
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and anticancer evaluation of [D-Ala]-nocardiotide A†
Rani Maharani *abc,
Muhamad Imam Muhajira,
Jelang Muhammad Dirgantaraad,
Ari Hardiantoa,
Tri Mayantiac,
Desi Harnetiac,
Nurlelasariac,
Kindi Farabiabc,
Ace Tatang Hidayatabc,
Unang Supratmanabc and
Teruna Siahaan*e
*abc,
Muhamad Imam Muhajira,
Jelang Muhammad Dirgantaraad,
Ari Hardiantoa,
Tri Mayantiac,
Desi Harnetiac,
Nurlelasariac,
Kindi Farabiabc,
Ace Tatang Hidayatabc,
Unang Supratmanabc and
Teruna Siahaan*e
aDepartment of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Jatinangor, West Java, Indonesia. E-mail: r.maharani@unpad.ac.id
bCentral Laboratory, Universitas Padjadjaran, Jalan Raya Bandung-Sumedang KM 21, Jatinangor 45363, West Java, Indonesia
cCentre of Natural Products and Synthesis Studies, Faculty of Mathematics and Natural Sciences, Universitas Padjadjaran, Jalan Raya Bandung-Sumedang KM 21, Jatinangor 45363, West Java, Indonesia
dDepartment of Chemistry, Graduate School of Science, Osaka University, Toyonaka, Osaka 560-0043, Japan
eDepartment of Pharmaceutical Chemistry, School of Pharmacy, The University of Kansas, 2095 Constant Avenue, Lawrence, Kansas 66047, USA. E-mail: siahaan@ku.edu
First published on 30th January 2024
Abstract
Cancer is currently one of the biggest causes of death in the world. Like some microorganisms, cancer cells also develop resistance to various chemotherapy drugs and are termed multidrug resistant (MDR). In this regard, there is a need to develop new alternative anticancer agents. Anticancer peptides (ACPs) with high selectivity and high cell penetration ability are a promising candidate, as well as they are easy to modify. A cyclohexapeptide called nocardiotide A was isolated from the marine sponge Callyspongia sp., which is cytotoxic towards several cancer cells such as MM, 1S, HeLa, and CT26 cells. Previously, nocardiotide A was synthesized with a very low yield owing to its challenging cyclization process. In this study, we synthesized [D-Ala]-nocardiotide A as a derivative of nocardiotide A using a combination of solid phase peptide synthesis (SPPS) and liquid phase peptide synthesis (LPPS). The synthesis was carried out by selecting a D-alanine residue at the C-terminus to give a desired cyclic peptide product with a yield of 31% after purification. The purified [D-Ala]-nocardiotide A was characterized using HR-ToF MS and 1H and 13C-NMR spectroscopy to validate the desired product. The anticancer activity of the peptide was determined against HeLa cancer cell lines with an IC50 value of 52 μM compared to the parent nocardiotide A with an IC50 value of 59 μM. In the future, we aim to mutate various L-amino acids in nocardiotide A to D-amino acids to prepare nocardiotide A derivatives with a higher activity to kill cancer cells with higher membrane permeation. In addition, the mechanism of action of nocardiotide A and its derivatives will be evaluated.
Introduction
Cancer is one of the most difficult diseases to treat and is the cause of high mortality worldwide.1 Unfortunately, finding chemotherapeutic agents for cancer treatment has been very challenging because they can have serious side effects such as anemia, gastrointestinal mucositis, and alopecia.2 In addition, it has been reported that long-term use of chemotherapeutic agents results in drug resistance in 50% of cancer cells in a patient.3,4 Cancer cells become resistant to chemotherapeutics due to the inactivation of the drug or efflux pump mechanisms that expel the drug from cancer cell membranes and prevent it from entering into the cytoplasm to exert drug activity.5,6 Therefore, the development of new anticancer agents as alternatives to the currently available therapies is necessary to resolve the side-effect problems.7Anticancer peptides (ACPs) are a promising group of compounds that have opened new avenues for future cancer therapies.8,9 ACPs are peptides with 5–50 amino acid residues with higher anticancer activity compared to small drug molecules because they have high selectivity.8 In addition, ACPs can be readily modified to increase their activity and selectivity to lower their side effects. Some ACPs show anticancer activity by disrupting cell membranes or by forming pores in cell membranes to induce cancer cell apoptosis; thus, ACPs can rapidly kill cancer cells via membrane-targeting mechanisms.5,8
Recently, nocardiotide A has been isolated from a culture broth of Nocardiopsis sp. from the marine sponge Callyspongia sp (Fig. 1, Left).10 The isolated nocardiotide A exhibits cytotoxicity against 1S, HeLa, and CT26 cells with IC50 values of 8, 11, and 12 μM, respectively.10 Thus, we previously synthesized nocardiotide A using a combination of the solid-phase and solution-phase peptide synthesis with the overall yield of nocardiotide A synthesis being 16%.11 This synthesis produced a high enough quantity to confirm nocardiotide A structure as well as to evaluate its biological activity and mechanism of action.
