
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganic pollutant degradation for micro-molecule product emission over SiO2 layers-coated g-C3N4 photocatalysts
Yongning Ma *a,
Peihan Wua,
Mengting Kua,
Mingyuan Guo*b,
Yuhao Yanga,
Xiaolong Lia and
Haiyong Chenc
*a,
Peihan Wua,
Mengting Kua,
Mingyuan Guo*b,
Yuhao Yanga,
Xiaolong Lia and
Haiyong Chenc
aShaanxi Key Laboratory of Chemical Additives for Industry, Shaanxi University of Science and Technology, Xi'an 710021, P. R. China. E-mail: ynma@sust.edu.cn
bCollege of Chemisty and Materials Science, Weinan Normal University, Weinan 714099, P. R. China. E-mail: claremy@sina.com
cGong Cheng Ji Shu Guan Li Bu, Changqing Oil Field Branch Company, PetroChina, Xi'an 710018, P. R. China
First published on 23rd February 2024
Abstract
In this study, a SiO2 layer-coated g-C3N4 catalyst was prepared by a sol–gel method to overcome the poor adsorption ability and high recombination rate of charge carriers of pristine g-C3N4. SEM and TEM images indicated that SiO2 nanoparticles were coated on the surface of g-C3N4 nanoparticles with a layered structure and the layers were tightly contacted with g-C3N4. XRD patterns, FTIR spectra, UV-vis spectra and XPS spectra revealed that the structure of g-C3N4 was not destroyed and its photoelectric catalytic properties were not suppressed by the coating of SiO2 layers. Adsorption experiments revealed that the SiO2 layers improved the adsorption performance of g-C3N4 and their ratios were adjusted. The molecular weights of the final products of the degradation of RhB and antibiotics were at the micro-molecule level while the amount of g-C3N4 reached 1.2% of the mass fraction, which were more suitable for pollutant degradation compared with those of g-C3N4 due to its poor adsorption ability. The reason for this was likely that the SiO2 layers were not only beneficial for the adsorption of pollutants and intermediate products but also for prolonging the life time of the separated electrons and holes. Finally, active trapping experiments confirmed that both the holes and superoxide radicals were the main factors in the degradation of RhB and antibiotics, with the superoxides being the most active species.
1. Introduction
Photocatalytic organic pollutant degradation is a feasible green process to solve the environmental problems caused by the dye industry and pharmaceutical industry.1 g-C3N4 is considered a promising catalyst in this regard due to its remarkable characteristics, such as environmental friendliness, low cost, and high chemical and thermal stability.2–4 Its unique two-dimensional structure with graphitic planes constructed from tri-s-triazine units is beneficial for the modification and establishment of heterojunctions.5,6 Additionally, its suitable band gap and redox potential makes it an ideal catalyst for splitting water for H2 generation and the degradation of organic pollutants.7 However, its low specific surface area and rapid electron–hole recombination, arising from the stacked layered structure and random charge transfer, limit its adsorption property and photocatalytic activity.8 Especially in the degradation of organic pollutants, the intermediate products are uncontrollable and also harmful to humans and animals.1Aiming at controlling the intermediate products, the adsorption behaviors of catalysts are important to consider, especially for the degradation of organic pollutants and pharmaceuticals due to the complex and changeable intermediate products.9 To solve the above-mentioned problems, the adsorption behaviors and catalytic properties can be improved by surface modification and multi-porous structure fabrication approaches, such as exfoliating g-C3N4 to obtain a single/few-layered structure,10 constructing a 3D multi-porous structure,11–13 and selectively breaking the hydrogen bonds of the layered structure.14 The essence of the above-mentioned approaches involves enlarging the reactive surface area,15 prolonging the active species lifetime,16 suppressing the recombination rate of the charge carriers,17 etc. Furthermore, noble metals (Au, Ag, Pt and Pd) are also commonly used to improve the catalytic performance of g-C3N4 due to their high specific surface area and unique electronic and optical properties.18,19 Besides, metal–organic framework materials (MOFs) are used to improve the adsorption behaviors and promote the reactive species at the same time. However, the high cost and aggregation property of the framework structure limit the application scenarios and chemical stability of the materials.20 Herein, developing a photocatalyst with good chemical stability, high universality and excellent control of the intermediate products is a feasible way to solve the above issues.21
Weighing the advantages and disadvantages of numerous co-catalysts and catalyst carriers, silicon dioxide (SiO2) is often considered a good option due to its high adsorption capacity, high chemical stability, modification property, and controllable pores and morphology.22 In particular, crystalline SiO2 is employed in electronics manufacturing as both a semiconductor and electrical insulator due to its large surface area, regular arrangement, and adjustable pores.23,24 The good adsorption ability of SiO2 means it can adsorb pollutants and intermediate products effectively, and also the improved recombination rate of charge carriers can prolong the fluorescence lifetime of the charge carriers. Herein, both the high adsorption capacity and prolonged lifetime of charge carriers can be used to absorb the intermediate products from the degradation of organic pollutants and pharmaceutical for photocatalytic performance improvement.22,25
Based on the above analysis, SiO2-loaded g-C3N4 (x-CN@SiO2) was designed via a sol–gel method to control the intermediate products from emissions, meanwhile, the combination states of g-C3N4 and SiO2 were analyzed by chemical states characterization. The adsorption properties of CN@SiO2 toward organic pollutants, and their intermediate products and final products were studied. Finally, degradation rate, and active species and intermediate products controlling mechanism were deduced from trapping experiments.
2. Experimental
2.1 Preparation of the photocatalysts
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :20:4, 100 mL EtOH was separated into two parts and a half of EtOH was mixed with TEOS (denoted as liquid A), while the other part was mixed with deionized water and adjusted the pH to 2–3 with hydrochloric acid (denoted as liquid B). The obtained solutions were stirred for 30 min with magnetic stirring, and then A and B were mixed in equal volume. Next, different weights of g-C3N4 (0.4 g, 0.8 g, 1.2 g, 1.6 g and 2.0 g) were introduced to the sol–gel system separately, and then ammonium was added to the mixture with about 10 min stirring until the solution turned to thicker pulp. The gels were washed with deionized water several times to remove the anhydrous ethanol in the gels and then freeze-dried for 24 h to obtain the SiO2 layers-coated g-C3N4 samples.26,27 The prepared samples were named as x-CN@SiO2, where x is the mass fraction of g-C3N4 in the composite catalyst. A schematic of the preparation process is presented in Fig. 1.
:20:4, 100 mL EtOH was separated into two parts and a half of EtOH was mixed with TEOS (denoted as liquid A), while the other part was mixed with deionized water and adjusted the pH to 2–3 with hydrochloric acid (denoted as liquid B). The obtained solutions were stirred for 30 min with magnetic stirring, and then A and B were mixed in equal volume. Next, different weights of g-C3N4 (0.4 g, 0.8 g, 1.2 g, 1.6 g and 2.0 g) were introduced to the sol–gel system separately, and then ammonium was added to the mixture with about 10 min stirring until the solution turned to thicker pulp. The gels were washed with deionized water several times to remove the anhydrous ethanol in the gels and then freeze-dried for 24 h to obtain the SiO2 layers-coated g-C3N4 samples.26,27 The prepared samples were named as x-CN@SiO2, where x is the mass fraction of g-C3N4 in the composite catalyst. A schematic of the preparation process is presented in Fig. 1.
