
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdsorption, dissociation and diffusion behavior of H2O on the PuO2(111) surface from DFT + U-D3: the role of hydrogen bonding
Huang Huang,
Min Zhu,
Fei Wu,
Longxian Li and
Yan Li *
*
Naval University of Engineering, Wuhan 430033, China. E-mail: yacareft@163.com
First published on 5th April 2024
Abstract
The interaction between H2O and plutonium oxide is an essential aspect of researching plutonium corrosion. We systematically studied the adsorption, dissociation, and diffusion of H2O molecules on the PuO2(111) surface with the DFT + U-D3 scheme. We find that the top of the Pu atom is the most stable adsorption site for H2O molecules on the PuO2(111) surface. When multiple H2O molecules are adsorbed, hydrogen bonding between molecules can increase the average adsorption energy. H2O molecules will dissociate into H atoms and O–H groups under certain conditions. We have paid special attention to the role of hydrogen bonds between H2O molecules. When the coverage of H2O molecules is low, hydrogen bonds can significantly promote the adsorption and dissociation of H2O molecules. And H2O tends to exist on the surface of plutonium oxide in dissociated and molecular mixed states. The H atoms produced by the dissociation of H2O molecules are not easily diffused, which may be related to the hydrogen bonding between O–H groups. This work has important theoretical significance for deepening the understanding of the corrosion mechanism of plutonium.
1 Introduction
Plutonium will quickly form a complex oxide layer on the surface when exposed to air. In environments with high humidity, the corrosion process significantly changes, and the corrosion rate increases exponentially. It is significant to study the interaction between H2O molecules and the surface of plutonium oxide to reveal this increasing effect.Despite the significant difficulty in conducting experimental research on plutonium, pioneers have established a basic understanding of the interaction between H2O molecules and plutonium oxides through unremitting efforts. Stackebake's1 research indicates that H2O vapor undergoes desorption within two ranges of 100 °C to 150 °C and 300 °C to 350 °C, with a single physical adsorption process accompanied by two chemical adsorption processes. Several years later, Stackebake2 conducted research on the characteristics of the reaction between binary plutonium oxide and H2O, obtained a series of conclusions, and proposed corresponding mechanisms that are still unclear. The Stackebake team's Haschke3 studied the strengthening effect of moisture on plutonium oxide corrosion. Their research showed that the corrosion rate is not significantly related to H2O pressure above 200 °C or below −25 °C. In comparison, the correlation between the two is high, near 100 °C. In subsequent studies, Haschke4 presented the adsorption process of plutonium oxide in H2O at 25 °C and its humidity dependence. They believed that H2O is regulated through a series of different steps, including five types of adsorbate oxide interactions. In his research on plutonium oxides, Haschke5 found that this non-stoichiometric oxide is formed through the reaction of carbon dioxide with H2O and carbon dioxide with oxygen catalyzed by H2O. Haschke6 also found that the enhanced corrosion of plutonium oxide by H2O is achieved through H2O catalytic cycling. Farr's7 research indicates that hydroxyl groups are commonly present on the surface of PuO2 exposed to H2O vapor or ambient air, and the active sites for H2O and other minor molecule reactions can be updated through thermal or radiation effects. Paffett8 studied the thermodynamic interactions between H2O and actinide oxide surfaces. This process was confirmed, and the estimated values were corrected. The adsorption energy values for the first layer dissociation adsorption and the second layer molecular adsorption at 371 K were reported to be −1.82 and −1.11 eV, respectively.
A more detailed mechanism study is conducted through theoretical calculations. Jomard9 studied the adsorption and dissociation processes of H2O molecules on the PuO2(110) surface, which resulted in two hydroxyl groups asymmetrically placed on the surface, and the dissociation was thermally activated. At low temperatures, molecular states can exist in a metastable configuration for a long lifetime. This surface hydration does not affect the oxidation state of Pu atoms. The research results of Tegner et al.10 on the adsorption of H2O molecules on various surfaces of PuO2 and UO2 indicate that the mixture of molecules and dissociated H2O adsorption is the most stable on the {111} surface. On the other hand, fully dissociated H2O adsorption is the most stable on the {110} and {100} surfaces, resulting in a completely hydroxylated monolayer. They also compared the adsorption of H2O molecules on the surface of PuO2 with oxygen vacancy defects.11 Results indicate that the vacancy creation substantially changes the most stable mode of H2O adsorption on the {111} surface, such that a strong preference for dissociative adsorption on the substoichiometric surface replaces the almost degenerate molecular and dissociative adsorptions on the pristine surface. Tegner12 also studied the adsorption of multilayer H2O molecules on the surface of PuO2. After several layers of adsorption, the adsorption energy gradually decreases and tends to stabilize. In this state, the adsorption mainly comes from hydrogen bonds between H2O molecules. Wellington's13 study on the adsorption of H2O molecules on various surfaces of PuO2 shows that the energy of molecules and dissociative adsorption is similar on the (111) surface, while on the (110) surface, there is a clear tendency towards dissociative adsorption. Zhang Cui14 analyzed the dissociation of PuO2(110) surface and found that the dissociation of H2O on the surface is a two-step hydroxylation process of monomers and clusters, with different mechanisms involved. For cluster H2O, hydrogen bonding interactions between H2O molecules promote dissociation and lead to a more stable dissociation state than individual molecules. Wang15 studied the adsorption, diffusion, and dissociation processes of individual H2O molecules on the surface of α-Pu2O3(111), and the results showed that H2O molecules were preferentially adsorbed on surface oxygen vacancies with shallow adsorption energy. Wang's team16 also analyzed the kinetic process of adsorption and dissociation of H2O molecules on the surface of Pu2O3(111), and confirmed the hydrogen bonding and hydrogen transfer reactions within clusters, which promoted the dissociation of H2O molecules on the surface of Pu2O3(111). The adsorbed H2O tends to dissociate into hydrogen atoms and hydroxyl groups in addition to dissociating into hydrogen molecules and oxygen atoms. Zhang Le et al.17–20 conducted a series of studies on the interaction between H2O and plutonium oxides. They first analyzed the modulation mechanism of H2O polarons adsorbed on the surface of PuO2(111), and found during the comparison with CO2 that electron polarons tend to promote H2O adsorption, while hole polarons tend to promote CO2 adsorption. In addition, studies on the adsorption thermodynamics of H2O and CO2 on PuO2(111) surfaces have found that as coverage increases, the adsorption form of H2O will change from a molecular state to a mixed adsorption state, but CO2 always adsorbs in a molecular state. The difference in adsorption behavior is mainly due to the hydrogen bonding and Coulomb interaction between the adsorbed molecules. They also believe that the two different O coordination numbers of Pu on the surface of Pu2O3(111) play a crucial role in the selective adsorption of H2O and CO2 on the surface sites. In recent work, they focused on the hydroxyl group promoting H2 release reaction after the decomposition of H2O on the surface of Pu oxide and found that both *H and *OH can promote the H2 release process, especially when the surface of PuO2 is reduced to Pu2O3, but the reaction mechanisms of the two are different. In the above theoretical research, people have focused on the adsorption configuration and energy issues between the surface of plutonium oxide and H2O molecules, as well as the effects of dissociation and O atom vacancy defects. However, a complete understanding of the adsorption dissociation and diffusion processes is still unclear. The understanding of the impact of hydrogen bonding between H2O molecules on adsorption, dissociation, and subsequent diffusion is still relatively vague.
In this work, a first principles approach based on DFT + U-D3 was used to systematically investigate the adsorption, dissociation, and diffusion processes of H2O molecules on the surface of PuO2(111). The rest of this paper is organized as follows. The details of the computational method are described in Section 2, and the results are discussed in Section 3. Section 4 contains the main conclusions.
2 Methods and computational details
The first-principles calculations were performed by the Vienna ab initio simulation package (VASP).21 The generalized gradient approximation (GGA) of Perdew, Burke, and Ernzerhof (PBE) is used for the exchange–correlation interaction of electrons.22,23 The Hubbard model is used within the DFT + U method in the Dudarev formalism to treat strong on-site Coulomb interaction.24 An effective U (Ueff = U − J; i.e., the difference between the Coulomb U and exchange J parameters, hereafter referred to as U) value of 4 eV is selected for the 5f electrons of Pu and U, according to our previous calculations25,26 and other computational experience.10,12,14,18,20,27–29 We use the widely accepted collinear 1-k antiferromagnetic (AFM) states along the (111) orientations for the magnetic orders.30,31 The Brillouin zone was sampled with a 3 × 3 × 1 k-points mesh for the surface models generated by the Monkhorst–Pack method.32 The cutoff energy is set to 520 eV. When optimizing the structure, the conjugate gradient method is used to search for the minimum energy value, the energy convergence is interpreted as 1 × 105 eV, and the force convergence criterion is set to 0.01 eV Å−1. The van der Waals correction is described by the DFT-D3 method.33,34PuO2 crystallizes in the cubic fluorite (spacegroup: Fm3![[m with combining macron]](https://www.rsc.org/images/entities/i_char_006d_0304.gif) ) structures with lattice parameters of 5.396 Å. Our calculated result is 5.432 Å, with an error of 6.6% in the experiment, which is within an acceptable range. We adopted a PuO2(111) surface slab model (shown in Fig. 1), consisting of five layers of plutonium atoms with 20 Pu atoms and 40 O atoms and a vacuum spacing of 20 Å, to avoid the interaction between periodic images. In the calculation of adsorption, the plutonium atoms in the top two layers (including nearby oxygen atoms) are allowed to relax together with the adsorbate, while the atoms in the other layers are frozen at optimized volume positions.
) structures with lattice parameters of 5.396 Å. Our calculated result is 5.432 Å, with an error of 6.6% in the experiment, which is within an acceptable range. We adopted a PuO2(111) surface slab model (shown in Fig. 1), consisting of five layers of plutonium atoms with 20 Pu atoms and 40 O atoms and a vacuum spacing of 20 Å, to avoid the interaction between periodic images. In the calculation of adsorption, the plutonium atoms in the top two layers (including nearby oxygen atoms) are allowed to relax together with the adsorbate, while the atoms in the other layers are frozen at optimized volume positions.
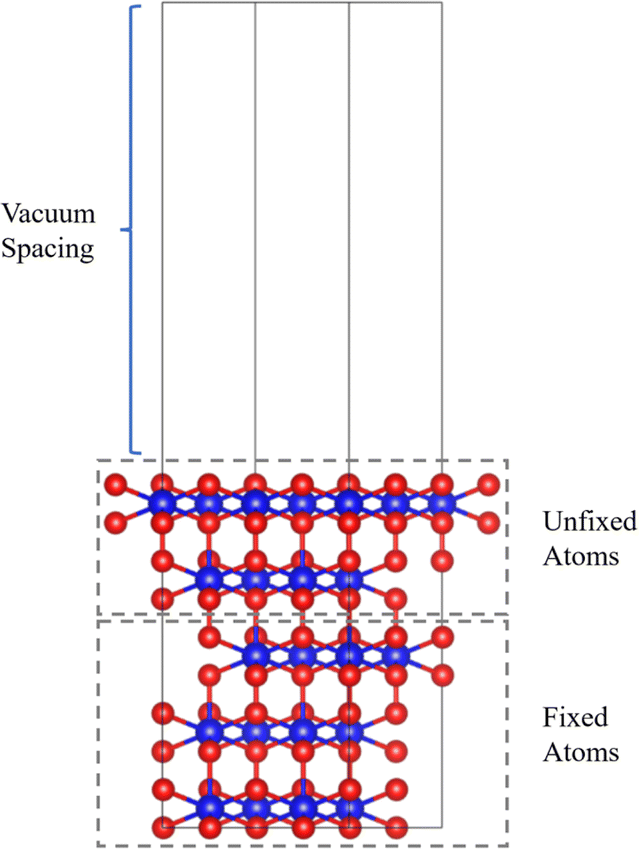 | ||
| Fig. 1 PuO2(111) surface model diagram. Red spheres are oxygen atoms, and blue spheres are plutonium atoms. | ||
The adsorption energy (Ea) of molecules on the surface is expressed as
| Ea = Eslab+P − Eslab − EP |
| Ea-ave = (Eslab+P − Eslab − nEP)/n |
A negative of Ea means heat release and spontaneous, and vice versa.
The H2O dissociation on PuO2(111) surfaces was explored using a climbing-image nudged elastic band (CI-NEB). The geometries of intermediate states and the activation energy were obtained via the simultaneous energy minimization of these discretized images between known initial and final states. In these CI-NEB calculations, relatively low accuracy is acceptable. The energy convergence is interpreted as 5 × 104 eV, and the force convergence criterion is set to 0.02 eV Å−1.
3 Results and discussion
3.1 Adsorption geometries and energetics of a single H2O molecule
After analysis, we found three adsorption sites for H2O molecules on the surface of PuO2(111): the Pu atomic top site, Pu atomic bridge site, and Pu atomic hole site (Fig. 2). H2O molecules can approach adsorption sites in three different postures: the molecule plane is parallel to the PuO2(111) surface (abbreviated as P), the molecule plane is vertical to the PuO2(111) surface with the H–H axis parallel to the PuO2(111) surface (abbreviated as V1), and the molecule plane is vertical to the PuO2(111) surface with the H–H axis vertical to the PuO2(111) surface (abbreviated as V2), as shown in Fig. 3.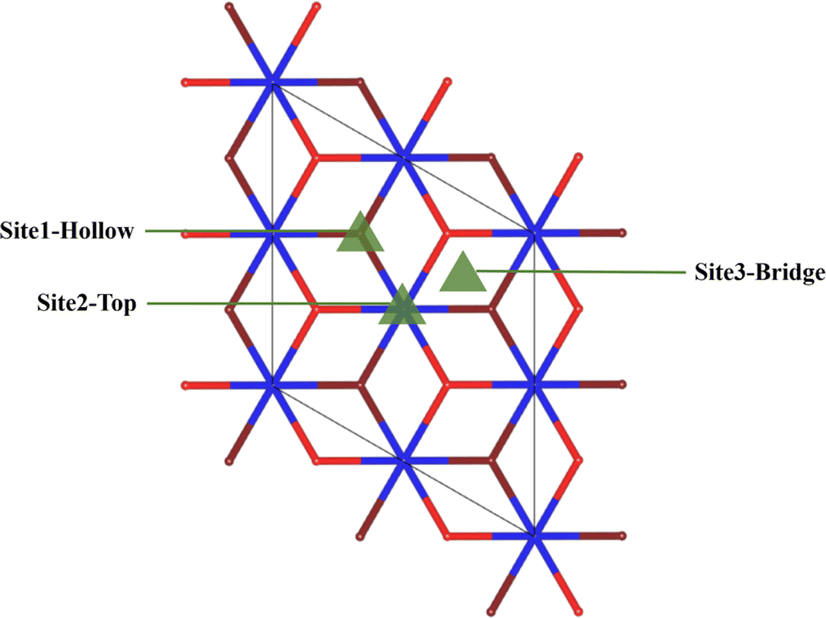 | ||
| Fig. 2 Schematic diagram of adsorption points. Red spheres are the surface oxygen atoms, brown spheres are the secondary subsurface oxygen atoms, and blue spheres are plutonium atoms. | ||
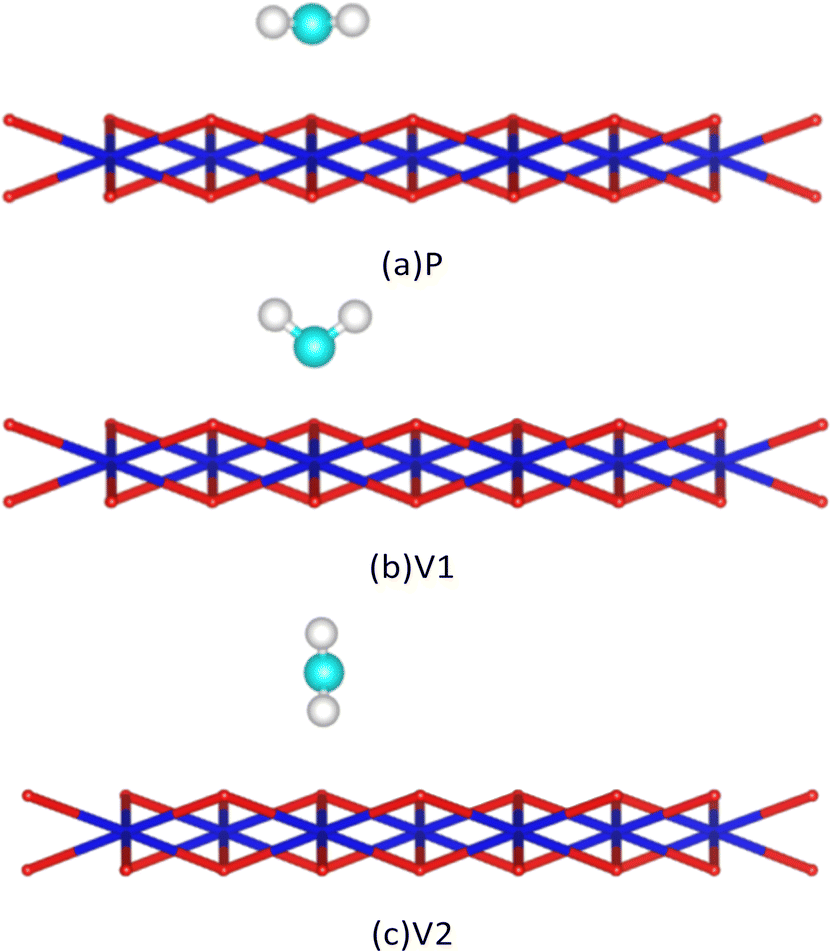 | ||
| Fig. 3 Schematic diagram of the posture of H2O molecules approaching the surface. Cyan spheres are oxygen atoms of H2O, and white spheres are hydrogen atoms of H2O. | ||
In a cubic box with side lengths of 10 Å, the free H2O molecule was simulated. The optimized O–H bond length and bond angle (∠H–O–H) are 0.97 Å and 104°, which is very close to the experimental values of 0.96 Å and 104.45°.
H–V1 represents H2O molecules approaching the surface at the hollow site in a V1 posture. The optimized adsorption energy for configurations is shown in Table 1.
| Configuration | H–P | H–V1 | H–V2 | T–P | T–V1 | T–V2 | B–P | B–V1 | B–V2 |
|---|---|---|---|---|---|---|---|---|---|
| Adsorption energy (eV) | −0.756 | −0.743 | −0.431 | −0.781 | −0.773 | −0.521 | −0.775 | −0.765 | −0.321 |
From the perspective of adsorption configuration, when the H–H axis of H2O molecules is parallel to the surface, regardless of the initial placement point, the optimized solution can always be at the top position of the surface Pu atom, and the H2O molecule plane is parallel to the surface. In the adsorption configuration, H2O molecules move slightly in the direction indicated by the two H atoms. The O–H bond is slightly elongated, which means H2O molecules tend to dissociate. The above situation is mainly due to the interaction between plutonium oxide and H2O, where the H atom in H2O molecules tends to interact with the O atom. In contrast, the O atom in H2O tends to interact with the Pu atom. Under these two interactions, H2O molecules will adsorb on the PuO2(111) surface in a relatively fixed posture and undergo strong interactions on the surface, resulting in higher adsorption energy. When the H–H axis of the H2O molecule is perpendicular to the surface, the H atom closest to the surface plays the leading role, while the O atom and another H atom are relatively far from the surface and cannot interact strongly with the surface atoms, resulting in smaller adsorption energy and different adsorption configurations.
3.2 Dissociation of a single H2O molecule
The CINEB method was used to explore the dissociation process of H2O molecules. This method has played an important role in surface research related to plutonium multiple times. The CINEB method requires determining a stable and reasonable initial state (IS) and final state (FS).In the initial state before dissociation, H2O stably adsorbs on the surface in molecular form. Based on the analysis of the adsorption of H2O molecules at various points on the Pu surface, we selected T–P as the IS. Compared to IS, the setting of FS needs to be carefully considered. Theoretical studies have shown that an H2O molecule adsorbed on the surface of plutonium oxide may dissociate into an H atom and an OH group under the influence of external energy. In previous research, we also found the dissociation trend of H2O molecules. For the H atom, the stable adsorption point is at the top position of the surface O atom, which has been validated in our previous work.35 We also conducted an adsorption analysis similar to H2O molecules for the OH group. The adsorption energy of each point is shown in Table 2. In terms of energy, the most stable adsorption site for OH is at the top of the Pu atom. After the dissociation of H2O molecules into H atoms and OH groups, the two types of particles will exist on their respective most stable sites on the surface of PuO2, forming the dissociation adsorption state of H2O molecules. After sufficient relaxation, the desired FS is obtained.
| Configuration | Hollow | Top | Bridge |
|---|---|---|---|
| Adsorption energy (eV) | −0.311 | −1.243 | −0.531 |
Fig. 4 shows the various steps of dissociation of a single H2O molecule on the surface of PuO2(111). The energy of the entire system increases from the IS to TS1, during which the O–H bonds of H2O molecules are stretched after the system absorbs energy. From TS1 to TS2, the energy continues to rise, requiring further absorption of energy to cross the energy barrier of 0.831 eV and achieve the breaking of O–H bonds. After crossing the energy barrier, the system energy gradually decreases until the H atom detached from the H2O molecule combines with the O atom on the surface of PuO2 to form a structure, and the O–H group of the H2O molecule combines with the Pu atom on the surface of PuO2.
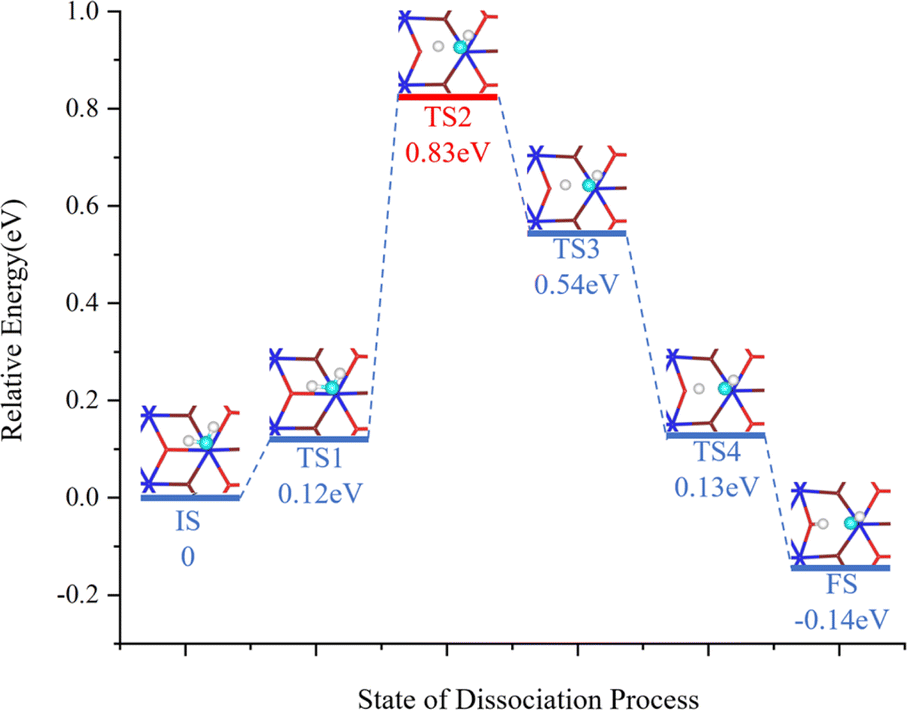 | ||
| Fig. 4 The minimum-energy path for H2O dissociation. The CINEB simulation consists of 6 images. Zero-point energy corrections are not included. | ||
3.3 Adsorption of multiple H2O molecule
There are hydrogen bonds (HBs) between H2O molecules within a specific range, and this special intermolecular force can have a particular impact on the adsorption process, resulting in apparent differences when individual H2O molecules and H2O molecule clusters interact with the surface of Pu oxide. To analyze this differences, multiple H2O molecule cluster adsorption models with different coverage were constructed based on the stable adsorption sites of a single H2O molecule (Fig. 5). There is a mutual correspondence between the O atom in H2O molecules and the Pu atom on the surface. Given this, we define coverage as the proportion of H2O molecules covering the top positions of surface Pu atoms.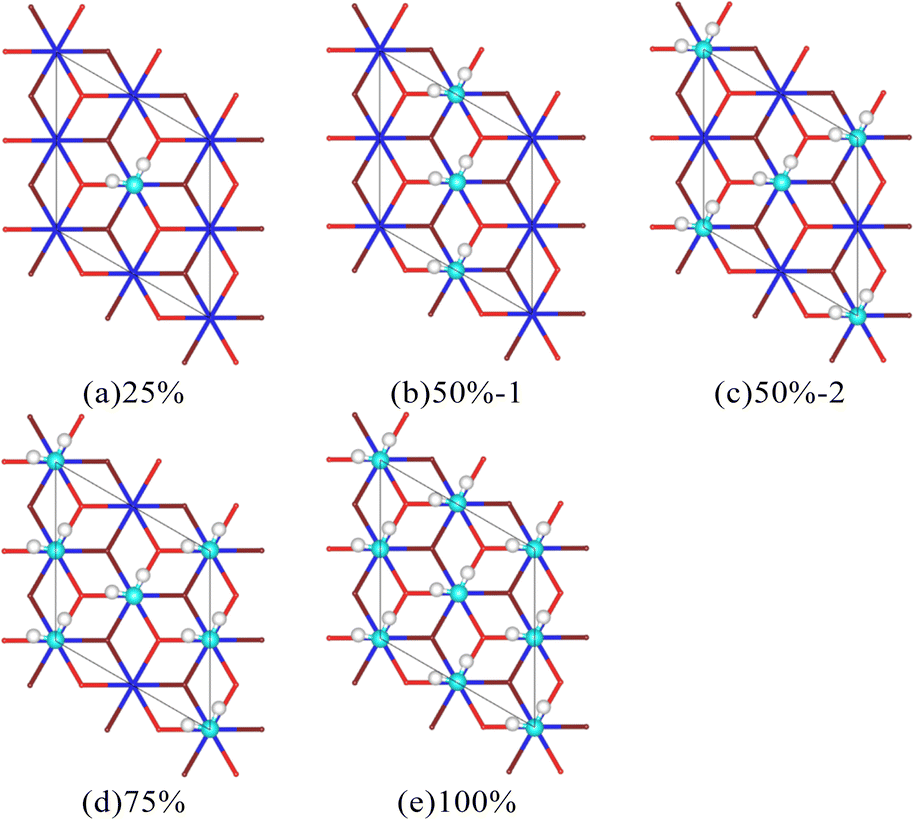 | ||
| Fig. 5 Multiple H2O molecules adsorption configurations. | ||
The average adsorption energy of each molecule and bond length of O–H are shown in Table 3. When multiple H2O molecules exist on the surface of Pu, the average adsorption energy is higher than that of a single H2O molecule, that is, the interaction between H2O molecules and the surface is strengthened. As the coverage increases, the average adsorption energy of each H2O molecule on the surface first increases and then decreases. The result is consistent with Tegner's.10 The increase in O–H bond length indicates a decrease in bond strength, indicating a more pronounced trend of bond breakage. Interestingly, with a coverage rate of 50%, different arrangements can also lead to differences in the adsorption energy of H2O molecules on the surface. The process of interaction between H2O molecules and the PuO2(111) surface can cause the plane of H2O molecules to no longer be parallel to the surface, as shown in Fig. 6. After relaxation, the distance between the donor and acceptor of hydrogen bonds in the 50%-1 system is 2.95 Å, while the distance between the two in the 50%-2 system is 3.19 Å. When H2O molecules are vertically arranged on the surface, the strong hydrogen bonding between molecules significantly promotes the adsorption of H2O molecules. When H2O molecules are horizontally arranged, the hydrogen bonding between molecules is weak, and the promoting effect on adsorption is not significant.
| Coverage | 25% | 50%-1 | 50%-2 | 75% | 100% |
|---|---|---|---|---|---|
| Adsorption energy/molecules (eV) | −0.781 | −0.814 | −0.793 | −0.801 | −0.791 |
| The bond length of O–H (Å) | 0.988 | 1.049 | 0.991 | 0.998 | 0.989 |
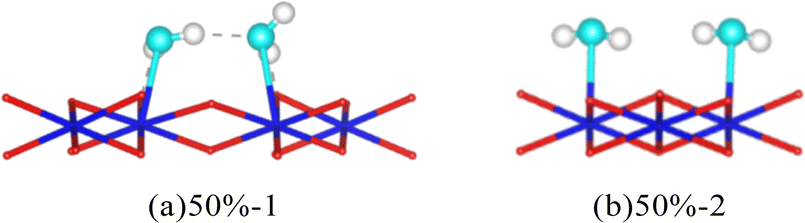 | ||
| Fig. 6 The configuration of H2O molecules arranged horizontally and vertically after relaxation. | ||
In addition, the O–H bond length also exhibits some variation characteristics. Therefore, it is worth further attention to how the dissociation process changes when multiple H2O molecules exist on the surface. We conducted a series of H2O molecule dissociation analyses using the CINEB method.
H2O adsorption in fully molecular (abbreviation as M), fully dissociative (abbreviation as D), mixed molecular, and dissociative (abbreviation as M-D) states on PuO2(111) surface, respectively. In our periodic model of the surface of PuO2(111), there are a total of four Pu atoms, corresponding to four H2O molecules with 100% coverage. The adsorption energies corresponding to each dissociation process are shown in Table 4. It can be observed that the decomposition of one of the two H2O molecules occurs very easily, and it does not require external energy input. As a comparison, if both molecules dissociate, it requires some external energy input. For the case of four H2O molecules with a coverage rate of 100%, when all three H2O molecules are in a dissociation state, the energy required for the dissociation of the last molecule is even higher than that of a single H2O molecule. In summary, when surface H2O molecules are not saturated, intermolecular hydrogen bonds can promote dissociation. When water molecules are saturated, the effect of hydrogen bonds cannot compensate for the energy loss caused by the mismatch in the number of oxygen (PuO2) and hydrogen (H2O atoms). The previous dissociation is prone to occur, while the energy required for subsequent dissociation gradually increases. The coexistence of hydroxyl groups and H2O molecules on the surface of plutonium oxide was also observed in the experiment.36
| State change | 1M → 1D | 2M → 1M1D | 1D1M → 2D | 3M → 2M1D |
| Energy barrier (eV) | 0.831 | 0 | 0.343 | 0.103 |
| State change | 2M1D → 1M2D | 1M2D → 3D | 4M → 3M1D | 3M1D → 2M2D |
| Energy barrier (eV) | 0.331 | 0.513 | 0.111 | 0.367 |
| State change | 2M2D → 1M3D | 1M3D → 4D | ||
| Energy barrier (eV) | 0.567 | 0.841 |
3.4 H atomic diffusion
For the hydrogenation corrosion of metals, hydrogen adsorbed in an atomic state on the surface may subsequently diffuse internally under certain conditions. In this section, we first analyze the diffusion of a single hydrogen atom adsorbed on the surface O atom of the PuO2(111) surface. Two diffusion paths were established, with diffusion path 1 being H atom diffusing from the top position of the surface oxygen atom (OT1) to the top position of the adjacent surface oxygen atom (OT2), and diffusion path 2 being H atom diffusing from the top position of the surface oxygen atom (OT1) to the top position of the adjacent subsurface oxygen atom (SOT1), as shown in Fig. 7. The energy barrier that diffusion path 1 needs to cross is 1.4 eV, and the energy barrier that diffusion path 2 needs to cross is 2.3 eV. From an energy perspective, H atoms tend to exist more on the surface of PuO2(111), indicating that the surface of PuO2(111) can act as a hydrogen barrier, which is consistent with Yu's37 judgment on PuO2(110).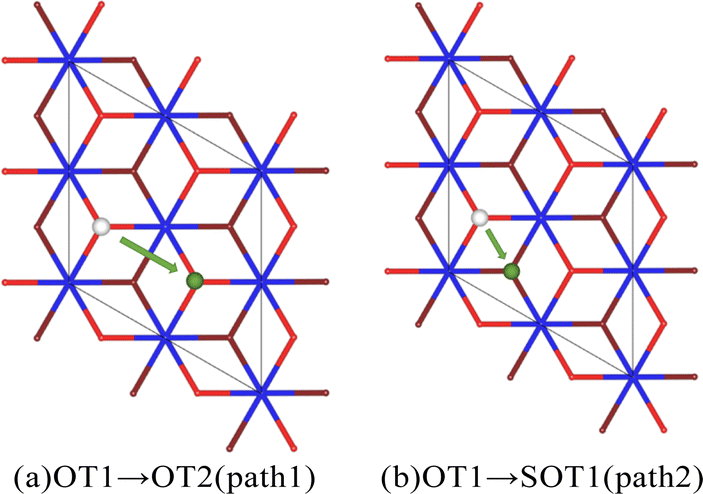 | ||
| Fig. 7 The diffusion path of the single H atom. | ||
Subsequently, we analyzed the diffusion of hydrogen atoms adsorbed on the surface of PuO2(111) in the dissociated state of a single H2O molecule. Three diffusion paths were also established. To distinguish them, define them as paths 3, 4, and 5, as shown in Fig. 8. The diffusion energy barriers of the three paths are 2.3, 3.1, and 4.3 eV, respectively. This indicates that whether diffusing to adjacent atoms on the surface or internally, the hydrogen atoms obtained from the dissociation of H2O molecules require more stringent conditions than individual hydrogen atoms. We speculate that this may be related to the hydrogen bonds formed between hydroxyl groups. The first hydroxyl group is composed of a hydrogen atom dissociated from a H2O molecule and a surface oxygen atom. The second hydroxyl group originates from the dissociation of a H2O molecule.
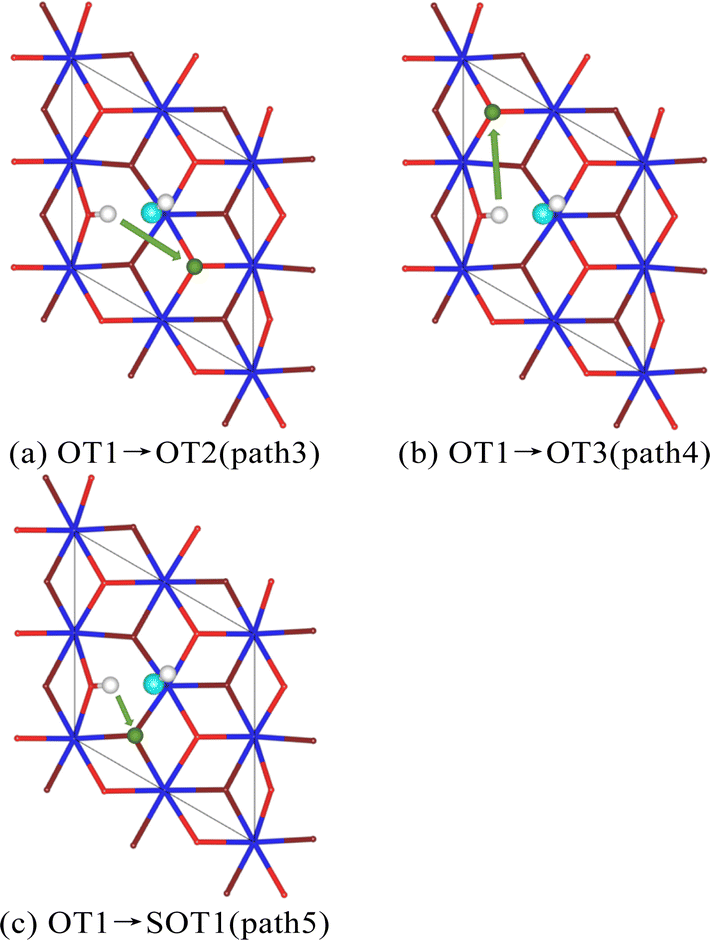 | ||
| Fig. 8 The diffusion path of the H atom from dissociation of H2O molecule. | ||
4 Conclusions
We have performed a series of first-principles calculations with DFT + U-D3 of H2O molecules on the PuO2(110) surface to investigate the mechanism of H2O adsorption, dissociation, and diffusion. The results indicate that hydrogen bonding has an impact on the surface behavior of H2O. The conclusions are as follow:(1) H2O molecules will adsorb at the top of Pu atoms on the surface of PuO2(111) in a fixed posture. Under certain external energy inputs, H2O molecules will detach a proton to produce O–H groups that combine with surface Pu atoms. The removed proton will then combine with surface O atoms to form new O–H groups.
(2) When H2O molecules do not fully cover the surface, hydrogen bonds between molecules can also promote the dissociation of H2O molecules. When H2O molecules cover the surface, the energy required for molecular dissociation gradually increases until the dissociation energy of the last molecule is equivalent to that of a single molecule.
(3) The H atom obtained from the dissociation of H2O molecules requires more energy for diffusion than a single H atom. Diffusion inward is particularly difficult. We speculate that this is related to the hydrogen bonding between the O–H groups formed after dissociation.
Conflicts of interest
There are no conflicts to declare.Notes and references
- J. L. Stakebake, J. Phys. Chem., 1973, 77, 581–586 CrossRef CAS.
- J. L. Stakebake, D. T. Larson and J. M. Haschke, J. Alloys Compd., 1993, 202, 251–263 CrossRef CAS.
- J. M. Haschke, T. H. Allen and J. L. Stakebake, J. Alloys Compd., 1996, 243, 23–35 CrossRef CAS.
- J. M. Haschke and T. E. Ricketts, J. Alloys Compd., 1997, 252, 148–156 CrossRef CAS.
- J. M. Haschke, T. H. Allen and L. A. Morales, Science, 2000, 287, 285–287 CrossRef CAS PubMed.
- J. M. Haschke, T. H. Allen and L. A. Morales, J. Alloys Compd., 2001, 314, 78–91 CrossRef CAS.
- J. D. Farr, R. K. Schulze and M. P. Neu, J. Nucl. Mater., 2004, 328, 124–136 CrossRef CAS.
- M. T. Paffett, D. Kelly, S. A. Joyce, J. Morris and K. Veirs, J. Nucl. Mater., 2003, 322, 45–56 CrossRef CAS.
- G. Jomard, F. Bottin and G. Geneste, J. Nucl. Mater., 2014, 451, 28–34 CrossRef CAS.
- B. E. Tegner, M. Molinari, A. Kerridge, S. C. Parker and N. Kaltsoyannis, J. Phys. Chem. C, 2017, 121, 1675–1682 CrossRef CAS.
- J. P. W. Wellington, B. E. Tegner, J. Collard, A. Kerridge and N. Kaltsoyannis, J. Phys. Chem. C, 2018, 122, 7149–7165 CrossRef CAS.
- B. E. Tegner and N. Kaltsoyannis, J. Vac. Sci. Technol., A, 2018, 36, 041402 CrossRef.
- J. P. W. Wellington, A. Kerridge, J. Austin and N. Kaltsoyannis, J. Nucl. Mater., 2016, 482, 124–134 CrossRef CAS.
- C. Zhang, Y. Yang and P. Zhang, J. Phys. Chem. C, 2018, 122, 371–376 CrossRef CAS.
- X.-X. Wang, S. Wang, C. Zhang, Y. Yang and P. Zhang, J. Phys.: Condens.Matter, 2019, 31, 265001 CrossRef CAS PubMed.
- S. X. Wang and P. Zhang, J. Nucl. Mater., 2022, 566, 153743 CrossRef CAS.
- L. Zhang, B. Sun, Q. L. Zhang, H. F. Liu, K. Z. Liu and H. F. Song, Appl. Surf. Sci., 2020, 516, 146164 CrossRef CAS.
- L. Zhang, B. Sun, Q. L. Zhang, H. F. Liu, K. Z. Liu and H. F. Song, Appl. Surf. Sci., 2021, 537, 147882 CrossRef CAS.
- L. Zhang, B. Sun, Q. L. Zhang, H. F. Liu, K. Z. Liu and H. F. Song, J. Alloys Compd., 2021, 870, 159371 CrossRef CAS.
- L. Zhang, B. Sun, Q. L. Zhang, H. F. Liu and H. F. Song, J. Nucl. Mater., 2023, 585, 154642 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- P. E. Blochl, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B, 1998, 57, 1505–1509 CrossRef CAS.
- G. K. Zheng, Q. Wang, J. F. Chen, R. Z. Qiu and M. Zhu, Inorg. Chem., 2023, 62, 16047–16058 CrossRef CAS PubMed.
- H. Huang, M. Zhu and Y. Li, Coatings, 2024, 14, 195 CrossRef CAS.
- L. Zhang, B. Sun, Q. Zhang, H. Liu, K. Liu and H. Song, ACS Omega, 2020, 5, 7211–7218 CrossRef CAS PubMed.
- L. Zhang, B. Sun, Q. Zhang, H. Liu, K. Liu and H. Song, Comput. Mater. Sci., 2020, 179, 109688 CrossRef CAS.
- L. Zhang, L. F. Wang, B. Sun, H. F. Liu, G. Li, H. L. Yu, Q. L. Zhang and H. F. Song, J. Solid State Chem., 2022, 313, 123314 CrossRef CAS.
- D. E. Peterson and M. E. Kassner, Bull. Alloy Phase Diagrams, 1988, 9, 261–267 CrossRef.
- M. Regulski, R. Przeniosło, I. Sosnowska, D. Hohlwein and R. Schneider, J. Alloys Compd., 2004, 362, 236–240 CrossRef CAS.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B, 1976, 13, 5188–5192 CrossRef.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- H. Huang, M. Zhu and Y. Li, Coatings, 2024, 14, 195 CrossRef CAS.
- A. Seibert, T. Gouder and F. Huber, Radiochim. Acta, 2010, 98, 647–654 CrossRef CAS.
- H. L. Yu, T. Tang, S. T. Zheng, Y. Shi, R. Z. Qiu, W. H. Luo and D. Q. Meng, J. Alloys Compd., 2016, 666, 287–291 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |