
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIntegration of O2-economised tumour-targeted photosensitive magnetic nanomaterials in the diagnosis and therapy of gastric cancer
JinRong Situab,
Yingying Yangab,
Lingle Zhangab,
Hongzhang Yanab and
Yingsheng Cheng*abc
aCollege of Fisheries and Life Science of Shanghai Ocean University, Shanghai, 201306, China. E-mail: Situ.JR@Outlook.com
bShanghai Jiao Tong University Affiliated Sixth People's Hosptial, China
cTongji Hospital Affiliated to Tongji University, Shanghai, China
First published on 25th March 2024
Abstract
Hypoxia in the tumour microenvironment is a major limiting factor in photodynamic therapy. The present study employed a novel O2-economised photosensitizer, ACSN, to effectively curtail oxygen consumption by impeding the aerobic respiration of tumour cells, thereby increasing the reactive oxygen species (ROS) production in photodynamic therapy. To enhance the efficacy of photodynamic therapy, the active targeting peptide iRGD was employed to facilitate drug accumulation in the tumour tissue. Therefore, we constructed a targeted drug platform, ACSN/Fe3O4@MSNs-iRGD, that integrates diagnosis and treatment. The drug exhibited excellent active targeting ability towards gastric cancer MGC-803 cells and can efficiently penetrate the mitochondria upon cellular internalisation. The photosensitizer ACSN, released from the drug, effectively suppressed mitochondrial aerobic respiration to conserve oxygen and exhibited robust ROS production upon laser excitation. The core–shell structure comprises Fe3O4, which offers excellent T2 dark contrast for real-time tumour monitoring through MRI imaging. By incorporating excellent photodynamic therapy and MRI imaging capabilities, this drug can serve as an effective platform for the integration of tumour diagnosis and treatment, thus addressing the limitations associated with conventional tumour therapies. It is anticipated that this approach will soon be clinically translated.
1. Introduction
Malignant tumours, which are formidable adversaries to human life and health, are some of the most prominent causes of mortality. Medically referred to as cancers, these diseases exhibit exceptionally high propensity for metastasis. Based on statistical data, it is projected that the USA will witness approximately 2.00 million new cancer cases and 0.61 million cancer-related fatalities by the year 2024.1 Among cancers, the incidence of gastric cancer ranks fifth, while it stands as the third leading cause of cancer-related mortality worldwide.2 The tumour microenvironment (TME) refers to the internal milieu in which tumours originate and grow, and comprises malignant tumour cells, diverse stromal cells, and the extracellular matrix.3,4 Compared with normal tissues, tumour tissues and other pathological sites are typically characterised by aberrant blood vessel formation, weak acidification (pH 6.8), abnormal temperature regulation, overexpression of specific enzymes, and hypoxia.5,6 The specificity of TME presents challenges and opportunities for cancer treatment. On one hand, the reciprocal interactions between cancer cells and TME contribute to increased malignancy and therapeutic resistance. The heterogeneity of the TME, such as hypoxic and acidic conditions, facilitates both tumour metastasis and drug resistance. On the other hand, leveraging the tumour-specific microenvironment as a viable target for drug delivery has immense potential for augmenting the therapeutic efficacy of drugs. Specifically, the utilisation of delivery systems that respond to the TME exhibits promising prospects for facilitating tumour-targeted delivery and controlled drug release.7Early cancer diagnosis and timely treatment play pivotal roles in enhancing patient survival. Magnetic Resonance Imaging (MRI),8 optical imaging,9 photoacoustic imaging,10 ultrasonic imaging,11 and computed tomography imaging,12 are predominantly used for tumour visualisation. MRI plays a pivotal role in the diagnosis and treatment of cancer, encompassing its applications in diagnostic procedures, staging assessments, determination of optimal therapeutic approaches, and identification of tumour recurrence. In recent years, the clinical application of MRI in gastric cancer diagnosis and treatment has witnessed a remarkable surge, owing to its exceptional soft-tissue contrast and advanced imaging capabilities, encompassing multi-angle, multi-directional, and multi-parametric approaches.13 At present, nanoparticulate iron oxide is a popular and unique nanoparticulate agent used in clinical practice. When applied during imaging, they reduce the intensity of the T2 signals inthe tissues which absorb the contrast agent.14
Drawing upon the functional design of magnetic nanomaterials, the integration of magnetic diagnosis and treatment using multifunctional nanomaterials holds great promise in addressing a critical clinical challenge of achieving timely tumor diagnosis and treatment. Conventional treatment modalities for malignant tumours, such as radiotherapy, chemotherapy, and surgical resection, are associated with certain limitations in terms of addressing tumour recurrence and metastasis, while also causing serious toxicity and side effects.15 Reactive oxygen species (ROS)-based tumour therapies such as photodynamic therapy (PDT) and chemodynamic therapy (CDT) have promising prospects for development. PDT involves the excitation of photosensitizers under light irradiation, inducing an energy transition to generate ROS and subsequently eliminating tumour cells. This non-invasive and targeted treatment approach exhibited enhanced efficacy. PDT is concomitant with an inflammatory response that facilitates the release of various inflammatory factors and the activation of the immune system, thereby achieving autologous tumour elimination.16,17 Chlorine6 (Ce6), a second-generation photosensitizer, with the ability to generate high levels of singlet oxygen upon laser stimulation for tumour cell eradication, has been approved. However, aberrant angiogenesis in tumour tissues and increased cellular oxygen consumption give rise to hypoxia within the TME, thereby attenuating ROS production and compromising the therapeutic efficacy of PDT.18 Therefore, augmenting oxygen production or reducing oxygen consumption could serve as a potent strategy to enhance the efficacy of PDT. Compared with traditional photosensitizers, oxygen-economised photosensitizers can reduce cancer cells' endogenous oxygen consumption and conserve oxygen to generate increased ROS, thereby facilitating efficient PDT for hypoxic solid tumours. To this end, Zhao19 proposed a novel oxygen-economised photosensitizer, ACSN, which is synthesised by combining the cellular aerobic respiration inhibitor Atovaquone (ATO) with the photosensitizer Ce6 through π–π bond stacking and hydrophobic interactions. Due to the inhibition of mitochondrial aerobic respiration by ATO, a compound found in ACSN, effective suppression of oxygen consumption occurs at the tumour site. This counteracted the low efficiency of PDT induced by hypoxia in the TME.
The efficacy of PDT also relies on the adequate accumulation of photosensitizers at the tumour site, which, upon laser irradiation, convert oxygen molecules into ROS at specific locations and subsequently induce selective damage to the tumour while sparing normal tissues. The transport of nanomaterials to tumour cells is facilitated by enhanced permeability and retention (EPR). However, studies have demonstrated that even in translocated tumour models exhibiting high EPR effects, the passive targeting of nanomedicine remains only 0.7%.20 Active-targeting drugs have been proposed to overcome the limitations of passive targeting and enhance nanomedicine transport. The disulfide bond-based loop RGD peptide-iRGD (CRCGDK/RGPDC) has garnered significant attention since its initial report by Kazuki21 in 2010. When chemically conjugated to a drug, iRGD facilitates the transportation of the drug into deep extravascular tumor tissue. The cell-penetrating peptide iRGD exhibits a single-ligand-multiple receptor-active targeting effect by specifically binding to multiple receptors on the surface of tumour cells. Through its interaction with the highly expressed integrin and neuropilin-1 receptors, iRGD effectively targets tumour cells and penetrates tumour tissues. Compared with traditional integrin-targeting RGD peptides, iRGD demonstrated superior tumour-targeting efficacy and enhanced penetration ability.22
In the present study, we aimed to develop an active-targeting nanomedicine, ACSN/Fe3O4@MSNs-iRGD. Notably, we demonstrated that the combination of Fe3O4 T2 high-contrast MRI and PDT with a novel O2-economised photosensitizing agent enabled real-time monitoring of tumour diagnosis and efficient treatment. We also tested the physicochemical properties of the drug and systematically investigated its MRI imaging and PDT therapeutic effects in vitro and in vivo. First, the drug's shell core comprised superparamagnetic Fe3O4 nanoparticles that exhibit a high T2-weighted relaxation rate owing to their exceptional chemical stability, non-toxicity, and biodegradability. Therefore, it represents a pioneering magnetic-nanoparticle-based contrast agent that has gained widespread use in MRI,23 which enables real-time tumour monitoring by MRI. Second, the design of O2-economised photosensitizer was used to deal with the limitation of hypoxia in TME, which can enhance the efficacy of PDT. Finally, iRGD was used for lactone. Above, ACSN/Fe3O4@MSNs-iRGD acts both as excellent real-time MRI diagnosis, and as a new strategy to overcome the limitations of low ROS production and non-targeting in PDT. The utilization of this drug significantly contributes to the seamless integration of tumor diagnosis and treatment.
2. Experimental section
2.1 Materials
Chlorin E6, Atovaquone, 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC HCl), and N-carboxythiosuccinimide (sulfo-NHS) were purchased from Shanghai Yuanye Biotechnology Co., Ltd. Dimethyl sulfoxide (DMSO) was purchased from Beijing LABGIC technology Co., Ltd; monodisperse carboxylic magnetic mesoporous silica nanomaterials (Fe3O4@MSNs) were purchased from Nanjing NANOEAST Biotech Co., Ltd; targeted peptide (iRGD) was purchased from Hefei Bank Peptide Biological technology Co., Ltd; 1,3-diphenylisobenzofuran (DPBF) was purchased from Shanghai Macklin Biochemical Co., Ltd; PBS buffer solution was purchased from Beijing Rangeke Technology Co., Ltd; DMEM medium, peptide bovine serum (FBS), trypsin, EDTA-free trypsin and double antibody (PS) were purchased from Thermo Fisher Scientific. 4′,6-Diamidino-2-phenylindole (DAPI), 4% paraformaldehyde, and electron microscope fixative were purchased from Wuhan Servicebio Technology Co., Ltd; cck8 kit and AnnexinV-FITC/PI double staining apoptosis detection kit were purchased from Jiangsu KeyGEN BioTECH Co., Ltd; 2′,7′-dichlorofluorescin diacetate (DCFH-DA) and calcein/PI cell activity and cytotoxicity detection kits were purchased from Shanghai Beyotime Biotechnology Co., Ltd.2.2 Preparation and characterization of ACSN/Fe3O4@MSNs-iRGD drugs
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min was performed to remove the supernatant, and the carboxyl-activated Fe3O4@MSNs were collected by resuspension in absolute ethanol. Next, 2 mg iRGD was added and allowed to react fully overnight at 4 °C in a rotary mixer to obtain Fe3O4@MSNs-iRGD.000 rpm for 10 min to remove the supernatant and the ACSN/Fe3O4@MSNs-iRGD samples were collected.
000 rpm for 10 min was performed to remove the supernatant, and the carboxyl-activated Fe3O4@MSNs were collected by resuspension in absolute ethanol. Next, 2 mg iRGD was added and allowed to react fully overnight at 4 °C in a rotary mixer to obtain Fe3O4@MSNs-iRGD.000 rpm for 10 min to remove the supernatant and the ACSN/Fe3O4@MSNs-iRGD samples were collected.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and N–H) present in the peptide bonds.
O and N–H) present in the peptide bonds.2.3 Cell culture
MGC-803 cells, purchased from Beijing Innochem Co., Ltd, were used for both the in vitro and in vivo experiments. The cells were cultured at 37 °C in a 5% CO2 incubator using DMEM medium supplemented with 10% foetal bovine serum and 1% double antibody.2.3.1.1 Laser confocal microscopy. The MGC-803 cells (1 × 105) were evenly distributed in a laser confocal dish. Once the cells adhered to the wall and proliferated, the medium was aspirated, and ACSN/Fe3O4@MSNs-iRGD and ACSN/Fe3O4@MSNs drugs (2 μg mL−1) were added respectively. After incubation for a specific duration (2 and 12 h), the medium was removed by aspiration, followed by washing with PBS thrice. Subsequently, 1 mL of 4% paraformaldehyde was added for fixation at room temperature in the dark for 30 min. Subsequently, the cells were washed thrice with PBS, followed by the addition of 1 mL of PBS for observation and imaging using a laser confocal microscope. The excitation wavelengths used were 405 nm and 633 nm for DAPI and ACSN, respectively.
2.3.1.2 Flow cytometry. MGC-803 cells (1 × 105) were evenly distributed in six-well plates. After the cells adhered and proliferated, ACSN/Fe3O4@MSNs-iRGD and ACSN/Fe3O4@MSNs (2 μg mL−1) were added respectively. Following incubation for a specific duration (2 and 12 h), the medium was aspirated, and the wells were washed thrice with PBS. Subsequently, 200 μL trypsin was added to each well and incubated at 37 °C for 2 min to facilitate digestion. The digestion was terminated by adding 1 mL of serum-free medium. Subsequently, the cells were collected in a 1.5 mL centrifuge tube and centrifuged at 1300 rpm for 3 min, followed by aspiration of the supernatant. After resuspending the cells in 500 μL PBS, flow cytometry was employed to detect the fluorescence intensity of ACSN in each group.
2.3.1.3 Observation of cells by transmission electron microscopy. MGC-803 cells (1 × 105) were seeded evenly into small culture dishes. After the cells adhered and proliferated, ACSN/Fe3O4@MSNs and ACSN/Fe3O4@MSNs-iRGD (2 μg mL−1), prepared using serum-free medium, were added. The cells were then incubated for 6 h. Subsequently, the medium was aspirated, and the cells were washed thrice with PBS before adding 200 μL of trypsin for digestion at 37 °C for 2 min. Finally, digestion was terminated by adding 1 mL of serum-free medium. The cells were then collected in a 1.5 mL centrifuge tube and centrifuged at 1300 rpm for 3 min to remove the supernatant. Subsequently, the cells were resuspended in 1 mL of electron microscopy fixative and sent to Wuhan Savier Biotechnology Co., Ltd for cell section preparation, followed by observation and photography using a transmission electron microscope.
2.3.3.1 Laser confocal microscopy. The 1 × 105 MGC-803 cells were evenly dispersed in a laser confocal dish. Upon cell attachment and growth, the medium was carefully removed and 2 μg mL−1 of drugs Ce6, ACSN, and ACSN/Fe3O4@MSNs-iRGD were sequentially added to each dish. In the control group, an equal volume of PBS was administered, and the cells were incubated for 24 h. The dish was subsequently aspirated and washed thrice with PBS before adding 1 mL serum-free medium. To investigate the effect of laser irradiation on ROS generation by photosensitizers in the drug, dark and laser treatment groups were established. The dark treatment group was excluded from laser irradiation, while the laser treatment group was exposed to a 633 nm (0.5 W cm−2) laser for 5 min. Subsequently, the dish was aspirated, and 1 mL of 10 mM DCFH-DA was added to the cells for 30 min. The cells were then washed thrice with PBS to remove excess extracellular DCFH-DA probes. Finally, 1 mL of serum-free medium was added, and the dish was placed in a laser confocal microscope for observation and imaging. The green fluorescence of 2′,7′-dichlorofluorescein (DCF), generated by the combination of ROS and DCFH-DA probe in the cells, was employed as an indicator, with the excitation wavelength set at 504 nm.
2.3.3.2 Flow cytometry. The 1 × 105 MGC-803 cells were evenly dispersed in a six-well plate. After they adhered to the wall and proliferated, the medium was removed and 2 μg mL−1 of drugs (Ce6, ACSN, and ACSN/Fe3O4@MSNs-iRGD prepared with serum-free medium) were added to each well. The cells in the control group were supplemented with equal amounts of PBS and incubated for 24 h. The medium was subsequently aspirated and washed thrice with PBS, followed by the addition of 1 mL of serum-free medium to each well. To investigate the effect of laser irradiation on the production of ROS by photosensitizers in the drug, dark and laser treatment groups were established, as previously mentioned. Subsequently, the medium was aspirated, 1 mL of 10 mM DCFH-DA was added, and the cells were incubated for 30 min. Finally, the cells were washed thrice with PBS to completely remove the excess extracellular DCFH-DA probe. An additional 200 μL of EDTA-free trypsin was added to each well and incubated at 37 °C for 2 min, followed by the addition of 1 mL of serum-free medium to terminate the digestion. The cells were collected in a 1.5 mL centrifuge tube, centrifuged at 1300 rpm for 3 min, and the supernatant was aspirated. The cells were resuspended in 500 μL PBS, and the green fluorescence intensity of DCF in each group was detected by flow cytometry, with the fluorescence channel being fluorescein isothiocyanate (FITC).
2.4 In vivo experiments
3. Results and discussion
3.1 Preparation and characterization of ACSN/Fe3O4@MSNs-iRGD materials
The distinctive advantage of mesoporous silica is that they have well-defined surface properties that allow easy functionalization of the silanol-containing surface. Moreover, the external surface can be conjugated with targeting ligands for efficient cell-specific drug delivery.24,25 This truly facilitates targeted peptide iRGD modification. The drug's morphology and size were observed using TEM (Fig. 1a–c). Sample ACSN/Fe3O4@MSNs-iRGD was a homogeneous mesoporous spherical structure with an Fe3O4 core coated with mesoporous silica, on which the photosensitizer ACSN was loaded. Thus, the permeability of the mesoporous layer was lower than that of the Fe3O4@MSNs. The size of Fe3O4@MSNs was approximately 117 nm, whereas that of ACSN/Fe3O4@MSNs-iRGD was 151 nm. Photosensitizer ACSN was self-assembled by Ce6 and ATO through π–π interaction. At pH 7.0, the zeta potentials of Ce6, ATO, and ACSN (Fig. 1d) were −27.3 mV, −15 mV, and −23.3 mV, respectively, with ACSN's negative potential between that of Ce6 and ATO. Zeta potentials (Fig. 1e) for Fe3O4@MSNs, Fe3O4@MSNs-iRGD, and ACSN/Fe3O4@MSNs-iRGD were 16.8 mV, −10.8 mV, and −21.7 mV, respectively. iRGD was conjugated to the carboxyl group on the surface of the Fe3O4@MSNs, and the photosensitizer ACSN was doped into the mesoporous silicon layer. Negatively charged iRGD and ACSN offset the positive charge of mesoporous silica, allowing easier absorption into the cells through the cell membrane, thereby improving the effectiveness of drug delivery. Meanwhile, the negative charge on the surface can attract ions with opposite charges and facilitate their dispersion. UV-visible spectrum analysis (Fig. 1f) further demonstrated a red-shifted absorption peak (approximately 30 nm) for ACSN/Fe3O4@MSNs-iRGD in the 600–700 nm range compared to that of Ce6, owing to the interaction between the Photosensitizer ACSN (Ce6) and the magnetic material, indicating that ACSN was doped into mesoporous silica Fe3O4. Fourier transform infrared spectroscopy (Fig. 1g) revealed characteristic absorption peaks of peptide bonds (CO, N–H) at 1645 cm−1 and 3375 cm−1, demonstrating that the target peptide, iRGD, dehydrates and condenses with the activated carboxyl group of the mesoporous silicon layer. Meanwhile, the absorption peaks at 1072 cm−1 (C–C) increased due to the addition of peptide, successfully demonstrating the incorporation of the drug into the target peptide.26 Finally, due to its rapid and accurate detection of oxygen concentration, DPBF exhibits an absorption peak at 420 nm, a decrease in this absorption peak was used to assess the photodynamic generation of ROS in ACSN/Fe3O4@MSNs-iRGD (Fig. 1h). After 5 min of 633 nm (0.5 W cm−2) laser irradiation, the absorbance at 420 nm decreased from 3.869 to 2.096 in 4 min, reaching 54.17%. These findings suggest that ACSN/Fe3O4@MSNs-iRGD can generate a substantial amount of ROS following laser excitation, thus exhibiting a promising PDT effect.
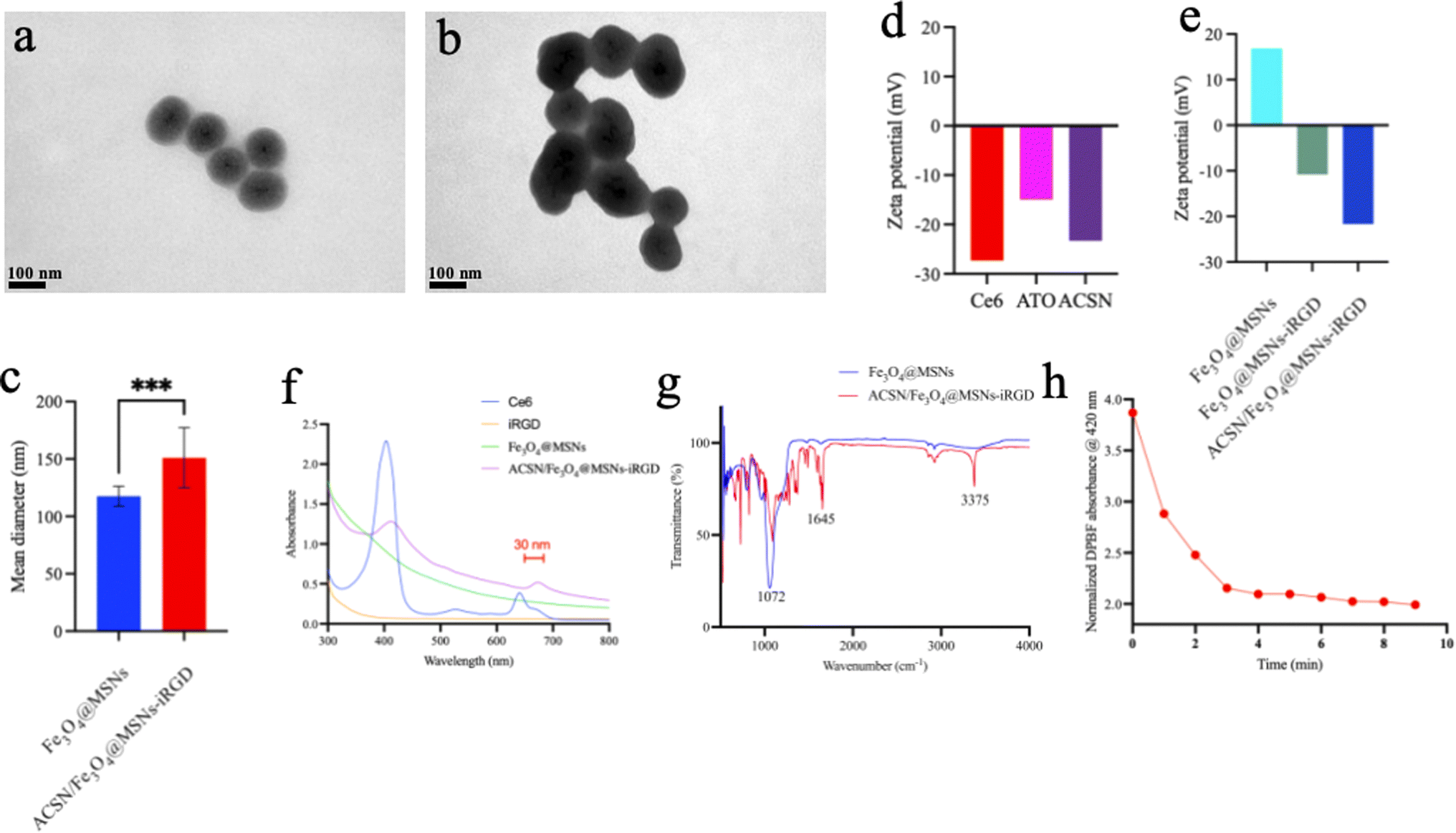 | ||
| Fig. 1 TEM images of Fe3O4@MSNs (a) and ACSN/Fe3O4@MSNs-iRGD (b); semi-quantitative analysis of Fe3O4@MSNs and ACSN/Fe3O4@MSNs-iRGD (c); the zeta potentials of Ce6, ATO, ACSN (d) and Fe3O4@MSNs, Fe3O4@MSNs-iRGD, ACSN/Fe3O4@MSNs-iRGD (e); UV-vis spectrum of Ce6, iRGD, Fe3O4@MSNs, ACSN/Fe3O4@MSNs-iRGD (f); the Fourier transform infrared of Fe3O4@MSNs and ACSN/Fe3O4@MSNs-iRGD (g). OD of ROS produced by ACSN/Fe3O4@MSNs-iRGD at 420 nm in vitro before and after laser irradiation (h). | ||
3.2 In vitro cell experiments
3.2.1.1 Laser confocal microscopy. ACSN (Ce6) emits excitable red light at 633 nm, whereas DAPI can bind to double-stranded DNA to produce excitable blue light at 488 nm. Consequently, the red fluorescence intensity could be quantified to reflect the amount of intracellular drug uptake. As observed using laser confocal microscopy (Fig. 2a), the blank control group supplemented with equal amounts of PBS did not exhibit red fluorescence. In contrast, the red fluorescence intensity increased with the incubation time in cells incubated with ACSN/Fe3O4@MSNs without targeted drugs and ACSN/Fe3O4@MSNs-iRGD with targeted drugs for 2 and 12 h, respectively. Cells were primarily concentrated in the nucleus. Semi-quantitative analysis of the red fluorescence (Fig. 2b) revealed that the uptake of the targeted drug ACSN/Fe3O4@MSNs-iRGD was significantly higher than that of the non-targeted drug ACSN/Fe3O4@MSNs at the same drug concentration and incubation time, indicating that the drug possesses potent targeting capabilities.
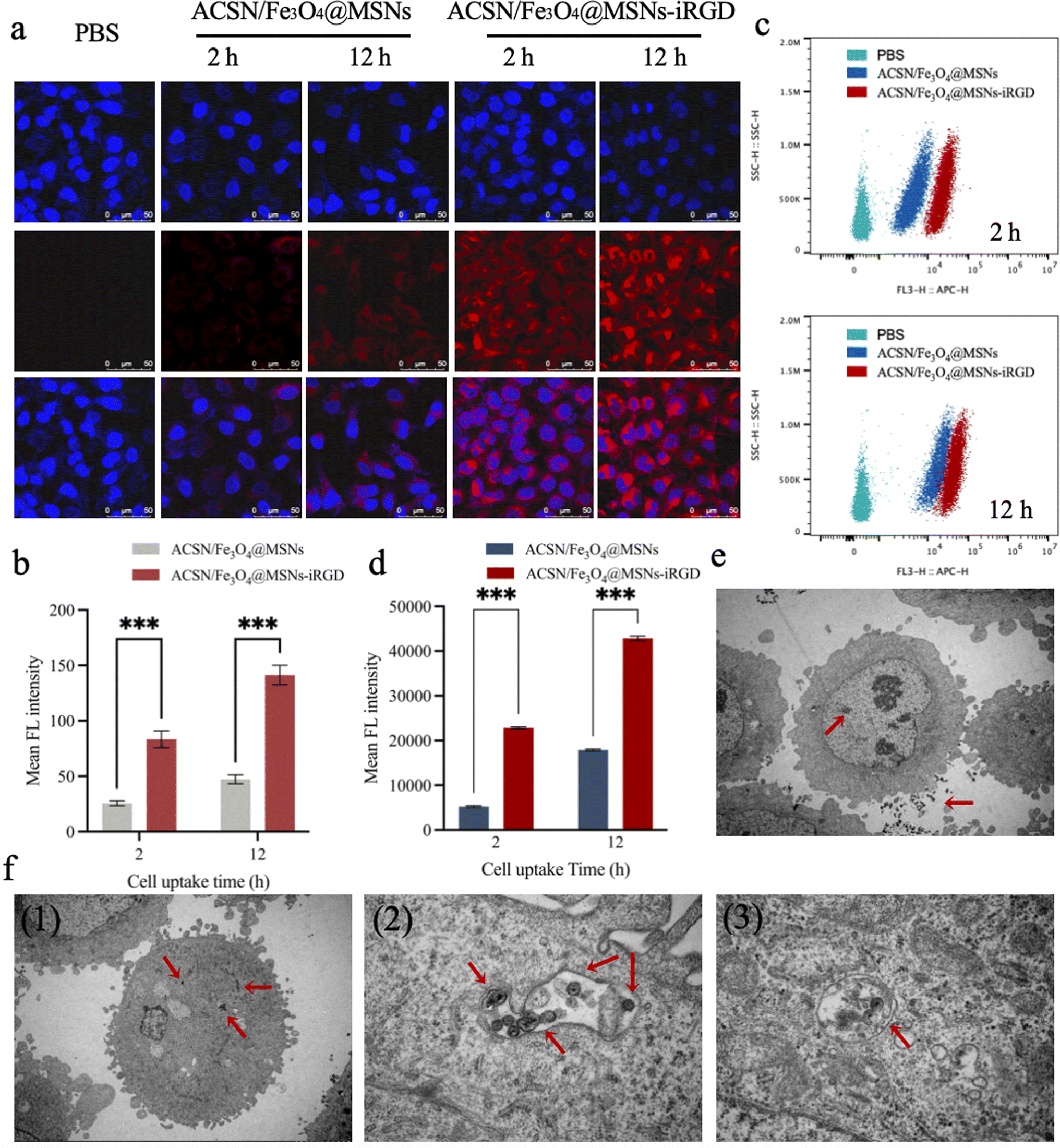 | ||
| Fig. 2 Results of cellular drug uptake. Laser confocal images (a) and semi-quantitative analysis (b) of MGC-803 cells incubated with non-targeted drug ACSN/Fe3O4@MSNs and targeted drug ACSN/Fe3O4@MSNs-iRGD for 2 and 12 h, respectively. Flow cytometry was used to measure the fluorescence intensity of cell uptake (c) and mean fluorescence intensity analysis (d). Biological image observation by transmission electron microscopy of cellular drug uptake, and the uptake of non-targeted drug ACSN/Fe3O4@MSNs (e) and targeted drug ACSN/Fe3O4@MSNs-iRGD (f) in MGC-803 cells. Distribution of the drug ACSN/Fe3O4@MSNs-iRGD in mitochondria after internalization (f 2, and 3), with red arrows pointing to the drug. | ||
3.2.1.2 Flow cytometry. The intracellular drug uptake was quantified using flow cytometry (Fig. 2c). The red fluorescence intensity of ACSN increased proportionally with the duration of drug incubation. The fluorescence intensity of ACSN/Fe3O4@MSNs-iRGD, administered with the same targeted drug concentration simultaneously, was notably superior to that of ACSN/Fe3O4@MSNs sans targeted drug. Furthermore, in the post-mean fluorescence intensity analysis (Fig. 2d), the drug uptake of ACSN/Fe3O4@MSNs-iRGD incubated for 2 h exceeded that of ACSN/Fe3O4@MSNs incubated for 12 h without targeted drug uptake. This validated the function of the targeted peptide, iRGD, demonstrating its ability to accelerate the uptake rate and enhance drug uptake.
The results of the flow cytometry-based quantitative detection corresponded with those of laser confocal microscopy, indicating that ACSN/Fe3O4@MSNs-iRGD has potent targeting ability towards MGC-803 cells. Consequently, damage to healthy tissues can be minimised upon drug entry into the body, ensuring sufficient accumulation of the drug at the tumour site, thereby guaranteeing the therapeutic efficacy of PDT and achieving accurate targeted therapy.
3.2.1.3 Biotype transmission electron microscopy. To elucidate the drug efficacy of PDT, it is crucial to ascertain the drug distribution in MGC-803 cells. Employing transmission electron microscopy to visualise the internalised drug distribution within cells demonstrated that most non-targeted drugs ACSN/Fe3O4@MSNs (Fig. 2e) were not taken up by cells, with only a few exceptions; conversely, a majority of the targeted drug ACSN/Fe3O4@MSNs-iRGD could enter cells and further translocate into mitochondria (Fig. 2f). Consequently, ACSN (ATO) released by ACSN/Fe3O4@MSNs-iRGD interacted with the mitochondrial electron transport chain and inhibited cellular aerobic respiration, thereby reducing oxygen consumption. Hence, the increased oxygen supply to tumour tissues can be utilised to generate ROS and enhance the lethality of PDT.
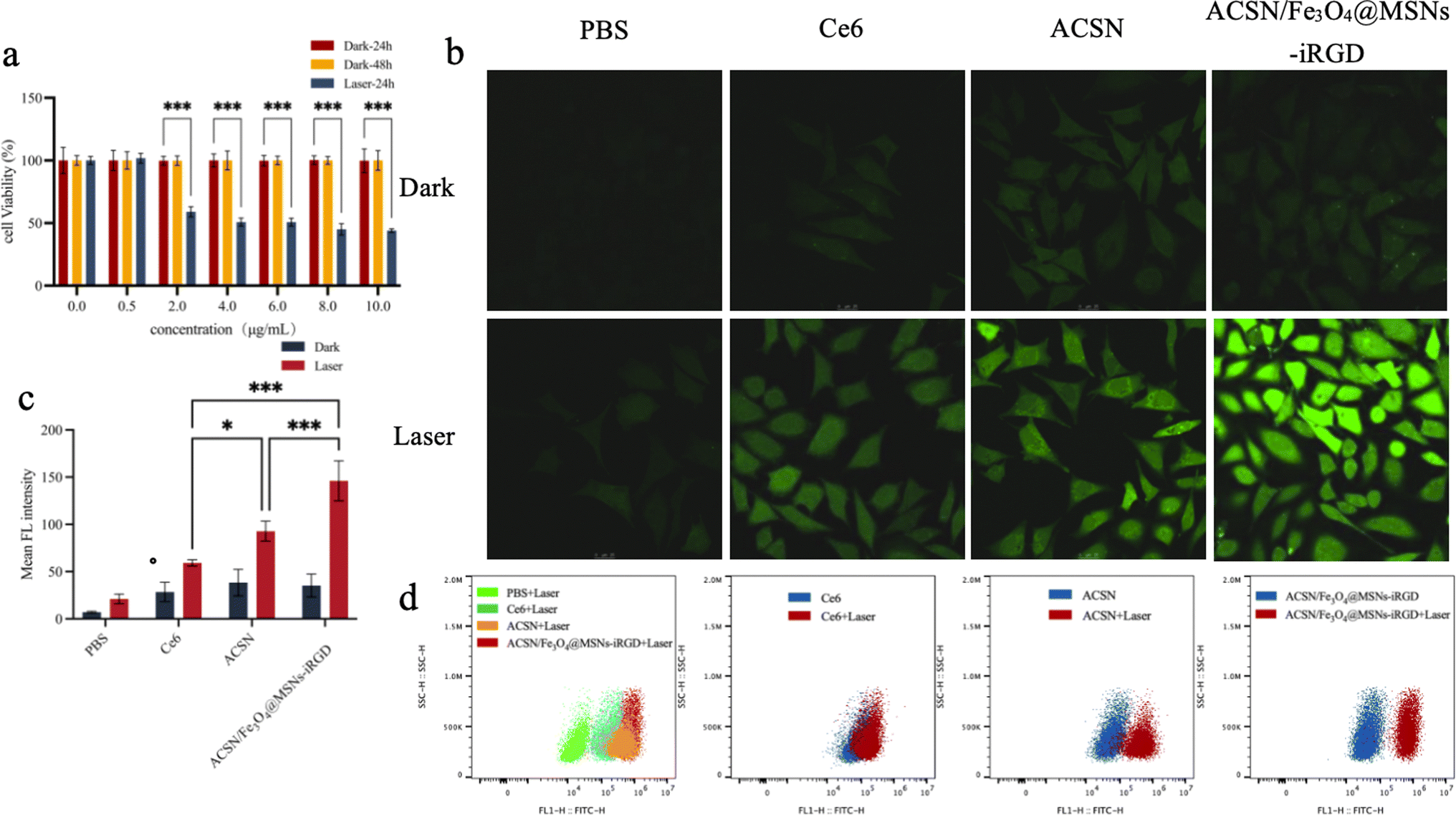 | ||
| Fig. 3 Graphs of the cytotoxicity assay and reactive oxygen species detection. Figure of cytotoxicity assay detection results (a), different concentrations of ACSN/Fe3O4-iRGD and MGC-803 cells incubated in the dark for 24 h and 48 h, and irradiated with 633 nm (0.5 W cm−2) laser for 24 h, respectively. Intracellular reactive oxygen species detection results, confocal laser images (b), and semi-quantitative analysis (c) of reactive oxygen species produced in MGC-803 cells incubated with the same concentrations of Ce6, ACSN, and ACSN/Fe3O4-iRGD in the dark and under laser treatment; fluorescence intensity of intracellular reactive oxygen species was measured by flow cytometry (d). | ||
3.2.3.1 Laser confocal microscope observation. The non-fluorescent probe DCFH-DA can enter cells through free diffusion, where it is hydrolysed by intracellular lipase to DCFH. This derivative is then oxidised by intracellular ROS to DCF, which exhibits green fluorescence and is loaded into cells. Consequently, the intensity of the intracellular green fluorescence can be used to reflect ROS levels. Laser confocal microscopy and semi-quantitative analysis of green fluorescence (Fig. 3b and c) showed no significant ROS generation in the blank control group and the three drug-treated groups (Ce6, ACSN, and ACSN/Fe3O4@MSNs-iRGD) without laser irradiation. However, after 633 nm (0.5 W cm−2) laser irradiation for 5 min, the green fluorescence intensity of ACSN at the same concentration was notably higher than that of Ce6. This was attributed to ACSN, an oxygen-economised photosensitizer that inhibits oxygen consumption in cells at similar oxygen levels, allowing for more oxygen accumulation in cells, thereby elevating ROS levels. These findings demonstrate a significantly more potent PDT effect for ACSN than for Ce6. The green fluorescence signal in the ACSN/Fe3O4@MSNs-iRGD group was the most robust following laser treatment, potentially because of the active targeting capability of the drug complex, which enabled greater accumulation of drugs in tumour cells under the same drug concentration incubation, thereby significantly enhancing PDT efficacy.
3.2.3.2 Flow cytometry. ROS production in the cells was quantitatively detected by flow cytometry (Fig. 3d). Under non-laser irradiation, a small amount of ROS was observed in the cells, with the lowest production observed in the ACSN/Fe3O4@MSNs-iRGD group, indicating the enhanced biological drug safety following modification. Pure laser irradiation did not induce ROS production. After 633 nm (0.5 W cm−2) laser irradiation for 5 min, the ROS production in the three drug groups (Ce6, ACSN, and ACSN/Fe3O4@MSNs-iRGD) significantly increased (ACSN/Fe3O4@MSNs-iRGD group > ACSN group > Ce6 group). The PDT effect of ACSN as a photosensitizer was superior to that of Ce6. Due to the targeting ability of Fe3O4@MSNs-iRGD-ACSN, cell uptake under the same drug concentration was greater, resulting in the highest amount of ROS produced.
This result is consistent with the findings of laser confocal microscopy. Under the same conditions, the ability of the drug to produce ROS after laser excitation was: ACSN/Fe3O4@MSNs-iRGD was significantly greater than that of ACSN and Ce6, thus demonstrating the excellent PDT effect of ACSN/Fe3O4@MSNs-iRGD in tumour treatment.
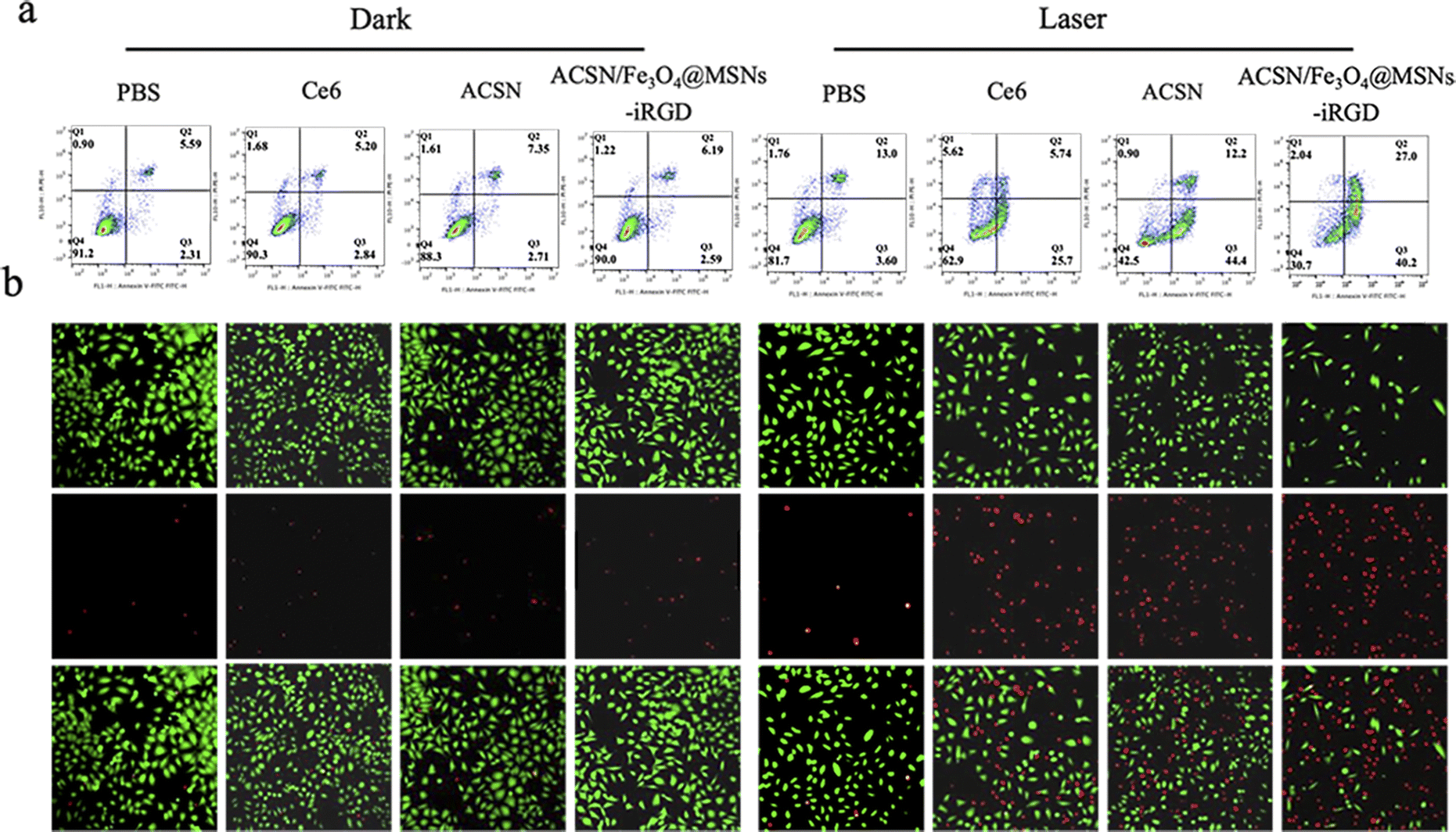 | ||
| Fig. 4 Graphs showing the results of apoptosis and staining of live and dead cells. Apoptosis results of MGC-803 cells treated with the same concentration of Ce6, ACSN, and ACSN/Fe3O4-iRGD in the dark and 633 nm (0.5 W cm−2) laser irradiation (a) and live–dead cell detection by calcein-Ca and PI staining (b). | ||
3.3 In vivo experiments
3.3.1.1 Fluorescence imaging. The use of distinct colours enables the display of varying fluorescence intensities, with the fluorescence intensity decreasing in the following order: red, yellow, green, and blue. As illustrated in Fig. 5a, following tail vein injection of the nontargeted drug ACSN/Fe3O4@MSNs, the fluorescence signal at the tumour site was weak, with the signal predominantly localised in the kidney and liver. In addition, the overall in vivo fluorescence of the drug was faint, suggesting a rapid metabolic rate with no significant aggregation at the tumour site. However, robust fluorescence signals were detected in the tumour area 6 h after the injection of the targeted drug ACSN/Fe3O4@MSNs-iRGD through the tail vein. A high fluorescence signal was maintained in the tumour at 8 and 12 h post injection, with significant metabolism observed in the kidney and liver at 24 h post injection. These findings imply that the iRGD-conjugated drug could effectively accumulate at the tumour site, ensuring the efficacy of PDT. By comparing the fluorescence signal intensities of ACSN/Fe3O4@MSNs and ACSN/Fe3O4@MSNs-iRGD in the tumour, kidney, and liver at the same concentration and time, it was evident that ACSN/Fe3O4@MSNs-iRGD exhibited greater aggregation in the tumour. The aggregation time of the targeted drug in tumours is longer, which could significantly delay metabolism. In contrast, the non-targeted drug ACSN/Fe3O4@MSNs accumulated less in tumours, was metabolised rapidly, and was primarily metabolised by the kidneys in vivo. The targeted drug ACSN/Fe3O4@MSNs-iRGD specifically targeted tumour cells, thereby reducing the impact on other normal tissues and facilitating a more efficient PDT effect.
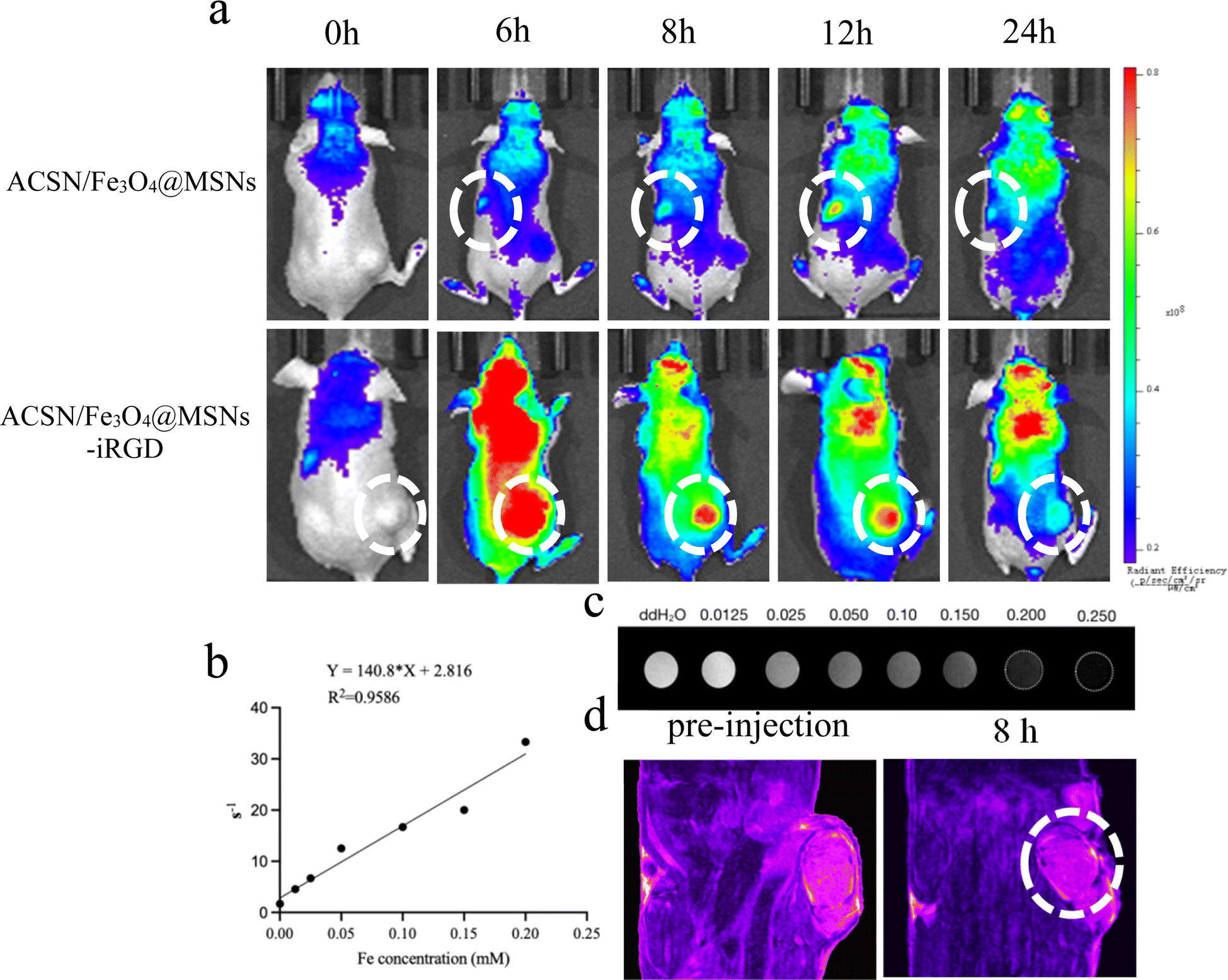 | ||
| Fig. 5 Results of dual-modality imaging in mice. (a) In vivo fluorescence imaging of tumour-bearing mice before and after tail vein injection of drugs (ACSN/Fe3O4@MSNs and ACSN/Fe3O4@MSNs-iRGD); white circles represent the kidney and tumour sites, respectively. Relaxation rates of ACSN/Fe3O4@MSNs-iRGD at different concentrations (b) and T2-weighted MRI images (c); in vivo T2-weighted MRI images (d) of tumour-bearing mice before and after injection of ACSN/Fe3O4@MSNs-iRGD via the tail vein; white circles represent the tumour sites. | ||
3.3.1.2 Magnetic resonance imaging. We examined the T2 relaxation rate (Fig. 5b) and T2-weighted MRI images (Fig. 5c) of ACSN/Fe3O4@MSNs-iRGD at varying drug concentrations. As the drug concentration increased, the T2 relaxation rate increased correspondingly, leading to progressively darker T2-weighted MRI images. Subsequently, following the injection of ACSN/Fe3O4@MSNs-iRGD into the tail vein of the tumour-bearing mice, T2-weighted MRI signals were analysed (Fig. 5d). With the accumulation of the targeted drug ACSN/Fe3O4@MSNs-iRGD in the tumour, significant darkening of the tumour area was observed after drug injection. Consequently, ACSN/Fe3O4@MSNs-iRGD can serve as an MRI contrast agent, facilitating the integration of drug diagnosis and treatment.
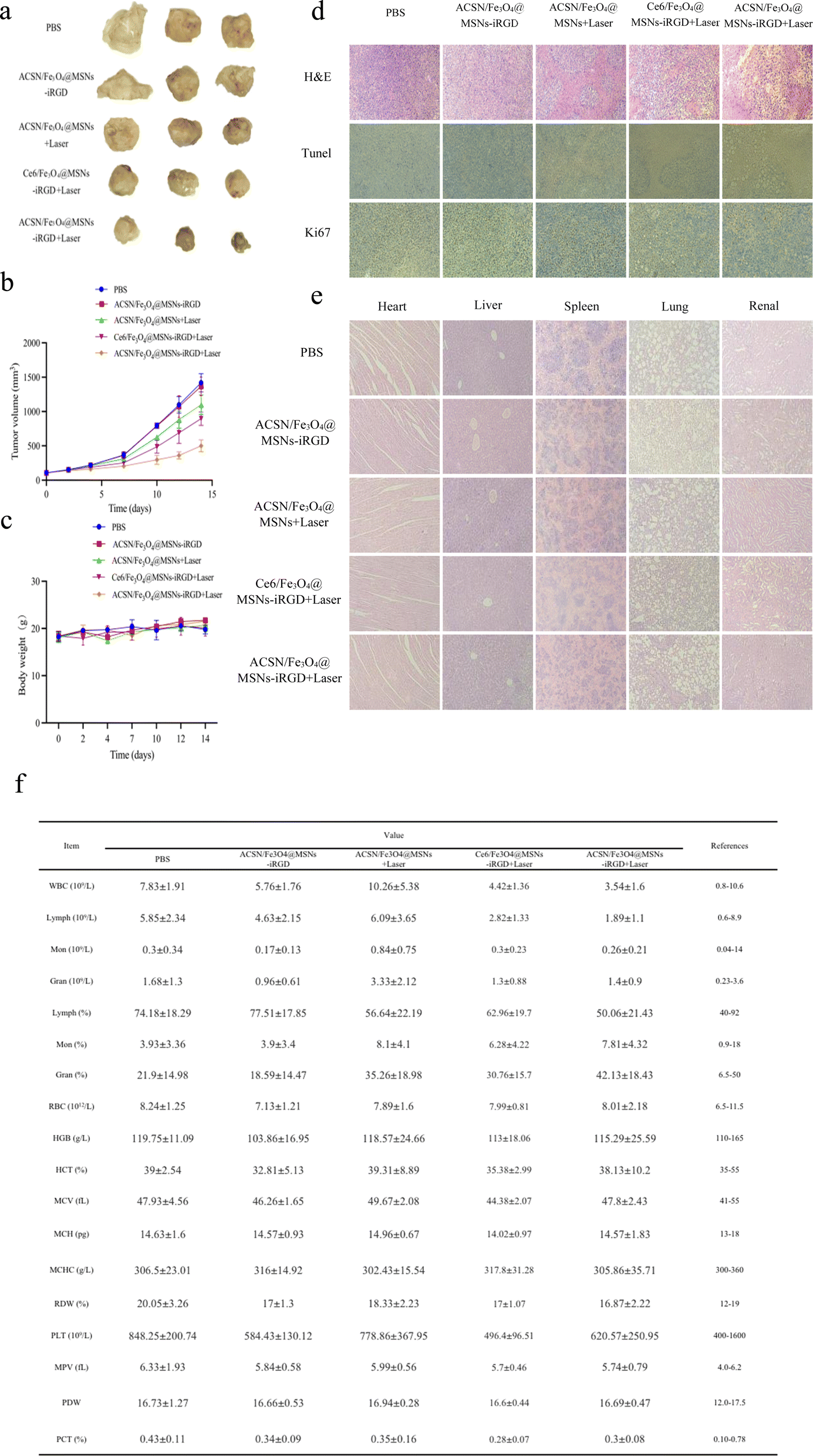 | ||
| Fig. 6 Tumours at the end of 14 days of treatment with PBS, ACSN/Fe3O4@MSNs-iRGD, ACSN/Fe3O4@MSNs + laser, Ce6/Fe3O4@MSNs-iRGD + laser, and ACSN/Fe3O4@MSNs-iRGD + laser (a); changes in tumour volume (b) and body weight (c) of tumour-bearing mice on day 0, 2, 4, 7, 10, 12, and 14 of treatment; immunohistochemical sections with H&E, TUNEL, and Ki67 staining of tumour tissue at the end of treatment (d); H&E staining of major organs (heart, liver, spleen, lung, and kidney) (e) and routine blood analysis (f). | ||
Compared with that in the ACSN/Fe3O4@MSNs + laser group, the drug in the ACSN/Fe3O4@MSNs-iRGD + laser group demonstrated superior tumour targeting guided by the targeted peptide iRGD. Moreover, a greater amount of the drug can accumulate at the tumour site within 6 h of injecting the same amount of drug. Consequently, the ACSN/Fe3O4@MSNs-iRGD + laser group exhibited a more effective PDT effect and prevented excessive drug damage in other tissues and organs. In comparison with the Ce6/Fe3O4@MSNs-iRGD + laser group, ACSN/Fe3O4@MSNs-iRGD + laser group utilised ACSN as a photosensitizer, which is better suited to the hypoxic characteristics of the TME. By inhibiting cellular aerobic respiration, this approach conserves oxygen in the tumour environment and generates more ROS during PDT, resulting in a stronger tumour-killing effect.
4. Conclusion
In summary, this study investigated an active targeted therapeutic drug based on a transmembrane peptide that can be used for MRI imaging and PDT treatment and its inhibitory effect on gastric cancer MGC-803 cells. Compared with the commonly used photosensitizer Ce6, the novel oxygen-economised tumour photosensitizer ACSN combined the superior characteristics of Ce6 and ATO. Ce6 exhibits a high singlet oxygen production rate and low dark toxicity, rendering it suitable for PDT development. The cell respiration inhibitor, ATO, can also damage the respiratory metabolism of tumour cells and induce oxygen retention. This can alleviate the limitations of TME hypoxia during PDT, to a certain extent, thereby enhancing the PDT effect of the photosensitizer Ce6. MRI employs high light–dark contrast images to achieve non-invasive, real-time monitoring using Fe3O4 as the shell core, effectively reducing the T2-weighted relaxation time in MRI and obtaining enhanced dark images, facilitating real-time monitoring of tumour changes through MRI during treatment. Both in vitro cellular uptake and in vivo fluorescence imaging demonstrated that the iRGD exhibited a potent targeting effect, capable of increasing drug uptake in tumour cells and prolonging the retention time of drugs in tumours in vivo, thereby promoting the full potential of PDT. This drug platform demonstrated promising PDT efficacy with significant tumour volume suppression. In PDT, tissue penetration of the laser plays a crucial role in determining the therapeutic efficacy against tumours. Although the 633 nm red laser exhibits excellent tissue penetration ability and holds significant promise for broadening the application prospects of tumour PDT across various anatomical sites, for large or deeply seated tumours, the effectiveness of PDT employing 633 nm laser may be limited. We propose further research, developing oxygen-saving photosensitizers with extended absorption wavelengths. This research is vital for broadening the scope of treating large solid- and deep-seated tumours.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (81971714).Notes and references
- R. L. Siegel, A. N. Giaquinto and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 12–49 CrossRef PubMed.
- E. C. Smyth, M. Nilsson, H. I. Grabsch, N. C. van Grieken and F. Lordick, Lancet, 2020, 396, 635–648 CrossRef CAS PubMed.
- T. Wu and Y. Dai, Cancer Lett., 2017, 387, 61–68 CrossRef CAS PubMed.
- S. Peng, F. Xiao, M. Chen and H. Gao, Adv. Sci., 2022, 9, e2103836 CrossRef PubMed.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- Y. Dai, C. Xu, X. Sun and X. Chen, Chem. Soc. Rev., 2017, 46, 3830–3852 RSC.
- Y. X. Fang, N. Zhang and Y. J. Liu, Chemistry of Life, 2020, vol. 40, pp. 1693–1699 Search PubMed.
- F. Bruno, F. Arrigoni, S. Mariani, A. Splendiani, E. Di Cesare, C. Masciocchi and A. Barile, Radiol. Med., 2019, 124, 243–252 CrossRef PubMed.
- C. Wang, Z. Wang, T. Zhao, Y. Li, G. Huang, B. D. Sumer and J. Gao, Biomaterials, 2018, 157, 62–75 CrossRef CAS PubMed.
- Q. Fu, R. Zhu, J. Song, H. Yang and X. Chen, Adv. Mater., 2019, 31, e1805875 CrossRef PubMed.
- D. Fischerova, G. Santos, L. Wong, V. Yulzari, R. J. Bennett, P. Dundr, A. Burgetova, P. Barsa, G. Szabo, N. Sousa, U. Scovazzi and D. Cibula, Ultrasound Obstet. Gynecol., 2023, 62, 727–738 CrossRef CAS PubMed.
- E. Seeram, Radiol. Technol., 2018, 89, 279CT–302CT Search PubMed.
- Y. Zhang and J. Yu, Diagn. Interv. Radiol., 2020, 26, 176–182 CrossRef PubMed.
- Y. D. Xiao, R. Paudel, J. Liu, C. Ma, Z. S. Zhang and S. K. Zhou, Int. J. Mol. Med., 2016, 38, 1319–1326 CrossRef CAS PubMed.
- J. H. Kim, S. J. Byun, S. G. Park, Y. K. Oh and S. K. Baek, Cancer Res. Treat., 2012, 44, 187–194 CrossRef PubMed.
- J. H. Correia, J. A. Rodrigues, S. Pimenta, T. Dong and Z. Yang, Pharmaceutics, 2021, 13, 1332 CrossRef CAS PubMed.
- S. Kwiatkowski, B. Knap, D. Przystupski, J. Saczko, E. Kedzierska, K. Knap-Czop, J. Kotlinska, O. Michel, K. Kotowski and J. Kulbacka, Biomed. Pharmacother., 2018, 106, 1098–1107 CrossRef PubMed.
- M. W. Dewhirst, Y. Cao and B. Moeller, Nat. Rev. Cancer, 2008, 8, 425–437 CrossRef CAS PubMed.
- L. P. Zhao, R. R. Zheng, H. Q. Chen, L. S. Liu, X. Y. Zhao, H. H. Liu, X. Z. Qiu, X. Y. Yu, H. Cheng and S. Y. Li, Nano Lett., 2020, 20, 2062–2071 CrossRef CAS PubMed.
- S. Wilhelm, A. J. Tavares, Q. Dai, S. Ohta, J. Audet, H. F. Dvorak and W. C. W. Chan, Nat. Rev. Mater., 2016, 1, 16014 CrossRef CAS.
- K. N. Sugahara, T. Teesalu, P. P. Karmali, V. R. Kotamraju, L. Agemy, D. R. Greenwald and E. Ruoslahti, Science, 2010, 328, 1031–1035 CrossRef CAS PubMed.
- X. Cun, J. Chen, S. Ruan, L. Zhang, J. Wan, Q. He and H. Gao, ACS Appl. Mater. Interfaces, 2015, 7, 27458–27466 CrossRef CAS PubMed.
- H. Shokrollahi, Mater. Sci. Eng., C, 2013, 33, 4485–4497 CrossRef CAS PubMed.
- Y. Zhou, G. Quan, Q. Wu, X. Zhang, B. Niu, B. Wu, Y. Huang, X. Pan and C. Wu, Acta Pharm. Sin. B, 2018, 8, 165–177 CrossRef PubMed.
- C. Bharti, U. Nagaich, A. K. Pal and N. Gulati, Int. J. Pharm. Invest., 2015, 5, 124–133 CrossRef CAS PubMed.
- Madhwi, R. Kumar, P. Kumar, B. Singh, G. Sharma, O. P. Katare and K. Raza, Int. J. Pharm., 2017, 519(1–2), 138–144 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2024 |