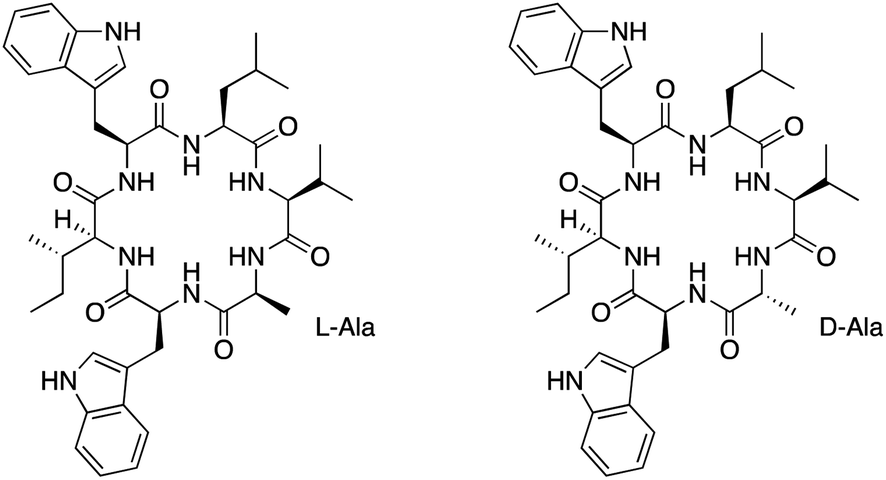 | ||
| Fig. 1 Chemical structures of nocardiotide A (left) and [D-Ala]-nocardiotide A (right). | ||
Davies had shown that the formation of cyclic peptides can be improved by having large and small amino acids at the N- and C-terminus amino acids, respectively.12 The presence of D-amino acid at the C-terminus could also increase the yield of the cyclization reaction.12 Therefore, we hypothesized that the substitution of the L-Ala residue on nocardiotide A to the D-Ala residue of [D-Ala]-nocardiotide A (Fig. 1, Right) could improve the cyclization reaction to produce a high synthetic yield of the peptide. In addition, the biological activity of [D-Ala]-nocardiotide A can be evaluated and compared to the parent nocardiotide A. In the future, we would like to evaluate the effects of different D-amino acids within the molecule on the cyclization reaction, biological activity, and mechanism of action.
Here, we synthesized [D-Ala]-nocardiotide A using a combination of the solid and solution phase methods. The linear peptide precursor was generated by solid phase peptide synthesis using 2-chlorotrityl chloride resin and Fmoc chemistry. The peptide on the resin was cleaved using mild conditions and the resulting linear peptide precursor was purified using a reversed-phase column chromatography. The cyclization of the peptide to produce [D-Ala]-nocardiotide A was accomplished in a high dilute solution followed by side chain deprotection and purification using a semi-preparative reversed-phase (RP) HPLC column. The pure [D-Ala]-nocardiotide A was characterized by mass spectrometry and NMR. The activities of both molecules (IC50) were evaluated for killing HeLa cancer cells, in comparison to positive and negative controls.
Materials and methods
Reagent for peptide synthesis
All amino acids, coupling reagents, solvents, and resin were purchased from GL-Biochem, Shanghai, China. They included 2-chlorotrityl chloride resin, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HBTU), 1-hydroxybenzotriazole (HOBt), dichloromethane (DCM), N-[(dimethylamino)-1H-1,2,3-triazolo-4,5-[b]pyridine-1-yl-methelene]-N-methylmethanaminium hexafluorophosphate N-oxide (HATU), dimethylformamide (DMF), N,N-diisopropylethylamine (DIPEA), Fmoc-D-Ala-OH, Fmoc-L-Val-OH, Fmoc-L-Leu-OH, Fmoc-L-Trp(Boc)-OH, trifluoroacetic acid (TFA), trifluoroethanol (TFE), piperidine, methanol, and n-hexane. The peptide synthesis was accomplished using an SPPS tube, rotatory suspension mixer, and rotatory evaporator.Peptide synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:16) for 15 min in two cycles. The reaction mixture was washed using DCM three times and then drained to obtain dry Fmoc-D-Ala resin. The dry resin was then determined for the amino acid loading and the resin loading was 0.42 mmol of Fmoc-D-Ala per gram resin.:1:3) was added to the hexapeptidyl-resin followed by shaking for 2 h. The filtrate was collected and the resin was washed with 5 mL of the cleavage reagent and 5 mL of dry DCM. The combined solutions were concentrated using rotatory evaporation to obtain the crude protected linear precursor [D-Ala]-nocardiotide A (82.8 mg; 83.4% yield). The white solid of [D-Ala]-nocardiotide A showed a TOF-ESI-MS m/z of 987.4915 with the calculated m/z for C52H74N8O11 is [M + H]+ 987.4913.
:1:16) for 15 min in two cycles. The reaction mixture was washed using DCM three times and then drained to obtain dry Fmoc-D-Ala resin. The dry resin was then determined for the amino acid loading and the resin loading was 0.42 mmol of Fmoc-D-Ala per gram resin.:1:3) was added to the hexapeptidyl-resin followed by shaking for 2 h. The filtrate was collected and the resin was washed with 5 mL of the cleavage reagent and 5 mL of dry DCM. The combined solutions were concentrated using rotatory evaporation to obtain the crude protected linear precursor [D-Ala]-nocardiotide A (82.8 mg; 83.4% yield). The white solid of [D-Ala]-nocardiotide A showed a TOF-ESI-MS m/z of 987.4915 with the calculated m/z for C52H74N8O11 is [M + H]+ 987.4913.Biological assay
000 cells per mL as determined using trypan blue exclusion. 196-well plates were used for the cell culture and the cells were incubated for 24 h or until they were at least 70% confluent at 37 °C under 5% CO2 gas. After 24 h, the peptide was added to the wells. After 96 h, cell viability was determined by measuring the metabolic conversion of resazurin substrate into pink fluorescent resorufin product as a result of the reduction in the viable cells. The absorbance was read using a multimode reader at 570 nm. All compounds were tested at eight concentrations of 3.91, 7.81, 15.63, 31.25, 62.50, 125.00, 250.00, and 500.00 μg mL−1 in 2% DMSO. Each concentration of the compound was tested in triplicate experiments; then, IC50 values were calculated by linear regression method using Microsoft Excel software.Result and discussion
Total synthesis of [D-Ala]-nocardiotide A
[D-Ala]-nocardiotide A was synthesized using a combination of solid- and solution-phase peptide synthesis methods (Fig. 2). The synthesis of linear hexapeptide of [D-Ala]-nocardiotide A was accomplished by Fmoc chemistry in the solid-phase method while cyclization of the linear peptide was performed using the solution phase.13 D-Alanine at the C-terminus and L-tryptophan at N-terminus as small and large amino acids, respectively, were used to provide advantage on the cyclization process14 because the presence of D-Ala residue induced a more favourable conformation cyclization, as reported by Davies et al.12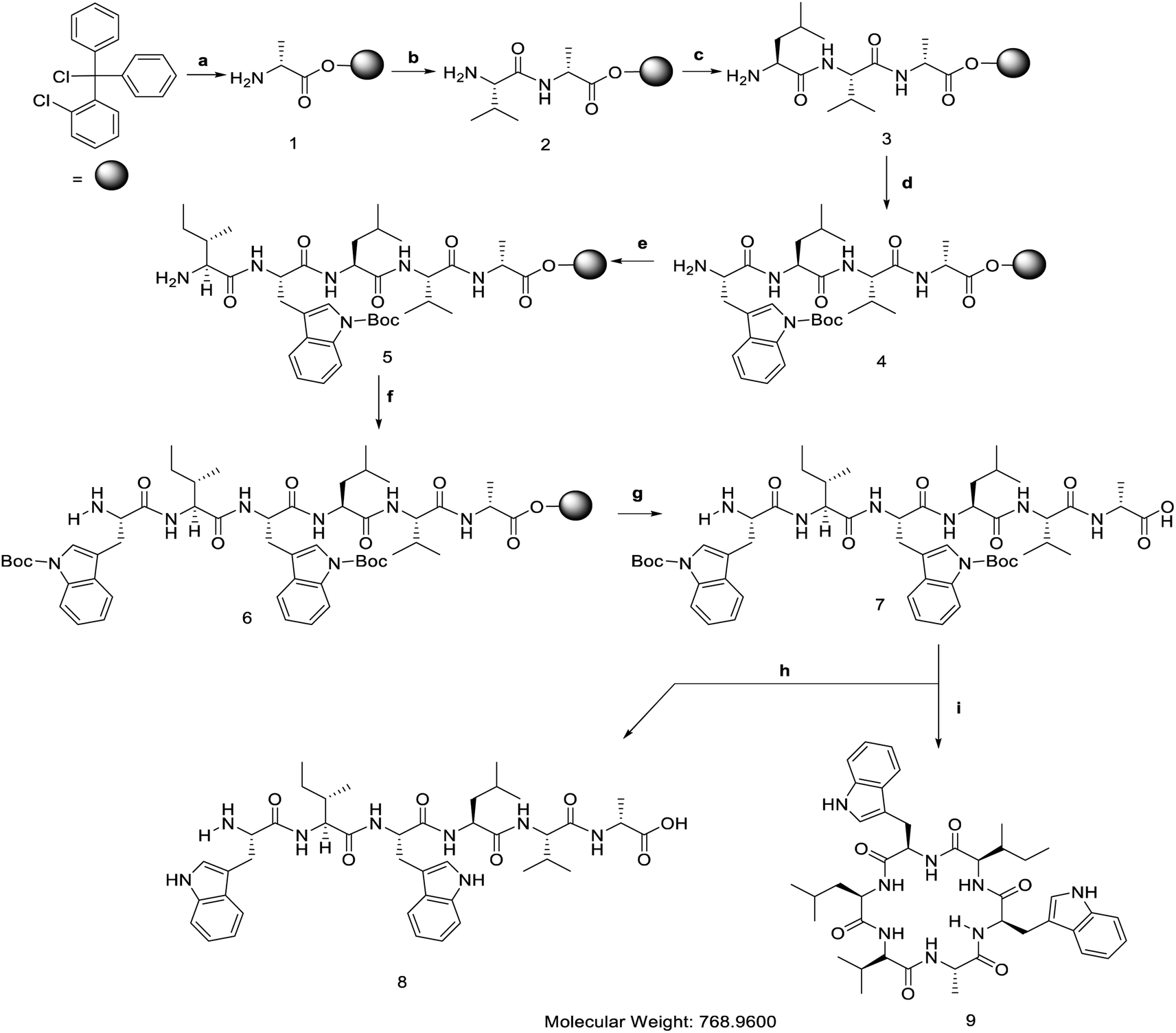 | ||
| Fig. 2 Solid phase peptide synthesis and macrocyclization of [D-Ala]-nocardiotide. (a).(1) Fmoc-D-Ala-OH (1 equiv.), DIEA (2 equiv.), 4 mL CH2Cl2, 4 h; (2) MeOH (15 equiv.), DIEA (5 equiv.), CH2Cl2 (80 equiv.), 15 min; (3) 20% piperidine in DMF, 10 min. (b). (1) Fmoc-L-Val-OH, (3 equiv.), HBTU (3 equiv.), HOBt (3 equiv.), DIEA (6 equiv.), 4 mL DMF, 4 h; (2) 20% piperidine in DMF, 10 min. (c). (1) Fmoc-L-Leu-OH (3 equiv.), HBTU (3 equiv.), HOBt (3 equiv.), DIEA (6 equiv.), 4 mL DMF, 4 h; (2) 20% piperidine in DMF, 10 min. (d). (1) Fmoc-L-Trp(Boc)-OH (3 equiv.), HBTU (3 equiv.), HOBt (3 equiv.), DIEA (6 equiv.), 4 mL DMF, 4 h; (2) 20% piperidine in DMF, 10 min. (e). (1) Fmoc-L-Ile-OH (3 equiv.), HBTU (3 equiv.), HOBt (3 equiv.), DIEA (6 equiv)., 4 mL DMF, 4 h; (2) 20% piperidine in DMF, 10 min. (f). (1) Fmoc-L-Trp-OH (3 equiv.), HBTU (3 equiv.), HOBt (3 equiv.), DIEA (6 equiv.), 4 mL DMF, 4 h; (2) 20% piperidine in DMF, 10 min. (g). TFE (1 equiv.), AcOH (1 equiv.), CH2Cl2 (3 equiv.), 1 h. (h). Trifluoroacetic acid (TFA) 95% in water, 1 h. (i). (1) HBTU (3 equiv.), DIEA (1%), 72 h; (2) TFA 95% in water, 1 h. | ||
The linear hexapeptide was prepared by the solid-phase peptide synthesis (SPPS) method using Fmoc chemistry (Fig. 2).15 The first amino acid, Fmoc-D-Ala-OH, was attached to 2-chlorotrityl chloride resin in DCM and in the presence of basic DIEA to provide 0.47 mmol D-Ala amino acid per gram resin. The unreacted sites on the resin were then capped by reacting them with MeOH:DIEA:DCM (15:5:85). The Fmoc group was deprotected using 20% piperidine in DMF to form a free amino group that was ready for the reaction with the next amino acid. The second amino acid, Fmoc-L-Val-OH, was then coupled to the D-Ala using a combination of HBTU and HOBt coupling reagents in the presence of DIEA. Three and six equivalents of HBTU/HOBt and DIEA were found to provide an effective coupling reaction to various types of peptides.15 The success of the coupling reaction was monitored by a chloranil test. The elongated peptide was then treated for the attachment of L-Leu, L-Trp(Boc), L-Ile, and L-Trp(Boc) to produce the desired linear peptide. The linear peptide was cleaved from the resin using a mixture of acetic acid and trifluroethanol (TFE) to yield a linear hexapeptide precursor with Boc protecting groups still attached on the side chains of both Trp residues. The linear precursor of [D-Ala]-nocardiotide A was obtained with 86.5% yield based on the loading resin value.
To prepare the linear peptide 8, both Boc protective groups on the Trp residues of peptide 7 were removed by treating it with 95% TFA in water for 15 min. The crude linear peptide 8 was dried using a rotary evaporator to produce a white powder. The crude product was purified using reversed-phase flash ODS column chromatography with methanol:water (7:3) as the eluent. The desired peptide 8 was found to be a single peak at a retention time of 4.32 min. The amount of pure linear peptide 8 was 15.3 mg or 95.8% yield. The HR-TOF ESI mass spectrometry result of peptide 8 showed [M + H]+ with m/z of 787.4507, which was consistent with the calculated [M + H]+ ion peak at m/z 787.4514 for C42H58N8O7 of compound 8. The structure of 8 was also confirmed using 1H-NMR and 13C-NMR (Table 1).
| Amino acid | Linear [D-Ala]-nocardiotide A (CD3OD, 500 MHz) | [D-Ala]-nocardiotide A (CD3OD, 500 MHz) | Nocardiotide A (CD3OD, 600 MHz) | |||
|---|---|---|---|---|---|---|
| 1H δ (ppm)(m, J in Hz) | 13C δ (ppm) | 1H δ (ppm)(m, J in Hz) | 13C δ (ppm) | 1H δ (ppm)(m, J in Hz) | 13C δ (ppm) | |
|
||||||
| Alanine | ||||||
| CO | 170.1 | 173.9 | 173.9 | |||
| α | 3.65 (q, J = 6.6) | 48.6 | 4.29 (q, J = 7.0) | 51.0 | 4.31 (q, J = 7.2) | 49.8 |
| β | 1.13 (d, J = 6.6) | 11.1 | 1.04 (d, J = 7.0) | 19.2 | 1.17 (d, J = 7.2) | 18.05 |
|
||||||
| Valine | ||||||
| CO | 171.4 | 174.8 | 175.3 | |||
| α | 4.08 (d, J = 7.8) | 57.2 | 4.09 (d, J = 7.5) | 60.5 | 4.22 (d, J = 7.7) | 60.7 |
| β | 2.77–2.80 (m) | 40.5 | 1.70 (m) | 31.8 | 1.97 (m) | 31.8 |
| γ | 0.82 (d, J = 7.4) | 17.8 | 0.86 (d, J = 3.0) | 21.3 | 0.88 (d, J = 3.1) | 19.8 |
| γ′ | 0.84 (d, J = 7.4) | 17.7 | 0.87 (d, J = 3.0) | 19.5 | 0.9 (d, J = 3.1) | 19.0 |
|
||||||
| Leucine | ||||||
| CO | 170.6 | 173.1 | 171.6 | |||
| α | 4.22–4.24 (m) | 56.4 | 3.89 (m) | 56.3 | 3.81 (t, J = 7.3) | 53.0 |
| β | 1.46 (t, J = 6.55) | 48.2 | 1.60–1.61 (m) | 45.8 | 1.59 (m) | 41.9 |
| γ | 1.87–1.90 (m) | 25.0 | 1.67–1.70 (m) | 25.7 | 1.62 (m) | 25.6 |
| δ | 0.85 (d, J = 6.55) | 21.8 | 1.05 (d, J = 7.0) | 23.9 | 0.92 (d, J = 6.2) | 22.6 |
| δ′ | 0.86 (d, J = 6.55) | 21.5 | 1.04 (d, J = 7.0) | 23.2 | 0.93 (d, J = 6.2) | 22.9 |
|
||||||
| Tryptophan | ||||||
| CO | 171.7 | 175.3 | 179.0 | |||
| α | 4.34 (t, J = 7.9) | 53.2 | 4.53 (dd, J = 5.0) | 57.8 | 4.37 (dd, J = 3.8) | 56.9 |
| β/β′ | 3.32–3.36 (m) | 29.6 | 3.03–3.04 (m) | 28.5 | 3.05 (dd, J = 14.7) | 29.1 |
| 2 | 7.02 (s) | 124.3 | 7.04 (s) | 124.5 | 7.03 (s) | 124.6 |
| 3 | 111.4 | 112.4 | 112.2 | |||
| 4 | 8.22 (dt, J = 7.9) | 118.5 | 7.55 (dt, J = 8.0) | 120.1 | 7.55 (dt, J = 7.8) | 119.6 |
| 5 | 7.61 (dt, J = 7.9) | 118.3 | 6.92 (m) | 119.9 | 6.88 (m) | 119.4 |
| 6 | 7.31 (dt, J = 7.9) | 121.0 | 6.99 (m) | 122.7 | 6.94 (m) | 122.1 |
| 7 | 7.13–7.18 (m) | 110.1 | 7.23 (dt, J = 6.5) | 110.9 | 7.18 (dt, J = 8.1) | 112.0 |
| 8 | 136.3 | 138.2 | 137.9 | |||
| 9 | 127.3 | 128.6 | 128.9 | |||
|
||||||
| Isoleucine | ||||||
| CO | 170.7 | 173.7 | 173.4 | |||
| α | 4.58–4.62 (m) | 54,3 | 4.02 (d, J = 7.5) | 59.8 | 4.20 (d, J = 4.6) | 59.0 |
| β | 1.21–1.25 (m) | 43.7 | 2.03 (m) | 37.8 | 2.03 (m) | 37.0 |
| γ | 0.73 (d, J = 7.4) | 19.15 | 0.83 (d, J = 7.0) | 15.8 | 0.84 (d, J = 7.0) | 12.1 |
| γ′ | 1.21–1.25 (m) | 27.5 | 1.28 (m) | 27.6 | 1.28 (m) | 27.4 |
| δ | 0.79 (t, J = 6.7) | 15.1 | 0.85 (d, J = 7.0) | 16.6 | 0.85 (d, J = 7.4) | 14.7 |
|
||||||
| Tryptophan | ||||||
| CO | 171.7 | 173.3 | 173.2 | |||
| α | 4.17 (t, J = 7.9) | 51.2 | 3.30 (t, J = 7.5) | 41.2 | 3.35 (t, J = 8.0) | 41.5 |
| β/β′ | 3.28–3.32 (m) | 30.8 | 2.98–3.01 (m) | 25.8 | 2.82 (td, J = 8.6) | 26.2 |
| 2′ | 7.03 (s) | 123.6 | 7.02 (s) | 124.4 | 6.96 (s) | 123.3 |
| 3′ | 111.2 | 112.5 | 113.3 | |||
| 4′ | 8.22 (dt J = 7.9) | 118.3 | 7.50 (dt, J = 8.0) | 119.4 | 7.45 (dt, J = 7.9) | 119.2 |
| 5′ | 7.61 (dt, J = 7.9) | 118.2 | 6.94 (m) | 119.5 | 6.90 (m) | 119.4 |
| 6′ | 7.31 (dt, J = 7.8) | 120.8 | 7.00 (m) | 122.7 | 6.98 (m) | 122.3 |
| 7′ | 7.13–7.18 (m) | 110.1 | 7.26 (dt, J = 7.0) | 111.5 | 7.22 (dt, J = 8.2) | 112.2 |
| 8′ | 136.0 | 138.3 | 138.7 | |||
| 9′ | 127.3 | 129.8 | 128.8 | |||
The cyclic [D-Ala]-nocardiotide 9 was synthesized from the precursor linear hexapeptide 7 by linking the amino group of the L-Trp with the carboxylic acid of the D-Ala residue. The peptide was diluted in DCM:DMF (9:1) at high dilution (1.25 mM) to avoid the formation of the intermolecular oligomerization products. A combination of three equivalents of HBTU/HOBt was employed as a coupling reagent in the presence of 1% DIEA. The reaction was carried out for 72 h at room temperature. The cyclization process was monitored by silica thin layer chromatography using 5% iso-propanol:n-hexane as the eluent. The reaction mixture was then concentrated using a rotary evaporator. The crude product was purified using semi-preparative RP-HPLC equipped with C18 column (5 μm, 10 mm × 250 mm, Phenomenex) using 100% H2O as solvent A as well as 100% acetonitrile as solvent B. A linear gradient from 0% to 80% acetonitrile within 60 min was used to purify the desired product with a flow rate of 2.0 mL min−1 to produce a 31% yield of [D-Ala]-nocardiotide. The analytical RP-HPLC showed that both nocardiotide A and [D-Ala]-nocardiotide A have purity of 95.16% and 97.49%, respectively, with the same retention time at 11.06 min in a C18 column (5.0 μm, 4.6 × 250 mm, COSMOSIL) (Fig. 3A and B).
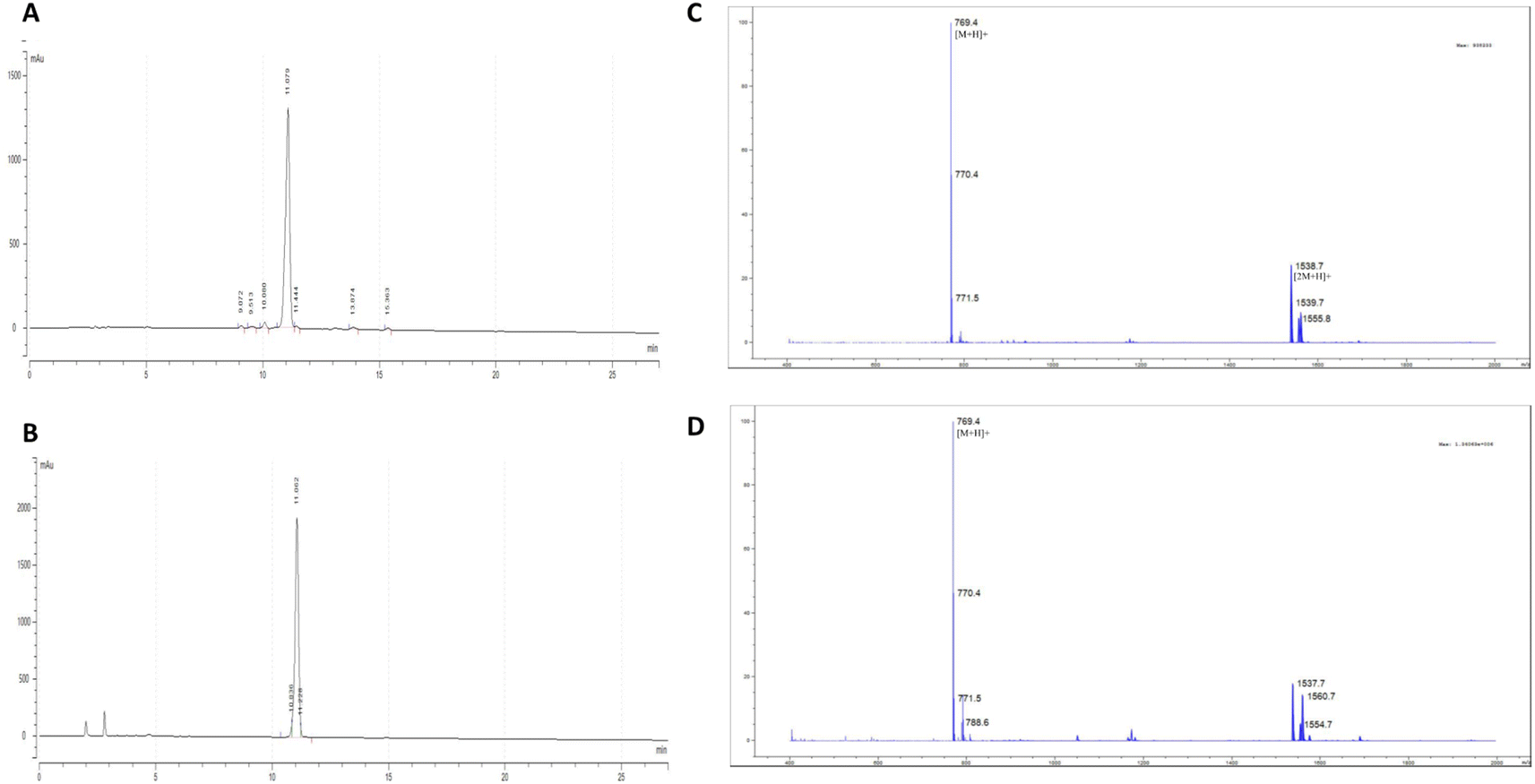 | ||
| Fig. 3 The purity analysis of each peptide using HPLC for (A) nocardiotide A and (B) [D-Ala]-nocardiotide A with a purity of 95.16% and 97.49%, respectively. The retention time for each peptide was 11.06 min. The mass spectrometry data for (C) nocardiotide A and (D) [D-Ala]-nocardiotide A show the same molecular weight at [M + H] at 769.4 amu. | ||
The identities of nocardiotide A and [D-Ala]-nocardiotide A 9 were characterized using HR-TOF ESI mass spectrometry (Fig. 3C and D) and NMR (Table 1). The HR-TOF ESI spectrum of [D-Ala]-nocardiotide A showed [M + H]+ at m/z 769.4403, which is consistent with the calculated molecular [M + H]+ ion peak at m/z 769.4401 for C42H57N8O6 (Fig. 3D). The product was further characterized using 1H- and 13C-NMR spectroscopy (Table 1). The 1H-NMR data of [D-Ala]-nocardiotide A 9 showed six alpha protons of the peptide with chemical shifts of 3.67, 4.08, 4.23, 4.34, 4.17, and 4.60 ppm. The 13C-NMR spectra showed the existence of six carbonyl signals of amide groups at 173.1, 173.9, 174.8, 173.3, 173.7, and 175.3 ppm. In addition, six alpha carbons were confirmed with chemical shifts of 51.0, 60.5, 56.3, 57.8, 59.8, and 41.2 ppm. Finally, 1H- and 13C-NMR data between the synthetic [D-Ala]-nocardiotide A and the isolated nocardiotide A were compared (Table 1). The chemical shifts of 13C atoms of [D-Ala]-nocardiotide A and the isolated nocardiotide A showed chemical shift differences in the range of 0.1–0.7 ppm; however, the Cβ of the Ala residue of [D-Ala]-nocardiotide A appeared at 19.2 ppm, while the Cβ of the Ala residue in the isolated nocardiotide A appeared at 18.05 ppm (Table 1).
Biological activity of nocardiotide A and [D-Ala]-nocardiotide A
The anticancer activities of nocardiotide A, [D-Ala]-nocardiotide A, CFSRM cyclic F5 (cyclo1,6 Ac-Cyc-Phe-Ser-Arg-Met-Cyc-NH2), and cisplatin were assessed in cervical cancer cells (HeLa) using MTT assay after 24 h incubation at various peptide (Fig. 4 and 5). As a positive control cisplatin has an IC50 of 2 μM. As a negative control, CFSRMC cyclic F5 has an IC50 of 522 μM. The results showed that nocardiotide A and [D-Ala]-nocardiotide A have IC50 of 59 μM and 52 μM, respectively, obtained through triplicate experiments. The results indicated that both peptides have a moderate level of anticancer activities but they are about 8-fold more active than the negative control CFSRMC cyclic F5.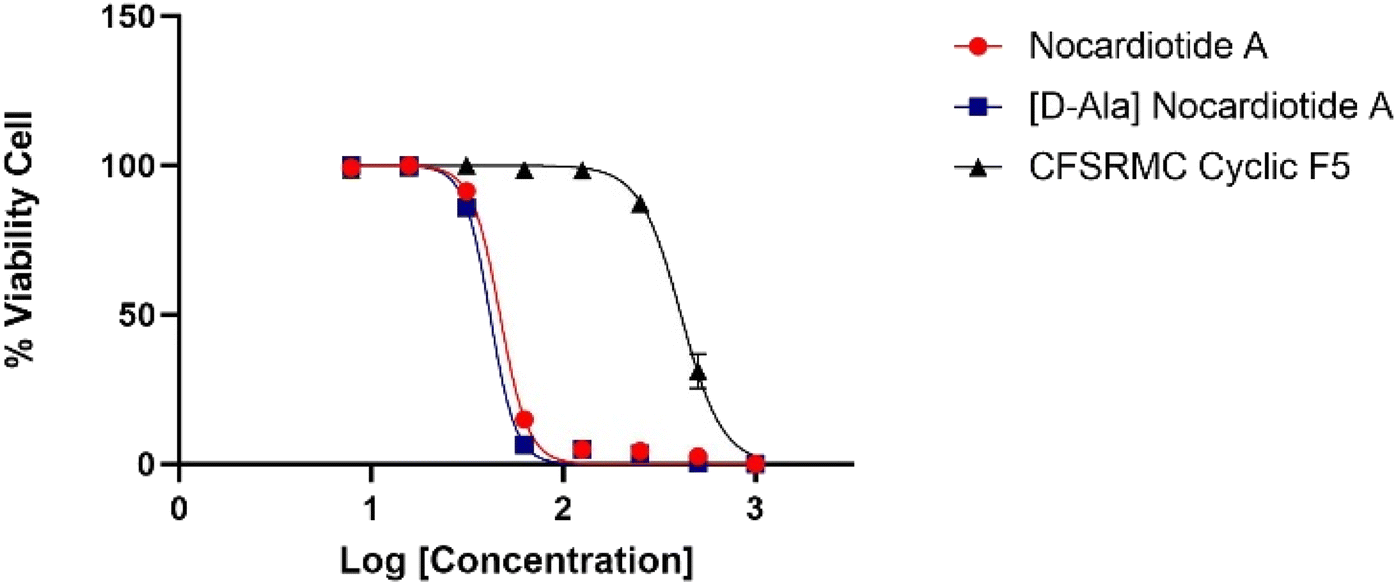 | ||
| Fig. 4 Graphic log[concentration] vs. % cell viability of nocardiotide A, [D-Ala]-nocardiotide, and CFSRMC cyclic F5 (cyclo(1,6)Ac-Cys-Phe-Ser-Arg-Met-Cys-NH2). | ||
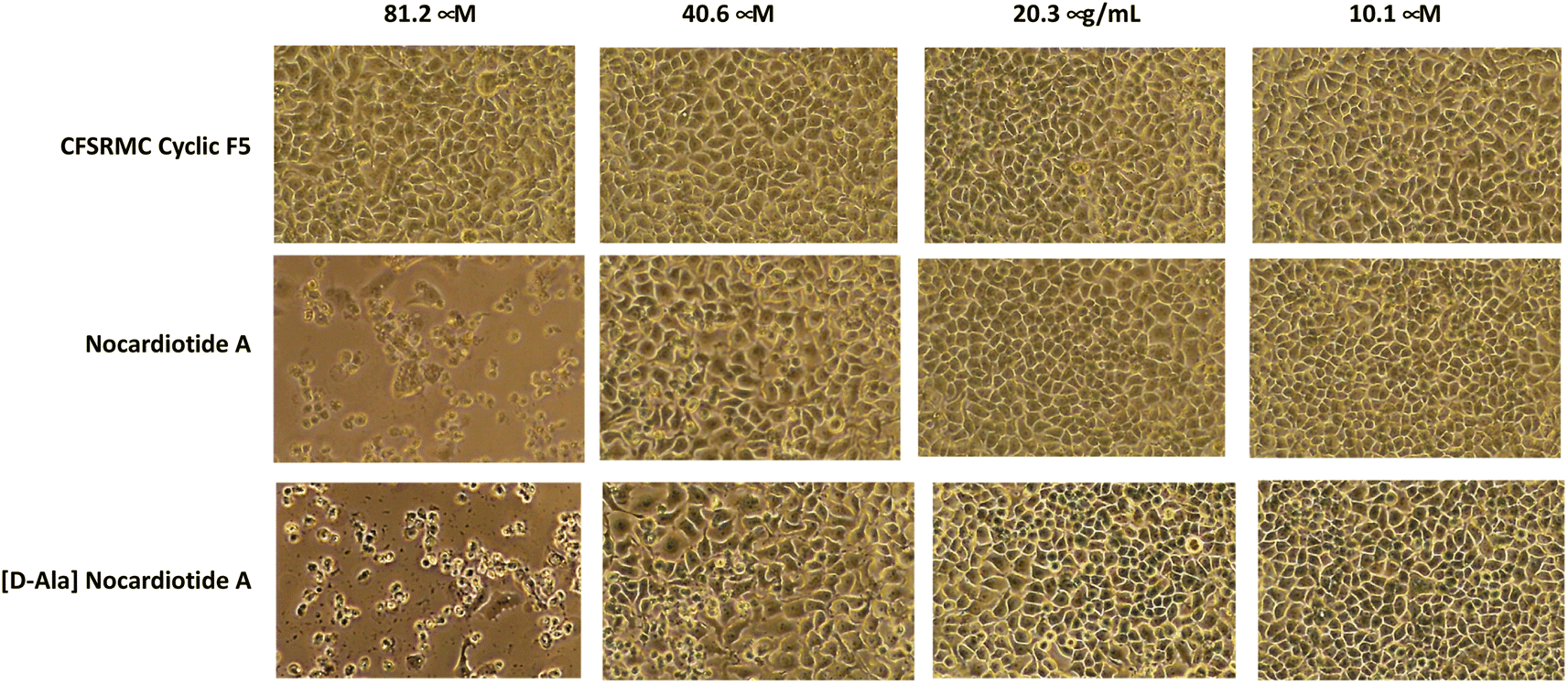 | ||
| Fig. 5 The effect of the concentrations of nocardiotide A, [D-Ala]-nocardiotide A, and CFSRMC cyclic F5 on the morphology of cervical cancer HeLa cells. Both nocardiotide A and [D-Ala]-nocardiotide A completely disrupted the monolayer integrity of HeLa cells at a concentration of 62.50 μg mL−1, while CFSRMC cyclic F5 as a negative control did not disrupt the monolayer integrity of HeLa cells. | ||
Both nocardiotide A and [D-Ala]-nocardiotide disrupted the cell monolayers at 81.2 μM while the negative control CFSRMC cyclic F5 did not (Fig. 5). We also found that the substitution of L-Ala into D-Ala did not change the anticancer activity of the peptide. It has been suggested previously that the presence of D-amino acid in a peptide increases the plasma stability of a peptide due to slowing down the enzymatic degradation of D-amino acid-containing peptide in plasma.16,17 Thus, our future study will be focused on evaluating the effects of substituting various L-amino acids to D-amino acids within the nocardiotide A on the anticancer activity of the peptide. It was interesting to find that the IC50 of 59 μM for our synthetic nocardiotide A was higher than that of the isolated nocardiotide A with IC50 of 11 μM in HeLa cells by Ibrahim et al.10 This difference in the IC50 could possibly be due to cell density used in both studies. The solubility of our synthetic peptides could also contribute to the toxicity of these synthetic peptides.
It is proposed that the anticancer activity of nocardiotide A and [D-Ala]-nocardiotide A is due to the disruption of cancer cell membranes. To test this hypothesis, we subjected both nocardiotide A and [D-Ala]-nocardiotide A to a 200 ns MD simulation in the presence of membranes to find out if both molecules could interact with membranes and disrupt the integrity of the cell membrane. Unfortunately, the results revealed that both peptides cannot disrupt cell the integrity of the membrane. Both cyclic peptides only transiently interact with the membrane.
Conclusions
We have successfully improved the cyclization reaction of nocardiotide A reaction by substituting the L-alanine residue with D-alanine residue. Thus, D-amino acid, especially D-alanine in this case promotes head-to-tail cyclization reaction of linear nocardiotide A. We also found that [D-Ala]-nocardiotide A is as active as nocardiotide A in killing cervical cancer (HeLa) cells; therefore, altering L-Ala with D-Ala did not change the biological activity of the peptide. Molecular dynamics simulations also indicated that both peptides did not disrupt the membrane along the 200 ns length of simulations. In the future, we will study the effects of D-amino acid mutations at various positions on the anticancer activity of the nocardiotide A derivatives. Furthermore, the mechanism of action of nocardiotide A and its derivatives in killing cancer cells will also be evaluated.Author contributions
The manuscript was written through the contributions of all authors. Conceptualisation, R. M., T. S., and M. I. M.; methodology, R. M., T. S., M. I. M, J. M. D., and A. H.; writing – original draft, R. M., and M. I. M.; writing – review and editing, R. M., T. S., M. I. M., T. M., D. H., N. N., and K. F.; supervision, A. T. H., and U.S.; funding acquisition, R. M., and A. T. H. All authors have read and agreed to the published version of the manuscript. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Authors would like to acknowledge Kemenristek DIKTI Indonesia (grant number 044/E5/PG.02.00.PL/2023) and World Class Professor Grant 2023-Kemendikbud Indonesia for financial support.References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. andBray, Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries CA Cancer, J. Clin., 2021, 71, 209–249, DOI:10.3322/caac.21660.
- G. Gabernet, T. Muller and G. Schneider, Membranolytic anticancer peptides, MedChemComm, 2016, 7, 2232–2245, 10.1039/C6MD00376A.
- J. F. Liang and V. C. andYang, Synthesis of doxorubicin-peptide conjugate with multidrug resistant tumor cell killing activity, Bioorg. Med. Chem. Lett., 2005, 15, 5071–5075, DOI:10.1016/j.bmcl.2005.07.087.
- M. R. Felicio, O. N. Silva, S. Goncalves, N. C. Santos and O. L. andFranco, Peptides with Dual Antimicrobial and Anticancer Activities, Front. Chem., 2017, 5, 5, DOI:10.3389/fchem.2017.00005.
- M. Singh, V. Kumar, K. Sikka, R. Thakur, M. K. Harioudh and D. P. Mishra, et al.) Computational Design of Biologically Active Anticancer Peptides and Their Interactions with Heterogeneous POPC/POPS Lipid Membranes, J. Chem. Inf. Model., 2020, 60, 332–341, DOI:10.1021/acs.jcim.9b00348.
- A. Sajid, H. Rahman and S. V. andAmbudkar, Advances in the structure, mechanism and targeting of chemoresistance-linked ABC transporters, Nat. Rev. Cancer, 2023, 23, 762–779, DOI:10.1038/s41568-023-00612-3.
- H. I. Farah, U. Supratman, A. T. Hidayat and R. Maharani, An overview of the synthesis of biologically active cyclodepsipeptides, ChemistrySelect, 2022, 7, e202103470, DOI:10.1002/slct.202103470.
- M. Xie, D. Liu and Y. andYang, Anti-cancer peptides: classification, mechanism of action, reconstruction and modification, Open Biol., 2020, 10, 200004, DOI:10.1098/rsob.200004.
- X. Liu, Y. Li, Z. Li, X. Lan, P. H. M. Leung, J. Li and L. Qin, Mechanism of anticancer effects of antimicrobial peptides, J. Fiber Bioeng. Inform., 2015, 8, 25–36 CrossRef.
- A. H. Ibrahim, E. Z. Attia, D. Hajjar, M. A. Anany, S. Y. Desoukey, M. A. Fouad, M. S. Kamel, H. Wajant, T. A. Gulder and U. R. Abdelmohsen, New Cytotoxic Cyclic Peptide from the Marine Sponge-Associated Nocardiopsis sp. UR67, Mar. Drugs, 2018, 16, 290, DOI:10.3390/md16090290.
- M. I. Muhajir, A. Hardianto, J. Al-Anshori, D. Sumiarsa, T. Mayanti, D. Harneti, A. T. Hidayat, U. Supratman and R. Maharani, Total Synthesis of Nocardiotide A by Using a Combination of Solid-and Solution-Phase Methods, ChemistrySelect, 2021, 6, 12941–12946, DOI:10.1002/slct.202103441.
- J. S. Davies, The cyclization of peptides and depsipeptides, J. Pept. Sci., 2003, 9, 471–501, DOI:10.1002/psc.491.
- A. Rahim, N. Nurlelasari, D. Harneti and U. Supratman, A total synthesis of cyclodepsipeptide [ Leu ] 6 -aureobasidin K using combination of solid-and solution-phase, Curr. Chem. Lett., 2019, 9, 97–104, DOI:10.5267/j.ccl.2019.9.002.
- X. M. Gao, Y. H. Ye, M. Bernd and B. andKutscher, Studies on the synthesis of cyclic pentapeptides as LHRH antagonists and the factors that influence cyclization yield, J. Pept. Sci., 2002, 8, 418–430, DOI:10.1002/psc.397.
- R. Maharani, R. T. C. Brownlee, A. B. Hughes and B. M. Abbott, A total synthesis of a highly N-methylated cyclodepsipeptide [2S,3S-Hmp]-aureobasidin L using solid-phase methods, Tetrahedron, 2014, 70, 2351–2358, DOI:10.1016/j.tet.2014.02.036.
- J. Lu, H. Xu, J. Xia, J. Ma, J. Xu, Y. Li and J. andFeng, D- and Unnatural Amino Acid Substituted Antimicrobial Peptides With Improved Proteolytic Resistance and Their Proteolytic Degradation Characteristics, Front. Microbiol., 2020, 11, 563030, DOI:10.3389/fmicb.2020.563030.
- R. Tugyi, K. Uray, D. Ivan, E. Fellinger, A. Perkins and F. andHudecz, Partial D-amino acid substitution: Improved enzymatic stability and preserved Ab recognition of a MUC2 epitope peptide, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 413–418, DOI:10.1073/pnas.0407677102.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00025k |
| This journal is © The Royal Society of Chemistry 2024 |