
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCopper-catalyzed synthesis of primary amides through reductive N–O cleavage of dioxazolones†
Hyeonwoong Baeab,
Jinhwan Parkab,
Rahyun Yoonb,
Seunghoon Lee *ab and
Jongwoo Son*ab
*ab and
Jongwoo Son*ab
aDepartment of Chemical Engineering (BK21 FOUR Graduate Program), Dong-A University, 37 Nakdong-Daero 550beon-gil, Saha-gu, Busan 49315, South Korea. E-mail: sonorganic@dau.ac.kr
bDepartment of Chemistry, Dong-A University, 37 Nakdong-Daero 550beon-gil, Saha-gu, Busan 49315, South Korea
First published on 20th March 2024
Abstract
A new method for the synthesis of primary amides is developed, in which dioxazolones are treated with a copper catalyst under mild reaction conditions. A broad scope of dioxazolones is exhibited as well as dioxazolones containing biologically active structural motifs. These robust and mild reaction conditions allow the transformation of dioxazolones to primary amides, in which sensitive functional groups such as hydroxyl, aldehyde, trialkylsilyl, and unsaturated carbon units are tolerated with excellent chemoselectivity.
Amides are among the most invaluable functional groups in organic chemistry and have been extensively utilized in drug discovery, functional materials, and natural products.1–5 In particular, primary amides and carbonyl groups directly bearing a free-NH2 unit, can serve as key synthetic intermediates in a wide range of N-substituted amides through catalytic C–N bond formation using transition metals such as copper and palladium.6–8 Several conventional strategies for the preparation of primary amides rely on the use of readily available carboxylic acid sources, ammonia treatment, activating agents such as thionyl chloride and oxalyl chloride, peptide-coupling agents, and phosphine-fluoride reagent system (Scheme 1A).9–15 Moreover, catalytic hydration of organic nitriles has been explored to avoid undesired hydrolysis to carboxylic acids using transition metal catalysts such as cobalt, ruthenium, palladium, rhodium, and platinum.16–18 However, these approaches are challenging because of their low functional group tolerance under harsh reaction conditions and reduced sustainability, which can produce toxic chemicals that may negatively impact environmental systems.19 Instead of carboxylic acids, hydroxamic acids and their derivatives are widely used to access primary amides (Scheme 1B). Several synthetic approaches have been developed from Weinreb amides to access primary amides, facilitated through weak N–O bond cleavage (55–65 kcal mol−1) using stoichiometric amounts of metal-based reducing agents such as Mo(CO)6,20–22 TiCl3,23–25 SmI2,26,27 Zn,28,29 and Li/NH3(l).30,31 Moreover, catalytic hydrogenation by the Raney-Ni32 and Pd/C33 protocols can further reduce other functional groups, such as carbonyls and unsaturated carbon motifs. More recently, organic photocatalytic systems have proven effective in cleaving N–O bonds through a single-electron transfer process, avoiding the use of stoichiometric reductants under mild conditions.34 In addition, O-acylated hydroxamic acids bearing weak N–O bonds (45 kcal mol−1) were effectively converted to primary amides by copper(II) salts35 and ruthenium catalysts.36 Moreover, a recent study showcased hydroxamic acids as amide sources, easily synthesized from carboxylic acids and hydroxylamine using 1,1′-carbonyldiimidazole, would be reduced to corresponding primary amides using thioacetic acid, NH4HCO3 in ethanol.37 In the past decade, dioxazolones, first invented by Beck and coworkers,38 have been widely studied in the research area of catalytic C–H amidations using precious transition metals such as iridium and rhodium.39–48 Despite the development of dioxazolones in transition metal catalysis, an alternative catalytic system is recognized to set the stage for a more convenient reaction regime that can be utilized under milder reaction conditions. Copper, one of the 3d transition metals, getting more attentions owing to its cost-effectiveness, low toxicity, and earth abundance.49 Recently, several decarboxylative N-functionalizations of dioxazolones using copper salts have been reported (Scheme 1C). First, the research group of de Bruin reported a preparation of N-acyl amidines from three-component reaction of dioxazolones, terminal alkynes, and amines using copper(I) catalyst.50 In addition, the enantioselective hydroamidation of styrene motifs was studied using a copper-hydride catalytic system.51 More recently, decarboxylative N-arylation of dioxazolones using boronic acids has been reported.52 All these transformations of dioxazolones using copper salts have been proposed to engage the formation of copper(III) species from terminal alkynes, alkenes, and arylboronic acids.
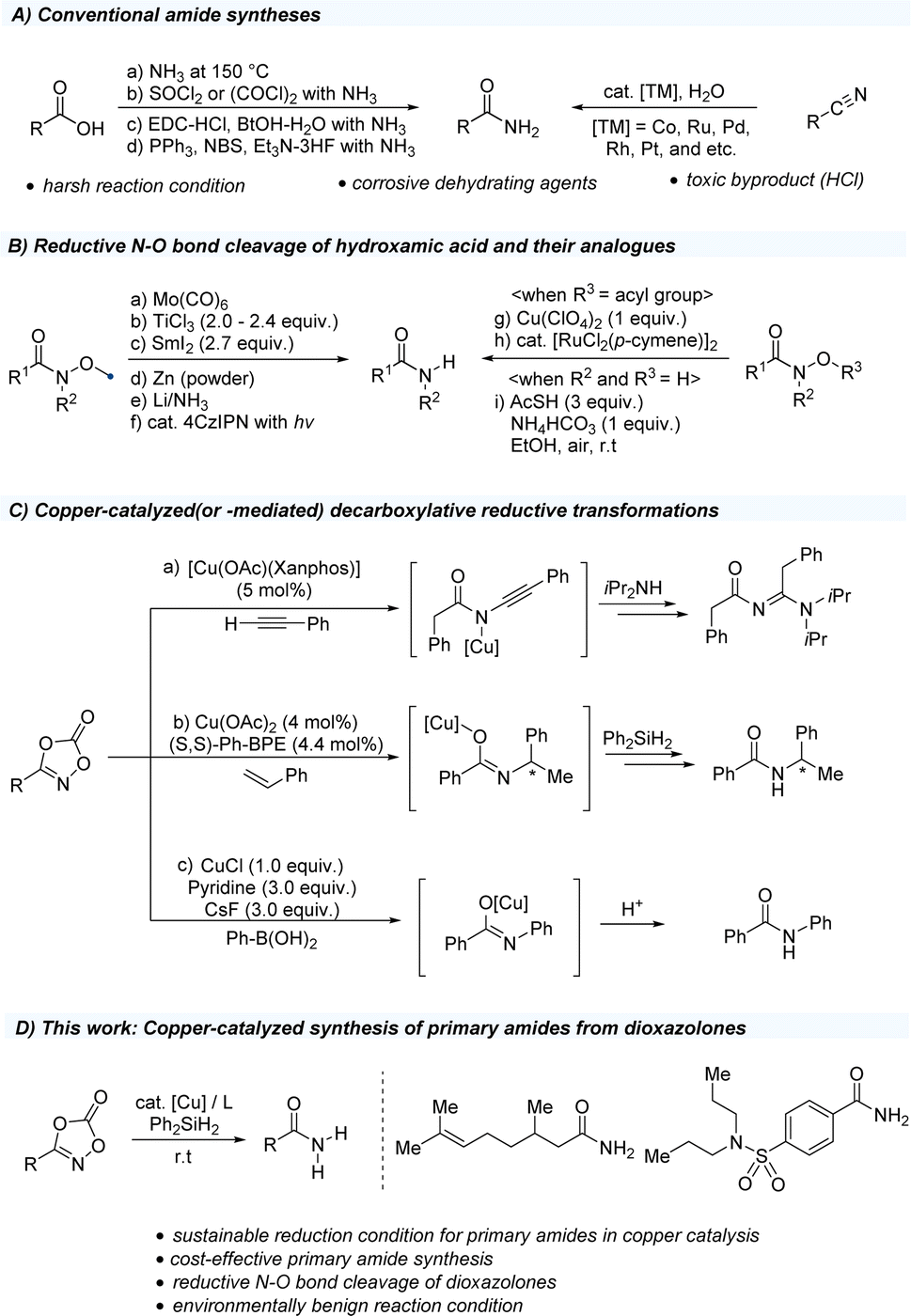 | ||
| Scheme 1 Synthetic methodology for primary amides. | ||
Based on these previously reported transformations of dioxazolones, we anticipated that if we use silane as a hydrogen source, the reaction might undergo N–O bond cleavage, resulting in the formation of primary amides (Scheme 1D). Herein, we report the first example of the synthesis of primary amides using dioxazolones as a convenient methodology to access a variety of primary amides.
We began the optimization screening using dioxazolone 1a as the starting material (Table 1). Optimization screening was commenced using γ-valerolactone (GVL) as a biomass-derived green solvent.53 Initial investigations using several copper salts indicated that the reaction with copper(II) acetate afforded the best performance (entry 1–3). Ligand screening showed that nitrogen-based ligands outweighed phosphine-based ligands, resulting in 1,10-phenanthroline being the best ligand system (entry 4–6). Further solvent screening revealed that GVL was the optimal solvent (entry 7–11). Several reductive N–O bond cleavage of isoxazolines and 2-oxa-3-azabicyclic compounds have been studied using copper(0) nanoparticle and heterogeneous copper-on-carbon in the absence of silane.54,55 It is noteworthy that the reaction without silane gave trace amount of amide, indicating the use of silane is essential (entry 9). To confirm that the reaction was not possible, several control experiments were further performed, which indicated that the copper catalyst was pivotal (entry 12 and 13). Based on these results, optimal reaction parameters were determined, demonstrating a catalytic copper(II) system using GVL as an environmentally benign solvent. Further screening of several N–O bond-containing scaffolds was explored under standard reaction conditions, revealing dioxazolone 1a was the optimal amide precursor in this transformation (see the ESI†).
|
||||
|---|---|---|---|---|
| Entry | [Cu] | Ligand | Solvent | Yield (%) |
| a Reaction conditions: 1a (0.2 mmol), Cu(OAc)2 (2 mol%), 1,10-phenanthroline (2 mol%), Ph2SiH2 (2.0 equiv.), and solvent (1 mL) under argon at room temperature for 18 h.b Ligand (4 mol%) was used.c Without Ph2SiH2. | ||||
| 1 | CuCl | 1,10-Phen | GVL | 41 |
| 2 | Cu(OAc)1 | 1,10-Phen | GVL | 92 |
| 3 | Cu(OAc)2 | 1,10-Phen | GVL | 98 |
| 4 | Cu(OAc)2 | DPPE | GVL | <5 |
| 5 | Cu(OAc)2 | PPh3 | GVL | <5b |
| 6 | Cu(OAc)2 | Pyridine | GVL | 95b |
| 7 | Cu(OAc)2 | 1,10-Phen | DMF | 45 |
| 8 | Cu(OAc)2 | 1,10-Phen | DCE | 95 |
| 9 | Cu(OAc)2 | 1,10-Phen | DCE | <5c |
| 10 | Cu(OAc)2 | 1,10-Phen | MeOH | 50 |
| 11 | Cu(OAc)2 | 1,10-Phen | Benzene | 57 |
| 12 | — | 1,10-Phen | GVL | N.R |
| 13 | Cu(OAc)2 | — | GVL | 26 |
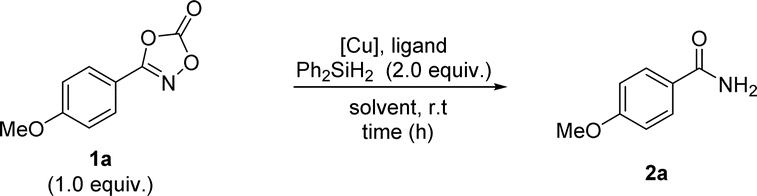
Under the optimized reaction conditions, the substrate scope of the dioxazolones was assessed to probe their reactivity, as shown in Scheme 2. Phenyl dioxazolones bearing methoxy, hydrogen, and tert-butyl substituents were transformed into the corresponding primary amides 2a–2d in excellent yields. Regioisomeric tolyl-dioxazolones were also successful, suggesting that the transformation was affected by steric factors (2e–2g). Dioxazolones bearing halogens were shown to be effective, affording the targeted primary amides 2h–2k in good to excellent yields. Notably, dioxazolones with electron-poor substituents, such as trifluoromethyl, nitrile, and nitro groups, were smoothly converted to the desired primary amides 2l–2n. Based on these results, the reduction system could be applied to both electron-rich and electron-poor dioxazolones. Fused and heteroaromatic motifs were also tolerated in this transformation (2o–2r). Excellent yields were obtained for dioxazolones containing aliphatic alkyl motifs (2s–2u). In addition, the trialkylsilyl protecting group remained intact in this transformation (2v). Surprisingly, the free OH-containing dioxazolone successfully underwent targeted reduction in high yield (2w). Notably, our catalytic reduction system represents a good chemoselective reduction process for the N–O bond of dioxazolone motifs rather than those of other possible unsaturated functional groups, such as aldehyde and alkene motifs (2x and 2y).
 | ||
| Scheme 2 Scope of dioxazolones 1. a 4 mol% of Cu(OAc)2 and 4 mol% of 1,10-phenanthroline are used. | ||
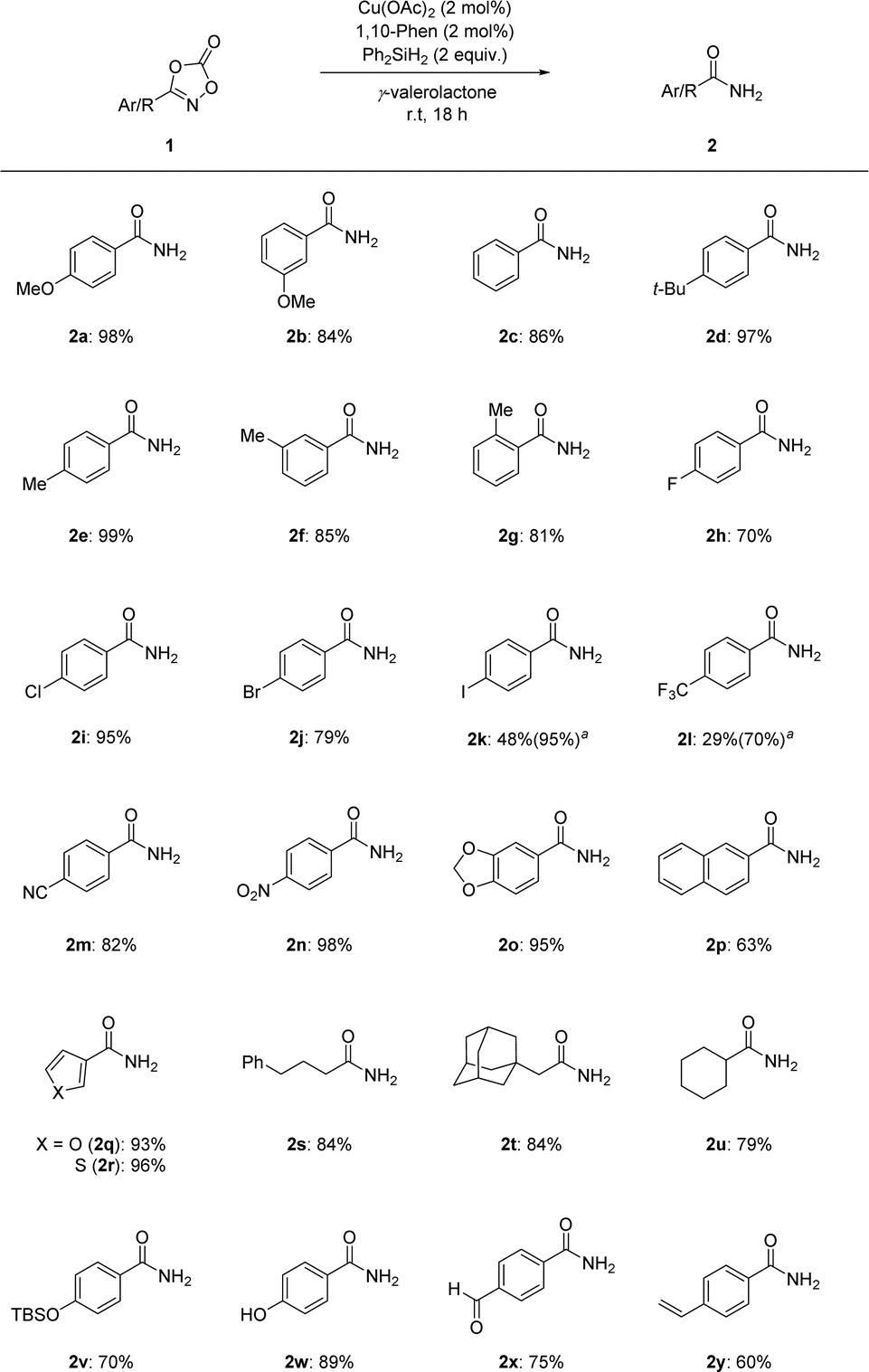
Motivated by this example of dioxazolone 1, we expanded our attention to dioxazolones bearing bioactive structural motifs (Scheme 3). Several dioxazolones derived from naturally occurring compounds such as glycine, citronellic acid, lauric acid, and cinnamic acid have been tested under standard reaction conditions and have shown promising reactivities (4a–4d). Moreover, dioxazolones bearing bioactive structural motifs, such as felbinac and probenecid, which are commercially available drugs, have been shown to be applicable to the corresponding primary amides (4e–4f). The poor yields of 4a and 4e were observed owing to the low conversion of dioxazolones. Based on these results, we anticipate that our reduction protocol for dioxazolones is more efficient for accessing primary amides, owing to the mild reaction conditions and convenient reaction setup.
 | ||
| Scheme 3 Scope of bioactive motif-containing dioxazolones 3. a 4 mol% of Cu(OAc)2 and 4 mol% of 1,10-phenanthroline are used. | ||
We also extended our reduction protocol to a large-scale process (Scheme 4). Several aryl- and aliphatic-substituted dioxazolones afforded the corresponding primary amides (2a, 2s, and 4c) in excellent yields. Based on these trials, it is anticipated that the catalytic reductive transformation of dioxazolones is amenable to large-scale processes (20 mmol scale of dioxazolones), highlighting the operationally simple procedure and mild reaction conditions without any significant loss of product formation.
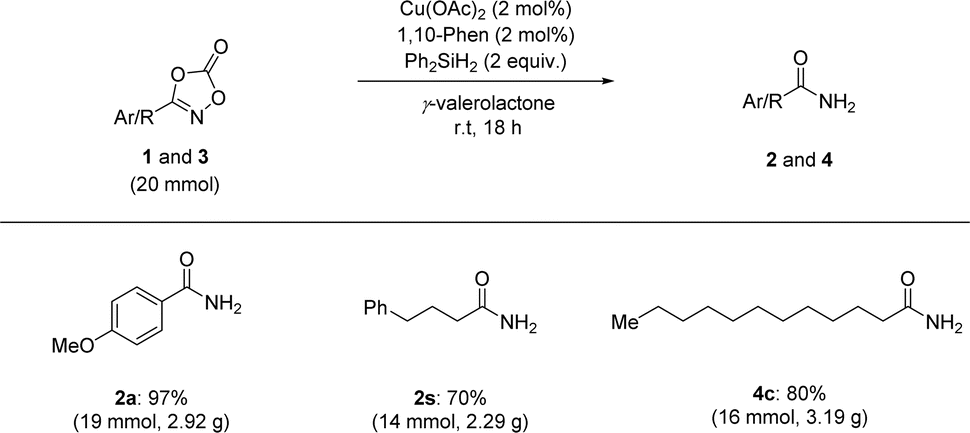 | ||
| Scheme 4 Gram-scale synthesis of primary amides 2 and 4. | ||
The robustness of the transformation was represented by practical copper catalysis in the presence of water (Scheme 5). We were excited to observe that the reaction was still amenable when 10 equivalent of water was present in reaction medium, implying the desired transformation was potentially nontoxic with excellent tolerance. It is noteworthy that the presence of 100 equivalent of water showed diminished reactivity presumably owing to lower solubility.
 | ||
| Scheme 5 Catalytic performance in the presence of H2O. | ||
To provide mechanistic insight into this transformation, several mechanistic studies were explored (Scheme 6). Stryker's reagent, a phosphine-based mild copper hydride source, resulted in no conversion of 1a (Scheme 6a). Interestingly, the same process with 1,10-phenanthroline ligand showed a promising reactivity, implying the use of nitrogen-based ligand is crucial in this transformation. In addition, a radical trapping experiment was performed using TEMPO (Scheme 6b). The addition of TEMPO to the reaction mixture for 10 min inhibited the reduction process. Moreover, the formation of silyl formate was not observed, implying the catalytic reduction of carbon dioxide byproduct was not possible in this transformation (see the ESI†).56–58 Based on this experiment, a radical-involving mechanism was envisioned.
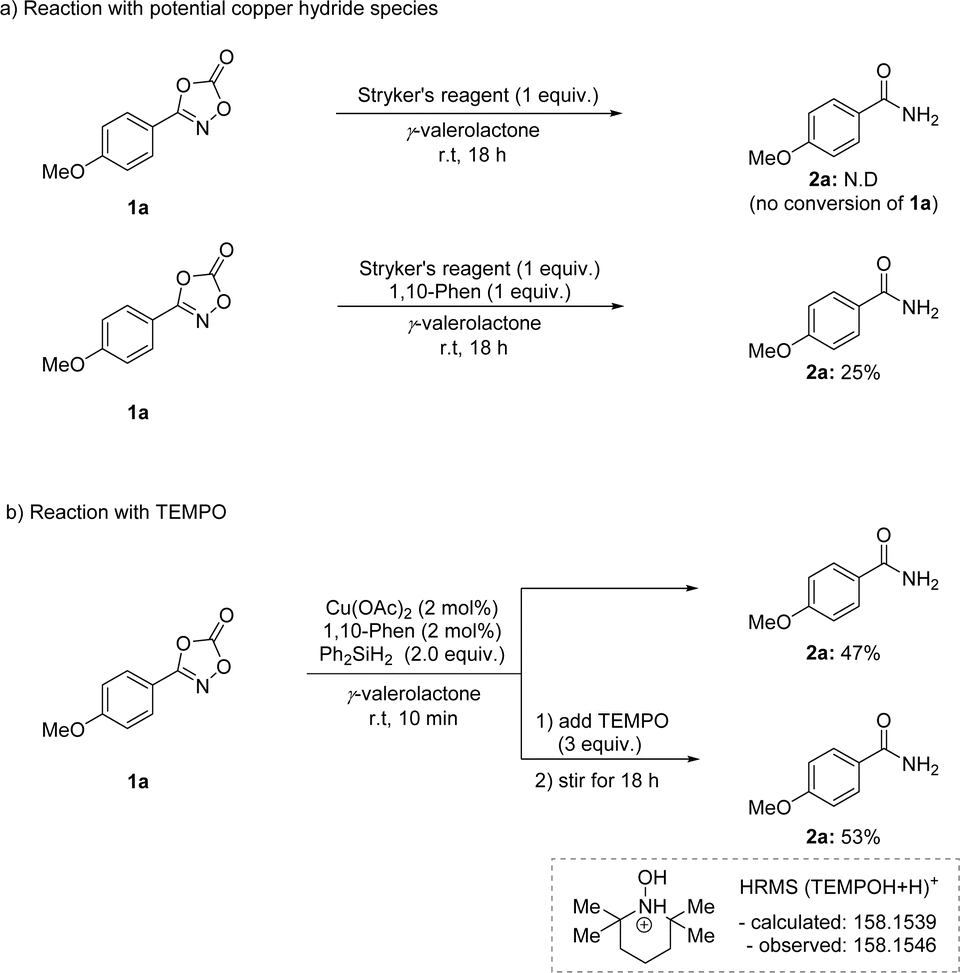 | ||
| Scheme 6 Mechanistic studies. | ||
Based on mechanistic observations and previous studies, a plausible reaction pathway was proposed (Scheme 7). The reaction commences with the association of dioxazolone 1 onto the copper catalyst to afford the complex A. Incorporation of silane as a reductant furnished decarboxylated amide–copper complex B, followed by hydrogen atom abstraction and protonation to afford a desired primary amide 2, which is supported by the experiment with radical trapping experiment. However, we cannot completely rule out the pathway based on the formation of copper hydride involving oxidative N–O bond insertion of dioxazolone, decarboxylative reductive elimination, and protonation processes.59,60
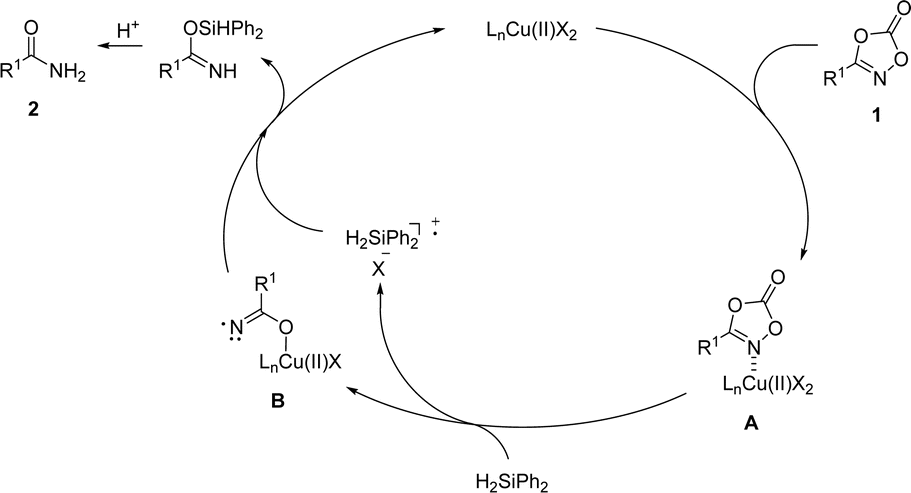 | ||
| Scheme 7 Proposed reaction pathway. | ||
Conclusions
In summary, we have investigated the Cu-catalyzed preparation of primary amides from dioxazolones using silanes. γ-valerolactone as a solvent was used in this transformation, featuring more sustainable reaction condition for primary amides. The scope of dioxazolones in this transformation was well tolerated with excellent chemoselectivity. In addition, we developed a sustainable copper catalytic system and synthesized several dioxazolones bearing biologically active structural motifs, highlighting a convenient synthetic approach to potentially bioactive primary amide analogs. The gram-scale test also demonstrated the possible large-scale production of primary amides in a more sustainable manner under mild reaction conditions. Further detailed mechanistic studies are underway in our laboratory to delineate the nature of catalytic reduction process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by a National Research Foundation of Korea grant (NRF-2021R1F1A1062822) funded by the South Korean government (MSIT). This work was also supported by the Korea Environment Industry & Technology Institute (KEITI) through the Technology Development Program for Safety Management of Household Chemical Products, funded by the Korea Ministry of Environment (MOE) (202200298).References
- X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943–9021 CrossRef CAS PubMed.
- D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- B. L. Bray, Nat. Rev. Drug Discovery, 2003, 2, 587–593 CrossRef CAS PubMed.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- J. M. Humphrey and A. R. Chamberlin, Chem. Rev., 1997, 97, 2243–2266 CrossRef CAS PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- A. Klapars, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 7421–7428 CrossRef CAS PubMed.
- A. Klapars, J. C. Antilla, X. Huang and S. L. Buchwald, J. Am. Chem. Soc., 2001, 123, 7727–7729 CrossRef CAS PubMed.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- A. El-Faham and F. Albericio, Chem. Rev., 2011, 111, 6557–6602 CrossRef CAS PubMed.
- E. Valeur and M. Bradley, Chem. Soc. Rev., 2009, 38, 606–631 RSC.
- S.-Y. Han and Y.-A. Kim, Tetrahedron, 2004, 60, 2447–2467 CrossRef CAS.
- M. Tryniszewski and M. Barbasiewicz, Synthesis, 2021, 54, 1446–1460 Search PubMed.
- S. B. Munoz, H. Dang, X. Ispizua-Rodriguez, T. Mathew and G. K. S. Prakash, Org. Lett., 2019, 21, 1659–1663 CrossRef CAS PubMed.
- R. P. Singh and T. Umemoto, J. Org. Chem., 2011, 76, 3113–3121 CrossRef CAS PubMed.
- Y. Xia, D. He and W. Wu, Chin. J. Org. Chem., 2021, 41, 969–982 CrossRef CAS.
- R. García-Álvarez, J. Francos, E. Tomás-Mendivil, P. Crochet and V. Cadierno, J. Organomet. Chem., 2014, 771, 93–104 CrossRef.
- T. J. Ahmed, S. M. M. Knapp and D. R. Tyler, Coord. Chem. Rev., 2011, 255, 949–974 CrossRef CAS.
- A. Isidro-Llobet, M. N. Kenworthy, S. Mukherjee, M. E. Kopach, K. Wegner, F. Gallou, A. G. Smith and F. Roschangar, J. Org. Chem., 2019, 84, 4615–4628 CrossRef CAS PubMed.
- A. R. Ritter and M. J. Miller, J. Org. Chem., 1994, 59, 4602–4611 CrossRef CAS.
- S. Cicchi, A. Goti, A. Brandi, A. Guarna and F. De Sarlo, Tetrahedron Lett., 1990, 31, 3351–3354 CrossRef CAS.
- M. Nitta and T. Kobayashi, J. Chem. Soc., Perkin Trans. 1, 1985, 1401–1406 RSC.
- L. E. Fisher, J. M. Caroon, Jahangir, S. R. Stabler, S. Lundberg and J. M. Muchowski, J. Org. Chem., 1993, 58, 3643–3647 CrossRef CAS.
- P. G. Mattingly and M. J. Miller, J. Org. Chem., 1980, 45, 410–415 CrossRef CAS.
- G. H. Timms and E. Wildsmith, Tetrahedron Lett., 1971, 12, 195–198 CrossRef.
- G. E. Keck, T. T. Wager and S. F. McHardy, Tetrahedron, 1999, 55, 11755–11772 CrossRef CAS.
- G. E. Keck, S. F. McHardy and T. T. Wager, Tetrahedron Lett., 1995, 36, 7419–7422 CrossRef CAS.
- G. Kresze and G. Schulz, Tetrahedron, 1961, 12, 7–12 CrossRef CAS.
- Y. A. Arbuzov, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1952, 1, 607–611 CrossRef.
- C. Taillier, V. Bellosta, C. Meyer and J. Cossy, Org. Lett., 2004, 6, 2145–2147 CrossRef CAS PubMed.
- M. Yus, G. Radivoy and F. Alonso, Synthesis, 2001, 2001, 0914–0918 CrossRef.
- D. Klamann, P. Weyerstahl, M. Fligge and K. Ulm, Chem. Ber., 1966, 99, 561–565 CrossRef CAS.
- S. D'Andrea, Z. B. Zheng, K. DenBleyker, J. C. Fung-Tomc, H. Yang, J. Clark, D. Taylor and J. Bronson, Bioorg. Med. Chem. Lett., 2005, 15, 2834–2839 CrossRef PubMed.
- J. Soika, C. McLaughlin, T. Neveselý, C. G. Daniliuc, J. J. Molloy and R. Gilmour, ACS Catal., 2022, 12, 10047–10056 CrossRef CAS.
- S. P. Wathen and A. W. Czarnik, Bioorg. Med. Chem. Lett., 1993, 3, 1245–1246 CrossRef CAS.
- T. You, M. Zhang, J. Chen, H. Liu and Y. Xia, Org. Chem. Front., 2021, 8, 112–119 RSC.
- R. Wang, Y. Chen, B. Fei, J. Hu, J. Chen, Y. Luo and Y. Xia, Org. Lett., 2023, 25, 2970–2974 CrossRef CAS PubMed.
- G. Beck, Chem. Ber., 1951, 84, 688–689 CrossRef CAS.
- M. Lee, J. Heo, D. Kim and S. Chang, J. Am. Chem. Soc., 2022, 144, 3667–3675 CrossRef CAS PubMed.
- S. Y. Hong, Y. Hwang, M. Lee and S. Chang, Acc. Chem. Res., 2021, 54, 2683–2700 CrossRef CAS PubMed.
- K. M. van Vliet and B. de Bruin, ACS Catal., 2020, 10, 4751–4769 CrossRef CAS.
- S. Y. Hong, Y. Park, Y. Hwang, Y. B. Kim, M.-H. Baik and S. Chang, Science, 2018, 359, 1016–1021 CrossRef CAS PubMed.
- S. Y. Hong, D. Kim and S. Chang, Nat. Catal., 2021, 4, 79–88 CrossRef CAS.
- J. Park, J. Lee and S. Chang, Angew. Chem., Int. Ed., 2017, 56, 4256–4260 CrossRef CAS PubMed.
- Y. Hwang, Y. Park and S. Chang, Chem.–Eur. J., 2017, 23, 11147–11152 CrossRef CAS PubMed.
- Y. Park, S. Jee, J. G. Kim and S. Chang, Org. Process Res. Dev., 2015, 19, 1024–1029 CrossRef CAS.
- J. Park and S. Chang, Angew. Chem., Int. Ed., 2015, 54, 14103–14107 CrossRef CAS PubMed.
- V. Bizet, L. Buglioni and C. Bolm, Angew. Chem., Int. Ed., 2014, 53, 5639–5642 CrossRef CAS PubMed.
- P. Gandeepan, T. Müller, D. Zell, G. Cera, S. Warratz and L. Ackermann, Chem. Rev., 2019, 119, 2192–2452 CrossRef CAS PubMed.
- K. M. van Vliet, L. H. Polak, M. A. Siegler, J. I. van der Vlugt, C. F. Guerra and B. de Bruin, J. Am. Chem. Soc., 2019, 141, 15240–15249 CrossRef CAS PubMed.
- Y. Zhou, O. D. Engl, J. S. Bandar, E. D. Chant and S. L. Buchwald, Angew. Chem., Int. Ed., 2018, 57, 6672–6675 CrossRef CAS PubMed.
- A. K. Adegboyega and J. Son, Org. Lett., 2022, 24, 4925–4929 CrossRef CAS.
- D. M. Alonso, S. G. Wettstein and J. A. Dumesic, Green Chem., 2013, 15, 584–595 RSC.
- K. S. Gayen, T. Sengupta, Y. Saima, A. Das, D. K. Maiti and A. Mitra, Green Chem., 2012, 14, 1589–1592 RSC.
- N. Yasukawa, Y. Miki, M. Kuwata, H. Sajiki and Y. Sawama, ChemSusChem, 2020, 13, 5632–5637 CrossRef CAS PubMed.
- C. M. Zall, J. C. Linehan and A. M. Appel, J. Am. Chem. Soc., 2016, 138, 9968–9977 CrossRef CAS PubMed.
- K. Motokura, D. Kashiwame, N. Takahashi, A. Miyaji and T. Baba, Chem.–Eur. J., 2013, 19, 10030–10037 CrossRef CAS.
- F. J. Fernández-Alvarez and L. A. Oro, ChemCatChem, 2018, 10, 4783–4796 CrossRef.
- C. Deutsch, N. Krause and B. H. Lipschutz, Chem. Rev., 2008, 108, 2916–2927 CrossRef CAS PubMed.
- A. J. Jordan, G. Lalic and J. P. Sadighi, Chem. Rev., 2016, 116, 8318–8372 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00320a |
| This journal is © The Royal Society of Chemistry 2024 |