
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOne-pot synthesis of 4-pyrimidone-2-thioether through base/acid-mediated condensation of S-alkylisothiourea and β-ketoester†
Zhichao Lu *a,
Tsung-Yun Wonga,
Yonghong Gana,
Guihui Chenb,
Dinesh J. Paymodea and
Cheng-yi Chen*a
*a,
Tsung-Yun Wonga,
Yonghong Gana,
Guihui Chenb,
Dinesh J. Paymodea and
Cheng-yi Chen*a
aMirati Therapeutics, San Diego, California 92121, USA. E-mail: luz@mirati.com
bPharmaBlock (USA), Inc., 777 Schwab Road, Unit D, Hatfield, Pennsylvania 19440, USA
First published on 13th February 2024
Abstract
4-Pyrimidone-2-thioethers can be useful synthetic precursors to densely functionalized pyrimidines, commonly encountered in bioactive molecules. A convenient one-pot access to 4-pyrimidone-2-thioethers is reported herein, which utilizes a sequential base- and acid-mediated condensation of alkylisothioureas with β-ketoesters. Owing to mild reaction conditions, good to excellent functional group tolerance and yields are achieved. The utility of this approach is demonstrated by the synthesis of the crucial adagrasib intermediate on a 200 gram scale.
Introduction
A pyrimidine ring is a staple of many naturally occurring, biorelevant molecules and a common feature in various other biologically active compounds.1,2 4-Pyrimidone-2-thioether serves as a versatile building block for the introduction of a pyrimidine ring and further chemical modifications in the syntheses of these biologically active compounds. In particular, it enables regioselective functionalization of the 2-position over the more reactive 4-position.3,4 For instance, with the 4-hydroxy group intact, the alkylthio at the 2-position can either serve as a linker to another moiety, or a latent sulfone for Nucleophilic Aromatic Substitution (SNAr). The 4-position can serve as a handle to introduce other moieties by ether formation or via an activation/SNAr sequence. One good example is the synthesis of adagrasib which is an oral, highly selective, and potent covalent KRASG12C inhibitor drug (Fig. 1) for patients with non-small cell lung cancer (NSCLC).4,5 Installation of a prolinol on the 2-position followed with the functionalized piperazine side chain on its 4-position was achieved, which avoided expensive palladium catalysis and protecting group manipulations.2 Selected examples with 2,4-functionalized pyrimidine, as shown in Fig. 1, include Rilpivirine as a non-nucleoside reverse transcriptase inhibitor for the prevention and treatment of HIV infection,6 Cerdulatinib as a SYK/JAK kinase inhibitor for the treatment of peripheral T-cell lymphoma;7 Revaprazan as an acid pump antagonist for the treatment of gastritis,8,9 Surufatinib as a tyrosine kinase inhibitor for the treatment of pancreatic and neuroendocrine tumor,10 and RK-287107 as a tankyrase inhibitor for colorectal cancer treatment.11 We envisioned that an efficient synthesis of 4-pyrimidone-2-thioether could significantly simplify access to these bioactive molecules and hence facilitate the discovery of new pyrimidine-containing drugs.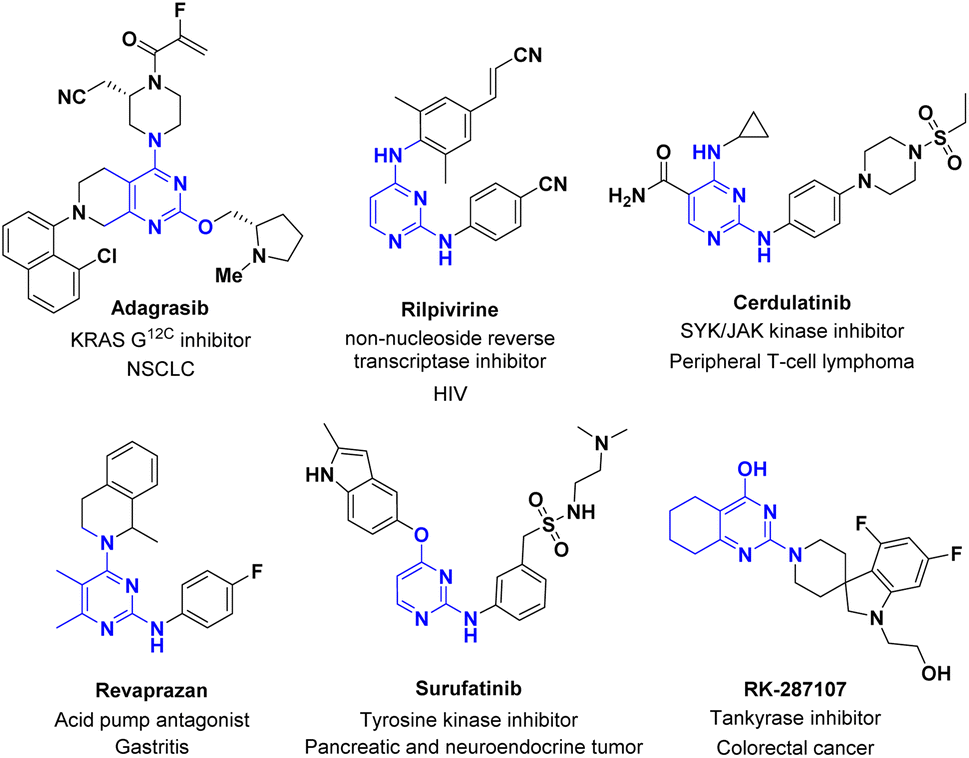 | ||
| Fig. 1 Approved or investigational drugs containing 2,4 functionalized pyrimidine core. | ||
The synthesis of 4-pyrimidone-2-thioether is typically achieved by a sequential β-ketoester condensation with thiourea and alkylation of thiol (Scheme 1A).12–15 This method usually affords high yields, but necessitates two chemical steps and is prone to overalkylation leading to the dialkyl impurity.4 Alternatively, the direct condensation of S-alkylisothiourea with β-ketoester leads to the desired 4-pyrimidone-2-thioether.4,11,16–19 This method takes advantage of commercial availability and easy access to many S-alkylisothioureas. However, the harsh reaction conditions with strong base and heating often cause S-alkylisothiourea degradation to side products and pungent thiol further complicates the workup (Scheme 1B). Herein, we wish to report a one-pot base/acid-promoted S-alkylisothiourea and β-ketoester condensation as an alternative method to circumvent the aforementioned issues with the existing approaches to 4-pyrimidone-2-thioether synthesis (Scheme 1C).
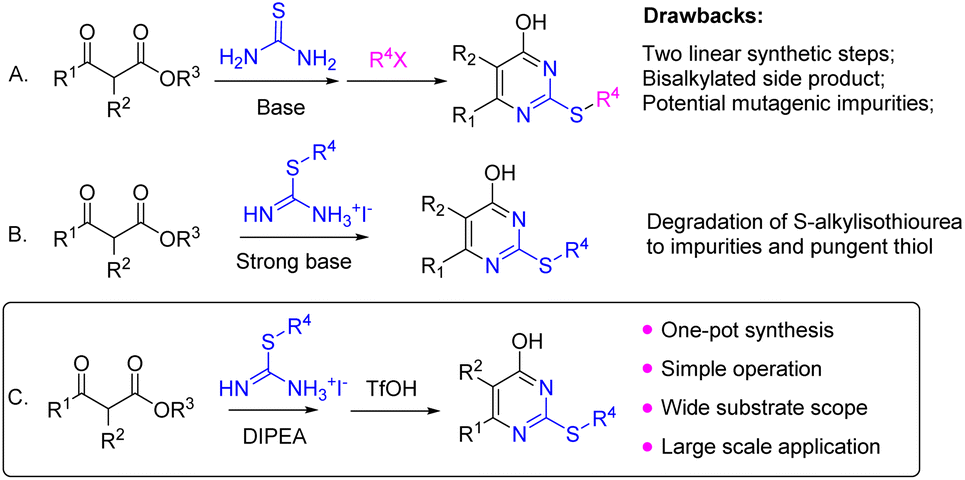 | ||
| Scheme 1 General methods for 4-pyrimidone 2-thioether synthesis. | ||
Results and discussion
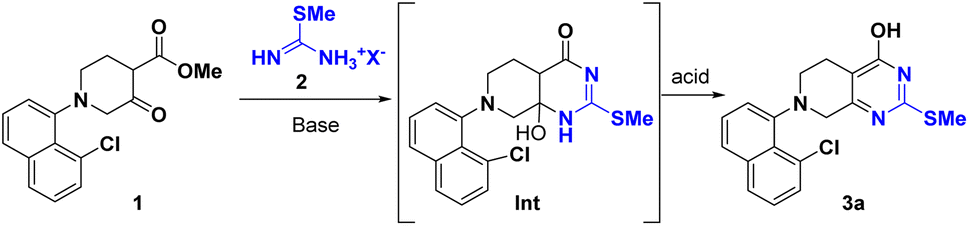
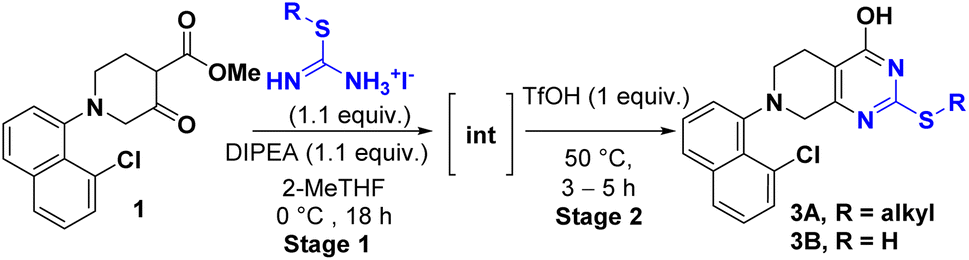
During our synthesis of the key intermediate 3 for adagrasib 2nd generation process,4 a conventional two-step β-ketoester/thiourea condensation/alkylation was initially pursued, resulting in 3 in 76% overall yield. Nevertheless, this approach generated 2,4-bis-isopropylated side-products (∼2%) which proved difficult to purge in the downstream process. To address these issues, a base-initiated condensation of S-isopropyl isothiouronium iodide 2 with β-ketoester 1 was explored, which led to discovery of a one-pot two-step sequence synthesis offering comparable yield (Scheme 2A). However, this route still suffered from the generation of a significant amount of impurities (∼10%) including pungent 2-propanethiol. In addition, this process calls for solvent swap between two stages. With continuous efforts to improve the synthesis of this key intermediate, we found that the issues can be addressed by simple substitution of the base with acid at the stage 2, this one-pot protocol helped to avoid the solvent swap and significantly improved the yield (Scheme 2B). Given the advantages of this new approach, we explored its generality and reported herein a more efficient protocol for the synthesis of a wide variety of 4-pyrimidone-2-thioethers. | ||
| Scheme 2 A case study of 4-pyrimidone-2-thioether derivative 3 synthesis. | ||
The optimization started with β-ketoester 1 and simple S-methylisothiouronium salts (Table 1). The counter anion of S-methylisothiouronium salts have substantial impact on the solubility. The most soluble iodide salt led to highest conversions (entries 1–5). Either TEA or DIPEA can be used as base (entries 6–8, base screen in ESI). To drive the reaction to completion, 1.1 equiv. DIPEA was sufficient (entry 1 vs. entries 9 and 10). Tetrahydrofuran (THF), dioxane, acetonitrile (ACN) and 2-methyltetrahydrofuran (2-MeTHF, entries 11–13) were all viable as the reaction solvents. 2-Methyltetrahydrofuran presented an advantage of a facile removal of water-soluble byproducts during work-up. On the other hand, undesirable oiling-out during workup was observed using methyl tert-butyl ether (MTBE) or toluene (see ESI†). Reactions proceeded well at −10 to −25 °C. The intermediate was readily formed in 3 h and found stable at 0 °C for at least 18 h, which grants operational flexibility between stages 1 and 2. For stage 2, weak acids such as acetic acid were not able to drive the reaction to completion. Strong acids such as TFA, HCl, MsOH, and TfOH substantially improved the conversion to the desired product. However, gummy material was observed upon working up all the reactions except the ones mediated by TfOH (1 equiv.). The reaction is optimally performed at 50 °C and reaches completion in 3–5 h (see ESI†).
