
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC(sp3)–H cyclizations of 2-(2-vinyl)phenoxy-tert-anilines†
Petra Dunkel *a,
Dóra Bogdána,
Ruth Demea,
Ádám Zimbera,
Veronika Ballayováab,
Eszter Csizmadiaa,
Bence Kontraac,
Eszter Kalydia,
Attila Bényeid,
Péter Mátyusae and
Zoltán Mucsi*cfg
*a,
Dóra Bogdána,
Ruth Demea,
Ádám Zimbera,
Veronika Ballayováab,
Eszter Csizmadiaa,
Bence Kontraac,
Eszter Kalydia,
Attila Bényeid,
Péter Mátyusae and
Zoltán Mucsi*cfg
aDepartment of Organic Chemistry, Semmelweis University, Hőgyes Endre utca 7, H-1092 Budapest, Hungary. E-mail: dunkel.petra@semmelweis.hu
bDepartment of Chemical Drugs, Masaryk University, Palackého 1946/1, 612 00 Brno, Czech Republic
cDepartment of Biological Chemistry, Brain Vision Center, Liliom utca 43-45, H-1094 Budapest, Hungary
dInstitute of Physical Chemistry, University of Debrecen, Egyetem tér 1, H-4010 Debrecen, Hungary
eUniversity of Veterinary Medicine, István utca 2, H-1078 Budapest, Hungary
fDepartment of Chemistry, Femtonics Ltd, Tűzoltó utca 59, H-1094 Budapest, Hungary. E-mail: zmucsi@femtonics.eu
gInstitute of Chemistry, University of Miskolc, Egyetem út 1, H-3515 Miskolc, Hungary
First published on 23rd May 2024
Abstract
1,5-hydride transfer-triggered cyclization reactions offering a robust method for C(sp3)–C(sp3) coupling and the synthesis of e.g. tetrahydroquinolines have been thoroughly investigated in the literature. Catalysts allowing milder reaction conditions or the development of enantioselective processes were important recent contributions to the field, as well as the studies on subtrates with oxygen or sulfur heteroatoms (besides the originally described nitrogen heterocycles). In a series of studies, we focused on expanded, higher order H-transfers/cyclizations by positioning the interacting substituents on distanced rings. Cyclizations of appropriately functionalized biaryl and fused bicyclic systems led to 7–9 membered rings. In the frame of this research, we set out to study the feasibility of the cyclization and the factors affecting it by in silico methods. The conclusions drawn from computational studies were complemented by cyclization screens on 2-(2-vinyl)phenoxy-tert-anilines and their CH2-expanded analogues, the results of which are presented here. Besides isolating the expected oxazonine products in several cases, we also observed a unique dimer formation, leading to an interesting 5-6-5 ring system.
Introduction
C(sp3)–H bond functionalization and C(sp3)–C(sp3)-cross coupling reactions (i.e. transformations involving inactivated C–H bonds) have long been at the forefront of interest in the field of synthetic organic chemistry. These methods allow access to complex structures, particularly in the field of total synthesis of natural products or medicinal chemistry.1–3 For instance, an elegant one-step approach leading to indolizidine derivatives, based on ‘inverse hydride shuttle catalysis’ was recently presented by the Maulide group.4 Of the various C–H bond functionalizations of amines,5 cyclizations of tertiary anilines with different double bonds in the ortho position were historically classified as variants of the ‘tert-amino effect’.6–9 A version of these reactions, the internal redox transformations involving [1,n]-H transfer and cyclization of tertiary anilines with an ortho-vinyl group (1) have been addressed in detail by several groups, particularly by Reinhoudt et al. (Scheme 1).10–15 In these reactions, typically a thermodynamically favoured 6-membered ring (often a tetrahydroquinoline; 2) is obtained with the formation of a new C–C bond. The reaction proceeds via several elementary steps and transition states (e.g. TS1), without the need for a transition metal catalyst. Consequently, this reaction type is of special interest in the field of synthetic chemistry.16–18 Besides detailed synthetic studies, the cyclization was judiciously used in medicinal chemistry as well, for the preparation of antibacterial DNA gyrase inhibitors.19 Moreover, the synthetic utility of the reaction and its significant potential was recently demonstrated also by the preparation of unsymmetric julolidines from 8-vinyl-tetrahydroquinolines via [1,5]-H transfer/cyclization.20 The tricyclic julolidine is a privileged scaffold in the design of fluorescent probes, photoswitches or photolabile protecting groups,21–24 therefore these fields could also benefit from straightforward synthetic approaches towards julolidine building blocks. Recently, Lewis- and Bronsted-acid catalysed variants of the cyclization were developed,25–27 allowing milder reaction conditions (of note, the first variants used typically higher temperatures). However, both the demand for an electron withdrawing group (EWG) and the preference of the six-member ring product formation limits the synthetic perspectives of this variant.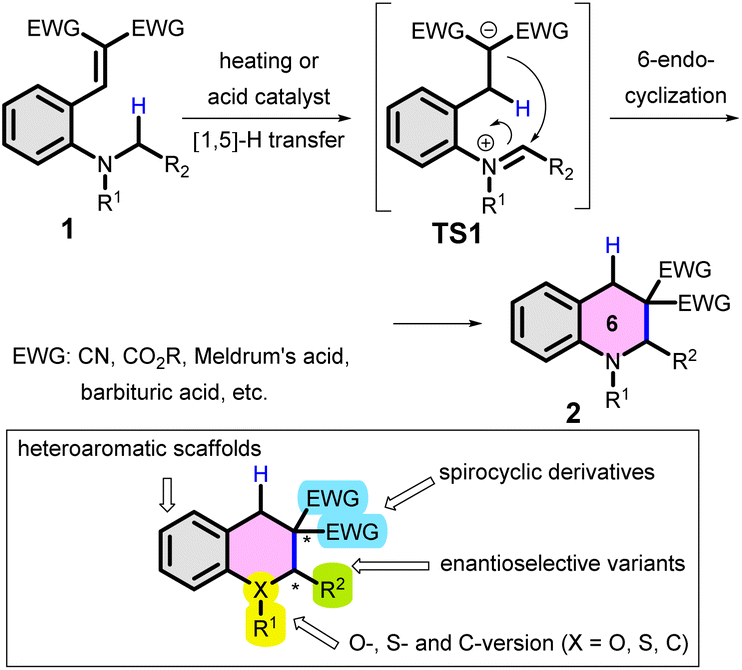 | ||
| Scheme 1 Typical [1,5]-H transfer/cyclization of 2-vinyl-N,N-dialkylanilines (1; “tert-amino effect”) and its potential variants. | ||
As further important developments, highly enantioselective cyclizations (as I → II → III) and sequential hydride transfer/cyclization protocols (as IV → V → VI) were disclosed (Scheme 2).27–32 The oxygen version of the reaction (cyclization of ortho-vinylaryl alkyl ethers) allows a straightforward access to dihydrobenzopyran derivatives.33,34 Of note, the cyclization is operating also with 2-(2-(alkylthio)benzylidene)malonates (i.e. with a sulphur analogue), leading to thiochromanes.35 Although the heteroatom has an essential role in the stabilization of the carbocation formed by the hydride transfer, recently the carbo-version of the reaction was developed. The [1,5]-hydride transfer/cyclization sequences of appropriately substituted aliphatic or benzylic substrates afford various tetrahydronaphthalene systems.36–38 Higher order H-transfers/cyclizations (as VII → VIII) could be envisaged by positioning the interacting substituents on different rings (Scheme 3). Cyclizations of fused bicyclic (3 → 4) and biaryl systems (5 → 6, 7 → 8) with the interacting dialkylamino and vinyl groups in ortho, ortho′ or ortho and peri positions furnished the respective 7–9 membered rings, besides the formation of novel zwitterionic phenantridium or benzo[d,e]quinolinium systems through a Michael type addition in several cases.39,40 Starting from triaryl derivatives, 10-membered azecine rings could be attained (12 → 13).41 As examples of nondirectly connected biaryl systems, where a sigmatropic hydrogen transfer could a priori be excluded, the cyclization of biphenyl derivatives bridged with a methylamino-N-methyl group (9 → 10) or an oxygen between the phenyl rings (14 → 15) were studied.42,43 Although in the former case the formation of a 6-membered ring was observed instead (9 → 11), the cyclization of the biaryl-ether provided the 9-membered oxazonine ring (14 → 15), the structure of which was confirmed by X-ray crystallography as well. Of note, several of these more complex ring systems have natural analogues and could serve as interesting scaffolds for medicinal chemistry. Therefore, finding novel approaches for their preparation could be also of significant practical value.44,45
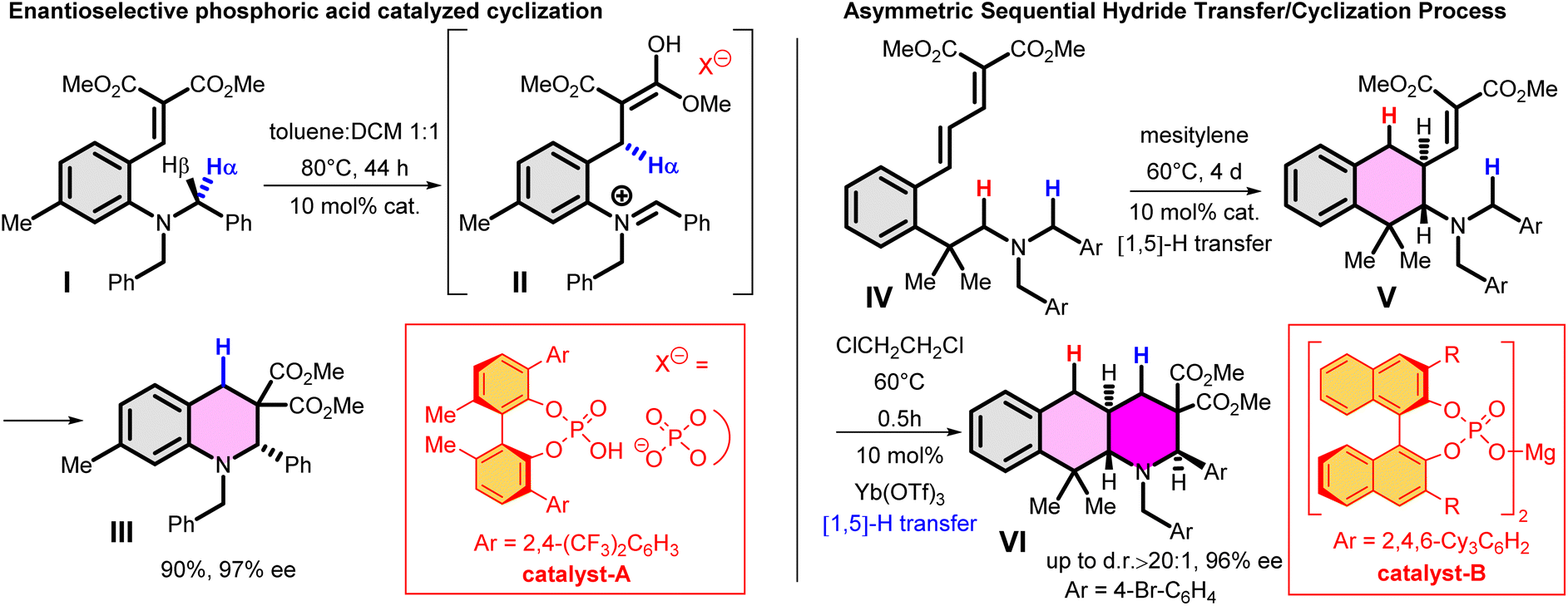 | ||
| Scheme 2 Examples for enantioselective and sequential [1,5] hydride transfer/cyclization processes.31,32 | ||
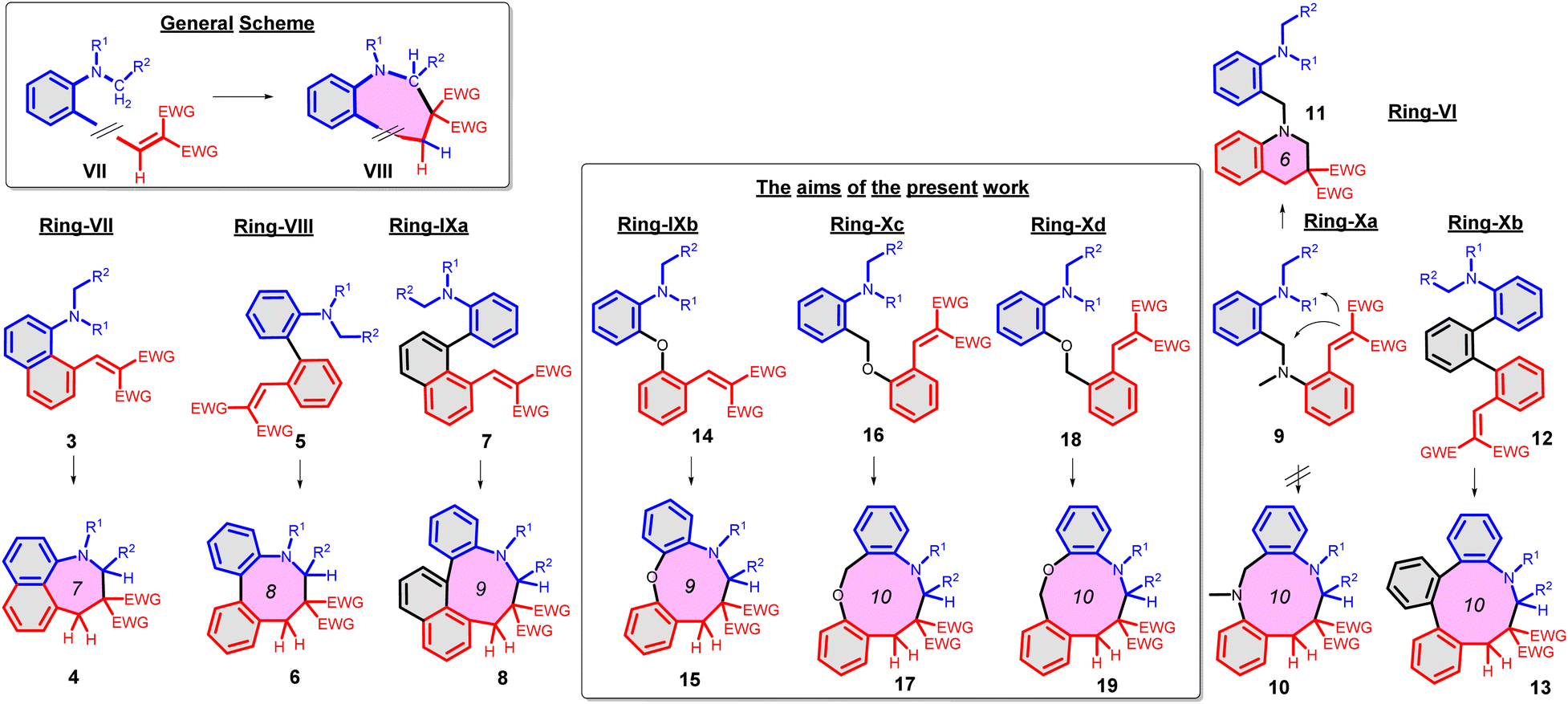 | ||
| Scheme 3 Higher order H-transfer/cyclization processes (ring-VII–X), resulting in larger ring sizes.39–43 | ||
The aim of the present work was to study in more detail the synthetic scope of the cyclization of biaryl ethers (14 → 15), potentially furnishing oxazonine derivatives via hydride transfer from an N-alkyl group to an electron-deficient alkene, facilitated in principle by the proximity of the groups and the electron-rich character of the N-alkyl group.46 To explore the frontiers of the cyclization, the respective CH2-analogues were tested as well (16 → 17 and 18 → 19). Moreover, we wished to gain a better understanding of our experimental results by computational chemistry calculations. We set out to study the influence of the conditions and the structural elements of the substrate 1/VII, helping to predict the mechanism and the feasibility of the transformation.
Results and discussion
Synthesis of biaryl ethers and the scope of their cyclization
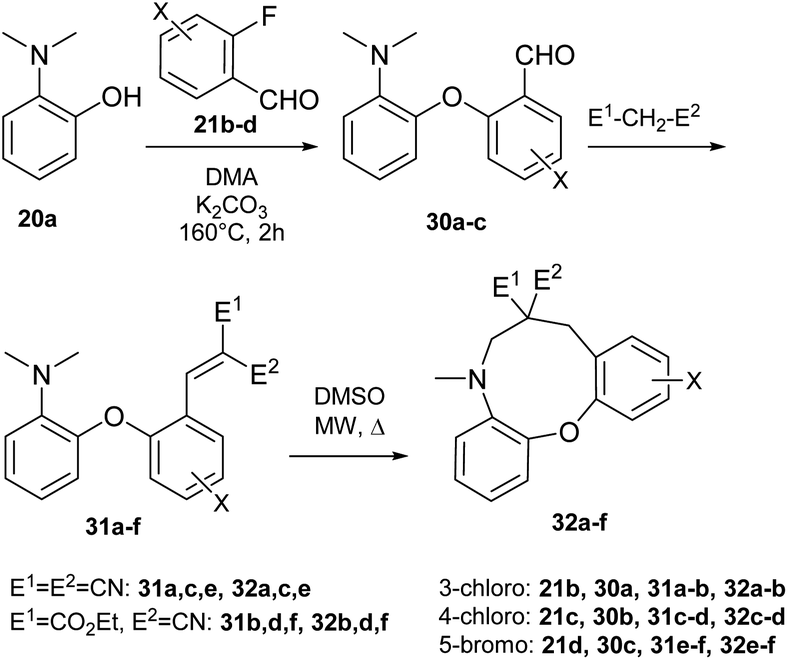
We previously described our first preliminary result for the cyclization of a biaryl ether (14 → 15, Scheme 3).42 In the present work, for studying the scope of the H-transfer/cyclization among biaryl ethers, a set of 2-(2-vinyl)phenoxy-tert-anilines (14a–i) were selected, with the most commonly used dialkylamino (dimethylamino, pyrrolidino and piperidino) and vinyl substituents (CN, CO2Et). These variants represent different reactivity, as shown by experimental data and our theoretical comparison of typical amine and EWG substituents (Scheme 4 and Fig. 5). The 22a–c aldehydes were prepared from commercially available 2-fluorobenzaldehyde (21a) and the corresponding 2-aminophenols47,48 (20a–c, Scheme 4). The vinyl precursors for the cyclization studies were prepared by the Knoevenagel condensation of 22a–c, with malononitrile, ethyl cyanoacetate or diethyl malonate as the active methylene agents. In the case of malononitrile and ethyl cyanoacetate, the 14a–f vinyl products were obtained with good yields under mild conditions. For diethyl malonate, refluxing in toluene was necessary (14g–i).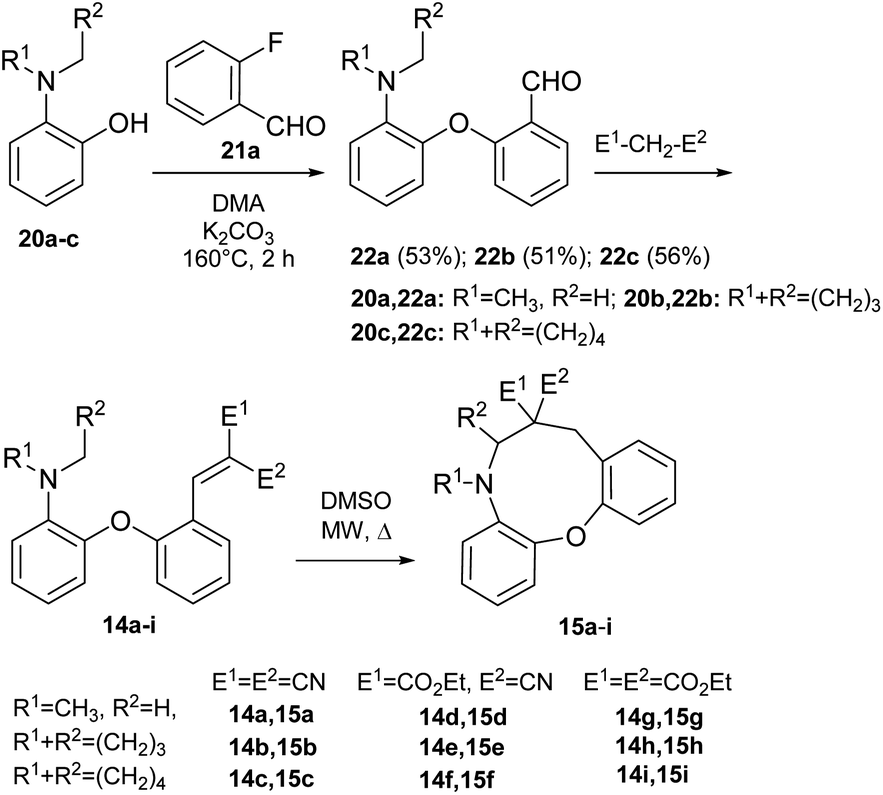 | ||
| Scheme 4 Preparation and cyclization of 2-(2-vinyl)phenoxy-tert-anilines (14a–i) to oxazonine products (15a–i). Reaction conditions: 14a–c: CH2(CN)2, EtOH, rt; 14d–f: CNCH2CO2Et, EtOH, rt; 14g–i: CH2(CO2Et)2, toluene, reflux; 14a (71%); 14b (65%); 14c (35%); 14d (73%); 14e (86%); 14f (80%); 14g (60%); 14h (65%); 14i (48%); 15a (27%); 15b (21%); 15d (28%); 15c,e–i (n.d). | ||
For the 14d–f derivatives, judged by the NMR spectra (δH of vinyl protons (in CDCl3): 14d: 8.92 ppm, 14e: 8.89 ppm, 14f: 8.96 ppm) exclusively the E isomers were formed (the less bulkier nitrile group being in cis position to the benzene ring), in accordance with former examples reported in the literature.12,49–51 Cyclizations of 14a–i were first attempted in DMSO solution with conventional heating (50/75/100 or 125 °C, 16 h). In the case of 14b,c,e and 14f, at higher temperatures complex mixtures were formed. Therefore, only the apparently thermally more stable 14a and 14d were subjected to heating at higher temperature in microwave reactor. For the latter, short irradiations (15/30 min) were screened (125, 150 and 175 °C) with LC-MS monitoring. In the dinitrile series, the expected oxazonine products were formed from 14a and 14b, albeit only in modest yields. For 14c no conversion was observed at lower temperatures, while higher temperatures led only to the decomposition of the vinyl precursor. Similar results were obtained for 14e and f from the ester series, having cyclic amine moieties. However, 14d could be cyclized, but a higher reaction temperature (175 °C) was required for the transformation. Of note, even for the successful attempts, a compromise between full conversion and decomposition was difficult to find, therefore shorter reaction times were selected for isolating the cyclic products. For the diester products (14g–i), a series of Lewis acids and catalysts previously tested [Yb(OTf)3, Gd(OTf)3, FeCl3, Mg(ClO4)2 and AlCl3] for cyclizations were first screened in MeCN solvent at 80 °C, 16 h. Heating in DMSO solution was attempted also in this series, without catalyst. Compound 14g proved inert under the conditions tested. For 14h and 14i, oxazonine products were not appearing in significant amount, however, in some cases the aldehyde was formed. Moreover for 14h, a higher molecular weight species was detected. (A summary of the reaction conditions tested is provided in the ESI†).
As an alternative activation method, we tested the cyclizations under photoirradiation on substrates already known to furnish the tetrahydoquinoline products [1: R1![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH3, R2H, R1 + R2(CH2)3 or (CH2)4].52 For the photoirradiation, the respective absorption maximum wavelengths were used in a dedicated PhotoCube instrument. A quick screen of conditions (solvent, irradiation time) showed traces of the products, assigned to thermal effects. Therefore, we did not pursue this direction for our present, more challenging substrates (see the ESI† for further details). Of note, successful photoinduced cyclization of the same substrates was recently reported following a more thorough screening of the conditions, such as significantly longer reaction times not tested by us.53
CH3, R2H, R1 + R2(CH2)3 or (CH2)4].52 For the photoirradiation, the respective absorption maximum wavelengths were used in a dedicated PhotoCube instrument. A quick screen of conditions (solvent, irradiation time) showed traces of the products, assigned to thermal effects. Therefore, we did not pursue this direction for our present, more challenging substrates (see the ESI† for further details). Of note, successful photoinduced cyclization of the same substrates was recently reported following a more thorough screening of the conditions, such as significantly longer reaction times not tested by us.53
The structures of 14c and 15a were confirmed by X-ray crystallography (structural data of 15b was reported previously)42 (Fig. 1). Studying the position of the interacting groups that might be relevant for cyclization, the distance between the tertiary amine nitrogen and the α vinyl carbon was found to be 4.37 Å in compound 14c, i.e. no closer proximity was observed. With the cyclization of 14b, a new stereogenic centre is formed. In chiral HPLC experiments, two peaks could be detected in an almost 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, presumably due to the formation of the two enantiomers. For 15b, upon X-ray crystallography, two molecules were observed in the asymmetric unit (R/S configuration at the stereogenic carbon, however no isomers resulting from inversion of the pyrrolidino nitrogen).
:1 ratio, presumably due to the formation of the two enantiomers. For 15b, upon X-ray crystallography, two molecules were observed in the asymmetric unit (R/S configuration at the stereogenic carbon, however no isomers resulting from inversion of the pyrrolidino nitrogen).
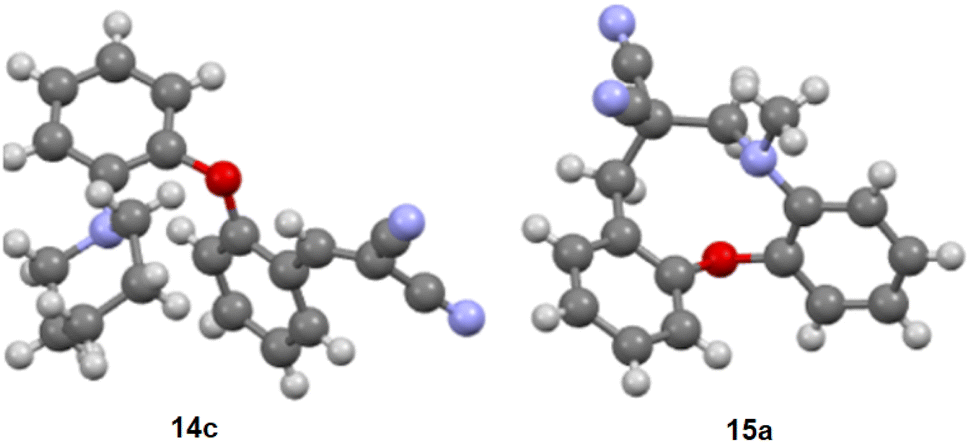 | ||
| Fig. 1 X-ray structure of 14c and 15a. | ||
Further optimization and scale-up studies were run with 14b (5 mg 14b in 0.5 mL solvent, NMR monitoring). On the one hand, changing the solvent to toluene, no cyclization was detected (MW, 100 °C, 0–120 min), whereas in n-BuOH a similar reaction pattern to that observed in DMSO was recorded.
The cyclization to 15b could be well monitored by NMR spectroscopy (Fig. 2), by the appearance of the characteristic NMR signals of the methylene hydrogens adjacent to the nitrogen and the carbon bearing the electron-withdrawing groups in the product.
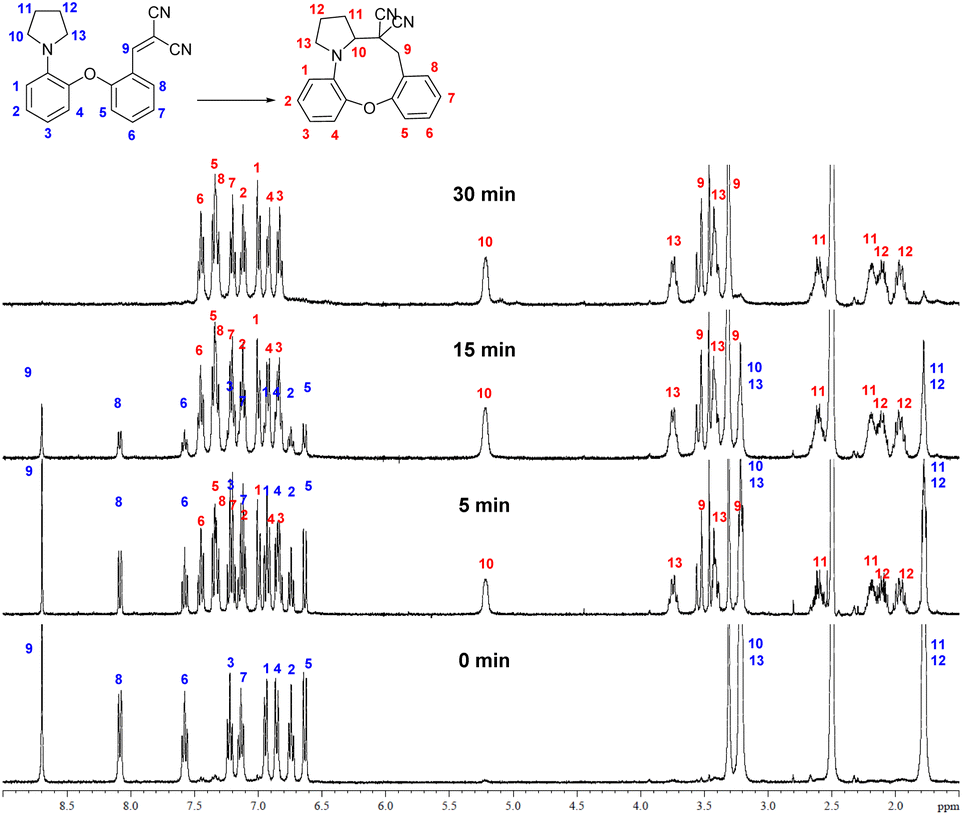 | ||
| Fig. 2 1H NMR monitoring of the cyclization (5 mg 14b in 0.5 mL DMSO-d6, 100 °C, NMR spectra taken at the indicated time points, the 30 min spectrum showing full conversion). | ||
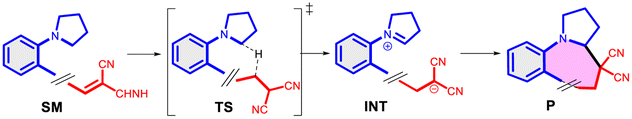
In a more concentrated sample (50 mg 14b in 0.5 mL solvent), besides the formation of 15b, two further products were detected. By NMR, the structure of 23 was identified as the result of a dimerization, confirmed also by the 631 Da peak detected in LC-MS (Scheme 5). The relative configuration of the octahydro-dipyrroloquinoline product was determined by the respective NOE interactions. To ascertain that 23 is not formed via 15b, in a control experiment an isolated sample of 15b was subjected to 100 °C heating in DMSO, however no change was observed. The NMR studies showed that the higher Mw product (24) obtained from 14h also corresponds to the respective octahydro-dipyrroloquinoline structure. A similar dimerization, leading to octahydro-dipyrroloquinoline derivatives was reported for carbo-analogues.54 The cascade mechanism suggested by the authors proceeds via dehydration and the formation of a carbocation, inducing a [1,5]-hydride transfer to an iminium cation. The reaction between the iminium cation and an enamine formed with the isomerization of the iminium cation leads to an intermediate suitable for an intramolecular Picter-Spengler type reaction, the result of which is an octahydro-dipyrroloquinoline derivative. Studying differently substituted diphenylmethanol derivatives, the authors observed, that the preferred reaction pathway is influenced by the electron density of the benzene ring (not carrying the amino group). The second pathway – via an intramolecular Pictet–Spengler reaction without the dimerization step – affords dihydrodibenzo[b,e]azepines in good yields. In our case, a similar process could be envisaged, involving a higher order [1,8]-H transfer, an intramolecular proton transfer, C–C bond formation between two pyrrolidine moieties followed by a SEAr step and finally a re-aromatization. The two possible reaction pathways (blue or orange), branching from the common 25 intermediate are shown in Scheme 6, while the corresponding calculated enthalpies on Fig. S10.† Interestingly, there is a negligible activation enthalpy difference between the 25 → 15b and the 25 → 26 steps (11.7 kJ mol−1 vs. 5 kJ mol−1). Noteworthy, that the formation of 23 is an exothermic process, in contrast to 15b.
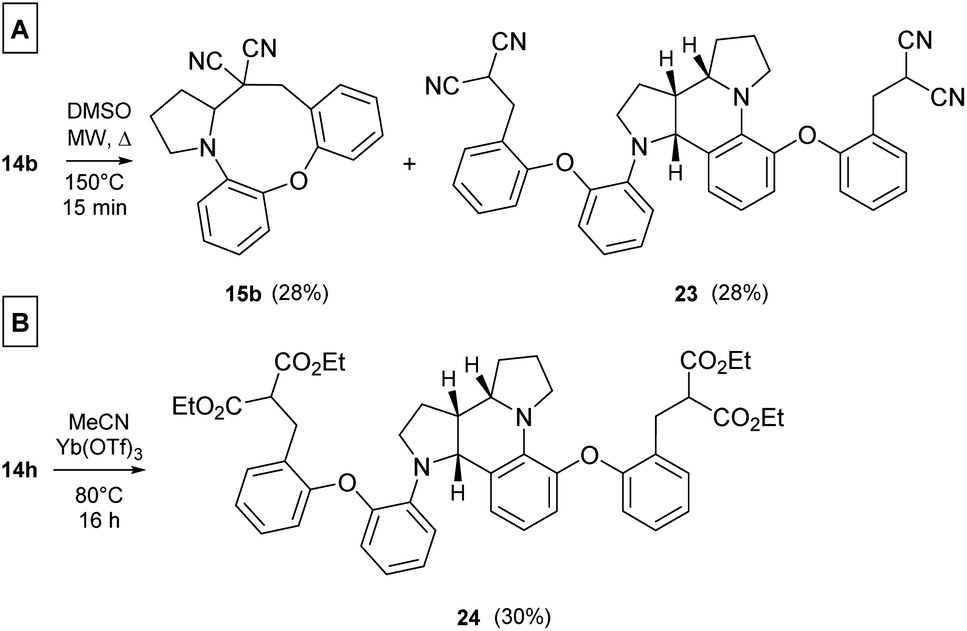 | ||
| Scheme 5 (A) Products 15b and 23 formed upon heating a more concentrated (1 g/10 mL) DMSO solution of 14b (MW, 100 °C, 15 min). (B) Dimer product obtained from 14h. | ||
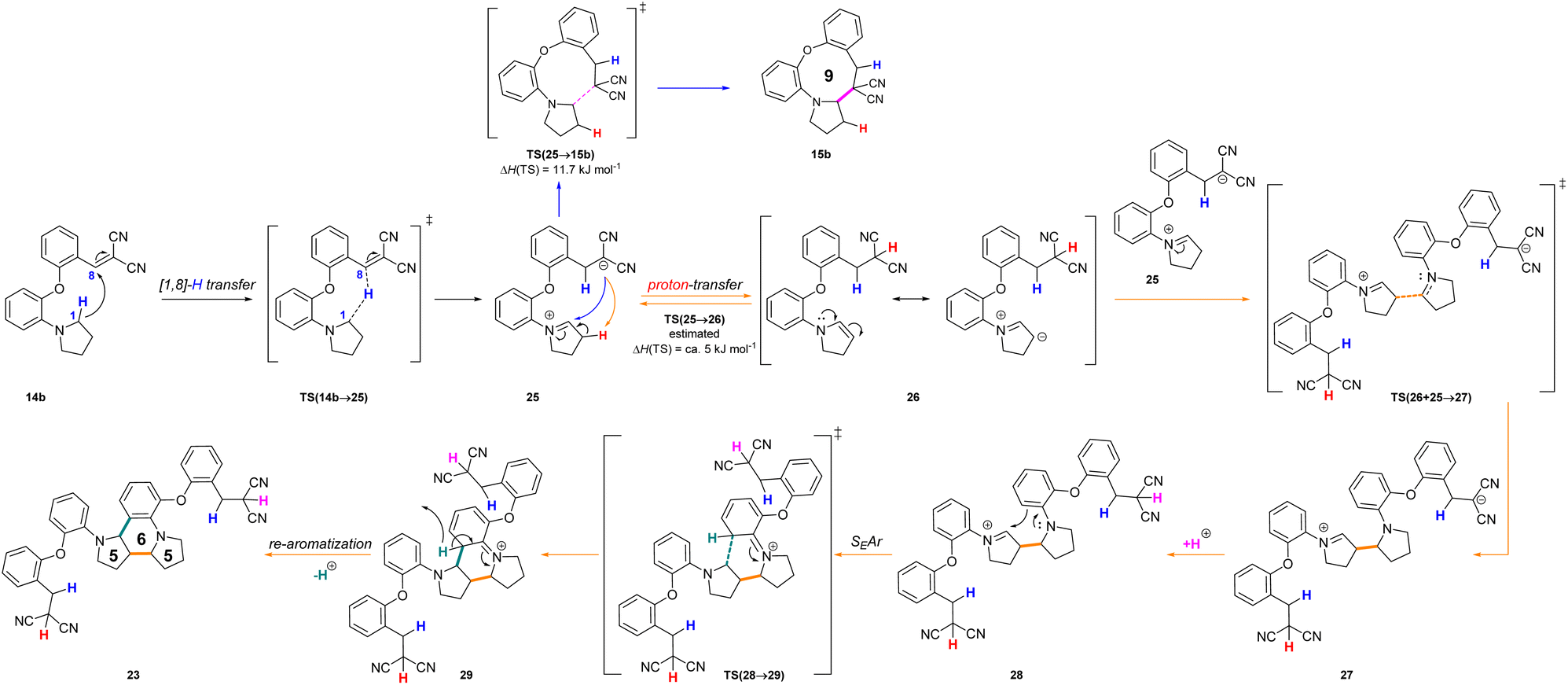 | ||
| Scheme 6 Reaction mechanism proposed for the formation of the octahydro-dipyrroloquinoline derivative 23 (orange) in comparison with the formation of 15b (blue) and the calculated enthalpies for the possible reaction pathways (14b → 15b or 14b → 23). The related enthalpy values can be seen in Fig. S10.† | ||
The [5-6-5] tetracyclic octahydro-dipyrroloquinoline scaffold is relatively rare among natural products, however there are some interesting biologically active alkaloids containing it, such as incargranine B and seneciobipyrrolidine (Scheme 7).55,56 For preparing the octahydro-dipyrroloquinoline skeleton several strategies were suggested in the literature, as (i) a chiral metal-phosphate-catalyzed asymmetric tandem hydroamination/formal Povarov reaction of aminoalkynes,57 (ii) a domino Mannich/electrophilic aromatic substitution sequence of aniline derivatives,58 (iii) oxidative annulation of aryl amines59 or a (iv) cycloaddition/arylation sequence of azomethine ylides.60 The operationally simple dimerization observed for 14b and 14h could serve as an interesting addition to this synthetic toolbox.
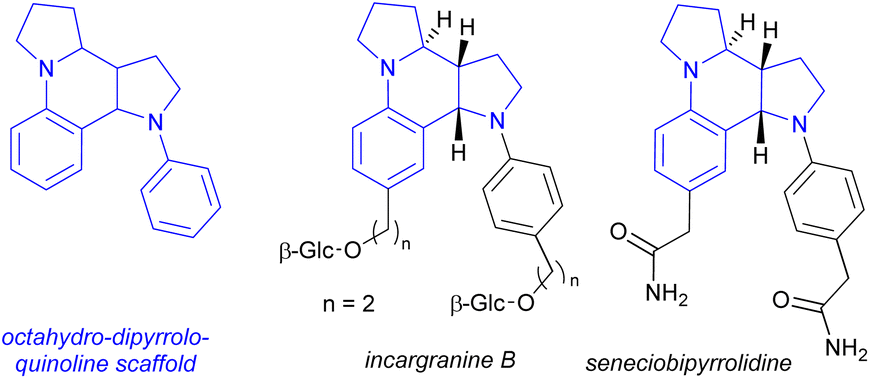 | ||
| Scheme 7 Natural products containing an octahydro-dipyrroloquinoline scaffold. | ||
To demonstrate the scope of the cyclization yielding oxazonine products, aldehydes 30a–c were prepared, substituted with halogens on the aryl ring carrying the vinyl moiety. For the cyclization a short series of test reactions were run, with 15/30 min MW irradiations at two temperatures (150/175 °C for the dinitrile or 175/200 °C for the CN–CO2Et derivatives). Of the six novel vinyl derivatives (31a–f), the expected oxazonine products were isolated in five cases (32a,c–f), with varying yields (Scheme 8). Although a further set of studies would be necessary to assess the effect of the aryl ring substituents on the cyclization, the halogens in our examples could serve as useful linchpins for further derivatizations of the ring systems.
 | ||
| Scheme 8 Preparation and cyclization of halo-substutited 2-(2-vinyl)phenoxy-tert-anilines (31a–f) to oxazonine products (32a–f). Reaction conditions: 31a,c,e: CH2(CN)2, EtOH, rt; 31b,d,f: CNCH2CO2Et, EtOH, rt; 31a (48%); 31b (46%); 31c (67%); 31d (60%); 31e (72%); 31f (65%); 32a (35%); 32b (n.d.); 32c (25%); 32d (30%); 32e (62%) 32f (21%). | ||
To discern the limits of the cyclization resulting in larger ring sizes, the –CH2- extended vinyl derivatives were prepared as well. The key intermediates were obtained by alkylation reactions of the appropriate phenol or benzyl alcohol (Scheme 9).61 For the cyclization the standard conditions were screened, i.e. heating in DMSO solution or in MeCN, in the presence of Lewis acid catalysts (see ESI† for a detailed list of the conditions tested).
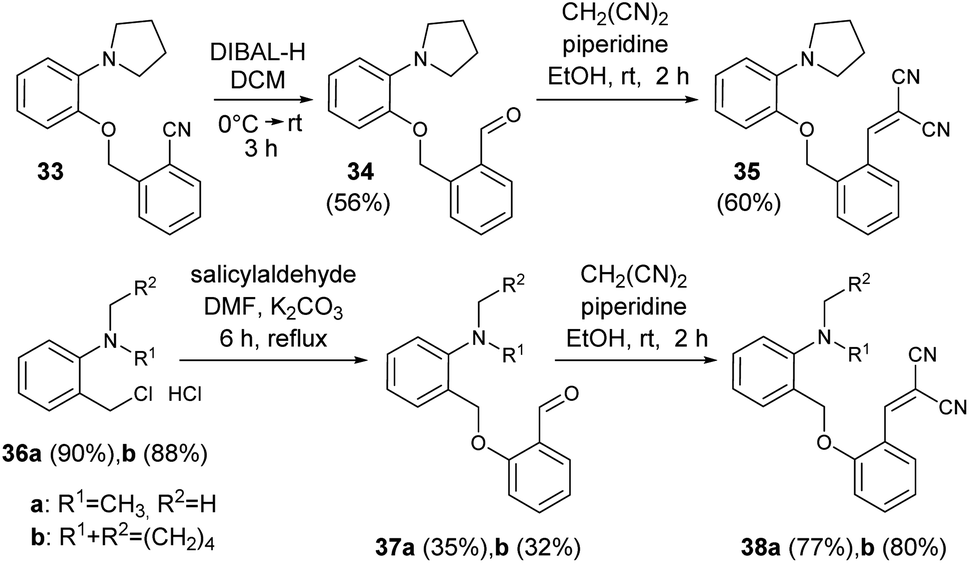 | ||
| Scheme 9 The synthesis of –CH2–O-bridged vinyl derivatives.61 | ||
In the case of 38a,b, only decomposition was observed and the expected 10-membered rings could not be isolated. Similarly for 35, mainly decomposition was observed for the different heating scenarios tested. However, in the presence of Gd(OTf)3 catalyst, the formation of three novel structures were observed by LC-MS, the Mw of which (660, 461, 461 Da) did not confirm the formation of the ten-membered ring. However, based on our previous results, the Mw 660 Da product could correspond to the formation of an octahydro-dipyrroloquinoline scaffold also in this case.
Theoretical investigations, comprehensive theoretical study
To get a better insight about the [1,5]-H transfer/cyclization reaction, we carried out a wide range of theoretical calculations. The first theoretical description dates back to 1988,12 however, later this direction did not attract too much interest. Therefore, a novel DFT study [B3LYP/6-31G(d,p)//PCM(DMSO)] is appropriate. Here, a comprehensive analysis is reported, involving several aspects. The Nucleus Independent Chemical Shifts (NICS in ppm)62 were calculated in the ring centre of TSs to measure the degree of aromaticity by the same level of theory. The classical method is not suitable to measure the aromaticity degree in a TS1 structure. The question arises, how can we classify this mechanism? Is this mechanism a pure sigmatropic rearrangement, involving a complete aromatic ring in the transition state (TS), or it is rather a hydride ion transfer, where only the H− jumps from the –NCH2– to the –CHC– (Scheme 10A)? Or could it be a mixture of the two? The main characteristics of the sigmatropic rearrangement, like 39 → TS(39), are that the process is solvent independent (no ionic character) and involves high aromatic ring current [δ(NICS) = −13.8 ppm]. In the case of a hydride ion transfer mechanism, like 40 → TS(40), one can observe high solvent dependence and a non-aromatic ring current, with low NICS values. In Scheme 10B, the most (41 → TS(41)) and the least aromatic (42 → TS(42)) models of hydride shift are considered at the theoretical level. In the case of 41, the highest NICS value [δ(NICS) = −7.6 ppm] is observed, due to the single aromatic ring forming in the TS. In the case of 42, there is no chance for any aromatic ring current, but the calculated NICS value is still unexpectedly significant [δ(NICS) = −5.3 ppm]. As one may expect, the general 1a → TS(1a) reaction exhibits a compromised NICS value of −6.0 ppm, which reveals the fact that this hydride shift is between the two sides. The calculated TS enthalpies are not too high, between 100–130 kJ mol−1, which is lowered by the quantum tunnelling effect by even 20 kJ mol−1.
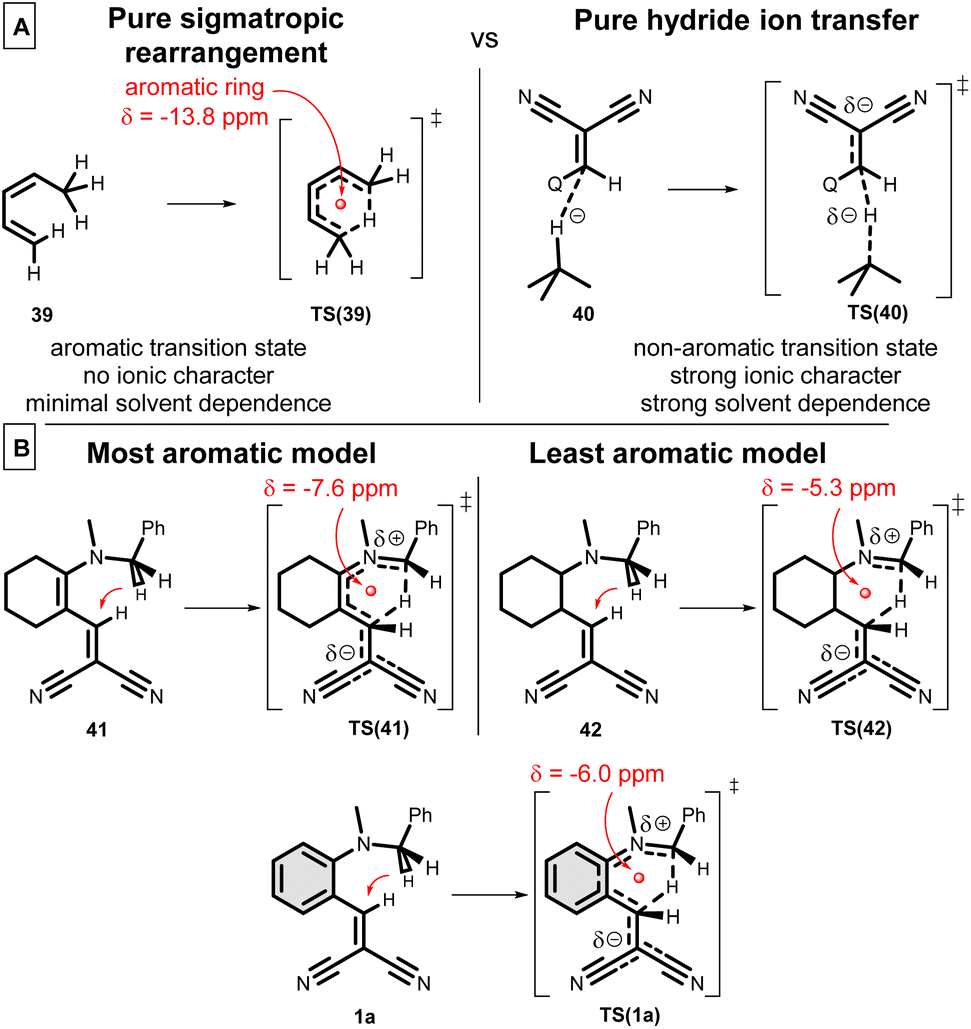 | ||
| Scheme 10 Comparison of sigmatropic rearrangement and ionic reactions. | ||
In the 3 next step, we studied the solvent dependence of 1 → TS(1) in comparison with 39 → TS(39), 41 → TS(41) and 42 → TS(42), by theoretical method in vacuo and in five solvents (water, DMSO, MeCN, dichloroethane, toluene). In the diagram in Fig. 3 we considered the (ε − 1)/(2ε + 1) as x axis, which gives a linear relationship with the ΔH or ΔG values of the TSs, when the formation of a dipole is the dominant process.63 The pure sigmatropic rearrangement, like model compound 39, exhibits complete solvent independence, as the horizontal red line in Fig. 3. Model compound 41 (green line) shows only a moderate slope, which represents its high sigmatropic characteristics. In contrast with that, the rather ionic rearrangement of 42 shows the deepest slope (blue), representing a strong dipole involvement in the course of the reaction.
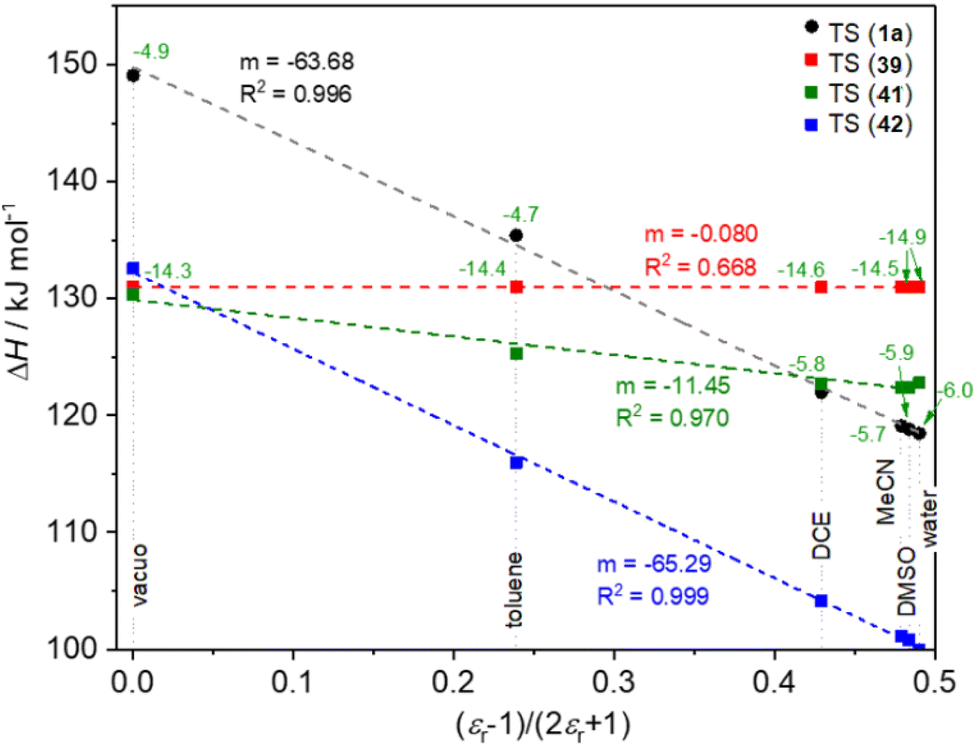 | ||
| Fig. 3 Calculated solvent dependence of the TSs and the formation of intermediate 43 in various solvents as a function of (ε − 1)/(2ε + 1) at B3LYP/6-31G(d,p)//PCM(solvent) level of theory. | ||
Compound 1a follows a slightly moderate solvent dependence compared to 41, but it is rather close to it, referring to a mixed mechanism between the sigmatropic and ionic mechanism.
In the third step, addressing the suitability of our substrates for the cyclization, the reaction parameters and the rate determining step were studied by computational chemistry. Three parameters were considered, the TS and the ΔH values of the intermediates [ΔH(1 → 43)] and the products [ΔH(1 → 2)]. According to general practise, if the ΔH(TS) is larger than 150 kJ mol−1, the reaction is extremely slow even in a MW reactor at high temperature, but above 180 kJ mol−1, the reaction can be considered as an unfeasible option. The negative enthalpy of product formation [ΔH(1 → 2)] indicates that the product formation is beneficial. Although in the cases of positive values, the formation is unbeneficial, however, with high temperature, it can provide some energy content (RT = ca 5–10 kJ mol−1, where R is the universal gas constant and T is the reaction temperature in K) to invest and proceed the product formation.
First, the amino part was varied with some typical functionalities applied earlier, such as N–Me-benzyl, N–Me-ethyl, pyrrolidinyl, piperidinyl or morpholinyl, as summarized in Fig. 4. In these series, the lowest gap and the most exothermic pathway belongs to the pyrrolidinyl (1b) derivative, but the N–Me-benzyl (1a) and piperidinyl (1c) are close to it. The morpholinyl (1d) and N–Me-ethyl (1e) are the least beneficial amines here, in agreement with the experimental findings. The study was extended to the EWG side as well, scanning several common functional groups (CN, COOMe, NO2, CF3, H, Me) in several combinations with each other with a constant N–Me-benzyl functionality (1a, 1f–q). The calculated enthalpies are collected in Fig. 5. In conclusion, in all the cases the product formations allow the cyclization. However, large differences can be observed in the TS enthalpies. Only two conjugative EWGs, such as CN, COOMe and NO2 can effectively decrease the energy gap to be close to 100 kJ mol−1. However, only one EWG and one EDG (like H or Me) results in too high energy values, the only exception may be one NO2 with one Me (1g). Two EDG groups do not allow the existence of the zwitterionic structures (43p,q), so neither the corresponding TS. Interestingly, the non-conjugative EWG CF3 (1o) has much less beneficial effect on the TS(1o) or the intermediate 43o, despite its strong electron withdrawing effect, indicating, that the conjugative characteristic is essential.
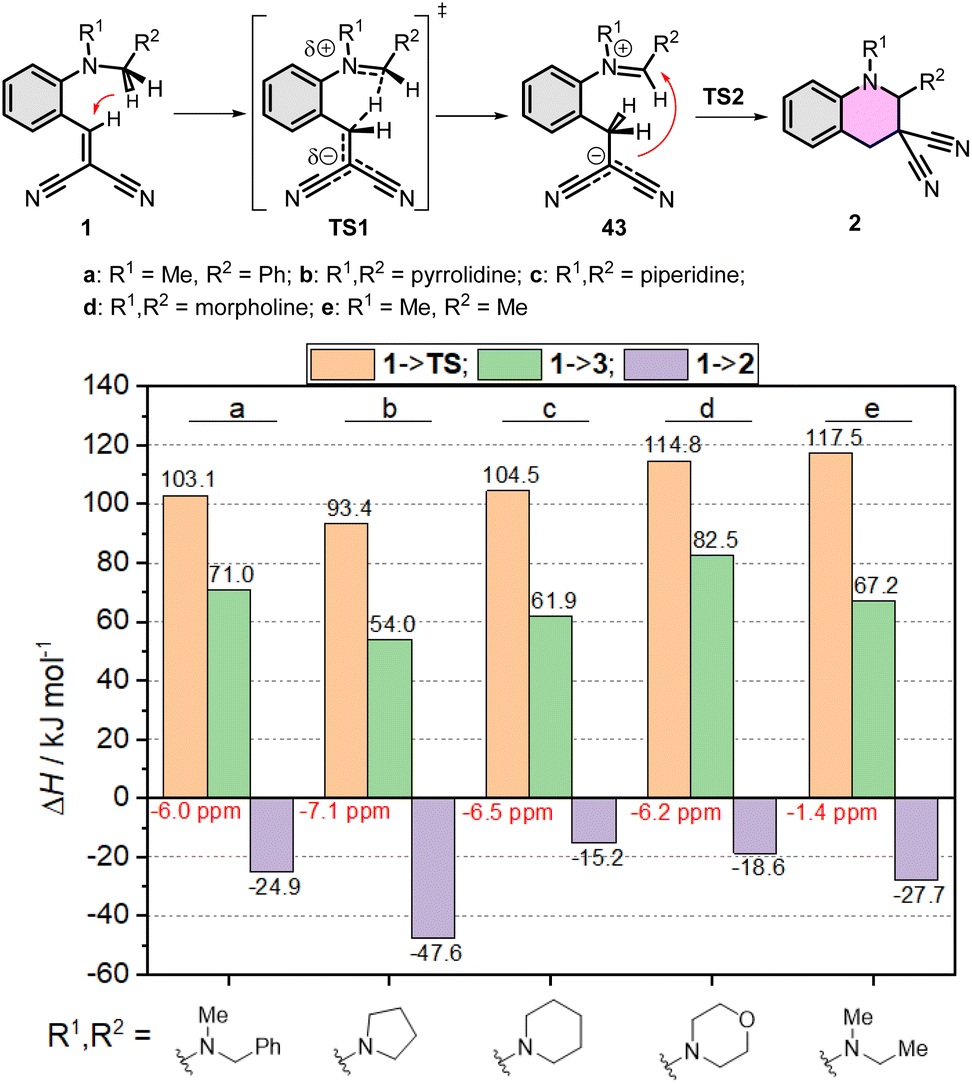 | ||
| Fig. 4 Simplified reaction mechanism of the cyclization of compounds 1a–e. Calculated enthalpies of TS(1a–e), the formation of intermediate 43a–e, and products 2a–e at B3LYP/6-31G(d,p)//PCM(DMSO) level of theory. | ||
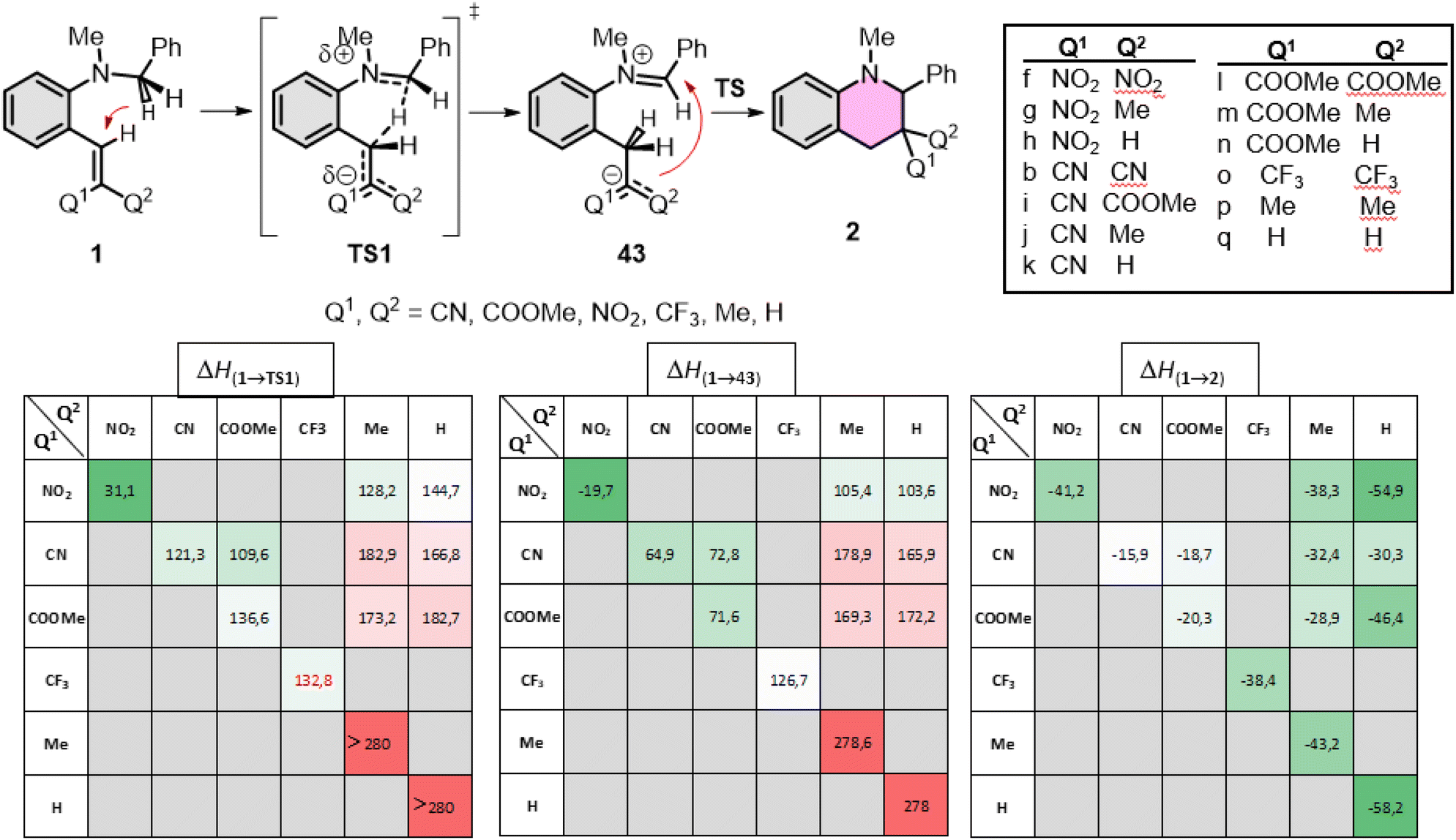 | ||
| Fig. 5 Calculated enthalpies of TS(1a) and TS(1f–q), the formation of intermediate 43a and 43f–q as well as 1a and 1f–q at B3LYP/6-31G(d,p)//PCM(DMSO) level of theory. The effect of the EWG group on the cyclization [B3LYP/6-31G(d,p)//PCM(DMSO)]. The colour code of the cells illustrates the potential of the reactions. Dark green and green cells are feasible, white cells are in the edge of the feasibility, while light red and red cells refer to the forbidden reactions. | ||
Finally, we studied the effect of the ring size from 6 to 10 members involving several model reactions, listed in Table 1. We analysed the number of participating π electrons, the computed ΔH, ΔG and ΔS values for TS, for the intermediate and the products. In the light of the results, one can understand, which parameters control the reaction, and which structural features may lead to cyclised products. The formation of the 6-member ring (entry 1) is very preferential, as was discussed earlier, however, surprisingly, the 7-member naphthalene product (entry 2) is more advantageous energetically. The aromatic character in the TS is verified exclusively in the first case (1b), as in the case of larger rings, the rings in the TSs are typically not planar. Maybe this is the reason that the anti-aromatic π-electron octet in entries 3, 4 and 5 cannot negatively dominate the hydride transfer and somewhat feasible energy gaps are involved in the TS. Consequently, compound 6b, 8b and 15b could be prepared in acceptable yields. As reported in the experimental section, due to the relatively high activation enthalpy, the reaction proceeds only at elevated temperature, which allows the formation of the competitive side product 23. Interestingly, despite the non-aromatic TS, compounds 17b and 19b (entries 6 and 7) exhibit moderate TS enthalpies. The ring closed products could not be isolated, due to the competitive reactions. Although entry 8b exhibits moderate TS, the formation of the product is quite endothermic. Taking into account the high reaction temperature (over 100 °C), it can perhaps be explained that the product appears, although in low yield.
| E | General scheme | SM → TS1 | n(πe−) | SM → intermed | SM → P | Conclusion |
|---|---|---|---|---|---|---|
| ΔH/kJ mol−1 | Planarity | ΔH/kJ mol−1 | ΔH/kJ mol−1 | |||
|
ΔG/kJ mol−1 | NICS | ΔG/kJ mol−1 | ΔG/kJ mol−1 | ||
| ΔS/J mol−1 K−1 | δ/ppm | ΔS/J mol−1 K−1 | ΔS/J mol−1 K−1 | |||
| a n.a. not available. | ||||||
| 1 | 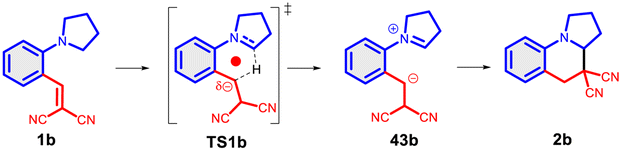 |
+93.4 | 6π | +54.0 | −47.8 | 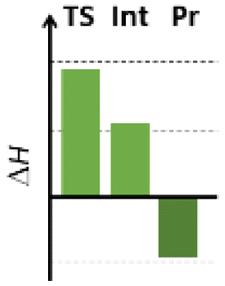 |
| +100.1 | Planar | +55.2 | −36.8 | |||
| −22.4 | −7.0 | −4.2 | −27.6 | |||
| Arom | ||||||
| 2 | 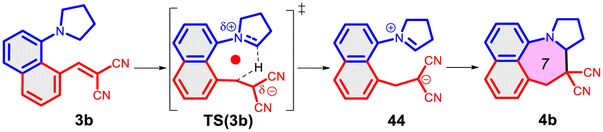 |
+84.5 | 8π | +70.6 | −43.4 | 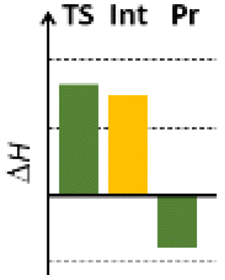 |
| +91.7 | Q-planar | +71.7 | −30.9 | |||
| −22.3 | −1.7 | −3.7 | −40.6 | |||
| Non-arom | ||||||
| 3 | 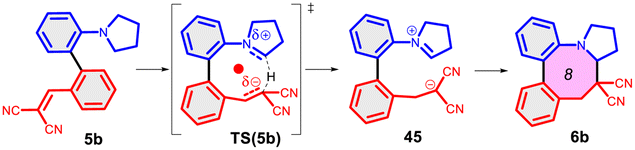 |
+103.7 | 8π | +55.6 | −19.0 | 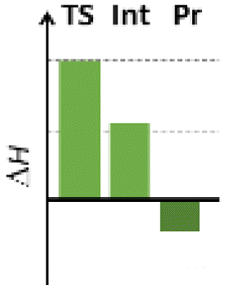 |
| +111.8 | Q-planar | +57.7 | −4.6 | |||
| −27.1 | −0.2 | −7.0 | −48.1 | |||
| Non-arom | ||||||
| 4 | 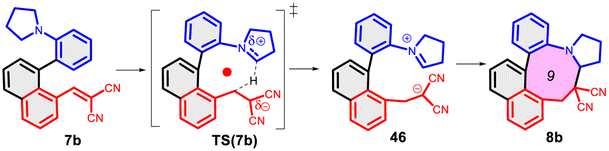 |
+136.5 | 8π | +68.4 | −2.0 | 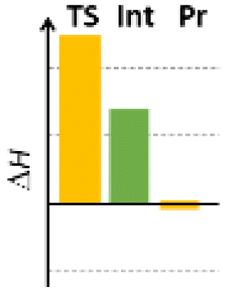 |
| +144.1 | Non-planar | +70.7 | +11.5 | |||
| −25.7 | +1.2 | −8.0 | −45.2 | |||
| Non-arom | ||||||
| 5 | 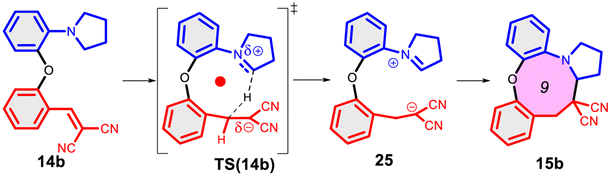 |
+114.7 | 8π | +54.8 | +10.1 | 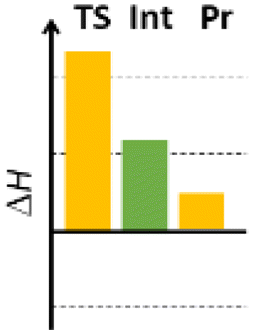 |
| +126.8 | Q-planar | +56.1 | +30.2 | |||
| −40.9 | +0.6 | −4.1 | −67.2 | |||
| Non-arom | ||||||
| 6 | 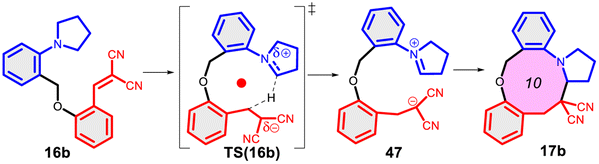 |
+112.5 | — | +70.7 | +20.2 | 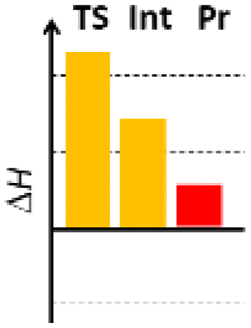 |
| +133.0 | Q-planar | +68.8 | +40.3 | |||
| −68.8 | n.a. | +6.4 | −67.3 | |||
| Non-arom | ||||||
| 7 | 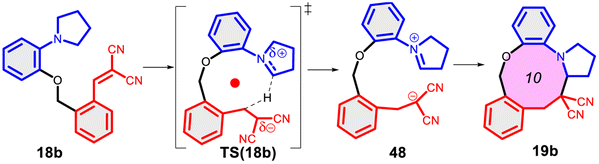 |
+90.7 | — | +48.7 | +6.9 | 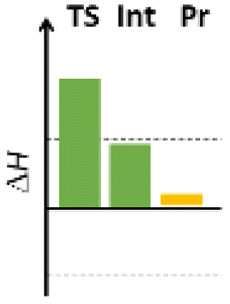 |
| +105.0 | Q-planar | +51.1 | +27.9 | |||
| −47.9 | n.a. | −8.0 | −70.3 | |||
| Non-arom | ||||||
| 8 | 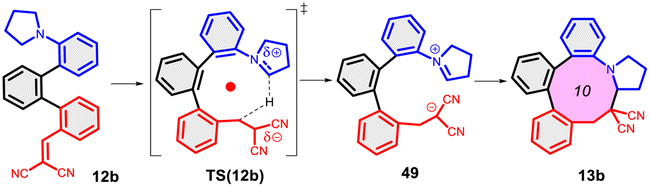 |
+104.6 | 10π | +67.8 | +27.6 | 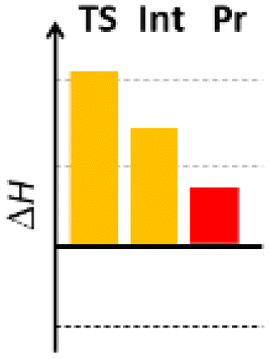 |
| +115.8 | Non-planar | +68.7 | +47.2 | |||
| −37.5 | +0.9 | −3.2 | −65.7 | |||
| Non-arom | ||||||
Conclusions
According to preliminary computational studies, biaryl ethers are less evident substrates for [1,8]-hydride transfer/cyclization reactions. Seeking an experimental verification, a set of biaryl ethers were prepared, with dialkylamino and vinyl substituents in ortho, ortho′ positions. Cyclizations under thermal conditions (MW, DMSO) afforded oxazonine derivatives in several cases. The structure of one vinyl and two oxazonine derivatives were confirmed also by X-ray crystallography. In the diester series, cyclizations were attempted also using Lewis-acid catalysts, however not resulting in the expected oxazonine products. An interesting dimerization was observed instead, leading to an octahydrodipyrrolo-quinoline derivative. The energy profile of the two possible reaction pathways was compared and a possible mechanism for the unexpected transformation was suggested. Theoretical studies show that the rate determining reaction step is the first hydride shift, which represents a mixed mechanism between the sigmatropic rearrangement and the ionic hydride transfer. Aromaticity of the TS plays an important role only in the formation of six membered products, but the weight of the ionic character is already significant. In the case of larger ring products, the aromatic character vanishes and only the pure ionic hydride transfer dominates the TSs, irrespective to the number of participating π-electrons.Materials and methods
General information
All reagents and solvents were purchased from commercial sources and were utilized without further purification. Melting points were determined on a Büchi-540 capillary melting point apparatus and are uncorrected. The 1H and 13C NMR spectra were recorded at ambient temperature, in the solvent indicated, with a Varian Mercury Plus spectrometer at a frequency of 400/600 or 100/150 MHz or with a Bruker 400 MHz spectrometer, at a frequency of 400 or 100 MHz, and are reported in ppm. Spectra were recorded at 400 MHz (1H) or 100 MHz (13C), if not indicated otherwise. Chemical shifts are given on the δ-scale relative to tetramethylsilane or the residual solvent signal as an internal reference. For structure elucidation, one-dimensional 1H, 13C, DEPT, two-dimensional 1H, 1H-COSY, 1H, 13C-HSQC, 1H, 13C-HMBC measurements were run. Mass spectra utilizing fast atom bombardment ionization were recorded on a VG-ZAB-2SEQ spectrometer. Elemental analyses were performed on a Carlo Erba 1012 apparatus. Microwave (MW) irradiation experiments were carried out in a monomode CEM-Discover MW reactor, using the standard configuration as delivered, including proprietary software.The experiments were executed either in 10 mL MW process vials or in open-vessel mode, with control of the temperature by infrared detection. After completion of the reaction, the vial/flask was cooled to 50 °C by air jet cooling. Reactions were monitored by a Shimadzu LC-MS 2020 system.
Preparative HPLC was applied for purification in several cases using an Armen SPOT Prep II instrument with UV detector (200–600 nm scan) equipped with a Phenomenex Gemini C18, 250 × 50.00 mm; 10 μm, 110A column. Gradient elution was employed using 0.08 g NH4HCO3 in 1 L water (A) and acetonitrile (B) or 2 mL TFA in 1 L water (A) and acetonitrile (B) as eluent systems, using the gradient method. For flash column chromatography purification, Kieselgel 60 (Merck, 0.040–0.063 mm) was used; for TLC analysis, Silica gel 60 F254 (Merck) plates were applied. Solvent mixtures used for chromatography are always given in a vol/vol ratio. The structures of all compounds were consistent with their analytical and spectroscopic data. Spectroscopic data are provided for compounds described previously but not characterized spectroscopically. For reporting NMR assignment, an arbitrary numbering was used.
X-ray diffraction studies
A good-looking single crystal of the compound was fixed on the top of a glass fiber using epoxy glue. Data were collected at 293(1) K, Enraf Nonius MACH3 diffractometer (Bruker Nonius, Delft, Netherlands), Mo Kα radiation λ = 0.71073 nm, ω motion. Raw data were evaluated using the XCAD4 software;64 the structure was solved using direct methods by the SIR-92 software65 and refined on F2 using SHELX-97 (ref. 66) program. Refinement was performed anisotropically for nonhydrogen atoms. Hydrogen atoms were placed into geometric position. Figures were prepared with the WINGX-97 suite.67 The PLATON program68,69 was used for crystallographic calculations.Deposition Numbers of 2253075 for 14c and 2253076 for 15a containing the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Theoretical calculations
Gaussian 16 program package (G16),70 using default convergence criteria was used, respectively. Computations were carried out at M06-2X/6-31G(d,p) level of theory.71 The method and basis sets were chosen for their reliability shown in earlier studies.72 The vibrational frequencies were computed at the same levels of theory as used for geometry optimization to properly confirm that all structures reside at minima on their potential energy hypersurfaces (PESs). Thermodynamic functions, such as energy (U), enthalpy (H), Gibbs free energy (G), and entropy (S) were computed for 398.15 K, using the quantum chemical, rather than the conventional thermodynamic reference state.General procedure for the synthesis biaryl ethers
To a solution of the appropriate phenol (15.00 mmol, for 22a, 30a–c: 2-(dimethylamino)phenol (2.06 g), for 22b: 2-(pyrrolidin-1-yl)phenol (2.45 g), for 22c: 2-(piperidin-1-yl)phenol (2.65 g)) in DMA (15 mL), the appropriate 2-fluorobenzaldehyde (for 22a–c: 2-fluorobenzaldehyde (21a, 1.58 mL, 15.00 mmol), for 30a: 3-chloro-2-fluorobenzaldehyde (21b, 1.76 mL, 15.00 mmol), for 30b: 4-chloro-2-fluorobenzaldehyde (21c, 2.38 g, 15.00 mmol), for 30c: 5-bromo-2-fluorobenzaldehyde (21d, 1.78 mL, 15.00 mmol)) and K2CO3 (4.15 g, 30.00 mmol) were added. The mixture was heated at 160 °C (oil temperature) under argon atmosphere for 1.5 h. To the cooled reaction mixture EtOAc (20 mL) was added. The organic phase was washed with H2O (1 × 20 mL) and aq saturated Na2CO3 solution (1 × 20 mL). The organic layer was dried (Na2SO4), filtered and evaporated to dryness. The crude product obtained was purified by flash column chromatography on silica gel (eluent for 22a: toluene → toluene:EtOAc 10:1 gradient, for 22b,c: toluene, for 30a–c: heptane → heptane:EtOAc 4:1 gradient).
Synthesis of 14a–f and 31a–f biaryl vinyl compounds via Knoevenagel condensation
To a solution of the aldehyde (22a–c, 30a–c) (2.00 mmol) in EtOH (2.50 mL), malononitrile (for 14a–c, 31a,c,e: 132 mg, 2.00 mmol) or ethyl cyanoacetate (for 14d–f, 31b,d,f: 0.21 mL, 2.00 mmol), and a few drops of piperidine were added. The mixture was stirred at room temperature until the starting material had been consumed (monitored by TLC, reaction time: 2–3 h). Work-up: (i) the reaction mixture was evaporated to dryness and the crude product obtained was purified by flash column chromatography on silica gel with toluene or heptane/EtOAc eluent (14a, 31a,b,c,f) or (ii) the precipitated crystals were filtered off and washed with 5 × 1 mL EtOH to afford the analytically pure product (14b–f, 31d,e).:EtOAc 9:1). 1H NMR (CDCl3) δ (ppm): 8.50 (s, 1H, H-23), 8.28 (dm, J = 8.0 Hz, 1H, H-15), 7.46–7.40 (m, 1H, H-13), 7.22–7.11 (m, 2H, H-2,14), 7.04 (dm, J = 8.0 Hz, 1H, H-1), 6.98–6.93 (m, 1H, H-3), 6.91 (dm, J = 8.0 Hz, 1H, H-4), 6.66 (dm, J = 8.0 Hz, 1H, H-12), 2.75 (s, 6H, H-8,9). 13C NMR (CDCl3) δ (ppm): 158.4 (C-11), 154.8 (C-17), 147.0 (C-5), 146.0 (C-6), 136.8 (C-13), 129.2 (C-15), 127.0 (C-2), 123.3 (C-14), 122.8 (C-4), 122.7 (C-3), 121.5 (C-16), 119.8 (C-1), 116.9 (C-12), 114.9 (C-19/21), 113.6 (C-19/21), 82.8 (C-18), 43.6 (C-8,9). Anal. calcd for C18H19NO2 (289.33): C, 74.72; H, 5.23; N, 14.52. Found: C, 74.82; H, 4.59; N, 13.86.:EtOAc 10:1). 1H NMR (CDCl3) δ (ppm): 8.53 (s, 1H, H-26), 8.26 (dm, J = 8.0 Hz, 1H, H-15), 7.43–7.38 (m, 1H, H-13), 7.23–7.18 (m, 1H, H-1/2/3/4), 7.14–7.09 (m, 1H, H-14), 7.08–7.04 (m, 1H, H-1/2/3/4), 7.03–7.00 (m, 2H, H-1/2/3/4), 6.63 (dm, J = 8.4 Hz, 1H, H-12), 2.92–2.87 (m, 4H, H-8,9), 1.45–1.32 (m, 6H, H-19,20,21). 13C NMR (CDCl3) δ (ppm): 158.7 (C-11), 154.8 (C-17), 147.6 (C-5/6), 146.2 (C-5/6), 136.5 (C-13), 128.9 (C-15), 127.2 (C-1/2/3/4), 123.7 (C-1/2/3/4), 123.0 (C-1/2/3/4), 121.2 (C-1/2/3/4), 121.0 (C-16), 116.5 (C-12), 114.9 (C-22/24), 113.7 (C-22/24), 82.4 (C-18), 52.7 (C-8,9), 26.8 (C-19,21), 24.7 (C-20). Anal. calcd for C21H19N3O (329.40): C, 76.57; H, 5.81; N, 12.76. Found: C, 76.33; H, 5.47; N, 12.22.:EtOAc 5:1). 1H NMR (CDCl3) δ (ppm): 8.92 (s, 1H, H-25), 8.38 (dm, J = 8.0 Hz, 1H, H-13), 7.40–7.35 (m, 1H, H-11), 7.19–7.10 (m, 2H, H-1/2/3/4,12), 7.04–7.01 (m, 1H, H-1/2/3/4), 6.96–6.89 (m, 2H, H-1/2/3/4), 6.65 (dm, J = 8.4 Hz, 1H, H-10), 4.37 (q, J = 7.2 Hz, 2H, H-19), 2.76 (s, 6H, H-21,22), 1.39 (t, J = 7.2 Hz, 3H, H-20). 13C NMR (CDCl3) δ (ppm): 163.3 (C-17), 158.6 (C-9), 150.1 (C-15), 147.5 (C-5), 146.0 (C-6), 135.3 (C-11), 129.8 (C-13), 126.5 (C-1/2/3/4), 123.2 (C-12), 122.7 (C-1/2/3/4), 122.5 (C-1/2/3/4), 122.2 (C-14), 119.6 (C-1/2/3/4), 116.7 (C-10), 116.5 (C-23), 103.7 (C-16), 63.2 (C-19), 43.6 (C-21,22), 14.8 (C-20). Anal. calcd for C20H20N2O3 (336.38): C, 71.41; H, 5.99; N, 8.33. Found: C, 71.31; H, 5.62; N, 8.28.Synthesis of biaryl vinyl compounds via Knoevenagel condensation 2
To a solution of the aldehyde (22a–c) (2.00 mmol) in toluene (5.0 mL), diethyl malonate (0.46 mL, 3.00 mmol) and a few drops of piperidine were added. The mixture was heated to reflux overnight. The reaction mixture was evaporated to dryness and the crude product obtained was purified by flash column chromatography on silica gel with hexane/ethyl acetate eluent (14h) or preparative HPLC (14g, 14i).:1). 1H NMR (400 MHz, CDCl3) δ 7.89 (s, 1H, H-17), 7.74 (d, J = 8.0 Hz, 1H, H-1), 7.55 (d, J = 7.8 Hz, 1H, H-12), 7.43 (t, J = 7.6 Hz, 1H, H-10), 7.33 (t, J = 8.0 Hz, 1H, H-3), 7.24 (t, J = 7.7 Hz, 1H, H-11), 7.19 (t, J = 7.7 Hz, 1H, H-2), 7.00 (d, J = 8.2 Hz, 1H, H-9), 6.84 (d, J = 8.3 Hz, 1H, H-4), 4.26 (q, J = 7.1 Hz, 2H, H-27), 4.25 (q, J = 7.1 Hz, 2H, H-23), 3.32 (s, 6H, H-15,16), 1.28 (t, J = 7.2 Hz, 3H, H-24), 1.24 (t, J = 7.5 Hz, 3H, H-28). 13C NMR (100 MHz, CDCl3) δ 165.8 (C-20), 163.5 (C-19), 153.0 (C-8), 150.3 (C-5), 136.2 (C-17), 133.2 (C-6), 132.2 (C-10), 130.5 (C-3), 129.7 (C-11), 128.9 (C-18), 125.9 (C-13), 125.5 (C-1), 124.6 (C-2), 123.5 (C-1), 119.4 (C-9), 119.0 (C-4), 61.9 (C-23/27), 61.7 (C-23/27), 44.8 (C-15,16), 14.0 (C-24), 13.9 (C-28). (ESI) m/z 384 (M+1)+.:1).1H NMR (400 MHz, CDCl3) δ 8.22 (s, 1H, H-15), 7.45 (dm, J = 7.8 Hz, 1H, H-13), 7.22 (tm, J = 8.7 Hz, 1H, H-11), 7.08 (tm, J = 8.5 Hz, 1H, H-3), 6.94 (tm, J = 7.6 Hz, 1H, H-12), 6.88 (dm, J = 7.9, 1H, H-1), 6.77 (dm, J = 8.2 Hz, 1H, H-4), 6.70 (tm, J = 7.6 Hz, 1H, H-2), 6.63 (dm, J = 8.4 Hz, 1H, H-10), 4.30 (q, J = 6.1 Hz, 2H, H-21), 4.28 (q, J = 7.0 Hz, 2H, H-26), 3.33–3.22 (m, 4H, H-22,23), 2.04–1.67 (m, 4H, H-29,30), 1.32 (t, J = 7.2 Hz, 3H, H-27), 1.23 (t, J = 7.2 Hz, 3H, H-28). 13C NMR (100 MHz, CDCl3) δ 166.6 (C-18), 164.1 (C-17), 157.4 (C-9), 142.1 (C-6), 142.0 (C-5), 137.8 (C-15), 131.7 (C-11), 128.9 (C-13), 126.6 (C-16), 125.6 (C-3), 122.7 (C-1), 122.7 (C-14), 121.4 (C-12), 117.5 (C-2), 115.3 (C-4), 115.3 (C-10), 61.4 (C-21), 61.3 (C-26), 49.7 (C-22,23), 25.2 (C-29,30), 14.0 (C-27), 13.7 (C-28). (ESI) m/z 410 (M+1)+.:1).1H NMR (400 MHz, CDCl3) δ 7.93 (s, 1H, H-15), 7.78 (d, J = 8.0 Hz, 1H, H-1), 7.53 (d, J = 7.8 Hz, 1H, H-13), 7.40 (t, J = 7.8 Hz, 1H, H-11), 7.30 (t, J = 7.8 Hz, 1H, H-3), 7.23–7.15 (m, 2H, H-2,12), 6.95 (d, J = 8.1 Hz, 1H, H-10), 6.87 (d, J = 8.3 Hz, 1H, H-4), 4.26 (q, J = 7.1 Hz, 4H, H-21,29), 3.74–3.61 (m, 4H, H-22,23), 2.08–1.94 (m, 4H, H-24,26), 1.70–1.58 (m, 2H, H-25), 1.28 (t, J = 7.1 Hz, 3H, H-31), 1.24 (t, J = 7.2 Hz, 3H, H-30). 13C NMR (100 MHz, CDCl3) δ 165.9 (C-18), 163.6 (C-17), 153.4 (C-9), 149.8 (C-5), 136.5 (C-15), 133.5 (C-6), 132.2 (C-11), 130.0 (C-3), 129.5 (C-13), 128.7 (C-16), 125.4 (C-14), 125.0 (C-2,12), 124.1 (C-1), 119.8 (C-4), 118.8 (C-10), 61.8 (C-29), 61.7 (C-21), 54.4 (C-22,23), 23.5 (C-24,26), 21.7 (C-25), 14.0 (C-31), 13.9 (C-30). (ESI) m/z 424 (M+1)+.MW-assisted isomerization of the biaryl vinyl compounds in DMSO
A solution of the vinyl precursor (14a,b,d, 31a–f) (2.00 mmol) in 1 mL dry DMSO was irradiated in a 10 mL MW process vial at the temperature and for the reaction time as indicated below. The reaction mixture was subsequently cooled to ambient temperature and poured into CH2Cl2 (15 mL). The organic layer was washed with H2O (3 × 15 mL), dried (MgSO4), filtered and evaporated to dryness. The residue obtained was purified by flash column chromatography on silica gel with hexane:EtOAc 4:1 eluent or by preparative HPLC.
:EtOAc 9:1). 1H NMR (CDCl3) δ (ppm): 7.39–7.33 (m, 2H, H-4,13), 7.24–7.13 (m, 2H, H-2,11), 7.12–7.08 (m, 1H, H-3), 7.05–7.01 (m, 1H, H-1), 6.83–6.78 (m, 1H, H-10), 3.77 (br s, 2H, H-16), 3.69 (br s, 2H, H-15), 2.79 (s, 3H, H-17). 13C NMR (CDCl3) δ (ppm): 157.2 (C-9), 150.3 (C-5), 145.5 (C-6), 131.8 (C-13), 130.6 (C-15), 126.3 (C-4), 124.5 (C-4), 124.5 (C-12), 124.5 (C-14), 123.8 (C-3), 119.9 (C-1), 118.3 (C-10), 115.6 (C-19), 115.6 (C-21), 64.8 (C-18), 40.8 (C-17), 39.3 (C-15), 36.4 (C-16). Anal. calcd for C18H15N3O (289.33): C, 74.72; H, 5.23; N, 14.52. Found: C, 75.17; H, 4.66; N, 13.85.:1). 1H NMR (500 MHz, CDCl3) δ 7.35 (dm, J = 6.8 Hz, 1H, H-33), 7.34 (dm, J = 7.1 Hz, 1H, H-15), 7.30–7.25 (m, 2H, H-13,31), 7.16 (tm, J = 7.8 Hz, 1H, H-20), 7.10 (td, J = 7.5, 1.2 Hz, 1H, H-32), 7.06 (td, J = 7.5, 1.1 Hz, 1H, H-14), 6.95–6.90 (m, 2H, H-2,19), 6.92 (dd, J = 7.9, 1.7 Hz, 1H, H-22), 6.86 (td, J = 7.6, 1.4 Hz, 1H, H-21), 6.82 (dd, J = 8.3, 1.1 Hz, 1H, H-30), 6.69 (dd, J = 8.4, 1.1 Hz, 1H, H-12), 6.66 (dd, J = 7.9, 1.6 Hz, 1H, H-4), 6.51 (t, J = 7.8 Hz, 1H, H-3), 5.31 (d, J = 7.4 Hz, 1H, H-26), 4.29 (t, J = 8.0 Hz, 1H, H-36), 4.26 (t, J = 8.0 Hz, 1H, H-18), 3.64–3.54 (m, 2H, H-8,9), 3.49–3.40 (m, 3H, H-9,17), 3.38–3.31 (m, 3H, H-27,35), 3.32–3.21 (m, 1H, H-27), 2.68–2.51 (m, 1H, H-40), 2.00–1.90 (m, 2H, H-37,39), 1.89–1.72 (m, 3H, H-38,39), 1.68–1.57 (m, 1H, H-37). 13C NMR (125 MHz, CDCl3) δ 157.1 (C-11), 156.2 (C-29), 144.5 (C-23), 141.3 (C-24), 141.2 (C-5), 138.9 (C-6), 131.9 (C-33), 131.6 (C-15), 130.5 (C-13), 130.4 (C-31), 127.0 (C-1), 126.0 (C-20), 125.5 (C-2), 123.2 (C-32), 122.7 (C-34), 122.6 (C-14), 122.1 (C-16), 121.6 (C-22), 121.3 (C-4), 119.8 (C-21), 118.5 (C-19), 117.6 (C-3), 117.0 (C-30), 116.3 (C-12), 112.6 (C-41/43/45/47), 59.8 (C-26), 58.7 (C-8), 50.6 (C-9), 49.1 (C-27), 41.0 (C-40), 32.9 (C-35), 32.8 (C-17), 28.8 (C-37), 24.0 (C-38), 22.8 (C-18), 22.7 (C-39), 22.5 (C-36). MS (ESI) m/z 631 (M+1)+.:EtOAc 5:1). 1H NMR (CDCl3) δ (ppm): 7.37–7.31 (m, 2H, H-8,15), 7.17–7.09 (m, 2H, H-6,13), 7.08–7.02 (m, 2H, H-7,14), 7.00 (dm, J = 8.0 Hz, 1H, H-5), 6.75 (dm, J = 8.0 Hz, 1H, H-12), 4.33 (q, J = 7.2 Hz, 2H, H-21), 4.01 (d, J = 13.2 Hz, 1H, H-17), 3.91 (d, J = 14.8 Hz, 1H, H-2), 3.37 (dd, J = 14.8, 1.8 Hz, 1H, H-2), 3.03 (dd, J = 13.2, 1.8 Hz, 1H, H-17), 2.73 (s, 3H, H-3), 1.37 (t, J = 7.2 Hz, 3H, H-22). 13C NMR (CDCl3) δ (ppm): 168.8 (C-20), 157.3 (C-11), 150.7 (C-9), 146.4 (C-4), 131.7 (C-15), 129.7 (C-13), 126.7 (C-16), 125.9 (C-6), 124.4 (C-8), 124.2 (C-14), 123.0 (C-7), 119.7 (C-5), 118.8 (C-19), 118.1 (C-12), 64.0 (C-2), 63.8 (C-21), 48.7 (C-18), 40.5 (C-3), 38.2 (C-17), 14.7 (C-22). Anal. calcd for C18H15N3O (289.33): C, 71.41; H, 5.99; N, 8.33. Found: C, 71.59; H, 5.29; N, 8.23.:EtOAc 4:1 gradient. White crystals. 1H NMR (CDCl3) δ (ppm): 7.35 (dd, J = 8.0, 1.7 Hz, 1H, H-10), 7.26 (d, J = 6.1 Hz, 1H, H-4), 7.24 (dd, J = 6.1, 1.4 Hz, 1H, H-12), 7.16–7.07 (m, 2H, H-2,11), 7.05–6.99 (m, 2H, H-1,3), 4.01 (s, 2H, H-16), 3.55 (s, 2H, H-18), 2.91 (s, 3H, H-15). 13C NMR (CDCl3) δ (ppm): 151.8 (C-8), 148.6 (C-5), 142.8 (C-6), 132.1 (C-10), 130.8 (C-12), 127.3 (C-13), 126.6 (C-9), 125.7 (C-2), 125.3 (C-11), 124.4 (C-4), 123.3 (C-3), 119.6 (C-1), 114.8 (C-20,22), 63.4 (C-16), 41.4 (C-15), 37.7 (C-18), 35.9 (C-19). (ESI) m/z 324 (M+1)+.:EtOAc 4:1 gradient. White crystals. 1H NMR (CDCl3) δ (ppm): 7.35 (dd, J = 7.8, 1.6 Hz, 1H, H-4), 7.30 (d, J = 8.1 Hz, 1H, H-12), 7.28 (s, 1H), 7.20 (td, J = 7.7, 1.6 Hz, 1H, H-2), 7.14–7.09 (m, 2H, H-3,11), 7.07 (dd, J = 8.0, 1.6 Hz, 1H, H-1), 6.78 (d, J = 2.1 Hz, 1H, H-9), 3.77 (s, 2H, H-16), 3.67 (s, 2H, H-17), 2.82 (s, 3H, H-15). 13C NMR (CDCl3) δ (ppm): 157.1 (C-8), 149.4 (C-5), 144.8 (C-6), 135.5 (C-10), 131.8 (C-12), 126.2 (C-2), 124.3 (C-11), 123.8 (C-4), 123.6 (C-3), 122.6 (C-13), 119.7 (C-1), 118.5 (C-19), 114.8 (C-20,22), 64.4 (C-16), 40.2 (C-15), 38.4 (C-17), 35.6 (C-18). (ESI) m/z 324 (M+1)+.:EtOAc 4:1 gradient. White crystals. 1H NMR (CDCl3) δ (ppm): 7.33 (dd, J = 7.8, 1.6 Hz, 1H, H-4), 7.30 (d, J = 8.2 Hz, 1H, H-12), 7.16 (td, J = 7.7, 1.6 Hz, 1H, H-2), 7.10–7.05 (m, 2H, H-3,11), 7.03 (dd, J = 8.0, 1.7 Hz, 1H, H-1), 6.75 (d, J = 2.1 Hz, 1H, H-9), 4.34 (qd, J = 7.2, 1.3 Hz, 2H, H-23), 3.97 (d, J = 13.5 Hz, 1H, H-17), 3.87 (dd, J = 14.7, 0.9 Hz, 1H, H-16), 3.37 (dd, J = 14.7, 1.9 Hz, 1H, H-16), 3.01 (dd, J = 13.5, 1.9 Hz, 1H, H-17), 2.76 (s, 3H, H-15), 1.38 (t, J = 7.2 Hz, 3H, H-24). 13C NMR (CDCl3) δ (ppm): 168.0 (C-19), 157.2 (C-8), 149.9 (C-5), 145.7 (C-6), 134.4 (C-10), 131.8 (C-12), 125.8 (C-2), 124.9 (C-13), 124.0 (C-11), 123.6 (C-4), 122.8 (C-3), 119.4 (C-1), 118.3 (C-9), 118.1 (C-25), 63.6 (C-16), 63.4 (C-23), 47.9 (C-18), 40.0 (C-15), 14.4 (C-24). (ESI) m/z 371 (M+1)+.:EtOAc 4:1 gradient. White crystals. 1H NMR (CDCl3) δ (ppm): 7.48 (d, J = 2.5 Hz, 1H, H-12), 7.33 (dd, J = 7.7, 1.7 Hz, 1H, H-4), 7.30 (dd, J = 8.7, 2.4 Hz, 1H, H-10), 7.17 (td, J = 7.7, 1.7 Hz, 1H, H-2), 7.10 (dd, J = 7.6, 1.7 Hz, 1H, H-1), 7.05 (td, J = 8.0, 1.7 Hz, 1H, H-3), 6.66 (d, J = 8.7 Hz, 1H, H-9), 3.76 (s, 2H, H-17), 3.69–3.57 (m, 2H, H-16), 2.80 (s, 3H, H-15). 13C NMR (CDCl3) δ (ppm): 155.8 (C-8), 149.4 (C-5), 144.8 (C-6), 133.7 (C-12), 133.0 (C-10), 126.0 (C-2,11), 123.8 (C-4), 123.4 (C-1), 119.5 (C-9), 119.4 (C-3), 116.2 (C-13), 114.7 (C-20,22), 64.3 (C-16), 40.2 (C-15), 38.2 (C-17), 35.5 (C-18). (ESI) m/z 369,371 (M+1)+.Synthesis of CH2-extended analogues
:EtOAc 4:1 eluent. Pale pink oil (0.97 g, 70%), Rf = 0.43 (hexane/EtOAc 4:1). 1H NMR (400 MHz, CDCl3) δ (ppm): 7.74–7.68 (m, 2H, H-15,18), 7.63 (t, J = 7.7 Hz, 1H, H-16), 7.43 (t, J = 7.6 Hz, 1H, H-17), 6.97–6.91 (m, 2H, H-3,5), 6.83–6.76 (m, 2H, H-2,4), 5.26 (s, 2H, H-13), 3.36–3.31 (m, 4H, H-9,12), 2.02–1.83 (m, 4H, H-10,11). 13C NMR (100 MHz, CDCl3) δ (ppm): 148.5 (C-6), 141.1 (C-14), 140.6 (C-1), 133.0 (C-16), 132.8 (C-18), 128.6 (C-15), 128.2 (C-17), 122.5 (C-3), 119.1 (C-4), 117.1 (C-20), 115.6 (C-2), 114.6 (C-5), 111.1 (C-19), 68.8 (C-13), 50.5 (C-9,12), 25.0 (C-10,11). MS (ESI) m/z 279 (M+1)+.:EtOAc 4:1 eluent. Pale yellow oil (1.28 g, 56%), Rf = 0.43 (hexane/EtOAc 4:1). 1H NMR (400 MHz, CDCl3) δ (ppm): 10.23 (s, 1H, H-20), 7.91 (d, J = 7.5 Hz, 1H, H-18), 7.79 (d, J = 7.7 Hz, 1H, H-15), 7.63 (t, J = 7.5 Hz, 1H, H-16), 7.52 (t, J = 7.5 Hz, 1H, H-17), 6.95–6.90 (m, 2H, H-3,5), 6.83–6.75 (m, 2H, H-2,4), 5.53 (s, 2H, H-13), 3.36–3.30 (m, 4H, H-9,12), 1.94–1.88 (m, 4H, H-10,11). 13C NMR (100 MHz, CDCl3) δ (ppm): 193.0 (C-20), 148.9 (C-6), 140.4 (C-1), 140.1 (C-14), 134.0 (C-16), 133.3 (C-18), 133.0 (C-19), 128.0 (C-15), 127.8 (C-17), 122.0 (C-3), 119.2 (C-4), 115.6 (C-2), 114.3 (C-5), 68.8 (C-13), 50.6 (C-9,12), 24.9 (C-10,11). MS (ESI) m/z 282 (M+1)+.:EtOAc 4:1 eluent. Orange crystals (0.28 g, 60%), Rf = 0.43 (hexane/EtOAc 4:1). 1H NMR (400 MHz, CDCl3) δ (ppm): 8.25 (s, 1H), 8.09 (d, J = 7.7 Hz, 1H), 7.63–7.50 (m, 3H), 6.97 (t, J = 7.2 Hz, 1H), 6.86 (d, J = 7.6 Hz, 1H), 6.84–6.75 (m, 2H), 5.10 (s, 2H), 3.27–3.21 (m, 4H), 1.94–1.86 (m, 4H). MS (ESI) m/z 330 (M+1)+.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
P. D. and Z. M. were recipients of the János Bolyai Research Scholarship of the Hungarian Academy of Sciences (BO/799/21/7, ÚNKP-23-ME4). The research was supported by the TKP2021-NVA-14 and ED-2022-00208. We thank the contribution of Tamás Gáti to expert NMR analyses and the assignment of the NMR spectra, of Gergő Riszter and Zoltán Kaleta to LC-MS analyses and of Zoltán Dobi and Gergely Hetényi to the synthetic work. We gratefully acknowledge our reviewer's suggestions for the mechanism in Scheme 6. This research was funded by the National Research, Development and Innovation Office (NKFIH) grant number SNN 135825. On behalf of the ‘Development and mechanistic study of DNA dyes’ (PI: Ervin Kovács) project we are grateful for the possibility to use ELKH Cloud which helped us achieve the computational results published in this paper.References
- W. R. Gutekunst and P. S. Baran, C–H functionalization logic in total synthesis, Chem. Soc. Rev., 2011, 40, 1976–1991 RSC.
- J. Yamaguchi, A. D. Yamaguchi and K. Itami, C–H Bond Functionalization: Emerging Synthetic Tools for Natural Products and Pharmaceuticals, Angew. Chem., Int. Ed., 2012, 51, 8960–9009 CrossRef CAS PubMed.
- M. C. Haibach and D. Seidel, C–H Bond Functionalization through Intramolecular Hydride Transfer, Angew. Chem., Int. Ed., 2014, 53, 5010–5036 CrossRef CAS PubMed.
- I. Klose, G. Di Mauro, D. Kaldre and N. Maulide, Nat. Chem., 2022, 14, 1306–1310 CrossRef CAS PubMed.
- S. Dutta, B. Li, D. R. L. Rickertsen, D. A. Valles and D. Seidel, C–H Bond Functionalization of Amines: A Graphical Overview of Diverse Methods, SynOpen, 2021, 5, 173–228 CrossRef CAS PubMed.
- O. Meth-Cohn and H. Suschitzky, Heterocycles by Ring Closure of Ortho-Substituted t-Anilines (The t-Amino Effect), Adv. Heterocycl. Chem., 1972, 14, 211–278 CrossRef CAS.
- O. Meth-Cohn, The t-Amino Effect: Heterocycles Formed by Ring Closure of ortho-Substituted t-Anilines, Adv. Heterocycl. Chem., 1996, 65, 1–37 CrossRef CAS.
- P. Mátyus, O. Éliás, P. Tapolcsányi, Á. Polonka-Bálint and B. Halász-Dajka, Ring-Closure Reactions of ortho-Vinyl-tert-anilines and (Di)Aza-Heterocyclic Analogues via the tert-Amino Effect: Recent Developments, Synthesis, 2006, 16, 2625–2639 CrossRef.
- W. Verboom and D. N. Reinhoudt, “Tert-amino effect” in heterocyclic synthesis. Ring closure reactions of N,N-dialkyl-1,3-dien-1-amines, Recl. Trav. Chim. Pays-Bas, 1990, 109, 311–324 CrossRef CAS.
- W. H. N. Nijhuis, W. Verboom, A. Abu El-Fadl, S. Harkema and D. N. Reinhoudt, Stereochemical aspects of the “tert-amino effect”. 1. Regioselectivity in the synthesis of pyrrolo[1,2-a]quinolines and benzo[c]quinolizines, J. Org. Chem., 1989, 54, 199–209 CrossRef CAS.
- W. H. N. Nijhuis, W. Verboom, A. Abu El-Fadl, G. J. Van Hummel and D. N. Reinhoudt, Stereochemical aspects of the “tert-amino effect”. 2. Enantio- and diastereoselectivity in the synthesis of quinolines, pyrrolo[1,2-a]quinolines, and [1,4]oxazino[4,3-a]quinolones, J. Org. Chem., 1989, 54, 209–216 CrossRef CAS.
- L. C. Groenen, W. Verboom, W. H. N. Nijhuis, D. N. Reinhoudt, G. J. Van Hummel and D. Feil, The tertiary amino effect in heterocyclic synthesis. Mechanistic and computational study of the formation of six-membered rings, Tetrahedron, 1988, 44, 4637–4644 CrossRef CAS.
- W. Verboom, D. N. Reinhoudt, R. Visser and S. Harkema, “Tert-Amino effect” in heterocyclic synthesis. Formation of N heterocycles by ring-closure reactions of substituted 2-vinyl-N,N-dialkylanilines, J. Org. Chem., 1984, 49, 269–276 CrossRef CAS.
- C. Rabong, C. Hametner, K. Mereiter, V. G. Kartsev and U. Jordis, Scope and limitations of the T-reaction employing some functionalized C-H-acids and naturally occurring secondary amines, Heterocycles, 2008, 75, 799–838 CrossRef CAS.
- N. Kaval, W. Dehaen, P. Mátyus and E. Van der Eycken, Convenient and rapid microwave-assisted synthesis of pyrido-fused ring systems applying the tert-amino effect, Green Chem., 2004, 6, 125–127 RSC.
- X. Xie, H. Huang, Y. Fan, Y. Luo, Q. Pang, X. Li and W. Huang, Assembly of spirocyclic pyrazolone-pyrrolo[4,3,2-de]quinoline skeleton via cascade [1,5] hydride transfer/cyclization by C(sp3)–H functionalization, Org. Biomol. Chem., 2023, 21, 7300–7304 RSC.
- T. Zhao, H. Zhang, L. Cui, J. Qu and B. Wang, Zinc Chloride Catalyzed Stereoselective Construction of Spiropyrazolone Tetrahydroquinolines via Tandem [1,5]-Hydride Shift/Cyclization Sequence, RSC Adv., 2015, 5, 86056–86060 RSC.
- E. R. Zaitseva, A. Yu. Smirnov, I. N. Myasnyanko, K. S. Mineev, A. I. Sokolov, T. N. Volkhina, A. A. Mikhaylov, N. S. Baleeva and M. S. Baranova, Imidazole-5-ones as a substrate for [1,5]-hydride shift triggered cyclization, New J. Chem., 2021, 45, 1805–1808 RSC.
- J. C. Ruble, A. R. Hurd, T. A. Johnson, D. A. Sherry, M. R. Barbachyn, P. L. Toogood, G. L. Bundy, D. R. Graber and G. M. Kamilar, Synthesis of (−)-PNU-286607 by Asymmetric Cyclization of Alkylidene Barbiturates, J. Am. Chem. Soc., 2009, 131, 3991–3997 CrossRef CAS PubMed.
- V. V. Shirokova, V. A. Ikonnikova, P. N. Solyev, V. A. Lushpa, A. A. Korlyukov, A. D. Volodin, N. S. Baleeva, M. S. Baranov and A. A. Mikhaylov, 1,5-Hydride-Shift-Triggered Cyclization for the Synthesis of Unsymmetric Julolidines, Synthesis, 2021, 53, 4689–4699 CrossRef CAS.
- A. M. Piloto, G. Hungerford, S. P. G. Costa and M. S. T. Gonçalves, Photoinduced Release of Neurotransmitter Amino Acids from Coumarin-Fused Julolidine Ester Cages, Eur. J. Org Chem., 2013, 34, 7715–7723 CrossRef.
- M. Bojtár, K. Németh, F. Domahidy, G. Knorr, A. Verkman, M. Kállay and P. Kele, Conditionally Activatable Visible-Light Photocages, J. Am. Chem. Soc., 2020, 142, 15164–15171 CrossRef PubMed.
- S. Wiedbrau, B. Maerz, E. Samoylova, A. Reiner, F. Trommer, P. Mayer, W. Zinth and H. Dube, Twisted Hemithioindigo Photoswitches: Solvent Polarity Determines the Type of Light-Induced Rotations, J. Am. Chem. Soc., 2016, 138, 12219–12227 CrossRef PubMed.
- S. Qiu, A. T. Frawley, K. G. Leslie and H. L. Anderson, How do donor and acceptor substituents change the photophysical and photochemical behavior of dithienylethenes? The search for a water-soluble visible-light photoswitch, Chem. Sci., 2023, 14, 9123–9135 RSC.
- M. Noguchi, H. Yamada and T. Sunagawa, A novel and regioselective pyridine-ring formation by Lewis acid induced cyclisation of 2-(N-allylbenzylamino)-3-[2,2-bis(ethoxycarbonyl)vinyl]pyrido[1,2-a]pyrimidin-4(4H)-one, J. Chem. Soc., Perkin Trans. 1, 1998, 3327–3329 RSC.
- S. Murarka, C. Zhang, M. D. Konieczynska and D. Seidel, Lewis Acid Catalyzed Formation of Tetrahydroquinolines via an Intramolecular Redox Process, Org. Lett., 2009, 11, 129–132 CrossRef CAS PubMed.
- S. Murarka, I. Deb, C. Zhang and D. Seidel, Catalytic Enantioselective Intramolecular Redox Reactions: Ring-Fused Tetrahydroquinolines, J. Am. Chem. Soc., 2009, 131, 13226–13227 CrossRef CAS PubMed.
- Y. K. Kang, S. M. Kim and D. Y. Kim, Enantioselective Organocatalytic C-H Bond Functionalization via Tandem 1,5-Hydride Transfer/Ring Closure: Asymmetric Synthesis of Tetrahydroquinolines, J. Am. Chem. Soc., 2010, 132, 11847–11849 CrossRef CAS PubMed.
- W. Cao, X. Liu, W. Wang, L. Lin and X. Feng, Highly Enantioselective Synthesis of Tetrahydroquinolines via Cobalt(II)-Catalyzed Tandem 1,5-Hydride Transfer/Cyclization, Org. Lett., 2011, 13, 600–603 CrossRef CAS PubMed.
- K. Mori, K. Ehara, K. Kurihara and T. Akiyama, Selective Activation of Enantiotopic C(sp3)-Hydrogen by Means of Chiral Phosphoric Acid: Asymmetric Synthesis of Tetrahydroquinoline Derivatives, J. Am. Chem. Soc., 2011, 133, 6166–6169 CrossRef CAS PubMed.
- K. Mori, R. Isogai, Y. Kamei, M. Yamanaka and T. Akiyama, Chiral Magnesium Bisphosphate-Catalyzed Asymmetric Double C(sp3)–H Bond Functionalization Based on Sequential Hydride Shift/Cyclization Process, J. Am. Chem. Soc., 2018, 140, 6203–6207 CrossRef CAS PubMed.
- K. Mori, C(sp3)–H Bond Functionalization Mediated by Hydride a Shift/Cyclization System, Bull. Chem. Soc. Jpn., 2022, 95, 296–305 CrossRef CAS.
- K. M. McQuaid, J. Z. Long and D. Sames, C-H Bond Functionalization via Hydride Transfer: Synthesis of Dihydrobenzopyrans from ortho-Vinylaryl Akyl Ethers, Org. Lett., 2009, 11, 2972–2975 CrossRef CAS PubMed.
- K. Mori, T. Kawasaki, S. Sueoka and T. Akiyama, Expeditious Synthesis of Benzopyrans via Lewis Acid-Catalyzed C-H Functionalization: Remarkable Enhancement of Reactivity by an Ortho Substituent, Org. Lett., 2010, 12, 1732–1735 CrossRef CAS PubMed.
- E. R. Zaitseva, A. Yu. Smirnov, V. I. Timashev, V. I. Malyshev, E. A. Zhigileva, A. A. Mikhaylov, M. G. Medvedev, N. S. Baleeva and M. S. Baranov, BF3 Mediated [1,5]-Hydride Shift Triggered Cyclization: Thioethers Join the Game, Eur. J. Org Chem., 2022, e202200547 CrossRef CAS.
- S. J. Mahoney, D. T. Moon, J. Hollinger and E. Fillion, Functionalization of Csp3–H bond—Sc(OTf)3-catalyzed domino 1,5-hydride shift/cyclization/Friedel–Crafts acylation reaction of benzylidene Meldrum's acids, Tetrahedron Lett., 2009, 50, 4706–4709 CrossRef CAS.
- K. Mori, S. Sueoka and T. Akiyama, Rapid Access to 3-Aryltetralin Skeleton via C(sp3)-H Bond Functionalization: Investigation on the Substituent Effect of Aromatic Ring Adjacent to C-H Bond in Hydride Shift/Cyclization Sequence, Chem. Lett., 2011, 40, 13861388 Search PubMed.
- K. Mori, S. Sueoka and T. Akiyama, Expeditious Construction of a Carbobicyclic Skeleton via sp3-C-H Functionalization: Hydride Shift from an Aliphatic Tertiary Position in an Internal Redox Process, J. Am. Chem. Soc., 2011, 133, 2424–2426 CrossRef CAS PubMed.
- Á. Polonka-Bálint, C. Saraceno, K. Ludányi, A. Bényei and P. Mátyus, Novel Extensions of the tert-Amino Effect: Formation of Phenanthridines and Diarene-Fused Azocines from ortho-ortho′-Functionalized Biaryls, Synlett, 2008, 18, 2846–2850 Search PubMed.
- Á. A. Földi, K. Ludányi, A. C. Bényei and P. Mátyus, tert-Amino Effect in peri-Substituted Naphthalenes: Syntheses of Naphthazepine and Naphthazonine Ring Systems, Synlett, 2010, 14, 2109–2113 Search PubMed.
- P. Dunkel, G. Túrós, A. Bényei, K. Ludányi and P. Mátyus, Synthesis of novel fused azecine ring systems through application of the tert-amino effect, Tetrahedron, 2010, 66, 2331–2339 CrossRef CAS.
- P. Bottino, P. Dunkel, M. Schlich, L. Galavotti, R. Deme, G. Regdon Jr, A. Bényei, K. Pintye-Hódi, G. Ronsisvalle and P. Mátyus, Study on the scope of tert-amino effect: new extensions of type 2 reactions to bridged biaryls, J. Phys. Org. Chem., 2012, 25, 1033–1041 CrossRef CAS.
- P. Dunkel, Novel Extensions of the Tert-Amino Effect: Synthesis of Azecine- and Oxazonine-Fused Ring Systems, PhD dissertation, Semmelweis University, 2012 Search PubMed.
- E. Kovács, B. Huszka, T. Gáti, M. Nyerges, F. Faigl and Z. Mucsi, Chemoselective Strategy for the Direct Formation of Tetrahydro-2,5-methanobenzo[c]azepines or Azetotetrahydroisoquinolines via Regio- and Stereoselective Reactions, J. Org. Chem., 2019, 84, 7100–7112 CrossRef PubMed.
- I. G. Molnár, Z. Mucsi, E. Kovács and M. Nyerges, Electrocyclization and Unexpected Reactions of Non-Stabilized α,β:γ,δ-Unsaturated Azomethine Ylides: Experimental and Theoretical Studies, Synthesis, 2022, 54, 3845–3857 CrossRef.
- J. O'Leary, X. Formosa, W. Skranc and J. D. Wallis, Structural studies of peri-interactions and bond formation between electron-rich atomic centres and N-phenylcarboxamides or nitroalkenyl groups, Org. Biomol. Chem., 2005, 3, 3273–3283 RSC.
- Y. Feng, H. Wang, F. Sun, Y. Li, X. Fu and K. Jin, A highly efficient and widely functional-group-tolerant catalyst system for copper(I)-catalyzed S-arylation of thiols with aryl halides, Tetrahedron, 2009, 65, 9737–9741 CrossRef CAS.
- K. C. Nicolau and S. T. Harrison, Total synthesis of abyssomicin C, atrop-abyssomycin C, and abyssomicin D: implications for natural origins of atrop-abyssomicin C, J. Am. Chem. Soc., 2007, 129, 429–440 CrossRef PubMed.
- P. Mátyus, K. Fuji and K. Tanaka, Efficient and facile syntheses of [4,5]-annelated pyridazines from 4-pyridazinecarbaldehydes, Heterocycles, 1994, 37, 171–174 CrossRef.
- J. Zabicky, The kinetics and mechanism of carbonyl-methylene condensation reactions. XI. Stereochemistry of the products, J. Chem. Soc., 1961, 683–684 RSC.
- P. C. Bell, M. Brameh, N. Hanly and J. D. Wallis, Interaction between a dimethylamino group and an electron-deficient alkene in ethyl (E)-2-cyano-3-(8-dimethylamino-1-naphthyl)propenoate, Acta Crystallogr., 2000, C56, 670–671 CAS.
- W. Verboom, D. N. Reinhoudt, R. Visser and S. Harkema, “Tert-Amino effect” in heterocyclic synthesis. Formation of N-heterocycles by ring-closure reactions of substituted 2-vinyl-N,N-dialkylanilines, J. Org. Chem., 1984, 49, 269–276 CrossRef CAS.
- N. S. Baleeva, A. Yu. Smirnov, E. R. Zaitseva, D. S. Ivanov, A. I. Sokolov, A. A. Mikhaylov, I. N. Myasnyanko and M. S. Baranov, Photoinduced [1,5]-hydride shift triggered cyclization, New J. Chem., 2023, 47, 12536–12540 RSC.
- S.-S. Li, L. Zhou, L. Wang, H. Zhao, L. Yu and J. Xiao, Organocatalytic C(sp3)–H Functionalization via Carbocation-Initiated Cascade [1,5]-Hydride Transfer/Cyclization: Synthesis of Dihydrodibenzo[b,e]azepines, Org. Lett., 2018, 20, 138–141 CrossRef CAS PubMed.
- Y.-H. Shen, Y.-Q. Su, J.-M. Tian, S. Lin, H.-L. Li, J. Tang and W.-D. Zhang, A Unique Indolo-[1,7]naphthyridine Alkaloid from Incarvillea mairei var. Grandiflora (WEHRH.) GRIERSON, Helv. Chim. Acta, 2010, 93, 2393–2396 CrossRef CAS.
- D. Tan, G. Chou and Z. Wang, Three New Alkaloids from Senecio scandens, Chem. Nat. Compd., 2014, 50, 329–332 CrossRef CAS.
- X.-L. Yu, L. Kuang, S. Chen, X.-L. Zhu, Z.-L. Li, B. Tan and X.-Y. Liu, Counteranion-Controlled Unprecedented Diastereo- and Enantioselective Tandem Formal Povarov Reaction for Construction of Bioactive Octahydro-Dipyrroloquinolines, ACS Catal., 2016, 6, 6182–6190 CrossRef CAS.
- P. D. Brown, A. C. Willis, M. S. Sherburn and A. L. Lawrence, Total Synthesis and Structural Revision of the Alkaloid Incargranine B, Angew. Chem., Int. Ed., 2013, 52, 13273–13275 CrossRef CAS PubMed.
- K. Matsumoto, R. Nakano, K. Yamada, T. Hirokane and M. Yoshida, Catalytic and Aerobic Oxidative C–H Annulation Reaction of Saturated Cyclic Amines for Synthesis of Dipyrroloquinolines, Adv. Synth. Catal., 2023, 365, 323–329 CrossRef CAS.
- M. Simic, P. Jovanovic, M. Petkovic, G. Tasic, M. Jovanovic and V. Savic, Toward the synthesis of incargranine B and seneciobipyrrolidine. Synthesis of octahydro-dipyrroloquinoline skeleton via dipolar cycloaddition/amination sequence, J. Heterocycl. Chem., 2021, 58, 1665–1674 CrossRef CAS.
- P. Bottino, Studies on extensions of tert-amino effect: Ring-fusion to bridged biaryls and steroids, PhD thesis, Universitá degli Studi di Catania, 2012, http://archivia.unict.it/handle/10761/1349 Search PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von Ragué Schleyer, Nucleus-Independent Chemical Shifts (NICS) as an Aromaticity Criterion, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- J. W. Morore and R. G. Pearson, Kinetics and Mechanism, Wiley, New York, 3rd edn, 1981, Ch. 7 Search PubMed.
- K. Harms and S. Wocadlo, XCAD-4-CAD-4 Data Reduction, University of Marburg, Germany, 1995 Search PubMed.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, Completion and refinement of crystal structures with SIR92, J. Appl. Crystallogr., 1993, 26, 343–350 CrossRef.
- G. M. Sheldrick, A short history of SHELX, Acta Crystallogr., 2008, A64, 112–122 CrossRef PubMed.
- L. J. Farrugia, WinGX suite for small-molecule single-crystal crystallography, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef CAS.
- A. L. Spek, PLATON, An Integrated Tool for the Analysis of the Results of a Single Crystal Structure Determination, Acta Crystallogr., 1990, A46, C34 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Netherlands, 1998 Search PubMed.
- M. J. Frisch et al. Gaussian Inc Wallingford CT, 2016.
- V. Barone and M. Cossi, Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS.
- E. Kovács, B. Rózsa, A. Csomos, I. G. Csizmadia and Z. Mucsi, Amide Activation in Ground and Excited States, Molecules, 2018, 23, 2859 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2253075 and 2253076. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3ra08974f |
| This journal is © The Royal Society of Chemistry 2024 |