
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and activity evaluation of arctigenin derivatives with HDAC inhibition activity†
Xinyue Jianga,
Yuchao Yana,
Huali Yanga,
Maosheng Cheng a,
Deqiang Dou*b and
Yang Liu*a
a,
Deqiang Dou*b and
Yang Liu*a
aKey Laboratory of Structure-Based Drug Design & Discovery, Ministry of Education, School of Pharmaceutical Engineering, Shenyang Pharmaceutical University, Shenyang, 110016, P.R. China. E-mail: y.liu@syphu.edu.cn
bDepartment of Chinese Medicine Chemistry, Liaoning University of Traditional Chinese Medicine, Dalian, 116000, P.R. China. E-mail: deqiangdou@126.com
First published on 20th March 2024
Abstract
Arctigenin, a natural product with diverse pharmacological activities, can inhibit cell proliferation and survival and has shown promising potential in cancer research. In this study, we designed a series of arctigenin derivatives with HDAC inhibitory activity based on the synergistic effects between HDAC inhibitors and arctigenin. Among them, compound B7 exhibited significantly higher antiproliferative activity in the MV411 cell line compared to the positive control, tucidinostat. Additionally, enzymatic activity testing was performed with compound B7. Further mechanistic studies indicated that compound B7 induced apoptosis through the Caspase-3 pathway in MV411 cells and enhanced histone acetylation levels in the MV411 cell line. These findings highlight the broad potential application of these arctigenin derivatives in cancer therapy.
Introduction
Arctigenin is a natural product primarily found in plants such as Arctium lappa (burdock). The molecular formula of arctigenin is C21H24O6, and the compound has been extensively studied and shown to possess certain anticancer activities.1–3 The anticancer effects of arctigenin are attributed to multiple mechanisms. Some studies have indicated that arctigenin can inhibit the proliferation and growth of tumour cells,4–7 induce apoptosis (programmed cell death),8 and impede the invasion and metastasis of cancer cells.9,10 Regarding haematologic malignancies, arctigenin has also demonstrated inhibitory effects. For instance, in vitro experiments have shown that arctigenin can inhibit the proliferation and growth of leukaemia cells, including acute lymphoblastic leukaemia and acute myeloid leukaemia cells, and induce apoptosis in these cells.11 Furthermore, arctigenin has shown inhibitory effects on the growth of lymphomas and multiple myeloma cell lines, among other haematologic malignancies.12Histone deacetylases (HDACs) are a class of enzymes that play a crucial role in regulating gene expression and epigenetic modifications13,14(Fig. 1). They are responsible for removing acetyl groups from lysine residues in histone proteins, leading to chromatin condensation and transcriptional silencing.15 Aberrant upregulation or excessive activity of HDACs frequently occurs in various malignancies, and as a result, tumour suppressor genes are silenced and tumour cells proliferate.16 Consequently, over the past 20 years, HDACs have been widely targeted for cancer therapy.17,18 To date, five HDAC inhibitors, including vorinostat (SAHA),19 romidepsin,20 belinostat,21 panobinostat,22 and tucidinostat,23 have been approved for the treatment of haematologic malignancies, and many others are currently in various stages of development.17
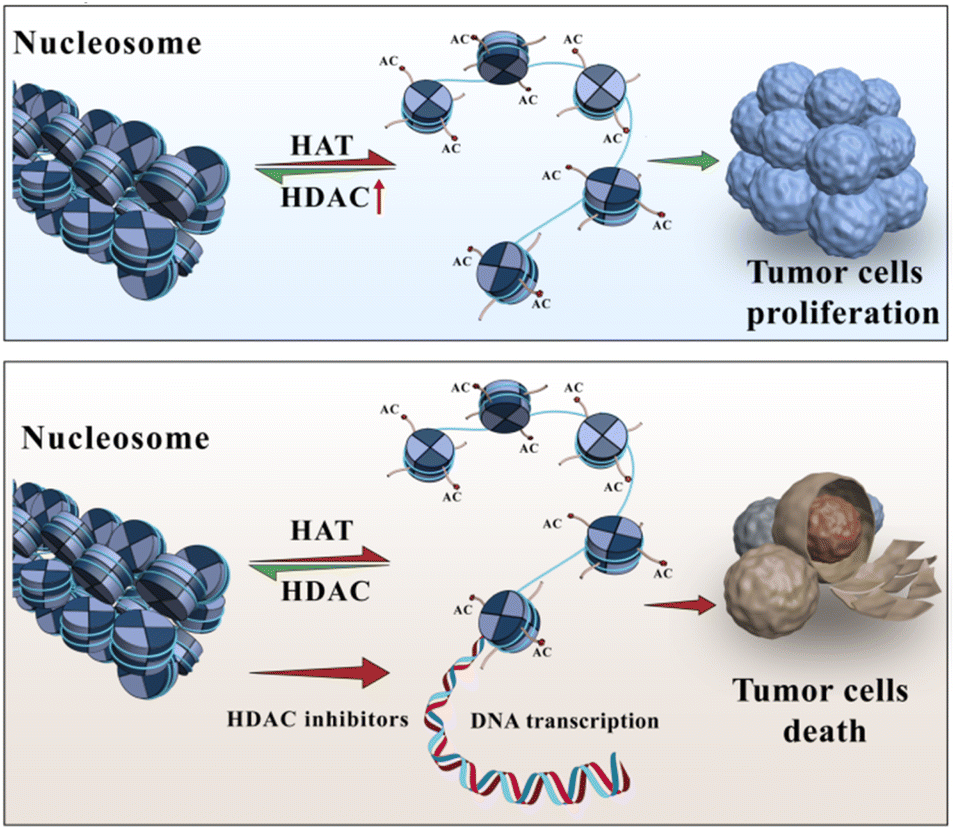 | ||
| Fig. 1 HATs and HDACs jointly regulate the acetylation pattern of histones. | ||
Combining histone deacetylase (HDAC) inhibitors with arctigenin, a natural product, is aimed at achieving anticancer activity, thereby potentially demonstrating synergistic antitumor effects. Importantly, by introducing HDAC inhibitory fragments, such as zinc ion-binding hydroxamic acid and ortho-benzamide moieties, the derivatives of arctigenin may demonstrate HDAC inhibitory activity, thereby enhancing the therapeutic efficacy of the natural product. Furthermore, by employing arctigenin as the cap group, it is anticipated to impact the selectivity towards different HDAC isoforms (Fig. 2).
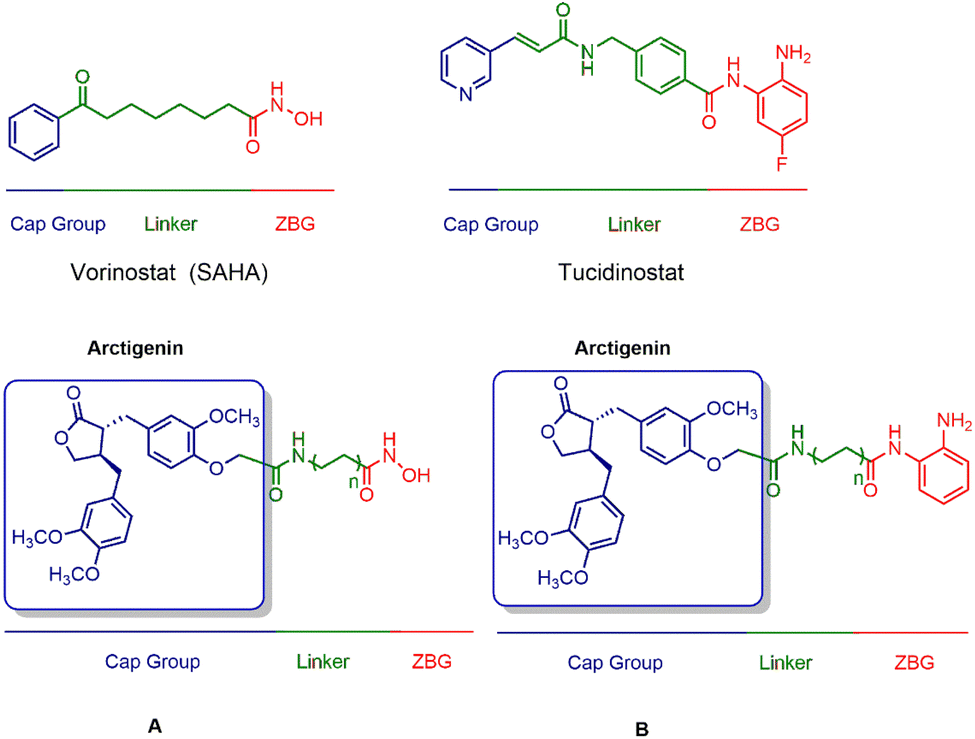 | ||
| Fig. 2 Design strategy of target compounds. | ||
Using arctigenin as our foundational compound in this study, we incorporated essential zinc-binding elements from the commercial drugs SAHA and tucidinostat. Consequently, we synthesized two series, A and B, that contained 18 distinct compounds. In vitro evaluation of their antitumour activity revealed that the optimized compound B7 displayed superior antiproliferative effects against the MV411 cell line. Further mechanistic investigations demonstrated that B7 induced cell apoptosis in MV411 cells in a dose-dependent manner mediated by Caspase-3. Caspase-3 plays a critical role in the process of programmed cell death or apoptosis. Additionally, compound B7 significantly increased cellular acetylation levels. Overall, compound B7 exhibited more potent antitumour activity than that of arctigenin and tucidinostat. The mechanism underlying the activity involves the induction of cell apoptosis and an increase in cellular acetylation levels. These findings provide valuable insights for the further development and potential clinical applications of this compound.
