
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStereoselective Shi-type epoxidation with 3-oxo-4,6-O-benzylidene pyranoside catalysts: unveiling the role of carbohydrate skeletons†
Daniela Imperio‡
 *a,
Filippo Vallonia,
Diego Caprioglioa,
Alberto Minassia,
Emanuele Casali‡*b and
Luigi Panzaa
*a,
Filippo Vallonia,
Diego Caprioglioa,
Alberto Minassia,
Emanuele Casali‡*b and
Luigi Panzaa
aDepartment of Pharmaceutical Sciences, Università del Piemonte Orientale, Largo Donegani 2, 28100, Novara, Italy. E-mail: daniela.imperio@uniupo.it
bDepartment of Chemistry, University of Pavia, Viale Taramelli, 12, 27100 Pavia, Italy. E-mail: emanuele.casali@unipv.it
First published on 23rd May 2024
Abstract
Asymmetric epoxidation represents a hot topic in organic synthesis. In recent years, organocatalysts based on sugar skeletons have been exploited in asymmetric epoxidation to achieve enantiomeric pure epoxides. In this work, two different endocyclic ketones derived from glucose and galactose protected with a 4,6-O-benzylidene group have been prepared and exploited for Shi-type epoxidation. The two carbohydrates show an opposite preferential stereoselective epoxidation on various olefins, affording the epoxides in high conversions and modest enantioselectivities. DFT calculations disclosed the reasons behind the inversion of selectivity achieved by the two catalysts, showing that a delicate balance between the catalyst conformation, its protecting groups, and the secondary interactions with the substrate govern the final observed results.
Introduction
Organocatalysis has started to be one of the most valuable fields of organic synthesis to help green chemistry.1,2 The increasing demand for scaffolds in the development of new organocatalysts has spurred researchers to explore carbohydrates with distinctive characteristics, making them promising candidates for organocatalysis.3,4 In the last years, several ketones based on sugar skeletons have been developed to be exploited in Shi-type epoxidation. In 1996, Shi et al. reported the use of a fructose-derived ketone as an organocatalyst for enantioselective epoxidation of trans-olefins with potassium peroxymonosulfate (Oxone) as oxidant.5,6 Since then, several other ketones have been prepared to achieve better selectivity and yields.7–11 Small ring heterocycles, such as epoxides, are present in several natural compounds12,13 and represent highly versatile building blocks, frequently involved in the synthesis of pharmaceuticals and bioactive products.14–16 In addition, carbohydrates possess several hydroxyl groups, which is a unique and valuable characteristic to serve as binding sites, being easily converted to other functional groups based on the structural requirements for catalyst development. In addition, the attachment of bulky groups to these hydroxyl moieties can also be used to block the space around the reactive site of the catalyst, which can control and direct the stereochemistry of the reaction. Moreover, the presence of free electron pairs on the endocyclic oxygen also provides an additional binding site and aids in the regulation of the selectivity of the reaction. Among bulky and rigid groups, benzylidene moiety has been largely exploited to modulate sugar reactivities for example to direct the stereoselectivity in glycosylation reactions.17–19 In the literature, keto derivatives of glucosamine20 or mannose21 containing a benzylidene group showed interesting results in terms of epoxidation stereoselectivity. With the aim of expanding the orienting effect of benzylidene groups, two novel catalysts based on glucose and galactose skeletons (Fig. 1, catalysts 1 and 2 respectively) have been synthesized and tested on several olefins. To our delight, the change from gluco to galacto series allows a reversal of stereoselectivity in the epoxidation reaction.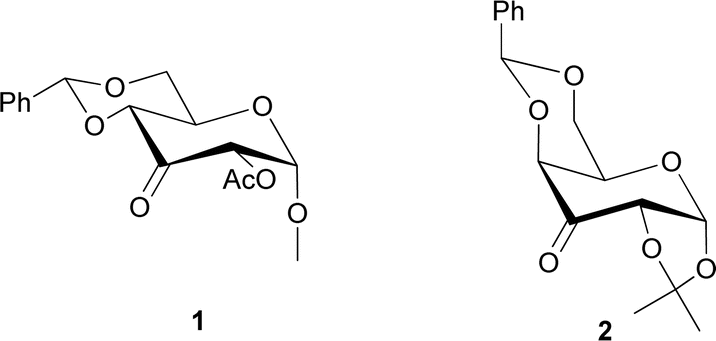 | ||
| Fig. 1 Structures of the two catalysts for asymmetric epoxidation. | ||
Results and discussion
The synthesis of catalyst 1 was achieved in three reaction steps (Scheme 1). The commercial methyl α-D-glucopyranoside (Me α-D-glu) was reacted with benzaldehyde dimethyl acetal in acetonitrile in the presence of camphorsulfonic acid (CSA) to give the crude compound 3, which crystallized from ethanol after quenching with triethylamine and solvent removal. The white solid was selectively acetylated by using a modified procedure from the literature.22 The benzylidene derivative 3 was dissolved in vinyl acetate and Lipase PS was added. The reaction was warmed at 45 °C and gently stirred for 7 days. After the filtration of the enzyme and evaporation of the solvent, compound 4 was purified by chromatography. The final oxidation with O-iodoxybenzoic acid (IBX) in dry acetonitrile at 85 °C afforded catalyst 1 in almost quantitative yields. The synthesis of catalyst 2 was performed starting from D-galactose (D-gal), which was reacted with benzaldehyde dimethyl acetal in dry DMF in the presence of camphorsulfonic acid. The reaction was quenched with triethylamine, the solvent was evaporated, and the crude was purified by flash chromatography obtaining compound 5 in satisfactory yields. The derivate was then suspended in a mixture of dimethoxypropane/acetone with a catalytic amount of camphorsulfonic acid and stirred for 1 h at 60 °C. After quenching with triethylamine, it was purified by flash chromatography and the obtained compound 6 was oxidized to final ketone 2 in the presence of IBX in dry acetonitrile at 85 °C.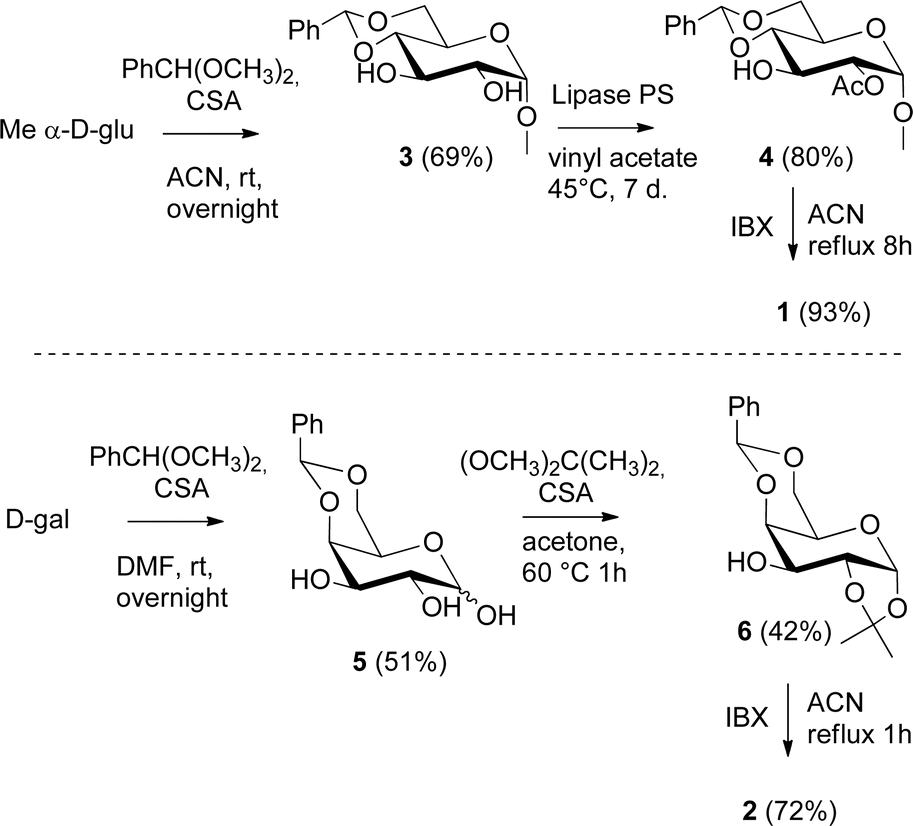 | ||
| Scheme 1 Synthesis of catalysts 1 and 2. | ||
Having in hand the two catalysts, we performed the epoxidation of several olefins at 0 °C exploiting Oxone as oxidant and keeping the pH at ∼10 by slowly adding an aqueous solution of K2CO3. The results are listed in Table 1. In all entries, excellent conversions and good yields were obtained with both of the two catalysts (80–100%). The epoxides were analyzed for the enantio- or diastereoselectivity by chiral HPLC (entries 1–5, 7–11) or by NMR (entries 6, 12). It is noteworthy that an inversion of epoxidation stereoselectivity was observed when employing the two catalysts, each possessing an opposite stereocenter at C4. Moreover, catalyst 2 resulted in better yield and stereoselectivity (see entries 4, and 10). Satisfactory results were also observed when the catalyst was used in 10% (entry 9b). When (R)-(+)-limonene was used as a substrate, a different outcome was obtained. Catalyst 1 showed to be effective on 1,2 epoxidation (entry 6), preferring the trans product.23 The two diastereomers cannot be separated by a chromatography column. Catalyst 2 performed better in terms of yields, by producing the four bis epoxide as an inseparable mixture (entry 12). Finally, considering the high conversions determined by 1H-NMR, the modest yields could be derived from the intrinsic instability of the epoxides during the purification step even if neutral silica was used.24
|
||||
|---|---|---|---|---|
| Olefin | Catalyst 1 | Catalyst 2 | ||
| Entrya (yield) | Convb erd or dr (confc; % ee) | Entrya (yield) | Convb erd or dr (confc; % ee) | |
| a All the reactions were carried out on 0.4 mol of the olefin.b Conversion was determined by 1H-NMR.c Configuration of the main stereoisomer.d Enantiomeric ratio was determined by chiral HPLC with a chiral stationary phase for enantiomers or by NMR for diastereomers.e 10% of catalyst 2 was used.f Determined by 1H NMR.g Determined by 13C-NMR. | ||||
 |
1 (75%) | 80% 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :49 :49 |
7 (65%) | 100% 31:69 (S; 38%) |
 |
2 (74%) | 100% 63:37 (S,R; 26%) |
8 (66%) | 100% 49:51 |
 |
3 (75%) | 100% 68:32 (R,R; 36%) |
9a (73%) | 100% 16:84 (S,S; 68%) |
| 9be (70%) | 98% 21:79 (S,S; 58%) |
|||
 |
4 (80%) | 100% 71:29 (R,R; 42%) |
10 (83%) | 100% 13:87 (S,S; 74%) |
 |
5 (61%) | 90% 69:31 (R,R; 38%) |
11 (71%) | 90% 21:79 (S,S; 58%) |
 |
6f (70%) | 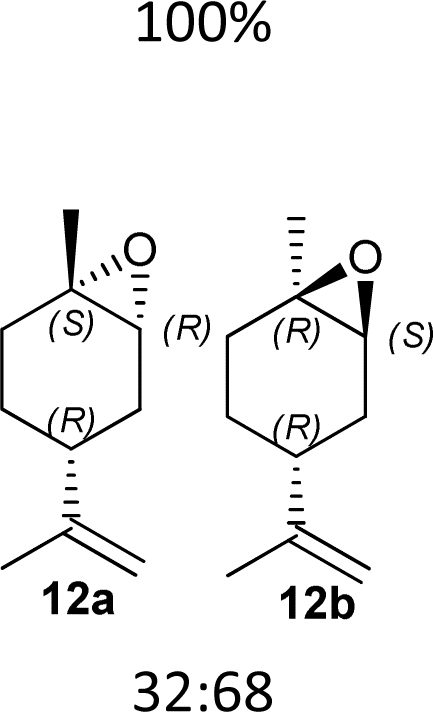 |
12g (74%) | 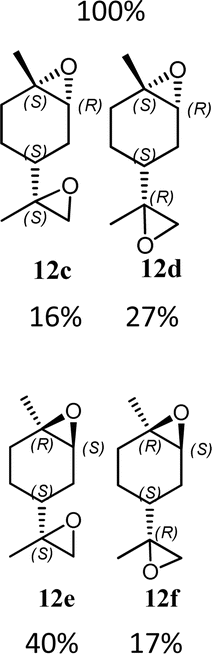 |
With the aim of providing an in-depth investigation on the reason governing the observed selectivity, extensive DFT calculations were performed (see ESI† for computational details). According to the results reported above, special attention was paid to the observed inversion in selectivity exerted by the two catalysts.
The main focus was thus applied to the interactions that regulate the observed selectivity on the epoxidation in response to the change in chirality on the C4 and protecting groups of the two sugar-based catalysts, using 9 as a model substrate.
The computational investigation started with the conformational analysis of all the possible transition states leading to the two enantiomeric products. Only the most stable for catalysts 1 and 2 will be reported and examined (Fig. 2; for a full analysis on the most stable structures for each catalyst, see the ESI file†).
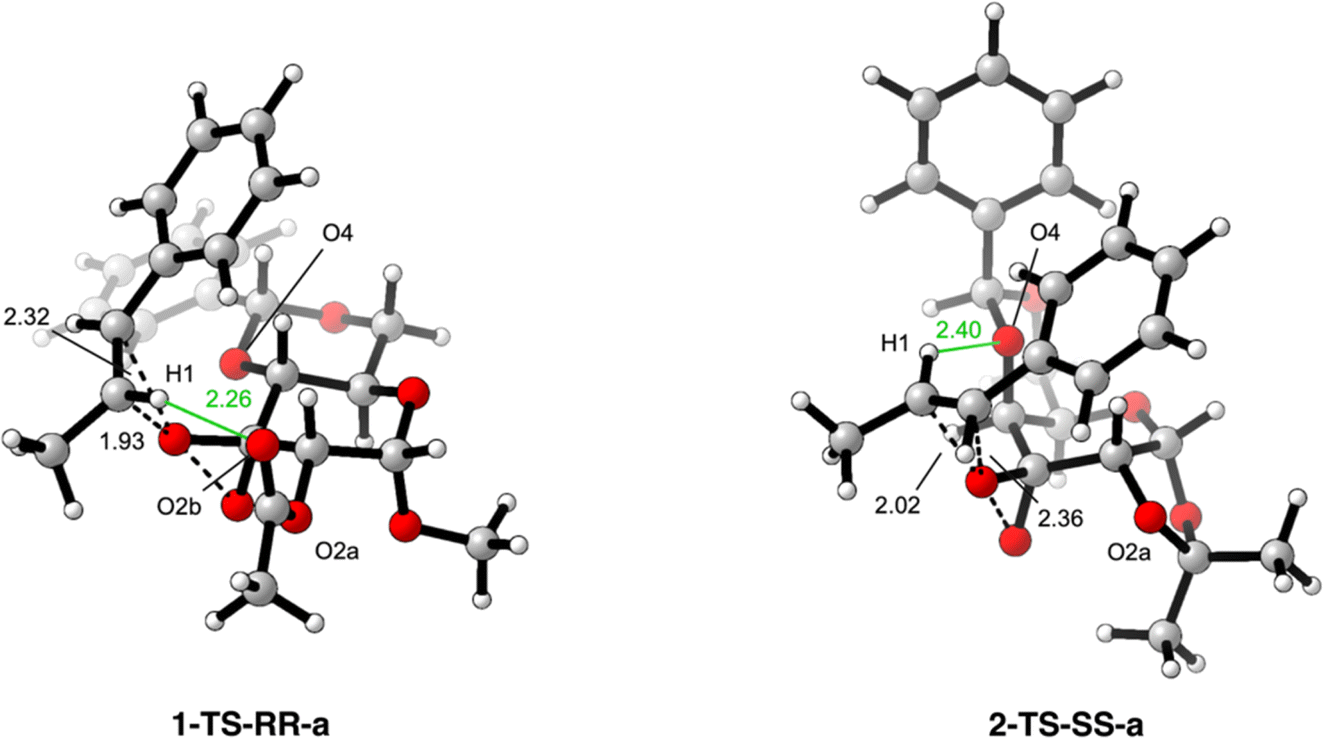 | ||
| Fig. 2 Most stable transition states for substate 9 displaying the attack on the two enantiotopic faces of the olefinic substrate, where the dashed bonds represent the ones broken or formed during the TS, while in green are highlighted the H-bonds. [SMD(Mix)-M06-2X/def2tzvp//B3LYP/6-31g(d)]. | ||
Starting from the overall analysis of both the transition state structures obtained with catalysts 1 and 2, a change in the face of the olefin where the epoxidation occurs can be spotted. In particular, while catalyst 1 prefers approaching the substrate along the Si-face (1-TS-RR-a), 2 favors the epoxidation to occur on the Re-face (2-TS-SS-a). The distances of the bonds directly involved in the epoxidation step are similar in the two cases (cfr., 1.93 Å and 2.32 Å for catalyst 1, 2.02 Å and 2.36 Å for catalyst 2) and thus cannot be used as an explanation for the obtained results. However, a closer look at the surrounding region showed an important difference between the two cases. Indeed, with catalyst 1 a non-covalent H-bond interaction can occur between the styrene olefinic hydrogen H1 and the carbonylic oxygen O2b of the acetate protecting group on C2. This H-bond is established at a distance of 2.26 Å and directs the highly polarizable hydrogen in styrene position towards the electron-rich oxygen of the nearby protecting group.
With the modification of the catalyst structure in 2, thanks to the change in chirality on C4 of the galactose ring, all the system has to accommodate the cis-decalin conformation of the benzylidene protecting group. This results in a consistent and non-innocent increase in steric encumbrance on to the upper face of the galactose ring. However, even if this strong modification in steric occupancy plays an important role in determining the approaching trajectory of the incoming substrate, another consideration has to be taken regarding the modification in the protecting group on C2. Differently from catalyst 1, 2 shows an acetonide protection which binds together the hydroxyl groups on C1 and C2. The huge difference has not to be searched in the O2a-which was present and similarly oriented also in 1, but in the absence of O2b in 2. This removes any possible opportunity for the styrene hydrogen H1 to establish and orient towards the carbonylic oxygen O2b, which was explicit in 1-but not present in 2. As a consequence of this structural modification, the styrene substrate prefers approaching the dioxirane moiety of 2 by facing the opposite side of the olefin (i.e., Re-face). The reason for this change in the olefin face can be also explained by looking at the new possible interactions that can arise between the styrene H1 and the benzylidene protecting group. Indeed, being now oriented upwards, the benzylidene group can provide the oxygen O4 to establish a new H-bond interaction with H1. This non-covalent bond occurs at 2.40 Å, a higher distance respect the one observed before, but it appears to be extremely favored by the intrinsic conformational shape of 2.
This infers that selectivity is probably impacted by the ability to establish secondary interactions with the hydrogen atoms on the olefin, along with alterations in the protecting groups and the initially assumed conformation of the catalyst. Consequently, the presence and arrangement of neighboring groups capable of engaging with the catalyst – considering the geometry assumed by the alkene in the substrate – govern the ultimate outcome.
Experimental
General
Reagents were purchased from Sigma-Aldrich, TCI, or Carlo Erba (Milan, Italy) and used without further purification. Thin-layer chromatography was performed on silica gel plates with fluorescent indicators. Visualization was accomplished by UV light (254 nm) and/or staining in ceric ammonium molybdate or sulfuric acid solution. Anhydrous solvents were obtained by utilizing activated molecular sieves (0.3 or 0.4 nm depending on the type of solvent). NMR spectra were recorded using a Bruker Avance Neo 400 MHz spectrometer (Bruker, Italy). Chemical shifts (δ) are quoted in parts per million referenced to the residual solvent peak. Melting points were determined using a Stuart Scientific SMP3 apparatus and remained uncorrected. Optical rotations were measured on a JASCO P1010 polarimeter at 20 °C. The instrument used for exact mass determination was an Orbitrap QExactive PLUS (Thermo Fischer Scientific, Bremen, Germany). Infrared Spectroscopy was performed using a Bruker Alpha II FTIR. The HPLC chromatograms were performed on a Shimadzu instrument equipped with a Lux® 5 μm cellulose-2, LC column 250 × 4.6 mm, at 25 °C with a photodiode array detector with an elution system of hexane:isopropanol at a flow rate of 0.5 mL min−1, in an isocratic way. Retention times were compared to racemic mixtures previously synthesized.10
Synthetic procedures
:5 as eluents. 650 mg of a white powder was obtained (80% yield). 1H NMR (400 MHz, CDCl3) δ 7.52 (m, 2H), 7.40 (m, 3H), 5.58 (s, 1H), 4.99 (d, J = 3.6 Hz, 1H), 4.84 (dd, J = 9.7, 3.6 Hz, 1H), 4.33 (dd, J = 9.9, 4.5 Hz, 1H), 4.22 (tapp, J = 9.5 Hz, 1H), 3.98–3.72 (m, 2H), 3.60 (t, J = 9.3 Hz, 1H), 3.44 (s, 3H), 2.43 (bs, 1H), 2.19 (s, 3H).:5 as eluents. A white powder was obtained (93% yield). Mp 152–154 °C, [α]D20: +60.6 (c = 1.0, CHCl3) 1H-NMR: (400 MHz, CDCl3) δ 7.58–7.45 (m, 2H, Ar) 7.43–7.34 (m, 3H, Ar), 5.60 (s, 1H, CH-Benz), 5,41 (dd, J = 4.0, 1H, H-2), 5.25 (d, J = 4.1, 1H, H-1), 4.44 (dd, J = 10.3, 4.6, 1H, H6a), 4.39 (d, J = 9.8, 1H, H-4), 4.14 (td, J = 9.9, 9.8, 4.6, 1H, H-5), 3.97 (t, J = 10.2, 1H, H6b), 3.49 (s, 3H, CH3–OMe) 2.25 (s, 3H, CH3–OAc). 13C-NMR: (101 MHz, CDCl3) δ 191.94 (CO), 169.66 (C OAc), 129.39 (C Ar), 128.32 (C Ar), 126.38 (C Ar), 101.99 (C Benz), 101.30 (C1), 82.14 (C4), 74.63 (C2), 69.37 (C6), 65.47 (C5), 55.66 (C OMe), 20.52 (C OAc). IR: 2940, 2914, 1751, 1740, 1235, 1122, 1051, 1014, 920, 752, 694, 462. HRMS (C16H18O7); found 345.09416 m/z [M + Na]+ (vs. calcd 345.09502).:1) and the phases were separated. The aqueous layer containing the product was evaporated and the crude was purified with a chromatographic column packed with flash silica using EtOAc/MeOH 9:1 as eluents. A yellow oil was obtained (yield 51%). 1H NMR (400 MHz, CD3OD) δ 7.58–7.53 (m, 2H), 7.40–7.34 (m, 3H), 5.62 (s, 1H), 5.25 (d, J = 3.6 Hz, 1H), 4.27 (d, J = 3.6 Hz, 1H), 4.15 (m, 2H), 3.97 (m, 2H), 3.87 (dd, J = 10.1, 3.6 Hz, 1H).:5 as eluents (yield 42%). 1H NMR (400 MHz, CDCl3) δ 7.52–7.37 (m, 5H), 5.88 (d, J = 4.3 Hz, 1H), 5.59 (s, 1H), 4.40 (dd, J = 12.7, 1.1 Hz, 1H), 4.34 (dd, J = 4.3, 2.3 Hz, 1H), 4.22–4.17 (m, 1H), 4.14 (dd, J = 12.7, 2.3 Hz, 1H), 4.10–4.04 (m, 1H), 3.88 (d, J = 1.4 Hz, 1H), 2.72 (d, J = 7.6 Hz, 1H), 1.57 (s, 3H), 1.42 (s, 3H).:5 as eluents. A waxy powder was obtained (72% yield). [α]D20: −10.3 (c = 1.0, CHCl3) 1H NMR (400 MHz, CDCl3) δ 7.52 (dd, J = 6.4, 2.8 Hz, 2H, Ar), 7.43–7.30 (m, 3H, Ar), 6.02 (d, J = 4.8 Hz, 1H, H-1), 5.63 (s, 1H, CH benz), 4.60 (d, J = 2.3 Hz, 1H, H-4), 4.54 (d, J = 4.8 Hz, 1H, H-2), 4.43 (dd, J = 12.9, 1.3 Hz, 1H, H-6a), 4.36 (m, 1H, H-5), 4.22 (dd, J = 12.9, 2.3 Hz, 1H, H-6b), 1.61 (s, 3H), 1.43 (s, 3H). 13C NMR (101 MHz, CDCl3) δ 198.14 (CO), 137.01 (C Ar), 129.32 (C Ar), 128.32 (C Ar), 100.46 (C Benz), 100.22 (C1), 76.75 (C4), 76.44 (C2), 68.86 (C6), 65.14 (C5), 26.38 (CH3), 25.96 (CH3). IR: 2986.72, 2938.37, 1738.78, 1377.86, 1295.42, 1255.90, 1213.14, 1061.34, 1025.24, 872.02, 753.05, 698.59. HRMS (C16H18O7); found 307.11755 m/z [M + H]+ (vs. calcd 307.11832).General procedure for epoxidation
In a jacketed flask cooled at 0 °C, the olefin (0.4 mmol) and the catalyst 1 or 2 (10–30 mol%) were dissolved in a mixture formed by ACN–dimethoxyethane (8 mL, 1:3). Tetrabutylammonium hydrogen sulfate (10 mg) was added followed by a buffer solution (4 mL) containing Na2(EDTA) (4 × 10−4 M) and CH3COOH–K2CO3 0.1 M (pH ∼ 9). Two solutions respectively of Oxone (1.28 mmol) (2.5 mL) and an aqueous solution of K2CO3 1 M (2.5 mL) were simultaneously added dropwise over 6 hours and then stirred for 18 hours. The mixture was diluted with water (10 mL) and extracted with the appropriate organic solvent (hexane or DCM, entry 5). The organic fraction was dried over Na2SO4 and evaporated at 25 °C. Conversion was determined by 1H NMR; enantioselectivity and the configuration of epoxides were established by comparison with those reported in the literature by using chiral HPLC10 or NMR for entry 6, 12 in Table 1. The crude material was purified by flash chromatography on neutral silica (SiO2·Et3N 2.5%).24
Products characterization
:5 hexane/2-propanol, 0.5 mL min−1, monitor at 220 nm): 10.1 min (R) and 10.7 min (S).:10 hexane/2-propanol, 0.5 mL min−1, monitor at 210 nm): 8.4 min (R,S) and 9.3 min (S,R).:10 hexane/2-propanol, 0.5 mL min−1, monitor at 220 nm): 8.6 min (R,R) and 10.2 min (S,S).:5 hexane/2-propanol, 0.5 mL min−1, monitor at 220 nm): 7.7 min (R,R) and 8.4 min (S,S).:20 hexane/2-propanol, 0.5 mL min−1, monitor at 210 nm): 13.1 min (R,R) and 15.1 min (S,S).13C NMR (101 MHz, CDCl3) δ 149.26 (12a), 149.05 (12 b), 109.09, 109.03, 60.56, 59.30, 57.53, 57.37, 40.76, 36.21, 30.76, 30.73, 29.88, 28.63, 25.92, 24.35, 24.29, 23.11, 21.10, 20.23.
Conclusions
In summary, the synthesis of two organocatalysts based on the skeleton of glucose and galactose both oxidized at carbon-3 and protected as 4,6-benzylidene was achieved shortly, in good yields starting from renewable and low-cost materials. The two catalysts showed great efficiency in epoxidation reactions, almost always achieving complete conversion and good yields. Although some stereoselectivities were modest, they demonstrated the ability to handle typically challenging terminal olefin substrates. (e.g., styrene, entry 1, 7). Furthermore, through DFT investigations, we discovered that by altering the protecting group on the C2 hydroxyl and reversing the C4 stereocenter, an important interaction could be eliminated, resulting in an opposite stereoselectivity in epoxidation and thus circumventing the need for the costlier enantiomer of the sugars.Data availability
Data will be made available on request.Author Contributions
Conceptualization: DI, LP; data curation: DI, EC; formal analysis: EC; investigation DI, EC, FV, DC; supervision: LP, AM; writing – original draft: DI, EC; writing – review & editing: all authors.Conflicts of interest
The authors declare that “There are no conflicts to declare.”Acknowledgements
This work was supported by the Italian Ministry for University and Research (MUR, PRIN 2020, 2020AEX4TA project). The authors want to express their gratitude to Professor Lucio Toma, for the insightful discussions.References
- A. Antenucci, S. Dughera and P. Renzi, ChemSusChem, 2021, 14, 2785–2853 CrossRef CAS PubMed.
- D. Krištofíková, V. Modrocká, M. Mečiarová and R. Šebesta, ChemSusChem, 2020, 13, 2828–2858 CrossRef PubMed.
- S. K. Singh, N. Mishra, S. Kumar, M. K. Jaiswal and V. K. Tiwari, ChemistrySelect, 2022, 7, e202201314 CrossRef CAS.
- F. Gallier and L. S. de M. e Miranda, Org. Biomol. Chem., 2022, 20, 919–933 RSC.
- Z.-X. Wang, Y. Tu, M. Frohn, J.-R. Zhang and Y. Shi, J. Am. Chem. Soc., 1997, 119, 11224–11235 CrossRef CAS.
- Y. Tu, Z.-X. Wang and Y. Shi, J. Am. Chem. Soc., 1996, 118, 9806–9807 CrossRef CAS.
- D. Goeddel, L. Shu, Y. Yuan, O. A. Wong, B. Wang and Y. Shi, J. Org. Chem., 2006, 71, 1715–1717 CrossRef CAS PubMed.
- C. P. Burke, L. Shu and Y. Shi, J. Org. Chem., 2007, 72, 6320–6323 CrossRef CAS PubMed.
- X. Feng and H. Du, Chin. J. Chem., 2021, 39, 2016–2026 CrossRef CAS.
- D. Imperio, E. Casali, E. Del Grosso, D. Caprioglio, A. Minassi and L. Panza, Eur. J. Org. Chem., 2024, 27, e20230116 CrossRef.
- F. E. Held, S. Wei, K. Eder and S. B. Tsogoeva, RSC Adv., 2014, 4, 32796–32801 RSC.
- J. Marco-Contelles, M. T. Molina and S. Anjum, Chem. Rev., 2004, 104, 2857–2900 CrossRef CAS PubMed.
- K. Miyashita and T. Imanishi, Chem. Rev., 2005, 105, 4515–4536 CrossRef CAS PubMed.
- F. Moschona, I. Savvopoulou, M. Tsitopoulou, D. Tataraki and G. Rassias, Catalysts, 2020, 10, 1117 CrossRef CAS.
- J. Li, T. Wu, X. Song, Y. Zheng, J. Meng, Q. Qin, Y. Liu, D. Zhao and M. Cheng, RSC Adv., 2020, 10, 18953–18958 RSC.
- K. Okuno, R. Nishiyori, K. Abe, T. Mori and S. Shirakawa, Chirality, 2022, 34, 915–924 CrossRef CAS PubMed.
- S. Figueroa-Pérez and R. R. Schmidt, Carbohydr. Res., 2000, 328, 95–102 CrossRef PubMed.
- L. Del Bino, I. Calloni, D. Oldrini, M. M. Raso, R. Cuffaro, A. Ardá, J. D. C. Codée, J. Jiménez-Barbero and R. Adamo, Chem.–Eur. J., 2019, 25, 16277–16287 CrossRef CAS PubMed.
- D. Imperio, L. Morelli, F. Compostella and L. Panza, Synlett, 2021, 32, 287–290 CrossRef CAS.
- S. E. Lewis, Annu. Rep., Sect. B: Org. Chem., 2010, 106, 34 RSC.
- J. M. Vega-Pérez, I. Periñán, M. Vega-Holm, C. Palo-Nieto and F. Iglesias-Guerra, Tetrahedron, 2011, 67, 7057–7065 CrossRef.
- L. Panza, S. Brasca, S. Riva and G. Russo, Tetrahedron: Asymmetry, 1993, 4, 931–932 CrossRef CAS.
- W. B. Cunningham, J. D. Tibbetts, M. Hutchby, K. A. Maltby, M. G. Davidson, U. Hintermair, P. Plucinski and S. D. Bull, Green Chem., 2020, 22, 513–524 RSC.
- N. Nieto, I. Munslow, H. Fernández-Pérez and A. Vidal-Ferran, Synlett, 2008, 2008, 2856–2858 CrossRef.
- A. V. Demchenko, P. Pornsuriyasak and C. De Meo, J. Chem. Educ., 2006, 83, 782 CrossRef CAS.
- B. Danieli, M. Luisetti, G. Sampognaro, G. Carrea and S. Riva, J. Mol. Catal. B: Enzym., 1997, 3, 193–201 CrossRef CAS.
- R. I. Duclos, Chem. Phys. Lipids, 2001, 111, 111–138 CrossRef CAS PubMed.
- H. Busse, M. Hakoda, M. Stanley and H. Streicher, J. Carbohydr. Chem., 2007, 26, 159–194 CrossRef CAS.
- Y. Tu, Z.-X. Wang and Y. Shi, J. Am. Chem. Soc., 1996, 118, 9806–9807 CrossRef CAS.
- H. Tanaka, H. Nishikawa, T. Uchida and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 12034–12041 CrossRef CAS PubMed.
- Z.-X. Wang, Y. Tu, M. Frohn, J.-R. Zhang and Y. Shi, J. Am. Chem. Soc., 1997, 119, 11224–11235 CrossRef CAS.
- P. C. B. Page, M. M. Farah, B. R. Buckley and A. J. Blacker, J. Org. Chem., 2007, 72, 4424–4430 CrossRef CAS PubMed.
- Y. Mahamat Ahmat and S. Kaliaguine, ACS Omega, 2022, 7, 31789–31800 CrossRef CAS PubMed.
- L. Schutz, F. Kazemi, E. Mackenzie, J. Bergeron, E. Gagnon and J. P. Claverie, J. Polym. Sci., 2021, 59, 321–328 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03011g |
| ‡ DI and EC contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |