
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of a selective and scalable N1-indazole alkylation†
Jimmy
Wang
 *a,
Aaron
Mccreanney‡
a,
Amelia
Taylor-Young‡
a,
Harriet A. M.
Fenton
a,
Rayyan
Miah
a,
Rebecca A.
Johnson
a,
James
Clarke
a,
Adam
Hopkins
a,
Ricky
Jones
a,
William
Waddington
a,
Steven J.
Fussell
a,
Matthew
Badland
a,
Benjamin
Pibworth
b and
Robert
Walton
*a
*a,
Aaron
Mccreanney‡
a,
Amelia
Taylor-Young‡
a,
Harriet A. M.
Fenton
a,
Rayyan
Miah
a,
Rebecca A.
Johnson
a,
James
Clarke
a,
Adam
Hopkins
a,
Ricky
Jones
a,
William
Waddington
a,
Steven J.
Fussell
a,
Matthew
Badland
a,
Benjamin
Pibworth
b and
Robert
Walton
*a
aPfizer R&D UK Limited, Ramsgate Road, Sandwich, Kent CT13 9NJ, UK. E-mail: Jimmy.Wang2@pfizer.com; Robert.Walton@pfizer.com
bConcept Life Sciences, Ramsgate Road, Sandwich, Kent CT13 9NJ, UK
First published on 20th February 2024
Abstract
N1-Alkyl indazoles are a ubiquitous and privileged motif within medicinal chemistry, yet methods to selectively furnish N1-alkyl indazoles with simple alkyl side chains remain sparse. Herein, negative data from high-throughput experimentation (HTE) enabled a confident pivot of resource from continued optimisation to the development of an alternative reaction. This workflow culminated in a methodology for the synthesis of N1-alkyl indazoles. The procedure is highly selective for N1-alkylation, practical, and broad in scope, with no N2-alkyl products detected at completion. Mechanistic understandings were consistent with attributing the high selectivity to thermodynamic control. Additional data-driven process development led to this reaction being safely demonstrated on a 100 g scale, with potential for further scale up. This study highlights pragmatic principles followed to develop a necessitated methodology, suitable for large scale manufacture.
Introduction
Regioselective N-functionalisation of ambidentate azoles represents a substantial challenge in organic synthesis and is of particular interest within the pharmaceutical industry due to the high frequency of nitrogen containing heterocycles.1,2 Indazoles and N1-alkyl indazoles compromise a significant proportion of these heterocycles and are often utilised as indole bioisosteres3 or pharmocophores4,5 (Scheme 1A). Whilst the reaction of indole or pyrrole anions with electrophiles in dipolar aprotic solvents occur predominantly at the lone nitrogen,6 the mesomeric nature of the indazole anion results in comparable conditions leading to highly variable N1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :N2 selectivity depending on substrate electronics (Scheme 1B).7,8 Whilst methods exist for the de novo construction of N1-alkyl indazoles,9 regioselective functionalisation of 1H-indazoles with simple, unfunctionalised alkyl groups remains challenging. Available methods are often not applicable to simple alkyl groups, typically relying on activated electrophiles such as oxoniums,10 α-halo carbonyls,11,12 and substituted alkenes.13–18 Recent advances in transition metal catalysis,19,20 photoredox catalysis,21–24 and electrosynthesis25,26 have provided elegant solutions to help address this issue, however these methodologies can be operationally complex and difficult to scale-up. Herein, the discovery and development of a selective synthesis of unfunctionalised N1-alkyl indazoles is described, with selectivity dictated by thermodynamics and a procedure suitable for large scale manufacture (Scheme 1C).
:N2 selectivity depending on substrate electronics (Scheme 1B).7,8 Whilst methods exist for the de novo construction of N1-alkyl indazoles,9 regioselective functionalisation of 1H-indazoles with simple, unfunctionalised alkyl groups remains challenging. Available methods are often not applicable to simple alkyl groups, typically relying on activated electrophiles such as oxoniums,10 α-halo carbonyls,11,12 and substituted alkenes.13–18 Recent advances in transition metal catalysis,19,20 photoredox catalysis,21–24 and electrosynthesis25,26 have provided elegant solutions to help address this issue, however these methodologies can be operationally complex and difficult to scale-up. Herein, the discovery and development of a selective synthesis of unfunctionalised N1-alkyl indazoles is described, with selectivity dictated by thermodynamics and a procedure suitable for large scale manufacture (Scheme 1C).
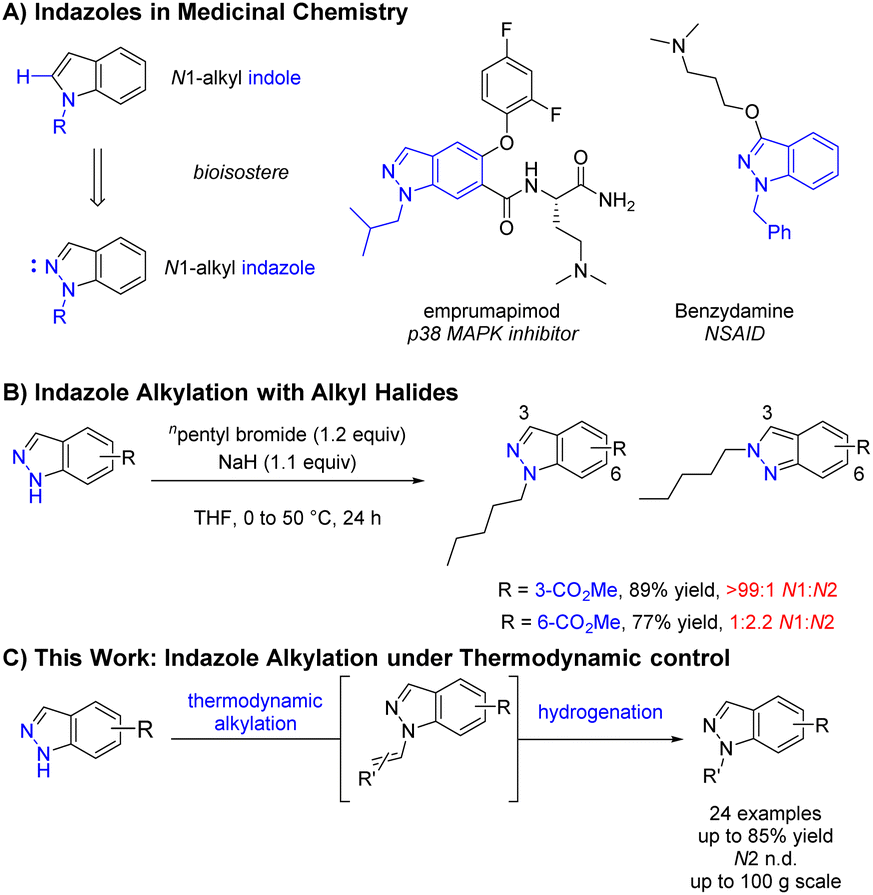 | ||
| Scheme 1 (A) N1-Alkyl indazoles as bioisosteres3 and pharmacophores4,5 in medicinal chemistry. (B) Selected example of substrate dependent indazole alkylation selectivity.7 (C) Thermodynamically controlled N1-selective indazole alkylation (this work). | ||
Results and discussion
During an internal development program, a selective and scalable27 transformation of indazole 1a to its N1-isobutyl variant 2a was required. This indazole featured an aryl-bromide sensitive to transition metal chemistry, a methyl ester prone to hydrolysis, no steric bias at C3 or C7, and was available to us on kilogram scale. Typical base mediated SN2 conditions to facilitate indazole 1a reacting with isobutyl bromide (K2CO3, DMF, 120 °C) resulted in full conversion but gave a 58:42 mixture of N1:N2 isomers 2a and 3a. Chromatography was required to separate and isolate desired product 2a in 47% yield; undesired isomer 3a was also obtained in 25% yield (Scheme 2A). The direct alkylation of 1a with an isobutyl group was the most attractive and synthetically straightforward method available to access the desired product 2a, however the low selectivity displayed was a concern.
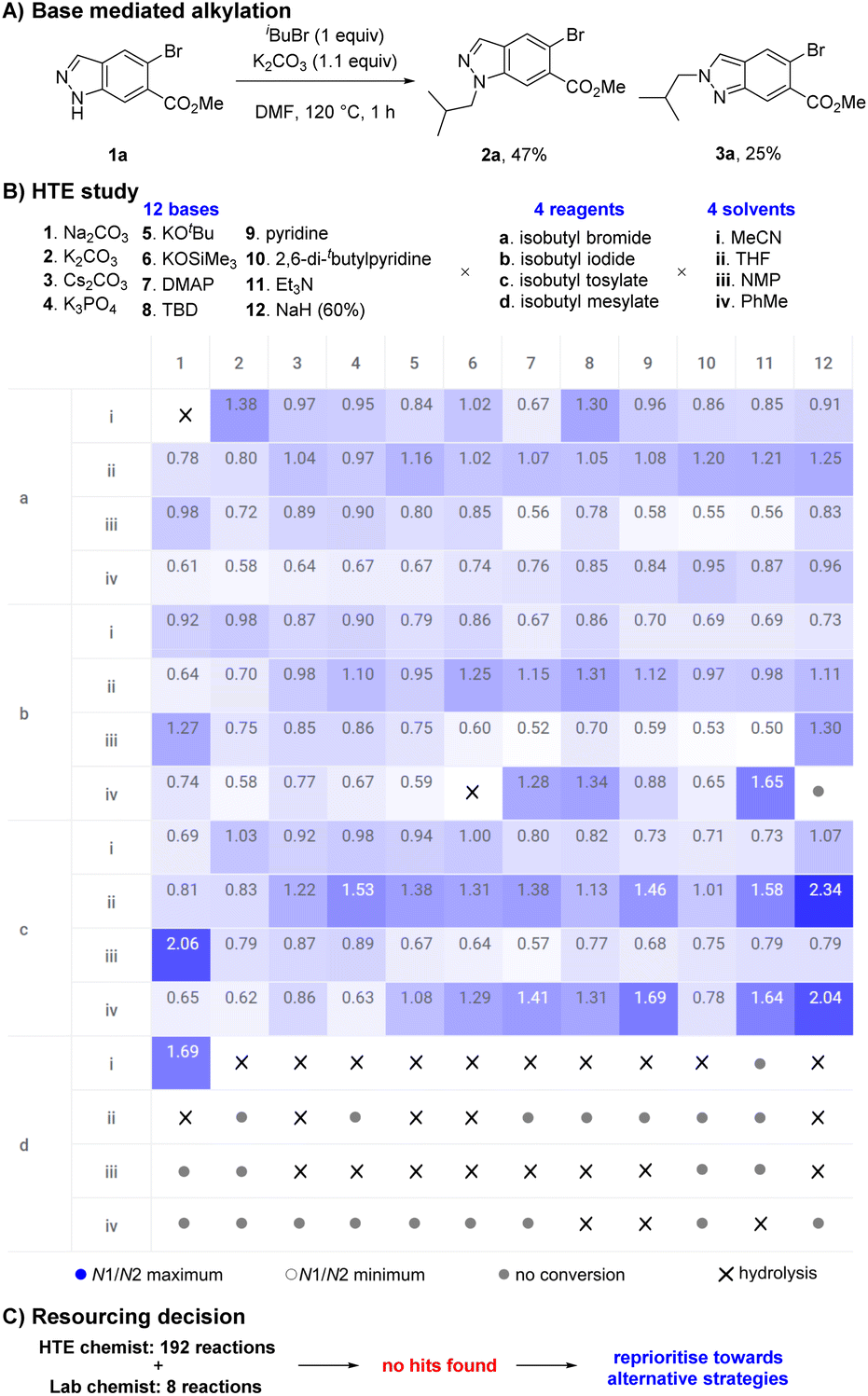 | ||
| Scheme 2 (A) Indazole alkylation using standard alkylation conditions. Crude 2a:3a ratio was 58:42 by LCMS (225 nm). (B) HTE study and selectivity results in a heat map. Standard conditions stirred 15 mg 1a, 1.1 equiv. base, 1 equiv. reagent, and 300 μL solvent at 50 °C for 16 h. DMAP = 4-dimethylaminopyridine; TBD = triazabicyclodecene. (C) Workflow followed leading to reprioritisation of resourcing. | ||
High-throughput experimentation (HTE) utilises technological advancements in automation and robotics to significantly accelerate the exploration of chemical space by executing numerous small-scale experiments in parallel.28–30 HTE has transformed the field of synthetic organic chemistry, rapidly increasing the rate of reaction discovery,31,32 reaction optimisation,33 and reaction scope evaluation.34,35 We initially leveraged our HTE capabilities to explore the influence of discrete variables to increase the selectivity in favour of desired product 2a. A study to explore diverse chemical space was designed involving 12 bases, 4 reagents, and 4 solvents, then executed using automated technologies and analysed with data visualisation techniques (Scheme 2B). Our results suggested the system could be tuned to favour either 2a or 3a in a limited fashion by selecting an appropriate combination of base, reagent, and solvent. However, the highest ratio of 2a:3a remained a moderate 2.34 (70:30) (NaH (60%), isobutyl tosylate, THF) and was insufficient to offer a useful preparative method.
Alongside the HTE study, data from 8 experiments from a lab chemist were also obtained, suggesting our HTE capabilities provided an approximate 24-fold time save (Scheme 2C). Unfortunately, these combined learnings failed to reveal suitable hits within the explored chemical space. We believed further optimisation would be highly unlikely to produce desired results and a decision to reprioritise resources was taken.
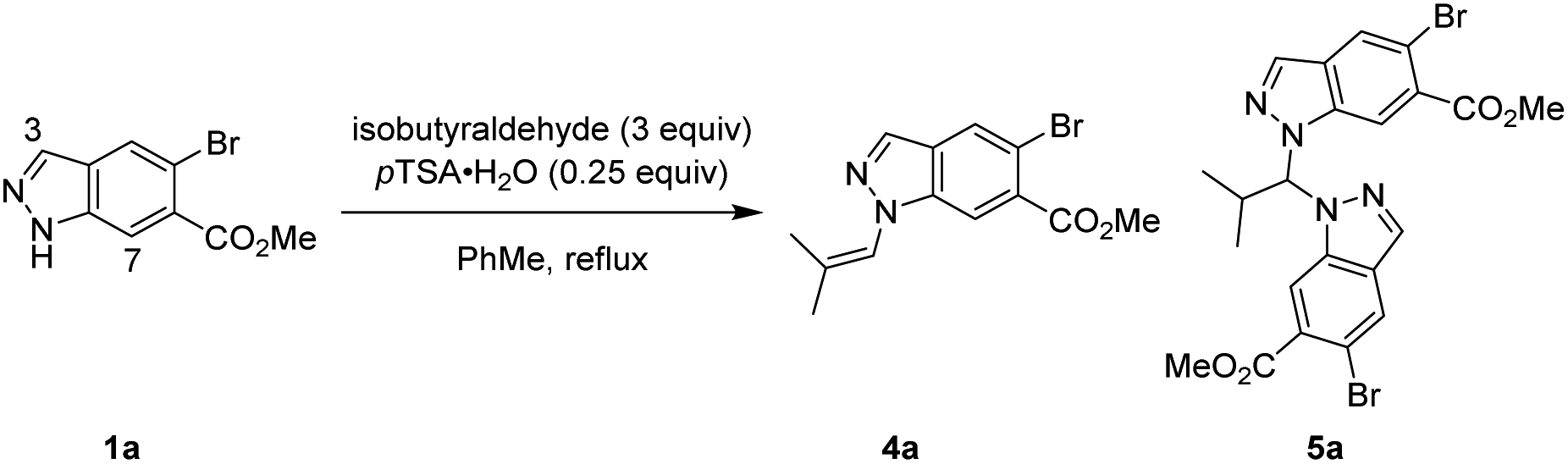
One strategy we explored focused on utilising isobutyraldehyde to introduce the isobutyl group through a formal reductive amination process. Reacting indazole 1a under standard reductive amination conditions36 resulted in negligible conversion and no detectable iminium formation (Table 1, entry 1). However, a two-step process of initial enamine condensation and subsequent hydrogenation resulted in success. Crucially, exclusive N1 selectivity was observed during the initial enamine formation, with the major side product found to be aminal pseudo-dimer 5a. To maximise conversion to 4a and minimise formation of 5a, an excess of aldehyde and effective water removal was essential. Dean–Stark conditions were highly effective compared to either a typical reflux or use of molecular sieves (Table 1, entries 2–4). Cooling the reaction below reflux resulted in some reversion of 4a to 5a, hence a quench with triethylamine was introduced at reflux to maintain high selectivity for 4a (Table 1, entries 5–7). Metal catalysed hydrogenations are highly scalable and 5% Pt/C was discovered to be the most active and efficient catalyst, providing full conversion whilst minimising dehalogenation (see ESI† for full details). Enamine 4a could be isolated crude and reacted further under an atmosphere of H2 at 40 psi and 30 °C with 5% Pt/C to afford 2a in high conversion whilst minimising overreduction of the aryl bromide. The transformation of indazole 1a to desired product 2a was achieved in two steps and a single purification in 76% yield, with no detectable N2 isomer produced (Table 1, entry 7).
|
|
||
|---|---|---|
| Entry | Variation from above | Conversion (ratio 4a:5a) |
| a isolated yield of 2a after hydrogenation of 4a (H2 (40 psi), 5% Pt/C (0.013 equiv.), PhMe, 30 °C), over two steps. MS = molecular sieves. | ||
| 1 | Na(OAc)3BH, 1,2-dichloroethane | n.d. |
| 2 | 4 Å MS, 24 h | 97% (52:48) |
| 3 | None | 95% (82:18) |
| 4 | Dean–Stark, sampled at reflux | >99% (>99:1) |
| 5 | Isobutyraldehyde (2 equiv.), Dean–Stark, sampled at reflux | 95% (90:10) |
| 6 | Dean–Stark, sampled at rt | 97% (96:4) |
| 7 | Dean–Stark, then Et 3 N (0.25 equiv.), sampled at rt |
>99% (>99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1) 76% yield
a :1) 76% yield
a
|
Following the development of a selective and robust transformation of indazole 1a to alkylated product 2a, the substrate scope of this reaction was explored to understand both its potential and limitations (Scheme 3). Using isobutyraldehyde as the alkylating partner our conditions were highly robust to changes in indazole electronics. 3-, 4-, 5-, and 6-carboxylate indazoles were all functionalised selectively at the N1 position (2c–f), whilst 7-carboxylate indazole remained unreactive (2g). As 7-bromoindazole reacted to form 2h in 27% yield, it is plausible the lack of reactivity with 7-carboxylate indazole was due to steric effects. In addition to the electron withdrawing esters and aryl bromides, the reaction also proceeded well with electronically neutral indazole (2b), fluorinated indazoles (2i–j), and electron rich methoxy (2k) and alkyl (2l) substituted indazoles.
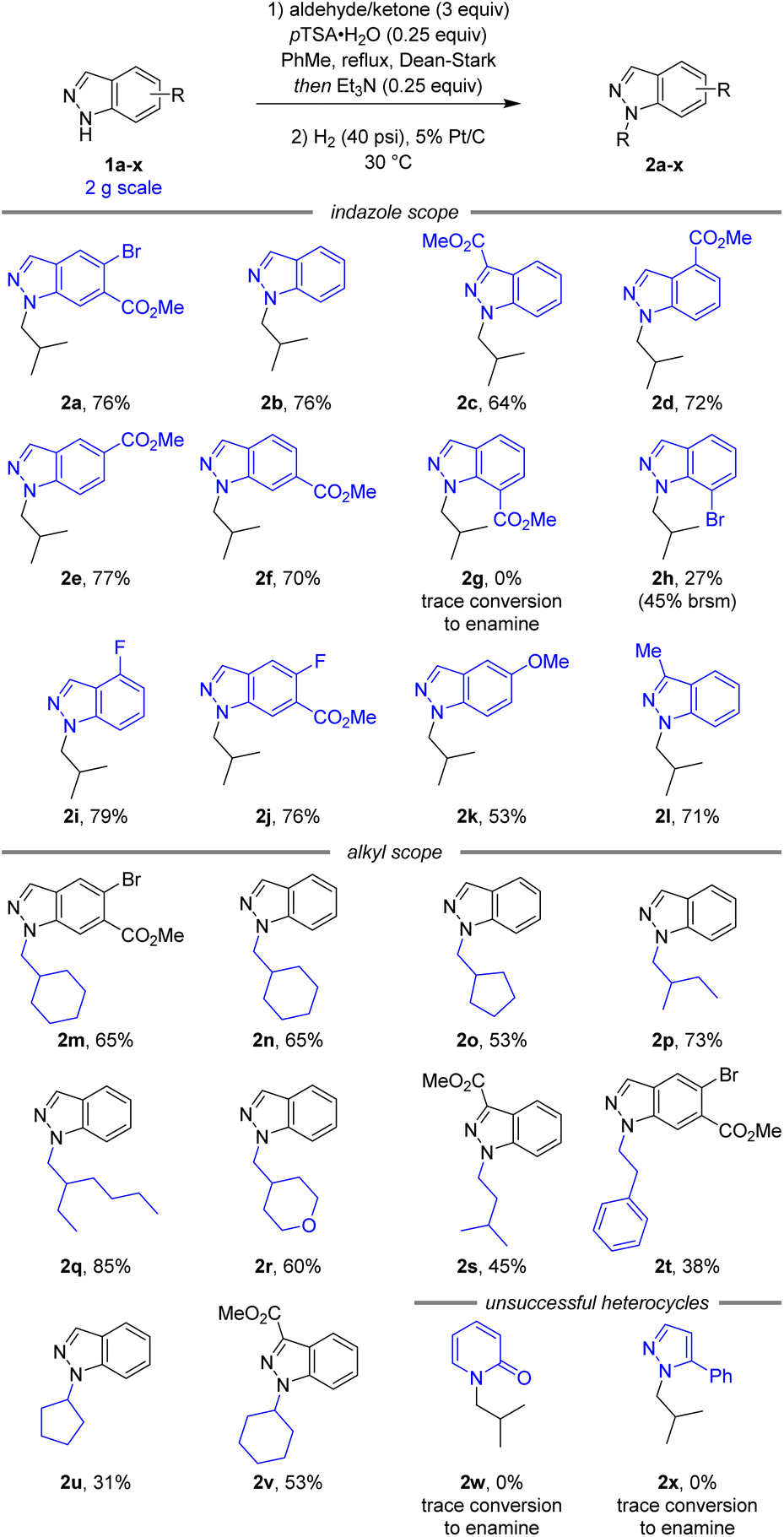 | ||
| Scheme 3 Reaction scope and limitations.a aAll yields are isolated yields over two steps. brsm = based on recovered starting material. | ||
A variety of simple alkyl reaction partners also reacted smoothly, maintaining N1 selectivity and high yields. Both cyclic (2m–o) and linear (2p–q) secondary aldehydes afforded the corresponding N1-alkyl-indazoles in good yields. An electronic variation in tetrahydro-2H-pyran-4-carbaldehyde continued to afford the corresponding product (2r) in good yield and selectivity. Reactions with primary aldehydes isovaleraldehyde (2s) and phenylacetaldehyde (2t) were also successful, although the products were obtained in slightly lower yield due to the decreased stability of the intermediate enamine. Additionally, this reaction was successful with simple ketones (2u,v), despite the increased steric hindrance and decreased reactivity of the alkylation reagent. Unfortunately, attempts to extend this methodology beyond indazoles to 2-pyridone (2w) and pyrazole (2x) heterocycles were unsuccessful.
Further investigations were enacted to understand the formation of aminal pseudodimer 5a and the high preference for enamine 4a. N1-selective indazole functionalisations often possess an element of reversibility, with selectivity attributed to thermodynamic control.10,11 Time-course data was gathered to explore this hypothesis. Potential intermediates including the N1 enamine 4a, N2 enamine 6a, N1,N1-aminal 5a, and N1–N2 aminal pseudodimers 7a were independently synthesised, isolated, and fully characterised.
Monitoring this reaction by LCMS with biphenyl as internal standard provided valuable insights into the reaction composition during its progression. In the standard reaction, large quantities of aminal pseudo-dimers 5a and 7a form at the start of the reaction, decreasing as the reaction progresses (Scheme 4A(i)). Whilst the N1,N2-aminal 7a depleted rapidly, the N1,N1-aminal 5a persists for longer, suggesting it is the more stable of the two. As discovered during reaction optimisation, continued heating leads to degradation of product 4a and reformation of aminal 5a. Commencing the standard reaction with 0.5 equiv. isobutyraldehyde resulted in a plateau containing a near equal mixture of starting material 1a, enamine product 4a, and 5a. An additional charge of 2.5 equiv. isobutyraldehyde resulted in a brief increase in 7a, before consumption of 1a, 5a, and 7a to form product 2a (Scheme 4A(ii)). Heating chromatographically purified 4a with pTSA·H2O in the absence of isobutyraldehyde resulted in noticeable increase of indazole 1a, aminal 5a, and small quantities of aminal 7a, whereas no reaction was observed in absence of pTSA·H2O (Scheme 4A(iii)).
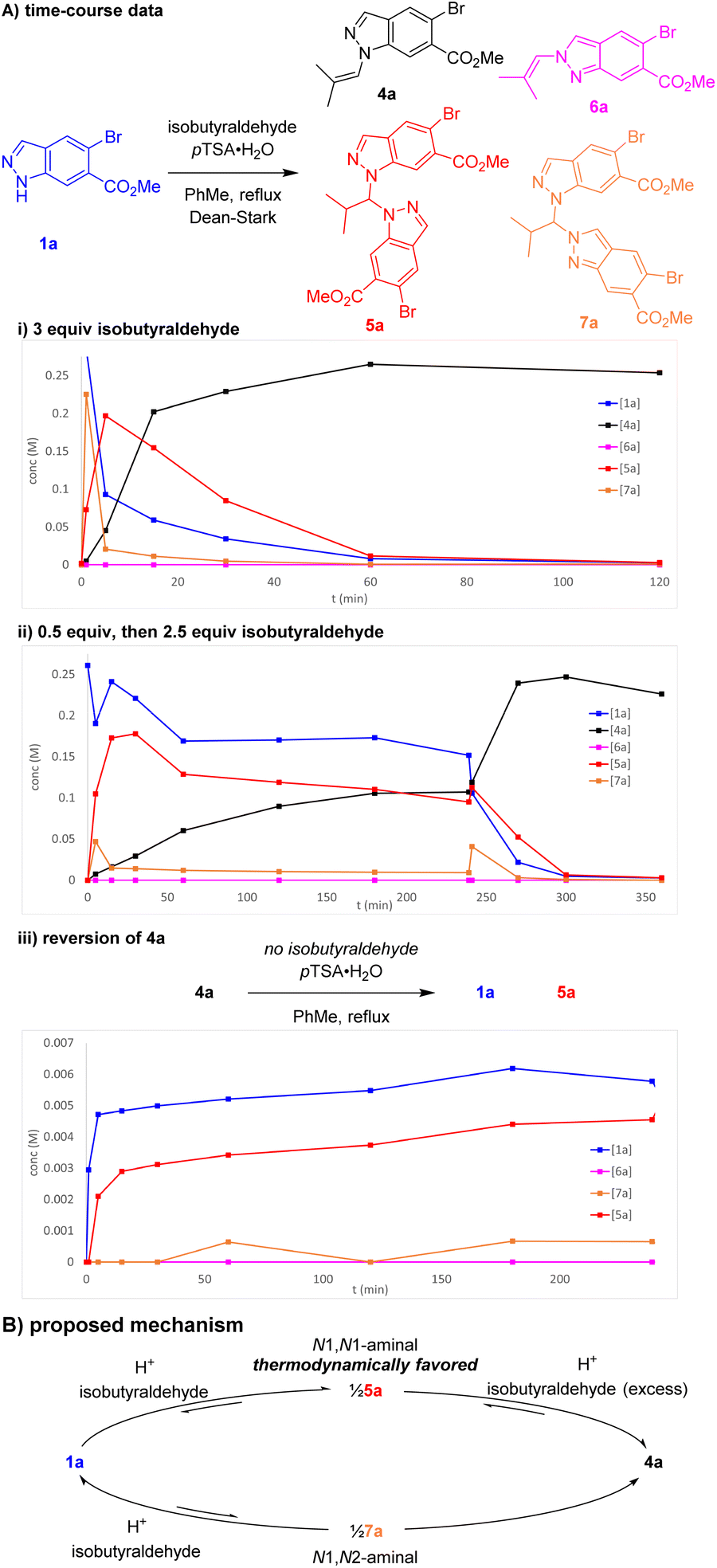 | ||
| Scheme 4 (A) Key time-course data of reaction of 1a and isobutyraldehyde. Concentrations determined by LCMS with biphenyl internal standard. (B) Proposed reaction mechanism for enamine condensation. | ||
Our proposed mechanism involves reversible equilibrium between all species involved, including indazole 1a, aminals 5a and 7a, and enamine 4a. Indazole 1a can condense rapidly with isobutyraldehyde to form either 5a or 7a. Whilst 7a appears unstable and likely reacts to form both 4a and 1a, 5a is evidently thermodynamically favoured, reacting with excess aldehyde to produce enamine 4a (Scheme 4B). The tendency of 4a to reform 5a can be attributed to a reduction in aldehyde excess owing to its inherent volatility (bp = 63 °C) and instability towards adventitious oxygen. This was further supported by observations of decreased conversion to 4a in reactions with inefficient condensers. These investigations were consistent with attributing the high regioselectivity to thermodynamic control and highlighted the importance of the Et3N addition to avoid reversion of product 4a to the aminal 5a.
From the outset of this development program, we aimed to produce N1-alkyl indazole 2a on a multi-kilogram scale. Despite the streamlined telescope procedure employed, undesirable processes such as use of MgSO4 as desiccant, multiple distillations to dryness, and chromatographic purification remained, resulting in a high PMI (process mass intensity)37 of >500 and necessitated additional process development (Fig. 1A). In addition, the nitrogen–nitrogen bond within the indazole raised process safety concerns and additional studies were required to de-risk potential issues upon scale-up.
 | ||
| Fig. 1 (A) Initial process followed for reaction scope. (B) Key process improvements including crystallisation discovery and development, workup improvements, and process safety testing. (C) Scalable process demonstration on 100 g scale. From left to right: starting materials and solvent; reaction in 2 L jacketed vessel; distillation and workup in 5 L jacketed vessel; charging of hydrogenation reaction; removal of hydrogenation catalyst by filtration over arbocel; crystallisation of 2a as the pTSA salt; isolated final product. aFinal product contained 1.25 equiv. pTSA by NMR. PMI = Process mass intensity. DSC = differential scanning calorimetry. TSU = thermal screening unit. ARC = accelerated rate calorimetry. | ||
The initial workup involved washes with H2O, 1 M HCl(aq) and saturated brine, before drying with MgSO4, filtration, evaporation to dryness, and redissolution in PhMe. Additional experimentation revealed removal of the HCl(aq) wash and replacing the MgSO4 desiccant with a controlled azeotropic distillation resulted in negligible impact to reaction yield or purity (Fig. 1B(i)). Although no instability of enamine towards the HCl(aq) wash was observed, its removal further derisked potential acid catalysed enamine hydrolysis. These modifications enabled both a reduction in PMI and unit operations, increasing throughput and resulting in a more streamlined process for manufacture.
Conditions to crystallise 2a were sought to avoid chromatographic purification and significantly reduce PMI (Fig. 1B(ii)). We once again leveraged our HTE resources to swiftly realise freebase 2a possessed high (>90 mg mL−1) solubility in a diverse set of 22 (binary)-solvent systems and would likely require isolation as a salt. Subsequently a HTE salt screen was performed, studying the mixture observed upon combining 2a with 24 acids in 4 process relevant solvents (MeCN, IPA, EtOAc, and PhMe). 9 conditions were found to produce slurries suitable for filtration, notably pTSA·H2O in the reaction solvent PhMe, and pTSA·H2O in MeCN. Gram scale synthesis and isolation of 2a·pTSA provided material to support further crystallisation development. Additional HTE solubility screens with 2a·pTSA in 46 (binary)-solvent systems highlighted the ability of MeCN to modulate the solubility of the 2a·pTSA salt in PhMe. Solvent systems containing <30% MeCN in PhMe provided the steepest solubility gradient, allowing for good solubility at high temperatures whilst maximising recovery at low temperatures. The initial slurries were typically slow to filter and optical microscopy revealed many fine particles. A heat-cool cycle was found to significantly improve the particle size distribution and filtration speed via Ostwald ripening.38 Lastly, temperature dependent solubility curves of 2a·pTSA in MeCN:PhMe mixtures were studied in a medium-throughput fashion using the Crystal16 benchtop crystallisation system. These studies led to a procedure in which a solution of crude 2a in PhMe taken directly after hydrogenation and catalyst removal would be concentrated to 6.7 mL g−1 (vs. initial 1a) and diluted with MeCN (2.2 mL g−1vs. initial 1a). Addition of pTSA·H2O and 2a·pTSA seed (0.01 equiv.) at 40 °C, followed by aging, heat-cool cycling, granulation, and collection by filtration at 10 °C yielded pure 2a·pTSA in 76% yield on 7.5 g scale.
Compounds containing functional groups with a nitrogen–nitrogen bond are known for their potential to release significant amounts of energy as they decompose,39 presenting the potential for process safety issues upon scale-up. To minimise process risks, it was essential to understand the thermal characteristics of isolated materials and reaction mixtures generated. These thermal characteristics were assessed using Differential Scanning Calorimetry (DSC), Thermal Screening Unit (TSU) and Accelerating Rate Calorimetry (ARC) for the reaction with 100 g 1a (Fig. 1B(iii)). Isolated solids of 1a, 4a, 2a and 2a·pTSA all displayed significant exotherms formation at temperatures above 200 °C in the initial DSC analysis, and exotherms and gas formation in subsequent ARC analysis. In all cases, the adiabatic temperature rise was found to be >200 °C, suggesting potential process risks relating to the thermal stability of the reaction mixtures. Fortunately, further thermal stability testing by TSU and ARC of the pre- and post-reaction mixtures for the enamine condensation to form 4a displayed no significant thermal events. Additional TSU testing of the pre- and post-reaction mixtures for the hydrogenation of 4a to 2a displayed some minor thermal events which were not associated with gas formation and were deemed negligible in the context of this process.
These improvements to work-up, purification, and process safety understanding enabled confidence in proceeding with a 100 g scale demonstration of an indazole alkylation. Our combined data-driven learnings culminated in a reproducible process for the isolation of 2a·pTSA as a crystalline solid in 72% yield (149.4 g product) over two steps and a significant reduced PMI of 22.8, down from >500 (Fig. 1C).
Conclusions
In summary, we have described the discovery and development of a thermodynamically driven N1-selective indazole alkylation. Throughout this study, we accelerated development by leveraging both positive and negative data from HTE. A broad scope was demonstrated, and mechanistic investigations corroborated our hypothesis that the high observed selectivity of this reaction was due to thermodynamic control. Process safety understanding and additional process optimisation lead to the successful completion of this reaction on a 100 g scale ready for multi-kilogram production in a pilot plant. During the preparation of this manuscript, the largest single batch used 321.6 kg of 1a to produce 408.4 kg of 2a·pTSA (67% yield, hydrogenation split into two batches); overall, >1 MT of 2a·pTSA has been manufactured with this process. We anticipate the core principles presented in this article will see further applications across both academia and industry.Author contributions
Conceptualization and supervision: RW, JW. Project administration and resources: RW. Investigation: JW, AM, ATY, HAMF, RM, RAJ, JC, AH, RJ, WSW, SJF, MB, BP. Visualisation and writing – original draft: JW, ATY, HAMF, JC, RAJ. Writing – review & editing: JW, JC, SJF, RW.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank numerous colleagues within Pfizer and Array BioPharma, as well as our external collaborators at CROs and CMDOs for their helpful ideas, discussions, and contributions to this research program. We thank Donald A. Clark, Ryan Osborne, and Shams T. A. Islam for assistance with analytical data collection. We thank Ian Moses, Philip Peach, and Phillip Greenwood for additional experimental support. We thank Michael Hawksworth, Caroline Chapman, and Heather Ingram for assistance with process safety data collection.Notes and references
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS.
- R. D. Taylor, M. MacCoss and A. D. Lawson, J. Med. Chem., 2014, 57, 5845–5859 CrossRef CAS PubMed.
- P. Fludzinski, D. A. Evrard, W. E. Bloomquist, W. B. Lacefield, W. Pfeifer, N. D. Jones, J. B. Deeter and M. L. Cohen, J. Med. Chem., 1987, 30, 1535–1537 CrossRef CAS PubMed.
- WO Pat., WO2004078116A2, 2004 Search PubMed.
- P. A. Quane, G. G. Graham and J. B. Ziegler, Inflammopharmacology, 1998, 6, 95–107 CrossRef CAS PubMed.
- H. Heaney and S. V. Ley, J. Chem. Soc., Perkin Trans. 1, 1973, 499–500 RSC.
- R. M. Alam and J. J. Keating, Beilstein J. Org. Chem., 2021, 17, 1939–1951 CrossRef CAS PubMed.
- H. He, J. Yan, J. Jin, Z. Yan, Q. Yan, W. Wang, H. Jiang, H. Wang and F. Chen, Chem. Commun., 2022, 58, 6429–6432 RSC.
- N. Cankařová, J. Hlaváč and V. Krchňák, Org. Prep. Proced. Int., 2010, 42, 433–465 CrossRef.
- D. J. Slade, N. F. Pelz, W. Bodnar, J. W. Lampe and P. S. Watson, J. Org. Chem., 2009, 74, 6331–6334 CrossRef CAS.
- K. W. Hunt, D. A. Moreno, N. Suiter, C. T. Clark and G. Kim, Org. Lett., 2009, 11, 5054–5057 CrossRef CAS.
- M. N. Vander Wal, A. K. Dilger and D. W. C. MacMillan, Chem. Sci., 2013, 4, 3075–3079 RSC.
- J. Moran, P. Dornan and A. M. Beauchemin, Org. Lett., 2007, 9, 3893–3896 CrossRef CAS PubMed.
- S. W. Kim, T. T. Schempp, J. R. Zbieg, C. E. Stivala and M. J. Krische, Angew Chem. Int. Ed. Engl., 2019, 58, 7762–7766 CrossRef CAS.
- W. S. Jiang, D. W. Ji, W. S. Zhang, G. Zhang, X. T. Min, Y. C. Hu, X. L. Jiang and Q. A. Chen, Angew Chem. Int. Ed. Engl., 2021, 60, 8321–8328 CrossRef CAS PubMed.
- Q. C. Xue, J. Xie, P. Xu, K. D. Hu, Y. X. Cheng and C. J. Zhu, ACS Catal., 2013, 3, 1365–1368 CrossRef CAS.
- A. M. Haydl, K. Xu and B. Breit, Angew Chem. Int. Ed. Engl., 2015, 54, 7149–7153 CrossRef CAS.
- J. Y. Yang, Y. F. Bao, H. Y. Zhou, T. Y. Li, N. N. Li and Z. Li, Synthesis, 2016, 48, 1139–1146 CrossRef CAS.
- B. Górski, A.-L. Barthelemy, J. J. Douglas, F. Juliá and D. Leonori, Nat. Catal., 2021, 4, 623–630 CrossRef.
- X. Y. Lv, R. Abrams and R. Martin, Angew Chem. Int. Ed. Engl., 2023, 62, e202217386 CrossRef CAS.
- Y. Liang, X. Zhang and D. W. C. MacMillan, Nature, 2018, 559, 83–88 CrossRef CAS PubMed.
- N. W. Dow, A. Cabre and D. W. C. MacMillan, Chem, 2021, 7, 1827–1842 CAS.
- V. T. Nguyen, V. D. Nguyen, G. C. Haug, N. T. H. Vuong, H. T. Dang, H. D. Arman and O. V. Larionov, Angew Chem. Int. Ed. Engl., 2020, 59, 7921–7927 CrossRef CAS.
- Y. Ge, Y. Shao, S. Wu, P. Liu, J. Li, H. Qin, Y. Zhang, X.-s. Xue and Y. Chen, ACS Catal., 2023, 13, 3749–3756 CrossRef CAS.
- L. Sun, X. Zhang, C. Wang, H. Teng, J. Ma, Z. Li, H. Chen and H. Jiang, Green Chem., 2019, 21, 2732–2738 RSC.
- J. W. Wu, Y. Zhou, Y. C. Zhou, C. W. Chiang and A. W. Lei, ACS Catal., 2017, 7, 8320–8323 CrossRef CAS.
- M. Butters, D. Catterick, A. Craig, A. Curzons, D. Dale, A. Gillmore, S. P. Green, I. Marziano, J. P. Sherlock and W. White, Chem. Rev., 2006, 106, 3002–3027 CrossRef CAS.
- J. R. Schmink, A. Bellomo and S. Berritt, Aldrichimica Acta, 2013, 46, 71–80 Search PubMed.
- S. M. Mennen, C. Alhambra, C. L. Allen, M. Barberis, S. Berritt, T. A. Brandt, A. D. Campbell, J. Castanon, A. H. Cherney, M. Christensen, D. B. Damon, J. E. de Diego, S. Garcia-Cerrada, P. Garcia-Losada, R. Haro, J. Janey, D. C. Leitch, L. Li, F. F. Liu, P. C. Lobben, D. W. C. MacMillan, J. Magano, E. McInturff, S. Monfette, R. J. Post, D. Schultz, B. J. Sitter, J. M. Stevens, J. I. Strambeanu, J. Twilton, K. Wang and M. A. Zajac, Org. Process Res. Dev., 2019, 23, 1213–1242 CrossRef CAS.
- M. Shevlin, ACS Med. Chem. Lett., 2017, 8, 601–607 CrossRef CAS.
- A. McNally, C. K. Prier and D. W. MacMillan, Science, 2011, 334, 1114–1117 CrossRef CAS PubMed.
- K. D. Collins, T. Gensch and F. Glorius, Nat. Chem., 2014, 6, 859–871 CrossRef CAS.
- S. M. Preshlock, B. Ghaffari, P. E. Maligres, S. W. Krska, R. E. Maleczka Jr and M. R. Smith 3rd, J. Am. Chem. Soc., 2013, 135, 7572–7582 CrossRef CAS.
- C. C. Wagen, S. E. McMinn, E. E. Kwan and E. N. Jacobsen, Nature, 2022, 610, 680–686 CrossRef CAS PubMed.
- K. Kang, N. L. Loud, T. A. DiBenedetto and D. J. Weix, J. Am. Chem. Soc., 2021, 143, 21484–21491 CrossRef CAS PubMed.
- A. F. Abdel-Magid, K. G. Carson, B. D. Harris, C. A. Maryanoff and R. D. Shah, J. Org. Chem., 1996, 61, 3849–3862 CrossRef CAS.
- C. Jimenez-Gonzalez, C. S. Ponder, Q. B. Broxterman and J. B. Manley, Org. Process Res. Dev., 2011, 15, 912–917 CrossRef CAS.
- I. M. Lifshitz and V. V. Slyozov, J. Phys. Chem. Solids, 1961, 19, 35–50 CrossRef.
- Bretherick's Handbook of Reactive Chemical Hazards, ed. P. G. Urben, Elsevier, 8th edn, 2017 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, including procedures, additional optimisation data, compound characterisation, process safety data, NMR spectra. See DOI: https://doi.org/10.1039/d4ra00598h |
| ‡ AM and ATY contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |