DOI:
10.1039/D4RA00666F
(Paper)
RSC Adv., 2024,
14, 9326-9338
Antimicrobial and alpha-glucosidase inhibitory flavonoid glycosides from the plant Mussaenda recurvata: in vitro and in silico approaches†
Received
26th January 2024
, Accepted 5th March 2024
First published on 19th March 2024
Abstract
Seven flavonoid glycosides were isolated from the aerial portions of Mussaenda recurvata during a phytochemical analysis. This comprised one novel component, ecurvoside, and six well-studied compounds, namely astragalin, isoquercitrin, nicotiflorin, rutin, hesperidin, and neohesperidin. The chemical structures of compounds were identified using spectroscopic techniques and a comparison with previously published studies. Alpha-glucosidase inhibition testing was carried out on all isolated compounds. The compounds evaluated have IC50 values between 35.6 and 239.1 g mL−1, indicating a moderate degree of inhibition. In vitro antimicrobial activities of compounds 1–7 have screened against the bacteria Pseudomonas aeruginosa (P. aeruginosa), methicillin-resistant Staphylococcus aureus (MRSA), Streptococcus faecalis (Strep. faecalis), and fungi: Candida albicans (C. albicans), Trichophyton mentagrophytes (T. mentagrophytes), and Microsporum gypseum (M. gypseum), where compound 6 showed excellent activity against fungi T. mentagrophytes with an MIC value of 12.5 μM. In accordance with the molecular docking study, ecurvoside (1) or pose 472 interacted well with the 3TOP enzyme: PDB and the molecular dynamic simulations proved that the complex of ecurvoside and 3TOP has a stable simulation time of 50–100 ns and the significant residual amino acids in 3TOP are relative to interactions more than one time such as Asp 960, Glu 961, Lys 1088, Glu 1095, Arg 1097, Gly 1102, Thr 1103, Gln 1109, Glu 1178: A chain and Glu 1095, Thr 1101, and Asp 1107: B chain. The docking studies of compounds 1–7 to the enzyme 2VF5 explain the general mechanism to inhibit bacteria and proved that compound 6 (pose 370) inhibited stronger than compound 7 (pose 362) and compound 5 (pose 280), and compounds 1 to 4 do not interact well with 2VF5.
Introduction
The molecular docking model is an in silico method that allows to evaluate many biological activities such as anticancer,1–3 enzyme α-glucosidase inhibition,4 antibacterial activity, and anti-inflammatory activity.5 This model needs to assess the validation based on RMSD values.6–8 Molecular dynamics (MD) simulations permit determining the accuracy of the molecular docking model. MD generates a more accurate biophysical simulation of protein–ligand complex binding to explain the model complex's stability.9
The genus Mussaenda (Rubiaceae) with approximately 160 species is native to tropical and subtropical regions.10 Some Mussaenda plants are used in folk medicine in China.11 The chemical data of this genus indicated the presence of saponins, terpenoids, iridoids, and flavonoids.12–23 Little is known about the chemical and biological data of Mussaenda recurvata Naiki, Tagane & Yahara. Our previous studies on the Vietnamese native plant indicated the presence of five saponins, nine ursane- and oleanane-type triterpenes.24,25 These compounds were believed to be active compounds with anti-inflammatory activity. A continuation on the polar fraction of the Vietnamese plant M. recurvata indicated the presence of a new compound ecurvoside, along with six known compounds astragalin, isoquercitrin, nicotiflorin, rutin, hesperidin, and neohesperidin. Isolated compounds were evaluated for their inhibitory effects on alpha-glucosidase. Molecular docking study and molecular dynamics simulations were conducted to determine the mechanism of biological activity.
Results and discussion
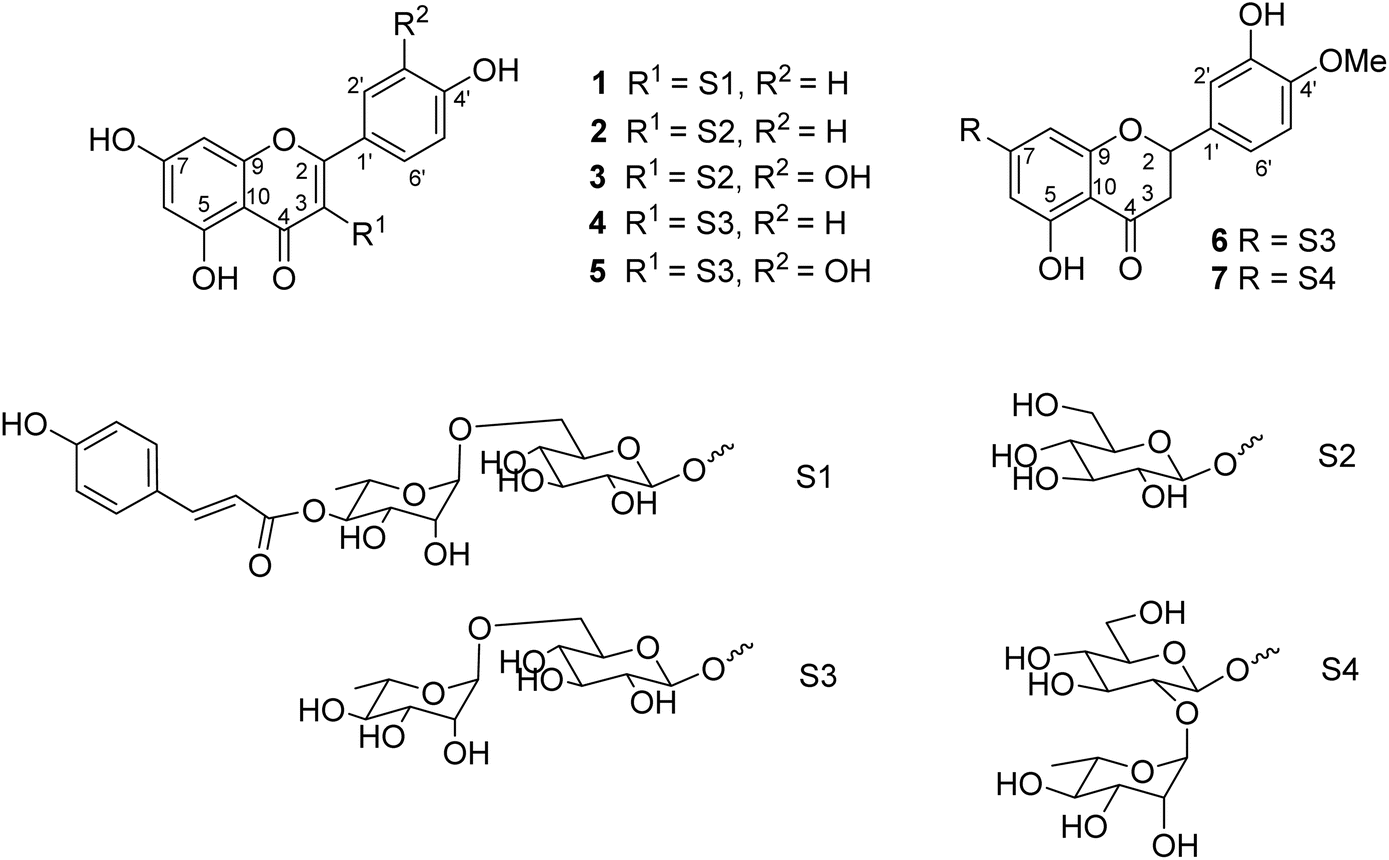
Seven flavonoid glycosides 1–7 were isolated from the EtOAc extract of M. recurvata. Six known compounds were elucidated as astragalin (2),26 isoquercitrin (3),26 nicotiflorin (4),27 rutin (5),27 hesperidin (6),28 and neohesperidin (7).29,30 The chemical structures of compounds were elucidated using NMR spectroscopy. The NMR data of 2–7 are shown in Tables S1 and S2.† Among the isolated compounds, compound 1 was a new compound, namely, ecurvoside. The chemical elucidation of compound 1 was described as follows.
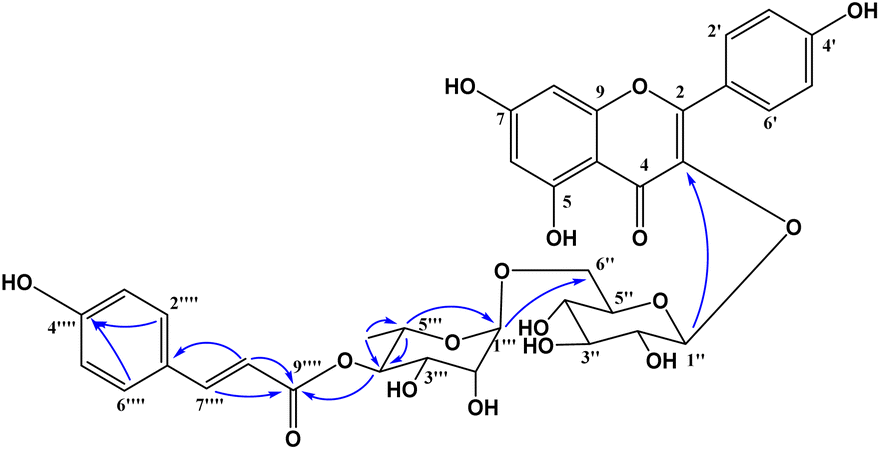
Compound 1 was obtained as a colorless gum. The molecular formula of 1 was deduced as C36H36O17 by the deprotonated molecular ion peak at m/z 739.1856 (calcd for [C36H36O17–H]−, 739.1874) in the HRESI mass spectrum (Fig. S1†). The 1H NMR spectrum showed eight aromatic protons of two 1,4-disubstituted benzene rings [B-ring: δH 7.96 (2H, d, J = 9.0, H-2′/H-6′) and 6.91 (2H, d, J = 9.0, H-3′/H-5′) and C-ring: δH 7.52 (2H, d, J = 8.5, H-2′′′/H-6′′′) and 6.83 (2H, d, J = 8.5, H-3′′′/H-5′′′)], two meta-coupled aromatic protons [δH 6.33 (1H, d, J = 1.5, H-8) and 6.18 (1H, brs, H-6)], two trans-coupled olefinic protons [δH 7.50 (1H, d, J = 15.5, H-2′′′) and 6.20 (1H, d, J = 16.0, H-2′′′)], and signals of two sugar units ranging from δH 0.75 to 5.40. The 13C NMR spectrum in accordance with the HSQC spectrum exhibited 36 carbon resonances attributable to two carbonyl carbons (δC 177.3 and 166.4), eight aromatic methine carbons [δC 130.2 (×2), 130.0 (×2), 115.9 (×2), 115.1 (×2), 98.4, and 93.6], two olefinic methine carbons (δC 144.6 and 114.4), two anomeric carbons (δC 101.1 and 100.3), a methyl carbon (δC 17.1), nine quaternary carbons (δC 159.3, 153.2, 150.0, 149.3, 129.1, and 123.5), and nine oxygenated carbons of two sugar units in the range of 65.9–76.5 ppm. These spectroscopic data indicated that 1 was a flavonoid glycoside bearing two sugar units and a 4-hydroxycinnamoyl moiety. The detailed comparison of the NMR data of 1 and nicotiflorin (4) gave the similarity, indicating that both compounds have a kaempferol-derived aglycone and two sugar units, D-glucose and L-rhamnose (Table 1). The nature of sugar units were strongly supported by NOESY correlations and the NMR comparison of 1 and co-isolates 2–7 (Fig. 2). The difference between them is the presence of an additional 4-hydroxycinnamoyl moiety in 1. The NMR data of 1 was highly similar to those of kaempferol-3-O-β-D-[4′′′-p-E-coumaroyl-α-L-rhamnosyl(1→6)]galactoside reported previously,31 supporting the chemical structure of 1. Combined, the chemical structure of 1 was elucidated as shown in Fig. 1, namely, ecurvoside.
Table 1 NMR data of 1 and 4 in DMSO-d6
| No. |
1 |
4 |
No. |
1 |
4 |
| δC |
δHa |
δC |
δHa |
δC |
δHa |
δC |
δHa |
| Multi, J in Hz. |
| 2 |
156.8 |
|
156.7 |
|
6′′-ORha |
| 3 |
133.0 |
|
133.1 |
|
1′′′ |
100.3 |
4.48 (1H, brs) |
100.7 |
4.38 (1H, d, 1.0) |
| 4 |
177.3 |
|
177.3 |
|
2′′′ |
68.3 |
3.56 (1H, m) |
70.3 |
3.41 (1H, m) |
| 5 |
161.2 |
|
161.1 |
|
3′′′ |
70.4 |
3.70 (1H, m) |
71.7 |
3.08 (1H, m) |
| 6 |
98.4 |
6.18 (1H, brs) |
98.7 |
6.20 (1H, d, 2.0) |
4′′′ |
73.4 |
4.77 (1H, t, 9.5) |
70.5 |
3.27 (1H, m) |
| 7 |
164.0 |
|
164.1 |
|
5′′′ |
65.9 |
3.48 (1H, m) |
68.1 |
3.25 (1H, m) |
| 8 |
93.6 |
6.33 (1H, d, 1.5) |
93.7 |
6.41 (1H, d, 2.5) |
6′′′ |
17.1 |
0.75 (d, 6.0) |
17.6 |
0.99 (1H, d, 6.0) |
| 9 |
156.5 |
|
156.4 |
|
2′′′-OH |
|
4.91 (1H, m) |
|
|
| 10 |
103.8 |
|
103.9 |
|
3′′′-OH |
|
4.65 (1H, m) |
|
|
| 5-OH |
|
12.56 (1H, brs) |
|
12.56 (1H, s) |
4′′′′-Cin |
| 1′ |
120.9 |
|
120.8 |
|
1′′′′ |
125.2 |
|
|
|
| 2′ |
130.2 |
7.96 (1H, d, 9.0) |
130.8 |
7.99 (1H, d, 8.0) |
2′′′′ |
130.0 |
7.52 (1H, d, 8.5) |
|
|
| 3′ |
115.1 |
6.91 (1H, d, 9.0) |
115.0 |
6.88 (1H, d, 8.0) |
3′′′′ |
115.9 |
6.83 (1H, d, 8.5) |
|
|
| 4′ |
159.9 |
|
159.8 |
|
4′′′′ |
159.8 |
|
|
|
| 5′ |
115.1 |
6.91 (1H, d, 9.0) |
115.0 |
6.88 (1H, d, 8.0) |
5′′′′ |
115.9 |
6.83 (1H, d, 8.5) |
|
|
| 6′ |
130.2 |
7.96 (1H, d, 9.0) |
130.8 |
7.99 (1H, d, 8.0) |
6′′′′ |
130.0 |
7.52 (1H, d, 8.5) |
|
|
| 3-OGlc |
7′′′′ |
144.6 |
7.50 (1H, d, 15.5) |
|
|
| 1′′ |
101.1 |
5.40 (1H, d, 8.0) |
101.3 |
5.31 (1H, d, 7.5) |
8′′′′ |
114.4 |
6.20 (1H, d, 16.0) |
|
|
| 2′′ |
74.2 |
3.19 (1H, m) |
74.1 |
3.18 (1H, d, 7.5) |
9′′′′ |
166.4 |
|
|
|
| 3′′ |
76.5 |
3.22 (1H, m) |
76.3 |
3.22 (1H, m) |
|
|
|
|
|
| 4′′ |
69.4 |
3.19 (1H, m) |
69.8 |
3.24 (1H, m) |
|
|
|
|
|
| 5′′ |
75.3 |
3.26 (1H, m) |
75.7 |
3.21 (1H, d, 7.5) |
|
|
|
|
|
| 6′′ |
66.2 |
3.48 (1H, m) |
66.8 |
3.29 (1H, m) |
|
|
|
|
|
| 3.68 (1H, d, 10.0) |
3.69 (1H, d, 10.0) |
| 2′′-OH |
|
5.36 (1H, d, 4.5) |
|
|
|
|
|
|
|
| 3′′-OH |
|
5.16 (1H, d, 5.5) |
|
|
|
|
|
|
|
| 4′′-OH |
|
5.09 (1H, d, 5.0) |
|
|
|
|
|
|
|
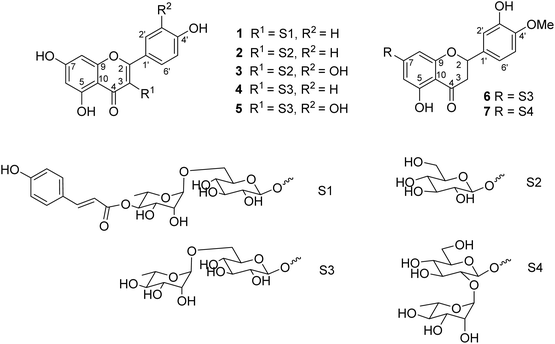 |
| | Fig. 1 Chemical structures of compounds 1–7. | |
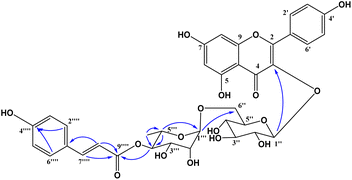 |
| | Fig. 2 Selected HMBC correlations of 1. | |
Flavonoid glycosides 1–7 were reported in the first time from M. recurvata. Astragalin (2), isoquercitrin (3), nicotiflorin (4), and rutin (5) were reported previously in M. arcuata leaves.32 Rutin was isolated from M. macrophylla.33,34 Flavanone glycosides such as hesperidin (6) and neohesperidin (7) rarely occurred in the genus Mussaenda.
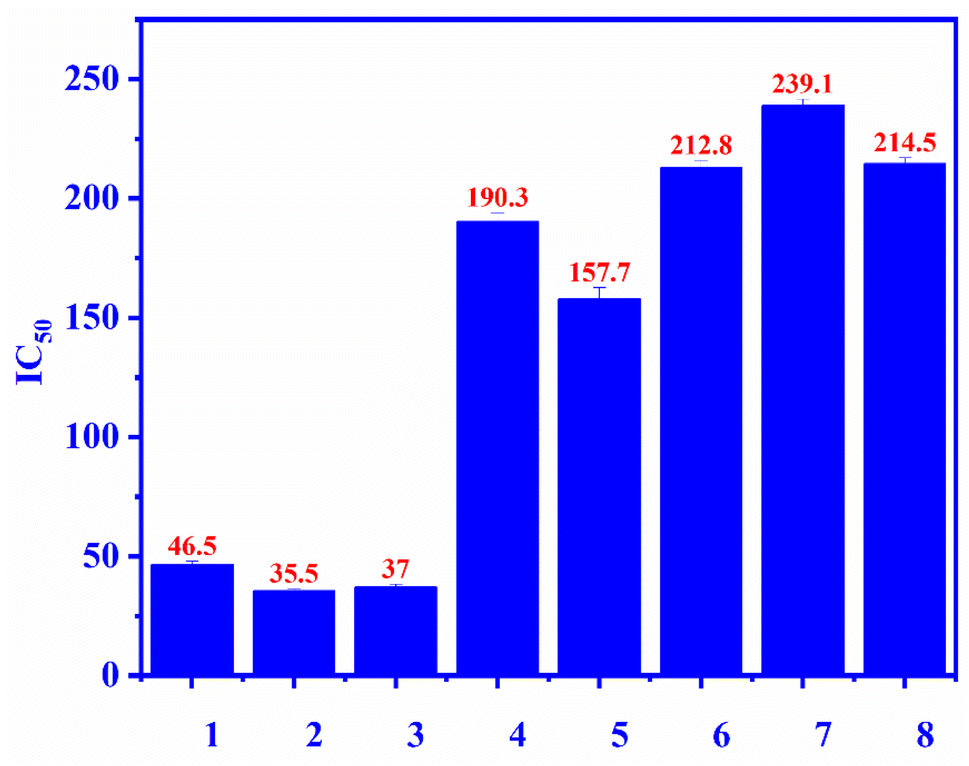
Compounds 1–7 were evaluated for the alpha-glucosidase inhibition, revealing the significant activity with IC50 values ranging from 35.5 to 239.1 μM (compared to the positive control acarbose IC50 214 μM), as seen in Fig. 3. Compounds 2 and 3 were stronger than compounds 4 and 5, indicating that the L-rhamnose moiety in 4 and 5 decreased the activity. The comparison of the inhibition of 1, 4, and 5 revealed that the 4-hydroxycinnamoyl group would enhance the activity. Flavanone glycosides 6 and 7 were weaker than flavonol glycosides 1–5, indicating the important role of the carbon skeleton in alpha-glucosidase inhibition. The alpha-glucosidase inhibition of compounds 2–7 has been reported previously. Astragalin (2) and isoquercitrin (3) strongly inhibited alpha-glucosidase, consistent with published results.35,36 Isoquercitrin (3) was in silico investigated, revealing its inhibitory mechanism.37 Rutin (5) is believed to have antidiabetic potential.38 This compound showed significant pancreatic amylase and alpha-glucosidase inhibition.38 In 2013, Shin and co-workers investigated the beta-glucosidase inhibition of flavonoid glycosides from Pyrococcus furiosus, revealing the inhibitory order: hesperidin > neohesperidin > rutin.39
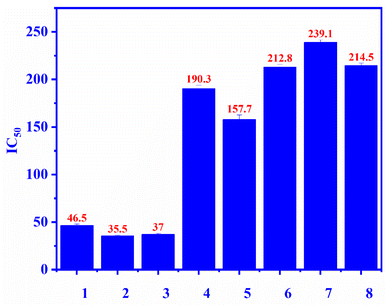 |
| | Fig. 3 The alpha-glucosidase inhibitory (IC50) of compounds 1–7 and positive control: ecurvoside (1), astragalin (2), isoquercitrin (3), nicotiflorin (4), rutin (5), hesperidin (6), neohesperidin (7), acarbose (8). | |
In vitro antimicrobial activity
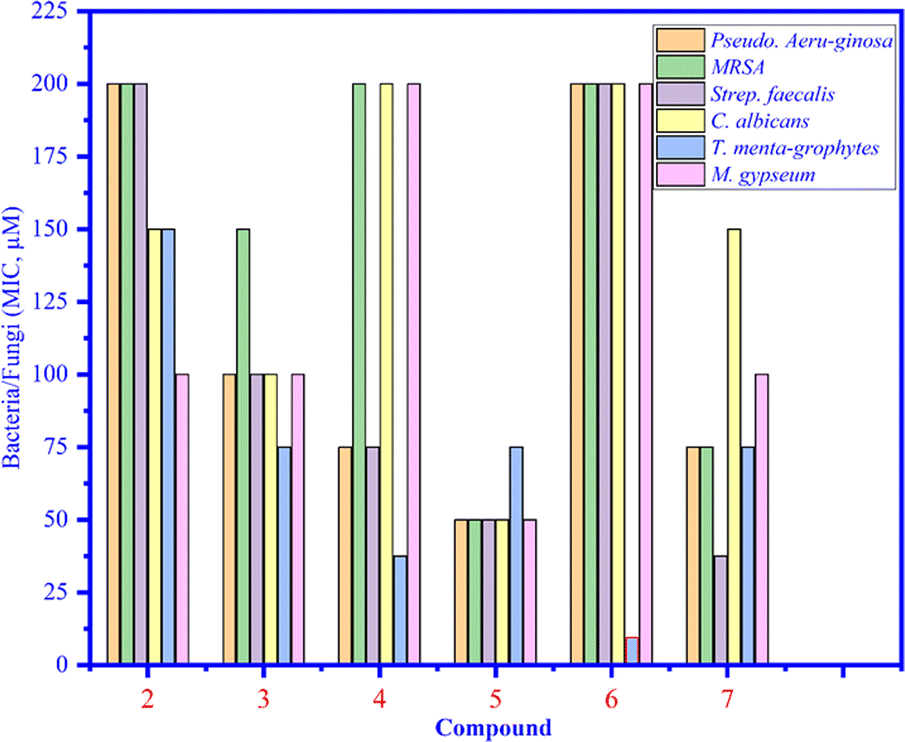
Compounds 1–7 were tested against three bacteria such as Pseudomonas aeruginosa, methicillin-resistant Staphylococcus aureus (MRSA), and Streptococcus faecalis (Strep. Faecalis) and three fungi, namely, Candida albicans (C. albicans), Trichophyton mentagrophytes (T. mentagrophytes), and Microsporum gypseum (M. gypseum). As seen in Fig. 4, the MIC values of compounds 1–7 were determined and among them, the MIC of compound 6 was the lowest (12.5 μM) (light blue bar), proving that compound 6 inhibited well against Trichophyton mentagrophytes fungus (Fig. 5).
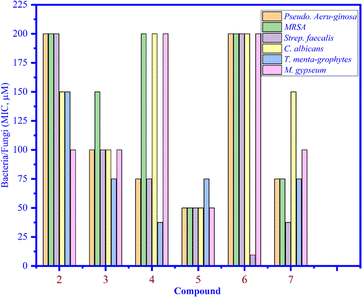 |
| | Fig. 4 The MIC values of compounds 1 to 7 that inhibited bacteria and fungi. | |
 |
| | Fig. 5 The inhibition zones at different concentrations of compound 6 (C1: 6.125, C2: 3.06, C3: 12.5, and C4: 25 μM) that inhibited against the fungus Trichophyton mentagrophytes. | |
In silico docking studies
Inhibition of the α-glucosidase enzyme.
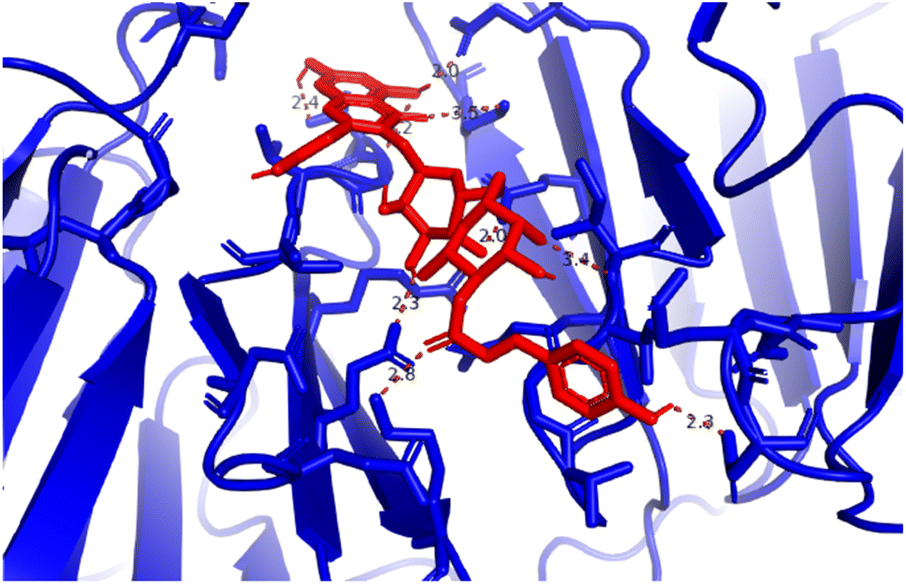
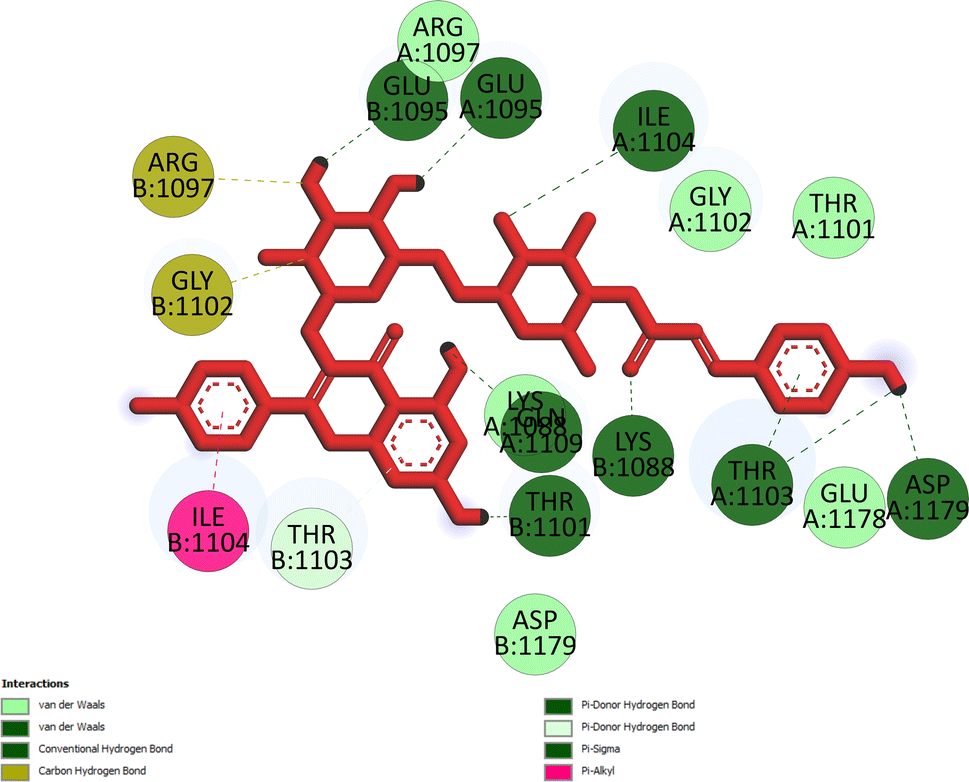
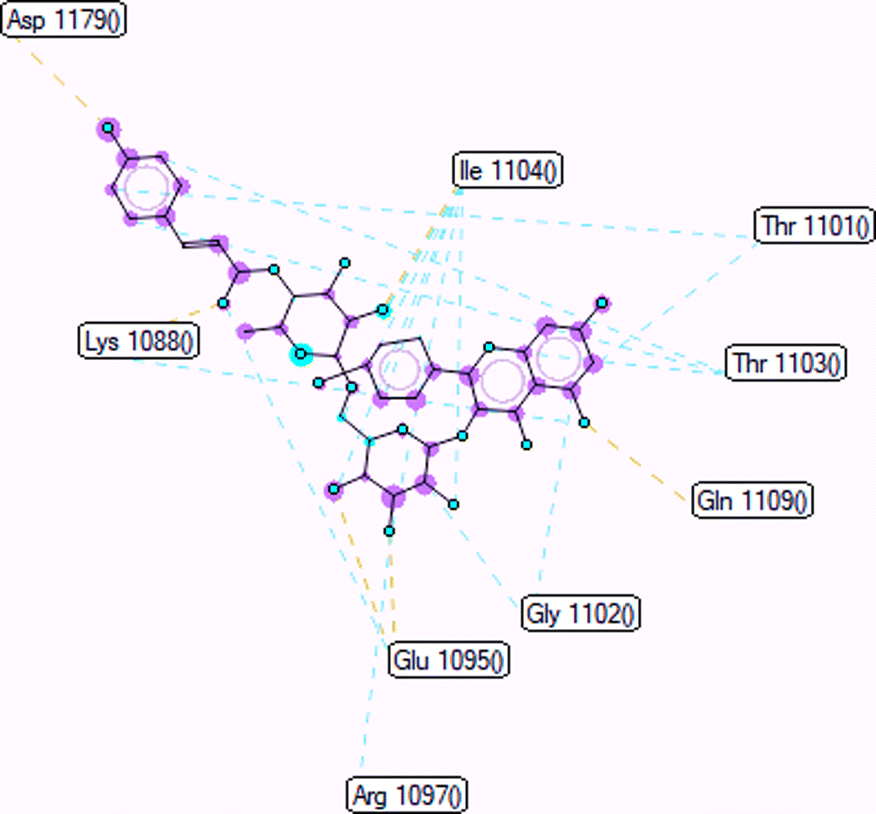
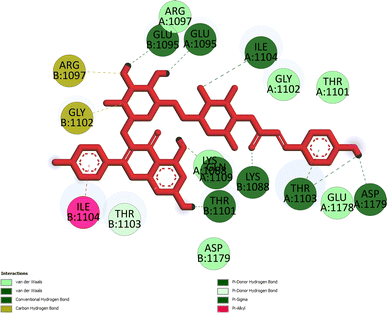
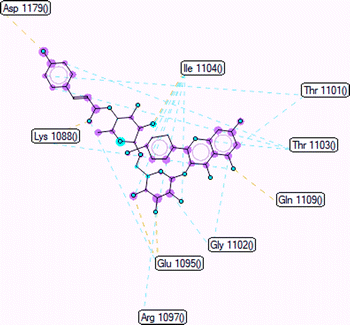
Pose 472. The best conformation of compound 1 among 500 docking conformations docked to 3TOP: PDB enzyme with values of free Gibbs energy and inhibition constant of −7.16 kcal mol−1 and 5.64 μM, respectively, as shown in Table 2. In this case, pose 472 formed 7 H-bonds, as seen in Table 2 and Fig. 6. Among the H-bonds, the two H-bonds from the hydrogen atoms on pose 472 to Glu 1095 and Gln 1109 are the strongest because of their shortest bond length, and they are listed as pose 472:H–A:Glu 1095:O (1.98 Å) and pose 472:H–A:Gln 1109:O (1.98 Å). As seen in Fig. 6, the H-bonds (red lines) formed from active atoms to active atoms of residual amino acids with 5 Å length and are demonstrated by the PyMOL software. As shown in Fig. 7, the functional group of this pose formed H-bonds from Glu 1095: B chain, Glu 1095: A chain, Ile 1104: A chain, Asp 1179: A chain, Gln 1109: A chain, Lys 1088: A chain, Thr 1101: B chain, and Thr 1103: A chain to hydrogen atoms and oxygen atoms of alcohol groups in the carbohydrate, hydroxyl phenolic groups, and carbonyl group. The capping group of this pose 472 are hydrophobic interactions such as pi–alkyl forming from Ile 1104: B chain to the pi-electron system of an aromatic ring. The connecting unit (CU) or the linker of the ligand was identified by hydrophobic interactions such as pi-donor H-bonds formed from Gly 1102: B chain and Arg 1097: B chain, and the Thr 1103: B chain to the pi-electron system of the phenyl ring, carbon, and oxygen atom on this pose. The ligand/pose 472 has three parts: functional group: H-bonding, capping group: protein identification via hydrophobic interactions, and connecting unit: detected via hydrophobic interactions that interacted well with residual amino acids on the enzyme.40 Pose 472 is a good ligand in interactions with enzyme 3TOP, and we say that this pose inhibited the enzyme well. As seen in Fig. 8, one ligand map indicated the secondary interactions formed from pose 472 to residual amino acids around this pose, which included H-bonds from Asp 1179, Ile 1104, Gln 1109, Glu 1095, and Lys 1088 (brown color lines) to atoms on pose 472, steric interactions (green lines) from Ile 1104, Thr 1101, Thr 1103, Gln 1109, Gly 1102, Glu 1095, Arg 1097, and Lys 1088 to atoms on pose 472, and overlap interactions (violet circles). The diameters of the circles are larger, and the overlap interactions are stronger. There are many steric interactions (many green lines) that formed around pose 472 that proved that the interaction between pose 472 and 3TOP is strong in conformation processing.
Table 2 The significant ligand interactions forming from pose 472, the best docking pose among 500 conformations of compound 1 to 3TOP enzyme: PDB
| Entry |
Posea |
Free Gibbs energyb |
Kic |
The number of hydrogend |
The character and bond lengthe |
| The calculation results from AutoDockTools-1.5.7 (ATD). The calculation results from AutoDockTools-1.5.7 (ATD). The calculation results from AutoDockTools-1.5.7 (ATD). Building from Discovery Studio 2021 Client software. Building from Discovery Studio 2021 Client software. |
| 1 |
Pose 472 |
−7.16 |
5.64 |
7 |
B:Lys 1088:N – pose 472:O (2.76 Å) |
| B:Thr 1101:O – pose 472:O (3.12 Å) |
| Pose 472:H–Asp 1179:O (2.31 Å) |
| Pose 472:H–A:Glu 1095:O (1.98 Å) pose 472:H–B:Glu 1095:O (2.26 Å) |
| Pose 472:H–B:Thr 1101:O (2.40 Å) |
| Pose 472:H–A:Gln 1109:O (1.98 Å) |
| Acarbose |
Pose 61 |
−8.81 |
0.351 |
0 |
— |
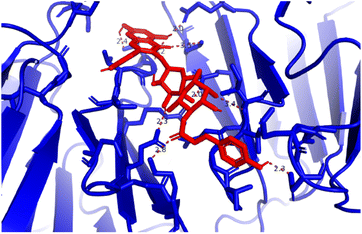 |
| | Fig. 6 H-bonds formed from active sites on pose 472, the best pose docking of compound 1, and residual amino acids on 3TOP by PyMOL software. | |
 |
| | Fig. 7 Pose 472, the best docking pose of compound 1 anchored to the 3TOP enzyme. | |
 |
| | Fig. 8 Ligand map indicated the secondary interactions between pose 472 and 3TOP such as H-bonding-brown color lines, steric interactions-green lines, and overlap interactions-violet circles. | |
 |
| | Fig. 9 One 2D diagram showing the most important ligand interactions between acarbose and 3TOP enzyme. | |
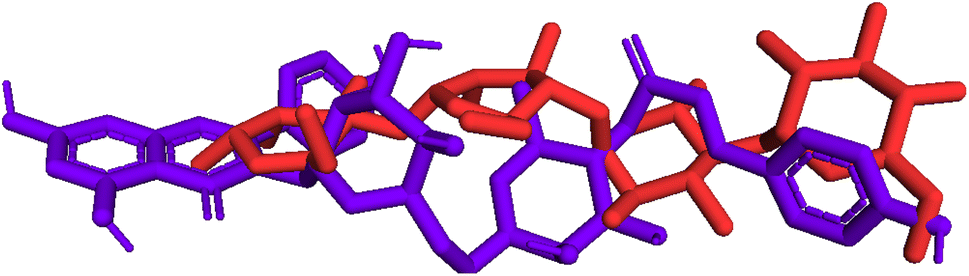
Model validation. As indicated in Fig. 10, pose 472 is aligned with pose 61 with an RMSD of 2.112 Å, indicating that the RMSD is about 2 Å, which was conducted by the PyMOL software. This value demonstrates that pose 472 lines up with pose 61, acarbose, and that there is not much variation in the conformation of pose 47241,42 as well as the confirmation of the model in docking parameters using docking calculation, active center on enzyme, docking orientation, docking processing, and prediction of good interactions with interesting ligands.
 |
| | Fig. 10 Pose 472, the best stable conformation ligand of 1 aligned to pose 61, the best stable conformation ligand of the acarbose ligand with the value of RMSD of 2.112 Å. | |
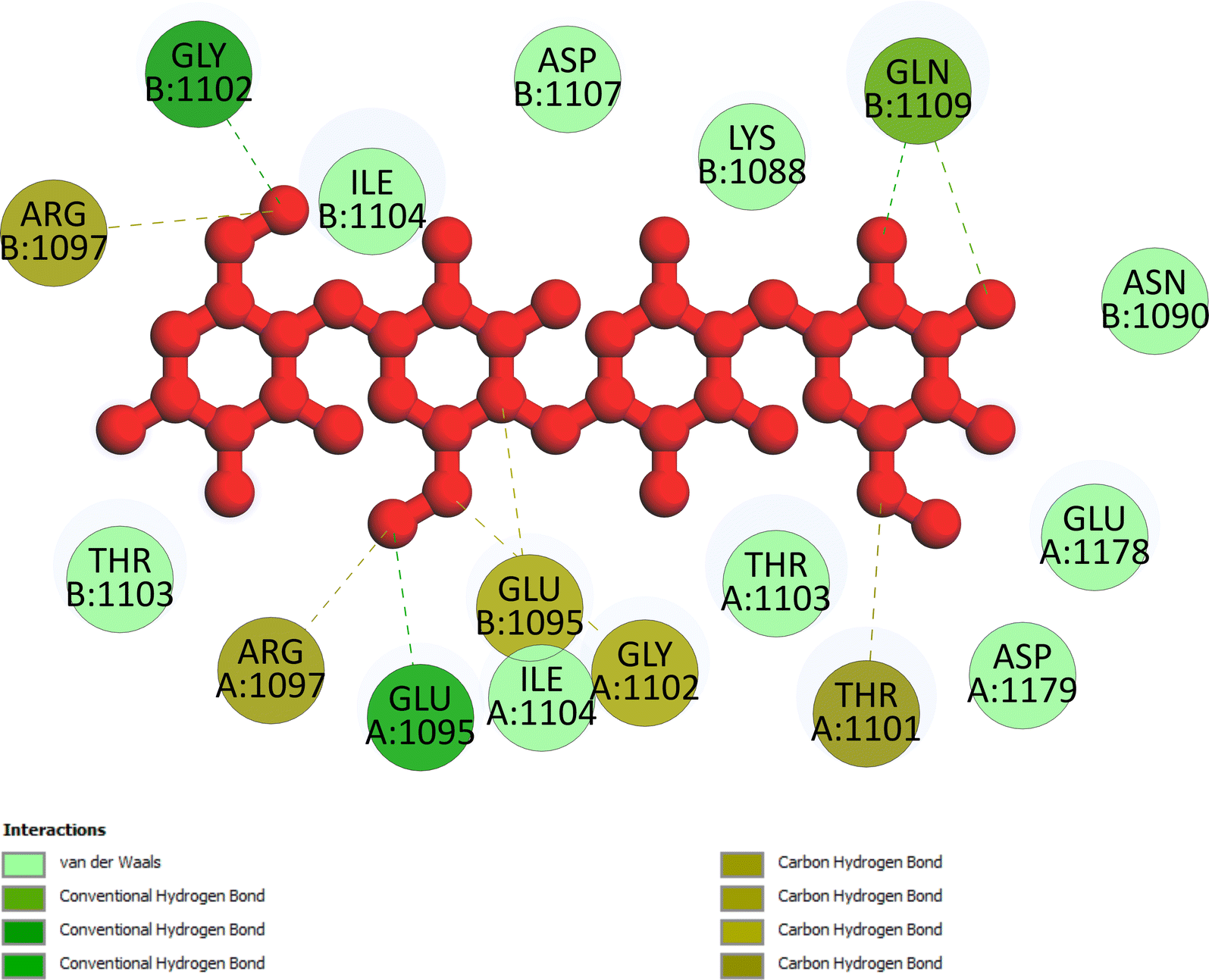
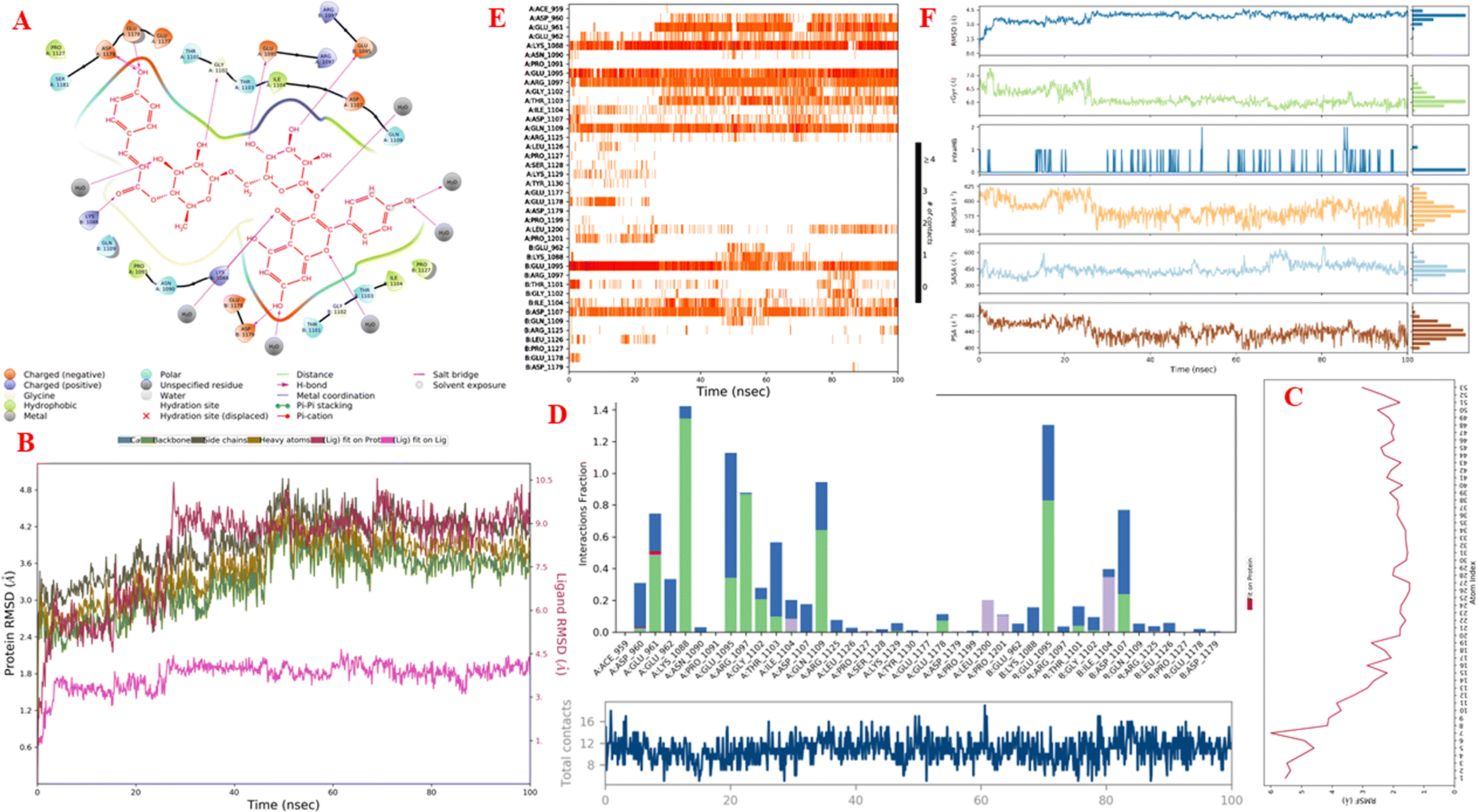
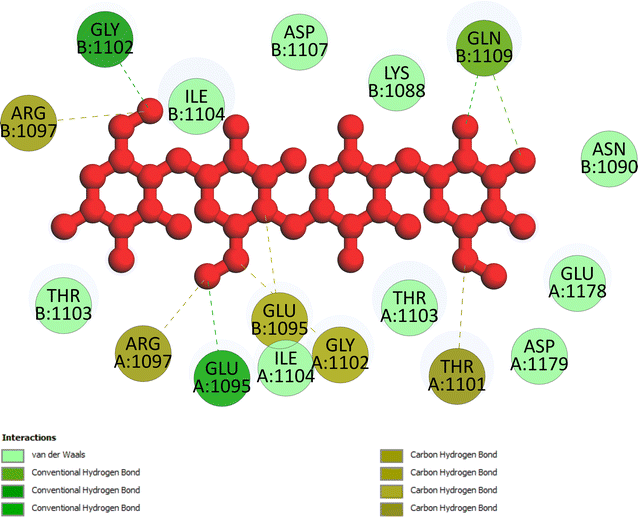
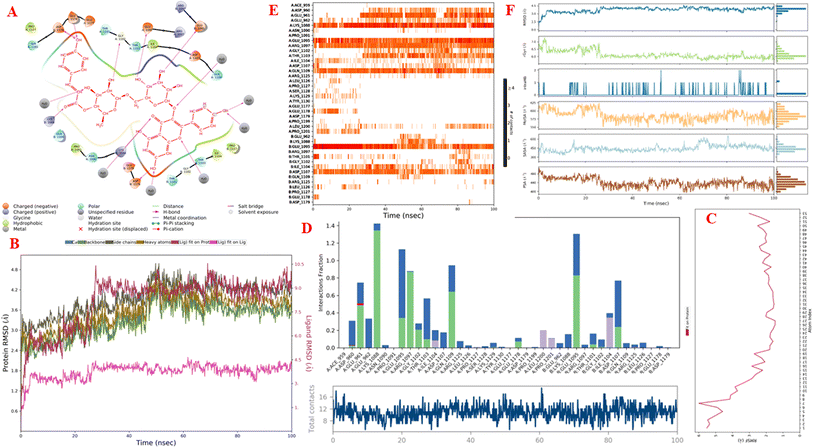
Molecular dynamics simulations. Fig. 11A–F and S9–S14† display the outcomes of the molecular dynamic simulations for the best pose docking 472 and 3TOP. The simulation course did not account for protein flexibility in all simulation courses from 0 to 100 ns. Molecular dynamic simulations were conducted using the Desmond program in Ubuntu to examine the process of ligand binding and the balance of the pose 772_3TOP complex. Pose 472 yields favorable outcomes in the ligand interaction model through docking. The simulation details are shown in Fig. S9† and the simulation system includes one CPU, Mdsim job type, NPT ensemble method, 300 K temperature, 100.102 ns time, 185![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 274 heavy atoms, 52285 water molecules, and 0 charge. The 3TOP enzyme consists of 1784 total residues, with A and B chains, 27992 atoms, 14324 heavy atoms, and a charge of −46. The data for pose 472 in Fig. S10† consists of 89 atoms, an atomic mass of 740.678 atomic units (au), 0 charge, and 19 rotational bonds. Fig. S11† displays the fragments and root mean square (RMSF) of 3TOP, which are valuable for analyzing local variations along the enzyme chain fluctuation and the residual amino acids (C-alpha). The enzyme undergoes significant conformational changes at 400, 500, and 1800 residual indexes due to large fluctuations. The secondary enzyme, as shown in Fig. S12,† consists of 15.36% helix, 24.13% strand, and 39.48% SSE elements of the 3TOP structure. The SSE plot ranges from 0 to 1800 index, showing the percentage of red color lines (15.36%) representing SEE and green color lines (24.13%) representing strand chain. Fig. S13† indicates that the contacts between pose 472 and 3TOP are exposed through hydrogen bonds. Specifically, the oxygen atom of phenolic hydroxyl group on pose 472 interacts with the Lys 1089: A chain (positively charged, violet color) for 73% of the simulation time, and with the oxygen atom of the carbonyl group on this pose for 61% of the simulation time. Arg 1097 formed a hydrogen bond with a hydroxyl group on the carbohydrate moiety of pose 472 for 86% of the simulation time. The hydrogen atom of the hydroxyl group from the carbohydrate part on pose 472 formed a hydrogen bond with B: Glu 1095 via the water bridge at 128% of the simulation time. At a specific time during the simulation, Glu 1095 formed a hydrogen bond with a hydroxyl group on the carbohydrate moiety via water bridge. One H-bonding forms between A: Arg 1097 and the hydrogen atom of the hydroxyl group of the carbohydrate part, occurring at 86% of the simulation time. At 75% of the simulation time, one of the hydroxyl phenolic groups linked to B: Asp 1107 through a water bridge. Fig. S14† displays the torsion profile of pose 471, showing a 10-torsion and two carbon–carbon bonds with large energies of 10.21 and 10.00 kcal mol−1 and torsion angles of −180 and +180 degrees, represented by pink and orange colors.
274 heavy atoms, 52285 water molecules, and 0 charge. The 3TOP enzyme consists of 1784 total residues, with A and B chains, 27992 atoms, 14324 heavy atoms, and a charge of −46. The data for pose 472 in Fig. S10† consists of 89 atoms, an atomic mass of 740.678 atomic units (au), 0 charge, and 19 rotational bonds. Fig. S11† displays the fragments and root mean square (RMSF) of 3TOP, which are valuable for analyzing local variations along the enzyme chain fluctuation and the residual amino acids (C-alpha). The enzyme undergoes significant conformational changes at 400, 500, and 1800 residual indexes due to large fluctuations. The secondary enzyme, as shown in Fig. S12,† consists of 15.36% helix, 24.13% strand, and 39.48% SSE elements of the 3TOP structure. The SSE plot ranges from 0 to 1800 index, showing the percentage of red color lines (15.36%) representing SEE and green color lines (24.13%) representing strand chain. Fig. S13† indicates that the contacts between pose 472 and 3TOP are exposed through hydrogen bonds. Specifically, the oxygen atom of phenolic hydroxyl group on pose 472 interacts with the Lys 1089: A chain (positively charged, violet color) for 73% of the simulation time, and with the oxygen atom of the carbonyl group on this pose for 61% of the simulation time. Arg 1097 formed a hydrogen bond with a hydroxyl group on the carbohydrate moiety of pose 472 for 86% of the simulation time. The hydrogen atom of the hydroxyl group from the carbohydrate part on pose 472 formed a hydrogen bond with B: Glu 1095 via the water bridge at 128% of the simulation time. At a specific time during the simulation, Glu 1095 formed a hydrogen bond with a hydroxyl group on the carbohydrate moiety via water bridge. One H-bonding forms between A: Arg 1097 and the hydrogen atom of the hydroxyl group of the carbohydrate part, occurring at 86% of the simulation time. At 75% of the simulation time, one of the hydroxyl phenolic groups linked to B: Asp 1107 through a water bridge. Fig. S14† displays the torsion profile of pose 471, showing a 10-torsion and two carbon–carbon bonds with large energies of 10.21 and 10.00 kcal mol−1 and torsion angles of −180 and +180 degrees, represented by pink and orange colors.
 |
| | Fig. 11 One simulation result course of complex between pose 472 and 3TOP enzyme (complex_pose_472_3TOP) from 0 to 100 ns in Linux or Ubuntu Environment including: (A) main interaction scheme of pose 472 and 3TOP; (B) protein–ligand RMSD indicated by Desmond; (C) the fluctuations of pose 472 on 3TOP enzyme; (D) the contacts between 3TOP enzyme and pose 472; (E) the contact maintained and shown by a darker shade of orange color in whole simulation time; and (F) pose 472 properties. | |
Fig. 11A illustrates that pose 472 exhibited strong interactions with the 3TOP enzyme through hydrophobic interactions involving specific amino acid residues such as Pro 109 in the A chain, Pro 1127 in the B chain, and Ile 1104 in the A chain. Additionally, polar interactions were observed with residues like Asn 1090 in the A chain, Thr 1101 in the B chain, and Gln 1109 in the A chain. Charge interactions were noted with residues like Asp 1179 in the B chain and Glu 1095 in the B chain. Positive charge interactions involve Lys 1088 in the B chain, while negative charge interactions are present. As indicated in Fig. 11A, pose 472 consisted of four regions: hydrophobic, hydrophilic, negative-charge interaction, positive-charge interaction, and the RMSD curves are shown on the left and right Y-axes in Fig. 11B. The pharmacokinetics of the 3TOP enzyme and pose 472 have been altered to glycine. The Root Mean Square Deviation (RMSD) of the 3TOP enzyme has stabilized to less than 30 ns, approximately 1.0. RMSD values exceeding 3 and a simulation time exceeding 30 ns indicate a change in the conformation (3.0 Å) of the 3TOP enzyme. The RMSD values for pose 472 indicate minimal conformational changes, reflecting the internal fluctuations of the ligand when aligned with the backbone frame curve of the 3TOP enzyme. The various configurations of pose 472 were analyzed by aligning it with the 3TOP enzyme (dark red line). The RMSD values exceed 3 Å when the simulation time exceeds 25 ns. Pose 472, previously confined, now expands beyond its original location. The root-mean-square fluctuations (RMSF) for each atom in the ligand are displayed in Fig. 11C, specifically in the RMSF of pose 472. The Root Mean Square Fluctuation (RMSF) calculated the average fluctuations of the ligand's heavy atoms squared deviation after aligning them with the reference frame over the entire simulation period. Pose 472's alignment on the enzyme showed variations in binding and values. The root mean square fluctuation (RMSF) of pose 472 when fitted onto the 3TOP enzyme ranges from less than 6 to 2 Å. The most prevalent atom is the oxygen atom located at pose 472 in the hydroxyl phenolic group of the seven atoms. The ligand contacts plot in Fig. 11D provides information on protein–ligand interactions. One bar chart displays the duration of ligand contact with particular amino acid residues during the simulation, while another chart illustrates how these contacts evolve over time. The diagram shows the H-bonds (green lines) between pose 472 and enzyme 3TOP, involving Asp 960, Glu 961, Kys 1088, Glu 1095, Arg 1097, Gly 1102, Thr 1103, Qln 1109, Glu 1178 in chain A, and Glu 1095, Thr 1101, and Asp 1107 in chain B. The residual amino acids of the 3TOp enzyme created two hydrogen bonds with the same atom at position 472: Lys 1088 in the A chain, Glu 1095 in the A chain, and Glu 1095 in the B chain. Fig. 11D illustrates an increase in the number of contacts between pose 472 and the 3TOP enzyme from 8 to 14 over a simulation period of 0–100 ns. Fig. 11E displays a simulation interaction diagram of the 3TOP enzyme, showing 472 contacts occurring between 0 and 100 ns during the simulation. Most interactions are identified through an orange color, including Asp 1107, Glu 1095: B chain, Gln 1109, Thr 1103, Arg 1097, Glu 1095, Lys 1088, Glu 961, and Asp 960: A chain. The properties of the ligand, such as polar surface area, solvent-accessible surface area, molecular surface area, number of intramolecular hydrogen bonds, radius of gyration, and ligand root-mean-square deviation from the initial conformation (RMSD), are visually represented in Fig. 11F. The mean values for RMSA, r-Gyr, intra-HB count, Mol SA, SASA, and PSA are 3 Å, 6 Å, 1 H-bond, 585 Å2, 450 Å2, and 440 Å2, respectively.
In silico antibacterial activity
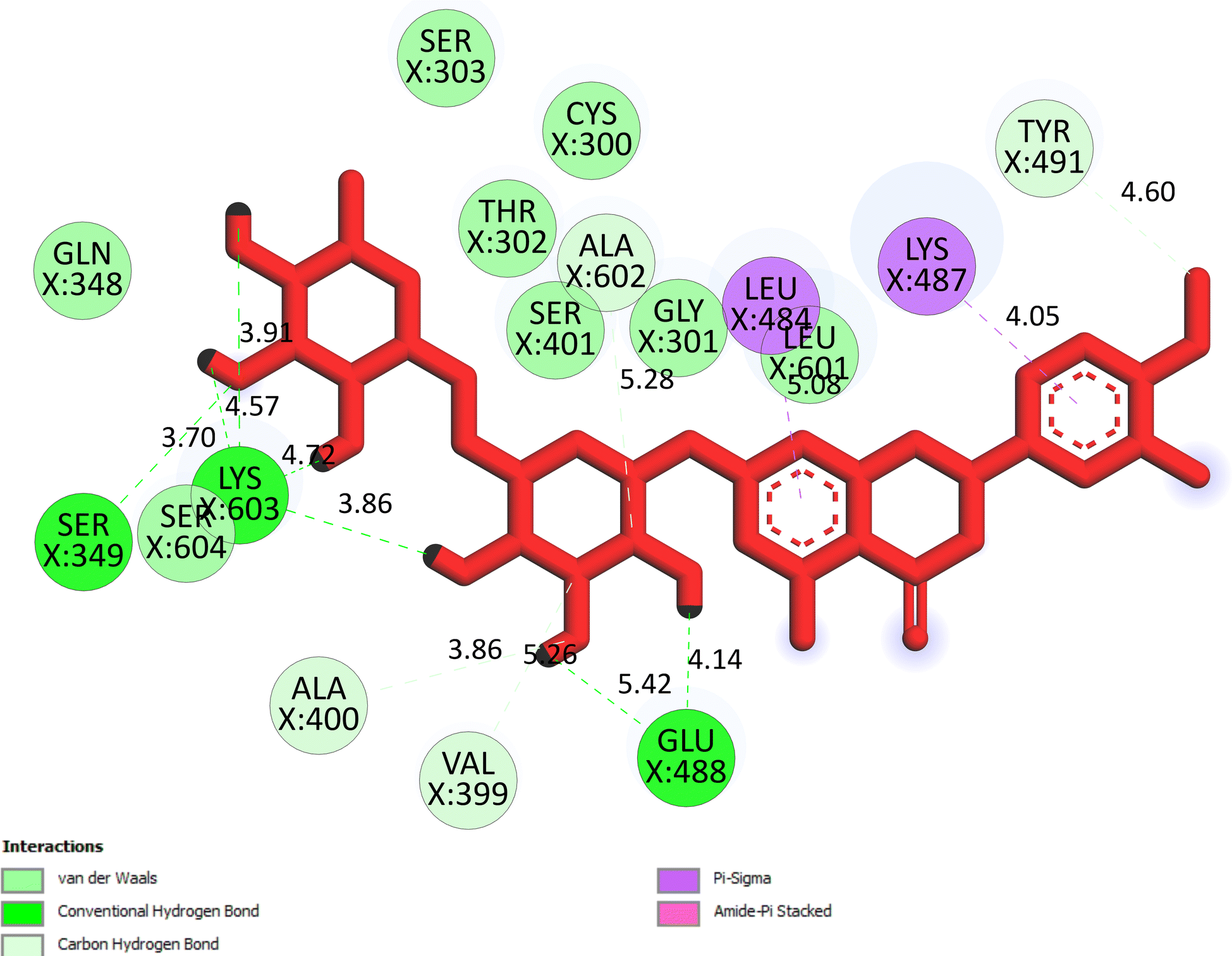
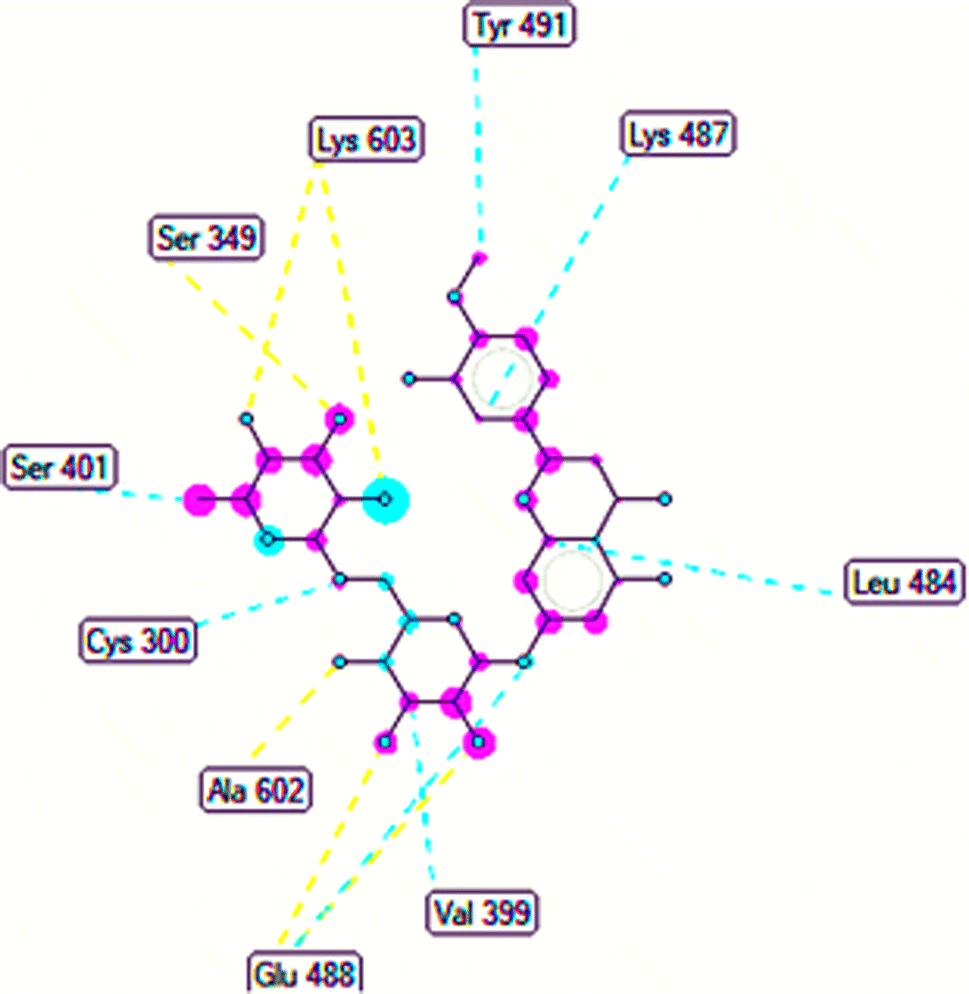
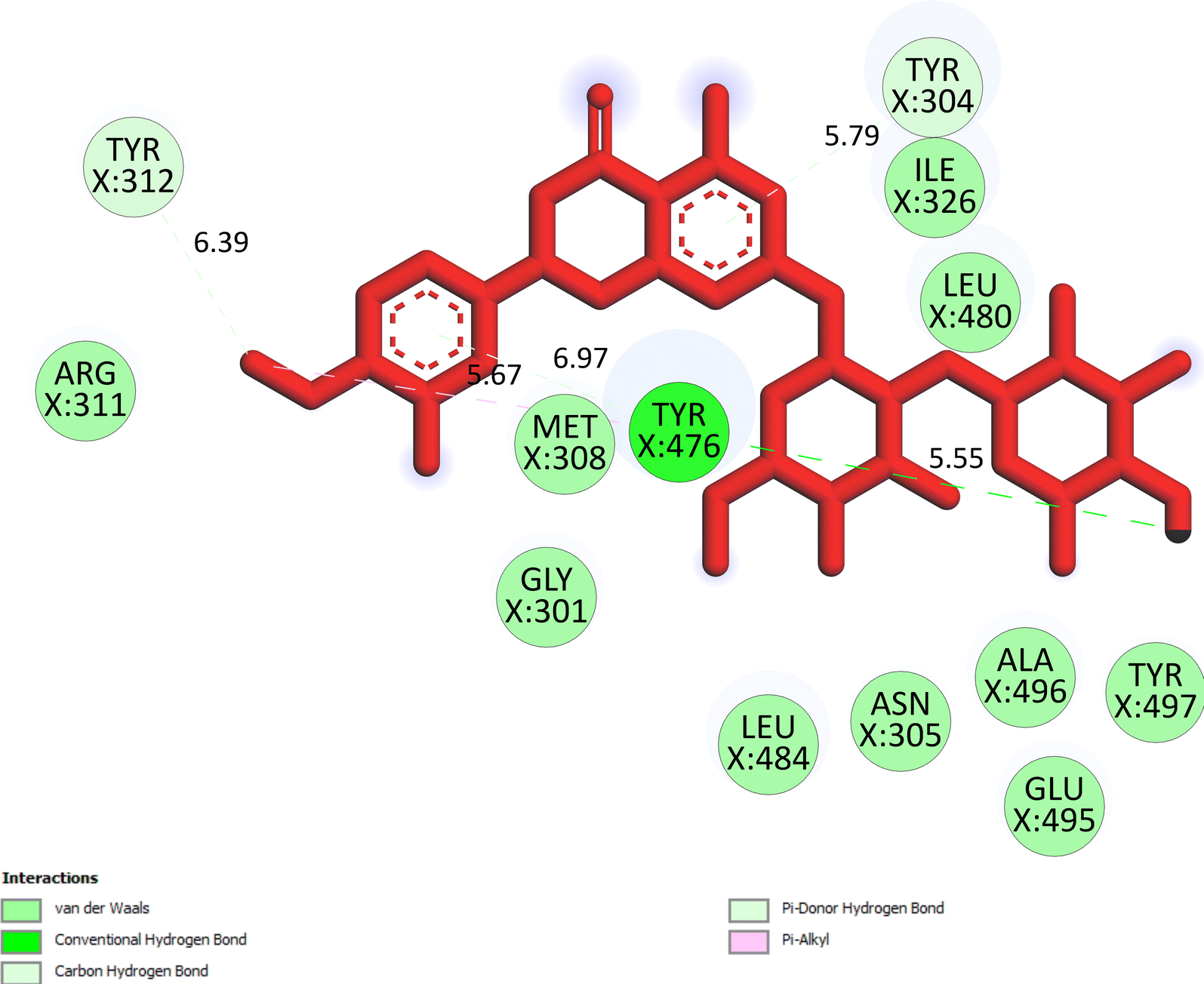
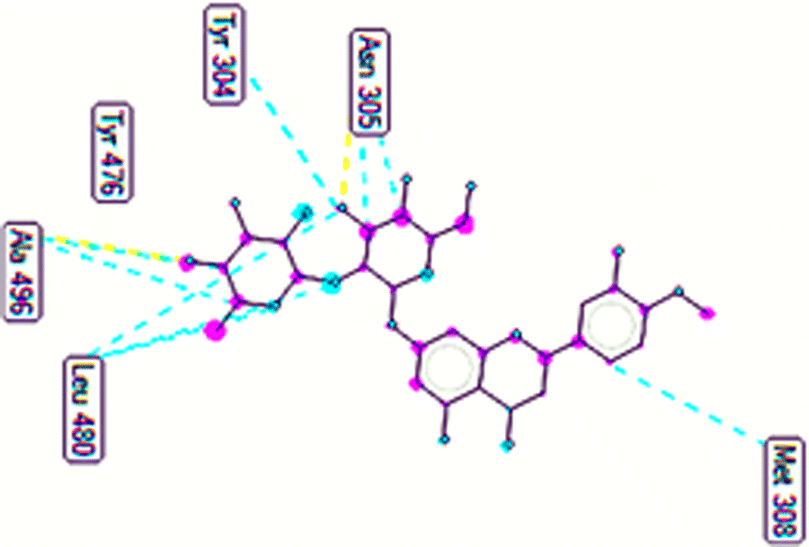
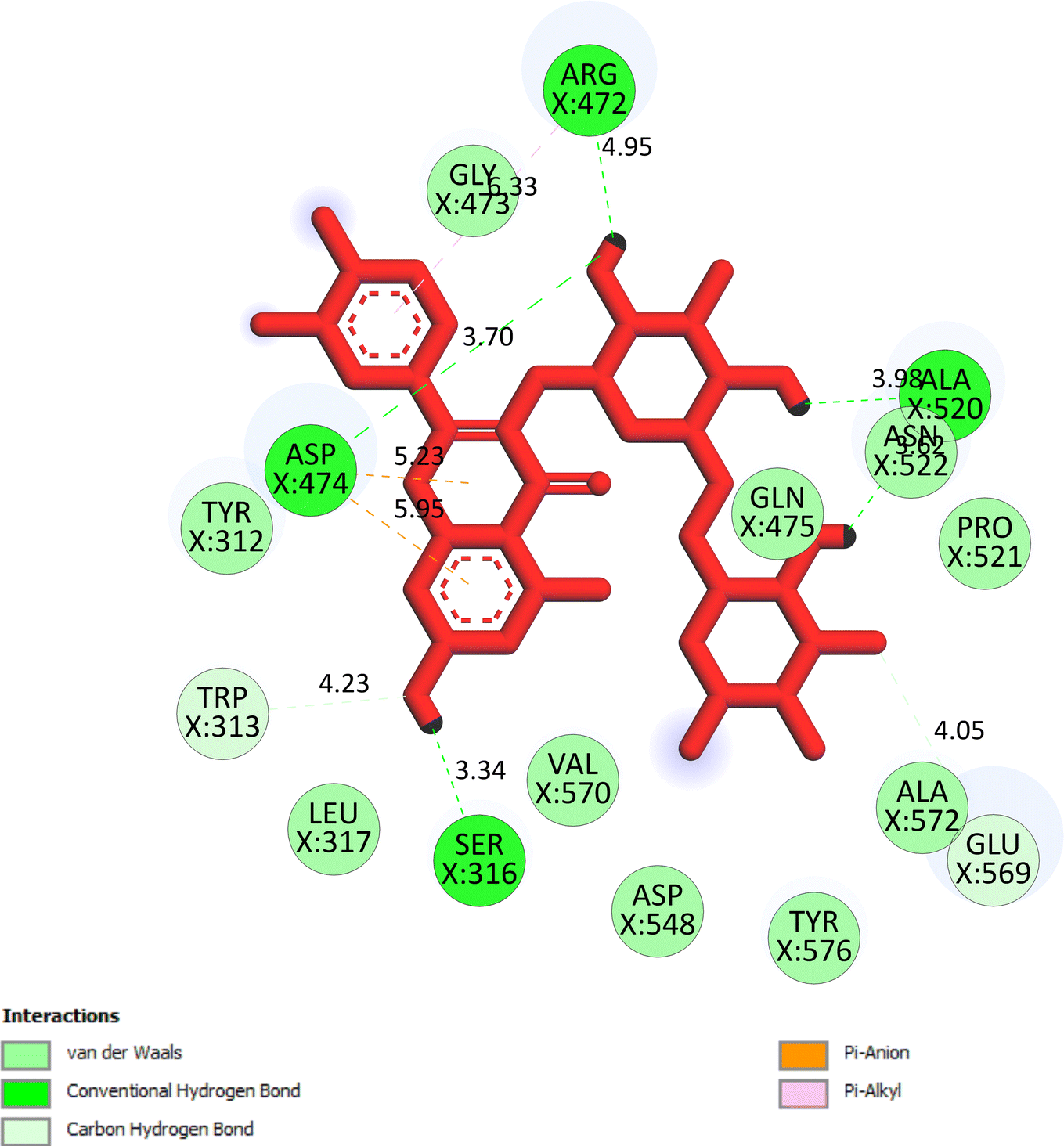
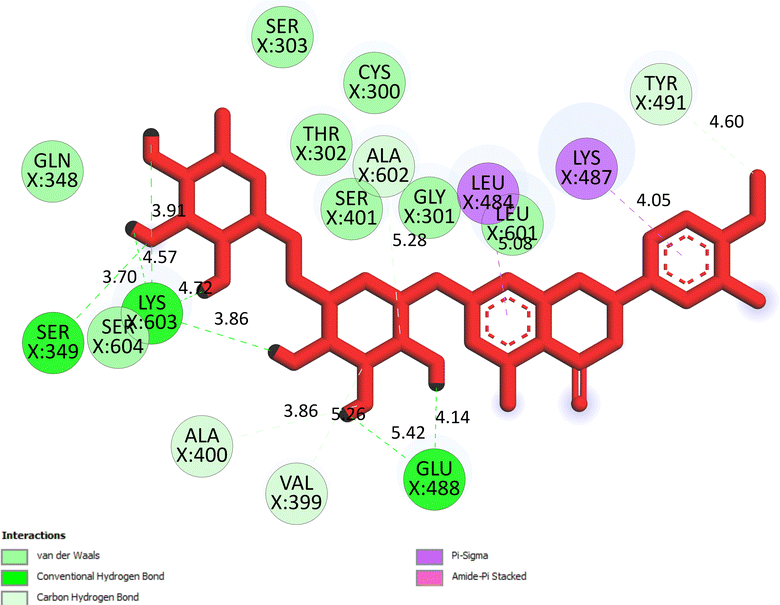
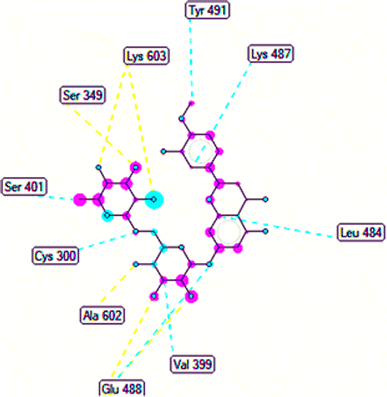
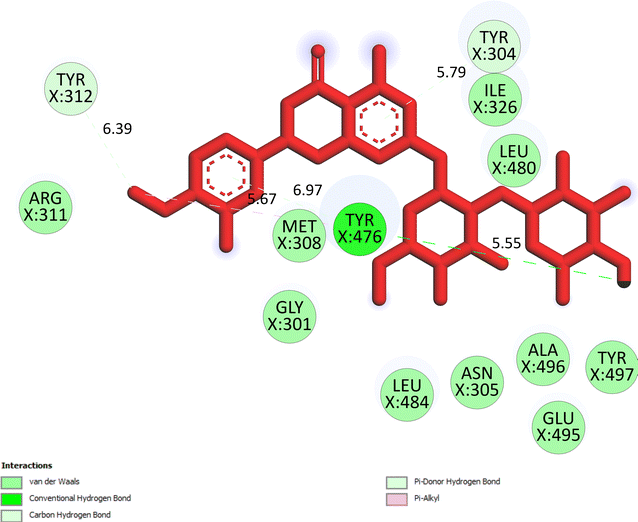
The important docking calculations for the best docking poses of entries 1–7 have been compiled in Table 3. Table 3 shows three compounds that have successfully docked to the crystal structure of the 2VF5 enzyme: compound 6 (pose 370), compound 7 (pose 362), and compound 5 (pose 280). Compound 370 exhibits the most favorable docking pose according to both the thermodynamic and ligand interaction models. Pose 370 is linked to the 2VF5 enzyme in the thermodynamic site with a free Gibbs energy of −7.57 kcal mol−1 and an inhibition constant of 2.83 μM. The pose also engaged in six hydrogen bonds with 2VF5, as shown in Table 3. The hydrogen bonding between pose 370:H–X:Lys 603:O is the strongest due to its shortest bonding length. The hydrogen bonds play a crucial role in the hydrophilic characteristics and metabolic processes. The interaction profile between poses 370 and 2VF5 has been documented in Fig. 12, 13 and Table 3. Fig. 12 illustrates the key interactions between pose 370 and 2VF5 on a single 2D diagram. Pose 370 had good interaction with 2VF5 due to the compatibility of the functional group, capping group (CA), and connecting unit (CU) with 2VF5. Hydrogen bonds are formed between the oxygen atoms of hydroxyl groups and hydroxyl methylene groups on pose 370 with Lys 603, Ser 349, and Glu 488 to identify the functional groups. Pi–sigma interactions between leucine 484 and lysine 487 aid in determining the capping group of this conformation. The linker in this pose is identified by carbon–hydrogen bonds from Tyr 491, Ala 602, and Val 399 to the carbon atoms of the methoxy group and the carbon atoms of the carbohydrate group in pose 370. Fig. 13 displays some of the secondary interactions occurring between pose 370 and 2VF5. The interactions consist of hydrogen bonds (Ser 349 and Lys 603 with atoms in pose 370), steric interactions (Tyr 491, Lys 487, Leu 484, Val 399, Cys 300, and Ser 401), and overlap interactions (violet circles) with atoms in pose 370. The greater diameters of violet circles indicate more intense overlap interactions between residual amino acids and atoms in pose 370. At pose 370, there are numerous steric, overlap, and hydrogen interactions. Altering the configuration of pose 370 without affecting the overall structure is challenging, and there are significant connections between pose 370 and 2VF5. Pose 370 is evaluated to have good interaction with 2VF5 in the ligand interaction model. Fig. 14, 15 and Table 3 display the interaction profile between pose 362 and 2VF5. It is anchored by pose 362 and the pocket enzyme 2VF5. The Gibbs free energy is −6.42 kcal mol−1, and the inhibition constant is 19.59 μM. It connected two hydrogen bonds from Tyr 476 to atomic oxygen on pose 362, as shown in Table 3 and Fig. 14 shows that pose 362 had favorable interactions with 2VF5, including hydrogen bonding between Tyr 476 and the oxygen atom of the hydroxyl methylene group on pose 362, pi–alkyl interactions between Met 308 and the carbon of the methoxy group on pose, and capping groups involving carbon–hydrogen bonds from Tyr 476 and Tyr 304 to aromatic rings for protein identification. Fig. 15 displays a ligand map illustrating the secondary interactions between the remaining amino acids on 2VF5 and atoms on pose 362. The interactions consist of hydrogen bonds between Asn 305 and Ala 496, steric interactions involving Asn 305, Met 308, Leu 480, Ala 496, and Tyr 304, and overlap interactions on pose 306 represented by violet circles. These interactions are weak due to the lower number of amino acids in this pose. The optimal docking conformation of compound 5, as shown in Table 3, pose 280, has successfully docked to 2VF5 with a free Gibbs energy value of −5.85 kcal mol−1 and an inhibition constant of 51.58 μM. Five hydrogen bonds have been established between active atoms on pose 280 and residual amino acids on 2VF5, with one of the hydrogen bonds specifically involving pose 280. H–X:Ser 316:O is the strongest due to its minimal bond length. Fig. 16 shows that pose 280 has effectively interacted with 2VF5 in the ligand interaction model. This interaction is facilitated by three components of pose 280: Asp 474, Ser 316, Arg 472, Ala 520, and Asn 522, which interact with 2VF5 through functional groups; Asp 474 forms pi–cation bonds with the aromatic and ketone groups on pose 280, acting as a capping group, and Arg 472 contributes a pi–alkyl interaction with the aromatic ring of pose 280 as part of the linker section. The ligand interaction model ranks the poses as follows: pose 370 (compound 6) > pose 362 (compound 7) > pose 280 (compound 5). Poses 305, 387, 136, and 43, representing compounds 2, 3, 4, and 1, respectively, demonstrated poor interactions with 2VF5 due to unfavorable donor–donor and acceptor–acceptor interactions, as observed in Fig. S14–S17.† The adverse interactions are located at Thr 302 (pose 305), Cys 300 (pose 387), Ala 602, Gly 301 (pose 36), and Gln 348 (pose 43), as depicted in Fig. S15–S18† and Table 3.
Table 3 The significant docking results of docking poses to 2VF5 enzyme: PDB
| Entry |
Posea |
Free Gibbs energyb |
Kic |
The number of hydrogend |
The character and bond lengthe |
Describe |
| The calculation results from AutoDockTools-1.5.7 (ATD). The calculation results from AutoDockTools-1.5.7 (ATD). The calculation results from AutoDockTools-1.5.7 (ATD). Building from Discovery Studio 2021 Client software. Building from Discovery Studio 2021 Client software. |
| 6 |
370 |
−7.57 |
2.83 |
6 |
X:Ser 349:O – pose 370:O, (2.92 Å); pose 370:H–X:Lys 603:O, (1.86 Å); pose 370:H–X:Glu 488:O, (2.14 Å); pose 370:H–X:Lys 603:O, (2.21 Å); pose 370:H–X:Lys 603:O, (2.08 Å); pose 370:H–X:Glu 488:O, (1.92 Å) |
Good interactions in ligand interaction model |
| 7 |
362 |
−6.42 |
19.59 |
2 |
X:Tyr 476:O – pose 362:O, (2.4 Å); pose 362:H–X:Tyr 476:O, (2.22 Å) |
Good interactions in ligand interaction model |
| 5 |
280 |
−5.85 |
51.58 |
5 |
X:Ser 316:O – pose 280:O, (2.82 Å); pose 280:H–X:Ser 316:O, (1.88 Å); pose 280:H–X:Arg 472:O, (2.01 Å); pose 280:H–X:Ala 520:O, (1.88 Å); pose 280:H–X:Ala 520:O, (2.44 Å) |
Good interactions in ligand interaction model |
| 2 |
305 |
−6.90 |
8.74 |
7 |
X:Thr 302:N – pose 305:O, (3.06 Å); X:Ser 349:N – pose 305:O, (3.20 Å); X:Ser 349:O – pose 305:O (2.87 Å); X:Ser 401:N – pose 305:O (2.78 Å); X:Lys 603:N – pose 305:O (3.02 Å); pose 305:H–X:Val 399:O (2.22 Å); pose 305:H–X:Val 399:O (2.11 Å) |
Unfavorable donor–donor and acceptor–acceptor: Thr 302 |
| 3 |
387 |
−5.92 |
45.57 |
8 |
X:Thr 302:N – pose 370:O (2.95 Å); X:Ser 347:O – pose 370:O (3.21 Å); X:Gln 348:N – pose 370:O (2.96 Å); X:Ser 349:N – pose 370:O (2.94 Å); X:Ser 349:O – pose 370:O (2.75 Å); X:Ser 401:N – pose 370:O (2.98 Å); pose 370:H–X:Ser 349:OG (2.04 Å); pose 370:H – X:Lys 603:O (1.93 Å) |
Unfavorable donor–donor and acceptor–acceptor: Cys 300 |
| 4 |
136 |
−5.90 |
47.64 |
8 |
X:Gly 301:N – pose 136:O (2.74 Å); X:Gln 348:N – pose 136:O (2.96 Å); X:Ser 349:N – pose 136:O (3.01 Å); X:Ser 349:O – pose 136:O (2.81 Å); X:Thr 352:O – pose 136:O (3.07 Å); pose 136:H–X:Glu 488:O (2.00 Å); pose 136:H – X:Glu 488:O (2.11 Å); pose 136:H–X:Ser 349:O (2.02 Å) |
Unfavorable donor–donor and acceptor–acceptor: Ala 602, and Gly 301 |
| 1 |
43 |
−5.40 |
110.8 |
7 |
X:Gly 301:N – pose 43:O (3.06 Å); X:Tyr 304:OH – pose 43:O (3.12 Å); X:Gln 348:N – pose 43:O (2.73 Å); X:Tyr 476:OH – pose 43:O (2.93 Å); X:Lys 487:N – pose 43:O (3.15 Å); pose 43:H–X:Gly 301:O (2.04 Å); pose 43:H–X:Tyr 476:OH (2.42 Å) |
Unfavorable donor–donor and acceptor–acceptor: Gln 348 |
 |
| | Fig. 12 The significant ligand interactions between pose 370 and 2VF5 in the 2D diagram. | |
 |
| | Fig. 13 The ligand map forming between pose 370 and 2VF5. | |
 |
| | Fig. 14 The significant ligand interactions between pose 362 and 2VF5 in the 2D diagram. | |
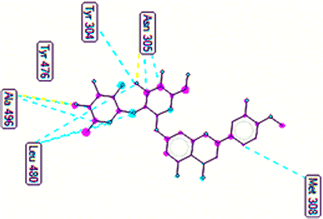 |
| | Fig. 15 The ligand map forming between pose 362 and 2VF5. | |
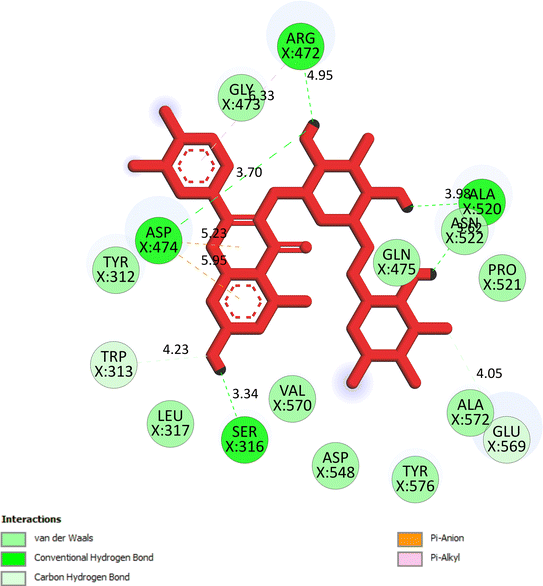 |
| | Fig. 16 The significant ligand interactions between pose 280 and 2VF5 in the 2D diagram. | |
Conclusions
Seven flavonoid glycosides have been isolated from the polar fraction of the aerial parts of Mussaenda recurvata. Ecurvoside (1) was a newly discovered compound among them. Six compounds identified as astragalin, isoquercitrin, nicotiflorin, rutin, hesperidin, and neohesperidin were analyzed for their structures. Individual compounds exhibited moderate inhibition against alpha-glucosidase. Ecurvoside was identified as an optimal candidate through molecular docking and molecular dynamics simulations, demonstrating strong interactions with the crystal structure of the 3TOP enzyme. The complex of 3TOP and pose 472 or compound 1 remained the most stable for a duration ranging from 40 to 100 ns. Compound 6 exhibited superior inhibition against the Trichophyton mentagrophytes fungus compared to other compounds. In the docking model, compound 6 exhibited better inhibition than compounds 7 and 5. Compounds 1–4 revealed poor in silico activity.
Experimental section
General
NMR spectra were recorded on a Bruker Avance III spectrometer (500 MHz for 1H NMR and 125 MHz for 13C NMR) with TMS as internal standard. HRESIMS was recorded using a MicrOTOF-Q mass spectrometer on an LC-Agilent 1100 LC-MSD Trap spectrometer. Thin-layer chromatography (TLC) was carried out using precoated silica gel 60 F254 or 60 RP-18 F254S (Merck). Spots were visualized by applying a 10% H2SO4 solution, followed by heating. Gravity column chromatography was performed on silica gel 60 (0.040–0.063 mm, Himedia). Saccharomyces cerevisiae α-glucosidase (E.C. 3.2.1.20) and acarbose were obtained from Sigma-Aldrich Co.
Plant material
Aerial parts of M. recurvata were collected from Khanh Hoa province, Vietnam in November 2019. The scientific name of the plant was authenticated by the botanist Dang Van Son, Institute of Tropical Biology, Vietnam Academy of Science and Technology. A voucher specimen (VNM_Dang356) was deposited in the VNM Herbarium, Institute of Tropical Biology, Vietnam Academy of Science and Technology.
Extraction and isolation of the EtOAc extract
The dried powder of M. recurvata aerial parts (6.0 kg) was extracted with MeOH (3 × 50 L) at room temperature by maceration method, which was further filtered, and solvents were removed under low pressure to obtain the crude extract (420 g). This extract was separated by liquid–liquid extraction procedures to yield n-hexane (95.0 g), CH2Cl2 (130.0 g), and EtOAc (80.0 g) extracts together with the water-soluble layer (110 g). The EtOAc extract was applied to silica gel column chromatography (CC) using the gradient system of CH2Cl2–MeOH (50:1–0:1, v/v), affording eight fractions E1–E8. Fraction E4 (16.0 g) was separated by silica gel CC and eluted with the gradient system of CH2Cl2–MeOH (20:1–0:1, v/v) to give six fractions E4.1–4.6. The fraction E4.3 (3.6 g) was rechromatographed by silica gel CC and eluted with solvent n-hexane–CH2Cl2–MeOH (1:5:1, v/v/v) to afford compounds 2 (9.0 mg) and 3 (11.0 mg). Fraction E6 (15.0 g) was chromatographed on a silica gel column eluting with a gradient of CH2Cl2–MeOH (10:1 → 0:1, v/v) to give five fractions (E6.1–6.5). Compound 1 (8.0 mg) was obtained from fraction E6.2 (120 mg) by two consecutive silica gel CC: a normal-phase column with n-hexane–CH2Cl2–MeOH (1:5:1 to 1:2:1, v/v/v) as the mobile phase and a C-18 reverse phase column using a solvent system of MeOH–H2O (3:1, v/v). Fraction E6.4 was chromatographed using a C-18 reverse phase silica gel CC eluted with MeOH–H2O (1:1, v/v) to obtain compounds 4 (12 mg) and 5 (9 mg). Fraction E7 (9.5 g) was subjected to silica gel CC, eluted with a gradient system of EtOAc–MeOH (10:1 to 0:10, v/v) to provide fractions E7.1–7.5. Subfraction E7.3 was chromatographed on silica gel CC with the eluent as EtOAc–MeOH–H2O (10:1:0.1 → 1:1:0.1, v/v/v) to give four fractions E7.3.1–7.3.4. Compounds 6 (6.0 mg) and 7 (8.0 mg) were obtained from fraction E7.3.3 on a C-18 reverse phase silica gel CC, eluted with MeOH–H2O (1:2, v/v).
Ecurvoside (1). Colorless gum; 1H NMR (DMSO-d6, 500 MHz) and 13C NMR (DMSO-d6, 125 MHz): see Table 1. HRESIMS m/z 739.1856 (calcd for [C36H36O17–H]−, 739.1874).
Alpha-glucosidase inhibition
The α-glucosidase inhibitory activity of compounds 1–7 was determined using a method adapted from a previous method.42 The sample was analyzed in triplicate at five different concentrations around the IC50 values, and the mean values were retained.
Antimicrobial activity
Antimicrobial activities of compounds 1–7 were screened against the bacteria Pseudomonas aeruginosa, methicillin-resistant Staphylococcus aureus (MRSA), and Streptococcus faecalis (Strep. faecalis) and fungi Candida albicans (C. albicans), Trichophyton mentagrophytes (T. mentagrophytes), and Microsporum gypseum (M. gypseum) according to the published method.43
Molecular docking study
The general docking of conformation of compound 1 and acarbose, one small ligand in 3TOP enzyme: PDB, were docked to 3TOP enzyme followed article.43 For docking, the optical energy of compound 1 or acarbose was determined by Avogadro software and saved as a *.pdb file, and 3TOP enzyme was collected from Protein Data Bank. From the enzyme, the small ligand acarbose and molecular waters were removed, hetero atoms and missing amino acids were checked using SPDBV_4.10 software, and the active center on the ligand was determined by Discovery Studio 2021 Client software (DSC).44 The grid parameters were saved in a dock.gpf file and set up by the numbers of grid points in X, Y, Z of 50, 50, 50, respectively, with a spacing of 0.400 Å and active center coordinates of (−41.385, 12.152, −16.311) corresponding to (X, Y, Z), respectively. AutoDockTools-1.5.7 package was used to calculate the docking of ligand compound to receptor or enzyme and build the best conformation ligand or the best pose to the 3TOP enzyme by DSC. The ligand map showed the secondary interactions between the best pose docking and 3TOP enzyme by Molegro Molecular Viewer (MMV) software version 2.5. The H-bonding interactions between the best docking pose and 3TOP were performed by PyMOL software version 2.5.4.
Molecular dynamic simulation
Maestro, Schrodinger Suite version 2018 version 4 was used to perform the molecular dynamic model45,46 in a Linux environment. Protein Preparation Wizard, Desmond system builder, and molecular dynamics are the three important parameters that should be configured. These parameters, such as assigning bond ordering, adding hydrogens, generating disulfide bonds, capping termini, and removing waters below 5 Å, must be chosen during protein manufacturing. All these steps were finished in protein wizard: interactive H-bond optimizer: selecting the interactive optimizer, assessing the network, and optimizing. We execute two items in the Desmond system builder menu, namely, solvation and ions. Predefined functions in the solvation panel include selecting SPC (water molecules as solvation), box form orthorombic, buffer, setting distances of 10 Å, selecting to show border box, clicking to minimize menu, and running. OPLS2005 is the force field parameter. In Desmond system builder's ion tab, the ion table is selected, the neutralize button is clicked, the salt concentration is entered as 0.15 M NaCl, and the ion tab function is run. Finally, three parameters are employed to start molecular dynamics in the menu: (1) choose “load from workspace”, (2) enter 100 ns in the simulation time box, (3) choose a temperature of 300 K and a pressure of 1.01325 bar, (4) select the number of CPUs or GPUs supported by the Desmond version, and (5) click run in the tab to start simulation and modelling.47–49
Author contributions
Conceptualization, methodology, investigation, writing – original draft preparation, writing – review and editing: Tran Thi Ngoc Mai, Tran Nguyen Minh An, Mai Dinh Tri; resources, data curation, software, formal analysis: Phan Nhat Minh, Nguyen Tan Phat, Thuc Huy Duong, Van Son Dang, Nguyen Van Hue. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conficts to declare.
Acknowledgements
This research is funded by Vietnam National Foundation for Science and Technology Development (NAFOSTED) under grant number 104.01-2018.353.
References
- B. L. Staker, M. D. Feese, M. Cushman, Y. Pommier, D. Zembower, L. Stewart and A. B. Burgin, J. Med. Chem., 2005, 48(7), 2336 CrossRef CAS PubMed.
- T. N. M. An, N. V. Cuong, N. M. Quang, T. V. Thanh and M. Alam, ChemistrySelect, 2020, 5(21), 6339 CrossRef CAS.
- T. N. M. An, T. P. Pham, N. M. Quang, V. S. Nguyen, N. V. Cuong, V. T. Le, D. T. Mai, M. Alam and V. T. Pham, Curr. Org. Synth., 2019, 17(2), 151 CrossRef PubMed.
- T. H. Duong, A. P. Devi, T. N. M. An, H. V. T. Phan, N. V. Huynh, J. Sichaem, H. D. Tran, M. Alam, T. P. Nguyen, H. H. Nguyen, W. Chavasiri and T. C. Nguyen, Bioorg. Med. Chem. Lett., 2020, 30(17), 127359 CrossRef CAS.
- F. U. Eze, U. C. Okoro, D. I. Ugwu and S. N. Okafor, Front. Chem., 2019, 7, 634 CrossRef CAS PubMed.
- N. Uwabagira, B. K. Sarojini, M. K. Shankar and R. S. Gani, SN Appl. Sci., 2019, 1(11), 1375 CrossRef CAS.
- E. W. Bell and Y. Zhang, J. Cheminf., 2019, 11(1), 40 Search PubMed.
- K. Sargsyan, C. Grauffel and C. Lim, J. Chem. Theory Comput., 2017, 13(4), 1518 CrossRef CAS PubMed.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindahl, SoftwareX, 2019, 1–2, 19 Search PubMed.
- P. Chantaranothai, Thai For. Bull. (Bot.), 2015, 43, 51 Search PubMed.
- M. Chowdury, M. Alam, S. Chowdhury, M. Biozid, M. Faruk, M. Mazumdar and A. Chowdhury, Eur. J. Med. Plants, 2015, 10, 1 CrossRef.
- Y. Takeda, H. Nishimura and H. Inouye, Phytochemistry, 1997, 16, 1401 CrossRef.
- W. Zhao, J. Xu, G. Qin and R. Xu, Phytochemistry, 1995, 39, 191 CrossRef CAS.
- W. Zhao, P. Wang, R. Xu, G. Qin, S. Jiang and H. Wu, Phytochemistry, 1996, 42, 827 CrossRef CAS PubMed.
- V. K. Thu, N. X. Bach, L. T. Anh, D. T. Trang, N. X. Nhiem, B. H. Tai, P. V. Kiem, C. V. Minh, S. Park, Y. Seo, W. Namkung and S. H. Kim, Phytochem. Lett., 2019, 33, 39 CrossRef CAS.
- W. Zhao, J. L. Wolfender, K. Hostettmann, K. Cheng, R. Xu and G. Qin, Phytochemistry, 1998, 45, 1073 CrossRef.
- V. K. Thu, N. X. Bach, C. T. Dung, N. T. Cuong, T. H. Quang, Y. C. Kim, H. Oh and P. V. Kiem, Nat. Prod. Res., 2021, 35, 4126 CrossRef CAS PubMed.
- W. Zhao, G. Yang, R. Xu and G. Qin, Phytochemistry, 1996, 41, 1553 CrossRef CAS.
- Y. Li, W. Qiao and K. Yuan, J. Med. Plants Res., 2011, 5, 1789 Search PubMed.
- B. Dinda, S. Debnath, S. Majumder, S. Arima, N. Sato and Y. Harigaya, Indian J. Chem., 2006, 44B, 2362 Search PubMed.
- B. Dinda, S. Majumder, S. Arima, N. Sato and Y. Harigaya, J. Nat. Med., 2008, 62, 447 CrossRef CAS PubMed.
- S. Shanthi and R. Radha, Phcog. J., 2020, 12(3), 630–635 CrossRef CAS.
- L. T. Huong, T. P. Thao and D. T. Trang, et al., Nat. Prod. Res., 2021, 36(18), 4591–4598 CrossRef.
- M. D. Tri, T. T. M. Tram, L. H. Ngoc, T. N. M. An, N. T. Phat, P. N. Minh, N. V. Kieu, D. V. Son, T. P. Nguyen, T. T. N. Mai and T. H. Duong, Nat. Prod. Res., 2023, 37, 2303–2310 CrossRef CAS PubMed.
- M. D. Tri, N. T. Phat, P. N. Minh, M. T. Chi, B. X. Hao, T. N. M. An, M. Alam, N. V. Kieu, V.-S. Dang, T. T. N. Mai and T. H. Duong, RSC Adv., 2023, 13, 5324–5336 RSC.
- K. N. Ha, T. V. A. Nguyen, D. T. Mai, N. M. A. Tran, N. H. Nguyen, G. V. Vo, T. H. Duong and H. T. Nguyen, Saudi J. Biol. Sci., 2022, 29, 1029–1042 CrossRef CAS PubMed.
- H. Cedeño, S. Espinosa, J. M. Andrade, L. Cartuche and O. Malagón, Molecules, 2019, 24(23), 4267 CrossRef.
- Y. S. Jadeja, K. M. Kapadiya, H. J. Jebaliya, A. K. Shah and R. C. Khunt, Magn. Reson. Chem., 2017, 55, 589–594 CrossRef CAS.
- N. Xia, W. Wan, S. Zhu and Q. Liu, Front. Bioeng. Biotechnol., 2020, 8, 1025 CrossRef.
- M. Zheng, S. Lu and J. Xing, Food Chem., 2021, 336, 127539 CrossRef CAS PubMed.
- S. Mussadiq, N. Riaz, M. Saleem, M. Ashraf, T. Ismail and A. Jabbar, J. Asian Nat. Prod. Res., 2013, 15, 708–716 CrossRef CAS PubMed.
- Y. Ranarivelo, A. L. Skaltsounis, M. Andriantsiferana and F. Tillequin, Ann. Pharm. Fr., 1990, 48(5), 273–277 CAS.
- P. Mala, G. A. Khan, R. Gopalan, D. Gedefaw and K. Soapi, Biosci. Rep., 2022, 42, BSR20220461 CrossRef CAS PubMed.
- M. A. A. Sikder, R. B. Rashid, F. Islam, A. K. M. N. Hossian, A. B. Siddique, S. Kabir, M. R. Haque, M. S. Rahman and M. A. Rashid, Orient. Pharm. Exp. Med., 2013, 13, 327–332 CrossRef.
- H. C. Hong, S. L. Li, X. Q. Zhang, W. C. Ye and Q. W. Zhang, Chin. Med., 2013, 8(1), 1–7 CrossRef PubMed.
- D. F. Pereira, L. H. Cazarolli, C. Lavado, V. Mengatto, M. S. R. B. Figueiredo, A. Guedes, M. C. Pizzolatti and F. R. M. B. Silva, Nutrition, 2011, 27(11–12), 1161–1167 CrossRef PubMed.
- Y. Cai, L. Wu, X. Lin, X. Hu and L. Wang, Ind. Crops Prod., 2020, 154, 112673 CrossRef CAS.
- S. Dubey, A. Ganeshpurkar, A. Ganeshpurkar, D. Bansal and N. Dubey, Future J.
Pharm. Sci., 2017, 3, 158–162 CrossRef.
- K. C. Shin, H. K. Nam and D. K. Oh, J. Agric. Food Chem., 2013, 61, 11532–11540 CrossRef CAS PubMed.
- G. Giannini, W. Cabri, C. Fattorusso and M. Rodriquez, Future Med. Chem., 2012, 4(11), 1439–1460 CrossRef CAS.
- G. V. Vo, T. H. T. Nguyen, T. P. Nguyen, T. H. T. Do, N. M. A. Tran, H. T. Nguyen and T. T. Nguyen, Saudi Pharm. J., 2022, 30, 1301–1314 CrossRef CAS PubMed.
- T. H. Do, T. H. Duong, H. T. Nguyen, T. H. Nguyen, J. Sichaem, C. H. Nguyen, H. H. Nguyen and N. P. Long, Molecules, 2022, 27, 2871–2879 CrossRef CAS PubMed.
- N. D. T. Ha, T. Phuong, N. V. Cuong and T. N. M. An, ChemistrySelect, 2023, 8, e202300246 CrossRef.
- S. U. Khan, N. Ahemad, L. H. Chuah, R. Naidu and T. T. Htar, Prog. Drug. Discov. Biomed. Sci., 2020, 3(1), a0000054 Search PubMed.
- J. Ash and D. Fourches, J. Chem. Inf. Model., 2017, 57, 1286–1299 CrossRef CAS PubMed.
- J. Bhachoo and T. Beuming, Methods Mol. Biol., 2017, 1561, 235–254 CrossRef CAS PubMed.
- D. E. Shaw Research, Desmond Version 3.0 Tutorial, 2011 Search PubMed.
- M. Aamir, V. K. Singh, M. K. Dubey, M. Meena, S. P. Kashyap, S. K. Katari, R. S. Upadhyay, A. Umamaheswari and S. Singh, Front. Pharmacol., 2018, 9, 1038 CrossRef CAS PubMed.
- N. Uwabagira, B. K. Sarojini, M. K. Shankar and R. S. Gani, SN Appl. Sci., 2019, 1, 1375 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence bc,
Thuc Huy Duong*d,
Tran Nguyen Minh An
bc,
Thuc Huy Duong*d,
Tran Nguyen Minh An