Results and discussion
Chemistry
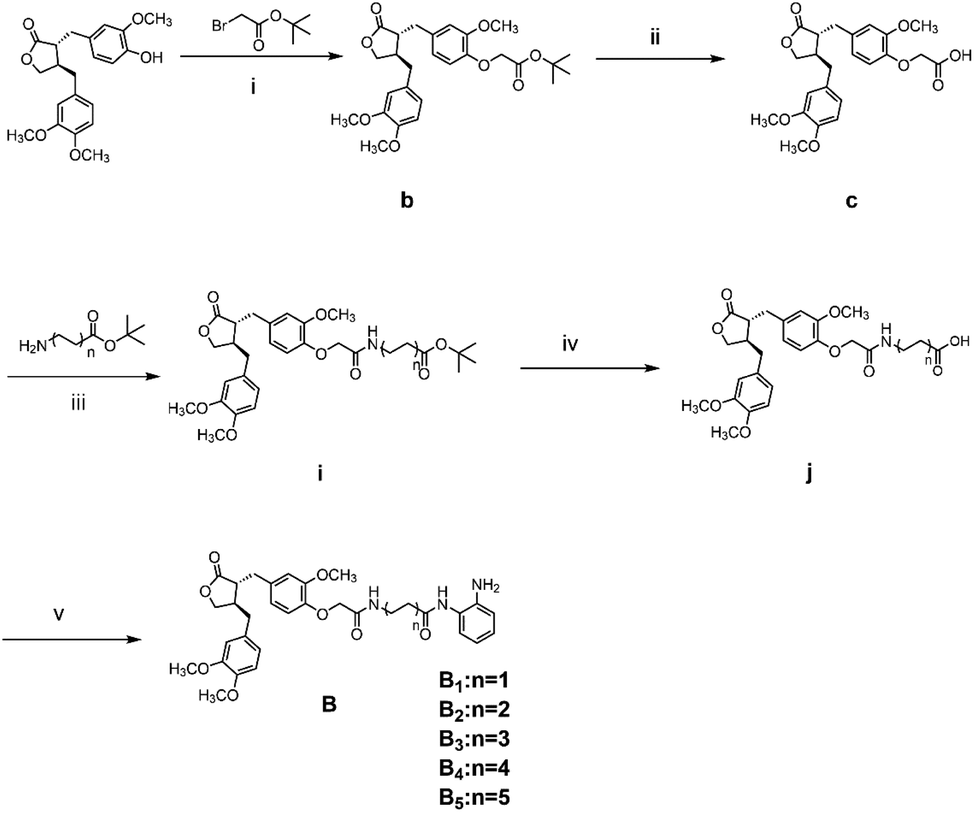
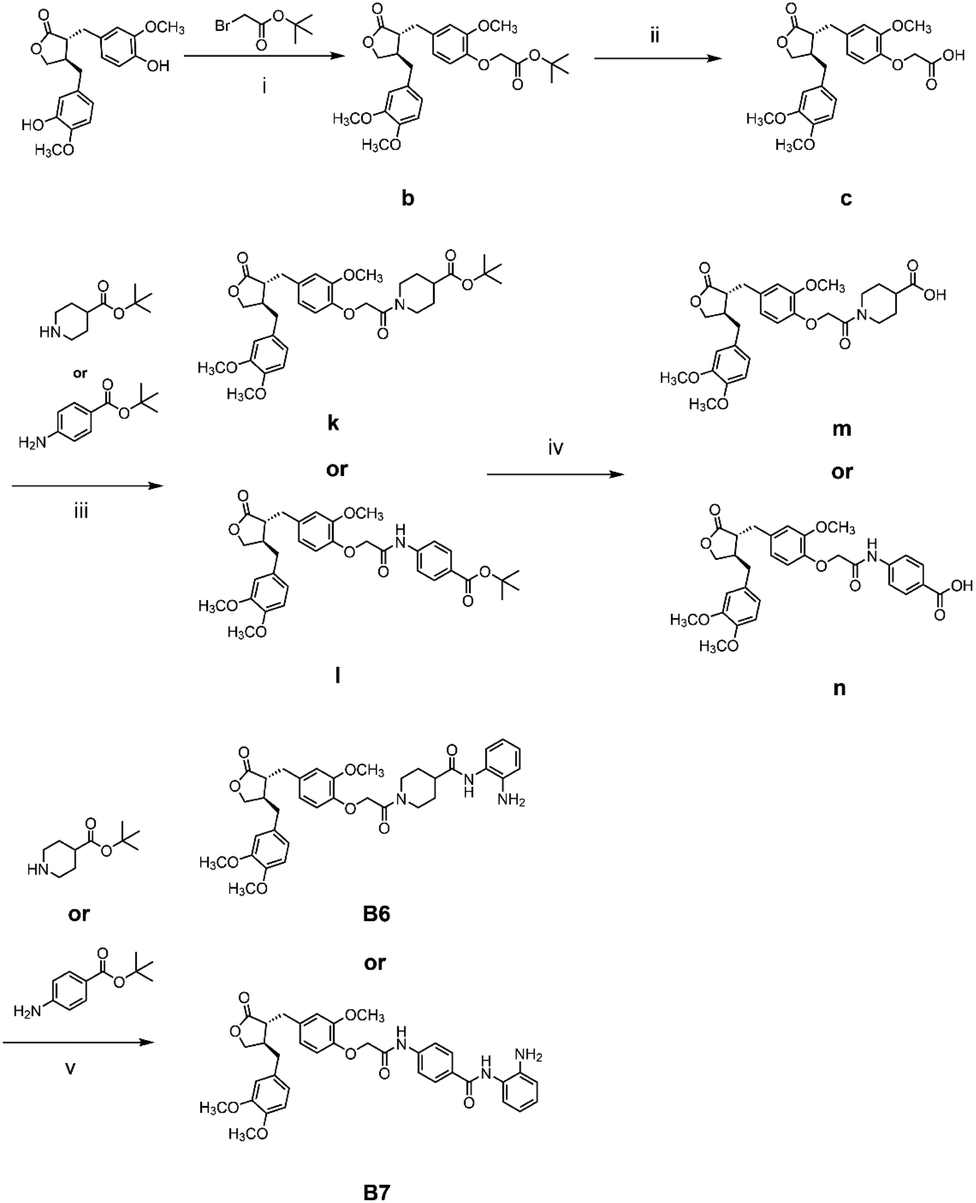
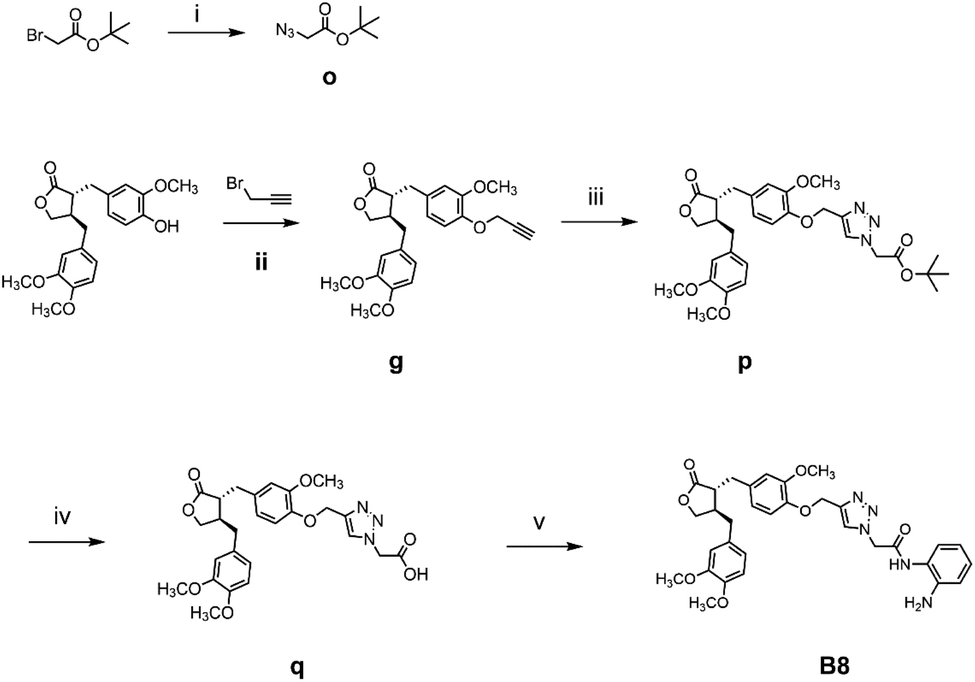
The general synthetic strategy for synthesizing a series of A–B target derivatives is outlined in Schemes 1–6. The synthesis route for the A-series derivatives begins with the natural product arctigenin. First, arctigenin undergoes a reaction with bromo-substituted alkyl esters of different carbon chain lengths. Afterwards, under basic conditions, the compound reacts with hydroxylamine hydrochloride to yield compounds A1–A7. Arctigenin, when reacted with tert-butyl bromoacetate, produces compound b. Following this, the protective group is removed in acidic conditions (using TFA), resulting in compound c. When coupled with ethyl 4-piperidinecarboxylate and ethyl 4-aminobenzoate under the influence of EDCI (1-ethyl-3-(3-dimethylaminopropyl)carbodiimide) and HOBt (1-hydroxy benzotriazole), this compound forms compounds d and e In basic conditions, compounds d and e react with hydroxylamine hydrochloride to produce compounds A8 and A9. Ethyl bromoacetate reacts with sodium azide in a reaction to form compound f. Compound f, when reacted with propargyl bromide, results in compound g. Subsequently, compound g undergoes a click reaction with compound f in the presence of copper(II) ions and sodium ascorbate, leading to compound h. Finally, under alkaline conditions, compound h reacts with hydroxylamine hydrochloride to produce compound A10.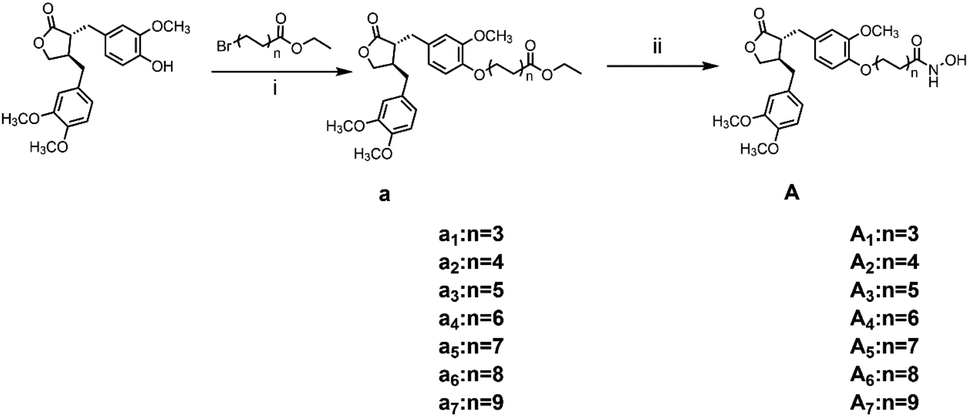 | ||
| Scheme 1 Reagents and conditions: (i) K2CO3, DMF, 60 °C, 6 h; (ii) hydroxylamine hydrochloride, KOH, MeOH, HCl, 0 °C – r.t., 1.5 h. | ||
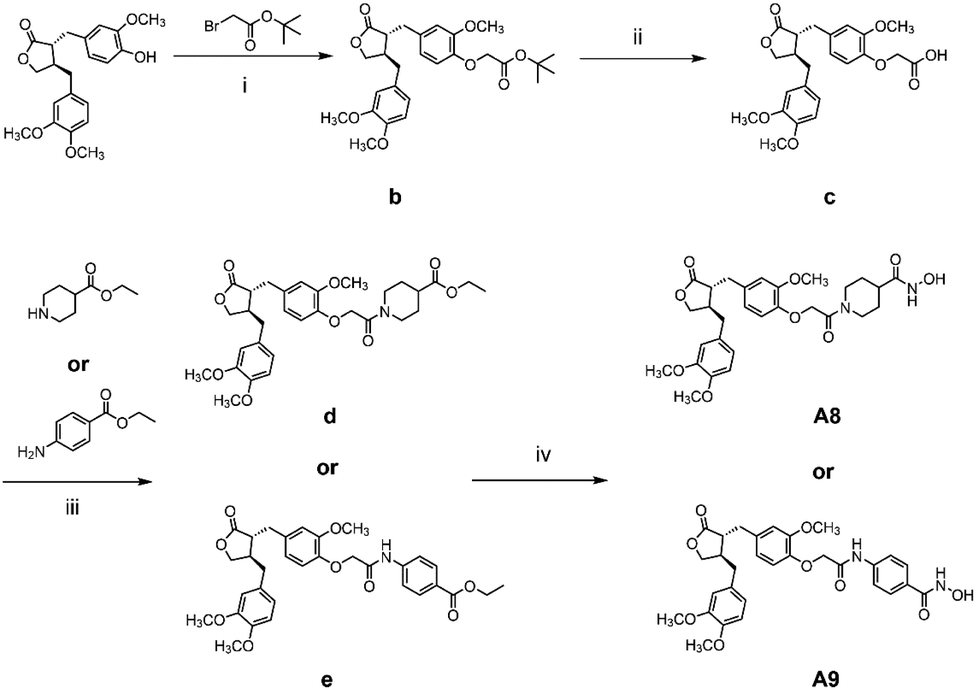 | ||
| Scheme 2 Reagents and conditions: (i) K2CO3, DMF, 60 °C, 6 h; (ii) TFA, DCM, r.t., 2 h; (iii) EDCI, HOBt, Et3N, DCM, r.t., 6 h; (iv) hydroxylamine hydrochloride, KOH, MeOH, HCl, 0 °C – r.t., 1.5 h. | ||
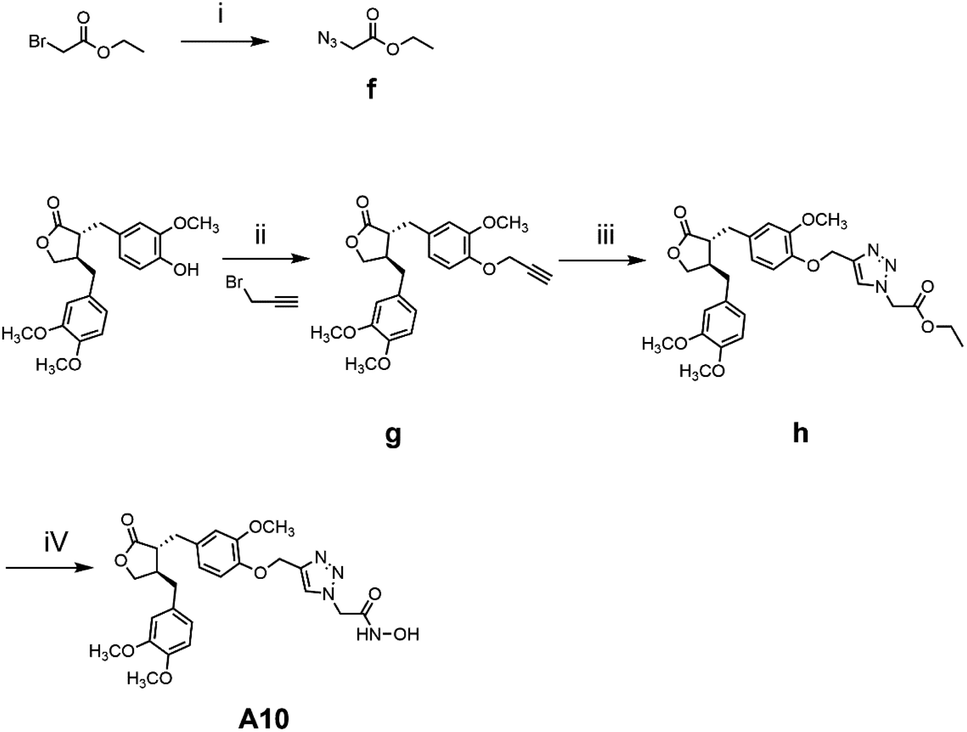 | ||
Scheme 3 Reagents and conditions: (i) DMF, NaN3, 80 °C, 6 h; (ii) K2CO3, DMF, r.t., 6 h; (iii) CuSO4·5H2O, sodium ascorbate, H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :t-BuOH = 1:1 (V/V), 60 °C, 4 h; (iv) hydroxylamine hydrochloride, KOH, MeOH, HCl, 0 °C – r.t., 1.5 h. :t-BuOH = 1:1 (V/V), 60 °C, 4 h; (iv) hydroxylamine hydrochloride, KOH, MeOH, HCl, 0 °C – r.t., 1.5 h. | ||
 | ||
| Scheme 4 Reagents and conditions: (i) K2CO3, DMF, 60 °C., 6 h; (ii) TFA, DCM, r.t., 2 h; (iii) EDCI, HOBt, Et3N, DCM, r.t., 6 h; (iv) TFA, DCM, r.t., 2 h; (v) o-phenylenediamine, HATU, DIPEA, DMF, r.t., 6 h. | ||
 | ||
| Scheme 5 Reagents and conditions: (i) K2CO3, DMF, 60 °C, 6 h; (ii) TFA, DCM, r.t., 2 h; (iii) EDCI, HOBt, Et3N, DCM, r.t., 6 h; (iv) TFA, DCM, r.t., 2 h; (v) o-phenylenediamine, HATU, DIPEA, DMF, r.t., 6 h. | ||
 | ||
| Scheme 6 Reagents and conditions: (i) NaN3, DMF, 80 °C, 6 h; (ii) K2CO3, DMF, 60 °C, 6 h; (iii) CuSO4·5H2O, sodium ascorbate, H2O:t-BuOH = 1:1 (V/V), 60 °C, 4 h; (iv) TFA, DCM, r.t., 2 h; (v) o-phenylenediamine, HATU, DIPEA, DMF, r.t., 6 h. | ||
The synthesis route for the B-series derivatives starts with the natural product arctigenin. Arctigenin initially undergoes a reaction with tert-butyl bromoacetate. Then, under acidic conditions provided by TFA (trifluoroacetic acid), the protective group is removed, resulting in compound c. This intermediate then reacts with amino acid tert butyl esters with different carbon chain lengths under the influence of EDCI and HOBt and then deprotects under acidic conditions with TFA to yield compounds j1–j5. Then, in the basic conditions provided by DIPEA (N,N-diisopropylethylamine), the compounds condense with o-phenylenediamine in the presence of the coupling agent HATU(o-(7-azabenzotriazol-1-yl)-N,N,N′,N′ tetramethyluronium hexafluorophosphate) to produce compounds B1–B5. Compound c undergoes condensation with ethyl 4-piperidinecarboxylate and ethyl 4-aminobenzoate in the presence of EDCI and HOBt, yielding compounds k and l After deprotection under acidic conditions provided by TFA, compounds m and n react in a condensation with o-phenylenediamine through the coupling agent HATU to produce compounds B6 and B7. Tert-butyl bromoacetate reacts with sodium azide to form compound o. Arctigenin reacts with propargyl bromide to yield compound g. This compound then undergoes a click reaction with compound o in the presence of copper(II) ions and sodium ascorbate, resulting in compound p. After deprotection under acidic conditions, compound p condenses with o-phenylenediamine under the influence of HATU to produce compound B8.
In vitro antiproliferative activity evaluation
The antiproliferative activities of two series of target compounds, A1 to A10 and B1 to B8, were screened in five human haematological malignancy cell lines (K562, MV411, KU812, Kasumi-1, CCRF-CEM) using a CCK8 assay. Arctigenin was used as the negative control, and tucidinostat was used as the positive control. Five optimized compounds were selected, and their IC50 values are presented in Table 1.| Compd. | Antiproliferative activitya (IC50/μM) | ||||
|---|---|---|---|---|---|
| K562 | MV411 | KU812 | Kasumi-1 | CCRF-CEM | |
| a IC50 value (means ± SD, n = 3, independent experiments). | |||||
| A1 | >5 | >5 | >5 | >5 | >5 |
| A2 | >5 | >5 | >5 | >5 | >5 |
| A3 | 2.12 ± 0.12 | 0.85 ± 0.17 | 3.88 ± 1.06 | >5 | 1.57 ± 0.13 |
| A4 | 1.19 ± 0.25 | 1.66 ± 0.26 | 1.41 ± 0.75 | >5 | 1.37 ± 0.11 |
| A5 | 1.82 ± 0.52 | 1.18 ± 0.44 | 2.27 ± 0.68 | >5 | 1.35 ± 0.21 |
| A6 | 3.03 ± 0.63 | 1.93 ± 0.13 | 3.72 ± 0.81 | >5 | 2.09 ± 0.17 |
| A7 | >5 | >5 | >5 | >5 | >5 |
| A8 | >5 | >5 | >5 | >5 | >5 |
| A9 | >5 | >5 | >5 | >5 | >5 |
| A10 | >5 | >5 | >5 | >5 | >5 |
| B1 | >5 | >5 | >5 | >5 | >5 |
| B2 | >5 | >5 | >5 | >5 | >5 |
| B4 | >5 | >5 | >5 | >5 | >5 |
| B5 | >5 | >5 | >5 | >5 | >5 |
| B6 | >5 | >5 | >5 | >5 | >5 |
| B7 | 1.96 ± 0.19 | 0.75 ± 0.09 | 2.09 ± 0.31 | 0.86 ± 0.04 | 2.21 ± 0.26 |
| B8 | >5 | >5 | >5 | >5 | >5 |
| Arctigenin | 24.22 ± 3.12 | 4.27 ± 1.68 | >30 | >30 | >30 |
| SAHA | 1.11 ± 0.05 | 1.00 ± 0.09 | 1.00 ± 0.11 | 0.47 ± 0.03 | 0.89 ± 0.03 |
| Tucidinostat | 2.68 ± 0.65 | 2.00 ± 0.15 | 4.26 ± 0.48 | >5 | 2.25.23 |
Notably, compound B7 exhibited higher antiproliferative activity than that of arctigenin and tucidinostat in the MV411 cell line, with an IC50 value of 0.75 ± 0.09 μM. Additionally, the negative control arctigenin also showed good antiproliferative activity in the MV411 cell line, with an IC50 value of 4.271 ± 1.68 μM. Therefore, the preliminary mechanism of MV411 cell antiproliferation was further explored using compound B7.
Due to the strong inhibitory effect of compound B7 on MV411 cells, we further tested its enzyme activity against different subtypes of HDACs (refer to Table 2). The results showed that compound B7 exhibited slightly better inhibition of HDAC2 than that of tucidinostat (with IC50 values of 130.6 and 160.1 nm). The compound also displayed mild inhibition of HDAC10 (with an IC50 of 1746.0 nm) but had no effect on HDAC4, HDAC5, and HDAC6 (IC50 > 1000 nm). Overall, the enzyme activity tests indicated that compound B7 selectively inhibits the activity of HDAC1, HDAC2, and HDAC3. The improved inhibition of HDAC2 by compound B7, emphasizes the significant role of arctigenin in enhancing the compound's selective inhibitory activity. The inclusion of arctigenin's structural motifs is instrumental in this efficacy, highlighting its utility in the strategic design of enzyme inhibitors.
| Compd. | HDACs isoforms | ||||||
|---|---|---|---|---|---|---|---|
| IC50 (nM) | |||||||
| HDAC1 | HDAC2 | HDAC3 | HDAC4 | HDAC5 | HDAC6 | HDAC10 | |
| Compound B7 | 261.1 | 130.7 | 298.7 | >10000 |
>10000 |
>10000 |
1746 |
| Tucidinostat | 95.4 | 160.1 | 733.2 | >10000 |
>10000 |
>10000 |
78.6 |
Analysis of apoptosis by annexin V-FITC/PI
To evaluate whether compound B7 can induce apoptosis in cancer cells, we conducted an annexin V-FITC apoptosis assay in MV411 cells, as shown in Fig. 3. The results demonstrate that B7 can induce apoptosis in MV411 cells. As the concentration of B7 increased (0.25, 0.5 and 1.0 μM) after 48 hours of treatment, the overall proportion of apoptotic cells (Q1-UR + Q1-LR) increased in a dose-dependent manner (21.84%, 37.90%, and 77.00%, respectively, at concentrations of 0.1, 0.5 and 1.0 μM). Additionally, compared to arctigenin and tucidinostat, B7 exhibits a stronger ability to induce apoptosis in the MV411 cell line.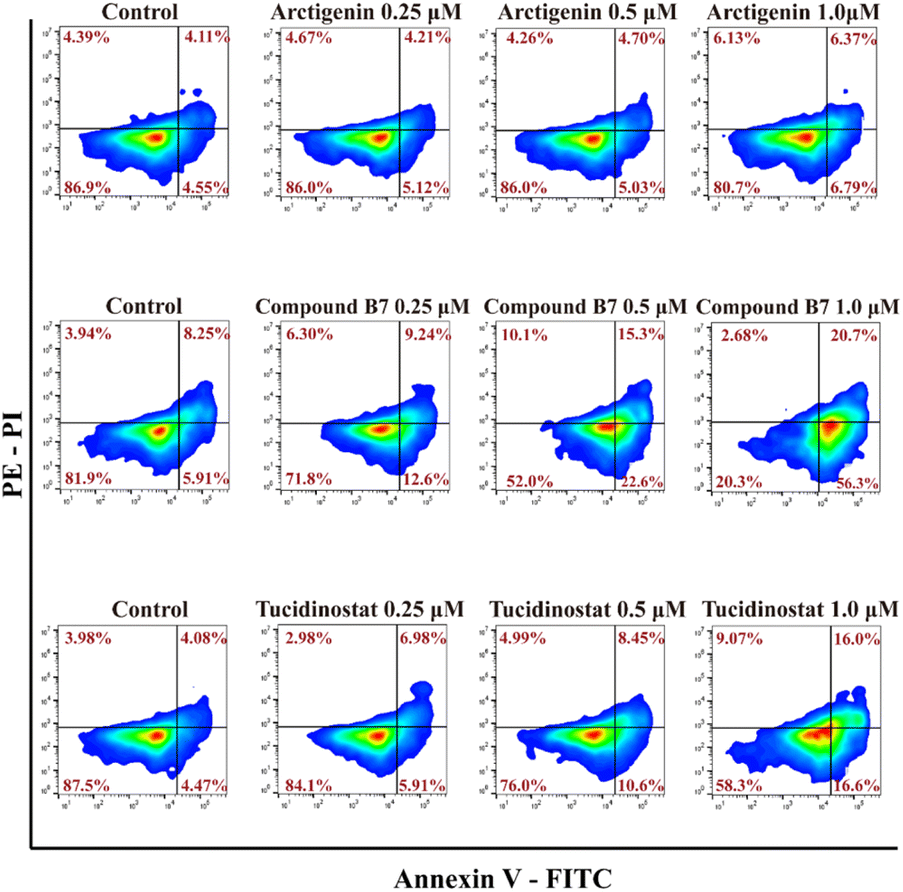 | ||
| Fig. 3 MV411 cells were treated with different concentrations of compounds, including arctigenin, compound B7, and tucidinostat (at levels of 0, 0.25, 0.5 and 1.0 μM). Subsequently, apoptosis was detected using flow cytometry after annexin V/PI staining. | ||
Analysis of the cell cycle
In the cell cycle analysis, in vitro antiproliferation experiments demonstrated that compound B7 can inhibit the proliferation of the MV411 cell line. To further investigate the mechanism of compound B7, flow cytometry (FCM) analysis was performed on the MV411 cell line using propidium iodide (PI) staining to study its effect on cell cycle progression. Under arctigenin and tucidinostat conditions, the proportion of cells arrested in the G0/G1 phase was increased with compound B7 compared to the control. Fig. 4 shows that as the concentration of compound B7 increased, the proportion of cells in the G0/G1 phase gradually increased, reflecting that compound B7 induced concentration-dependent cell cycle arrest in the G0/G1 phase.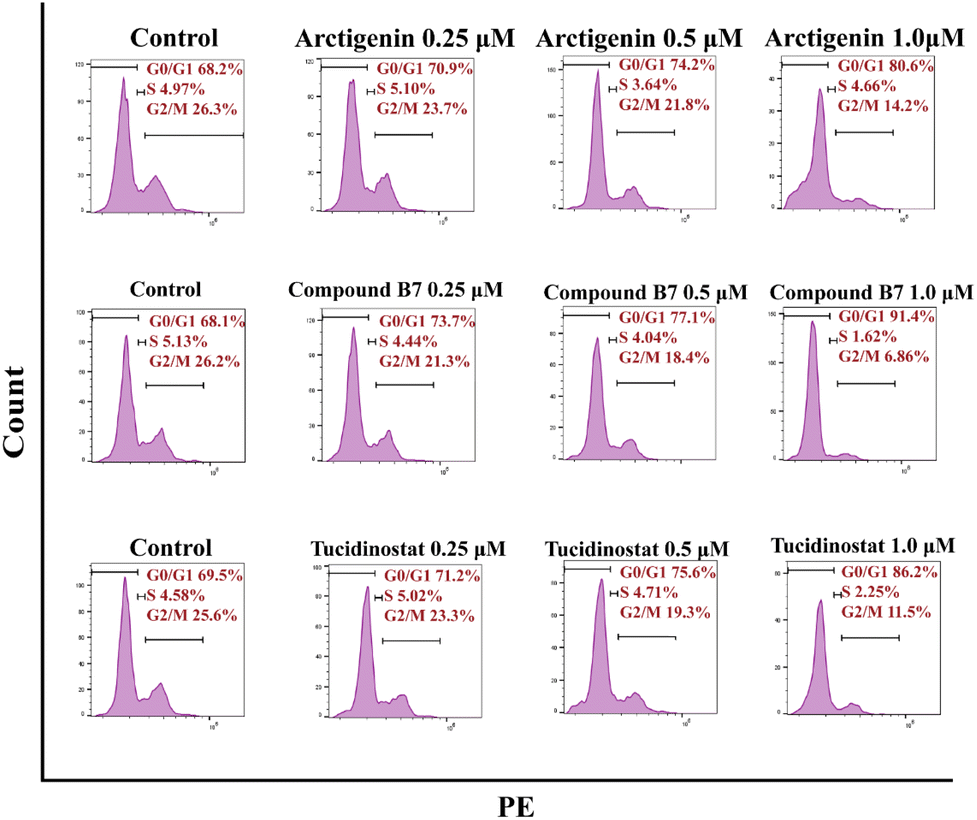 | ||
| Fig. 4 MV411 cells were exposed to varying concentrations of several compounds, namely arctigenin, compound B7, and tucidinostat (0, 0.25, 0.5 and 1.0 μM). Following this, an analysis of the cell-cycle distribution was conducted utilizing flow cytometry. The panel showcases the distribution percentages of cells throughout different stages of the cell cycle. | ||
Caspase 3 activity assay
First, we used Beyotime Biotechnology's GreenNuc™ Caspase-3 activity assay kit to monitor the activity of Caspase-3 in real-time. Flow cytometric analysis results showed that under different concentrations of compound B7 treatment, Caspase-3 activity exhibited a clear concentration-dependent increase, with a more pronounced trend compared to the arctigenin and tucidinostat conditions. This result indicates that compound B7 enhances the activity of Caspase-3 in the MV411 cell line, thereby promoting cell apoptosis.Next, we observed the activity of Caspase-3 using fluorescence microscopy. Through live cell staining, we directly observed the fluorescence signals of Caspase-3. In the experiment, we similarly observed a concentration-dependent increase in Caspase-3 fluorescence signals under different concentrations of compound B7 treatment, with a more pronounced trend compared to the arctigenin and tucidinostat conditions. This further confirmed the results obtained from flow cytometry, demonstrating that compound B7 significantly increases caspase-3 activity in the MV411 cell line, leading to the induction of cell apoptosis.
In summary, the results obtained from both monitoring methods consistently indicate that compound B7 promotes caspase-mediated apoptosis in the MV411 cell line (Fig. 5).
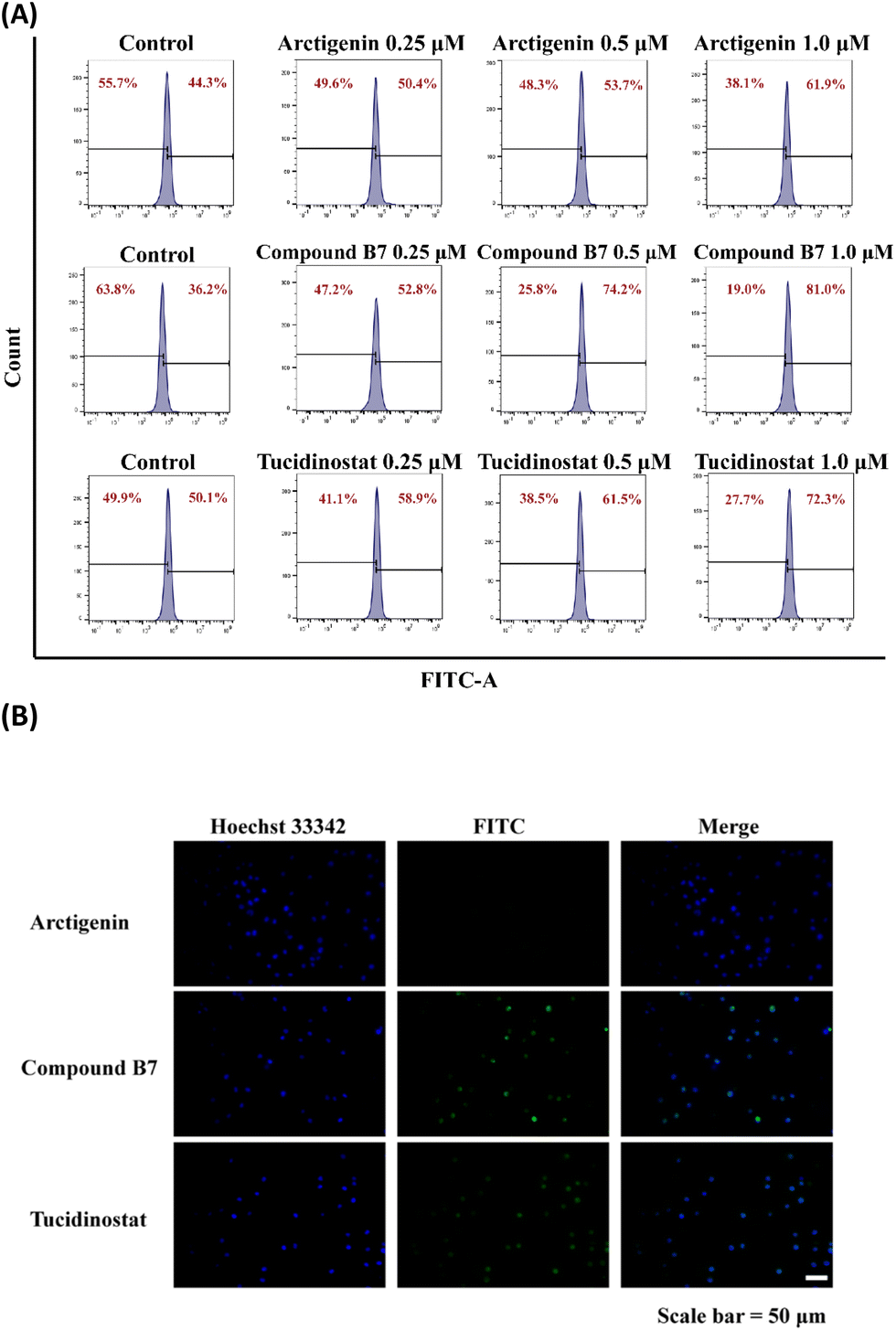 | ||
| Fig. 5 (A) Flow cytometric analysis: flow cytometric analysis of MV411 cells treated with increasing concentrations of arctigenin, compound B7, and tucidinostat (0, 0.25, 0.5 and 1.0 μM) revealed a concentration-dependent increase in Caspase-3 activity. As shown in the figure, the percentage of cells with high Caspase-3 activity increased in a dose-dependent manner, with the highest proportion observed at 1.0 μM B7 treatment. (B) Fluorescence microscopy: fluorescence microscopy images of MV411 cells stained with the GreenNuc™ Caspase-3 assay kit further confirmed the induction of apoptosis by compound B7. The control group (0 μM B7) showed minimal Caspase-3 activity, indicated by weak fluorescence. In contrast, cells treated with increasing concentrations of compound B7 (1.0 μM) displayed intensified fluorescence signals, affecting Caspase-3 activity and, hence, increased apoptosis. The scale bar denotes 50 μM. | ||
Western blotting
Given the outstanding antiproliferative activity of compound B7 in MV411 cells, we conducted western blotting experiments to verify its intracellular mechanism of HDAC inhibition. The results showed that at different doses, compound B7 significantly increased the level of acetylated H3 in MV411 cells in a dose-dependent manner. This result indicates that compound B7 strongly inhibits the HDAC1/2/3 signalling pathway, and its HDAC inhibitory activity is significantly superior to that of arctigenin and slightly superior to that of tucidinostat. Thus, the HDAC inhibitory activity of compound B7 were confirmed (Fig. 6).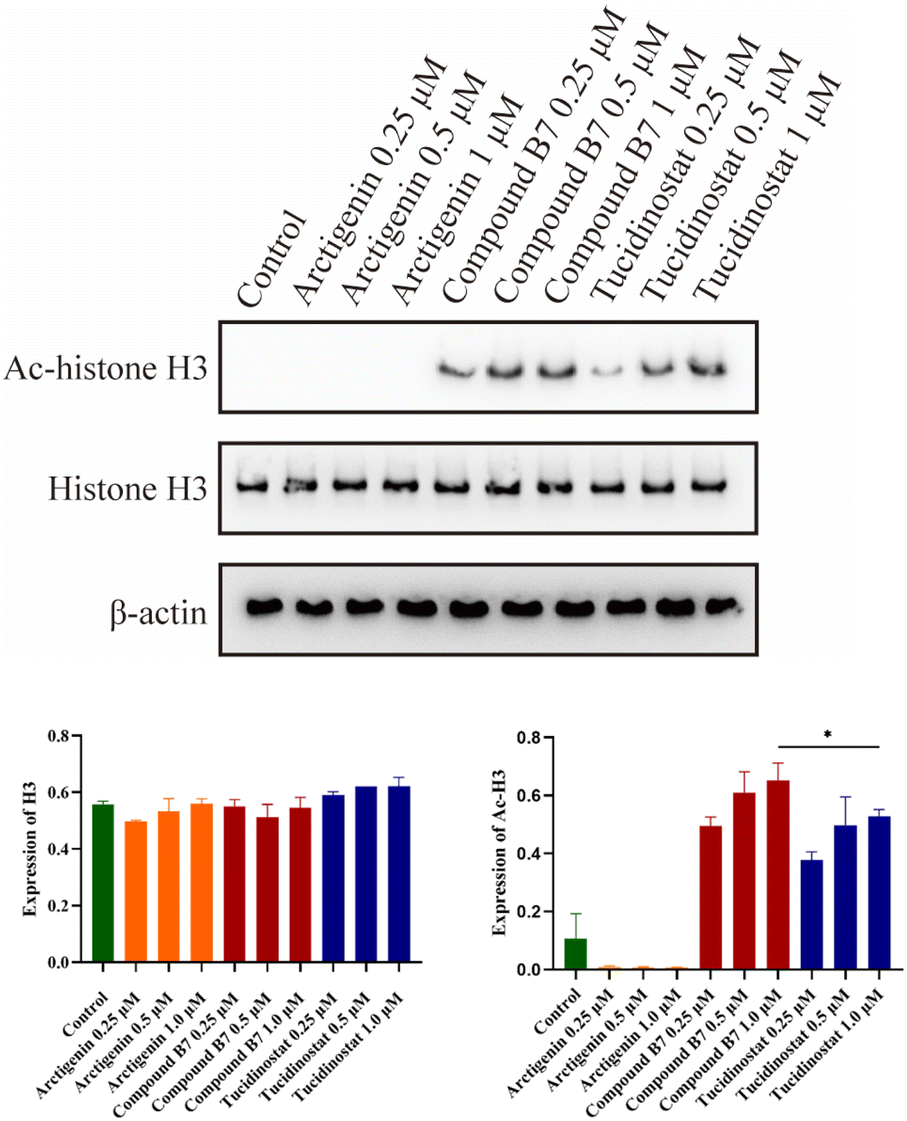 | ||
| Fig. 6 MV411 cells were treated with arctigenin, compound B7, and tucidinostat at concentrations of 0, 0.25, 0.5 and 1.0 μM for 48 hours. The levels of H3 and acetylated H3 (Ac-H3) were determined using western blot analysis. β-Actin was used as the loading control. | ||
Conclusions
The objective of this study was to design and synthesize arctigenin derivatives that can inhibit HDAC, thoroughly evaluate their antiproliferative effects on the leukaemia cell line MV411, and investigate their underlying mechanisms. We successfully synthesized a series of compounds and meticulously assessed their potential as antitumour agents. Our findings revealed that compound B7 exhibited exceptional antiproliferative activity among all the designed derivatives, outperforming the standards arctigenin and tucidinostat. Further enzymatic assays demonstrated that compound B7 showed comparable levels of HDAC inhibitory activity to tucidinostat, the positive control. After thoroughly investigating the mechanism of action, we discovered that compound B7 induced cell apoptosis in MV411 cells through the Caspase-3 pathway, effectively inhibiting cell proliferation. Caspase-3 plays a crucial role in driving cell apoptosis, and its increased activity contributes to enhanced cell death.Moreover, compound B7 significantly increased the levels of acetylation within MV411 cells, a key protein modification that regulates various cellular functions. This result provided further evidence for the potent HDAC inhibitory activity of compound B7, aligning with the characteristics of commercially available HDAC inhibitors.
In summary, through designing and synthesizing arctigenin derivatives with HDAC inhibitory properties, we discovered a novel antileukaemia compound, namely, compound B7. This compound exhibited significant antiproliferative effects and demonstrated its antitumour properties by inducing cell apoptosis and enhancing cellular acetylation levels. Furthermore, our research paves the way for further investigations on arctigenin derivatives in the field of cancer treatment, offering valuable insights that could aid in the discovery of additional potential anticancer drugs.
Experimental section
Chemistry
:1, V/V) to afford compounds A1–A7.
4-(4-(((3R,4R)-4-(3,4-Dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)-N-hydroxypentanamide (A1). White solid, yield: 72.7%. m.p. 102.1–103.4 °C, 1H NMR (600 MHz, DMSO-d6) δ 10.41 (s, 1H), 8.70 (s, 1H), 6.85–6.81 (m, 2H), 6.77 (d, J = 1.7 Hz, 1H), 6.68 (dd, J = 8.2, 1.7 Hz, 1H), 6.65–6.58 (m, 2H), 4.09 (t, J = 8.0 Hz, 1H), 3.91–3.86 (m, 3H), 3.71 (s, 3H), 3.70 (s, 3H), 3.34 (s, 3H), 2.85–2.80 (m, 1H), 2.77 (dd, J = 13.7, 6.9 Hz, 1H), 2.74–2.69 (m, 1H), 2.50–2.44 (m, 3H), 2.12 (t, J = 7.4 Hz, 2H), 1.90 (dd, J = 13.9, 7.1 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 178.90, 169.14, 149.28, 149.11, 147.76, 147.11, 131.67, 131.27, 121.77, 120.84, 113.83, 113.59, 112.80, 112.25, 71.18, 68.11, 55.93, 55.91, 55.80, 46.06, 41.26, 37.36, 34.12, 29.25, 25.42. FT-MS: calcd for C25H32NO8, [M + H]+, 474.2128, found 474.2122. HPLC purity: 98.24% (tR = 7.06 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
5-(4-(((3R,4R)-4-(3,4-Dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)-N-hydroxypentanamide (A2). White solid, yield: 57.3%. m.p. 104.3–104.5 °C, 1H NMR (600 MHz, DMSO-d6) δ 10.36 (s, 1H), 8.70 (d, J = 0.8 Hz, 1H), 6.85–6.83 (m, 1H), 6.82 (d, J = 8.2 Hz, 1H), 6.77 (d, J = 1.6 Hz, 1H), 6.68 (dd, J = 8.2, 1.6 Hz, 1H), 6.64 (d, J = 1.5 Hz, 1H), 6.60 (dd, J = 8.1, 1.4 Hz, 1H), 4.09 (t, J = 8.0 Hz, 1H), 3.91–3.89 (m, 1H), 3.89–3.86 (m, 2H), 3.71 (s, 3H), 3.70 (d, J = 1.9 Hz, 3H), 3.70 (s, 3H), 3.34 (s, 1H), 2.84 (dd, J = 13.7, 5.4 Hz, 1H), 2.77 (dd, J = 13.7, 6.9 Hz, 1H), 2.74–2.69 (m, 1H), 2.48 (t, J = 8.2 Hz, 1H), 2.43 (dd, J = 18.2, 10.1 Hz, 1H), 2.01 (t, J = 7.0 Hz, 2H), 1.66 (dd, J = 12.0, 6.3 Hz, 2H), 1.65–1.60 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 178.90, 169.42, 149.20, 149.11, 147.76, 147.22, 131.67, 131.06, 121.75, 120.85, 113.79, 113.35, 112.80, 112.25, 71.18, 68.24, 55.91, 55.80, 55.39, 46.07, 41.25, 37.37, 34.11, 32.41, 28.80, 22.35. FT-MS: calcd for C26H34NO8, [M + H]+, 488.2277, found 488.2278. HPLC purity: 99.27% (tR = 5.81 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
6-(4-(((3R,4R)-4-(3,4-Dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)-N-hydroxyhexanamide (A3). Yellow oil, yield: 60.5%. 1H NMR (600 MHz, DMSO-d6) δ 10.34 (s, 1H), 8.67 (s, 1H), 6.84 (d, J = 8.2 Hz, 1H), 6.81 (t, J = 8.7 Hz, 1H), 6.77 (d, J = 1.2 Hz, 1H), 6.68 (d, J = 8.1 Hz, 1H), 6.63 (d, J = 14.0 Hz, 1H), 6.60 (d, J = 8.0 Hz, 1H), 4.09 (t, J = 7.9 Hz, 1H), 3.90–3.88 (m, 1H), 3.87 (d, J = 6.1 Hz, 2H), 3.71 (s, 3H), 3.70 (s, 3H), 3.70 (s, 3H), 3.34 (s, 1H), 2.84 (dd, J = 13.7, 5.3 Hz, 1H), 2.77 (dd, J = 13.6, 7.0 Hz, 1H), 2.73–2.68 (m, 1H), 2.48 (d, J = 8.0 Hz, 1H), 2.44 (dd, J = 12.7, 5.4 Hz, 1H), 1.96 (t, J = 7.3 Hz, 2H), 1.68 (dd, J = 13.9, 6.9 Hz, 2H), 1.53 (dd, J = 15.0, 7.5 Hz, 2H), 1.37 (dd, J = 16.9, 9.7 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 178.90, 169.49, 149.22, 149.11, 147.76, 147.26, 131.67, 131.05, 121.76, 120.84, 113.79, 113.38, 112.80, 112.25, 71.18, 68.52, 55.91, 55.79, 55.39, 46.07, 41.27, 37.37, 34.13, 32.70, 29.07, 25.68, 25.39. FT-MS: calcd for C27H36NO8, [M + H]+, 502.2433, found 502.2435. HPLC purity: 99.87% (tR = 2.71 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
7-(4-(((3R,4R)-4-(3,4-Dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)-N-hydroxyheptanamide (A4). Yellow oil, yield: 42.8%. 1H NMR (600 MHz, DMSO-d6) δ 10.33 (s, 1H), 8.66 (s, 1H), 6.85–6.83 (m, 1H), 6.80 (d, J = 17.5 Hz, 1H), 6.75 (d, J = 26.9 Hz, 1H), 6.68 (d, J = 7.9 Hz, 1H), 6.63 (d, J = 13.7 Hz, 1H), 6.58 (t, J = 14.4 Hz, 1H), 4.09 (t, J = 7.7 Hz, 1H), 3.89 (d, J = 3.1 Hz, 1H), 3.87 (s, 2H), 3.73 (s, 9H), 3.34 (s. 1H), 2.84 (dd, J = 13.6, 5.0 Hz, 1H), 2.77 (dd, J = 13.5, 7.0 Hz, 1H), 2.71 (d, J = 5.9 Hz, 1H), 2.49–2.44 (m, 2H), 1.94 (t, J = 7.1 Hz, 2H), 1.69–1.64 (m, 2H), 1.52–1.47 (m, 2H), 1.37 (d, J = 6.3 Hz, 2H), 1.28 (d, J = 6.9 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 178.90, 169.56, 149.25, 149.11, 147.76, 147.27, 131.67, 131.05, 121.76, 120.84, 113.82, 113.43, 112.80, 112.25, 71.18, 68.57, 55.93, 55.91, 55.79, 46.07, 41.28, 37.37, 34.14, 32.69, 29.20, 28.83, 25.76, 25.56. FT-MS: calcd for C28H38NO8, [M + H]+, 516.2594, found 516.2591. HPLC purity: 98.01% (tR = 6.15 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
8-(4-(((3R,4R)-4-(3,4-Dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)-N-hydroxyheptanamide (A5). Yellow oil, yield: 49.2%. 1H NMR (600 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.65 (s, 1H), 6.84 (d, J = 8.2 Hz, 1H), 6.82 (d, J = 8.1 Hz, 1H), 6.77 (d, J = 1.3 Hz, 1H), 6.68 (d, J = 8.1 Hz, 1H), 6.64 (d, J = 1.2 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 4.08 (t, J = 7.9 Hz, 1H), 3.88 (t, J = 6.5 Hz, 3H), 3.71 (s, 3H), 3.70 (s, 3H), 3.70 (s, 3H), 3.33 (s, 1H), 2.84 (dd, J = 13.7, 5.2 Hz, 1H), 2.77 (dd, J = 13.7, 7.0 Hz, 1H), 2.71 (dd, J = 13.2, 7.2 Hz, 1H), 2.47 (d, J = 6.3 Hz, 1H), 2.44 (dd, J = 11.2, 6.0 Hz, 1H), 1.93 (t, J = 7.3 Hz, 2H), 1.66 (dd, J = 13.8, 6.4 Hz, 2H), 1.48 (dt, J = 14.5, 7.3 Hz, 2H), 1.39–1.35 (m, 2H), 1.31–1.28 (m, 2H), 1.24 (d, J = 7.2 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 178.90, 169.57, 149.25, 149.11, 147.76, 147.27, 131.67, 131.03, 121.76, 120.84, 113.81, 113.43, 112.80, 112.25, 71.18, 68.60, 55.93, 55.91, 55.79, 46.06, 41.28, 37.37, 34.15, 32.71, 29.25, 29.01, 28.95, 25.92, 25.54.FT-MS: calcd for C29H40NO8, [M + H]+, 530.2754, found 530.2748. HPLC purity: 98.04% (tR = 6.82 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
9-(4-(((3R,4R)-4-(3,4-Dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)-N-hydroxyheptanamide (A6). Yellow oil, yield: 50.3%. 1H NMR (600 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.65 (s, 1H), 6.84 (d, J = 8.2 Hz, 1H), 6.82 (d, J = 8.1 Hz, 1H), 6.77 (d, J = 1.3 Hz, 1H), 6.68 (d, J = 8.1 Hz, 1H), 6.64 (d, J = 1.2 Hz, 1H), 6.59 (d, J = 8.1 Hz, 1H), 4.08 (t, J = 7.9 Hz, 1H), 3.89 (d, J = 4.0 Hz, 1H), 3.88 (d, J = 6.3 Hz, 2H), 3.71 (s, 3H), 3.70 (s, 3H), 3.70 (s, 3H), 3.33 (s, 1H), 2.84 (dd, J = 13.7, 5.2 Hz, 1H), 2.77 (dd, J = 13.7, 7.0 Hz, 1H), 2.73–2.69 (m, 1H), 2.48 (s, 2H), 2.47–2.39 (m, 2H), 1.93 (t, J = 7.3 Hz, 2H), 1.67 (dd, J = 14.0, 7.0 Hz, 2H), 1.48 (dd, J = 14.4, 7.2 Hz, 2H), 1.38–1.34 (m, 2H), 1.30–1.27 (m, 2H), 1.24 (d, J = 7.2 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 177.81, 168.47, 148.16, 148.02, 146.67, 146.18, 130.58, 129.94, 120.67, 119.75, 112.72, 112.35, 111.71, 111.16, 70.09, 67.53, 55.21, 54.84, 54.82, 54.70, 44.97, 40.20, 36.27, 33.07, 31.63, 28.98, 28.19, 28.09, 24.92, 24.48. FT-MS: calcd for C30H42NO8, [M + H]+, 544.2911, found 544.2904. HPLC purity: 99.45% (tR = 1.94 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
10-(4-(((3R,4R)-4-(3,4-Dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)-N-hydroxyheptanamide (A7). Yellow oil, yield: 32.1%. 1H NMR (600 MHz, DMSO-d6) δ 10.32 (s, 1H), 8.68 (d, J = 37.2 Hz, 1H), 6.86–6.82 (m, 1H), 6.82–6.78 (m, 1H), 6.78–6.70 (m, 1H), 6.70–6.65 (m, 1H), 6.62 (d, J = 20.0 Hz, 1H), 6.57 (t, J = 15.4 Hz, 1H), 4.09 (dd, J = 16.2, 8.6 Hz, 1H), 3.89 (d, J = 5.0 Hz, 1H), 3.88 (d, J = 6.1 Hz, 2H), 3.71 (s, 3H), 3.70 (s, 3H), 3.70 (s, 3H), 3.55–3.46 (m, 1H), 3.33 (s, 1H), 2.84 (dt, J = 24.4, 12.3 Hz, 1H), 2.76 (dd, J = 13.6, 7.1 Hz, 1H), 2.74–2.66 (m, 1H), 2.48 (s, 1H), 2.46 (d, J = 7.2 Hz, 1H), 2.43 (dd, J = 18.4, 5.5 Hz, 1H), 1.93 (t, J = 7.3 Hz, 2H), 1.66 (dd, J = 14.0, 6.8 Hz, 2H), 1.49–1.45 (m, 2H), 1.37 (d, J = 6.3 Hz, 2H), 1.24 (s, 6H). 13C NMR (151 MHz, DMSO-d6) δ 177.82, 168.50, 148.19, 148.04, 146.69, 146.20, 130.60, 129.96, 120.69, 119.76, 112.75, 112.37, 111.72, 111.18, 70.11, 67.54, 54.86, 54.84, 54.72, 44.99, 40.23, 36.30, 34.61, 33.10, 31.66, 28.31, 28.22, 28.16, 28.10, 24.95, 24.53. FT-MS: calcd for C31H44NO8, [M + H]+, 558.3062, found 558.3061. HPLC purity: 98.53% (tR = 3.44 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
:TFA = 1:1 (V/V) and stirred for 2 h at room temperature, and then the mixture was extracted with EtOAc and concentrated under reduced pressure, yielding compound c. Compound c was dissolved with ethyl 4-piperidinecarboxylate or ethyl 4-aminobenzoate, EDCI (30.6 mg, 0.16 mmol), and HOBt (21.6 mg, 0.16 mmol) in dry DMF and stirred for 6 h at room temperature. The mixture was extracted with EtOAc and concentrated under reduced pressure, yielding compounds d or e; hydroxylamine hydrochloride (90.0 mg, 1.3 mmol), anhydrous sodium sulfate and potassium hydroxide (72.9 mg, 1.3 mmol) were added at 0 °C and stirred for 0.5 h at room temperature. The resulting mixture was then extracted and filtered, and the obtained filtrate was added dropwise to the methanol solution of compounds d or e. Potassium hydroxide (0.65 mmol) was added to the mixture, and after 1.5 hours, 20 mL of water was added; the aqueous solution was acidified to pH 4–5 with dilute hydrochloric acid (1 M) and extracted with dichloromethane (50 mL × 3). The dichloromethane layer was concentrated and dried in a vacuum, and the residue was purified by flash chromatography (silica gel, 200–300 mesh, DCM/MeOH = 50:1, V/V) to afford compounds A8 or A9.
1-(2-(4-(((3R,4R)-4-(3,4-Dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)acetyl)-N-hydroxypiperidine-4-carboxamide (A8). White solid, yield: 42.1%. m.p. 105.0–105.6 °C. 1H NMR (600 MHz, DMSO-d6) δ 10.47 (s, 1H), 8.74 (s, 1H), 6.83 (d, J = 8.1 Hz, 1H), 6.78 (d, J = 8.2 Hz, 2H), 6.72–6.62 (m, 2H), 6.60 (d, J = 8.0 Hz, 1H), 4.72 (d, J = 4.4 Hz, 2H), 4.30 (d, J = 11.9 Hz, 1H), 4.10 (t, J = 8.0 Hz, 1H), 3.73 (s, 3H), 3.70 (s, 3H), 3.70 (s, 3H), 3.37 (s, 2H), 3.02 (t, J = 11.1 Hz, 1H), 2.83 (dd, J = 13.7, 5.3 Hz, 1H), 2.77 (dd, J = 13.7, 6.8 Hz, 1H), 2.72 (dd, J = 8.4, 6.2 Hz, 1H), 2.61 (t, J = 12.1 Hz, 1H), 2.49–2.42 (m, 2H), 2.25 (t, J = 10.9 Hz, 1H), 2.12 (s, 1H), 1.63 (s, 3H), 1.44–1.37 (m, 1H). 13C NMR (151 MHz, DMSO-d6) δ 207.76, 177.79, 170.23, 165.06, 148.09, 148.01, 146.66, 145.50, 130.66, 130.56, 120.48, 119.78, 112.80, 111.73, 111.15, 70.09, 67.89, 66.37, 54.90, 54.81, 54.71, 44.95, 36.26, 33.00, 31.49, 30.07, 28.97, 28.04, 27.40. FT-MS: calcd for C29H37N2O9, [M + H]+ 557.2502, found 557.2493. HPLC purity: 98.39% (tR = 6.90 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
1-4-(2-(4-(((3R,4R)-4-(3,4-Dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)acetamido)-N-hydroxybenzamide (A9). Yellow solid, yield: 28.3%. m.p. 118.1–118.3 °C. 1H NMR (600 MHz, DMSO-d6) δ 9.23 (s, 1H), 7.17 (d, J = 6.8 Hz, 1H), 6.90–6.86 (m, 1H), 6.83 (s, 1H), 6.82 (s, 1H), 6.78 (d, J = 1.8 Hz, 1H), 6.78 (s, 1H), 6.76 (s, 1H), 6.71 (dd, J = 8.0, 1.1 Hz, 1H), 6.66 (d, J = 1.7 Hz, 1H), 6.61–6.59 (m, 2H), 6.55–6.51 (m, 1H), 4.71 (s, 2H), 4.10 (s, 1H), 3.88 (s, 1H), 3.73 (s, 3H), 3.72 (s, 3H), 3.70 (s, 3H), 3.07 (d, J = 9.4 Hz, 2H), 2.7–2.77 (m, 2H), 2.48 (d, J = 5.8 Hz, 1H), 2.46 (s, 1H). 13C NMR (151 MHz, DMSO-d6) δ 177.81, 172.24, 164.99, 148.08, 148.02, 146.68, 145.53, 141.30, 130.62, 130.58, 125.11, 124.71, 122.74, 120.51, 119.79, 115.48, 115.23, 112.83, 112.76, 111.75, 111.17, 70.10, 66.39, 54.92, 54.83, 54.73, 44.97, 42.30, 36.28, 33.02. FT-MS: calcd for C30H33N2O9, [M + H]+, 565.2171, found 565.2180. HPLC purity: 99.32% (tR = 4.61 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
:t-BuOH = 1:1 (V/V) and stirred at 60 °C for four hours. The mixture was poured into water and extracted with DCM (20 mL × 3). The combined organic layers were concentrated to obtain compound h. Hydroxylamine hydrochloride (90.0 mg, 1.3 mmol), anhydrous sodium sulfate and potassium hydroxide (72.9 mg, 1.3 mmol) were added at 0 °C and stirred for 0.5 h at room temperature. The mixture was then extracted and filtered, and the obtained filtrate was added dropwise to the methanol solution of compound h. Potassium hydroxide (0.65 mmol) was added to the mixture, and after 1.5 hours, 20 mL of water was added. The aqueous solution was acidified to pH 4–5 with dilute hydrochloric acid (1 M) and extracted with DCM (50 mL × 3). The dichloromethane layer was concentrated and dried under vacuum, and the residue was purified by flash column chromatography (silica gel, 200–300 mesh, DCM/MeOH = 50:1, V/V) to obtain compound A10.
2-(4-((4-(((3R,4R)-4-(3,4-Dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-hydroxyacetamide (A10). White solid, yield: 38.3%. m.p. 98.3–99.1 °C. 1H NMR (600 MHz, DMSO-d6) δ 8.15 (s, 1H), 7.05–6.83 (m, 1H), 6.82 (d, J = 8.2 Hz, 1H), 6.78 (d, J = 1.5 Hz, 1H), 6.72–6.69 (m, 1H), 6.65 (d, J = 1.6 Hz, 1H), 6.60 (dd, J = 8.1, 1.6 Hz, 1H), 5.06 (s, 2H), 4.95 (d, J = 46.5 Hz, 2H), 4.10 (t, J = 8.0 Hz, 1H), 3.89 (t, J = 8.4 Hz, 1H), 3.70 (s, 6H), 3.69 (s, 3H), 2.83 (dd, J = 18.1, 12.9 Hz, 1H), 2.81–2.72 (m, 2H), 2.55–2.52 (m, 1H), 2.49–2.43 (m, 2H), 1.23 (s, 2H). 13C NMR (151 MHz,DMSO-d6) δ 178.91, 162.66, 149.22, 149.10, 147.76, 146.67, 142.97, 132.01, 131.65, 131.58, 129.14, 126.36, 121.63, 120.86, 113.71, 112.81, 112.26, 71.19, 65.50, 62.03, 55.92, 55.81, 50.51, 46.06, 37.38, 34.14. FT-MS: calcd for C26H31N4O8, [M + H]+, 527.2130, found 527.2136. HPLC purity: 98.69% (tR = 7.21 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
:TFA = 1:1 (V/V) and stirred for 2 h at room temperature, and then the mixture was extracted with EtOAc and concentrated under reduced pressure, yielding compound c. Compound c was dissolved with amino acid t-butyl esters of different carbon chain lengths (0.16 mmol), EDCI (30.6 mg, 0.16 mmol), and HOBt (21.6 mg, 0.16 mmol) in dry DMF and stirred for 6 h at room temperature. The mixture was extracted with EtOAc and concentrated under reduced pressure, yielding compounds d1–d5. Compound i was dissolved in a solution of DCM:TFA = 1:1 (V/V) and stirred for 2 h at room temperature, and then the mixture was extracted with EtOAc and concentrated under reduced pressure, yielding compounds j1–j5. o-Phenylenediamine (17.3 mg, 0.26 mmol), DIPEA (2.8 mL, 0.26 mmol), and HATU (60.8 mg, 0.26 mmol) were added to a solution of acid compound j1–j5 anhydrous DMF. The mixture was stirred at room temperature for 6 h. Water was added, and the mixture was extracted with ethyl acetate. The organic layers were combined, concentrated, and purified with flash column chromatography (DCM/MeOH = 50:1, V/V) to afford compounds B1–B5.
N-(2-Aminophenyl)-2-(2-(4-(((3R,4R)-4-(3,4-dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)acetamido)acetamide (B1). White solid, yield: 53.1%. m.p. 104.2–104.9 °C. 1H NMR (600 MHz, DMSO-d6) δ 9.21 (s, 1H), 8.17–8.15 (m, 1H), 7.12 (d, J = 7.4 Hz, 1H), 6.91 (t, J = 7.0 Hz, 2H), 6.82 (d, J = 8.1 Hz, 2H), 6.7–6.66 (m, 2H), 6.65 (d, J = 1.7 Hz, 1H), 6.59 (dd, J = 8.1, 1.7 Hz, 1H), 6.53 (dd, J = 10.9, 4.1 Hz, 1H), 4.90 (s, 2H), 4.51 (s, 2H), 4.10 (t, J = 8.0 Hz, 1H), 4.00 (d, J = 5.5 Hz, 2H), 3.88 (t, J = 8.5 Hz, 1H), 3.75 (s, 3H), 3.70 (d, J = 1.0 Hz, 6H), 3.33 (s, 1H), 2.85 (dd, J = 13.7, 5.4 Hz, 1H), 2.75 (ddd, J = 20.9, 13.2, 6.8 Hz, 2H), 2.51 (d, J = 8.0 Hz, 1H), 2.49–2.46 (m, 1H). 13C NMR (151 MHz, DMSO-d6) δ 178.88, 168.83, 167.85, 149.48, 149.10, 147.76, 146.39, 143.01, 132.67, 131.64, 126.71, 126.36, 123.02, 121.75, 120.87, 116.40, 115.99, 115.09, 113.95, 112.81, 112.25, 71.19, 68.93, 56.03, 55.91, 55.81, 46.02, 42.65, 41.31, 37.36, 34.15. FT-MS: calcd for C31H36N3O8, [M + H]+, 578.2495, found 578.2496. HPLC purity: 99.22% (tR = 7.07 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
N-(2-Aminophenyl)-3-(2-(4-(((3R,4R)-4-(3,4-dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)acetamido)propanamide (B2). White solid, yield: 44.1%. m.p. 105.1–106.3 °C, 1H NMR (600 MHz, DMSO-d6) δ 9.27 (s, 1H), 8.04 (t, J = 5.6 Hz, 1H), 7.18 (d, J = 7.7 Hz, 1H), 6.90–6.84 (m, 2H), 6.83–6.80 (m, 2H), 6.70 (dd, J = 8.0, 1.2 Hz, 1H), 6.68–6.64 (m, 2H), 6.59 (dd, J = 8.1, 1.8 Hz, 1H), 6.54–6.49 (m, 1H), 4.97 (d, J = 67.1 Hz, 2H), 4.42 (s, 2H), 4.10 (dd, J = 8.6, 7.4 Hz, 1H), 3.88 (t, J = 8.5 Hz, 1H), 3.74 (s, 3H), 3.70 (d, J = 2.4 Hz, 6H), 3.43 (q, J = 6.6 Hz, 2H), 2.84 (dd, J = 13.7, 5.3 Hz, 1H), 2.74 (ddd, J = 20.9, 13.1, 6.9 Hz, 2H), 2.56 (t, J = 6.8 Hz, 2H), 2.47 (dd, J = 15.7, 5.5 Hz, 2H). 13C NMR (151 MHz, DMSO-d6) δ 178.87, 169.88, 168.32, 149.50, 149.09, 147.75, 146.40, 142.41, 132.67, 131.63, 126.19, 125.84, 123.71, 121.74, 120.86, 116.40, 116.19, 115.15, 113.96, 112.82, 112.25, 71.18, 69.06, 56.01, 55.91, 55.82, 46.01, 41.32, 37.34, 35.98, 35.50, 34.17. FT-MS: calcd for C32H37N3NaO8, [M + H]+, 614.2480, found 614.2472. HPLC purity: 99.89% (tR = 6.49 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
N-(2-Aminophenyl)-3-(2-(4-(((3R,4R)-4-(3,4-dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)acetamido)propanamide (B3). White solid, yield: 34.1%. m.p. 106.2–106.8 °C. 1H NMR (600 MHz, DMSO-d6) δ 9.09 (s, 1H), 7.91 (t, J = 5.5 Hz, 1H), 7.16 (d, J = 7.0 Hz, 1H), 6.88 (dd, J = 11.1, 3.9 Hz, 1H), 6.85 (d, J = 8.2 Hz, 1H), 6.82 (d, J = 8.1 Hz, 2H), 6.73–6.67 (m, 2H), 6.65 (d, J = 1.5 Hz, 1H), 6.60–6.57 (m, 1H), 6.53 (t, J = 7.0 Hz, 1H), 4.86 (s, 2H), 4.41 (s, 2H), 4.09 (t, J = 7.7 Hz, 1H), 3.88 (t, J = 8.2 Hz, 1H), 3.75 (s, 3H), 3.70 (d, J = 1.4 Hz, 6H), 3.33 (s, 2H), 3.15 (dd, J = 12.7, 6.5 Hz, 2H), 2.79–2.70 (m, 2H), 2.46 (dd, J = 13.2, 5.3 Hz, 2H), 2.32 (t, J = 7.3 Hz, 2H), 1.57 (dd, J = 14.9, 7.5 Hz, 2H), 1.50–1.45 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 178.87, 171.46, 168.20, 149.53, 149.10, 147.76, 146.47, 142.24, 132.69, 131.64, 126.14, 125.70, 124.05, 121.74, 120.85, 116.67, 116.37, 115.22, 113.94, 112.82, 112.25, 71.18, 69.22, 55.98, 55.91, 55.81, 46.02, 41.35, 38.52, 37.35, 35.83, 34.21, 29.18, 23.14. FT-MS: calcd for C32H38N3O8, [M + H]+, 606.2819, found 606.2809. HPLC purity: 99.42% (tR = 6.89 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
N-(2-Aminophenyl)-5-(2-(4-(((3R,4R)-4-(3,4-dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)acetamido)pentanamide (B4). Yellow oil, yield: 29.3%. 1H NMR (600 MHz, DMSO-d6) δ 9.09 (s, 1H), 7.91 (t, J = 5.5 Hz, 1H), 7.16 (d, J = 7.0 Hz, 1H), 6.88 (dd, J = 11.1, 3.9 Hz, 1H), 6.85 (d, J = 8.2 Hz, 1H), 6.82 (d, J = 8.1 Hz, 2H), 6.73–6.67 (m, 2H), 6.65 (d, J = 1.5 Hz, 1H), 6.60–6.57 (m, 1H), 6.53 (t, J = 7.0 Hz, 1H), 4.86 (s, 2H), 4.41 (s, 2H), 4.09 (t, J = 7.7 Hz, 1H), 3.88 (t, J = 8.2 Hz, 1H), 3.75 (s, 3H), 3.70 (d, J = 1.4 Hz, 6H), 3.33 (s, 2H), 3.15 (dd, J = 12.7, 6.5 Hz, 2H), 2.79–2.70 (m, 2H), 2.46 (dd, J = 13.1, 5.3 Hz, 2H), 2.32 (t, J = 7.3 Hz, 2H), 1.60–1.55 (m, 2H), 1.50–1.46 (m, 2H). 13C NMR (151 MHz, DMSO-d6) δ 178.86, 171.20, 168.38, 149.55, 149.10, 147.76, 146.47, 142.48, 131.64, 126.22, 125.89, 123.88, 121.74, 120.86, 116.53, 116.24, 115.26, 113.97, 112.83, 112.26, 71.18, 69.24, 55.98, 55.91, 55.82, 46.02, 41.34, 38.37, 37.34, 34.19, 33.59, 29.49, 25.86. FT-MS: calcd for C34H41N3O8, [M + H]+, 620.2969, found 620.2966. HPLC purity: 98.44% (tR = 6.86 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
N-(2-Aminophenyl)-5-(2-(4-(((3R,4R)-4-(3,4-dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)acetamido)pentanamide (B5). Yellow oil, yield: 56.2%. 1H NMR (600 MHz, DMSO-d6) δ 9.13 (s, 1H), 7.89 (d, J = 5.8 Hz, 1H), 7.16 (dd, J = 7.8, 1.0 Hz, 1H), 6.90–6.80 (m, 4H), 6.73–6.67 (m, 2H), 6.65 (d, J = 1.8 Hz, 1H), 6.59 (dd, J = 8.1, 1.8 Hz, 1H), 6.55–6.50 (m, 1H), 4.83 (s, 2H), 4.40 (s, 2H), 4.09 (d, J = 1.3 Hz, 1H), 3.88 (s, 1H), 3.75 (s, 3H), 3.70 (d, J = 2.2 Hz, 6H), 3.12 (dd, J = 13.1, 6.7 Hz, 2H), 2.86 (dd, J = 13.6, 5.2 Hz, 1H), 2.79–2.70 (m, 2H), 2.51 (d, J = 1.7 Hz, 1H), 2.47 (dd, J = 13.6, 5.6 Hz, 2H), 2.30 (t, J = 7.5 Hz, 2H), 1.61–1.56 (m, 2H), 1.48–1.43 (m, 2H), 1.29 (d, J = 7.0 Hz, 2H), 0.85 (ddd, J = 16.5, 9.7, 5.2 Hz, 3H). 13C NMR (151 MHz, DMSO-d6) δ 178.87, 171.56, 168.16, 149.50, 149.09, 147.76, 146.45, 142.31, 132.67, 131.63, 126.10, 125.71, 124.06, 121.74, 120.85, 116.60, 116.34, 115.16, 113.93, 112.81, 112.24, 71.18, 69.18, 55.99, 55.91, 55.81, 46.02, 41.35, 38.65, 37.34, 36.17, 34.20, 29.35, 26.50, 25.49. FT-MS: calcd for C35H43N3O8, [M + H]+, 634.3133, found 634.3122. HPLC purity: 98.67% (tR = 7.01 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
:TFA = 1:1 (V/V) and stirred for 2 h at room temperature, and then the mixture was extracted with EtOAc and concentrated under reduced pressure, yielding compound c. Compound c was dissolved with t-butyl piperidine-4-carboxylate or t-butyl 4-aminobenzoate, EDCI, and HOBt in dry DMF and stirred for 6 h at room temperature. The mixture was extracted with EtOAc and concentrated under reduced pressure, yielding compounds k or l. Dissolved compound k or l in a solution of DCM:TFA = 1:1 (V/V) was stirred for 2 h at room temperature, and then the mixture was extracted with EtOAc and concentrated under reduced pressure, yielding compounds m or n. o-Phenylenediamine (17.3 mg, 0.26 mmol), DIPEA (2.8 mL, 0.26 mmol), and HATU (60.8 mg, 0.26 mmol) were added to a solution of acid compound m or n, anhydrous DMF. The mixture was stirred at room temperature for 6 h. Water was added, and the mixture was extracted with ethyl acetate. The organic layers were combined, concentrated, and purified with flash column chromatography (DCM/MeOH = 50:1, V/V) to afford compounds B6 or B7.
4-(2-(4-(((3R,4R)-4-(3,4-Dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)acetamido)-N-hydroxybenzamide (B6). White solid, yield: 52.0%. m.p. 109.4–110.2 °C. 1H NMR (600 MHz, DMSO-d6) δ 9.26 (s, 1H), 7.18 (d, J = 7.0 Hz, 1H), 6.90–6.86 (m, 1H), 6.82 (d, J = 8.1 Hz, 2H), 6.80 (d, J = 8.3 Hz, 2H), 6.78 (s, 2H), 6.76 (s, 1H), 6.71 (d, J = 7.9 Hz, 1H), 4.71 (s, 2H), 4.10 (s, 2H), 3.87 (d, J = 8.4 Hz, 2H), 3.73 (s, 3H), 3.72 (s, 3H), 3.70 (s, 6H), 3.08 (s, 2H), 2.84–2.81 (m, 2H), 2.78–2.75 (m, 2H), 2.49–2.46 (m, 2H), 1.23 (s, 4H). 13C NMR (151 MHz, DMSO-d6) δ 178.89, 173.33, 166.20, 166.07, 149.16, 149.10, 147.76, 146.60, 142.37, 131.65, 126.17, 125.77, 123.83, 121.59, 120.87, 116.55, 116.30, 113.90, 113.84, 112.82, 112.25, 71.18, 70.25, 67.47, 55.99, 55.91, 55.80, 46.05, 45.47, 44.46, 43.37, 42.35, 41.27, 37.36, 34.10. FT-MS: calcd for C35H41N3NaO8, [M + H]+, 631.2933, found 654.2780. HPLC purity: 98.87% (tR = 7.45 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
N-(2-Aminophenyl)-4-(2-(4-(((3R,4R)-4-(3,4-dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)acetamido)benzamide (B7). Yellow solid, yield: 45.3%. m.p. 118.1–118.3 °C. 1H NMR (600 MHz, DMSO-d6) δ 10.29 (s, 1H), 9.59 (s, 1H), 7.97 (d, J = 8.5 Hz, 2H), 7.75 (d, J = 8.7 Hz, 2H), 7.17 (t, J = 10.8 Hz, 1H), 7.00–6.94 (m, 1H), 6.90 (d, J = 8.2 Hz, 1H), 6.85 (d, J = 1.8 Hz, 1H), 6.80 (t, J = 6.2 Hz, 1H), 6.78 (dd, J = 8.0, 1.2 Hz, 1H), 6.71 (dd, J = 8.2, 1.7 Hz, 1H), 6.65 (d, J = 1.8 Hz, 1H), 6.60 (td, J = 8.6, 1.5 Hz, 2H), 5.05–4.73 (m, 2H), 4.68 (s, 2H), 4.11 (dd, J = 8.5, 7.4 Hz, 1H), 3.89 (t, J = 8.4 Hz, 1H), 3.78 (s, 3H), 3.69 (s, 3H), 3.69 (s, 3H), 2.94–2.84 (m, 1H), 2.76 (ddd, J = 20.9, 13.1, 6.9 Hz, 2H), 2.50–2.42 (m, 3H). 13C NMR (151 MHz, DMSO-d6) δ 178.87, 167.67, 149.48, 149.10, 147.76, 146.49, 143.62, 141.63, 135.34, 132.66, 131.64, 129.90, 129.20, 127.14, 126.87, 123.91, 121.76, 120.86, 119.05, 117.78, 116.74, 116.61, 115.03, 114.91, 114.02, 112.81, 112.23, 71.19, 69.11, 56.03, 55.90, 55.80, 46.02, 41.36, 37.36, 34.18. FT-MS: calcd for C33H38N3NaO8, [M + H]+, 640.2653, found 640.2653. HPLC purity: 99.87% (tR = 6.33 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
:t-BuOH = 1:1 (V/V) and stirred at 60 °C for four hours. The mixture was poured into water and extracted with DCM (20 mL × 3). The combined organic layers were concentrated to obtain compound p. Compound p was dissolved in a solution of DCM:TFA = 1:1 (V/V) and stirred at room temperature for two hours. The mixture was then extracted with ethyl acetate and concentrated under reduced pressure to obtain compound q. Compound q was mixed with o-phenylenediamine (17.3 mg, 0.26 mmol), DIPEA (2.8 mL, 0.26 mmol), and HATU (60.8 mg, 0.26 mmol) in anhydrous DMF. The mixture was stirred for 6 h at room temperature. Water was added, and the mixture was extracted with ethyl acetate. The combined organic layers were concentrated and purified by flash column chromatography (DCM/MeOH = 50:1, V/V) to obtain compound B8.
N-(2-Aminophenyl)-2-(4-((4-(((3R,4R)-4-(3,4-dimethoxybenzyl)-2-oxotetrahydrofuran-3-yl)methyl)-2-methoxyphenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (B8). White solid, yield: 33.9%. m.p. 130.0–130.3 °C. 1H NMR (600 MHz, DMSO-d6) δ 9.80 (s, 1H), 8.23 (s, 1H), 7.20 (dd, J = 7.8, 1.1 Hz, 1H), 7.06 (d, J = 8.3 Hz, 1H), 6.93–6.89 (m, 1H), 6.83–6.81 (m, 1H), 6.79 (d, J = 1.7 Hz, 1H), 6.74–6.69 (m, 2H), 6.65 (d, J = 1.6 Hz, 1H), 6.60 (dd, J = 8.1, 1.6 Hz, 1H), 6.55–6.50 (m, 1H), 5.39 (s, 2H), 5.09 (s, 2H), 5.07 (s, 2H), 4.10 (t, J = 8.0 Hz, 1H), 3.88 (t, J = 8.4 Hz, 1H), 3.70 (s, 9H), 2.85 (dd, J = 13.7, 5.3 Hz, 1H), 2.79 (dd, J = 13.7, 6.9 Hz, 1H), 2.74–2.70 (m, 1H), 2.49 (d, J = 8.3 Hz, 2H), 2.47–2.45 (m, 1H). 13C NMR (151 MHz, DMSO-d6) δ 178.92, 164.84, 149.25, 149.11, 147.76, 146.69, 143.00, 142.71, 131.66, 131.60, 126.75, 126.71, 125.94, 122.66, 121.64, 120.87, 116.35, 116.06, 113.76, 113.71, 112.82, 112.26, 71.20, 70.25, 62.09, 55.92, 55.81, 52.42, 46.07, 41.25, 37.38, 34.15. FT-MS: calcd for C32H35N5NaO7, [M + H]+, 624.2423, found 624.2428. HPLC purity: 99.56% (tR = 7.58 min, 10% MeCN/90% H2O/0.1% TFA), 1.0 mL min−1 in 15 min.
Pharmacology
342 for 5 minutes at room temperature, followed by a PBS wash. The stained cells were resuspended in PBS and subsequently observed and photographed under a fluorescence microscope.000 g at 4 °C for a duration of 15 minutes. Protein levels in the clear supernatants were determined with the BCA protein assay kit. Following this, the protein samples underwent 12% SDS-PAGE separation, were moved onto a PVDF sheet, and then blocked using 5% non-fat milk in TBS with 0.10% Tween 20 for an hour at ambient temperature. The sheets were later treated with primary antibodies within 5% non-fat milk for 6 hours at 4 °C, then with secondary antibodies for an additional hour at ambient conditions. Antibody interactions were displayed using an ECL detection solution.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and its ESI.†Author contributions
Compound design and synthesis: Xinyue Jiang, Yuchao Yan; in vitro activity testing: Xinyue Jiang, Huali Yang; writing: Xinyue Jiang; review and editing: Yang Liu; arctigenin supply and subject cooperation: Deqiang Dou; funding and supervision: Yang Liu, Maosheng Cheng.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This project was financially supported by National Natural Science Foundation of China (No. 22177079). Dr Y. Liu wishes to express his thanks for the support by the Program for Liaoning Innovative Talents in University (LR2017043). Dr D. Dou wishes to express his gratitude for the support provided by the Key Scientific Research Projects of Intercollegiate Cooperation in Colleges and Universities of Liaoning Province (2020JYT005). The figure in Entry for the Table of Contents was made by Figdraw.Notes and references
- Q. C. Li, Y. Liang, Y. Tian and G. R. Hu, J. BUON, 2016, 21, 87–94 Search PubMed.
- N. Kudou, A. Taniguchi, K. Sugimoto, Y. Matsuya, M. Kawasaki, N. Toyooka, C. Miyoshi, S. Awale, D. F. Dibwe, H. Esumi, S. Kadota and Y. Tezuka, Eur. J. Med. Chem., 2013, 60, 76–88 CrossRef CAS PubMed.
- Y. Tezuka, K. Yamamoto, S. Awale, F. Lia, S. Yomoda and S. Kadota, Nat. Prod. Commun., 2013, 8, 463–466 CrossRef CAS PubMed.
- S. Su, X. Cheng and M. Wink, J. Pharm. Pharmacol., 2015, 67, 1316–1323 CrossRef CAS PubMed.
- S. Susanti, H. Iwasaki, M. Inafuku, N. Taira and H. Oku, Phytomedicine, 2013, 21, 39–46 CrossRef CAS PubMed.
- S. Yang, J. Ma, J. Xiao, X. Lv, X. Li, H. Yang, Y. Liu, S. Feng and Y. Zhang, Anat. Rec., 2012, 295, 1260–1266 CrossRef CAS PubMed.
- K. Kang, H. J. Lee, J. H. Yoo, E. H. Jho, C. Y. Kim, M. Kim and C. W. Nho, DNA Cell Biol., 2011, 30, 623–629 CrossRef CAS PubMed.
- C. J. Hsieh, P. L. Kuo, Y. C. Hsu, Y. F. Huang, E. M. Tsai and Y. L. Hsu, Free Radical Biol. Med., 2014, 67, 159–170 CrossRef CAS PubMed.
- J. H. Yoo, H. J. Lee, K. Kang, E. H. Jho, C. Y. Kim, D. Baturen, J. Tunsag and C. W. Nho, Food Chem. Toxicol., 2010, 48, 2247–2252 CrossRef CAS PubMed.
- Y. Gu, C. Scheuer, D. Feng, M. D. Menger and M. W. Laschke, Anticancer Drugs, 2013, 24, 781–791 CrossRef CAS PubMed.
- S. Kuehnl, S. Schroecksnadel, V. Temml, J. M. Gostner, H. Schennach, D. Schuster, S. Schwaiger, J. M. Rollinger, D. Fuchs and H. Stuppner, Phytomedicine, 2013, 20, 1190–1195 CrossRef CAS PubMed.
- J. J. Tong, J. Liu, N. R. Bertos and X. J. Yang, Nucleic Acids Res., 2002, 30, 1114–1123 CrossRef CAS PubMed.
- T. Eckschlager, J. Plch, M. Stiborova and J. Hrabeta, Int. J. Mol. Sci., 2017, 18, E1414 CrossRef PubMed.
- D. Li, Z. Zhang, Y. Li, X. Wang, H. Zhong, H. Yang, Y. Xi, H. Liu, A. Shen and Y. Hu, J. Med. Chem., 2023, 66, 7016–7037 CrossRef CAS PubMed.
- M. Brindisi, A. P. Saraswati, S. Brogi, S. Gemma, S. Butini and G. Campiani, J. Med. Chem., 2020, 63, 23–39 CrossRef CAS PubMed.
- J. Roche and P. Bertrand, Eur. J. Med. Chem., 2016, 121, 451–483 CrossRef CAS PubMed.
- Y. Huang, N. Liu, Z. Pan, Z. Li and C. Sheng, J. Med. Chem., 2023, 66, 1239–1253 CrossRef CAS PubMed.
- T. C. S. Ho, A. H. Y. Chan and A. Ganesan, J. Med. Chem., 2020, 63, 12460–12484 CrossRef CAS PubMed.
- P. A. Marks and R. Breslow, Nat. Biotechnol., 2007, 25, 84–90 CrossRef CAS PubMed.
- L. P. Yang, BioDrugs, 2011, 25, 393–395 CrossRef CAS PubMed.
- R. M. Poole, Drugs, 2014, 74, 1543–1554 CrossRef CAS PubMed.
- K. P. Garnock-Jones, Drugs, 2015, 75, 695–704 CrossRef CAS PubMed.
- R. Parveen, D. Harihar and B. P. Chatterji, Cancer, 2023, 129, 3372–3380 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI contains additional figures, compound characterization including 1H and 13C NMR spectra, ultra-high resolution mass spectrometry, and western blot experimental images. See DOI: https://doi.org/10.1039/d4ra00050a |
| This journal is © The Royal Society of Chemistry 2024 |