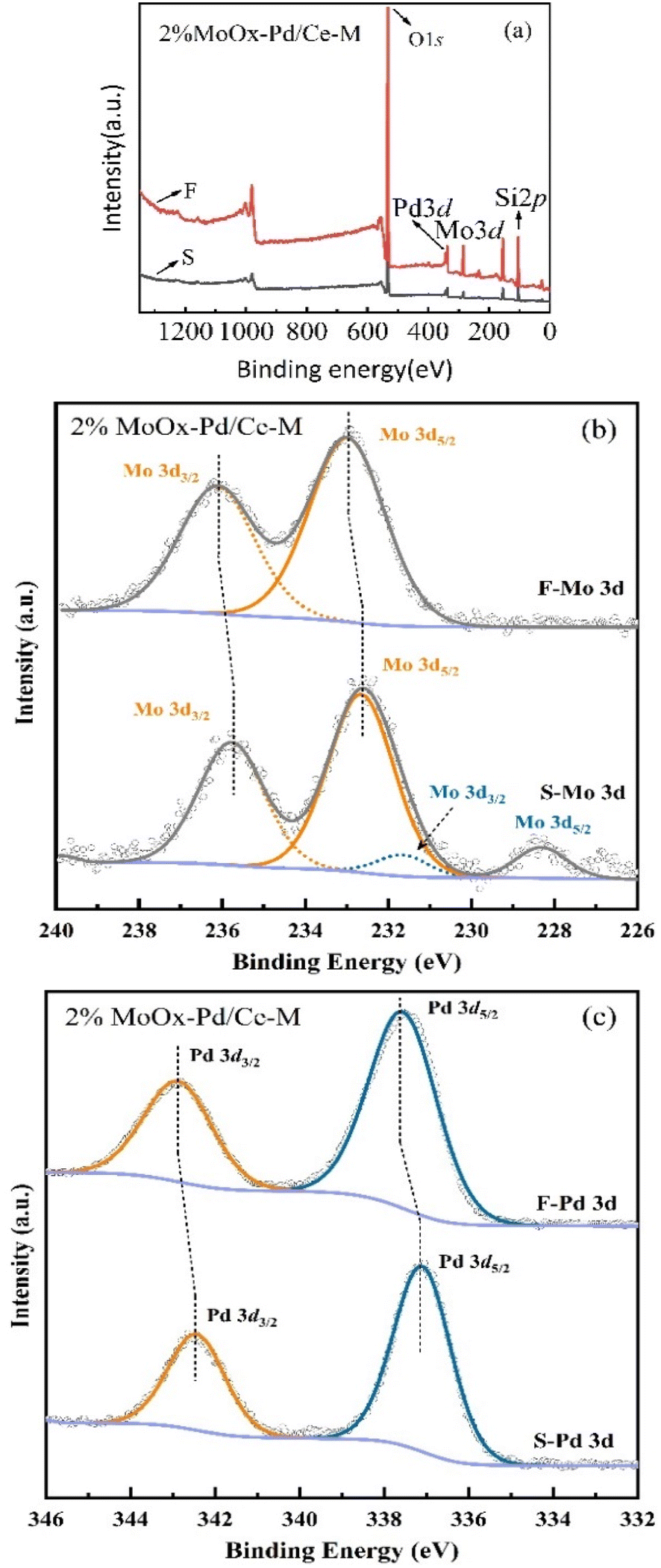
3.2.3 XPS analysis. In order to study the chemical states of Mo and Pd in the reaction, X-ray photoelectron spectroscopy (XPS) was used to analyze the elemental composition and valence states of 2% MoOx Pd/Ce-M catalyst samples before use (F represents) and after use for 48 h (S represents). As shown in Fig. 10(a), the full spectrum of two samples confirmed the coexistence of Mo and Pd elements in the material. As shown in Fig. 10(b), the fitted Mo 3d spectra of two samples showed two strongly symmetric peaks of approximately 232.65 eV (3d5/2) and 235.85 eV (3d3/2), with the binding energy allocated to Mo6+.15 There were some Mo(0) present in the waste catalyst, and the symmetric peak shifted towards a lower angle. The sample was continuously reduced by hydrogen, resulting in complex Mo valence states in the Mo (3d) energy region (Fig. 1(d)). And a bifunctional MoO2x(OH)y phase may have been generated on the catalyst surface for catalytic reactions. Once this active phase was formed, which exhibited a certain degree of stability and exhibited a certain ability to resist carbon poisoning.37 As shown in Fig. 10(c), the fitted Pd 3d spectra of two samples showed two peaks, Pd 3d3/2 and Pd 3d5/2, for both fresh and waste catalysts,38 whose binding energies were 337.4 eV and 342.8 eV, respectively, and can be allocated as Pd2+ in PdOx particles or clusters located on the catalyst support. The chemical state of palladium on the catalyst surface was clearly related to the shape.39 After being reduced at 400 °C, both peaks of the used catalyst shifted towards lower binding levels. After the introduction of Mo, a unique metal support interaction was generated, resulting in the unique electronic properties of Pd atoms. X-ray electron spectroscopy confirmed that this unique electronic state was the reason for the high catalytic activity.40
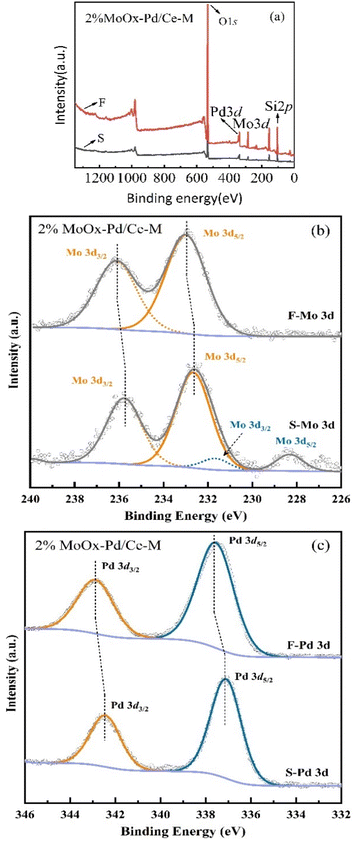 |
| | Fig. 10 XPS spectra of fresh and spent 2%MoOx-Pd/Ce-M catalyst: (a) XPS full spectra, (b) Mo 3d, and (c) Pd 3d of 2%MoOx-Pd/Ce-M. | |
3.3 Product distribution of n-heptane isomerization reaction of 2%MoOx-Pd/Ce-M
Table 4 showed the product distribution in the catalytic reaction of Pd/Ce-MCM-48 and 2%MoOx-Pd/Ce-M for n-heptane isomerization. When the reaction temperature was 300 °C, and the conversion rates were 43.8% and 58.7%, respectively. The product included single branched heptane isomers and multi branched heptane isomers, with single branched isomers including 2-methylhexane (2-MH) and 3-methylhexane (3-MH). The multi branched isomers include 2,2-dimethylpentane (2,2-dMP), 2,3-dimethylpentane (2,3-dMP), 2,4-dimethylpentane (2,4-DMP), 3,3-dimethylpentane (3,3-dMP), and trimethylbutane (2,2,3-TMB). For 2% MoOx-Pd/Ce-M, the content of 2,3-dMP in the multi branched isomers of n-heptane was 23.5%. 2,3-dMP was a compound with a high research octane number (RON = 92), which directly helped to improve the octane number of the product mixture.31 The ratio of multi branched isomers to single branched isomers (represented by R) in the product composition remained ST, SMH, SDMP, STMB and Sc correspond to the selectivities to isoheptane, MH, DMP, TMB and the cracking products, respectively.
Table 4 Product distribution of n-heptane isomerization reaction of 2%MoOx-Pd/Ce-M
| Catalyst |
Pd/Ce-MCM-48 |
2%MoOx-Pd/Ce-M |
| ST (%) |
74.8 |
91.2 |
| SMH (%) |
40.7 |
46.9 |
| 2-MH |
33.7 |
23.3 |
| 3-MH |
27.9 |
23.6 |
| SDMP (%) |
34.1 |
44.3 |
| 2,2-DMP |
7.8 |
9.4 |
| 2,3-DMP |
15.4 |
23.5 |
| 2,4-DMP |
7.2 |
5.7 |
| 2,2,3-TMB |
3.7 |
6.0 |
| STMB (%) |
1.2 |
1.7 |
| SC (%) |
25.2 |
8.8 |
| n-C4H10 |
7.9 |
2.9 |
| i-C4H10 |
6.6 |
2.7 |
| C3H8 |
6.1 |
2.5 |
| Others |
4.6 |
0.7 |
Around 1.0, very close to the thermodynamic equilibrium value, which was similar to the distribution of n-heptane isomerization products catalyzed by Pt/HPW/Zr MCM-41 prepared by J. A. Wang's research group31 and different from the research results of catalysts prepared by Justine Harmel and Zhang Wei et al.1,16 The main feature of the 2% MoOx Pd/Ce-M catalyst was the difference in pore size. Before cracking, the primary reaction product of n-heptane passed through larger mesoporous pores and exhibited good diffusion. The molecular size of multi branched isoheptane was larger than that of single branched isoheptane, resulting in cracking. Due to reasonable acidity, macropores, appropriate Si/Mo ratio, and high metal dispersion, it had a high yield of multi branched isomers.3 The cracking products mainly include n-butane (n-C4H10), isobutane (i-C4H10), and propane (C3H8), which were obtained by breaking a single C–C bond.44 There were also small amounts of C1, C2, C5, and C6 products, and there were no components ≥C7 in the isomerization reaction products.
The mechanism of n-heptane isomerization reaction has been studied in many literature,3,16,43,45 Based on the experimental results and literature, we proposed that the isomerization reaction mechanism of n-heptane on MoOx Pd/Ce-M catalyst followed a classical bifunctional mechanism (Fig. 11). During the isomerization reaction process on the metal acid support bifunctional catalyst, the dehydrogenation–hydrogenation of hydrocarbons occurred at the metal center, while the acid center served as the backbone isomerization or cracking center. The n-heptane molecule dehydrogenated at the Pd site to form a carbocation intermediate, which transferred to the acidic site through diffusion for skeleton rearrangement, and hydrogenated to form isoheptane at the metal active site. Metal Pd was the active site for dehydrogenation–hydrogenation, Ce-MCM-48 was the active site for isomerization, and the introduction of MoOx enriched the metal and acidic sites. The process of hydrogenation isomerization of heptane was shown in Fig. 12.
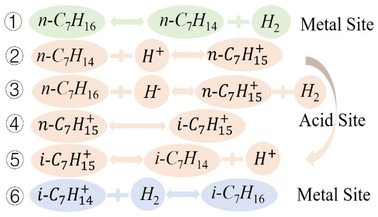 |
| | Fig. 11 Schematic diagram of reaction mechanism. | |
 |
| | Fig. 12 Schematic diagram of n-heptane isomerization process. | |
4. Conclusions
The Ce-MCM-48 molecular sieve prepared by hydrothermal synthesis method in this article had a highly ordered mesoporous material. The Ce-MCM-48 molecular sieve had a suitable specific surface area and mesoporous pore size, a spherical particle morphology, and a regularly arranged long-range ordered pore structure. Ce, as a heteroatom source, was introduced into the framework of mesoporous silicates, generating certain weak acid site, which could serve as an acidic support for alkane isomerization.
MoOx was used as a structural aid to prepare MoOx modified yMoOx-Pd/Ce-M catalyst by impregnation method. Compared with the Pd/Ce-MCM-48 catalyst prepared by the same method, the specific surface area and pore size decreased, but yMoOx-Pd/Ce-M still remained a mesoporous material. The spherical particles were uniform and compact in aggregation, and the metal Pd had good dispersion on the surface of the support. And MoOx increased the acid sites of the support, the acidity of Brønsted acid and Lewis acid. The MoOx group affected the local environment of the molecular sieve skeleton.
In the catalytic isomerization of n-heptane, the effect of MoOx supported catalyst on the performance of n-heptane hydroisomerization was investigated. The 2% MoOx-Pd/Ce-M catalyst showed good catalytic activity, and the appropriate addition of Mo could improve the conversion rate and selectivity of the catalyst. The selectivity for multi branched isoheptane was the highest, increasing the octane number of gasoline. MoOx modified Pd/Ce-MCM-48 was a promising catalyst for improving octane number through alkane isomerization. The combined effect of Pd and MoOx was necessary to enhance the isomerization of n-heptane.
Author contributions
Yanhong Cui: writing – original draft, writing – review and editing, methodology, conceptualization, software, and investigation; Xiuli Dong: project administration and formal analysis; Zhen Jiang: formal analysis and data analysis; Yanhua Suo: validation, visualization, and supervision; Wei Zhang: formal analysis, writing – review and editing; Yingjun Wang: project administration, funding acquisition, and resources.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work greatly acknowledges the financial support from the China Postdoctoral Science Foundation (2023MD744180), the Heilongjiang Postdoctoral Fund (LBH-Z23100), the Support Plan for Outstanding Scientific Research Talents of Provincial Advantageous and Characteristic Disciplines of Chemical Engineering and Technology of Northeast Petroleum University. The authors acknowledge the support of Analysis and Test Center of Northeast Petroleum University.
Notes and references
- W. Zhang, R. Liu, S. Ma, K. Dimitriyet, Y. Suo and Y. Wang, China Pet. Process. Petrochem. Technol., 2022, 24(01), 68–80 Search PubMed.
- L. P. Teh, H. D. Setiabudi, S. M. Sidik, N. H. R. Annuar and A. A. Jalil, Mater. Chem. Phys., 2021, 263, 124406 CrossRef CAS.
- Z. Ghaderi, M. H. Peyrovi and N. Parsafard, J. Porous Mater., 2023, 30(5), 1–7 CrossRef.
- Y. Zheng, M. Ma and H. Shao, Carbon Neutrality, 2023, 2, 23 CrossRef.
- N. S. Ali, Z. T. Alismaeel, H. S. Majdi, H. G. Salih, M. A. Abdulrahman, N. M. Cata Saady and T. M. Albayati, Heliyon, 2022, 8(6), 1–6 CAS.
- R. Sotelo-Boyás, L. O. Alemán-Vázquez and J. R. Ramírez-Segundo, Ind. Eng. Chem. Res., 2023, 62(25), 9607–9618 CrossRef.
- M. B. Bahari, A. A. Jalil, C. R. Mamat, N. S. Hassan, M. H. Razak, N. M. Izzudin, M. A. Aziz, N. F. Khusnun, M. A. H. Aziz, A. F. A. Rahman, W. Nabgan and S. Rajendran, Int. J. Hydrogen Energy, 2022, 252, 653–665 Search PubMed.
- T. Pantita, J. Amnart, J. Wuttithep, K. Yoottapong, J. Bunjerd and P. Piyasan, Microporous Mesoporous Mater., 2023, 354, 1112556 Search PubMed.
- W. Li, X. Lu, K. Xu, J. Qu and Z. Qiang, Water Res., 2015, 86, 1–7 CrossRef PubMed.
- S. Li, J. Wang, Y. Ye, Y. Tang, X. Li, F. Gu and L. Li, Appl. Catal., B, 2020, 263, 118311 CrossRef CAS.
- C. Lin, H. Pan, Z. Yang, X. Han, P. Tian, P. Li, Z. Xiao, J. Xu and Y. Han, Ind. Eng. Chem. Res., 2020, 59(14), 1–37 CrossRef.
- W. Liang, J. Fu, H. Chen, X. Zhang and G. Deng, Mater. Lett., 2021, 283(1), 128857 CrossRef CAS.
- M. D. Smolikov, V. A. Shkurenok, L. I. Bikmetova, I. P. Prosvirin, T. I. Gulyaeva, A. V. Bukhtiyarov, E. A. Paukshtis and V. I. Bukhtiyarov, Mol. Catal., 2022, 112527 CrossRef CAS.
- M. B. Bahari, A. A. Jalil, C. R. Mamat, N. S. Hassan, N. F. Khusnun, A. R. Herrynaldi, A. F. A. Rahman, A. H. Hatta, M. A. H. A. Aziz, N. N. M. Huzaimi and M. H. Razak, Mol. Catal., 2023, 112873 CrossRef CAS.
- Q. Li, H. Guo, R. Xue, M. Wang, M. Xu, W. Yang, J. Zhang and W. Yang, Int. J. Hydrogen Energy, 2020, 45(41), 1–15 Search PubMed.
- J. Harmel, T. Roberts, Z. Zhang, G. Sunley, P. Jongh and K. P. Jongh, J. Catal., 2020, 390, 161–169 CrossRef CAS.
- J. Oenema, J. Harmel, R. Pérez Vélez, M. K. Meijerink, W. Eijsvogel, A. Poursaeidesfahani, T. J. H. Vlugt, J. Zečević and K. P. de Jong, ACS Catal., 2020, 10(23), 14245–14257 CrossRef CAS PubMed.
- J. Wang, G. Wang, Z. Zhang, G. Ouyang and Z. Hao, RSC Adv., 2021, 11, 36577 RSC.
- S. Li, X. Li, H. Wu, X. Sun, F. Gu, L. Zhang, H. He and L. Li, ACS Appl. Mater. Interfaces, 2019, 11(27), 1–49 CAS.
- W. Zhan, G. Lu and Y. Guo, J. Rare Earths, 2008,(04), 515–522 CrossRef.
- M. Benaissa, A. M. Alhanash, M. Eissa, A. Aldalbahi, S. Alzahly, M. Rahaman, G. Periyasami and M. S. Hamdy, J. Porous Mater., 2020, 27(6), 1–9 CrossRef.
- Y. Wang, X. Dong, Y. Cui, S. Ma, Y. Suo and W. Zhang, J. Porous Mater., 2019, 1–8 Search PubMed.
- A. Boro and A. K. Talukdar, J. Porous Mater., 2019, 26(4), 1–12 CrossRef.
- Z. Li, X. Dong, M. Zhang, L. Leng, W. Chen, J. H. Horton, J. Wang, Z. Li and W. Wu, ACS Appl. Mater. Interfaces, 2020, 12(51), 57569–57577 CrossRef CAS PubMed.
- X. Pan, X. Wang, X. Wang, Y. Li, G. Liu and H. Lin, CrystEngComm, 2019, 21(42), 6472–6481 RSC.
- Z. Yang, Y. Liu, J. Zhao, J. Guo, K. Sun and C. Liu, Chin. J. Catal., 2017, 38(3), 509–517 CrossRef CAS.
- H. Chen, J. Yi, C. Ma, X. Gao, S. Liu, Z. Tao, B. Wu, H. Xiang, Y. Yang and Y. Li, Fuel, 2020, 268, 117241 CrossRef CAS.
- N. A. A. Fatah, S. Triwahyono, A. A. Jalil, A. Ahmad and T. A. T. Abdullah, Appl. Catal., A, 2016, 1–40 Search PubMed.
- Z. Ghaderi, M. H. Peyrovi and N. Parsafard, J. Iran. Chem. Soc., 2022, 1–7 Search PubMed.
- L. Gao, Z. Shi, U. J. Etim, P. Wu, W. Xing, Y. Zhang, P. Bai and Z. Yan, Fuel, 2019, 653–665 CrossRef CAS.
- J. A. Wang, L. F. Chen, L. E. Noreña, J. Navarrete, M. E. Llanos, J. L. Contreras and O. Novaro, Microporous Mesoporous Mater., 2008, 112(1–3), 61–76 CrossRef CAS.
- N. Parsafard, M. H. Peyrovi and Ni. Parsafard, React. Kinet., Mech. Catal., 2016, 1–16 Search PubMed.
- H. Qin, N. Feng, Q. Lv, H. Wan and G. Guan, Fuel Process. Technol., 2023, 241, 107604 CrossRef CAS.
- N. Parsafard, A. Garmroodi and A. S. Mirzaei, Int. J. Chem. Kinet., 2021, 1–11 Search PubMed.
- Y. Li, H. Wu, B. Zhang, Z. Yang, G. Han and S. Pan, Inorg. Chem., 2018, 57(15), 9376–9384 CrossRef CAS.
- J. Ling, H. Zhang, K. Yuan, D. Burgess, J. Hu and M. Hu, J. Solid State Chem., 2020, 287, 121317 CrossRef CAS.
- H. Al-Kandari, F. Al-Kharafifi and A. Katrib, Appl. Catal., A, 2010, 383, 141–148 CrossRef CAS.
- S. Ganji, P. Bukya, V. Vakati, K. S. R. Rao and D. R. Burri, Catal. Sci. Technol., 2013, 3(2), 409–414 RSC.
- Z. Hu, X. Liu, D. Meng, Y. Guo, Y. Guo and G. Lu, ACS Catal., 2016, 6(4), 2265–2279 CrossRef CAS.
- Q. Shang, N. Tang, H. Qi, S. Chen, G. Xu, C. Wu, X. Pan, X. Wang and Y. Cong, Chin. J. Catal., 2020, 41(12), 1812–1817 CrossRef CAS.
- Z. Ghaderi, M. H. Peyrovi and N. Parsafard, BMC Chem., 2021, 15(1), 1–8 CrossRef PubMed.
- Z. Ghaderi, M. H. Peyrovi and N. Parsafard, React. Kinet., Mech. Catal., 2022, 135(6), 3099–3111 CrossRef CAS.
- K. Guo, A. Ma, Z. Wang, J. Li, B. Wu, T. Liu and D. Li, New J. Chem., 2022, 46, 16752–16763 RSC.
- T. Matsuda, F. Uchijima, S. Endo and N. Takahashi, Appl. Catal., A, 1999, 176(1), 91–99 CrossRef CAS.
- M. Fedyna, M. Sliwa, K. Jaroszewska and J. Trawczyński, Fuel, 2020, 118607 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Xiuli Donga,
Zhen Jianga,
Yanhua Suoa,
Wei Zhang
ab,
Xiuli Donga,
Zhen Jianga,
Yanhua Suoa,
Wei Zhang