
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of hydrazone-based polyhydroquinoline derivatives – antibacterial activities, α-glucosidase inhibitory capability, and DFT study†
Muhammad Ismailab,
Rashid Ahmad *a,
Sobia Ahsan Halimc,
Adnan Ali Khanab,
Saeed Ullahc,
Abdul Latifa,
Manzoor Ahmada,
Ajmal Khanc,
Fethi Ahmet Ozdemird,
Asaad Khalide,
Ahmed Al-Harrasi*c and
Mumtaz Ali*a
*a,
Sobia Ahsan Halimc,
Adnan Ali Khanab,
Saeed Ullahc,
Abdul Latifa,
Manzoor Ahmada,
Ajmal Khanc,
Fethi Ahmet Ozdemird,
Asaad Khalide,
Ahmed Al-Harrasi*c and
Mumtaz Ali*a
aDepartment of Chemistry, University of Malakand, P.O. Box 18800, Dir Lower, Khyber Pakhtunkhwa, Pakistan. E-mail: mumtazphd@gmail.com; rashmad@gmail.com
bCentral for Computational Materials Science, P.O. Box 18800, Dir Lower, Khyber Pakhtunkhwa, Pakistan
cNatural and Medical Sciences Research Center, University of Nizwa, Nizwa 616, Oman. E-mail: aharrasi@unizwa.edu.om
dDepartment of Molecular Biology and Genetics, Faculty of Science and Art, Bingol University, Bingol, Turkey
eSubstance Abuse and Toxicology Research Center, Jazan University, P.O. Box: 114, Jazan 45142, Saudi Arabia
First published on 4th April 2024
Abstract
In recent years, polyhydroquinolines have gained much attention due to their widespread applications in medicine, agriculture, industry, etc. Here, we synthesized a series of novel hydrazone-based polyhydroquinoline derivatives via multi-step reactions. These molecules were characterized by modern spectroscopic techniques (1H-NMR, 13C NMR, and LC-HRMS) and their antibacterial and in vitro α-glucosidase inhibitory activities were assessed. Compound 8 was found to be the most active inhibitor against Listeria monocytogenes NCTC 5348, Bacillus subtilis IM 622, Brevibacillus brevis, and Bacillus subtilis ATCC 6337 with a zone of inhibition of 15.3 ± 0.01, 13.2 ± 0.2, 13.1 ± 0.1, and 12.6 ± 0.3 mm, respectively. Likewise, compound 8 also exhibited the most potent inhibitory potential for α-glucosidase (IC50 = 5.31 ± 0.25 μM) in vitro, followed by compounds 10 (IC50 = 6.70 ± 0.38 μM), and 12 (IC50 = 6.51 ± 0.37 μM). Furthermore, molecular docking and DFT analysis of these compounds showed good agreement with experimental work and the nonlinear optical properties calculated here indicate that these compounds are good candidates for nonlinear optics.
1. Introduction
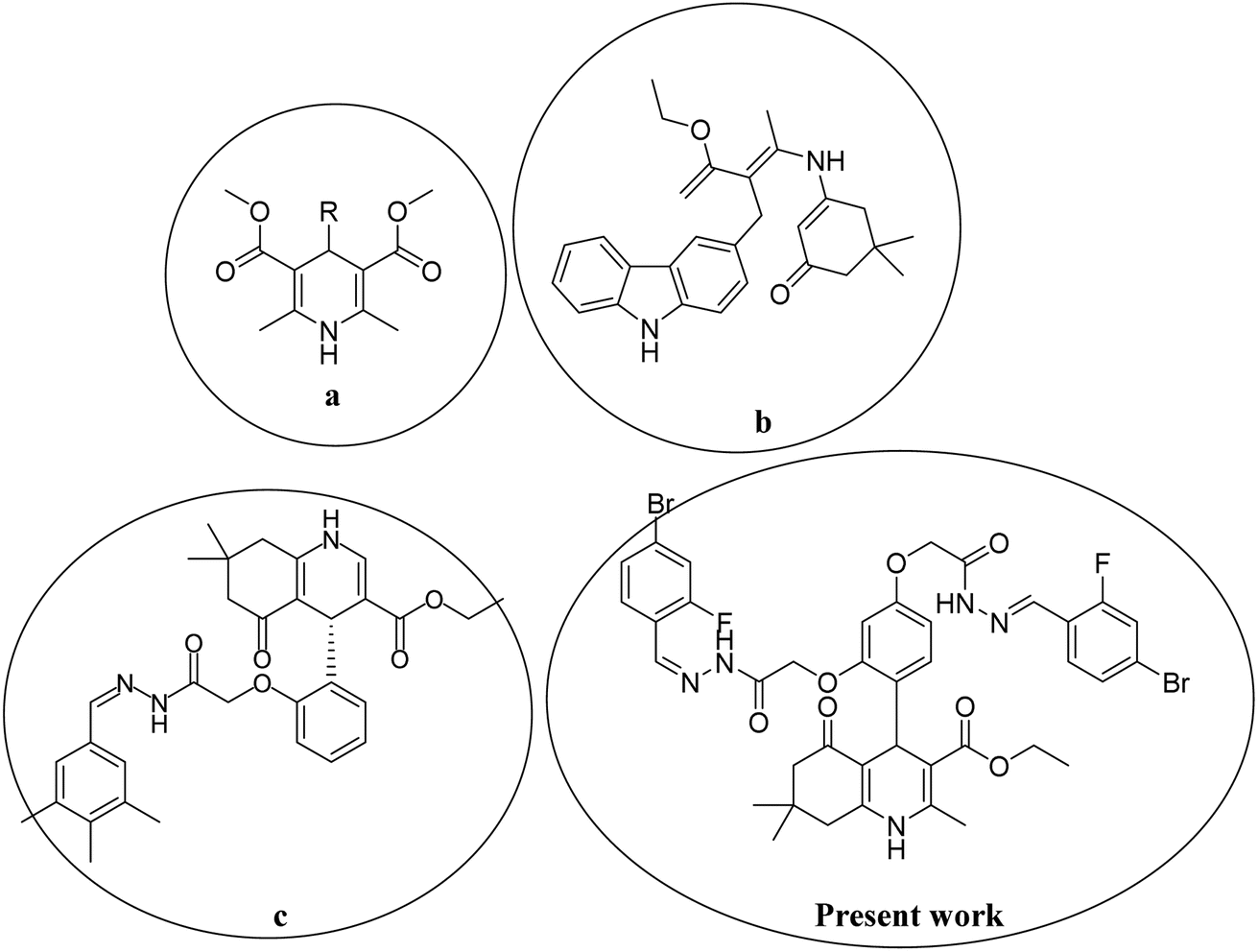
In the last decade, 1,4-dihydropyridine (1,4-DHP) and polyhydroquinolines (PHQ) composites have received much attention for scientific study because of their significant biological activities.1,2 Polyhydroquinoline is structurally correlated with 1,4-DHP, which is synthetically prepared by multi-component (MCRs) via a one-pot reaction. One-pot MCR reactions have important advantages compared to bimolecular reactions due to their structural diversity, atom economy, convergence, operational simplicity, and short duration of the synthetic process.3 Usually, the synthesis of PHQ derivatives involves the coupling of three or four components, such as dimedone, aldehyde, ethyl acetoacetate, and ammonia reflux in ethanol or acetic acid.4 In drug discovery, the multi-component reaction plays a pivotal role. Therefore, for synthetic purposes, multi-component reactions are a more favored approach by younger researchers in academic and industrial realms.5,6 Cardiovascular drugs, such as nifedipine, and nicardipine, and also some derivatives of 1,4-DHPs, such as amlodipine, isradipine, felodipine, nimodipine, nitrendipine, and lacidipine, are more effective in the treatment of hypertension.7,8 Polyhydroquinolines also show properties similar to different bioactive compounds, i.e., vasodilation, anti-tumor, anti-convulsant, antianxiety, antidiabetic,9 bronchodilation, antianginal, anti-atherosclerosis, antidiabetic, and anti-asthmatic activity to cure the Alzheimer's disease.10,11 Several methods have been reported for the synthesis of PHQ and its derivatives to enhance their yields.12–15 The literature review indicates that in different synthetic methods, various kinds of catalysts are used in the synthesis of polyhydroquinolines, for instance, mesoporous vanadium ion-doped titania nanoparticles,14 sulfonic acid-functionalized SBA-15 (SBA-Pr-SO3H),16 FSM-16-SO3H, cobalt(II) complex,17 Fe3O4/SiO2–OSO3H nanostructure catalyst,18 an acidic ionic liquid immobilized onto magnetic Fe3O4 as an efficient heterogeneous catalyst,19 L-proline,20 Zn-MOF microspheres,21 HY-zeolite,22 HClO4–SiO2,23 and chitosan-decorated copper nanoparticles.24,25 However, these methods have some limitations, such as the requirement for expensive catalysts, toxicity of catalysts, harsh reaction conditions, and low product yields. Chemists are trying to develop methods that are milder and require hazardless reaction conditions.Moreover, in recent years, several 1,4-DHPs and PHQ derivatives with different biological activities have been reported. Malhi D.S. et al. synthetically prepared several compounds and screened them for antibacterial activities; among them, compound (a) displayed prevalent activity against E. Coli.26 Venkatapathy K. et al., synthesized several carbozlyl polyhydroquinoline derivatives and evaluated their antibacterial activities; among them, compound (b) was found as the most active against Escherichia coli, Streptococcus pneumonia, Shigella dysenteriae, Staphylococcus aureus, Pseudomonas aeruginosa, and Salmonella typhi.27 Similarly, Shahab N. et al. prepared several derivatives of polyhydroquinoline and investigated their α-glucosidase inhibitory activity; among them, compound (c) was observed as the most potent inhibitor compared to standard drugs.28 Thus, in the present study, we developed a procedure to rapidly synthesize hydrazone-based polyhydroquinoline derivatives under mild reaction conditions with excellent yields and without the use of any catalyst. We investigated their antibacterial and α-glucosidase inhibitory activities. The chemical structures of synthesized compounds were optimized and their various electronic properties were studied using the DMol3 code and Gaussian 09 package. The active compounds that showed antibacterial and α-glucosidase inhibitory activities are shown in Fig. 1.
 | ||
| Fig. 1 Active compounds reported here against various bacterial strains and α-glucosidase. | ||
2. Experimental work
All the chemicals were bought from Aldrich (USA) and TCI (Japan) and used without further purgation. The melting points of targeted compounds were noted on the capillary tube with the help of a digital melting apparatus [FALC]. During the reaction, thin-layer chromatography (TLC) was used to check the reaction completion. The TLC was performed in n-hexane and EtOAc (70![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30 (v/v)) solution. The novel derivatives were transferred to confirm their chemical structures using 1H NMR, 13C NMR, and LC-HRMS spectroscopic techniques.
:30 (v/v)) solution. The novel derivatives were transferred to confirm their chemical structures using 1H NMR, 13C NMR, and LC-HRMS spectroscopic techniques.
2.1. General procedure for the synthesis of hydrazone-based derivatives
A mixture of aromatic aldehyde and bis-hydrazide of PHQ 2.0 and 1.0 mmol were added in a round bottom flask (R.B.), respectively, and 10 mL of EtOH was poured into R.B. through a pipet as a solvent. The reactant mixture was stirred over a hot plate for 45 minutes. The full conversion of reactants into the products was confirmed through TLC. Each reaction mixture was added to an ice bath containing cold water, and as a result, precipitates were formed and separated by a simple filtration process. The resultant precipitates were further washed with hot n-hexane and water several times to remove impurities. Finally, each product was dried by air and stored in a pin-drop.2.2. Physical and spectroscopic data
Compound 3, 1H NMR (DMSO deuterated δ ppm, 600 MHz): 11.93 (s, 1H, O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–NH), 11.54 (s, 1H, OC–NH), 10.44 (s, 2H, CH = N), 9.11 (br. s, 1H, NH), 8.15 (s, 2H, OH), 7.82 (m, 5H, Ar–H), 6.42 (m, 2H, Ar–H), 5.23 (s, 1H, CH), 4.6 (s, 4H, –CH2), 3.88 (m, 2H, CH2), 2.27 (s, 4H, CH2), 1.02 (s, 6H, –CH3), 0.84 (t, J = 6.6 Hz, 3H, –CH3). Melting point: 245 °C, yield: 82%.
C–NH), 11.54 (s, 1H, OC–NH), 10.44 (s, 2H, CH = N), 9.11 (br. s, 1H, NH), 8.15 (s, 2H, OH), 7.82 (m, 5H, Ar–H), 6.42 (m, 2H, Ar–H), 5.23 (s, 1H, CH), 4.6 (s, 4H, –CH2), 3.88 (m, 2H, CH2), 2.27 (s, 4H, CH2), 1.02 (s, 6H, –CH3), 0.84 (t, J = 6.6 Hz, 3H, –CH3). Melting point: 245 °C, yield: 82%.
Compound 5, 1H NMR (DMSO deuterated δ ppm, 600 MHz): 12.03 (s, 1H, HN–CO), 11.69 (s, 1H, H–N–CO), 9.15 (s, 1H, –N–H), 8.61 (s, 2H, –CHN), 8.11 (s, 1H, –Ar–H), 7.79 (m, 2H, Ar–H), 7.65 (d, J = 9 Hz, 2H, Ar–H), 7.46 (d, J = 8.4 Hz, 2H, Ar–H), 5.25 (s, 1H, –CH), 6.41 (s, 2H, Ar–H), 5.04 (s, 4H, –CH2), 4.80 (m, 2H, CH2), 2.27 (s, 4H, CH2), 2.25 (m, –CH3, 3H), 1.04 (s, CH3, 6H), 0.84 (s, –CH3, 3H). Melting point: 200 °C, yield: 86%.
Compound 7, 1H NMR (DMSO deuterated δ ppm, 600 MHz): 12. 09 (s, 1H, OC–NH), 11.75 (s, 1H, NH–CO), 9.12 (br.s, 1H, N–H), 8.16 (s, 2H, HCN), 8.21 (m, 4H, Ar–H), 8.10 (m, 4H, Ar–H), 7.74 (m, 2H, Ar–H), 7.73 (m, 2H, Ar–H), 7.02 (m, 1H, Ar–H), 5.26 (s, 1H, –C–H), 4.64 (s, CH2, 4 H), 3.88 (m, CH2, 2H), 2.29 (s, CH2, 4H), 2.18 (s, CH3, 3H), 1.04 (s, –CH3, 6H), 0.85 (s, 3H, CH3). Melting point: 183 °C, yield: 90%.
Compound 8, 1H NMR (DMSO deuterated δ ppm, 600 MHz): 11.70 (s, 1H, OC–NH), 11.48 (s, 1H, OC–NH), 10.71 (s, OH, 1H), 10.49 (s, OH, 1H), 9.32 (s, 1H, NCH), 9.13 (s, 1H, NCH), 8.55 (s, 1H, NH), 7.29 (d, J = 7.8 Hz, 2H, Ar–H), 6.97 (m, 4H, Ar–H), 6.95 (m, Ar–H, 2H), 6.51 (s, Ar–H, 1H), 5.26 (s, CH, 1H), 4.69 (s, –CH2, 4H), 3.87 (m, –CH2, 2H), 3.79 (s, –CH3, 6H), 2.49 (s, –CH3, 3H), 2.289 (s, 4H, –CH2), 1.05 (s, –CH3, 3H), 1.03 (s, –CH3, 6H), 0.86 (t, J = 6 Hz, –CH3, 3H). Melting point: 190 °C, yield: 78%.
Compound 9, 1H NMR (DMSO deuterated δ ppm, 600 MHz): 11.36 (s, OC–NH, 1H), 11.34 (s, OC–NH, 1H), 9.12 (s, NCH, 1H), 9.11 (s, NCH, 1H), 8.55 (s, 1H, –NH), 7.74 (s, 2H, Ar–H), 6.55 (m, 4H, Ar–H), 6.37 (m, 2H, Ar–H), 6.45 (s, 1H, Ar–H), 5.24 (s, 1H, –CH), 4.71(s, –CH2, 4H), 3.87 (m, –CH2, 2H), 3.79 (s, –CH3, 12H), 2.28 (m, 4H, –CH2), 2.27 (s, –CH3, 3H), 1.04 (m, –CH3, 6H), 0.87 (s, –CH3, 3H). Melting point: 203 °C, yield: 79%.
Compound 10, 1H NMR (DMSO deuterated δ ppm, 600 MHz): 11.92 (s, 1H, OC–H), 11.85 (s, 1H, OC–H), 11.61 (s, 2H, OC–NH), 10.01 (br.s, 1H, –NH), 9.13 (s, 2H, NCH), 8.31 (s, 1H, Ar–H), 7.89 (m, 4H, Ar–H), 7.79 (m, 4H, Ar–H), 6.50 (m, 2H, Ar–H), 5.26 (s, –CH, 1H), 4.61 (s, –CH2, 4H), 3.50 (m, –CH2, 2H), 2.29 (s, –CH2, 4H), 1.10 (s, –CH3, –H), 1.06 (t, J = 7.2 Hz, –CH3, 3H), 1.03 (s, CH3, 3H), 0.85 (s, –CH3, 3H). Melting point: 170 °C, yield: 84%.
Compound 11, 1H NMR (DMSO deuterated δ ppm, 600 MHz): 12.53 (s, 1H, OC–NH), 12.40 (s, 1H, OC–NH), 12.22 (s, 1H, –OH), 11.79 (s, 1H, –OH), 9.42 (s, 1H, NCH), 9.25 (s, 1H, NCH), 9.21 (s, –NH, 1H), 8.20 (m, Ar–H, 2H), 7.91 (m, Ar–H, 2H), 7.87 (m, Ar–H, 2H), 7.52 (m, Ar–H, 1H), 7.50 (m, Ar–H, 1H), 7.21 (m, Ar–H, 2H), 7.08 (s, Ar–H, 1H), 6.58 (m, Ar–H, 2H), 5.35 (s, CH, 1H), 4.69 (s, 4H, –CH2), 3.89 (m, 2H, –CH2), 2.40 (s, 3H, –CH3), 2.29 (s, –CH2, 4H), 1.07 (m, –CH3, 6H), 0.90 (s, –CH3, 3H). Melting point: 239 °C, yield: 82%.
Compound 12, 1H NMR (DMSO deuterated δ ppm, 600 MHz): 11.67 (s, 1H, OC–NH), 11.66 (s, 1H, OC–NH), 9.20 (s, 1H, NCH), 9.12 (s, 1H, NCH), 9.11 (s, 2H, –OH), 8.23 (br.s, 1H, –NH), 7.15 (s, Ar–H, 2H), 6.95 (m, Ar–H, 6H), 6.44 (s, Ar–H, 1H), 5.26 (s, 1H, –CH), 4.60 (s, –CH2, 4H), 3.89 (m, –CH2, 2H), 2.24 (s, –CH3, 3H), 2.38 (s, –CH3, 6H), 2.09 (m, 4H, –CH2), 1.02 (s, –CH3, 6H), 0.85 (s, –CH3, 3H). Melting point: 168 °C, yield: 81%.
13C NMR: compound 3, (DMSO deuterated) δ 13.9 (C-23), 18.2 (C-13), 26.5 (C-12), 26.6 (C-11), 28.5 (C-7), 29.0 (C-6), 32.2 (C-1), 50.3 (C-8), 64.8 (C-22), 66.6 (C-2′′), 66.7 (C-2′), 98.6 (C-16), 105.1 (C-2), 106.6 (C-18), 110.8 (C-10′), 110.8 (C-11′′), 112.2 (C-14), 112.2 (C-10), 128.7 (C-9′′), 128.7 (C-9′), 129.9 (C-12′), 130.1 (C-8′), 130.6 (C-12′′), 130.7 (C-8′′), 140.9 (C −7′′), 144.5 (C-7′), 145.1 (C-6′′), 145.1 (C-6′), 145.9 (C-19), 150.9 (C-5), 150.9 (C-3), 152 (C-15), 152.3 (C-17), 157.5 (C-10′′), 164.7 (C-10′), 164.7 (C-20), 167.0 (C-3′), 169 (C-3′′), 195.5 (C-9). LC-HRMS (ESI+): [M + H]+ exact mass calculated for C39H37Br4N5O9: 1034.93, found: 1035.94.
13C NMR: compound 5, (DMSO deuterated) δ 13.92 (C-23), 18.19 (C-13), 26.61 (C-12), 26.70 (C-11), 28.32 (C-7), 32.26 (C-6), 32.26 (C-6), 50.43 (C-1), 59.11 (C-8), 64.79 (C-22), 66.36 (C-2′′), 66.59 (C-2′), 98.19 (C-2), 106.53 (C-14), 106.53 (C-16), 106.61 (C-14), 119.39 (C-7′′), 119.39 (C-7′), 119.56 (C-19), 127.79 (C-9′′), 127.93 (C-9′), 128.21 (C-11′), 128.35 (C-11′′), 130.08 (C-12′), 130.21 (C-12′′), 135.64 (C-6′′), 139.92 (C-6′′), 153.14 (C-3), 154.43 (C-17), 156.83 (C-5), 157.43 (C-15), 159.69 (C-8′′), 161.38 (C-8′), 164.97 (C-20), 166.95 (C-3′), 169.23 (C-3′′), 196.05 (C-9). HRMS (ESI+): [M + H]+ exact mass calculated for C39H37Br2F2N5O7: 883.10, found: 884.11.
13C NMR: compound 7, (DMSO deuterated) δ 13.9 (C-23), 18.2 (C-13), 26.6 (C-12), 26.6 (C-11), 28.6 (C-7), 29.0 (C-1), 39.7 (C-6), 50.4 (C-8), 59.1 (C-22), 66.6 (C-2′), 66.6 (C-2′′), 98.6 (C-2), 106.6 (C-16), 106.7 (C-18), 121.0 (C-14), 121.4 (C-10), 124.4 (C-8′′), 124.4 (C-8′), 130.0 (C-10′′), 130.2 (C-10′), 130.5 (C-12′′), 130.7 (C-11′′), 132.9 (C-11′), 133.4 (C-12′′), 133.6 (C-19), 141.5 (C-7′′), 145.6 (C-7′), 145.7 (C-6′′), 145.8 (C-6′), 151.1 (C-5), 156.5 (C-9′), 156.9 (C-9′′), 157.5 (C-17), 164.6 (C-15), 167.0 (C-3′′), 169.4 (C-3′), 196.0 (C-9). HRMS (ESI+): [M + H]+ exact mass calculated for C39H39N7O11: 781.2708, found: 782.2815.
13C NMR: compound 8, (DMSO deuterated) δ 13.9 (C-23), 18.2 (C-13), 26.6 (C-12), 26.7 (C-11), 28.4 (C-1), 28.9 (C-7), 50.3 (C-6), 55.8 (C-8), 59.1 (C-13′), 64.8 (C-13′′), 66.2 (C-22), 66.3 (C-2′), 66.6 (C-2′′), 98.8 (C-2), 106.5 (C-16), 112.9 (C-18), 113.8 (C-10), 113.9 (C-14), 117.8 (C-10′), 117.8 (C-10′′), 119.1 (C-11′′), 120.0 (C-11′), 120.1 (C-7′), 120.7 (C-7′′), 130.0 (C-12′), 141.1 (C-19), 147.8 (C-6′′), 147.8 (C-6′′), 148.0 (C-3′), 148.3 (C-8′′), 151.2 (C-9′), 153.3 (C-9′′), 156.9 (C-5), 157.5 (C-3), 164.1 (C-15), 164.5 (C-17), 167.0 (C-20), 167.0 (C-3′), 168.7 (C-3′′), 196.2 (C-9). HRMS (ESI+): [M + H]+ exact mass calculated for C41H45N5O11: 783.3116, found: 784.32311.
13C NMR: compound 9, (DMSO deuterated) δ 13.90 (C-12), 18.20 (C-13), 26.82 (C-12), 29.94 (C-11), 28.70 (C-7), 32.1 (C-1), 40.09 (C-6), 50.39 (C-8), 55.46 (C-14′′), 55.66 (C-13′′), 55.79 (C-14′), 57.09 (C-13′), 64.80 (C-22), 66.31 (C-2′′), 66.55 (C-2′), 98.27 (C-16), 105.26 (C-18), 106.22 (C-9′), 106.42 (C-9′′), 106.49 (C-2), 110.95 (C-11′′), 114.85 (C-11′) 114.92 (C-7′′), 126.74 (C-7′), 130.03 (C-10), 130.35 (C-14), 139.48 (C-19), 143.53 (C-12′′), 144.41 (C-12′), 153.22 (C-6′′), 156.91 (C-6′), 157.48 (C-5), 159.09 (C-3), 162.53 (C-8′′), 163.71 (C-8′), 164.24 (C-10′′), 164.28 (C-10′), 168.64 (C-3′), 168.98 (C-20), 168.98 (C-3′′), 195.89 (C-9). HRMS (ESI+): [M + H]+ exact mass calculated for C43H49N5O11: 811.3429, found 812.3594.
HRMS (ESI+): [M + H]+ exact mass calculated for C41H45N5O11: 783.3116, found: 784.32311.
13C NMR: compound 10, (DMSO deuterated) δ 13.9 (C-23), 18.2 (C-13), 26.6(C-12′), 26.6 (C-12′′), 26.7 (C-11), 28.5 (C-7), 28.9 (C-1), 50.4 (C-6), 52.4 (C-8), 64.8 (C-22), 66.5 (C-2′′), 66.6 (C-2′), 98.5 (C-2), 101.4 (C-16), 105.4 (C-18), 106.6 (C-10), 110.8 (C-14), 127.0 (C-8′′), 127.5 (C-7′), 127.7 (C-8′′), 127.7 (C-8′), 129.9 (C-10′′), 129.9 (C-10′), 130.0 (C-19), 130.2 (C-7′′), 130.2 (C-7′), 136.7 (C-12′′), 136.9 (C-12′), 139.5 (C-11′′), 142.4 (C-11′), 144.4 (C-6′′), 146.7 (C-7′), 151.1 (C-5), 153.3 (C-3), 156.9 (C-15), 157.5 (C-17), 165.1 (C-20), 167.0 (C-3′′), 169.4 (C-3′), 192.7 (C-13′′), 192.7 (C-13′), 195.9 (C-9). HRMS (ESI+): [M + H]+ exact mass calculated for C41H41N5O9: 747.2904, found: 748.3004.
13C NMR: compound 11, (DMSO deuterated) δ 13.96 (C-23), 18.19 (C-13), 26.83 (C-12), 28.76 (C-1), 32.36 (C-7), 50.70 (C-8), 59.10 (C-22), 66.11 (C-2′′), 66.77 (C-2′), 98.39 (C-2), 105.58 (C-18), 106.82 (C-16), 108.45 (C-10), 108.85 (C-7′′), 108.85 (C-7′), 108.42 (C-8), 108.45 (C-10), 118.82 (C-14), 118.86 (C-15′′), 118.86 (C-15′), 120.79 (C-9′′), 120.79 (C-9′), 120.86 (C-13′′), 123.58 (C-13′), 123.65 (C-14′′), 127.56 (C-14′), 127.79 (C-12′′), 127.82 (C-12′), 127.88 (C-11′′), 128.96 (C-11′), 129.05 (C-19), 130.36 (C-10′′), 130.06 (C-10′), 131.61 (C-16′′), 131.69 (C-16′), 132.88 (C-6′′), 133.80 (C-6′), 144.30 (C-5), 147.42 (C-31), 151.83 (C-17), 153.22 (C-15), 156.89 (C-17′′), 157.97 (C-17′), 158.01 (C-20), 164.40 (C-3′), 166.89 (C-3′′), 196.34 (C-9). HRMS (ESI+): [M + H]+exact mass calculated for C47H45N5O9: 823.3217, found: 824.4217.
13C NMR: compound 12, (DMSO deuterated) δ13.9 (C-13), 18.2 (C-23), 26.7 (C-12), 26.7 (C-12), 28.4 (C-7), 28.9 (C-1), 50.5 (C-8), 55.6 (C-15′′), 59.5 (C-15′), 64.7 (C-22), 66.6 (C-2′′), 66.5 (C-2′′), 105.2 (C-10), 106.3 (C-3), 106.9 (C-14), 111.8 (C-18), 111.9 (C-16), 120.0 (C-8′′), 120.3 (C-8′), 120.4 (C-12′′), 130.0 (C-12′), 130.1 (C-19), 130.3 (C-7′′), 130.8 (C-17′), 149.9 (C-5), 151.0 (C-3), 153.3 (C-9′), 157.0 (C-9′′), 157.5 (C-10′′), 163.9 (C-10′), 164.3 (C-15), 164.4 (C-17), 167.0 (C-3′′), 168.7 (C-3′), 195.9 (C-9). HRMS (ESI+): [M + H]+ exact mass calculated for C41H45N5O11: 783.8229, found: 784.32076.
2.3. Antibacterial assay
The disc diffusion method was used to determine the zone of inhibition of microbial growth. A dilution of 10 μL of each synthesized compound, along with dimethyl sulfoxide (DMSO), was used to sterilize the disc with a diameter of 6 millimeters (mm). DMSO was used as the negative control. Media were spread in the Petri plates using a sterile swab containing the microbial suspension, Petri plates were placed at 4 °C for 2 h. They were then incubated at 37 °C for 24 h. The diameter of the clear zone around the disc was measured in mm to determine the antibacterial activity. All the tests were repeated thrice.2.4. In vitro α-glucosidase inhibition assay
To perform an α-glucosidase (E.C.3.2.1.20) enzyme inhibition assay, 0.1 M phosphate buffer (PH 6.8) was used at 37 °C. The enzyme (0.2 μ mL−1) in the phosphate buffer saline was incubated at 37 °C along with each synthesized compound for 15 min.29 After that, p-nitrophenyl-α-D-glucopyranosid (0.7 mM) was added and variation in the absorbance at 400 nm was observed for 30 min by using a spectrophotometer (xMark microplate spectrophotometer, BIO-RAD). Evaluated scaffolds were substituted with DMSO-d6 (7.5% final concentration) in the control. Acarbose was used as a standard inhibitor. The following equation was used to determine percent inhibition.| % Inhibition = 100 (OD test well/OD control) × 100 |
3. Molecular docking
For molecular docking, we utilized the homology model of Saccharomyces cerevisiae α-glucosidase enzyme from the AlphaFold Protein Structure Database [Entry code: P38158 (https://alphafold.ebi.ac.uk/entry/P38158)]. The Molecular Operating Environment (MOE version 2020.0901) was used to add missing hydrogens, atom types, and other forcefield parameters (e.g., van der Waals, angles and bond stretch parameters, residues chirality) regarding the protein by MOE's QuickPrep tool with Amber14:EHT (Amber ff14SB combined with EHT) forcefield. The modified structure was then refined by molecular dynamics simulations via AMBER22. The detail of this method is reported in our previous publication.30 The structures of compounds optimized in Section 2.2 were imported into the MOE database and minimized with MMFF94x forcefield and an RMS gradient of 0.1 kcal mol−1 Å−1. The refined protein model and the database of the three compounds were docked with the Triangle Matcher Docking Algorithm and AMBER14:EHT forcefield with 30 docking conformations. The generated conformations of all three ligands were scored by the London dG scoring function. Each docked conformation was visualized by the MOE interface. The docking interactions are illustrated by MOE.3.1. Computational analysis
The simulation for optimization and various electronic properties of the newly synthesized compounds was carried out based on DMol3 code using the material studio. Generalized Gradient Approximation (GGA) was used in tools of DFT as an approximation. The Double Numerical Polarization (DNP) was employed as a basis set with an energy cutoff of 4.5 Å. The optimized structures of each moiety were further utilized to gain insight into the frontier molecular orbitals (FMOs), density of state (DOS), natural population analysis (NPA), and molecular electrostatic potential map (MEP).31 The Nonlinear optics (NLO), and correlation between experimental and computed 1H NMR values were calculated on the Gaussian 09 package.4. Results and discussion
4.1. General findings
The sketch for the synthesis of the targeted compounds is given in Scheme 1. The starting material (bis-hydrazide of polyhydroquinoline) was prepared via a multi-step reaction. In the first step, substituted aromatic aldehyde, 2,4-dihydroxy benzaldehyde, was esterified with chloroethylacetate in DMF, while K2CO3 was used as the catalyst. In the second step, the PHQ nucleus was synthesized by a multi-component one-pot Hantzsch condensation reaction (esterified aldehyde, ethyl acetoacetate, dimedone, and ammonium acetate were taken in the R.B., and ethanol was added as a solvent without any catalyst. The contents were agitated over the hot plate for 4 hours). After reaction completion, the reaction mixture was put into an ice bath comprising a beaker with cold water. The product appeared as yellow precipitates, which were filtered, washed with water, and then dried. After drying, PHQ was treated with hydrazine hydrate in ethanol as the solvent to synthesize bis-hydrazide of PHQ. Bis-hydrazide of PHQ was treated with three different aldehydes (2,5-fibromo-4-hydroxy benzaldehyde, 4-bromo-2-fluro benzaldehyde, and 3-nitro benzaldehyde) independently to synthesize the target compounds with good yields (82–90%). The chemical structures of synthesized compounds were confirmed by their 1H NMR, 13C NMR, and LC-HRMS spectroscopic data.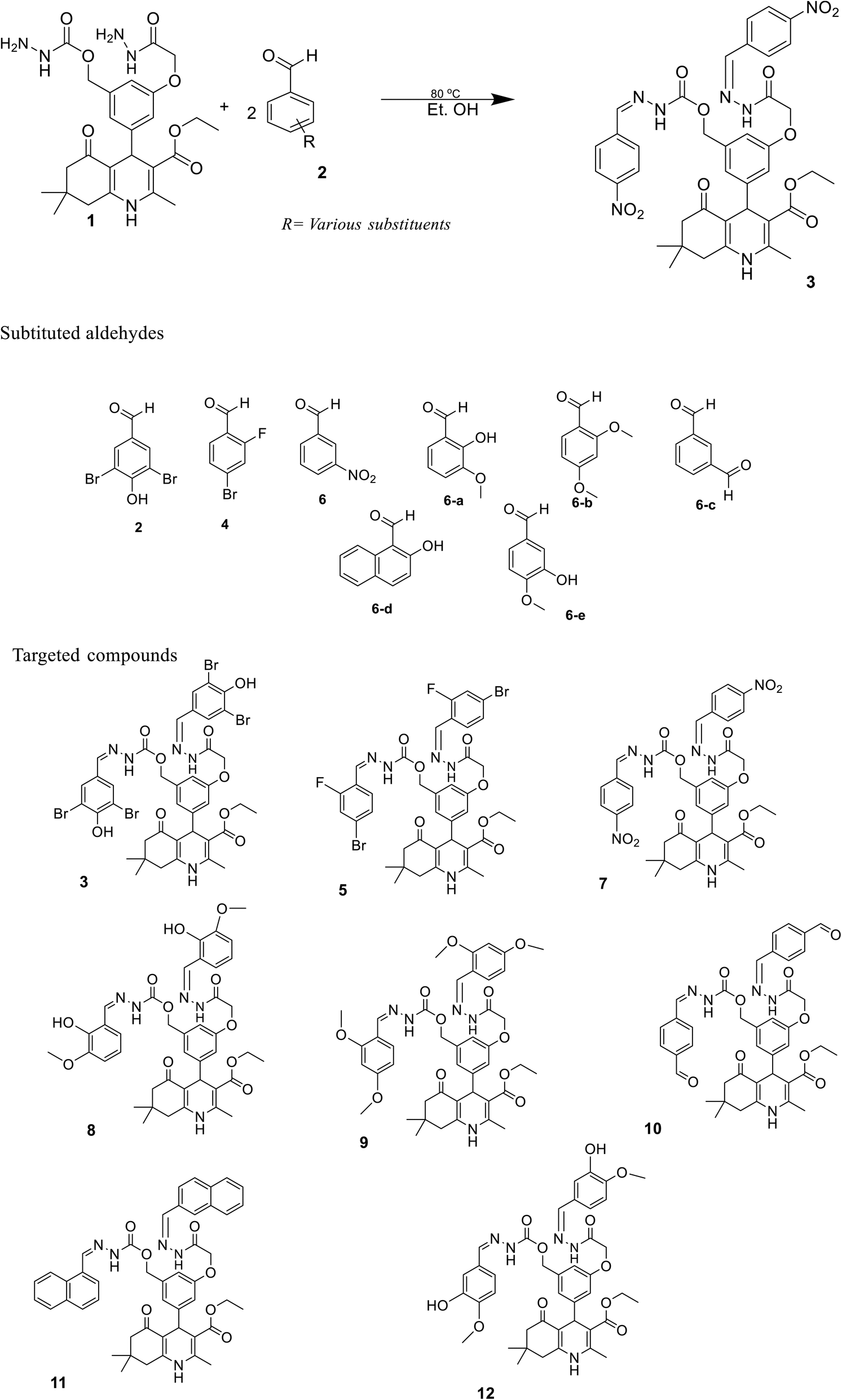 | ||
| Scheme 1 Synthesis of bis-Schiff's bases with polyhydroquinoline derivatives. | ||
4.2. Antibacterial activities
In the current study, all compounds were evaluated for their anti-bacterial and anti-diabetic potential because people with type-2 diabetes (T2DM) have an increased susceptibility to infectious illnesses. More glucose is accessible in T2DM patients, which increases the rate at which invasive bacteria proliferate by providing them with a source of metabolic energy. Furthermore, there is a connection between diabetes and bacterial infections as diabetic people are far more likely to develop bacterial infections, including malignant otitis externa, periodontitis, emphysematous pyelonephritis, and emphysematous cholecystitis. Streptococci, pneumococci, and enterobacteria are the most prevalent types of bacteria; it is essential to gain insight into more explanations for this association.32–34 Sixteen different strains of Gram-positive (Listeria monocytogenes NCTC 5348, Staphylococcus aureus 6538 P, Bacillus cereus EMC 19, Bacillus subtilis IM 622, Bacillus megaterium DSM 32, Brevibacillus brevis, Bacillus subtilis ATCC 6337) and Gram-negative (Pseudomonas fluorescens, Proteus vulgaris FMC II, Salmonella typhimurium NRRLE 4413, Enterobacter aerogenes CCM 2531, Escherichia coli ATCC 25922, Proteus vulgaris, Pseudomonas aeruginosa DSM 50070, Salmonella enterica ATCC 13311, and Klebsiella pneumoniae EMCS) bacteria were used to evaluate the antibacterial properties of the synthesized target compounds by measuring their zone of inhibition.It was observed that compound 8 has the highest efficacy against Gram-positive strains, including L. monocytogenes NCTC 5348, B. subtilis IM 622, B. brevis, and B. subtilis ATCC 6337, with a maximum zone of inhibition (z. o. i) of 15.3 ± 0.01, 13.2 ± 0.2, 13.1 ± 0.1, and 12.6 ± 0.3 mm, respectively. Compound 3 was found to be the most effective against Gram-positive bacteria, including L. monocytogenes NCTC 5348, B. subtilis IM 622, B. subtilis ATCC 6337, and B. cereus EMC 19, with the z. o. i of 11.6 ± 0.6, 12.6 ± 0.3, 13.2 ± 0.01, and 13.4 ± 0.03 mm, respectively. Compound 5 showed maximum inhibition against B. megaterium DSM 32, B. cereus EMC 19, and B. subtilis ATCC 6337 with the z. o. i of 11.3 ± 0.3, 10.2 ± 0.01 and 9.2 ± 0.01 mm, whereas compound 7 showed maximum inhibition against B. megaterium DSM 32, B. cereus EMC 19, and L. monocytogenes NCTC 5348, with z. o. i of 9.2 ± 0.2, 8.6 ± 0.3, and 8.1 ± 0.6 mm, respectively. The maximum zone of inhibition for compounds 8–10 was observed against Gram-positive bacteria, such as B. subtilis IM 622, B. cereus EMC 19, and B. brevis with the z. o. i of 13.2 ± 0.2, 9.3 ± 0.1, and 8.1 ± 0.01 mm, respectively. In contrast, compounds 11–12 showed maximum z. o. i (11.5 ± 0.01 and 10.6 ± 0.01 mm) against S. aureus 6538 P and B. subtilis IM 622, respectively. Compound 9 showed minimum inhibition against Gram-negative bacteria strains, such as P. aeruginosa DSM 50070, S. typhimurium NRRLE 4413, and P. fluorescens, with z. o. i of 1.3 ± 0.6, 2.1 ± 0.2, and 2.1 ± 0.1 mm, respectively. The results are summarized in Table 1.
| Bacteria | Gram (+/−) | Zone of inhibition (diameter in mm) | |||||||
|---|---|---|---|---|---|---|---|---|---|
| 3 | 5 | 7 | 8 | 9 | 10 | 11 | 12 | ||
| Bacillus megaterium DSM 32 | (+) | 10.6 ± 0.1 | 11.3 ± 0.3 | 9.2 ± 0.2 | 10.3 ± 0.2 | 8.3 ± 0.2 | 5.5 ± 0.1 | 8.2 ± 0.1 | 8.3 ± 0.1 |
| Brevibacillus brevis | (+) | 11.4 ± 0.3 | 8.1 ± 0.2 | 6.1 ± 0.1 | 13.1 ± 0.1 | 6.5 ± 0.6 | 8.1 ± 0.01 | 10.3 ± 0.3 | 7.4 ± 0.1 |
| Bacillus subtilis ATCC 6337 | (+) | 13.2 ± 0.01 | 9.2 ± 0.01 | 7.3 ± 0.3 | 12.6 ± 0.3 | 7.1 ± 0.4 | 4.3 ± 0.3 | 9.5 ± 0.6 | 7.9 ± 0.3 |
| Bacillus subtilis IM 622 | (+) | 12.6 ± 0.3 | 7.6 ± 0.1 | 6.0 ± 0.01 | 13.2 ± 0.2 | 8.0 ± 0.3 | 6.6 ± 0.3 | 7.6 ± 0.6 | 10.6 ± 0.01 |
| Staphylococcus aureus 6538 P | (+) | 10.9 ± 0.2 | 7.1 ± 0.3 | 7.3 ± 0.3 | 11.1 ± 0.3 | 6.1 ± 0.1 | 7.3 ± 0.1 | 11.5 ± 0.01 | 6.4 ± 0.6 |
| Bacillus cereus EMC 19 | (+) | 13.4 ± 0.03 | 10.2 ± 0.01 | 8.6 ± 0.3 | 10.3 ± 0.2 | 9.3 ± 0.1 | 4.1 ± 0.01 | 10.0 ± 0.1 | 9.7 ± 0.1 |
| Listeria monocytogenes NCTC 5348 | (+) | 11.6 ± 0.6 | 8.2 ± 0.2 | 8.1 ± 0.6 | 15.3 ± 0.01 | 6.4 ± 0.01 | 5.6 ± 0.2 | 9.1 ± 0.3 | 8.5 ± 0.3 |
| Pseudomonas fluorescens | (−) | 4.3 ± 0.2 | 6.6 ± 0.3 | 5.3 ± 0.1 | 4.1 ± 0.1 | 2.2 ± 0.6 | 2.1 ± 0.1 | — | — |
| Klebsiella pneumoniae EMCS | (−) | 3.2 ± 0.3 | 5.3 ± 0.1 | 4.6 ± 0.2 | 7.3 ± 0.6 | 4.7 ± 0.3 | 3.0 ± 0.3 | — | — |
| Enterobacter aerogenes CCM 2531 | (−) | 7.3 ± 0.01 | 4.1 ± 0.2 | 3.1 ± 0.01 | 6.1 ± 0.3 | 5.5 ± 0.1 | 3.1 ± 0.6 | — | — |
| Salmonella typhimurium NRRLE 4413 | (−) | 5.3 ± 0.01 | 3.3 ± 0.1 | 3.6 ± 0.2 | 5.6 ± 0.6 | 2.1 ± 0.2 | 1.0 ± 0.1 | — | — |
| Escherichia coli ATCC 25922 | (−) | 6.6 ± 0.01 | 3.6 ± 0.6 | 2.3 ± 0.3 | 8.2 ± 0.2 | 3.5 ± 0.5 | 4.0 ± 0.01 | — | — |
| Proteus vulgaris FMC II | (−) | 5.6 ± 0.3 | 4.2 ± 0.03 | 3.3 ± 0.1 | 4.1 ± 0.01 | 5.6 ± 0.4 | 2.6 ± 0.3 | — | — |
| Proteus vulgaris | (−) | 5.6 ± 0.1 | 6.3 ± 0.3 | 5.1 ± 0.6 | 6.3 ± 0.3 | 4.7 ± 0.3 | 3.5 ± 0.6 | — | — |
| Pseudomonas aeruginosa DSM 50070 | (−) | 6.1 ± 0.01 | 5.0 ± 0.1 | 4.2 ± 0.3 | 3.3 ± 0.3 | 1.3 ± 0.6 | 2.3 ± 0.1 | — | — |
| Salmonella enterica ATCC 13311 | (−) | 4.3 ± 0.2 | 3.1 ± 0.01 | 3.0 ± 0.1 | 5.0 ± 0.1 | 3.4 ± 0.3 | 2.6 ± 0.2 | — | — |
| DMSO | Control | — | — | ||||||
4.3. α-Glucosidase inhibitory activities of compounds and their molecular docking analysis
The synthesized compounds (3, 5, 7–12) were scrutinized in vitro to determine their inhibitory potential for α-glucosidase because of their key importance in diabetes mellitus. Moreover, their modes of binding to the active site of α-glucosidase were predicted through computational molecular docking. These compounds exhibited significant inhibitory activities against α-glucosidase as compared to the standard drug (acarbose, IC50 = 873.34 ± 1.67 μM) (Table 2). These compounds have different substituents attached to phenyl rings which are responsible for the differing binding affinities of these compounds with α-glucosidase. The structure–activity relationship (SAR) analysis explained that different substituents are attached at benzene rings of PHQ at either the same or different positions which have a great influence on inhibitory activities. For example, compound 3 (IC50 = 8.89 ± 0.37 μM) with the substitution of Br at the meta position and –OH groups at the para position demonstrate significant inhibitory activity against α-glucosidase as compared to acarbose. The docking result indicates that the –OH group at one of its phenyl rings mediates hydrogen bonding with the side chain of Asp215 at 1.57 Å, however, the other substituted phenyl ring does not interact with any residues. The nitrogen on the quinoline ring of this molecule forms a strong hydrogen bond with the side chain of Asp305 at 1.71 Å. Moreover, one of the hydrazide nitrogens of compound 3 also mediates a hydrogen bond with the side chain of Lys156 at 2.56 Å. Due to these interactions, compound 3 exhibits a docking score of −4.99 Kcal mol−1. The inhibitory activity of compound 5 (IC50 = 9.26 ± 0.34 μM) was slightly lower than compound 3 with the substitution of F at the ortho position and Br at the para position. The binding mode of 5 shows slight variation in its docked conformation, due to which the quinoline nitrogen forms a hydrogen bond with the carbonyl oxygen of Pro312 (1.68 Å) at the entrance loop of the active site, whereas hydrazide oxygen interacts with the side chain of Arg315 at 2.25 Å. We observed that one of its phenyl rings mediates a hydrophobic interaction with the side chain of Tyr72. The substituted groups of compound 5 do not participate in protein-ligand binding which may be the reason for its lower activity than compound 3. This is also confirmed by the docking score of compound 5 (−4.82 Kcal mol−1) which is less than the docking score of compound 3. The inhibitory potency of compound 7 (IC50 = 17.14 ± 0.39 μM) was even lower than that of 3 and 5 upon substitution of –NO2 at the meta position of phenyl rings. The docking analysis revealed that the hydrazide-substituted phenyl rings of compound 7 are bent towards each other and form hydrogen bonds with the side chains of His351 (1.96 Å and 2.26 Å) and Gln353 (2.21 Å), however, the hydrazide and quinoline groups of this molecule are involved in binding with any residue. Therefore, compound 7 exhibited a docking score of −3.65 Kcal mol−1 which is the least docking score among all the compounds studied here.| Compound | Percent inhibition (0.5 mM) | IC50 ± μM (SEM) | Docking score (kcal mol−1) | Ligand atom | Receptor atom | Type of interaction | Distance (Å) |
|---|---|---|---|---|---|---|---|
| a SEM = standard error mean, not active = N/A, HBA = hydrogen bond acceptor, HBD = hydrogen bond donor. | |||||||
| 3 | 92.71 | 8.89 ± 0.37 | −4.99 | N20 | OD1-ASP307 | HBD | 1.71 |
| O81 | OD1-ASP215 | HBD | 1.54 | ||||
| N91 | OD1-ASN415 | HBD | 2.56 | ||||
| 5 | 91.57 | 9.26 ± 0.34 | −4.82 | N20 | O-PRO312 | HBD | 1.68 |
| O45 | NH1-ARG315 | HBA | 2.25 | ||||
| 6-Ring | 6-Ring-TYR72 | π–π | 3.99 | ||||
| 7 | 90.59 | 17.14 ± 0.39 | −3.65 | O92 | NE2-GLN353 | HBA | 2.21 |
| O95 | NE2-HIS351 | HBA | 1.96 | ||||
| O96 | NE2-HIS351 | HBA | 2.26 | ||||
| 8 | 93.27 | 5.31 ± 0.25 | −6.15 | N20 | OD2-ASP242 | HBD | 1.73 |
| O40 | N-ARG315 | HBA | 2.30 | ||||
| O52 | NE2-GLN353 | HBA | 2.00 | ||||
| C10 | 5-Ring-HIS280 | H–π | 4.01 | ||||
| 9 | 91.38 | 10.58 ± 0.34 | −4.66 | N106 | OD1-ASP69 | HBD | 1.72 |
| O52 | NH1-ARG446 | HBA | 1.82 | ||||
| O52 | NH2-ARG446 | HBA | 2.46 | ||||
| N108 | NH1-ARG446 | HBA | 1.87 | ||||
| 10 | 92.50 | 6.70 ± 0.38 | −5.35 | O89 | NH2-ARG213 | HBA | 1.82 |
| O89 | NE2-HIS351 | HBA | 2.05 | ||||
| N96 | ND2-ASN350 | HBA | 2.65 | ||||
| O40 | N-ARG315 | HBA | 1.79 | ||||
| 6-Ring | 6-Ring-TYR347 | π–π | 3.24 | ||||
| 11 | 91.58 | 7.18 ± 0.26 | −5.02 | N20 | OE1-GLN279 | HBD | 3.32 |
| O34 | NH1-ARG442 | HBA | 2.86 | ||||
| O40 | NH2-ARG213 | HBA | 2.76 | ||||
| O52 | NH1-ARG446 | HBA | 2.58 | ||||
| 12 | 92.49 | 6.51 ± 0.37 | −5.89 | O83 | OD2-ASP352 | HBD | 2.53 |
| O52 | OH-TYR347 | HBA | 2.55 | ||||
| O83 | NE2-HIS351 | HBA | 2.76 | ||||
| 6-Ring | 6-Ring-PHE301 | π–π | 3.29 | ||||
The inhibitory potency of compound 8 (IC50 = 5.31 ± 0.25 μM) was drastically enhanced with the addition of –OCH3 at ortho and para positions, and it was identified as the most active inhibitor of α-glucosidase. Compound 8 showed excellent interactions at the active site with key residues, including Asp242, Arg315, and Gln353. We observed that the hydroquinoline-carboxylate moiety of the compound formed hydrogen bonds with the side chains of Asp242 and Arg315, whereas one of the hydrazide moieties interacted with the side chain of Gln353 at 2.30 Å. The docking score of 8 was −6.15 Kcal mol−1 which is the highest docking score as compared to the rest of the compounds, which further correlates with our in vitro results. The binding mode of compound 8 is shown in Fig. 2.
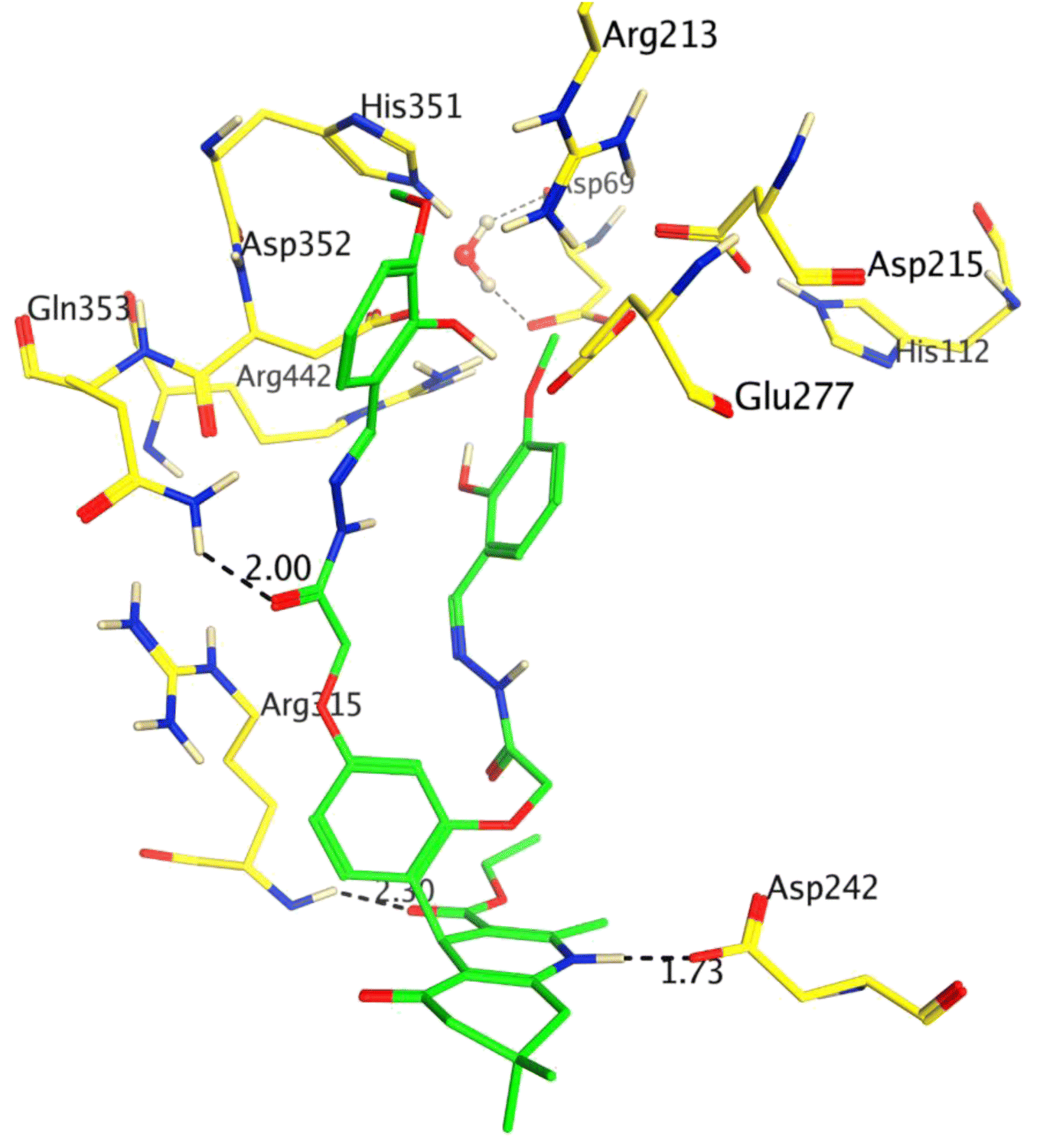 | ||
| Fig. 2 The binding mode of the most active compound (8) is shown in the active site of the enzyme. Ligand is presented in green sticks, interacting residues are shown in yellow sticks. H-bonds and hydrophobic interactions are displayed as black and green dashed lines, respectively. | ||
However, compound 9 demonstrated lower activity (IC50 = 10.58 ± 0.34 μM) than compound 8 when –OH and –OCH3 were added at the ortho and para positions, respectively. The binding mode of compound 9 revealed that only one of the hydrazide moieties of this molecule is engaged in interaction with the side chains of Asp69 and Arg446 through hydrogen bonds. The –R group and the hydroquinoline moiety do not interact within the active site which may be the reason for its lower activity than compound 8. This is also clear in the docking score of compound 9 (−4.66 Kcal mol−1) which is lower than that of compound 8. Interestingly, the addition of OC–H at meta positions in compound 10 has favorable effects on its inhibitory activity (IC50 = 6.70 ± 0.38 μM). The docking results show that one of the OC–H moieties participates in hydrogen bonding with the side chains of Arg213 and His351. Whereas similar to the carboxylate and hydrazide groups, hydroxyquinoline also forms a hydrogen bond with Arg315 and Asn350, respectively. Moreover, compound 10 also exhibits an improved docking score (−5.35 Kcal mol−1) as compared to compounds 3, 5, 7, 9, and 11. Compound 11 (IC50 = 7.18 ± 0.26 μM) bears an –OH group at the ortho position, while 12 (IC50 = 6.51 ± 0.37 μM) has –OH at the meta and –OCH3 at the para positions, which has a positive impact on the inhibitory activities of these molecules. In compound 11, both the hydrazide and hydroquinoline carboxylate moieties contribute to protein-ligand binding, while in compound 12, a hydrazide group and a substituted –OH group interact with the surrounding residues, including Asp352, Tyr347, and His351, through hydrogen bonds. Additionally, Phe301 provides hydrophobic interactions to 12. These molecules also exhibit good docking scores (11: −5.02 Kcal mol−1 and 12: −5.89 Kcal mol−1). Compounds 8, 12, 10, and 11 have significant inhibitory potential, followed by 3, 5, 9, and 7. Therefore, these molecules can serve as drug-like candidates for diabetes mellitus by specifically blocking the function of α-glucosidase. The docking scores of all the compounds and their interactions with the α-glucosidase residues are tabulated in Table 2. The docking scores of all the compounds show excellent correlation with their inhibitory concentrations. The binding orientation of all the compounds is given in Fig. S1 (ESI†).
4.4. Molecular geometry optimization
The optimized molecular geometries of compounds 3, 5, and 7–12 are given in Fig. 3. The parameters used for optimization are mentioned in the computational method. The minimum self-consistent field energy of compounds 3, 5, 7–12 are −12713.1979660, −7386.9705133, −2720.1630889, −2687.518103, −2766.273963, −2535.517684, −2766.382650, and −2688.250328. Hartree (Ha), respectively. The geometry optimization reveals that compound 11 is more stable among all the compounds due to the more negative energy in SCF. The optimized geometries of these compounds were further used to investigate their different electronic properties.35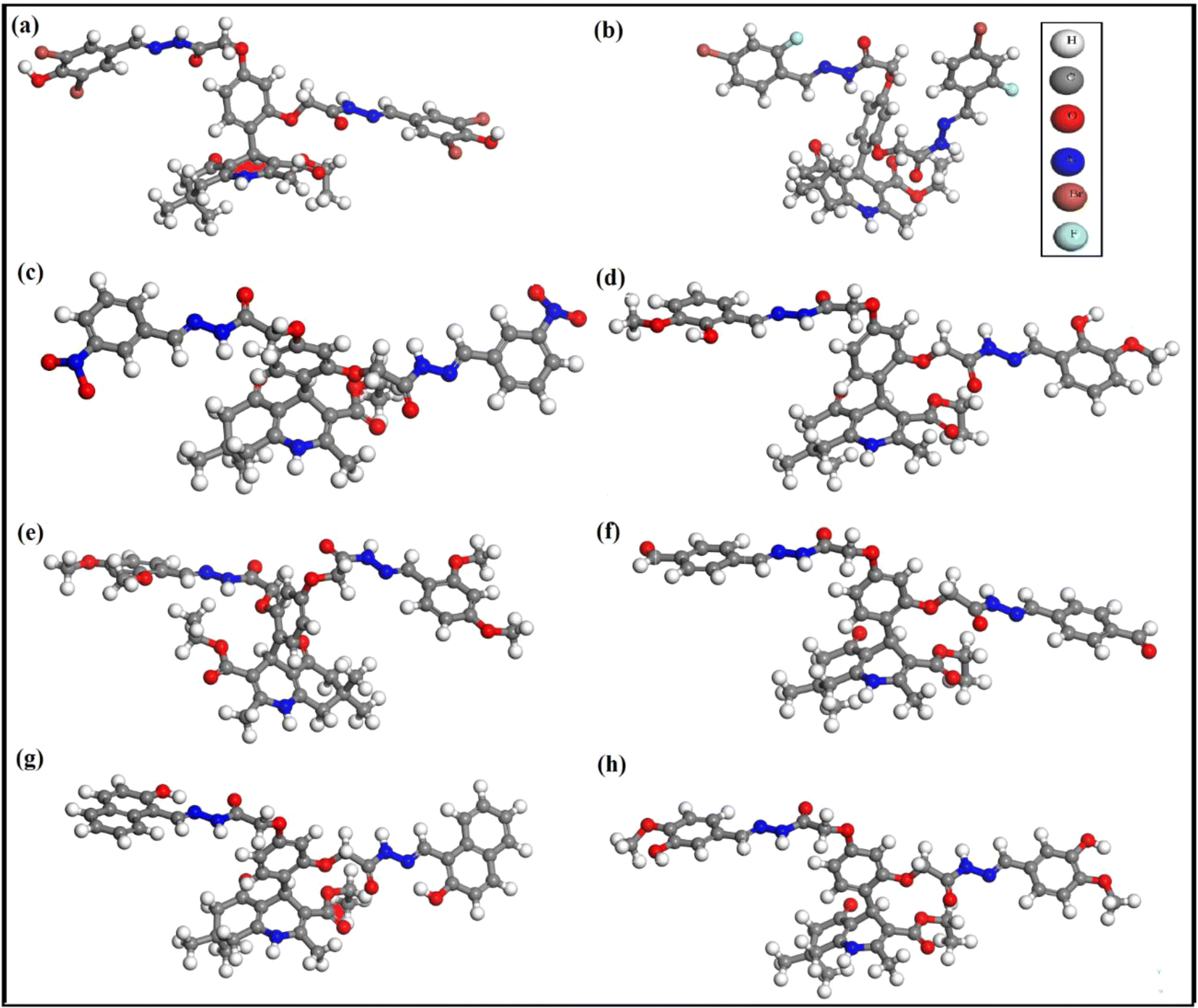 | ||
| Fig. 3 Optimized geometry of compounds 3 (a), 5 (b) 7 (c) 8 (d), 9 (e), 10 (f), 11 (g), and 12 (h). | ||
4.5. Natural population analysis (NPA)
Mulliken and Hirshfeld's atomic charges of all atoms of synthesized compounds were found out on DMole3 code in the framework of GGA and PBE as functional and DNP as a basis set. Here, only the Mulliken and Hirshfeld atomic charges of most electronegative atoms, such as fluorine, oxygen, nitrogen, and bromine, are given in Table S1† and depicted in Fig. 4.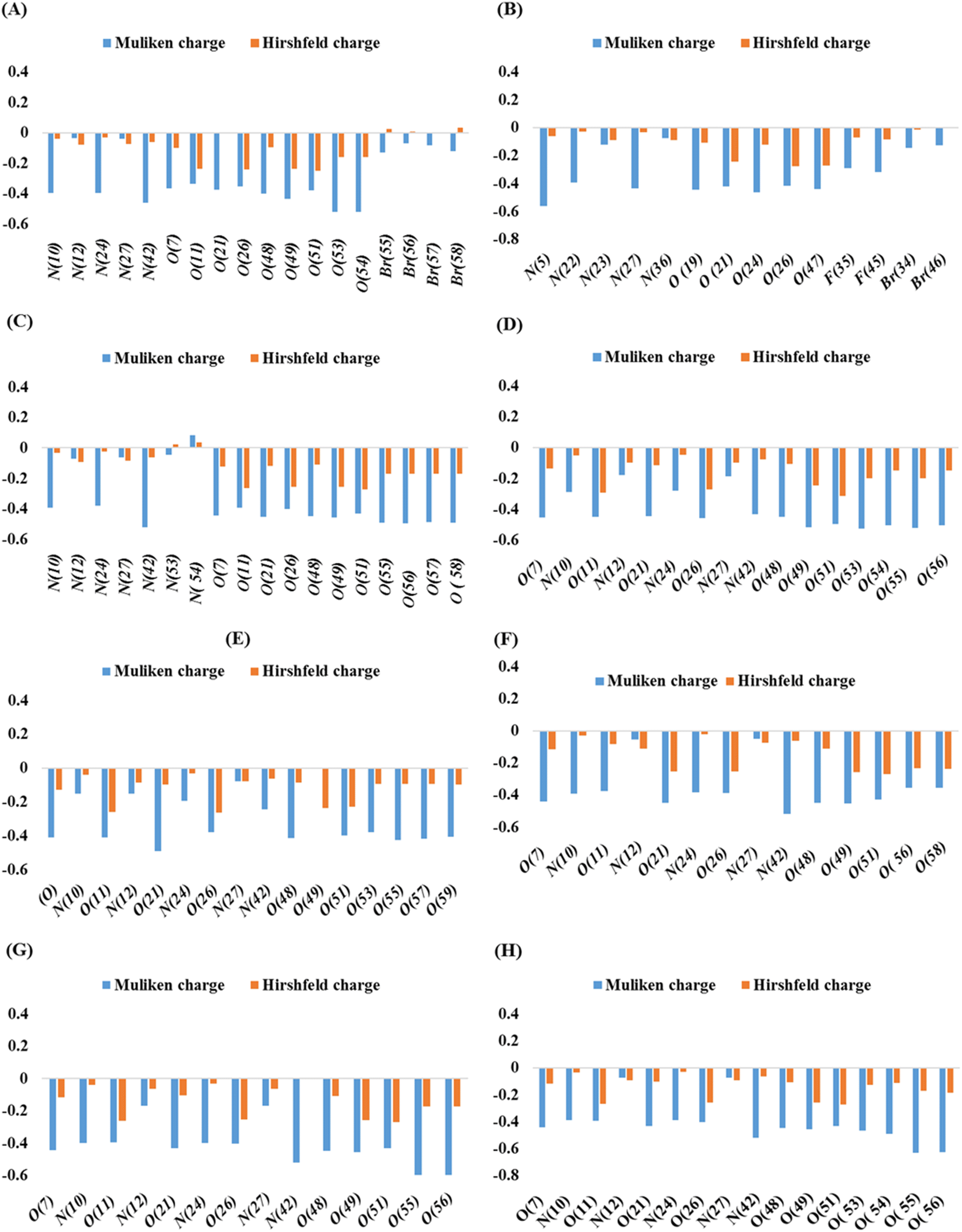 | ||
| Fig. 4 Mulliken and Hirshfeld atomic charges in compounds 3 (A), 5 (B), 7 (C), 8 (D), 9 (E), 10 (F), 11 (G), and 12 (H). | ||
In compounds 3, 5, and 7, the highest Mulliken atomic negative charge among the N atoms was observed on N(42), N(10), N(24), N(5), N(27), N(42), and N(10) with the values of −0.46, −0.398, and −0.398, respectively. For O atoms, it was on O(54), O(53), O(49), O(24), O(19), O(56), and O(57) with the values of −0.522, −0.52, and −0.433, 0.563, −0.437, −0.52, −0.391, −0.463, −0.446, −0.495, and −0.489, respectively. The highest Mulliken atomic negative charges among the N atoms of compounds 8, 9, and 10 were found on N(42), N(10), N(42), N(24), N(10), and N(24) with the values of −0.434, −0.287, −0.244, −0.194, −0.393, and −0.383, respectively, while for O atoms, they were present on O(53), O(55), O(21), O(49), O(49), and O(21) with the values of −0.526, −0.523, −0.489, O(49), −0.454, and −0.449, respectively. Similarly, for compounds 11 and 12, the highest Mulliken atomic negative charge among N atoms was noted on N(42), N(24), N(42), and N(10) with values of −0.519, −0.397, −0.521, and −0.391, respectively, and for O atoms, it was on O(55), O(56), O(55), and O(56), with values of −0.595, −0.595, −0.631, and −0.626, respectively.
Among the Br atoms of compound 3, the highest negative Mulliken atom charge was monitored on Br(55), i.e., −0.13. In contrast, the highest Mulliken atomic negative charge on F(45) and Br(34) atoms of compound 5 was found to be −0.32 and −0.144, respectively. All the heteroatoms of investigated compounds possessed a negative Mulliken atomic charge, except N(54), of compound 7 which bore a positive charge of +0.085.31
4.6. Molecular electrostatic potential (MEP) map
The MEP map is one of the best ways to describe the reactivity of a compound. Therefore, MEP of organic compounds is plotted to find out the desirable sites for electrophilic and nucleophilic attack. It also helps to understand the interaction with other species and estimate the reactive behavior. MEP maps of the target molecules in this study were prepared based on electron density versus the electrostatic potential in the framework of GGA and PBE. The MEP diagrams are shown in Fig. 5 (with a range of colors from deepest red to deepest blue). The surface with green color is neutral, while red and yellow are high electron density (nucleophilic center) and the blue region indicates low electron density (electrophilic center). The red and yellow colors are mostly observable on F, O, N, Cl, and Br atoms and the benzene rings, while blue is mostly on the H atoms. Overall, high electron density is found on the surface of the most electronegative atoms and benzene rings.36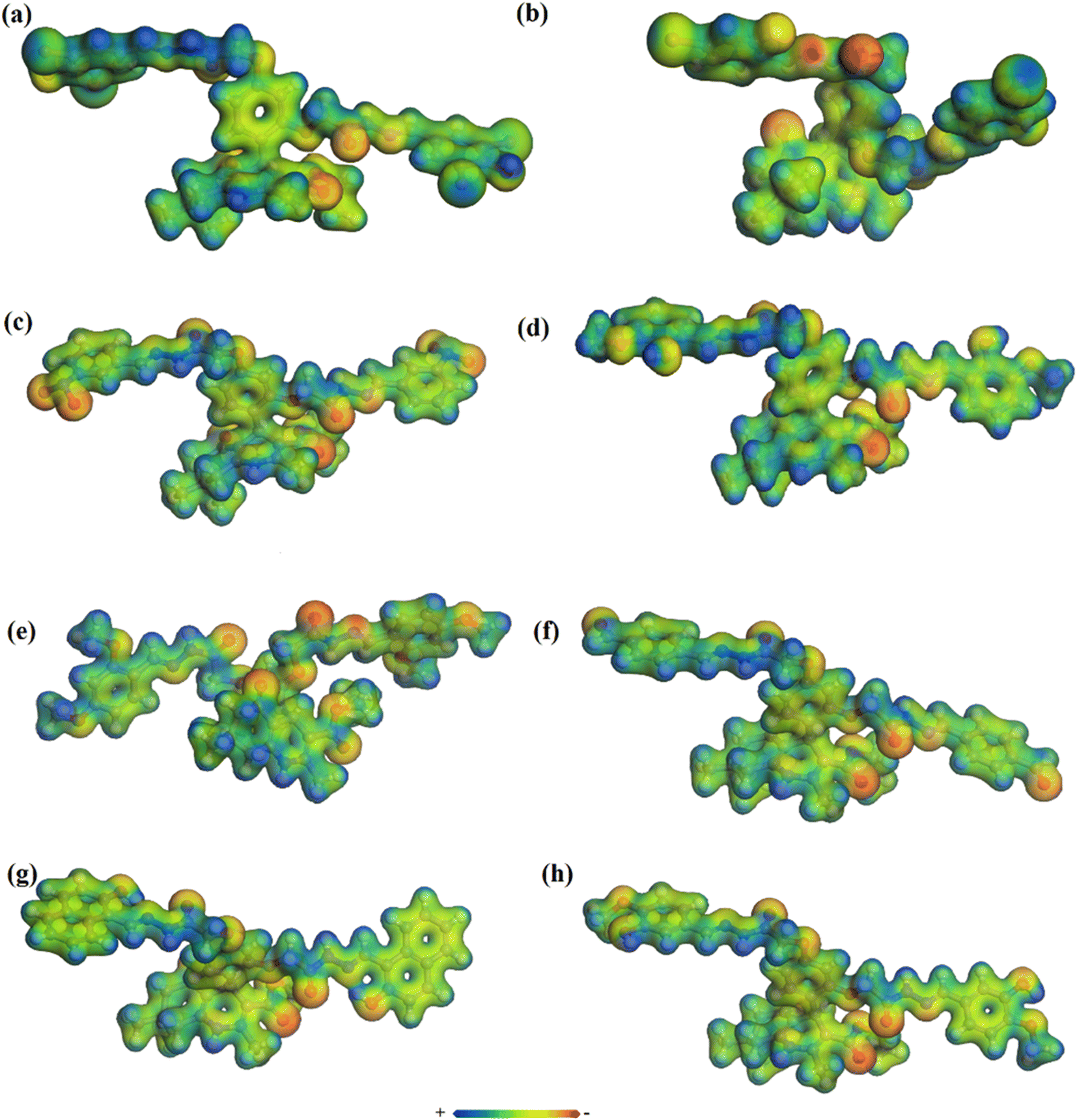 | ||
| Fig. 5 Molecular electrostatic potential maps of compounds 3 (a), 5 (b), 7 (c), 8 (d), 9 (e), 10 (f), 11 (g), and 12 (h). | ||
4.7. Frontier molecular orbitals (FMOs) and density of state (DOS)
FMOs are the highest molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) which predict the charge transfer and the reactivity of compounds. FMOs of synthesized compounds are shown in Fig. 6 which indicates that HOMOs are found on polyhydroquinoline, while LUMOs are found on any one side of benzene rings. The global chemical reactivity descriptors were found from the HOMO–LUMO gap within the framework of GGA and PBE (Table 3). The high negative value of electronic chemical potential (μ) and the low value of chemical hardness (η) indicate the compound to be a good electrophile. The global chemical reactivity descriptors are presented in Fig. 7. The energy gap (Eg) shows the stability and chemical activity of a compound. If the compound has the highest energy gap, it will be very stable with lower reactivity. Herein, compound 12 has the highest energy gap, while compound 7 has the lowest energy gap, thus, compound 12 is more stable and 7 is less stable and vice versa. The DOS of synthesized compounds has been plotted in Fig. 8, which represents the number of molecular orbitals available at distinct energy values in the range of 40 eV.37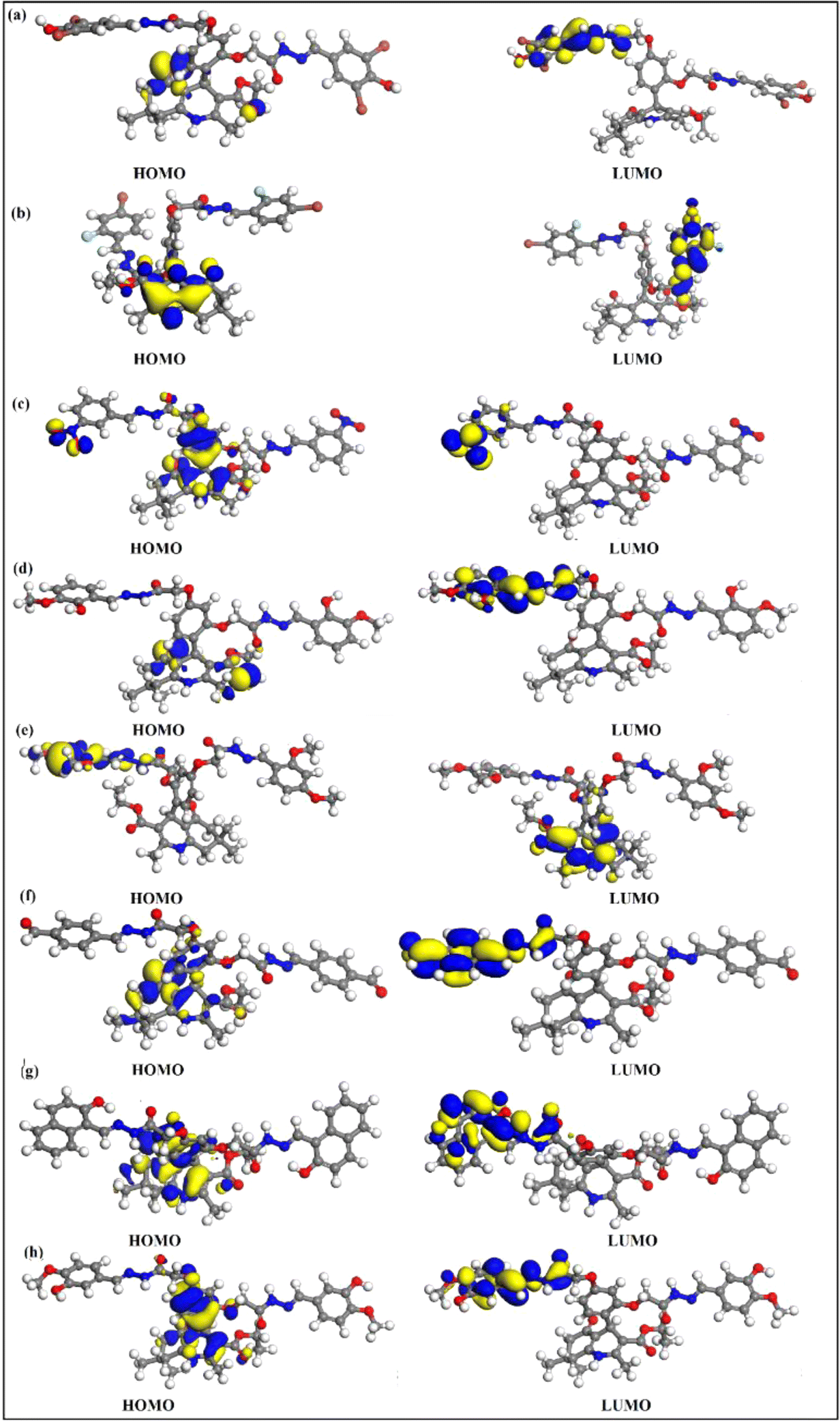 | ||
| Fig. 6 HOMOs–LUMOs of compounds, 3 (a), 5 (b), 7 (c), 8 (d), 9 (e), 10 (f), 11 (g), and 12 (h). | ||
| Compound | HOMO (eV) | LUMO (eV) | Eg (eV) | μ (eV) | η (eV) | ω (eV) |
|---|---|---|---|---|---|---|
| 3 | −4.85711 | −3.87539 | 0.98172 | −4.36625 | 0.49086 | 19.41913 |
| 5 | −4.55261 | −3.01342 | 1.539189 | −3.78301 | 0.769595 | 9.297863 |
| 7 | −5.60331 | −4.8995 | 0.703809 | −5.25141 | 0.351905 | 39.18287 |
| 8 | −4.6456 | −3.3459 | 1.29969 | 0.64984 | −3.99579 | 12.284651 |
| 9 | −4.58353 | −2.52834 | 2.05518 | 1.02759 | −3.55594 | 6.15258 |
| 10 | −4.98869 | −3.19116 | 1.79753 | 0.89876 | −4.08992 | 9.30582 |
| 11 | −4.81388 | −2.51825 | 2.29563 | 1.14781 | −3.66606 | 5.85461 |
| 12 | −4.73530 | −2.17798 | 2.55731 | 1.27865 | −3.45664 | 4.67222 |
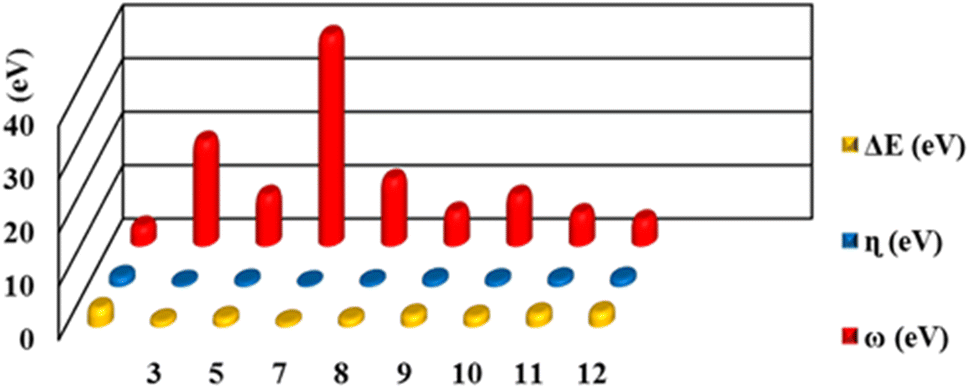 | ||
| Fig. 7 Global reactivity descriptors of compounds 3, 5, 7–12. | ||
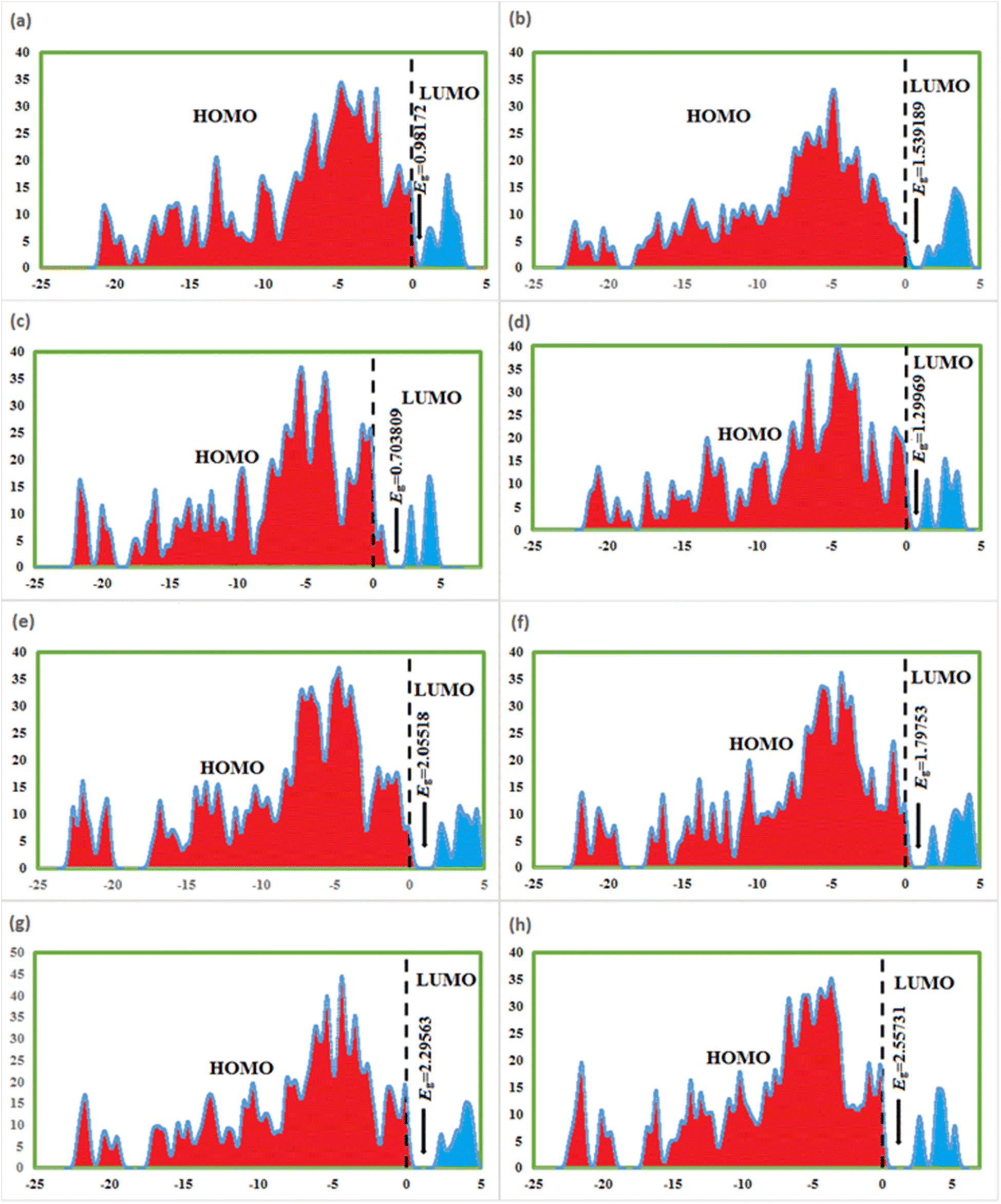 | ||
| Fig. 8 DOS of 3 (a), 5 (b), 7 (c), 8 (d), 9 (e), 10 (f), 11 (g), and 12 (h). | ||
4.8. Correlation of 1H NMR
In the DFT study, the gauge invariance atomic orbitals (GIAO) approach is used to compute chemical shifts (δ, ppm), relative to tetramethylsaline (TMS, considered an internal standard). The chemical shifts have been calculated at the CAM-B3LYP/631-G (d,p) level of theory; this was done in the solvent dimethylsulfoxide (DMSO) and the gas phase using the SMD model. The δ31 was computed with the help of eqn (1), and results are given in Table S2.†| δ = σTMS – σcal | (1) |
The computed 1H NMR values were compared with experimental chemical shifts and were found to be in good agreement (Fig. 9), particularly in the solvent phase due to the same solvent effect. The signals of 1H NMR have been observed for newly synthesized compounds between 0.84 and 12.09 ppm. Two main factors cause 1H NMR signals (in ppm); one is electron density over the atom in the molecule and the other is the anisotropy-induced magnetic field. The attachment of more electronegative atoms among the neighboring atoms causes a decrease in electron density over the nucleus of an atom, therefore, the chemical shift of the nucleus increases the deshielding and vice versa.
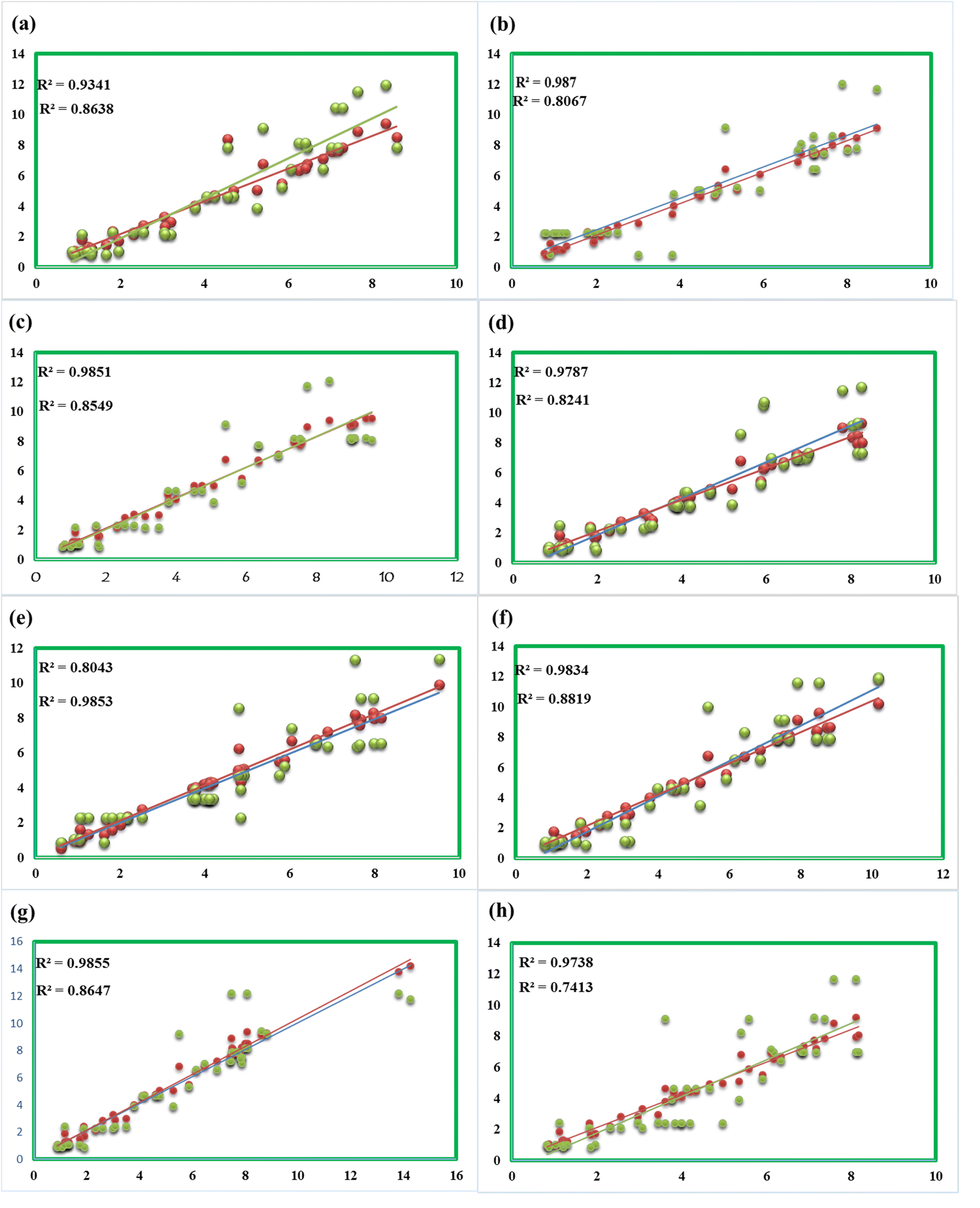 | ||
| Fig. 9 Correlation of computed and experimental δ of compounds 3 (a), 5 (b), 7 (c), 8 (d), 9 (e), 10 (f), 11 (g), and 12 (h). | ||
4.9. Hyperpolarizability and NLO (nonlinear optical properties) analysis
The statistical average of microscopic-level polarizability (P′) is known as macroscopic polarization (P) of a material. The efficiency of a material for NLO response and whether it has the potential for NLO can be estimated from its average polarizability and first hyper-polarizability values.The ab initio technique correlated with the finite field approach that can be utilized by quantum chemical investigation to calculate average hyperpolarizability (α0) and first-hyperpolarizability (βtot). These values are interrelated with the origins of macroscopic second-order NLO properties of the material. The average polarizability (α0) and first-hyperpolarizability (βtot) are defined as:38
| α0 = 1/3(αxx+αyy+αzz) | (2) |
| βtot = [(βxxx + βxyy + βxzz)2 + (βyyy + βyzz + βyxx)2 + (βzzz + βzxx + βzyy)2]1/2 | (3) |
The magnitude of the dipole moment of compounds 3, 5, 7, 8, 9, 10, 11, and 12 is 8.7087, 9.8433, 8.0252, 4.7580, 11.1103, 6.7245, 6.9360, and 9.0013 Debye, respectively. Initially, the average hyperpolarizability values of compounds were calculated (Table 4) in a.u. using eqn (2) and then converted into esu (1 a.u. = 8.6393 × 10−33 esu) using a conversion factor. Among these molecules, compound 8 has the highest average polarizability value (−2.26 × 10−22 esu), while compounds 3 and 7, 9–12 have −3.16 × 10−22, −3.14 × 10−22, −2.47 × 10−22, −2.83 × 10−22, −2.78 × 10−22, and −2.50 × 10−22 esu, respectively. Similarly, the first-hyperpolarizability of compounds 3, 5, 7–12 was calculated using eqn (3), and higher first-hyperpolarizability values were found along a direction of β_xxx for compounds 3, 9, 10, and 11, β-yyy for compounds 8 and 12, β-yyz for compound 7, and β-xzz for compound 5; these observations show the direction of electron charge transfer. First-hyperpolarizability of these compounds is given in Table 5. Compound 5 has a higher value of first-hyperpolarizability, i.e., 4.01 × 10−27 esu. While compounds 3, 7, 8, 9, 10, 11, and 12 have 1.08 × 10−27, and 9.3 × 10−28, 8.65 × 10−28, 3.6 × 10−27, 6.82 × 10−28, 1.17 × 10−27, and 6.71 × 10−28 esu, respectively. The dipole moment average hyperpolarizability and first-hyperpolarizability values of urea were calculated to be 4.56 Debye and α0 = −6.8264 × 10−24, β0 = 0.83 × 10−30 esu, respectively, which is used as standard. However, the dipole moment, average hyperpolarizability, and first-hyperpolarizability values of investigated compounds are almost two times greater than those of the standard. Therefore, these compounds are good agents for NLO.
| Compound | α_xx | α_yy | α_zz | α0 |
|---|---|---|---|---|
| 3 | −373.88 | −350.572 | −371.8834 | −3.16 × 10−22 |
| 5 | −320.388 | −346.642 | −327.8479 | −2.87 × 10−22 |
| 7 | −400.458 | −309.677 | −335.757 | −3.14 × 10−22 |
| 8 | −165.284 | −299.906 | −320.7021 | −2.26 × 10−22 |
| 9 | −198.876 | −308.841 | −348.5798 | −2.47 × 10−22 |
| 10 | −376.933 | −292.823 | −313.2808 | −2.83 × 10−22 |
| 11 | −289.729 | −324 | −351.1406 | −2.78 × 10−22 |
| 12 | −224.007 | −318.478 | −326.5189 | −2.50 × 10−22 |
| Cpd | β-xxx | β-xxy | β-xyy | β-yyy | β-xxz | β-yyz | β-xzz | β-yzz | β-zzz | β-tot = 10−33 esu |
|---|---|---|---|---|---|---|---|---|---|---|
| 3 | 103.531 | −27.2876 | −74.621 | 91.2567 | 23.6445 | 48.6206 | 26.180 | −36.075 | 41.2186 | 1.08 × 10−27 |
| 5 | −591.52 | −27.5735 | −61.981 | −35.478 | −440.202 | −85.8671 | 34.566 | −54.420 | −36.622 | 4.01 × 10−27 |
| 7 | −218.27 | −25.4466 | −84.231 | −17.871 | −10.3593 | 108.722 | 3.9509 | −26.604 | 46.833 | 9.3 × 10−28 |
| 8 | −113.69 | −108.831 | −93.504 | 97.4609 | −203.346 | −106.683 | −111.15 | 1.4509 | −44.890 | 8.65 × 10−28 |
| 9 | 693.592 | −127.591 | 79.833 | 26.0823 | 105.1097 | −67.6839 | −45.095 | −8.8995 | −55.640 | 3.6 × 10−27 |
| 10 | 358.618 | −252.986 | 174.97 | 222.661 | −21.0241 | 30.2823 | 57.380 | 1.5163 | −36.576 | 6.82 × 10−28 |
| 11 | 381.470 | −48.7487 | 34.343 | 45.5244 | 35.2695 | 76.0531 | −60.925 | −13.457 | 57.5254 | 1.17 × 10−27 |
| 12 | −147.66 | −174.394 | 43.679 | 135.877 | 5.4399 | −62.8392 | −58.406 | −12.095 | −65.054 | 6.71 × 10−28 |
5. Conclusion
In this work, hydrazone-based polyhydroquinoline derivatives were prepared and their chemical structures were confirmed by modern spectroscopic techniques. The target compounds were evaluated for antibacterial and α-glucosidase inhibitory activities. Among all compounds, compound 8 exhibited potent inhibition of all strains of Gram-positive and Gram-negative bacteria, while featuring excellent α-glucosidase inhibitory activity. Additionally, molecular docking was employed to predict the binding mechanism of these inhibitors with α-glucosidase, which was in good agreement with our in vitro observations. Moreover, our DFT analysis showed that compound 7 has the lowest Eg, hence higher reactivity, while compound 12 bears the highest Eg indicating it to be more stable. All the compounds have first-hyperpolarizability values greater than the standard, and thus, can be good applicants for NLO. The correlation of computed and experimental chemical shifts was studied and showed reasonable agreement.Author contributions
Muhammad Ismail: conceptualization, validation, writing – original draft, Rashid Ahmad: software, validation, writing – review & editing. Abdul Latif: formal analysis and review. Adnan Ali Khan: conceptualization, software, validation & writing – original draft. Manzoor Ahmad: formal analysis and review. Fethi Ahmet Ozdemir: antibacterial activities. Sobia Ahsan Halim: software, molecular docking, formal analysis and writing – review & editing. Saeed Ullah: α-glycosidase inhibitory activity. Ajmal Khan: writing – review & editing and α-glycosidase inhibitory activity. Asaad Khalid: formal analysis, software, validation. Ahmed Al-Harrasi: supervision & project administration, characterization and α-glycosidase inhibitory activity. Mumtaz Ali: supervision & project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors extend their appreciation to the Deputyship for Research and Innovation, Ministry of Education in Saudi Arabia, for funding this research work through project number ISP23-81. This study is based on the HED (Higher Education Department), HEREF project number 041, titled “Synthesis and Antimicrobial activities of polyhydroquinoline derivatives: Journey towards lead Antibiotics” which is highlighy aknowledged.References
- R. Mathur, K. S. Negi, R. Shrivastava and R. Nair, RSC Adv., 2021, 11, 1376–1393 RSC.
- M. Mashaly, S. El-Gogary and T. Kosbar, J. Heterocycl. Chem., 2014, 51, 1078–1085 CrossRef CAS.
- A. Maleki, R. Firouzi-Haji and Z. Hajizadeh, Int. J. Biol. Macromol., 2018, 116, 320–326 CrossRef CAS PubMed.
- M. M. Heravi and V. Zadsirjan, ChemistrySelect, 2022, 7, e202104032 CrossRef CAS.
- A. Shaabani, R. Mohammadian, R. Afshari, S. E. Hooshmand, M. T. Nazeri and S. Javanbakht, Mol. Diversity, 2021, 25, 1145–1210 CrossRef CAS PubMed.
- R. M. Borik, N. M. Fawzy, S. M. Abu-Bakr and M. S. Aly, Molecules, 2018, 23, 1398 CrossRef PubMed.
- R. Karthick, G. Velraj, M. Pachamuthu and S. Karthikeyan, J. Mol. Model., 2022, 28, 1–15 CrossRef PubMed.
- P. Ioan, E. Carosati, M. Micucci, G. Cruciani, F. Broccatelli, B. S. Zhorov, A. Chiarini and R. Budriesi, Curr. Med. Chem., 2011, 18, 4901–4922 CrossRef CAS PubMed.
- M. Rahmati, H. Ghafuri, N. Ghanbari and Z. Tajik, Polycyclic Aromat. Compd., 2022, 42, 3019–3035 CrossRef CAS.
- M. Nasr-Esfahani, S. J. Hoseini, M. Montazerozohori, R. Mehrabi and H. Nasrabadi, J. Mol. Catal. A: Chem., 2014, 382, 99–105 CrossRef CAS.
- P. Talwan, S. Chaudhary, K. Kumar and R. K Rawal, Curr. Bioact. Compd., 2017, 13, 109–120 CrossRef CAS.
- M. Sepahvand, F. Buazar and M. H. Sayahi, Appl. Organomet. Chem., 2020, 34, e6000 CrossRef CAS.
- M. G. Dekamin, Z. Karimi, Z. Latifidoost, S. Ilkhanizadeh, H. Daemi, M. R. Naimi-Jamal and M. Barikani, Int. J. Biol. Macromol., 2018, 108, 1273–1280 CrossRef CAS PubMed.
- G. D. Rao, S. Nagakalyan and G. Prasad, RSC Adv., 2017, 7, 3611–3616 RSC.
- N. Taheri, F. Heidarizadeh and A. Kiasat, J. Magn. Magn. Mater., 2017, 428, 481–487 CrossRef CAS.
- Z. G. Mohammadi, A. R. Badiei, Y. Khaniania and M. Hadadpour, 2010.
- S. Hashemi-Uderji, M. Abdollahi-Alibeik and R. Ranjbar-Karimi, Main Group Met. Chem., 2018, 41, 91–101 CrossRef CAS.
- M. A. Ghasemzadeh, J. Appl. Chem. Res., 2019, 13, 8–23 Search PubMed.
- H. T. Nguyen, V. A. Truong and P. H. Tran, RSC Adv., 2020, 10, 25358–25363 RSC.
- N. N. Karade, V. H. Budhewar, S. V. Shinde and W. N. Jadhav, Lett. Org. Chem., 2007, 4, 16–19 CrossRef CAS.
- S. M. Ramish, A. Ghorbani-Choghamarani and M. Mohammadi, Sci. Rep., 2022, 12, 1479 CrossRef CAS PubMed.
- B. Das, B. Ravikanth, R. Ramu and B. V. Rao, Chem. Pharm. Bull., 2006, 54, 1044–1045 CrossRef CAS PubMed.
- B. Das, K. Venkateswarlu, K. Suneel and A. Majhi, Tetrahedron Lett., 2007, 48, 5371–5374 CrossRef CAS.
- K. S. Alghamdi, N. S. Ahmed, D. Bakhotmah and M. M. M. Mostafa, Preprints, 2018, 2018100123 Search PubMed.
- H. Alhazmi, W. Ahsan, B. Mangla, S. Javed, M. Hassan, M. Asmari, M. Al Bratty and A Najmi, Nanotechnol. Rev., 2022, 11, 96–116 CrossRef CAS.
- D. S. Malhi, H. S. Sohal, K. Singh, Z. M. Almarhoon, A. B. Bacha and M. I. Al-Zaben, ACS Omega, 2022, 7, 16055–16062 CrossRef CAS PubMed.
- K. Venkatapathy, C. J. Magesh, G. Lavanya, P. T. Perumal and S. Prema, J. Heterocycl. Chem., 2020, 57, 1936–1955 CrossRef CAS.
- N. Shahab, D. Kong, M. Ali, A. Alam, N. Ur Rehman, S. Ullah, Z. Zainab, M. Khan, A. Latif and M. Shah, Res. Chem. Intermed., 2023, 1–24 Search PubMed.
- N. U. Rehman, R. Maqsood, S. Ullah, S. A. Halim, M. U. Anwar, A. Khan, A. Hussain, J. Hussain and A. Al-Harrasi, S. Afr. J. Bot., 2022, 148, 88–92 CrossRef CAS.
- S. Ullah, M. Waqas, S. A. Halim, I. Khan, A. Khalid, A. N. Abdalla, H. A. Makeen, A. Ibrar, A. Khan and A. Al-Harrasi, Int. J. Biol. Macromol., 2023, 250, 126227 CrossRef CAS PubMed.
- M. Ali, A. Latif, S. Bibi, S. Ali, A. Ali, M. Ahmad, R. Ahmad, A. A. Khan, A. Khan and A. I. Ribeiro, ChemistrySelect, 2022, 7, e202102715 CrossRef CAS.
- M. A. Tienda-Vázquez, E. M. Melchor-Martínez, J. H. Elizondo-Luévano, R. Parra-Saldívar, J. S. Lara-Ortiz, B. Luna-Sosa and C. Q. Scheckhuber, Processes, 2023, 11, 1299 CrossRef.
- J. Casqueiro, J. Casqueiro and C. Alves, Indian J. Endocrinol. Metab., 2012, 16, S27 CrossRef PubMed.
- R. W. Thomsen, A. H. Riis, S. Kjeldsen and H. C. Schønheyder, J. Infect., 2011, 63, 8–16 CrossRef PubMed.
- Y. Y. Cai, L. Y. Xu, L. Q. Chai and Y. X. Li, J. Mol. Struct., 2020, 1204, 127552 CrossRef CAS.
- M. A. Mumit, T. K. Pal, M. A. Alam, M. A.-A.-A.-A. Islam, S. Paul and M. C. Sheikh, J. Mol. Struct., 2020, 1220, 128715 CrossRef CAS PubMed.
- M. Ismail, R. Ahmad, A. Latif, A. A. Khan, M. Ahmad, A. Khan, A. Al-Harrasi and M. Ali, J. Mol. Struct., 2023, 136059 CrossRef CAS.
- R. Thirumurugan, B. Babu, K. Anitha and J. Chandrasekaran, J. Mol. Struct., 2018, 1171, 915–925 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information related to Hirshfeld and Mulliken atomic charges (Table S1), correlation of simulated and experimental 1H NMR (Table S2), 1H and 13C NMR and LC-HRMS spectra, and the binding modes of compounds in the α-glucosidase active site (Fig. S1). See DOI: https://doi.org/10.1039/d4ra00045e |
| This journal is © The Royal Society of Chemistry 2024 |