|
|||||
|---|---|---|---|---|---|
| Entry | Stage | X− | Base/acid | Solvent | int![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:3ab (%) :1:3ab (%) |
| a Unless otherwise noted, stage 1 were conducted with ketoester (0.5 mmol), S-methylisothiourea (0.55 mmol), and base (0.55 mmol) in 2-MeTHF (1.5 mL) at 0 °C for 3 hours; for stage 2, acid was added to the stage 1 reaction mixture, using 1.1 equiv. DIPEA as the base and 2-MeTHF as the solvent, stirred at 50 °C for 5 hours.b HPLC area percentage.c isothiourea (1.5 equiv.) was applied.d DIPEA (2.0 equiv.) was applied.e DIPEA (3.0 equiv.) was applied. | |||||
| 1 | Stage 1 | I− | DIPEA | 2-MeTHF | 94.4:0.5:5.1 |
| 2 | Br− | DIPEA | 25.2:71:3.6 |
||
| 3 | Cl− | DIPEA | 5.6:94.3:0 |
||
| 4 | MsO− | DIPEA | 0:100:0 |
||
| 5 | SO42− | DIPEA | 0:100:0 |
||
| 6c | I− | DIPEA | 92.2:1.3:6.6 |
||
| 7 | TEA | 89.9:7.3:2.7 |
|||
| 8 | Pyridine | 0:100:0 |
|||
| 9d | DIPEA | 87.6:7.6:5.4 |
|||
| 10e | DIPEA | 65.6:28:6.7 |
|||
| 11 | DIPEA | THF | 92.6:2.6:4.8 |
||
| 12 | DIPEA | Dioxane | 87.0:6.8:6.3 |
||
| 13 | DIPEA | ACN | 90.5:2.0:7.3 |
||
| 14 | Stage 2 | AcOH | 2-MeTHF | 93:1.6:5.4 |
|
| 15 | TFA | 0:22.6:46.9 |
|||
| 16 | HCl | 0:0:61 |
|||
| 17 | MsOH | 0:8.2:65.5 |
|||
| 18 | TfOH | 0:0:78.4 |
|||
With the optimized reaction conditions in hand, we explored the reaction scope of S-alkylisothiourea (Table 2). Demethylation at stage 2 was observed when S-methylisothiourea was employed (entry 1). We envisioned that increasing the size of the alkyl group in S-alkylisothiourea would prevent the dealkylation. Hence, a series of S-alkylisothiouronium iodides were prepared by reaction of thiourea and alkyl halides in the corresponding alcohols under reflux conditions (see ESI†). As expected, 0° (entry 1), 1° (entries 2, 3, 5, 8), and 2° S-alkylisothiourea (entries 4, 6) displayed similar reactivity at stage 1, while dealkylation decreases significantly with increased bulkiness of the alkyl group (entry 1 vs. entries 2–4). However, dealkylation of 3° S-alkylisothiourea (entry 7) still occurred via E2 elimination at stage 1 and E1 elimination at stage 2, respectively, which is in accordance with literature reports.20,21 It should be noted that reaction with S-benzylisothiouronium chloride, a readily available compound, also works given the stage 1 temperature was raised to room temperature. S-Isopropylisothiouronium iodide can be prepared in high yield and good quality. Based on these results, we selected S-isopropylisothiouronium iodide to further explore its condensation with β-ketoesters (entries 4).
|
|||
|---|---|---|---|
| Entry | R | Stage 2 (3A:3B) |
Isolated yield (%) |
| a Unless otherwise noted, reactions were conducted as follows: to a solution of S-alkylisothiouronium iodide (1.1 equiv.) and β-ketoester (3.0–6.0 mmol, 1.0 equiv.) in 2-MeTHF (0.33 M with respect to ketoester) at 0 °C was added DIPEA (1.1 equiv.) dropwise. The reaction stirred at 0 °C for 18 hours and then was added with TfOH (1.0 equiv.) dropwise. The reaction was heated to 50 °C in 0.5 h and then stirred for 3–5 hours.b S-Cyclohexylisothiouronium bromide was applied.c Phenylisothiouronium degraded to urea at stage 1.d S-Benzylisothiouronium chloride was applied, and stage 1 was stirred at room temperature for 18 h. | |||
| 1 | Me | 78.4:14.7 |
69 |
| 2 | Et | 98.6:0.9 |
84 |
| 3 | n-Pr | 99.0:0.6 |
85 |
| 4 | i-Pr | 98.8:0 |
94 |
| 5 | n-Bu | 99.3:0.4 |
73 |
| 6 | sec-Bu | 97.2:0 |
91 |
| 7 | t-Bu | 34.9:62.9 |
n/a |
| 8 | Dodecyl | 91.4:0 |
85 |
| 9 | Cyclopentyl | 98.7:0 |
87 |
| 10b | Cyclohexyl | 83.9:0 |
60 |
| 11c | Ph | 0:0 |
0 |
| 12d | Bn | 74.0:0 |
75 |
Having identified S-isopropylisothiourea and the optimal reaction conditions, we next explored the scope of the β-ketoester. As shown in Scheme 3, all the β-ketoesters afforded the corresponding products in good to excellent yields. Substrates with less bulky alkoxyl (OR3) and alkyl (R1 and R2) gave much higher yields (entries 5a–5d; entries 5e–5h; entries 5i–5n). Aryl ketoester (R1 = phenyl) did not give the desired product probably due to the bulkiness of the phenyl group. A wide range of functional groups such as ester (5t), ether (5q), amine (5r, 5s), alcohol (5y) were well tolerated in this protocol. When di-β-ketoester was applied (5u), double cyclization afforded bi-4-pyrimidone 2-thioether in 61% yield. Several cyclic β-ketoester were also tested and gave acceptable to excellent yields (5w, 5x, 5y). Obviously, acid-labile group like Boc- was deprotected under acidic condition at stage 2 (5aa), whereas other protecting groups such as Cbz- (5ac), Bn- (5ab, 5ad), remained intact.
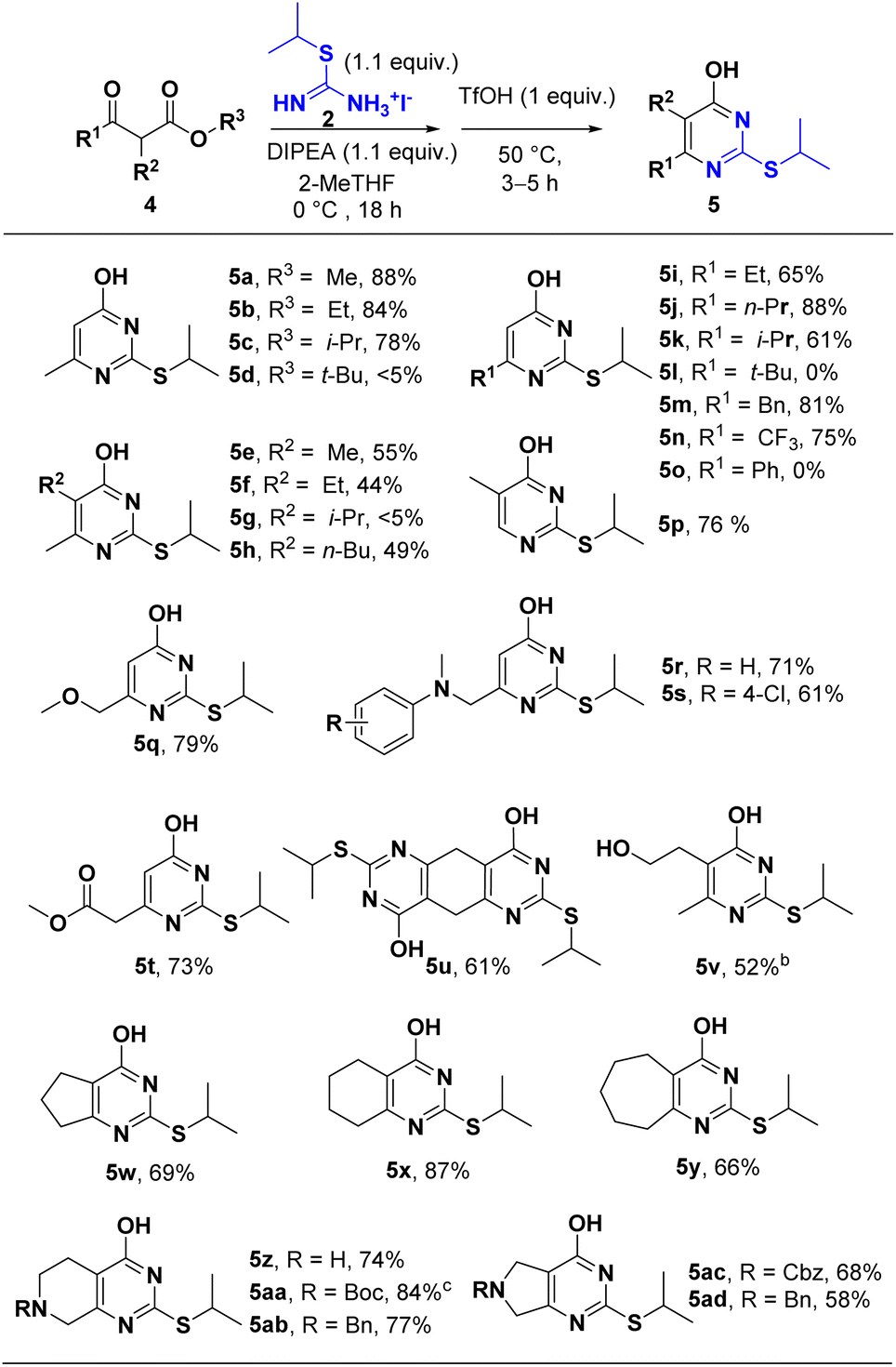 | ||
| Scheme 3 Substrate Scope of β-ketoestera. a Unless otherwise noted, ethyl β-ketoester substrate were applied and reactions were conducted as follows: to a solution of S-alkylisothiouronium iodide (1.1 equiv.) and β-ketoester (3.0–6.0 mmol, 1.0 equiv.) in 2-MeTHF (0.33 M with respect to β-ketoester) at 0 °C was added DIPEA (1.1 equiv.) dropwise. The reaction stirred at 0 °C for 18 hours and then was added with TfOH (1.0 equiv.) dropwise. The reaction was heated to 50 °C in 0.5 h and then stirred for 3–5 hours. b 2-Acetylbutyrolactone was used as the starting material. c deBoc product was obtained. | ||
The practicality of this novel method was tested in a large-scale synthesis of 3, a key intermediate in the synthesis of adagrasib. Delightfully, the compound 3 was obtained in 94% yield and 99.7% purity in the 200 gram scale reaction (Scheme 4A). Furthermore, the S-alkylisothiourea preparation and cyclization were telescoped to accomplish the one-pot synthesis of 3 in 92% yield (Scheme 4B).
 | ||
| Scheme 4 Large-scale synthesis and telescope. | ||
As shown in Scheme 5, a plausible reaction mechanism was proposed. At stage 1, the condensation of isothiourea with β-ketoester affords int A, which readily cyclizes to give int B. The structure of int B was definitively confirmed by NMR analysis (see ESI†). At stage 2, cyclic aldol int B dehydration/aromatization mediated by acid, instead of base,22,23 leads to the desired 4-pyrimidone-2-thioether.
 | ||
| Scheme 5 Plausible mechanism. | ||
Conclusions
In summary, a one-pot, two-stage base/acid-mediated reaction of isothiourea and β-ketoester provided various 4-pyrimidone 2-thioether in good to excellent yields with broad functional group compatibility. The broad substrate scope takes advantage of readily available starting materials and mild reaction conditions. Importantly, this methodology has been applied to the large-scale syntheses of KRASG12C inhibitor adagrasib and can be readily extended to the synthesis of other bioactive molecules containing pyrimidine core.Author contributions
ZCL, CYW, CYC directed the project. The experiments were conducted and characterized by ZCL, CYW, YHG, GHC, and DP. ZCL and CYC prepared the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to Pharmablock US for the chemistry support. To Todd Baumgartner (Mirati Therapeutics) and Stanley Yu (Mirati Therapeutics) for the analytical support, To Aastha Chadha for the melting point test, and to Michał Achmatowicz (Mirati Therapeutics) for the helpful discussion during the preparation of the manuscript.Notes and references
- A. M. Chiacchio, D. Iannazzo, R. Romeo, V. S. Giofrè and L. Legnani, Curr. Med. Chem., 2019, 26, 7166–7195 CrossRef PubMed.
- S. Nadar and T. Khan, Chem. Biol. Drug Des., 2022, 100, 818–842 CrossRef CAS PubMed.
- C. Li, F. Haeffner, S. Wang, C. Yuan, D. Shang, X. Shi, B. Ma, B. T. Hopkins and E. M. O'Brien, Org. Process Res. Dev., 2022, 26, 137–143 CrossRef CAS.
- C.-y. Chen, Z. Lu, T. Scattolin, C. Chen, Y. Gan and M. McLaughlin, Org. Lett., 2023, 25, 944–949 CrossRef CAS PubMed.
- J. B. Fell, J. P. Fischer, B. R. Baer, J. F. Blake, K. Bouhana, D. M. Briere, K. D. Brown, L. E. Burgess, A. C. Burns, M. R. Burkard, H. Chiang, M. J. Chicarelli, A. W. Cook, J. J. Gaudino, J. Hallin, L. Hanson, D. P. Hartley, E. J. Hicken, G. P. Hingorani, R. J. Hinklin, M. J. Mejia, P. Olson, J. N. Otten, S. P. Rhodes, M. E. Rodriguez, P. Savechenkov, D. J. Smith, N. Sudhakar, F. X. Sullivan, T. P. Tang, G. P. Vigers, L. Wollenberg, J. G. Christensen and M. A. Marx, J. Med. Chem., 2020, 63, 6679–6693 CrossRef CAS PubMed.
- I. Usach, V. Melis and J.-E. Peris, J. Int. AIDS Soc., 2013, 16, 18567 CrossRef PubMed.
- C. Ishikawa, M. Senba and N. Mori, Int. J. Oncol., 2018, 53, 1681–1690 CAS.
- K.-S. Yu, K.-S. Bae, J.-H. Shon, J.-Y. Cho, S.-Y. Yi, J.-Y. Chung, H.-S. Lim, I.-J. Jang, S.-G. Shin, K.-S. Song and B.-S. Moon, J. Clin. Pharmacol., 2004, 44, 73–82 CrossRef CAS PubMed.
- H.-K. Kim, S.-H. Park, D.-Y. Cheung, Y.-S. Cho, J.-I. Kim, S.-S. Kim, H.-S. Chae, J.-K. Kim and I.-S. Chung, J. Gastroenterol. Hepatol., 2010, 25, 1618–1625 CrossRef CAS PubMed.
- A. Koumarianou and G. Kaltsas, Nat. Rev. Endocrinol., 2021, 17, 9–10 CrossRef CAS PubMed.
- F. Shirai, T. Tsumura, Y. Yashiroda, H. Yuki, H. Niwa, S. Sato, T. Chikada, Y. Koda, K. Washizuka, N. Yoshimoto, M. Abe, T. Onuki, Y. Mazaki, C. Hirama, T. Fukami, H. Watanabe, T. Honma, T. Umehara, M. Shirouzu, M. Okue, Y. Kano, T. Watanabe, K. Kitamura, E. Shitara, Y. Muramatsu, H. Yoshida, A. Mizutani, H. Seimiya, M. Yoshida and H. Koyama, J. Med. Chem., 2019, 62, 3407–3427 CrossRef CAS PubMed.
- J.-B. Wang, M.-D. Ma, N. Lu, Y.-Z. Yang, J.-X. Yang, Y.-M. Li, C.-Q. Xie, N.-Y. Ma, R.-H. Luo, Y.-P. Wang, L.-M. Yang, H.-B. Zhang, Y.-T. Zheng and Y.-P. He, Drug Dev. Res., 2023, 84, 465–474 CrossRef PubMed.
- R.-M. Rui, C.-R. Tang, C.-T. Zhang, W.-K. Pan, K. Gan, R.-H. Luo, Z.-Q. Wei, F.-S. Jing, S.-M. Huang, L.-M. Yang, Y.-M. Li, Y.-P. Wang, W.-L. Xiao, H.-B. Zhang, Y.-T. Zheng and Y.-P. He, Bioorg. Chem., 2022, 119, 105494 CrossRef CAS PubMed.
- D. Wu, Y. Feng, H. Wang, J. Yang, X. Chen, Y. Wang, C. Cong Lai, Y. Zhang, C. Li, X. Xia and Y. He, Med. Chem. Res., 2017, 26, 1388–1396 CrossRef CAS.
- C. Tintori, A. Brai, M. C. Dasso Lang, D. Deodato, A. M. Greco, B. M. Bizzarri, L. Cascone, A. Casian, C. Zamperini, E. Dreassi, E. Crespan, G. Maga, G. Vanham, E. Ceresola, F. Canducci, K. K. Ariën and M. Botta, J. Med. Chem., 2016, 59, 2747–2759 CrossRef CAS PubMed.
- S. Pochampally, K. L. Hartman, R. Wang, J. Wang, M.-K. Yun, K. Parmar, H. Park, B. Meibohm, S. W. White, W. Li and D. D. Miller, ACS Pharmacol. Transl. Sci., 2023, 6, 526–545 CrossRef CAS PubMed.
- Z. Wang, W. A. Zalloum, W. Wang, X. Jiang, E. De Clercq, C. Pannecouque, D. Kang, P. Zhan and X. Liu, J. Med. Chem., 2021, 64, 13658–13675 CrossRef CAS PubMed.
- B. Selvakumar, N. Gujjar, M. Subbiah and K. P. Elango, Med. Chem. Res., 2018, 27, 512–519 CrossRef CAS.
- V. M. Alford, A. Kamath, X. Ren, K. Kumar, Q. Gan, M. Awwa, M. Tong, M. A. Seeliger, J. Cao, I. Ojima and N. S. Sampson, ACS Chem. Biol., 2017, 12, 2788–2803 CrossRef CAS PubMed.
- N. F. Haley and M. W. Fichtner, J. Org. Chem., 1980, 45, 175–177 CrossRef CAS.
- Y. El-Ahmad and P. Reynaud, J. Heterocycl. Chem., 1988, 25, 711–714 CrossRef CAS.
- H. L. Wheeler and H. F. Merriam, Am. Chem. J., 1903, 29, 478–492 CAS.
- J. B. Campbell and J. W. Firor, J. Org. Chem., 1995, 60, 7687–7689 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00039k |
| This journal is © The Royal Society of Chemistry 2024 |