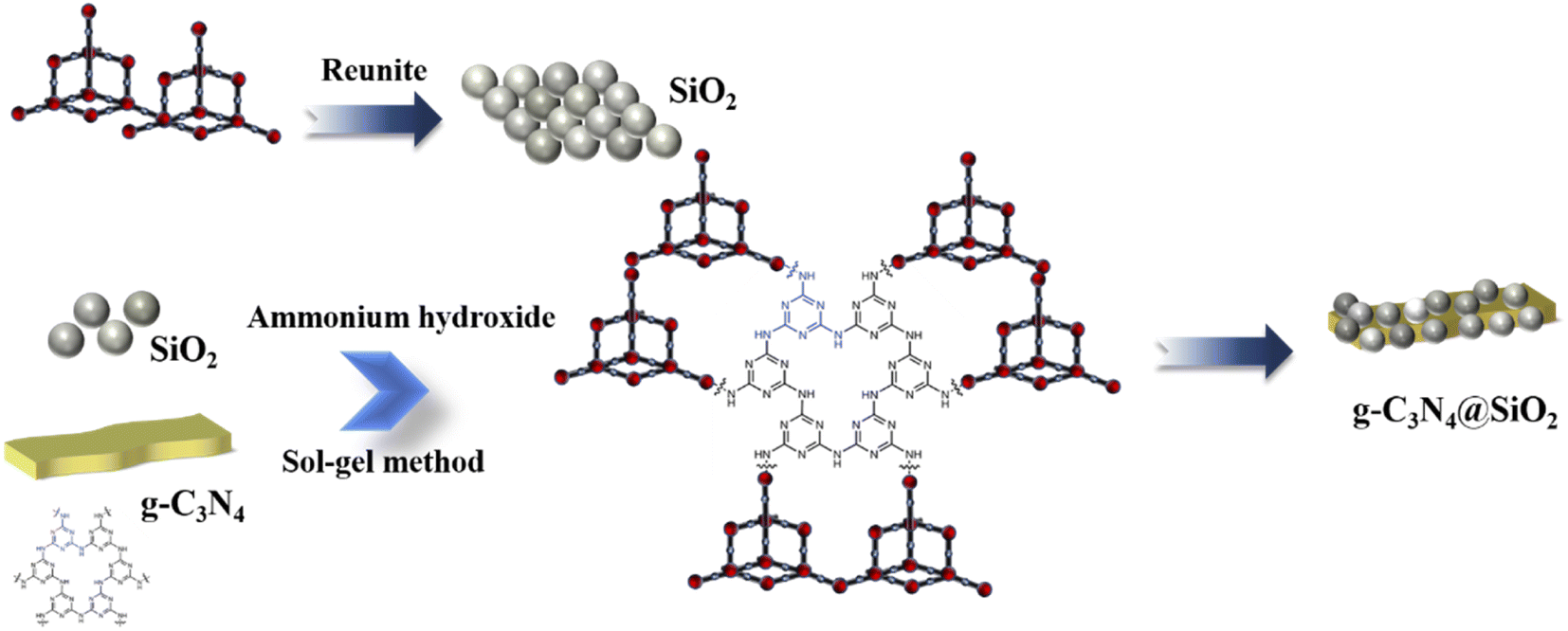 | ||
| Fig. 1 Schematic showing the preparation process for the photocatalyst samples. | ||
2.2 Characterization
The crystalline phase was determined by X-ray diffraction (XRD) using a Bruker D8 diffractometer powder diffractometer (Cu K radiation). Fourier transform infrared spectra (FTIR) were recorded using INVEIO R in the mid IR range with the ATR accessory. The morphologies were acquired using a Tecnai G2F20 transmission electron microscopy system (TEM). X-Ray photoelectron spectroscopy (XPS) measurements were obtained with a Thermo Kalpha instrument. The Brunauer–Emmett–Teller (BET) surface area was measured using an OUADRASORB SI sorption analyzer based on nitrogen adsorption–desorption isotherms at 77 K. Besides, the UV-vis diffuse reflectance spectra were recorded on a UV-2600 UV/vis/NIR spectrophotometer, with BaSO4 used as a reference. Also, the photoluminescence (PL) spectra were obtained using a Hitachi-F7000 fluorescence spectrophotometer.Photoelectrochemical measurements were performed on a CHI660E electrochemical workstation using a standard three-electrode cell with Pt wire as the counter electrode and Hg/Hg2Cl2 (in saturated KCl) as the reference electrode. A solar simulator with a 300 W Xe lamp served as a light source, and the irradiation intensity was 100 mW cm−2 at the reaction liquid level with an irradiance meter, and 0.5 M Na2SO4 solution was used as the electrolyte. The working electrode was prepared as follows: 20 mg of the ground sample was mixed with 1 mL N,N-dimethylformamide (DMF), and then 0.15 mL of the resultant suspension was uniformly dropped onto an ITO glass and then calcined at 300 °C for 1 h under a nitrogen atmosphere.
2.3 Photocatalytic organic pollutants degradation
The photocatalytic organic pollutants degradation tests were performed at ambient temperature in a thermostatic reactor.28 For each sample, 100 mg catalyst was dispersed in 100 mL (10 mg L−1) pollutant solutions (RhB or antibiotic) in a Pyrex photocatalytic reactor. The sample was kept in the dark for 20 min prior to irradiation for achieving adsorption–desorption equilibrium. After reaching equilibrium, a 300 W xenon lamp (Micro-solar 300) was used as the simulated light source, and the irradiation intensity was 100 mW cm−2 at the reaction liquid level as measured with the irradiance meter. The concentration of RhB or antibiotic was measured using a UV-2600 spectrophotometer at 10 min intervals. The degradation rate (η) of RhB or antibiotic was calculated as: η(%) = (1 − C/C0) × 100%, where C is the tested concentration of the pollutants after reaction, and C0 represents the initial concentration of the pollutants.29,302.4 Cyclic tests for the photocatalytic degradation of organic pollutants
The cyclic tests for the photocatalytic degradation of RhB and antibiotics were performed under visible light. Specifically, the experiments were carried out in a photocatalytic reactor irradiated by a solar simulator with a 300 W xenon lamp as the light source, and the irradiation intensity at the reaction liquid surface was 100 mW cm−2. RhB and antibiotics were selected as the target contaminant, and the initial concentration was 10 mg L−1. In a typical photocatalysis experiment, 100 mg photocatalyst, 100 mL RhB and antibiotics (10 mg L−1) were dispersed into a photocatalytic reactor to form a suspension. Before xenon lamp irradiation, the suspension was stirred in the dark for 10 min to establish an adsorption–desorption balance, and were then exposed to visible light. To determine the concentration of the solution, 1 mL suspension was removed at a predetermined interval and put to rest to obtain the clarified solution. The degradation products were identified using a UV-2600 spectrophotometer.2.5 Active species trapping experiments
The active species for the pollutants (RhB or antibiotics) degradation were evaluated by capture experiments.31,32 Typically, triethanolamine (TEOA), benzoquinone (BQ) and isopropanol (IPA) were used to capture the hydroxyl radicals (˙OH), superoxide radicals (˙O2−) and holes (h+), respectively. The process followed for the active species trapping experiments was similar to that for the RhB and antibiotic degradation experiments. Before adding the photocatalyst, scavengers were introduced into the pollutant solutions, respectively.3. Results and discussion
3.1 Morphological analysis
In order to study the morphology of the prepared samples, SEM, TEM and EDS mapping images of SiO2, g-C3N4 and 1.2-CN@SiO2 were obtained and are shown in Fig. 2. Fig. 2a–c are the SEM images of SiO2, g-C3N4 and 1.2-CN@SiO2, respectively. In Fig. 2a, it can be seen that the morphology of SiO2 was spherical like, with many pores in the stacked planes and a particle size less than 500 nm, which are beneficial for the adsorption of reactants.33 Fig. 2b indicates that the morphology of g-C3N4 showed a structure with layers of materials stacked on the surface and almost no pores in the two-dimensional plane, suggesting a poor adsorption ability compared with that of SiO2.34 As shown in Fig. 2c, the surfaces of the stacked g-C3N4 were covered by SiO2 nanoparticles and the covered SiO2 nanoparticles could improve the adsorption ability. Fig. 2d and e show enlarged TEM images of the 1.2-CN@SiO2 and SiO2 nanoparticles, respectively. It can be seen that the stacked g-C3N4 had no pores and SiO2 nanoparticles were coated on the surface of g-C3N4 (Fig. 2d). The TEM image of SiO2 nanoparticles (Fig. 2e) also showed that many pores existed in the planes of the coated layers. Based on the above analysis, it could be inferred that SiO2 nanoparticles covered the surface of g-C3N4 with many pores existing, which could enhance the adsorption ability of the SiO2 layers-coated g-C3N4. | ||
| Fig. 2 SEM images of (a) SiO2, (b) g-C3N4 and (c) 1.2-CN@SiO2. TEM images of (d) 1.2-CN@SiO2 and (e) SiO2, EDS mapping images of 1.2-CN@SiO2 corresponding to the (f) SEM image, and (g) element distribution maps. | ||
Furthermore, the EDS mapping images of 1.2-CN@SiO2 and corresponding C, N, O and Si elements were investigated and are shown in Fig. 2f and g, from which it could be seen that C and N elements were distributed uniformly with that of O and Si elements, which further illustrated that g-C3N4 was covered by SiO2 nanoparticles uniformly. Herein, it could be concluded that the SiO2 nanoparticles coating on g-C3N4 was successfully and the nanoparticles were distributed uniformly.
3.2 Crystal phase analysis
The XRD patterns of SiO2, g-C3N4 and x-CN@SiO2 were used to analyze the crystal phases of the samples, and the results are shown in Fig. 3a. It could be seen that g-C3N4 displayed a strong peak at 27.4°, which was generated from diffraction from the interaction of the layers, while the weak peak at 13.5° was from the interaction of the triazine rings. The wide diffraction peak at 22.5° was generated from the amorphous SiO2 nanoparticles.35 For the SiO2 layers-coated g-C3N4 samples, a peak for g-C3N4 (27.4°) could be detected due to the existence of g-C3N4, and the intensity of this peak gradually increased with the increase in g-C3N4 amounts. These results suggest g-C3N4 was successfully composited with SiO2 nanoparticles, and the crystals of g-C3N4 were not destroyed in the coating process.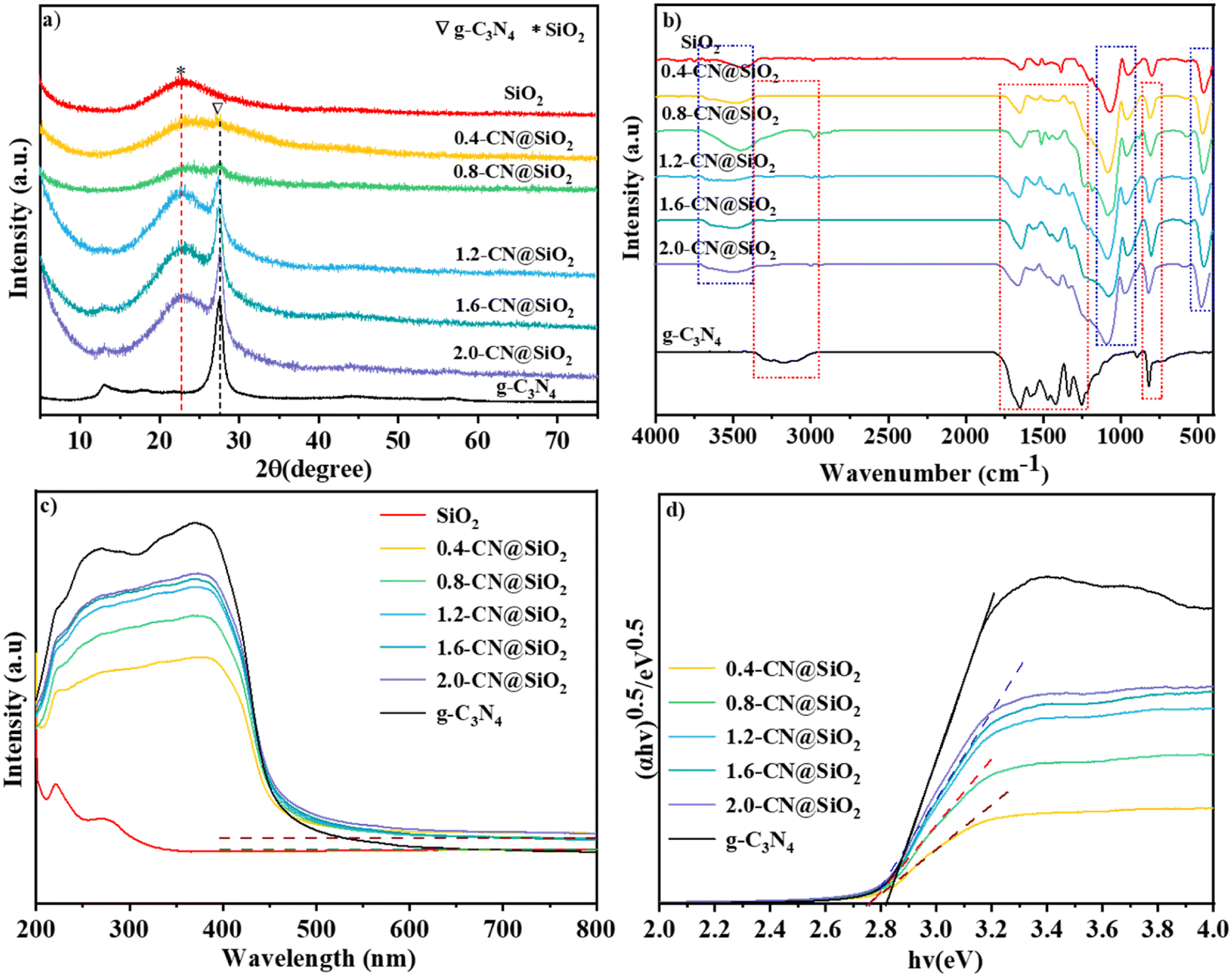 | ||
| Fig. 3 XRD patterns of (a) SiO2, g-C3N4 and composite samples, FTIR patterns of (b) SiO2, g-C3N4 and x-CN@SiO2. (c) UV-Vis and (d) bandgap patterns of SiO2, g-C3N4 and x-CN@SiO2. | ||
The FTIR spectra of SiO2, g-C3N4 and x-CN@SiO2 were used to determine the structures of the coated samples and the results are shown in Fig. 3b. For g-C3N4, the broad peak between 2800 and 3400 cm−1 corresponded to the stretching vibration of N–H at the end of the triazine ring, while the peaks between 1634 and 1230 cm−1 were related to the stretching vibration of the C–N triazine ring, and the peak around 807 cm−1 was the binding vibration of the triazine ring.36,37 For SiO2 nanoparticles, the broad peak at 3400 cm−1 corresponded to the anti-symmetric stretching vibration of the –OH group, while the peak at 1630 cm−1 was due to the –OH vibration of water adsorbed on the SiO2 surface, and the peak around 967 cm−1 was the Si–OH vibration of the SiO2 surface and that at 800 cm−1 was the Si–O vibration of SiO2. The peaks for the vibrations of C–N, triazine rings, and those of Si–O, Si–OH, and –OH vibrations of SiO2 nanoparticles were not changed in the composite catalyst, also indicating that the main molecular structures of g-C3N4 and SiO2 were not destroyed. The main difference was that the broad peak of g-C3N4 at 3000–3500 cm−1 in the composite catalyst had disappeared, which was mainly due to the connection of the amino group and SiO2 layers, leading to the disappearance of the broad peak at the edge of g-C3N4. These results show that the coated catalyst still reserved the typical features of SiO2 and g-C3N4.
The UV-vis spectra were used to identify the light adsorption abilities of SiO2, g-C3N4 and x-CN@SiO2 and the results are shown in Fig. 3c. For the SiO2 nanoparticles, almost no obvious absorption edge was observed and these showed a poor adsorption ability in the UV and visible regions, which means the introduced SiO2 nanoparticles had no effect on irradiated light. For g-C3N4, the adsorption edge was located at about 460 nm, which exhibited an intense adsorption ability in the ultraviolet regions and weak one in visible regions. For the X-CN@SiO2 samples, the absorption intensity in the visible-light region was lower than that of g-C3N4 because of the existence of SiO2 reflected irradiated light. From the band gap patterns (Fig. 3b), it could be seen the energy band of the composite was smaller than that of the pristine g-C3N4.
The XPS spectra of g-C3N4 and 1.2-CN@SiO2 were used to identify the chemical composition and chemical state of the catalyst, and the results are shown in Fig. 4. In the survey spectra (Fig. 4a), strong peaks were observed at 290, 400, and 530 eV corresponding to the typical peaks of the C 1s, N 1s, and O 1s spectra. Compared with that of g-C3N4, the new peaks at 105 and 160 eV could be attributed to the 2p spectra and 2 s spectra of the Si atom, respectively.38 Besides, the intensity of the O 1s peak was enhanced while that of N 1s peak was decreased, indicating the g-C3N4 was composited with SiO2 nanoparticles by chemical bonding and not just a simple physical contact, which accorded with the FTIR analysis in Fig. 4b. The C 1s spectrum (Fig. 4b) of g-C3N4 was similar to that of 1.2-CN@SiO2, whereby the peaks at 284.6, 287.9, and 286.0 eV could be attributed to C–C, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and N–CN, respectively, and indicated that the Si atoms did not form chemical bond with C atoms in the composite process. For the N 1s spectrum of 1.2CN@SiO2 (Fig. 4c), the peaks at 398.5, 400.1, and 404.0 eV could be attributed to CN–C, N–(C)3, and C–N–H bonds, respectively. Moreover, the peaks at 533.2 and 531.3 eV were attributed to the O atoms and oxygen vacancies in the siloxane group, respectively.
N and N–CN, respectively, and indicated that the Si atoms did not form chemical bond with C atoms in the composite process. For the N 1s spectrum of 1.2CN@SiO2 (Fig. 4c), the peaks at 398.5, 400.1, and 404.0 eV could be attributed to CN–C, N–(C)3, and C–N–H bonds, respectively. Moreover, the peaks at 533.2 and 531.3 eV were attributed to the O atoms and oxygen vacancies in the siloxane group, respectively.
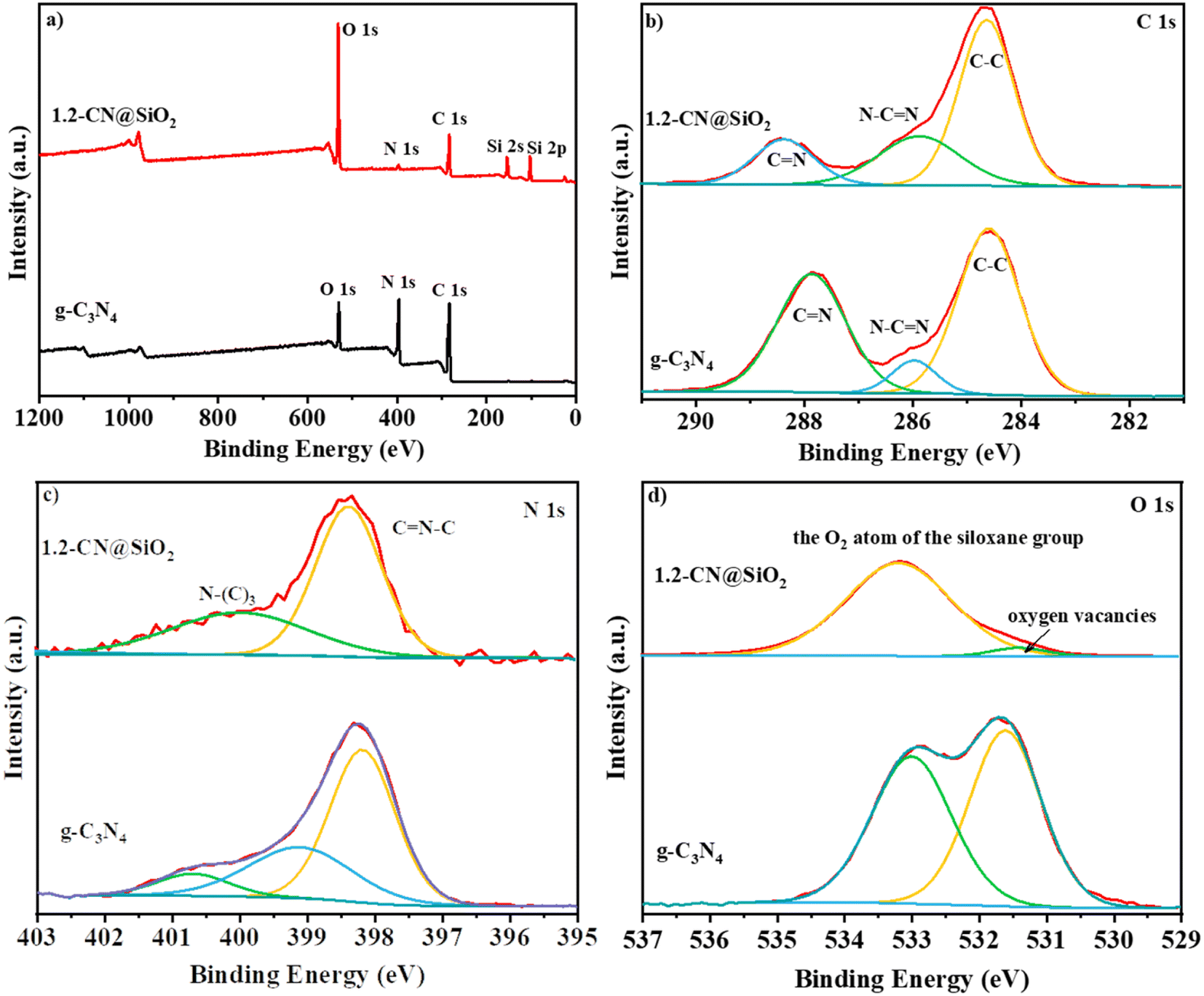 | ||
| Fig. 4 XPS (a) survey spectra, (b) C 1s spectra, (c) N 1s spectra and (d) O 1s spectra of g-C3N4 and 1.2-CN@SiO2. | ||
3.3 Photocatalytic performance analysis
Tests into the photocatalytic degradation of RhB and antibiotics were conducted to study the performance of the composite catalyst, and the results are shown in Fig. 5.39,40 Fig. 5a presents the series of adsorption and degradation curves for the photocatalysts, where it could be seen that the adsorption efficiency (−20 min to 0 min) of g-C3N4 was the worst among the investigated samples, and that the adsorption properties were improved with the increase in SiO2 layers. The highest adsorption efficiency of bare SiO2 was equal to that of 0.4-CN@SiO2 and was superior to that of the other samples, which may have been due to the SiO2 nanoparticles, as more nanoparticles could adsorb more reactants. However, the photocatalytic degradation rate of SiO2 nanoparticles could be neglected and the performance was enhanced with the increase in the amount of g-C3N4. To balance the adsorption property and degradation property, the most suitable amount of g-C3N4 was optimized at 1.2-CN@SiO2, and the ideal removal rate of RhB was within 30 min. We used a first-order kinetics model to evaluate the degradation rate of RhB, as shown in Fig. 5b. For 1.2-CN@SiO2, the rate constant was calculated to be 0.01918 min−1, which was numerically lower than that for g-C3N4. It is worth noting that the presence of SiO2 nanoparticle layers improved the adsorption performance of the composite catalyst,41,42 and helped realize the coordination between the adsorption and degradation processes in the photocatalytic degradation of organic pollutants. The intermediate products of RhB degradation on g-C3N4 and 1.2-CN@SiO2 were investigated by liquid chromatography-mass spectrometry and the results are shown in Fig. 5c and d, respectively. After 30 min degradation, the charge mass ratio of the intermediate products on g-C3N4 (Fig. 5c) ranged from about 200 to 720, meanwhile, there was a much higher charge mass ratio generation of intermediate products. Compared with that of 1.2-CN@SiO2 (Fig. 5d), the charge mass ratio of the intermediate products was much lower (ranging from about 200 to 370).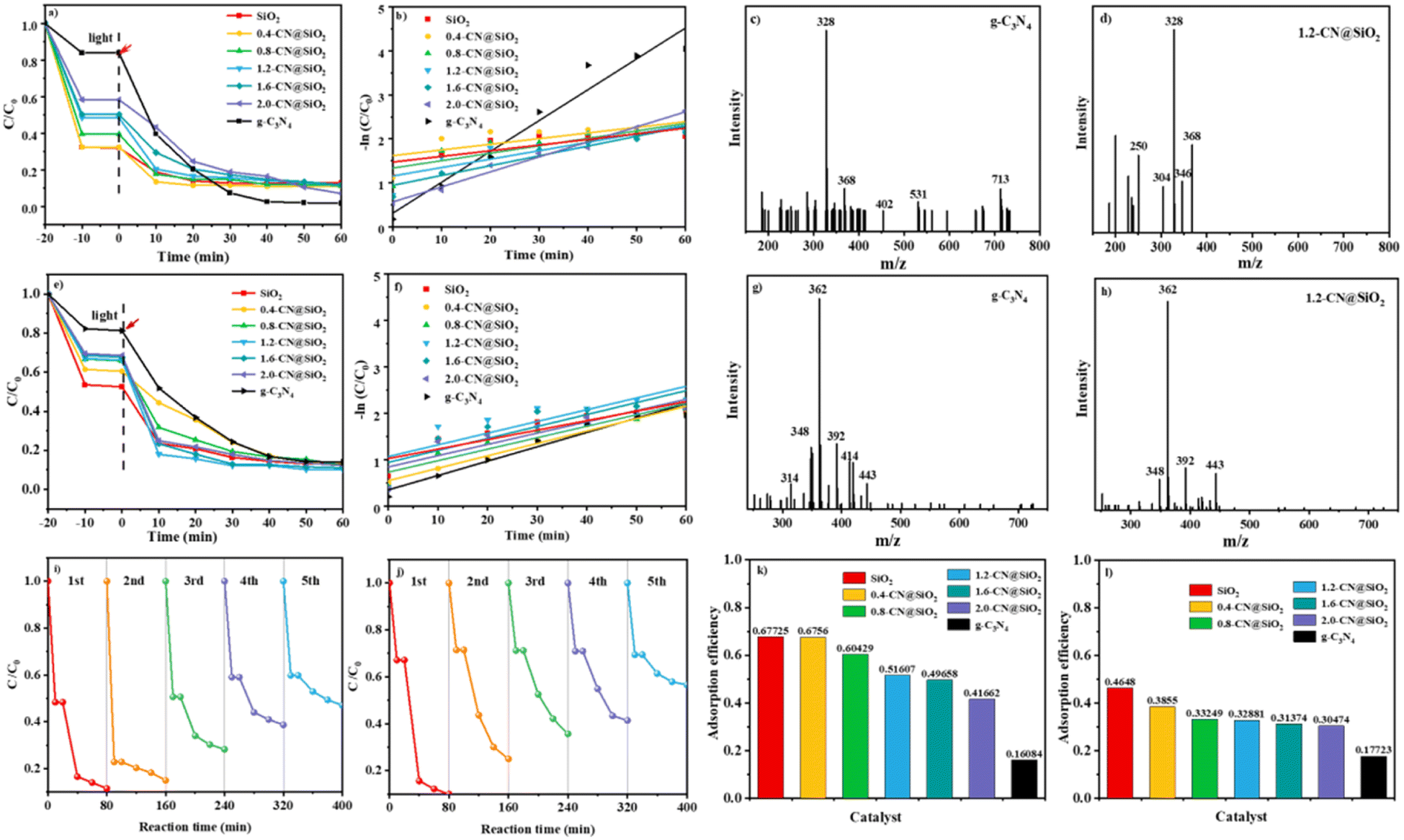 | ||
| Fig. 5 RhB degradation of the (a) instantaneous concentration, and (b) kinetic plot of RhB dye degradation by the composite photocatalysts. (c and d) Mass spectra of the degradation products with g-C3N4 and 1.2-CN@SiO2 as catalysts and 30 min irradiation; antibiotic degradation showing the (e) instantaneous concentration, (f) kinetic plot for antibiotic dye degradation by the composite photocatalysts (g and h) mass spectra of the degradation products with g-C3N4 and 1.2-CN@SiO2 as catalysts and 30 min irradiation. (i and j) Recycled photodegradation of RhB and antibiotic dyes over the optimal 1.2-CN@SiO2 photocatalysts. (k and l) Adsorption efficiency of the catalyst for RhB and antibiotics in 10 minutes. | ||
The performances of the composite catalysts were further studied in the degradation of antibiotics based on the excellent degradation performance of RhB (Fig. 5e and g).43 It was obvious that 1.2-CN@SiO2 still exhibited the best photocatalytic degradation efficiency, reaching 89% within 30 min, and the Kap was 0.02523 min−1 for 1.2-CN@SiO2. According to the liquid chromatography-mass spectrometry results, the intermediate products from antibiotics degradation on g-C3N4 and 1.2-CN@SiO2 were investigated, and the results are shown in Fig. 5g and h, respectively. After degradation for 30 min, 1.2-CN@SiO2 had a much smaller charge mass ratio than g-C3N4.
The stability of the 1.2-CN@SiO2 photocatalyst was tested by cyclic experiments. As shown in Fig. 5i and j, after cycling for 5 cycles under the same conditions, the degradation efficiency of 1.2-CN@SiO2 decreased slightly, indicating that the 1.2-CN@SiO2 photocatalyst had favorable stability and reusability. The removal of organic pollutants by SiO2 nanoparticles was mainly through adsorption, and the removal efficiency of the pollutants reached 46–68% after the end of the dark adsorption (Fig. 5k and l); whereas pristine C3N4 had a small specific surface area (10.24 m2 g−1) (Table 1) and poor molecular adsorption of organic pollutants, by contrast. Due to the introduction of SiO2, the composite catalyst showed an excellent adsorption effect. After the photocatalytic experiments, the composite catalyst displayed an 85% degradation efficiency for organic pollutants, realizing the adsorption–photocatalytic collaborative regulation of organic pollutants and their intermediate products.44
| Sample | Surface area (m2 g−1) | Total pore volume (cm3 g−1) | Average pore diameter (nm) |
|---|---|---|---|
| SiO2 | 775.01 | 0.53 | 3.94 |
| 1.2-CN@SiO2 | 529.17 | 0.38 | 4.00 |
| g-C3N4 | 10.24 | 0.07 | 3.90 |
3.4 Photocatalytic mechanism analysis
The particle size is a key factor in catalysis and from the results shown in Fig. 6a it can be seen the sizes of SiO2, g-C3N4 and 1.2-CN@SiO2 were different. The particle size of SiO2 was centered around 300 nm while that of g-C3N4 was about 600 nm, and the particle size even increased to 600–1200 nm. To study the improved adsorption properties of the samples, the specific surface area and pore size of the photocatalysts are other factors to consider for photocatalysts. Consequently, nitrogen adsorption–desorption isotherms and the hysteresis loops of SiO2, g-C3N4 and 1.2-CN@SiO2 were investigated and the results are shown in Fig. 6b. The isothermal curve of g-C3N4 belonged to a H3 hysteresis loop, indicating that it was a slit formed by the accumulation of lamellar particles, while the isothermal curve of SiO2 belonged to an H4 hysteresis loop, indicating it was due to the mutual accumulation of microspheres on the surface of SiO2. Further, 1.2-CN@SiO2 displayed an H4 hysteresis loop in the low and medium pressure regions, while it showed an H3 hysteresis loop in the high pressure region.45,46 The accumulation of slit holes on the surface of microspheres, and then the accumulation of slit holes between microspheres and lamellar particles. The specific surface areas of each catalyst calculated through the BET equation are shown in Table 1. Obviously, the specific surface area of SiO2 was 33 times higher than that of g-C3N4 due to its porous structure. The specific surface area of the composite catalyst exhibited a regular decrease due to the possibility of incomplete filling of the SiO2 pores with g-C3N4. However, the specific surface area of the composite catalyst was greater than that of pristine g-C3N4, which provides the possibility for adsorbing macromolecular organic pollutants. For SiO2 and 1.2-CN@SiO2, the pore-size distribution was between 2 and 5 nm, due to the fact that SiO2 particles were uniformly sized microspheres, and so the cumulative pore-size distribution was calculated according to the BET characterization with the BJH fitting method (Fig. 6c and Table 1). The main pore-size distribution of the catalysts was between 3.5 and 4.5 nm for the composite samples, where the average pore volume of g-C3N4 was larger due to its uneven sheet size and wide pore-size distribution. It was shown that the composite catalyst had a greater adsorption capacity for nitrogen with the same quality catalyst, so the composite catalyst had a better adsorption capacity for organic pollutants than pristine g-C3N4.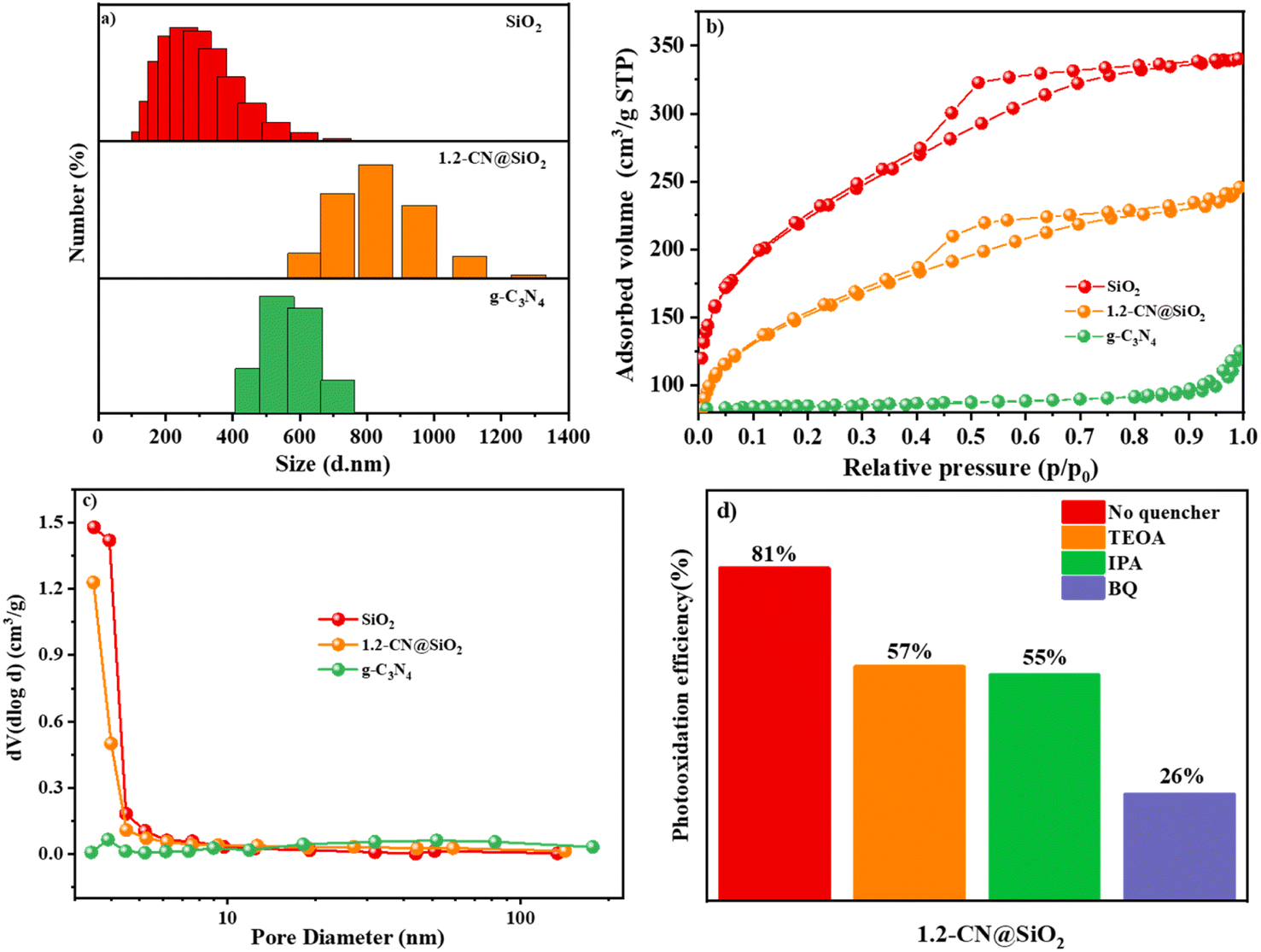 | ||
| Fig. 6 Grain size distribution map (a), N2 adsorption–desorption curves (b), pore-size distributions (c) of SiO2, g-C3N4 and 1.2-CN@SiO2. Photooxidation efficiency of RhB by radical trapping reagents treated with 1.2-CN@SiO2 (d). | ||
To further evaluate the contribution of various radicals, 1.2-CN@SiO2 was treated with 1,4-benzoquinone (BQ), isopropyl alcohol (IPA), and disodium triethanolamine (TEOA) to remove ˙O2−, ˙OH, and photogenerated holes (h+) before measuring the photooxidation efficiency, respectively. As shown in Fig. 6d, the ˙O2− scavenger had the most significant inhibitory effect, with the efficiency dropping about 70%, while the second best inhibitor was h+, which reduced the efficiency about 45%. Although the photooxidation efficiency fell substantially for 1.2-CN@SiO2, the ˙OH radical scavenger caused the lowest decrease (43%). These results indicated that ˙O2− was the most active species for RhB degradation while ˙OH and h+ were had equal effects.
According to the double-layer capacitance (Cdl) analysis, the results for the electrochemically active surface area (ECSA) analysis from the cyclic voltammetry (CV) tests are shown in Fig. 7a and d, where the ECSA reflects the number of catalytic sites during the reaction, and a higher value of Cdl means more catalytic sites are exposed in the reaction solution system.47,48 The CV curves under different scanning rates for SiO2, 1.2-CN@SiO2 and g-C3N4 of 10–140 mV s−1 are shown in Fig. 7(a)–(c). From Fig. 7d, it can be seen that the Cdl of 1.2-CN@SiO2 was lower than that of SiO2 but greater than that of g-C3N4, indicating that 1.2-CN@SiO2 had more sites exposed than g-C3N4. The linear slope was equivalent to 10−3 times Cdl based on the linear fitting of the scanning rate by the capacitance current density of the non-Faraday interval double layer. As shown in Fig. 7d, 1.2-CN@SiO2 had a Cdl of 25.75 μF cm−2, which was much higher than that of g-C3N4 (20.42 μF cm−2). The above analysis shows that the SiO2-coated g-C3N4 photocatalysts could induce a greater ECSA, resulting in a higher exposed active surface area for reaction with the electrolyte, and enabling an improved mineralization rate for the degradation of organic pollutants.
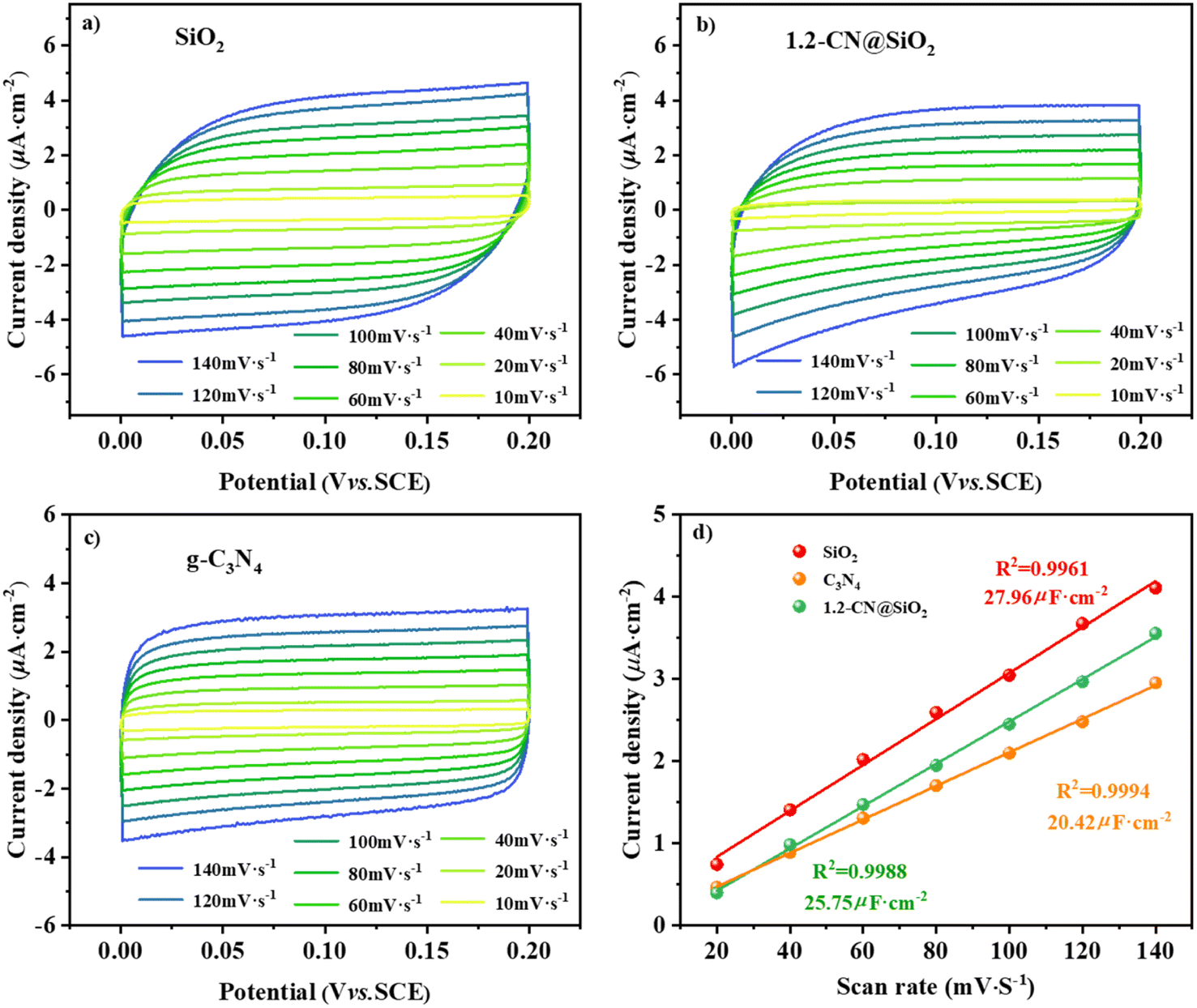 | ||
| Fig. 7 CV curves at different scan rates for (a) SiO2, (b) C3N4, and (c) 1.2-CN@SiO2. (d) Linear relation between the current density and scan rate. | ||
The current densities generated from pristine g-C3N4, SiO2 and x-CN@SiO2 under simulated illumination are shown in Fig. 8. For x-CN@SiO2, the composite catalyst still exhibited a certain current intensity and did not lose current intensity due to the loading of SiO2, indicating that the excitation electrons generated by the composite catalyst under illumination were mainly provided by g-C3N4. Also, the electrons of the composite catalyst could still be transported in the internal plane of the material (Fig. 8a).49,50 The impedance spectra further illustrated the resistance to internal electron migration between the composite catalysts. The radius is slightly larger of spectra in arc radius between the EIS Nyquist diagram of pristine g-C3N4, which is due to the SiO2 layers coated on surface of g-C3N4 consistent with pristine g-C3N4, which suppressed the migration rate of charge carriers (Fig. 8b).
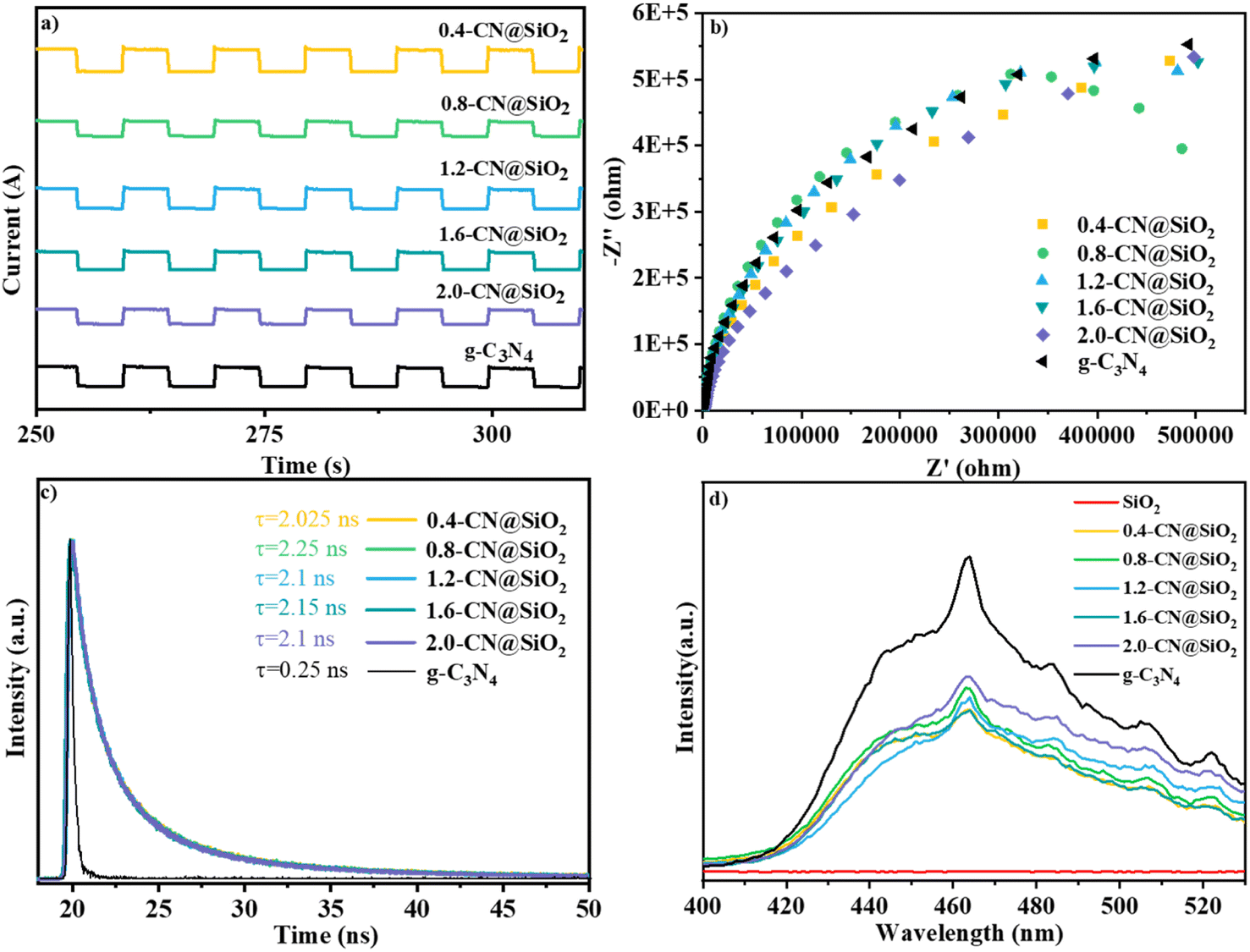 | ||
| Fig. 8 (a) Transient photocurrent curves of g-C3N4 and x-CN@SiO2, (b) EIS spectra of g-C3N4 and x-CN@SiO2, (c) transient fluorescence spectra of g-C3N4 and x-CN@SiO2, (d) PL spectra of g-C3N4 and x-CN@SiO2. | ||
The fluorescence lifetime is the average residence time of electrons for returning from the excited state to the ground state. Fig. 8c shows the fluorescence lifetime curves of g-C3N4 and the composite catalysts, and the corresponding average lifetime values. The fluorescence lifetime of pristine g-C3N4 was 0.25 ns, while the fluorescence lifetime of the composite catalyst was 8.4 times higher. This was due to the fact that under the excitation of light, the photogenerated electrons migrated to the surface of SiO2 and were consumed by water, resulting in an extended time for the electrons to return to the ground state. Therefore, the average lifetime for the composite catalyst was longer than for the pristine g-C3N4.
According to the PL spectra, the composite catalyst had wide and strong fluorescence emission peaks. Under light excitation, the composite catalyst could generate electrons and holes, and the peak strengths of the composite catalyst were lower than those for the pristine g-C3N4, indicating that the recombination rate of photogenerated electrons and holes was lower for the composite catalyst than for the pristine g-C3N4, indicating that the loading of SiO2 caused its electrons and holes to undergo catalytic reactions (Fig. 8d). The figure shows that SiO2 had no fluorescence emission peak, indicating that the composite catalyst could generate photogenerated electrons and holes mainly due to g-C3N4.
Based on the above experimental data and analysis, a reasonable process for the degradation of organic pollutants by the composite catalysts is summarized in Fig. 9. After the composite catalyst is exposed, the organic pollutants (RhB, antibiotic drugs) are adsorbed onto the porous surfaces of the SiO2 spheres due to the large specific surface area. Second, the adsorbed organic pollutants permeate into the internal section and contact with g-C3N4. Under light irradiation, the photogenerated electrons and oxidation holes are separated and the electrons migrate to the surface of g-C3N4 to participate in redox reactions. The migrated electrons contact with oxygen for superoxide radical generation, and the adsorbed organic pollutants react with superoxide radicals leading to the formation of complex macromolecular intermediate products. Fortunately, the coated SiO2 layers can adsorb the intermediate products to further react with the superoxide radicals that circulate until small molecular products are generated and escape from the pores of the composite catalysts. This process can be described by the formulas below.
| g-C3N4 + hv = e− + h+ | (1) |
| e− + O2 = ˙O2− | (2) |
| h+ + OH− → ˙OH | (3) |
| RhB + h+ + ˙O2− → CO2 + H2O | (4) |
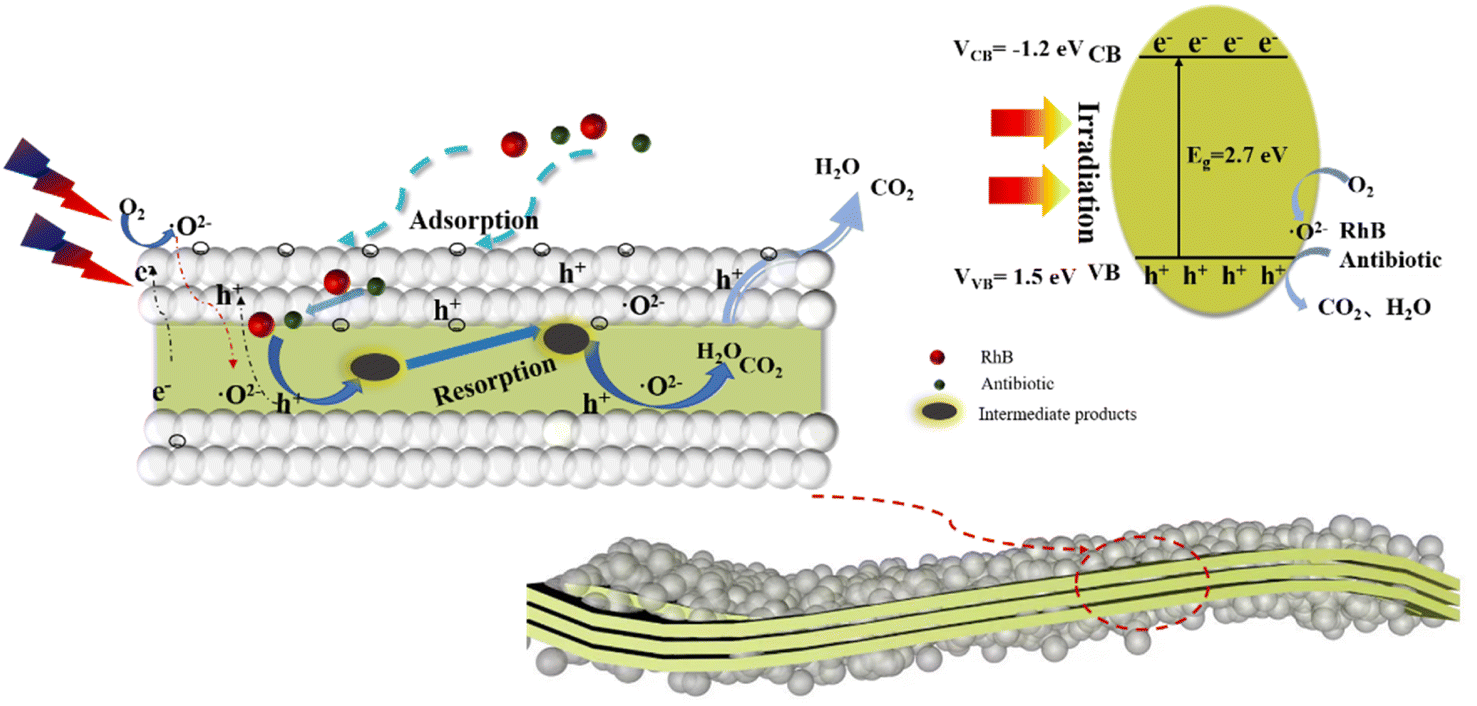 | ||
| Fig. 9 Photocatalytic mechanism of micro-molecule products emission over the SiO2 layers-coated g-C3N4 photocatalyst. | ||
4. Conclusion
A SiO2 layers-coated g-C3N4 composite photocatalyst was successfully prepared by a sol–gel method, which demonstrated an increased specific surface area and good adsorption property. The improved performance of the as-fabricated 1.2-CN@SiO2 could be ascribed to the SiO2 layers, which were beneficial for not only the adsorption of pollutants and intermediate products but also for prolonging the life time of the charge carriers. In the photooxidation mechanism, ˙O2− played a crucial role in pollutants degradation and the effects of ˙OH and h+ equally contributed as well. The coated SiO2 layers could adsorb the intermediate products to further react with superoxide radicals that circulate until small molecular products were generated and escaped from the pores of the composite catalysis.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the National Natural Science Foundation of China [No. 22208199 and 22008147]; Natural Science Basic Research Program of Shaanxi Province [No. 2021JQ-826]; Key Laboratory Fund of Education Department of Shaanxi [No. 20JS048]; Scientific Research Program Project of Science and Technology Association of Shaanxi [No. 20200611] and Scientific Research Program Project of Weinan Normal University [No. 20RC+9].References
- D. Masekela, N. C. Hintsho-Mbita, B. Ntsendwana and N. Mabuba, ACS Omega, 2022, 7, 24329–24343 CrossRef CAS PubMed.
- Y. Fan, Y.-N. Yang and C. Ding, J. Inorg. Organomet. Polym. Mater., 2021, 31, 4722–4730 CrossRef CAS.
- H. Zhang, M. Liu, X. Zhang, B. Yuan, P. Wu, C. Liu and W. Jiang, Chem. Eng. J., 2022, 438, 135644 Search PubMed.
- Z.-J. Chen, H. Guo, H.-Y. Liu, C.-G. Niu, D.-W. Huang, Y.-Y. Yangand and J.-C. Li, Chem. Eng. J., 2022, 438, 135471 CrossRef CAS.
- X. Jiang, Q. Li, W. Xia, Z. Huang, X. Wu, H. Zhaoand and H. Shen, Appl. Surf. Sci., 2022, 589, 153048 CrossRef CAS.
- B. Zhu, L. Zhang, B. Cheng and J. Yu, Appl. Catal., B, 2018, 224, 983–999 CrossRef CAS.
- M. Ren, X. Zhang, Y. Liu, G. Yang, L. Qin, J. Mengand and Y. Yang, ACS Catal., 2022, 12, 5077–5093 CrossRef CAS.
- X.-J. Wang, W.-Y. Yang, F.-T. Li, Y.-B. Xue, R.-H. Liu and Y.-J. Hao, Ind. Eng. Chem. Res., 2013, 52, 17140–17150 CrossRef CAS.
- R. Rajendran, S. Vignesh, V. Raj, B. Palanivel, A. M. Ali, M. A. Sayed and M. Shkir, J. Alloys Compd., 2022, 894, 162498 CrossRef CAS.
- X. Yuan and W. Li, Appl. Clay Sci., 2017, 138, 107–113 CrossRef CAS.
- H. Gao, R. Cao, S. Zhang, H. Yang and X. Xu, ACS Appl. Mater. Interfaces, 2019, 11, 2050–2059 CrossRef CAS PubMed.
- W. Li, X.-S. Chu, F. Wang, Y.-Y. Dang, X.-Y. Liu, X.-C. Wang and C.-Y. Wang, Appl. Catal., B, 2021, 288, 120034 CrossRef CAS.
- X. Yuan, C. Zhou, Y. Jin, Q. Jing, Y. Yang, X. Shenand and A. K. Du, J. Colloid Interface Sci., 2016, 468, 211–219 CrossRef CAS PubMed.
- C. Yang, S. Zhang, Y. Huang, K. Lv, S. Fang, X. Wuand and J. Fan, Appl. Surf. Sci., 2020, 505, 144654 CrossRef CAS.
- J. Shi, L. Mao, C. Cai, G. Li, C. Cheng, B. Zhengand and G. Żyła, Catal. Sci. Technol., 2020, 10, 5896–5902 RSC.
- M. Qiao, L. Fu and D. Barcelo, Process Saf. Environ. Prot., 2022, 159, 376–381 CrossRef CAS.
- K. Kočí, M. Reli, I. Troppová, M. Šihor, J. Kupková, P. Kustrowski and P. Praus, Appl. Surf. Sci., 2017, 396, 1685–1695 CrossRef.
- J. Xue, S. Ma, Y. Zhou, Z. Zhang and M. He, ACS Appl. Mater. Interfaces, 2015, 7, 9630–9637 CrossRef CAS PubMed.
- M. Li, J. Zhang, Y. He, X. Zhang, Z. Cui, P. Fuand and X. Pang, Polym. Chem., 2022, 13, 1022–1030 RSC.
- J. Burner, L. Schwiedrzik, M. Krykunov, J. Luo, P. G. Boyd and T. K. Woo, J. Phys. Chem. C, 2020, 124, 27996–28005 CrossRef CAS.
- H. Huang, B. Pradhan, J. Hofkens, M. B. J. Roeffaers and J. A. Steele, ACS Energy Lett., 2020, 5, 1107–1123 CrossRef CAS.
- H. Gomaa, M. T. Hussein, M. M. Motawea, A. M. Aboraia, M. F. Cheira, M. T. Alotaibiand and H. M. Ali, Colloids Surf., A, 2022, 644, 128811 CrossRef CAS.
- Q. Zhao, S. Xie, H. Wang, L. Yang, X. Mei and Y. He, Mater. Sci. Semicond. Process., 2022, 146, 106702 CrossRef CAS.
- E. Nadimi, A. Rahimi, S. Masoumi and M. Schreiber, Thin Solid Films, 2022, 746, 139116 CrossRef CAS.
- A. G. El-Shamy, Polymer, 2022, 242, 124579 CrossRef CAS.
- L. Peng, R.-R. Zheng, D.-W. Feng, H. Yu and X.-T. Dong, Arabian J. Chem., 2020, 13, 4275–4285 CrossRef CAS.
- B. Jiang, T. Wang, Y. Cheng, F. Liao, K. Wu and M. Shao, ACS Appl. Mater. Interfaces, 2018, 10, 39161–39167 CrossRef CAS PubMed.
- Z. Ma, P. Zhou, L. Zhang, Y. Zhong, X. Sui, B. Wangand and Z. Mao, Chem. Phys. Lett., 2021, 766, 138335 CrossRef CAS.
- D. Wang, Y. Li, B. Yu, H. Li, W. Jiang, X. Dengand and G. Che, Adv. Powder Technol., 2021, 32, 1653–1662 CrossRef CAS.
- X. Yang, L. Zhang, D. Wang, Q. Zhang, J. Zeng and R. Zhang, RSC Adv., 2021, 11, 30503–30509 RSC.
- J. Shi, C. Ju, J. Nie, H. Wang, T. Yang, K. Puand and T. Zhao, ECS J. Solid State Sci. Technol., 2023, 11, 123012 CrossRef.
- T. Chankhanittha and S. Nanan, J. Colloid Interface Sci., 2021, 582, 412–427 CrossRef CAS PubMed.
- B. Pan, Y. Wu, B. Rhimi, J. Qin, Y. Huang, M. Yuan and C. Wang, J. Energy Chem., 2021, 57, 1–9 CrossRef CAS.
- Y. Yu and H. Huang, Chem. Eng. J., 2023, 453, 123011 Search PubMed.
- V. V. Shanbhag, S. C. Prashantha, C. R. Ravi Kumar, P. Kumar, B. S. Surendra, H. Nagabhushanaand and Y. G. Krupanidhi, Inorg. Chem. Commun., 2021, 134, 108960 CrossRef CAS.
- Y. Yang and Z. Bian, Sci. Total Environ., 2021, 753, 141908 CrossRef CAS PubMed.
- S. Lu, T. Wu, Y. Liu, H. Luo, F. Jiang, X. Nie and H. Chen, J. Alloys Compd., 2022, 911, 164980 CrossRef CAS.
- M. Guo, Y. Ma, Z. Liu, D. Wang, Y. Yang, X. Li and E. Liu, J. Catal., 2024, 430, 115332 CrossRef CAS.
- T. Yu, W. Wu, L. Liu, C. Gao and T. Yang, Ceram. Int., 2020, 46, 9567–9574 CrossRef CAS.
- V. V. Shanbhag, S. C. Prashantha, T. R. S. Shekhar, H. Nagabhushana, R. Naik, K. M. Girishand and D. S. Prasanna, Ceram. Int., 2021, 47, 10346–10354 CrossRef CAS.
- L. Li, W. Cao, C. Liang, X. Shi and C. Wang, J. Alloys Compd., 2023, 960, 170580 CrossRef CAS.
- S. Ji, Y. Yang, Z. Zhou, X. Li and Y. Liu, J. Water Process Eng., 2021, 40, 101804 CrossRef.
- T. Bi, Z. Du, S. Chen, H. He, X. Shen and Y. Fu, Appl. Surf. Sci., 2023, 614, 156240 CrossRef CAS.
- L. Meng, C. Zhao, T. Wang, H. Chu and C.-C. Wang, Sep. Purif. Technol., 2023, 313, 123511 CrossRef CAS.
- J. Yu, J. Lei, L. Wang, C. Guillard, J. Zhang, Y. Liu and M. Anpo, Res. Chem. Intermed., 2019, 45, 4237–4247 CrossRef CAS.
- D. Lu, H. Fan, K. K. Kondamareddy, H. Yu, A. Wang, H. Haoand and J. Shen, ACS Sustain. Chem. Eng., 2018, 6, 9903–9911 CrossRef CAS.
- Q. Zhang, Z. Wang, Y. Song, J. Fan, T. Sun and E. Liu, J. Mater. Sci. Technol., 2024, 169, 148–157 CrossRef.
- T. Sun, C. Li, Y. Bao, J. Fan and E. Liu, Acta Phys.-Chim. Sin., 2023, 39, 2212009 Search PubMed.
- W. Li, W. Li, Z. Guo, Y. Song, S. Tang, Y. Maand and Q. Wang, ACS Appl. Mater. Interfaces, 2021, 13, 52560–52570 CrossRef CAS PubMed.
- M. Jafarpour, F. Feizpour, A. Rezaeifard, N. Pourmorteza and B. Breit, Inorg. Chem., 2021, 60, 9484–9495 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |