
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhase behavior of colloidal nanoparticles and their enhancement effect on the rheological properties of polymer solutions and gels
Liu Yanga,
Jiiang Gea,
Hao Wub,
Hongbin Guob,
Jingling Shanb and
Guicai Zhang *a
*a
aSchool of Petroleum Engineering, China University of Petroleum, Qingdao, Shandong 266580, China. E-mail: zhanggc@upc.edu.cn
bSinopec Shengli Oilfield Petroleum Engineering Technology Research Institute, Dongying, Shandong 257091, China
First published on 12th March 2024
Abstract
The interaction between nanoparticles and polymers has been of great interest in colloidal theory and novel materials. For example, the properties of polyacrylamide solutions and gels, which are usually used for conformance control and water shut-off in oilfields, can be improved with the addition of nanoparticles. This underlying mechanism and its applicability are investigated in this paper. A strong relationship between the phase behaviors of nanoparticles in polymer solutions and their enhancement effect on the rheology of the nanocomposite polymer solutions and gels was observed. Experiment results showed that the stability of nanoparticles was dependent on several factors, including pH, salinity, and polymer type. At neutral pH conditions, the tendency of the aggregation of nanoparticles was strengthened upon increasing the salinity, polymer concentration, and electronegativity of the polymers. Rheological measurements showed that nanoparticles could improve the viscosity of polymer solutions or the fracture stress of gels only if nanoparticles were aggregated in the corresponding systems. In addition, these rheological parameters significantly increased with increasing salinity and nanoparticle concentration. As a result, the mobility ratio of polymer solutions may be increased several times by the addition of nanoparticles. Referring to the gels, their rupture pressure gradient in the ideal model was also found to increase with nanoparticle concentration. In particular, if the nanoparticle concentration was sufficiently high (reaching 2%), the formed gels would not be destroyed by the injected water, but rather functionally act as a porous medium for permeation.
1 Introduction
In recent decades, the interactions between nanoparticles and polymers have been of great interest in both research and industrial applications, including water treatment,1,2 nanoparticle printing,3 and nanocomposite polymers4,5/gels.6–8 Studies have shown that nanoparticles in polymer solutions can display various phase behaviors depending on their physicochemical interactions and the environment (e.g., temperature, pH, and salinity).9–14 Meanwhile, the rheology of polymer-based materials may be affected, even significantly improved.15–19 Therefore, understanding the underlying mechanism has received increasing interest to solve industrial problems.For example, polyacrylamide and its derivatives are the most widely used polymers for conformance control in oilfields. These polymers have the potential to improve the mobility ratio of the displacing phase and displaced phase by increasing the viscosity, thereby leading to better sweep efficiency.20–22 On the other hand, if these polymers react with multivalent ions (e.g., Al3+, Cr3+, and Cd4+) or several organic crosslinkers (e.g., phenol-formaldehyde resin and polyethyleneimine), the formed polymer-based gels are able to block the advantageous channels of the formation. However, the properties of these polymers are sensitive to the condition of the reservoir.23–25 In high-salinity environments, the viscosity of the polymer solution would be reduced, as the polymer chains tend to adopt curled conformations. Moreover, the polyacrylamide molecules face risks of severe oxidative degradation and hydrolysis reaction at elevated temperatures.
Recently, attempts to utilize nanotechnology in the petroleum industry have been widely reported. Elhaei et al.26 found that a silica suspension could increase the viscosity of a polymer solution above a certain concentration, and the critical concentration was related to the identity of the polymer. Ansari27 observed that nanocomposite polymer solutions exhibited stronger elastic response in creep and creep-recovery experiments. Besides, several papers claimed that the strength and thermal stability of polymer-based gels may be significantly improved by the addition of nanoparticles.28–33 Referring to the underlying mechanism, most of the previous studies focused on the interactions, particularly the hydrogen bonds, between the polymer chains and the nanoparticle surface. However, others argued that the weak interactions were inadequate to make such improvements.
In nanoparticle–polymer mixed solutions, several contributions from non-DLVO force (e.g., hydration, depletion, and hydrophobicity) also play essential roles in the stability of nanoparticles.34 Polymer chains may either absorb on the nanoparticle surface or remain free in the solution, and the phase behaviors of the mixture are affected by both the absorbed and non-absorbed polymers. As the polymer concentration is far below the coverage saturation of the nanoparticle surface, bridging flocculation occurs, resulting in the destabilization of the colloidal suspensions. When the polymer concentration is increased to the coverage saturation, the nanoparticles might be stabilized by the steric repulsion. Furthermore, as the polymer concentration increases beyond saturation, the non-absorbed polymers lead to depletion interaction, exhibiting both short-range attraction and long-range repulsion. As a result, the nanoparticles are flocculated at a relatively lower polymer concentration (higher than the saturation concentration) and re-stabilized at a higher polymer concentration, known as depletion flocculation and depletion stabilization, respectively. In conclusion, the phase behaviors of nanoparticles in polymer solutions are supposed to change as the nanoparticle surface transitions from the undersaturated regime to the oversaturated regime of adsorption.
In practical cases, the above interactions may co-exist and not be distinguished clearly. A reentrant phase behavior has been commonly observed in nanoparticle–polymer systems. For example, Kumar et al.35 investigated the phase behaviors of colloidal silica nanoparticles in polyethylene glycol solutions. In the absence of electrolyte, the mixtures remained clear irrespective of the polymer concentration, even at polymer concentrations of 10 wt%. In contrast, mixtures with electrolyte were transformed from clear to turbid and back to clear with increasing polymer concentration, meaning that nanoparticle aggregation (two-phase system) took place at the intermediate polymer concentration. Besides, the two-phase regime was broadened with the addition of electrolyte. In other words, the addition of electrolyte allowed the transformation from one-phase to two-phases to take place at a lower polymer concentration, while reentrant stabilization occurred at a higher polymer concentration.
Nowadays, scattering methods have been used to analyze microscopic structures. In Kawaguchi's review,36 the structure of aggregates in SiO2 nanoparticle-polymer systems fell mainly into two classifications: (i) aggregates with self-similar structures, in which the particles are in direct contact and caused by the addition of strongly charged polymer at a low pH or a high ionic strength; and (ii) aggregates with structures displaying strong short-range order but heterogeneity at large distances, in which the silica particles are prevented from direct contact by weakly charged or neutral polymers. More specially, Kim et al.34 proved that the structures of aggregates are significantly distinctive in each phase behavior (from bridging flocculation to depletion stabilization) by small angle X-ray scattering.
In our recent study, a strong relationship between the phase behavior of nanoparticles in polymer solutions or gels and their rheology was found. In this article, the dispersion stability of colloidal silica nanoparticles in polymer solutions was studied. The rheology of the nanocomposite polymer solutions and polymer-based gels corresponding to each phase behavior were also investigated. The results showed that the solutions in two-phase exhibited an enhancement in rheology, which is assumed to be due to the transition of the nanoparticles from individual to ordered aggregates. We hope our work helps to clarify the mechanism of nanocomposite polymers and provide ideas for system designs.
2 Materials and methods
2.1. Materials
Two kinds of polymer, i.e., polyacrylamide (PAM) and partially hydrolyzed polyacrylamide (HPAM), were kindly donated by SNF (China) Flocculant and used as received. Both polymers had molecular weights of 12 × 106 g mol−1 and their hydrolysis degree was <0.3% and 10%, respectively. Colloidal silica nanoparticles (Ludox AS-30, 30 wt%, specific surface area = 220 m2 g−1, and density = 2.37 × 103 kg m−3, according to the supplier) were purchased from Sigma-Aldrich. Unless otherwise specified, the nanoparticles were used after diluting to a certain concentration. Besides, three analytically pure materials were purchased from Sinopharm Chemical Reagent: chromium acetate was used as a crosslinker, hydrochloric acid (HCl) and sodium chloride (NaCl) were used to adjust the pH and salinity, respectively.2.2. Preparation and phase behavior
NaCl brine of three different salinities (1 wt%, 2 wt%, and 4 wt%) were used to prepare the following solution. Stock solutions of polymer and crosslinker were firstly prepared with the corresponding brines. Polymer powders were gradually added to the brine and stirred for 12 h at 100 rpm. To avoid pH variation due to the addition of nanoparticles, the colloidal suspensions were diluted and adjusted to neutral pH (6.9–7.1) before mixing. Finally, the nanocomposite polymer solutions (or gellants) were obtained by mixing the stock solutions and brines proportionally, and then stirred for another hour. For the inorganic polymer-based gels, the concentration of the crosslinker (chromium acetate) was fixed at 0.0417 wt%. All solutions were fully degassed by a vacuum pump before further experiments took place.After preparation, the polymer solutions were transferred into sample bottles and aged in a 60 °C bath for a certain number of hours. After cooling down to room temperature, the phase behavior of the solutions was determined by measuring their transmittance at 633 nm using a spectrophotometer. Distilled water was used as the reference solution.
2.3. Rheological measurements
Rheological measurements of polymer solutions and gels were performed on an Anton Paar MCR 92 rheometer, using a concentric cylinder with bob diameter and cup diameter of 38.731 mm and 42.008 mm, respectively.For polymer solutions, dynamic viscosity of the samples was measured at room temperature, at shear rate ranging from 0.1 s−1 to 20 s−1. After the measurements, the zero-shear viscosity (η0) of the solutions was obtained from the software, based on the Carreau–Yauda model.37
Rheological measurements of the gels were conducted in two stages. The temperature was fixed at 60 °C at both stages, and a thin layer of mineral oil was added over the samples to prevent evaporation. Firstly, the gelation reactions were monitored by time sweep measurements at an oscillation amplitude of 1% and a frequency of 1 rad s−1. The dynamics of storage modulus G′ and loss modulus G′′ were recorded as a function of time. The gelation process usually took 4–6 h, after which the moduli reached a plateau. The “in situ gelling” method aims to avoid the wall-slip effect during measurements at large deformations. Afterward, the mechanical properties of the gels were measured by amplitude sweep experiments, in which the frequency was fixed at 1 rad s−1, and the strain (γ) ranged from 1% to 2000%. Parameters stress (σ), G′, and G′′ were recorded as a function of strain. The gels exhibited a typical elastic response, where G′ was much larger than G′′. Thus, G′′ was neglected while analyzing the rheological data for simplicity. It is worth noting that the stress response in non-linear deformation is no longer sinusoidal. The apparent modulus GA, calculated as stress/strain, is introduced to indicate the elastic response of the polymer gels. Therefore, GA is equal to the plateau modulus G0 (approximated by G′) in the linear viscoelastic region. The fracture point was defined as the strain at which a decrease in GA occurred.
2.4. Rupture behaviors of bulk gels
Fig. 1 provides a flowchart of the rupture experiments. The rupture process of the bulk gels was studied in an ideal model, which consisted of two polymethyl methacrylate plates and was tightly bolted without spacers. As shown in Fig. 1, a main channel and a connected U-shaped channel were etched on the inner surface of one plate. The main channel was linear, with a section size of 3 mm × 3 mm and a length of 120 mm, and was used to store the gels. The U-shaped channel acted as a three-way valve to remove air before the rupture experiments.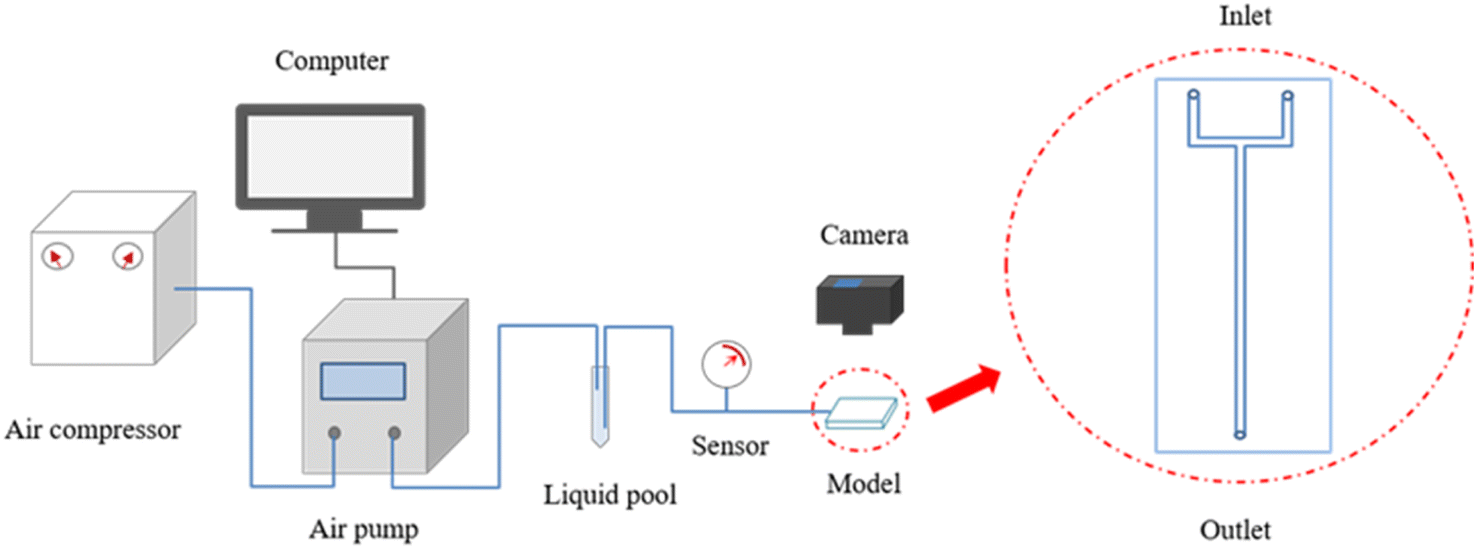 | ||
| Fig. 1 Flowchart of rupture experiments. | ||
First, approximately 0.7 mL of gellant (8 cm in length) was slowly injected into the main channel through the outlet, and the model was slightly tilted until the gellant flowed to the junction of the channels. It should be noted that it was necessary to have a slug of air at the end of the main channel to balance the air pressure during heating. Second, the model was placed in an oven at 60 °C for 12 h and then cooled down. Experiments were conducted at room temperature. A brine of 1 wt% NaCl was dyed red and injected through an inlet of the U-shaped channel by a Fluigent air pump. The output pressure of the pump was set to rise linearly at a rate of 0.1 kPa s−1, and the inlet pressure of the model was recorded by a pressure sensor. Finally, the rupture behavior of the bulk gels was recorded by a digital camera at 50 Hz.
3 Results and discussion
3.1. Phase behaviors of nanoparticles in polymer solutions
First, the stability of the nanoparticles in the brines was evaluated. As displayed in Fig. 2(a), the SiO2 nanoparticles were stable in the 1 × 104 mg L−1 NaCl brine. All the suspensions remained clear without precipitate after aging for 24 h at 60 °C. In contrast, the stability of the nanoparticles varied depending on their concentration in the 2 × 104 mg L−1 NaCl brine (Fig. 2(b)). It was observed that the suspensions became turbid as the nanoparticle concentration increased. For example, the transmittance of the 0.25% nanoparticle suspension was close to 90%, which decreased to <10% at a concentration of 1.0%, meaning large aggregates were formed at the higher nanoparticle concentration. As the salinity increased to 4 × 104 mg L−1, flocculation was observed at relatively lower nanoparticle concentrations (<1.0%). However, if the nanoparticle concentration was above 1.50%, the suspensions became turbid but homogenous from a macro perspective. | ||
| Fig. 2 Phase behaviors of nanoparticles in NaCl brines of different concentrations: (a) 1 × 104 mg L−1 NaCl, (b) 2 × 104 mg L−1 NaCl, and (c) in 4 × 104 mg L−1 NaCl. | ||
Most of the phase behaviors of nanoparticles in aqueous phase are governed by the DLVO theory, which mainly accounts for van der Waals attraction and electrostatic repulsion. In a stable system, the above two interactions are in balance and dynamic with the Brownian motion of nanoparticles. However, excessive salt may compress the diffused electric double layer of the nanoparticles and reduce their electrostatic repulsion, thus resulting in flocculation or gelation of the nanoparticles. It should be noted that only flocculation was observed in this paper because the nanoparticle concentration was not sufficiently high.
Fig. 3 displays the phase behavior of nanoparticles in PAM solutions and HPAM solutions, in which all nanoparticle concentrations were constant at 0.5%, and the polymer concentration ranged from 0.01% to 0.8%. Similar to the phenomenon in brine, the nanoparticles remained stable in both the PAM and HPAM solutions at a relatively low salinity of 1 × 104 mg L−1 (Fig. 3(a)). The transmittance of these solutions gradually decreased from 98% to 80% as the polymer concentration increased. When the salinity was 2 × 104 mg L−1, the phase behavior of the nanoparticles was divided into three types based on polymer concentration (Fig. 3(b)). The mixtures were homogenous at the low polymer concentration regime (0.01–0.08%). However, flocculation was found to be promoted by polymer concentration in the intermediate polymer concentration regime (0.1–0.4%). Meanwhile, the transmittances of the supernatants became higher, implying that bridge flocculation occurred between the polymer and the nanoparticles. In particular, it is noted that HPAM exhibited a stronger tendency of bridge flocculation than PAM, based on the higher transmittance and larger amount of flocculate of the former. As the polymer concentration increased to 0.6%, the mixtures re-stabilized and became homogenous again. At a salinity of 4 × 104 mg L−1, both polymers and nanoparticles flocculated at polymer concentrations of <0.4%, as shown in Fig. 3(c). Furthermore, when the polymer concentration increased to 0.6%, both the combined PAM solutions and HPAM solutions became homogenous again due to steric repulsion, similar to the phenomenon observed in the 2 × 104 mg L−1 brine. It is suggested that the re-stabilization regime strongly depended on the polymer concentration.
 | ||
| Fig. 3 Phase behaviors of nanoparticles in polymer solutions of different concentrations: (a)–(c) HPAM solutions at salinities of 1 × 104 mg L−1, 2 × 104 mg L−1, and 4 × 104 mg L−1, respectively. (e)–(f) PAM solutions at salinities of 1 × 104 mg L−1, 2 × 104 mg L−1, and 4 × 104 mg L−1, respectively. | ||
Fig. 4 shows the phase behavior of nanoparticles with different concentrations in 0.4% HPAM solutions and 0.4% PAM solutions, both with salinities of 1 × 104 mg L−1 NaCl. Significant differences were found between these two kinds of polymer solutions. Referring to the HPAM solutions, transmittance dramatically decreased to 2.7% when the nanoparticle concentration increased to 1.0%, suggesting that higher amounts of nanoparticles strongly exacerbated their aggregating. In contrast, few aggregations formed in the PAM solutions even at nanoparticle concentrations of 3.0%; their transmittance only slightly decreased from 91.9% to 89.4%, as seen in Fig. 4(b), indicating that the particles remained at nanometer scale during the aging time.
 | ||
| Fig. 4 Phase behaviors of nanoparticles with different concentrations in polymer solutions: (a) 0.4% HPAM solutions and (b) 0.4% PAM solutions. | ||
The results implied that the flocculation of nanoparticles in polymer solutions occurred at only certain nanoparticle concentrations, so that aggregate might be formed. Therefore, either increasing the polymer concentration or the nanoparticle concentration may be conducive to flocculation in some cases (e.g., nanoparticles in HPAM solutions at a relatively low concentration). Besides, the difference of phase behaviors in HPAM solutions and PAM solutions was unexpected. In theory, the HPAM molecules should not exhibit a stronger interaction with SiO2 nanoparticles than PAM molecules. Firstly, the carboxyl groups on HPAM molecules do not easily form hydrogen bonds with the surface of nanoparticles, as compared with the amide groups on the PAM molecules. Secondly, the negatively charged carboxyl groups would provide electrostatic repulsion with the SiO2 nanoparticles, which also bear negative charge, thus weakening the interaction between polymers and nanoparticles. However, the experimental results indicated that stronger interaction occurred in the HPAM solutions. This phenomenon is assumed to be related to the microstructure of the polymer network. Our previous study38 showed that HPAM exhibited a smaller entanglement concentration than PAM, meaning that HPAM polymers were more entangled and heterogeneous than PAM. As Fig. 5 illustrates, the polymer network of PAM is relatively homogenous, and the nanoparticles are uniformly distributed on the polymer molecules. On the other hand, physical entanglements exist around the uncharged amide groups, and the negatively charged segments repel each other, making the HPAM polymer network heterogeneous. As a result, the nanoparticles accumulate around the entangled junctions, as they also bear negative charge. Consequently, aggregations of the nanoparticles are easily formed in the HPAM solutions.
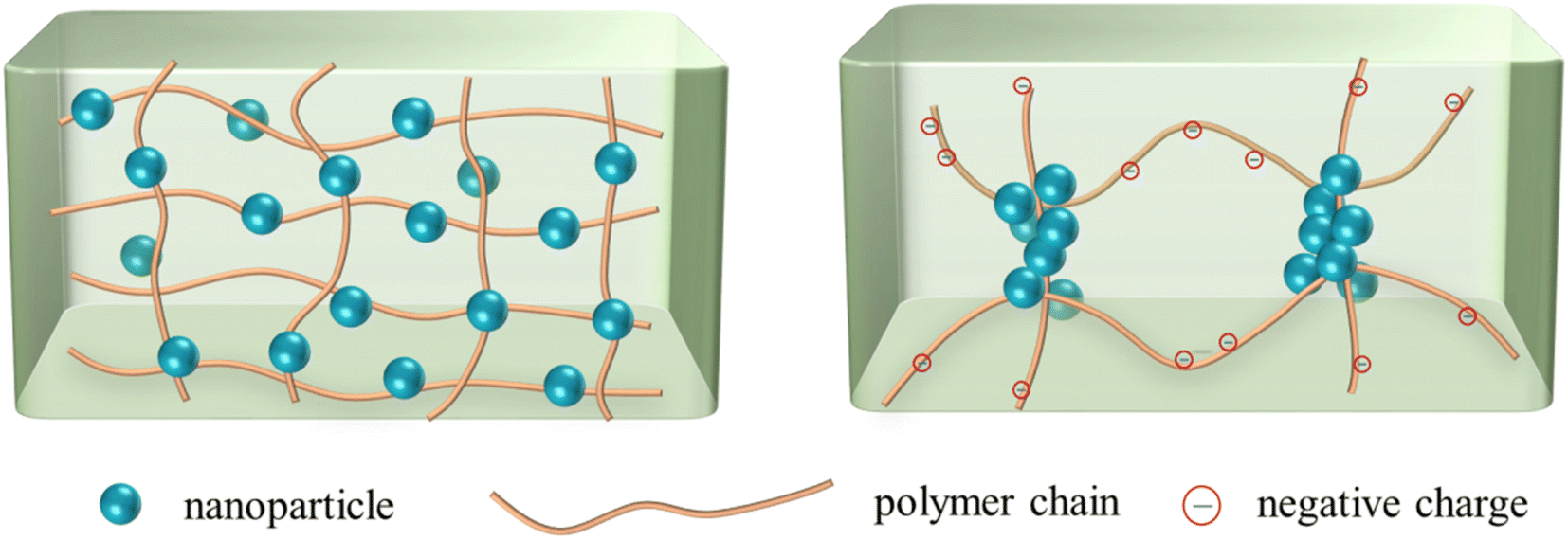 | ||
| Fig. 5 Sketch map of nanoparticles in homogenous PAM solution (left) and entangled HPAM solution (right). | ||
3.2. Rheology of polymer solutions and gels with nanoparticles
Rheological experiments were conducted referring to the two typical systems of Fig. 4. The results are displayed in Fig. 6 and 7, where the dots in the figure represent the measured data, and the dashed lines represent the fitting curves based on the Carreau–Yasuda model. The zero-shear-viscosity η0 is obtained from the fitting curves at a shear rate of 0.001 s−1.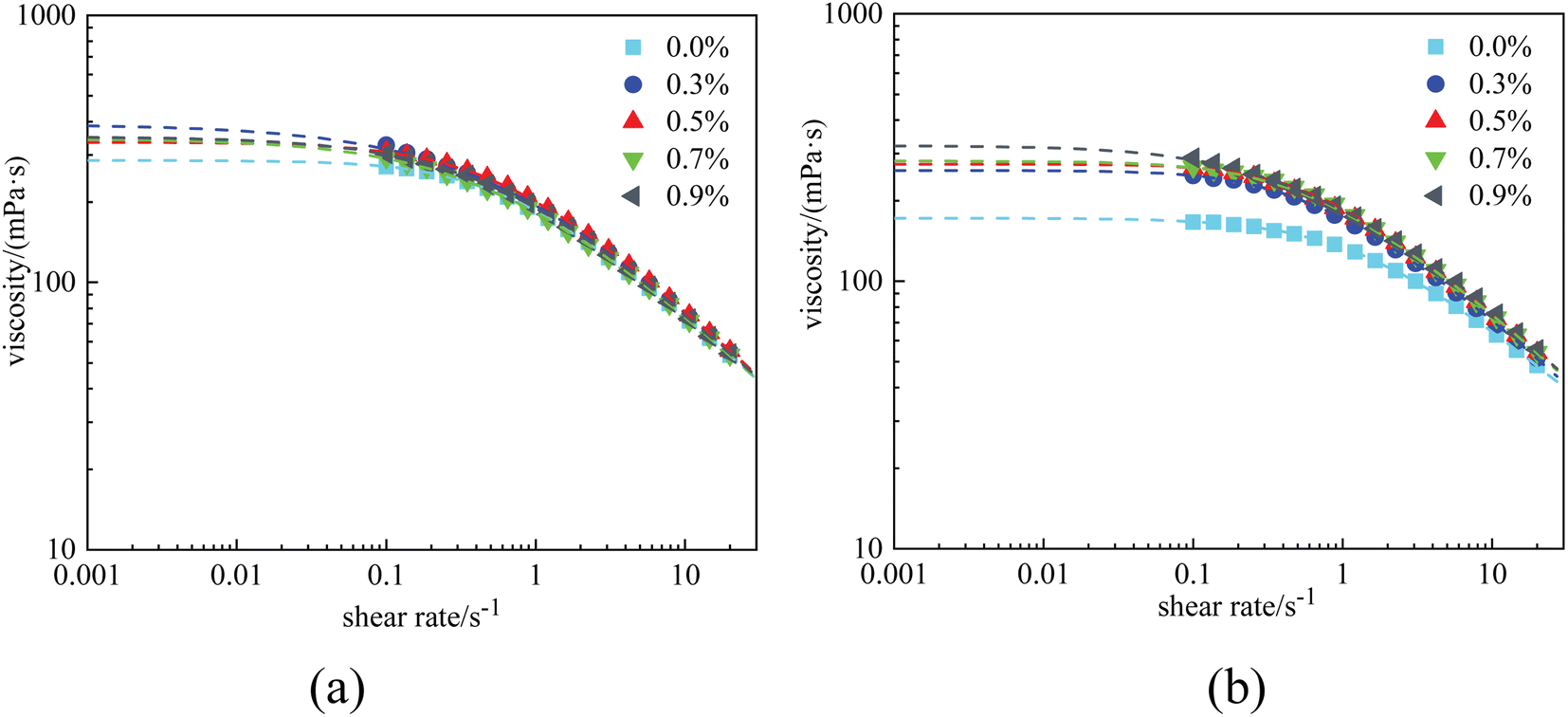 | ||
| Fig. 6 Viscosity curves of HPAM solutions at 1 × 104 mg L−1 with different nanoparticle concentrations: (a) before aging and (b) after aging. | ||
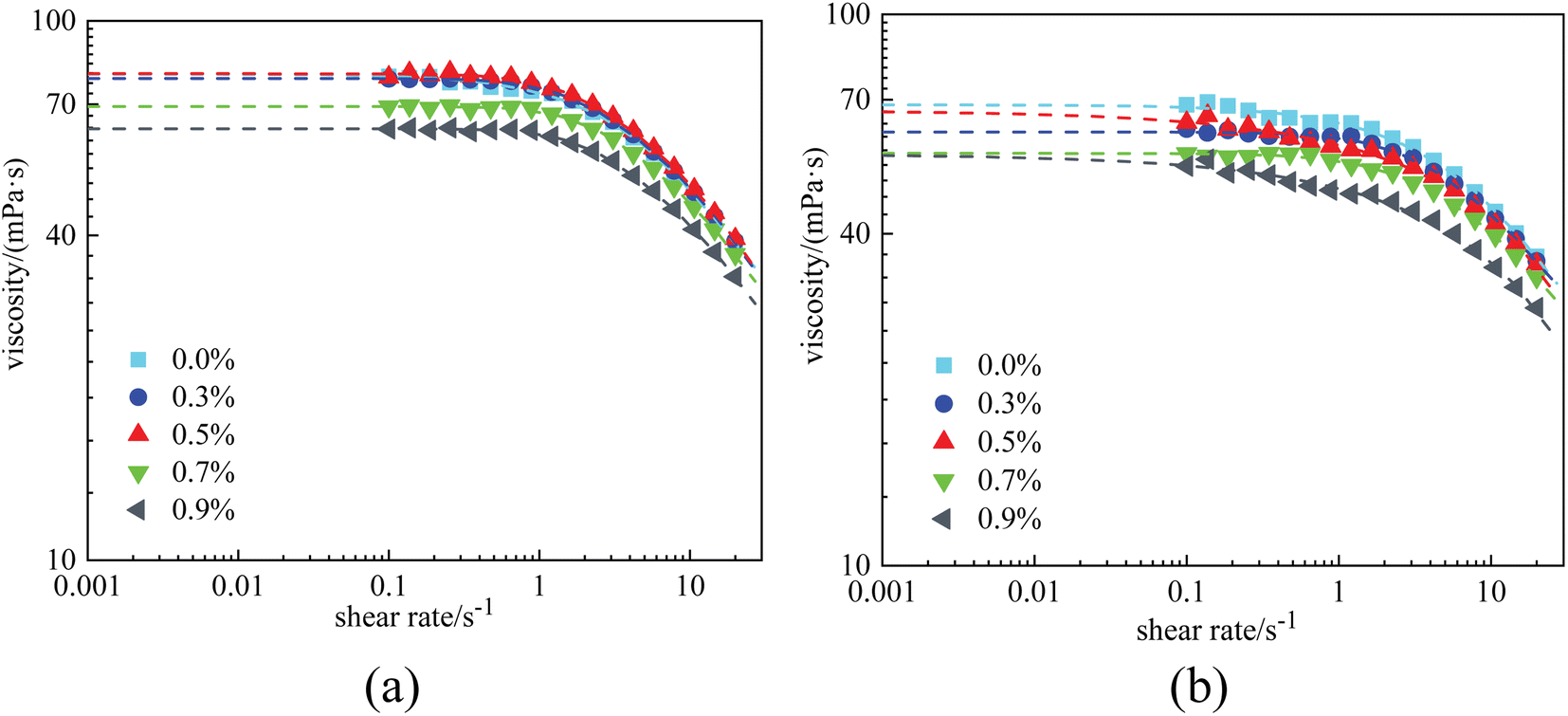 | ||
| Fig. 7 Viscosity curves of PAM solutions at 1 × 104 mg L−1 with different nanoparticle concentrations: (a) before aging and (b) after aging. | ||
Fig. 6(a) shows that the presence of nanoparticles slightly increased the viscosity of the HPAM solutions. Before aging, the η0 of the 0.4% HPAM solution without nanoparticles was 286.3 mPa s, whereas that of the combined solutions with 0.3–0.9% nanoparticles fluctuated in the range of 334.8 mPa s to 349.4 mPa s. Due to the oxidative degradation of polymers, the η0 of the HPAM solution decreased to 172 mPa s after aging for 12 h. In contrast, the reduction in the viscosity of the combined solutions was less significant, ranging from 269.6 mPa s to 320.4.8 mPa s. Besides, the η0 of the combined solutions was found to increase with nanoparticle concentration (Fig. 6(b)). As a conclusion, the addition of nanoparticles enhanced the rheological properties of the HPAM polymer solutions. This experimental result corresponds to that of previous papers, which claimed that there existed hydrogen bonding interactions between –Si–O– and –Si–OH on the surface of the nanoparticles, and amide groups and carboxylic groups on the polymer chains. Therefore, the thermal motion of polymer chains is restricted, resulting in an increased viscosity. In other words, the nanoparticles act as a physical crosslinker in these combined solutions.
Fig. 7(a) displays the viscosity curves of the combined PAM solutions before aging. It was observed that the nanoparticles had almost no impact on viscosity at low nanoparticle concentrations. The zero-shear-viscosity was almost constant (80 mPa s) when the nanoparticle concentration was ≤0.5%. However, as the nanoparticle concentration increased to 0.7% and 0.9%, the η0 of the combined solutions decreased to 55.9 mPa s and 55.4 mPa s, respectively. More significantly, after aging for 12 h, η0 was found to gradually decrease with nanoparticle concentration. This unexpected phenomenon revealed that in a stable combined solution, that is, when no aggregation of the nanoparticles was formed, the interaction between nanoparticles and polymers was adverse to the viscosity of the polymer solutions.
In the last decade, nanoparticles have been widely used as additives in polymer gels for conformance control and water shut-off in oilfields. Many papers reported that nanoparticles could improve the plugging efficiency and the thermal stability of polymer gels. Referring to rheology, current studies mainly focus on the linear deformation properties of polymer gels. In our opinion, linear rheology is related to the stability of polymer gels, rather than the plugging efficiency. At the microscopic scale, the modulus of materials is determined by the distance between neighboring junctions in the gel network, commonly known as the mesh size, ξ.39 As for polymer gels, both the crosslinking density and the polymer entanglements contribute to the densification of mesh size. As a consequence, an S-shaped growth curve of modulus versus time is detected during the gelation. However, once the 3D network of polymer gels encounters instability (i.e., breaking of polymer chains or crosslinking bonds), the modulus subsequently decreases.
Rupture pressure reflects the maximum pressure that a gel slug can endure before rupture and is usually measured to evaluate the plugging efficiency of a gel treatment. Several models have been proposed to relate the rheological parameters of polymer gels with their rupture pressure. For example, Ganguly40,41 regarded the deformation of gels in regular tubes as shear deformation, and derived the following equation:
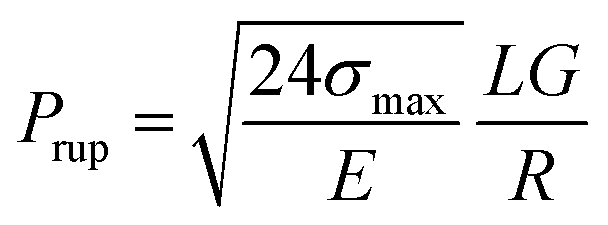 | (1) |
| σmax/(G0γmax) = exp(γmax/γ*), | (2) |
Fig. 8 displays the deformation curves of HPAM gels with 0.4% polymer and different nanoparticle concentrations, plotting the apparent modulus GA as a function of strain, where a strain hardening behavior is observed. Specifically, the measured GA was found to increase with strain after the boundary of the linear viscoelastic region. This behavior, which has been sufficiently investigated in our previous studies, is mainly the result of temporary junctions that form at large deformation (i.e., temporary entanglement of polymer chains at large deformation). Effect of the nanoparticles on the deformation curves of HPAM gels mainly consists of two aspects: Firstly, G0 increases with the amount of nanoparticles, indicating that the crosslinking density is increased by the physical interaction between nanoparticles and polymers. Secondly, the stiffening tendency is also promoted with the crosslinking density, which is resulted from the increase of nanoparticle concentration. As a result, the measured fracture stress is sharply increased by the addition of nanoparticles. According to eqn (2), the second mechanism is considered to play an essential role in the increase in fracture stress, as the γ* is an exponential term.
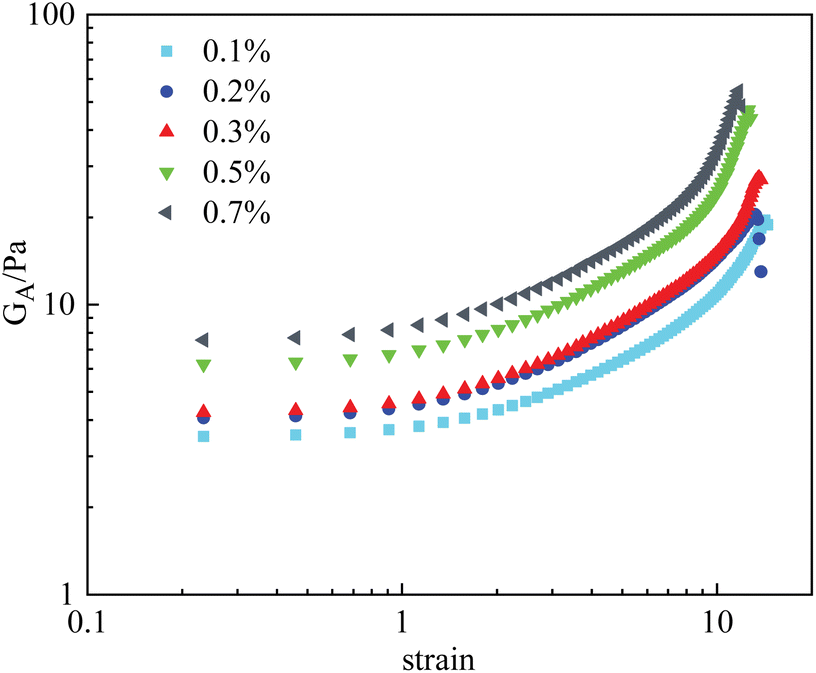 | ||
| Fig. 8 Measured strain vs. GA curves of HPAM gels with different nanoparticle concentrations. | ||
By comparison, the effect of nanoparticle concentration on the rheological properties of the PAM gels was less significant. Fig. 9 displays the γ vs. GA curves of PAM gels with different nanoparticle concentrations. When the nanoparticle concentration was <0.5%, the deformation curve was barely influenced by nanoparticle concentration. When the nanoparticle concentration was ≥0.7%, the mechanical properties of the PAM gels were obviously enhanced, similar to the HPAM gels. However, based on the experimental results shown in Fig. 7, increasing the nanoparticle concentration was not able to increase the viscosity of the polymer solution under the same experimental conditions. It is indicated that aggregation of nanoparticles were formed during the gelation reaction of the PAM gels.
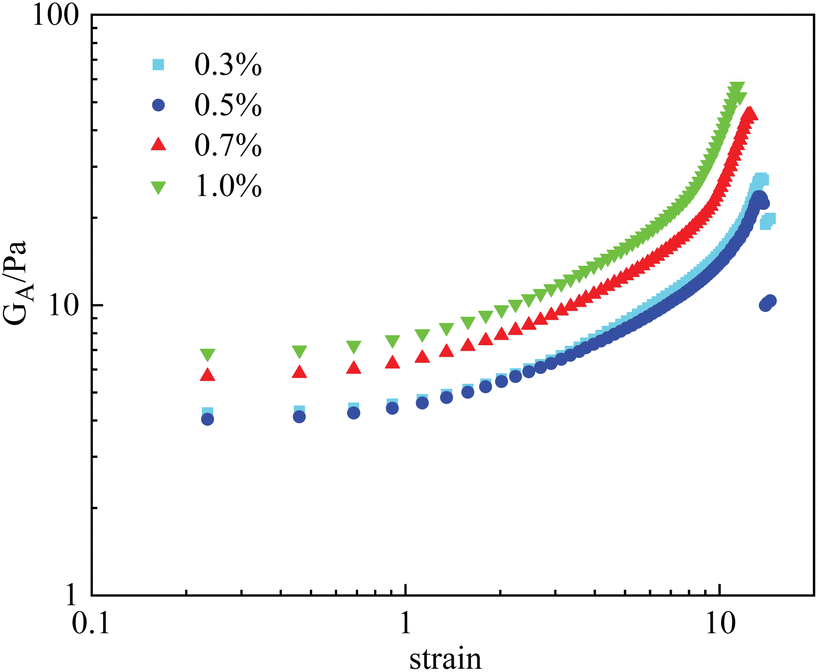 | ||
| Fig. 9 Measured strain vs. GA curves of PAM gels with different nanoparticle concentrations. | ||
To further investigate the effect of nanoparticles on the rheological properties of gels, Fig. 10 summarizes the G0 and fracture stress of the HPAM and PAM gels with different nanoparticle concentrations. It was observed that the G0 of both the HPAM and PAM gels gradually increased with nanoparticle concentration. However, the growth of fracture stress did not synchronize with, but rather lagged behind, the moduli. In other words, although the modulus of the gels was higher at lower nanoparticle concentrations, their toughness was not influenced until the nanoparticle concentration reached a certain value. For the PAM gel, the critical concentration of nanoparticles sufficient to improve the toughness was 0.5%, whereas it was 0.3% for the HPAM gels. Therefore, the nanoparticles exhibited a stronger tendency to improve the fracture stress of the HPAM gels compared to the PAM gels, which agrees with the results of the phase behavior experiments.
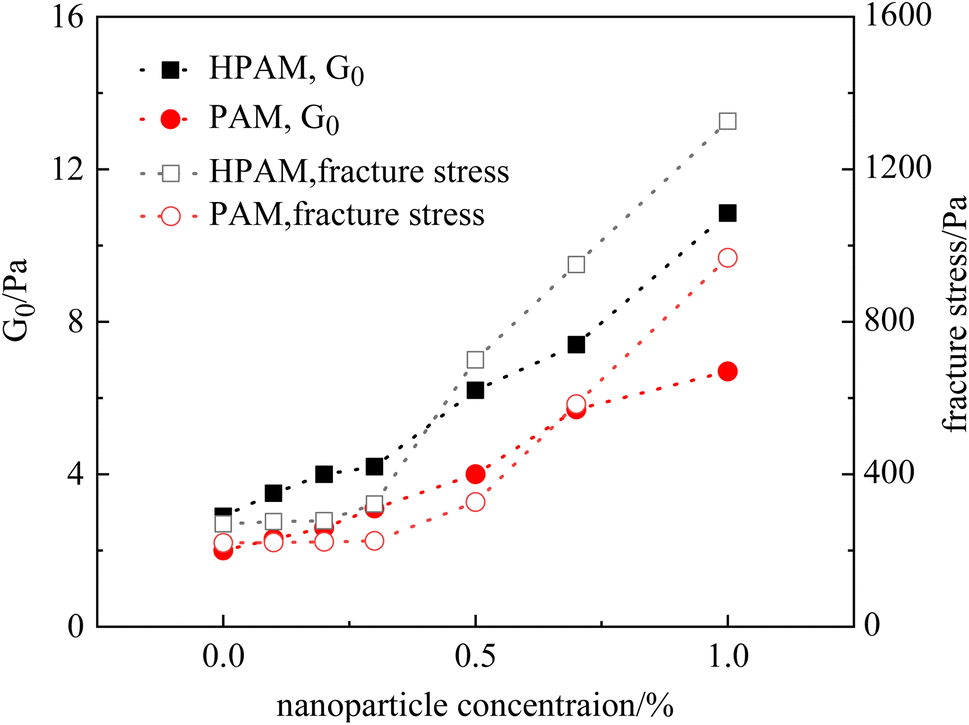 | ||
| Fig. 10 Summary of the measured rheological parameters of nanocomposite gels. | ||
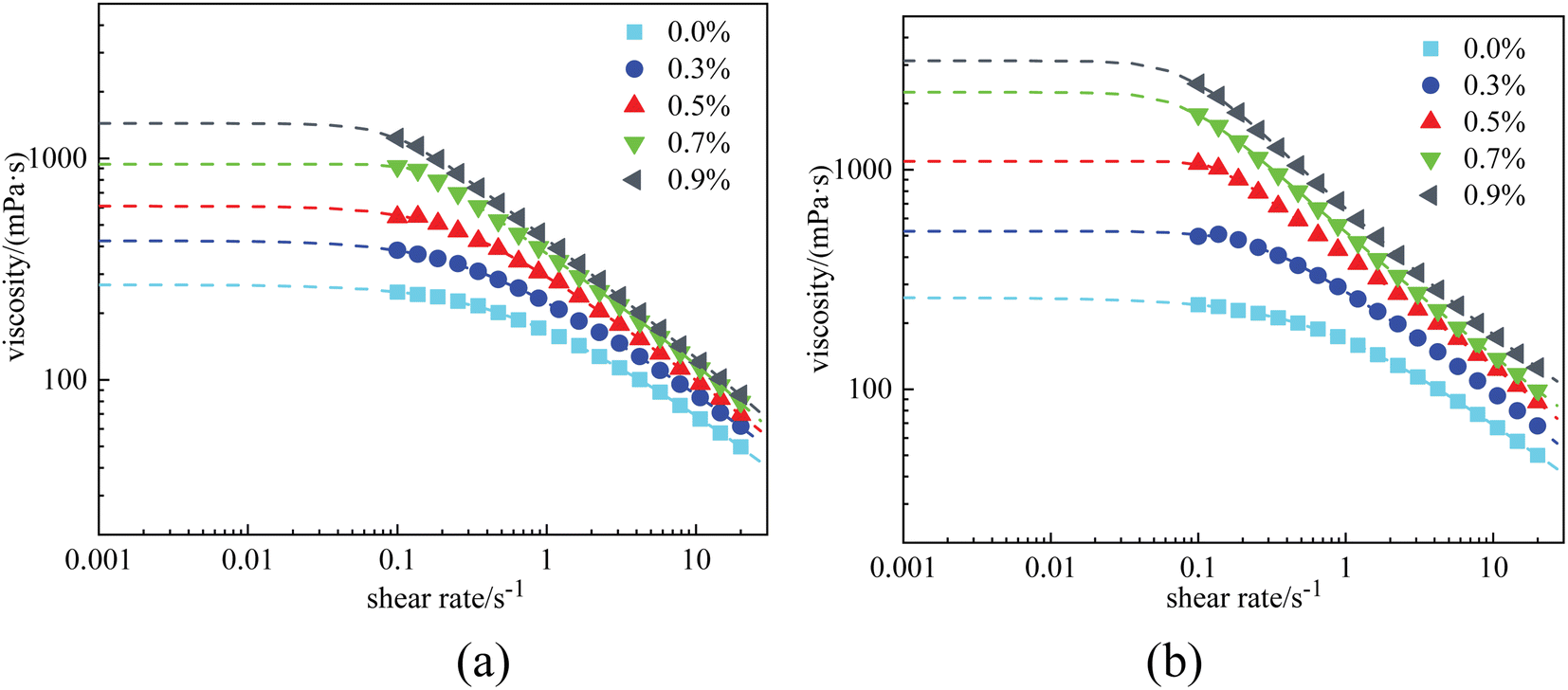 | ||
| Fig. 11 Viscosity curves of 0.4% HPAM solutions with different nanoparticle concentrations after aging: (a) at a salinity of 2 × 104 mg L−1, and (b) at a salinity of 4 × 104 mg L−1. | ||
A similar phenomenon was also observed in the HPAM gels. Fig. 12 plots the fracture stress of the gels vs. nanoparticle concentration at salinities ranging from 1 × 104 mg L−1 to 4 × 104 mg L−1. At a salinity of 2 × 104 mg L−1, the growth trend of the curve coincided with that measured at 1 × 104 mg L−1, staying constant at nanoparticle concentrations of <0.3%, and increasing at nanoparticle concentrations of >0.3%. However, the fracture stress of the gels was relatively higher at 2 × 104 mg L−1 than that at 1 × 104 mg L−1. In contrast, at a higher salinity of 4 × 104 mg L−1, fracture stress continuously increased with nanoparticle concentration, even at low concentrations. In general, nanoparticles effectively improved the mechanical properties of the HPAM gels. Within the scope of our experiments, the fracture stress of the gels could be increased to 3–5 times with the addition of nanoparticles.
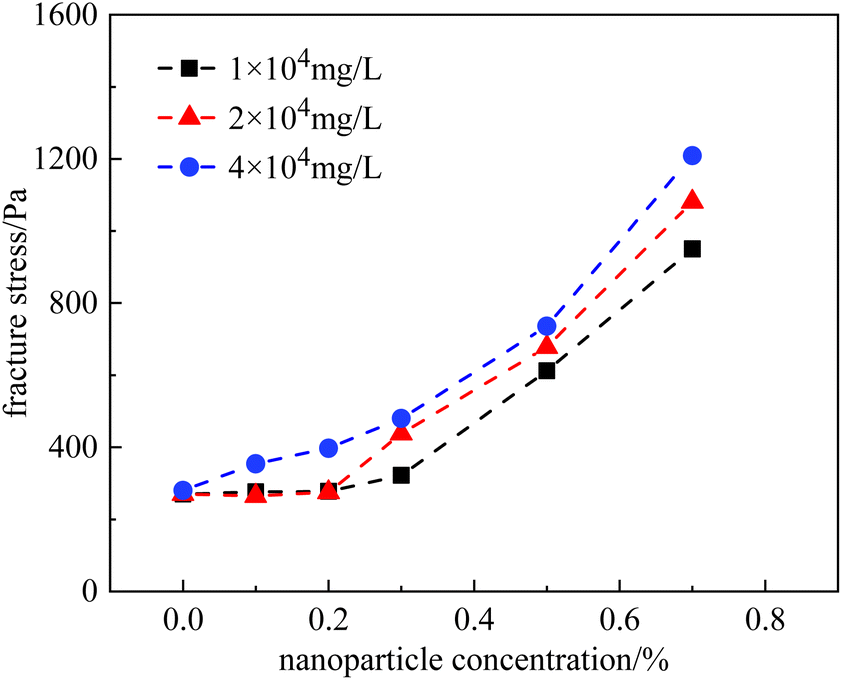 | ||
| Fig. 12 Fracture stress of HPAM with different nanoparticle concentrations. | ||
3.4. Effect of pH on phase behavior and rheology
In the gel system, a certain amount of acidic material is usually added to improve performance. For example, lowering the pH can delay the gelation time of inorganic gels crosslinked by chromium ions. On the other hand, for organic gels crosslinked by phenolic resins, the gelation reaction must be catalyzed by an acid when the reservoir temperature is low. Therefore, it is necessary to investigate the effect of nanoparticles on the rheological properties of gels under acidic conditions. Interfacial properties of nanoparticles are very sensitive to pH. For colloidal SiO2, its isoelectric point is generally around 2–3. Under acidic conditions, groups such as –Si–O– on the surface of the particles are converted to –Si–OH, accompanied by a reduction of the zeta potential.First, the stability of nanoparticles in HPAM solution and PAM solution under pH = 3 was investigated at a salinity of 2 × 104 mg L−1. In this case, the nanoparticle concentration was 0.5%, and the polymer concentration ranged from 0.1% to 0.8%. The experimental results are displayed in Fig. 13.
 | ||
| Fig. 13 Phase behaviors of nanoparticles in polymer solutions with different concentrations at pH = 3: (a) HPAM solutions and (b) in PAM solutions. | ||
The results showed that the stability of the nanoparticles in polymer solutions near the isoelectric point was higher than that under neutral conditions. As seen in Fig. 13, the mixed solutions were clarified and precipitation-free, and the transmittances were all >80%. The above phenomenon is related to the stabilization mechanism of SiO2 nanoparticles in brine. Under neutral conditions, the surface of SiO2 nanoparticles is enriched with a large number of –Si–O– and other structures, which makes it negatively charged. According to the DLVO theory, the particles repel each other due to electrostatic repulsion. Increasing the salinity compresses the diffuse double electric layer on the surface of SiO2 nanoparticles, and the electrostatic repulsion is weakened, resulting in the aggregation of different particles into large particles or gels. Under normal conditions, the stability of nanoparticle suspensions becomes more stable with their increasing surface charge. However, near the isoelectric point of SiO2 nanoparticles, a large number of –Si–O– structures are replaced by electrically neutral –Si–OH, which decreases the electronegativity of the particles. Water molecules in the solvent can form hydrogen bonds with –Si–OH and generate a hydrated layer of a certain thickness on the surface of the particles; thus, inter-particle aggregation is hindered. More importantly, the stability of nanoparticles near the isoelectric point is insensitive to salinity. According to the above results, this phenomenon is not favorable for the improvement of the rheological properties of polymer solutions and gels.
The effect of SiO2 nanoparticles on the rheological properties of gels at different pH values was investigated using HPAM as an example. For comparison, a gel composed of 0.5% HPAM and 0.0417% chromium acetate was used as the base system, and three experimental conditions were designed: (1) adding 0.3% acetic acid and adjusting the pH of the gallant to 3; (2) adding 0.5% SiO2 nanoparticles; and (3) simultaneously adding 0.3% acetic acid and 0.5% SiO2 nanoparticles. It should be noted that the addition of acetic acid would not influence the formation of gel at a macroscopic scale. The measured G0 and fracture stress of these three samples are shown in Fig. 14.
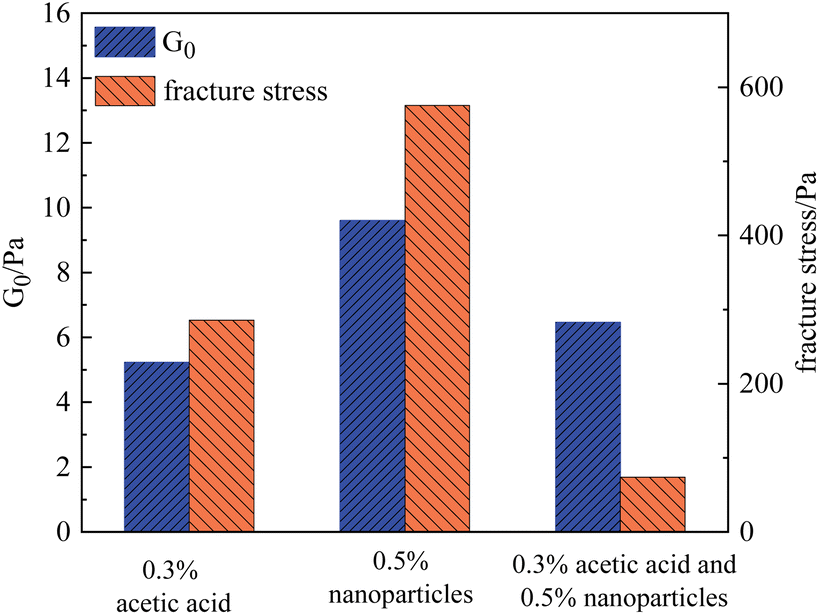 | ||
| Fig. 14 Rheological parameters of HPAM gels with different compositions. | ||
Under condition (1), the initial modulus G0 and the fracture stress of the gel were 5.6 Pa and 285.6 Pa, respectively. Under condition (2), after the addition of 0.5% SiO2 nanoparticles, the initial modulus increased from 5.23 Pa to 9.60 Pa, whereas the fracture stress increased to 575.7 Pa. Therefore, either the addition of acid or nanoparticles can improve the properties of gels to varying degrees. However, when both components were added simultaneously, the nonlinear rheological parameters of the gel decreased substantially, while the strength of the gel increased slightly. Specifically, under condition (3), G0 increased to 6.46 Pa, but the corresponding fracture stress decreased to 74.1 Pa, which was lower than that of the base system. This result indicated that nanoparticles in the sol state adversely affected the performance of the gels. Similar results were also found for the PAM gels. Combined with the experimental results, it is speculated that the SiO2 nanoparticles at the isoelectric point tended to remain in a sol state, which will enhance the non-homogeneity of the polymer solution. Once the polymer chains are crosslinked to the 3D network, this inhomogeneous structure becomes a potential crack nucleation region, which leads to the fracture of the gel at low stress.
 | ||
| Fig. 15 Rupture process of a conventional gel over pressure. | ||
When the nanoparticle concentration was relatively low (<1.0%), the rupture behavior of the composite gel was similar to that of the conventional gel, differing only in the degree of fingering and the rupture pressure gradient. Fig. 16 summarizes the rupture pressure of gels with different nanoparticle concentrations. The rupture pressure increased significantly when the nanoparticle concentration was >0.3%. For example, under a nanoparticle concentration of 1.0%, the rupture pressure gradient in the 3 mm × 3 mm channel reached 3.44 MPa m−1. According to eqn (1), the rupture pressure gradient of the gel is inversely proportional to the channel size. It could be hypothesized that at the scale of pore media, nanoparticle-enhanced gel is sufficient for most of plugging treatment needs.
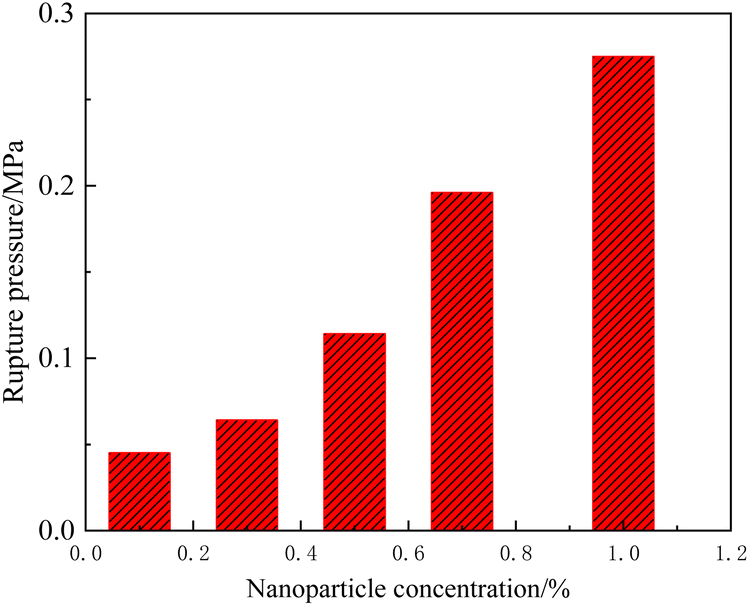 | ||
| Fig. 16 Rupture pressure of HPAM gels with different nanoparticle concentrations in a 3 mm × 3 mm ideal model. | ||
It was also observed that if the nanoparticle-enhance gels were sufficiently strong, the above rupture process would be replaced by permeation of the injected water into bulk gels. As Fig. 17 illustrates, the dyed water fingered into the bulk gel at low pressure, similar to the conventional gels. As the pressure difference increased, the injected dyed water gradually permeated towards the outlet end. Meanwhile, water in the network of gels was squeezed out, and the first drop of water was produced from the outlet at 0.08 MPa. However, gels in the channel were hardly moved or deformed, as their apparent modulus was quite large due to their significant strain hardening properties. Finally, the injected water dyed the whole bulk gel red at 0.4 MPa and subsequently produced from the outlet end. The structure of the gel remained complete until the experiment was finished at 0.7 MPa, which is the rated pressure of our pump.
 | ||
| Fig. 17 Rupture process of the HPAM gel with 2.0% nanoparticles over pressure. | ||
4 Conclusion
(1) The phase behaviors of nanoparticles in polymer solutions are influenced by several factors, including salinity, polymer, and pH. Increasing the salinity or polymer concentration induces aggregation of nanoparticles. The aggregation tendency of nanoparticles is stronger in HPAM solutions than in PAM. Around the isoelectric point, SiO2 nanoparticles are significantly more stable in brine than in neutral conditions.(2) The enhancement of nanoparticles on the rheological properties of polymer solutions and gels exhibits a strong correspondence with phase behavior. The occurrence of aggregation is a necessary condition for the improvement of rheological properties. In addition, in the presence of nanoparticles, the viscosity of polymer solutions and the fracture stress of gels are increased with salinity.
(3) Nanoparticles could improve the plugging properties of gels. At relatively low nanoparticle concentrations, the rupture pressure gradient increases with increasing nanoparticle concentration. If the nanoparticle concentration is sufficiently high (e.g., 2%), the injected water will no longer be able to rupture through the gel but gradually penetrate it instead.
Author contributions
All authors disclosed no relevant relationships.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
No potential conflict of interest was reported by the authors.Acknowledgements
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.References
- A. Ebeling, V. Hartmann and A. Rockman, et al., Silver Nanoparticle Adsorption to Soil and Water Treatment Residuals and Impact on Zebrafish in a Lab-scale Constructed Wetland, Comput. Water Energy Environ. Eng., 2017, 2(3B), 16–25 Search PubMed.
- Y. P. Sun, X. Q. Li and J. Cao, et al., Characterization of zero-valent iron nanoparticles, Adv. Colloid Interface Sci., 2006, 120(1–3), 47–56 CrossRef CAS PubMed.
- C. R. Barry, Printing nanoparticle building blocks from the gas phase using nanoxerography, Appl. Phys. Lett., 2003, 83(26), 5527–5529 CrossRef CAS.
- X. Cheng and L. J. Guo, Electrostatic self-assembly of nanocomposite polymers in grating structures, J. Vac. Sci. Technol., B: Microelectron. Nanometer Struct.--Process., Meas., Phenom., 2001, 19(6), 2736–2740 CrossRef CAS.
- Y. Tomita, Y. Endoh, N. Suzuki, et al., Nanocomposite photopolymers doped with nanoparticles for volume holographic recording, in Photonics North 2006, International Society for Optics and Photonics, 2006 Search PubMed.
- K. Haraguchi, Nanocomposite Gels: New Advanced Functional Soft Materials, Macromol. Symp., 2007, 256(1), 120–130 CrossRef CAS.
- L. Song, M. Zhu and Y. Chen, et al., Temperature- and pH-Sensitive Nanocomposite Gels with Semi-Interpenetrating Organic/Inorganic Networks, Macromol. Chem. Phys., 2010, 209(15), 1564–1575 CrossRef.
- L. Wu, L. Zeng and H. Chen, et al., Preparation and Characterization of Poly(Acrylamide)/Silica Nanocomposite Gels, Adv. Mater. Res., 2011, 306–307, 1252–1256 CAS.
- W. K. Lim and A. R. Denton, Polymer crowding and shape distributions in polymer-nanoparticle mixtures, J. Chem. Phys., 2014, 141(11), 114909 CrossRef PubMed.
- T. Chen, H. J. Qian and Z. Y. Lu, Note: Chain length dependent nanoparticle diffusion in polymer melt: Effect of nanoparticle softness, J. Chem. Phys., 2016, 145(10), 118001 CrossRef PubMed.
- J. L. Byung, A. S. Mark and T. Erik, et al., Competition between kaolinite flocculation and stabilization in divalent cation solutions dosed with anionic polyacrylamide, Water Res., 2012, 46(17), 5696–5706 CrossRef PubMed.
- Y. Samoshina, A. Diaz and Y. Becker, et al., Adsorption of cationic, anionic and hydrophobically modified polyacrylamides on silica surfaces, Colloids Surf., A, 2003, 231(1–3), 195–205 CrossRef CAS.
- C. O. Metin, R. T. Bonnecaze and L. W. Lake, et al., Aggregation kinetics and shear rheology of aqueous silica suspensions, Appl. Nanosci., 2012, 4(2), 169–178 CrossRef.
- H. Yousefvand and A. Jafari, Enhanced oil recovery using polymer/nanosilica, Procedia Mater. Sci., 2015, 11, 565–570 CrossRef CAS.
- R. Elhaei, R. Kharrat and M. Madani, Stability, flocculation, and rheological behavior of silica suspension-augmented polyacrylamide and the possibility to improve polymer flooding functionality, J. Mol. Liq., 2021, 322, 114572 CrossRef CAS.
- P. Nath, R. Mangal and F. F. E. Kohle, et al., Dynamics of nanoparticles in entangled polymer solutions, Langmuir, 2017, 34(1), 241–249 CrossRef PubMed.
- A. Bera, S. Shah and M. Shah, et al., Mechanistic study on silica nanoparticles-assisted guar gum polymer flooding for enhanced oil recovery in sandstone reservoirs, Colloids Surf., A, 2020, 598, 124833 CrossRef CAS.
- L. Chen, J. Wang and Y. Long, et al., Experimental Investigation on the Nanosilica-Reinforcing Polyacrylamide/Polyethylenimine Hydrogel for Water Shutoff Treatment, Energy Fuels, 2018, 32(6), 6650–6656 CrossRef CAS.
- L. Chen, G. Li and Y. Chen, et al., Thixotropy research of laponite-hydrogel composites for water shutoff in horizontal wells, J. Pet. Sci. Eng., 2022, 208, 109600–109611 CrossRef CAS.
- D. A. Z. Wever, F. Picchioni and A. A. Broekhuis, Polymers for enhanced oil recovery: A paradigm for structure–property relationship in aqueous solution, Prog. Polym. Sci., 2011, 36(11), 1558–1628 CrossRef CAS.
- J. C. Jung, K. Zhang and B. H. Chon, et al., Rheology and polymer flooding characteristics of partially hydrolyzed polyacrylamide for enhanced heavy oil recovery, J. Appl. Polym. Sci., 2013, 22, 42 Search PubMed.
- L. Chen, F. Huang and G. Li, et al., Experimental Study on Fiber Balls for Bridging in Fractured-Vuggy Reservoir, SPE J., 2023, 28, 1880–1894 CrossRef CAS.
- J. R. Gales, T. S. Young and G. P. Willhite, et al., Equilibrium swelling and syneresis properties of Xanthan gum-Cr (III) gels, SPE Adv. Technol., 1994, 2(02), 190–198 CrossRef.
- R. D. Sydansk and G. P. Southwell, More than 12 Years' Experience with a Successful Conformance-Control Polymer-Gel Technology, SPE-66558-PA, 2000 Search PubMed.
- R. D. Sydansk and R. S. Seright, When and where relative permeability modification water-shutoff treatments can be successfully applied, SPE Prod. Oper., 2007, 22(2), 236–247 CAS.
- R. Elhaei, R. Kharrat and M. Madani, Stability, flocculation, and rheological behavior of silica suspension-augmented polyacrylamide and the possibility to improve polymer flooding functionality, J. Mol. Liq., 2021, 322, 114572 CrossRef CAS.
- E. Aliabadian, M. Kamkar and Z. Chen, et al., Prevention of network destruction of partially hydrolyzed polyacrylamide (HPAM): Effects of salt, temperature, and fumed silica nanoparticles, Phys. Fluids, 2019, 31, 013104 CrossRef.
- S. Pérez-Robles, F. B. Cortés and C. A. Franco, Effect of the nanoparticles in the stability of hydrolyzed polyacrylamide/resorcinol/formaldehyde gel systems for water shut-off/conformance control applications, J. Appl. Polym. Sci., 2019, 136(21), 1–12 CrossRef.
- A. A. Adewunmi, S. Ismail and A. S. Sultan, Study on strength and gelation time of polyacrylamide/polyethyleneimine composite gels reinforced with coal fly ash for water shut-off treatment, J. Appl. Polym. Sci., 2015, 132(5), 1–8 CrossRef.
- L. F. Chen, J. J. Wang and L. Yu, et al., Experimental investigation on the nanosilica-reinforcing polyacrylamide/polyethylenimine hydrogel for water shutoff treatment, Energy Fuels, 2018, 32(6), 6650–6656 CrossRef CAS.
- A. Yudhowijoyo, R. Rafati and A. S. Haddad, Developing Nanocomposite Gels from Biopolymers for Leakage Control in Oil and Gas Wells, SPE-195765, 2019 Search PubMed.
- Z. Camellia, R. B. Ahmad and M. V. Seftia, Network-gel strength relationship and performance improvement of polyacrylamide hydrogel using nano-silica with regards to application in oil wells conditions, J. Mol. Liq., 2019, 278, 512–520 CrossRef.
- D. Z. Azimi, A. Ghaffarkhah and S. Sadeghnejad, et al., Effect of silica nanoparticle size on the mechanical strength and wellbore plugging performance of SPAM/chromium (III) acetate nanocomposite gels, Polym. J., 2019, 51(7), 693–707 CrossRef.
- S. Kim, K. Hyun and J. Y. Moon, et al., Depletion stabilization in nanoparticle–polymer suspensions: Multi-length-scale analysis of microstructure, Langmuir, 2015, 31(6), 1892–1900 CrossRef CAS PubMed.
- S. Kumar, V. K. Aswal and J. Kohlbrecher, Small-angle neutron scattering study of interplay of attractive and repulsive interactions in nanoparticle-polymer system, Langmuir, 2016, 32(6), 1450–1459 CrossRef CAS PubMed.
- M. Kawaguchi, Rheological properties of silica suspensions in polymer solutions, Adv. Colloid Interface Sci., 1994, 53, 103–127 CrossRef CAS.
- Y. Zare, P. P. Sang and K. Y. Rhee, Analysis of complex viscosity and shear thinning behavior in poly (lactic acid)/poly (ethylene oxide)/carbon nanotubes biosensor based on Carreau–Yasuda model, Results Phys., 2019,(13), 102245 CrossRef.
- H. Wu, J. Ge and Y. Liu, New insight into the rheological and fracture properties of PAM gels: effect of entanglements, J. Pet. Sci. Eng., 2022, 212, 110260 CrossRef CAS.
- K. Bertula, L. Martikainen and P. Munne, et al., Strain-Stiffening of Agarose Gels, ACS Macro Lett., 2019, 8, 670–675 CrossRef CAS PubMed.
- S. Ganguly, Rupture of Polyacrylamide Gel in a Tube in Response to Aqueous Pressure Gradients, Soft Mater., 2009, 7(1–4), 37–53 CrossRef CAS.
- S. Ganguly, Displacement of Cr(III)–Partially Hydrolyzed Polyacrylamide Gelling Solution in a Fracture in Porous Media, Transp. Porous Media, 2010, 84(1), 201–218 CrossRef CAS.
- J. E. van der Hoek, W. Botermans, et al., Full Blocking Mechanism of Polymer Gels for Water Control, SPE68982, 2001 Search PubMed.
- G. A. Al-Muntasheri, I. A. Hussein and H. A. Nasr-El-Din, et al., Viscoelastic properties of a high temperature cross-linked water shut-off polymeric gel, J. Pet. Sci. Eng., 2007, 55(1–2), 56–66 CrossRef CAS.
- K. A. Erk, K. J. Henderson and K. R. Shull, Strain Stiffening in Synthetic and Biopolymer Networks, Biomacromolecules, 2010, 11(5), 1358–1363 CrossRef CAS PubMed.
- K. A. Erk, J. D. Martin and Y. T. Hu, et al., Extreme strain localization and sliding friction in physically associating polymer gels, Langmuir, 2012, 28(9), 4472–4478 CrossRef CAS PubMed.
- K. A. Erk and K. R. Shull, Rate-Dependent Stiffening and Strain Localization in Physically Associating Solutions, Macromolecules, 2011, 44(4), 932–939 CrossRef CAS.
- H. Wu, J. Ge and L. Yang, et al., Effect of entanglement on rheological and ultimate properties of inorganic HPAM gels, J. Mol. Liq., 2022, 351, 118669 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |