
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAchieving high molecular weight alternating copolymers of 1-octene with methyl acrylate via Lewis acid catalyzed copolymerization†
Jiefan
Wan
 ,
Yi
Dan
,
Yun
Huang
and
Long
Jiang
*
,
Yi
Dan
,
Yun
Huang
and
Long
Jiang
*
State Key Laboratory of Polymer Materials Engineering of China (Sichuan University), Polymer Research Institute of Sichuan University, Chengdu 610065, China. E-mail: jianglong@scu.edu.cn
First published on 20th February 2024
Abstract
The radical (co)polymerization of long-chain α-olefins (C4+) to produce high molecular weight (Mw) polymers is of great importance. However, this process is currently faced with significant challenges due to the presence of less reactive allylic radicals during radical (co)polymerization, leading to oligomers or polymers with extremely low Mw (less than 1 × 104 g mol−1). Using copolymerization of 1-octene with methyl acrylate (MA) as a proof-of-concept for addressing this challenge, we present a feasible method for synthesizing high Mw α-olefin copolymers via scandium trifluoromethanesulfonate (Sc(OTf)3)-mediated radical copolymerization. In this case, copolymers of 1-octene and MA (poly(1-octene-alt-MA)) with a Mw exceeding 3 × 104 g mol−1 were successfully synthesized in the presence of Sc(OTf)3. Meanwhile, the presence of alternating 1-octene–MA sequential structures was observed. To further enhance the Mw of poly(1-octene-alt-MA), a difunctional comonomer, 1,7-octadiene, was introduced to copolymerize with 1-octene and MA. The results indicate that the incorporation of difunctional comonomer leads to a significant increase in the Mw of the copolymers synthesized. The addition of 1 mol% of 1,7-octadiene resulted in a copolymer with a remarkably high Mw of up to 13.45 × 104 g mol−1 while still maintaining a high degree of the alternating 1-octene–MA sequence (41%). The influence of polymerization parameters on the molecular weight were also investigated. Increasing the monomer concentration, reducing the dosage of initiator, and lowering the polymerization temperature have been found to be advantageous in enhancing the molecular weight. This approach has also been successfully applied to the synthesis of high molecular weight alternating copolymers of other long-chain α-olefins, including 1-hexene, 1-decene and 1-tetradecane, with methyl acrylate. In summary, this study provides a feasible method for converting “less activated” α-olefins into high Mw olefin copolymers. This approach holds significant potential for the production of value-added polyolefins, thus offering promising prospects for future applications.
1 Introduction
In recent decades, the production of polyethylene has consistently accounted for approximately 30% of the overall global plastic production due to the continuous growth in demand for PE products.1–5 Consequently, there has been a substantial increase in the production capacity of ethylene, which currently reaches a total of 120 million tons per year.6,7 However, the ethylene cracking process also results in the generation of a significant quantity of low-value by-products, including hexene, octene, and other long chain α-olefins (C4+). Although these α-olefins are valuable non-renewable petrochemical resources,8–12 their chemical inertness poses a significant challenge when attempting to broaden their applications.13,14 In fact, a portion of these olefins are presently being combusted, leading to a substantial waste of precious petroleum resources and a rise in carbon emissions.2,15 To enhance the efficient utilization of non-renewable petrochemical resources and achieve carbon neutrality, it is crucial for the petrochemical industry to develop value-added strategies for the utilization of these low-value by-products. Radical copolymerization of α-olefins with functional monomers to produce high-performance polymers has been proposed as a promising strategy to reduce the depletion of non-renewable petrochemical resources and enables their efficient and valuable utilization.High molecular weight is a crucial determinant for high-performance polymer materials. However, the synthesis of high molecular weight α-olefin (co)polymers through radical polymerization is challenging, mainly attributed to the existence of allyl hydrogens.16 The allyl hydrogens are readily abstracted by the active radicals during the chain growth, leading to the formation of allylic radicals with extremely low reaction activity. The consequence of this is the incapacity to generate high molecular weight polymers. Although poly(α-olefins) can be synthesized through coordination polymerization, such as homo-polymerization for isotactic polypropylene or copolymerization for functional polyolefins,17,18 the simultaneous improvement of catalytic efficiency and copolymer molecular weight remains an unattained goal.13,19 Furthermore, the incorporation of functional monomers, particularly polar monomers, into olefin copolymers is restricted by the limited tolerance of coordination polymerization catalysts towards polar monomers.20,21 Unlike coordination polymerization, free radical polymerization exhibits less stringent limitation regarding the suitability of monomer types. However, the differences in reactivity ratios of non-polar α-olefins (0.0016) and polar acrylates (92) are large, leading to notably low integration ratio of α-olefin units in the copolymer produced through radical copolymerization.22,23 In recent decades, considerable efforts have been undertaken to address the issue of the relatively low reactivity exhibited radical polymerization of α-olefins.16,17,24,25 Techniques such as “living” radical polymerization have been devised to improve the activity of α-olefins by maintaining a dynamic equilibrium of free radicals in the reaction system.26,27 The molecular weight of polymers is typically controlled by polymerization parameters such as initiator dosage, monomer concentration, and branching structure, etc.28,29 Furthermore, it is essential to account for the impact of changes in the reaction environment on the polymerization mechanism. For instance, the inhibition of cationic polymerization is advantageous for the production of polymers with higher molecular weight polymers, particularly in the presence of Lewis acid.30
In addition to increasing the molecular weight, there has been significant research interest in controlling the sequence structure of α-olefin copolymers,31 since well-defined sequence structures promise great potential for developing advanced polymer materials, such as self-healing,32 molecular data storage and cryptography,33 superior adhesion performance,34 and high ion conduction.35 Up to now, various state-of-the-art methods have been developed to synthesize alternating copolymers, including introducing of specific recognition sites,36 iterative coupling through repeated cyclization,37 and using bulky protection groups.38 While the sequence controlled radical copolymerization of commodity monomers including acrylate, styrene, and acrylamide has made great progress,39–41 the synthesis of sequence-controlled α-olefin copolymers still presents enormous challenges. This is due to the extremely low reactivity of α-olefins and the significant difference in reactivity ratios between α-olefins and commodity co-monomers such as acrylates and styrene. The most promising protocol for synthesizing sequence-defined α-olefin copolymers is the Lewis acid-catalyzed alternating copolymerization of α-olefins with acrylates.42–44 In this protocol, Lewis acids (LA), including BF3,45 ethylaluminum dichloride,46 ZnCl2,47 and scandium triflate (Sc(OTf)3)48etc., exhibit unique property for altering the reactivity of the acrylates through the formation of acrylate-LA complexes. The acrylate-LA complex exhibits extremely low propensity toward homo-polymerization but has the capability to copolymerize with ethylene49 and propylene,50 leading to the formation of alternating copolymers. Inspired by these pioneering works, our group has succeeded in the synthesis of alternating copolymers containing long-chain α-olefin units by using AlCl3 as Lewis-acid catalyst.51,52 However, for LA catalyzed alternating copolymerization, a higher than stoichiometric dosage of LA was essential for forming nearly alternating copolymers, leading to a high-cost purification process and a large amount of waste water containing toxic ions.
In the study, a feasible radical copolymerization technique was developed to synthesize alternating α-olefin copolymer with high molecular weight by using a catalytic dosage of Sc(OTf)3 as the Lewis acid catalyst and 1-octene as a representative of α-olefins to copolymerize with methyl acrylate. Although Sc(OTf)3 have been well applied in catalyzing (meth)acrylate/nonpolar alkene radical copolymerizations,44,48,53 few efforts have been made to quantitatively demonstrate the influence of the polymerization parameters on the molecular weight of the resulting copolymers. In addition, strategy to further enhance the molecular weight of the copolymers is lacking. Herein, the impact of initiator dosage, solvent, monomer concentration and polymerization temperature on copolymer sequence, molecular weight, and molecular weight distribution were studied. To further improve the molecular weight, a difunctional comonomer was introduced to copolymerize with 1-octene and methyl acrylate, and the impact of the difunctional comonomers on the sequence structure were also investigated.
2 Experimental section
2.1 Materials and chemicals
Methyl acrylate (MA, 99%) and 1-octene (99%) from Shanghai Titan Technology Co. Ltd (Shanghai, China) were purified by distillation under reduced pressure and stored under N2 before use. 2,2′-Azobis(2-methylpropionitrile) (AIBN, 99%), 2,2′-azobis-(2,4-dimethylvaleronitrile) (ABVN, 98%), scandium triflate (Sc(OTf)3, 99%), methylene chloride (CH2Cl2, 99.5%), 1,4-dioxane (99.7%) and chlorobenzene (PhCl, 98.0%) purchased from Shanghai Titan Technology Co. Ltd (Shanghai, China) and 1,7-octadiene (98.5%) purchased from Thermo Fisher Scientific (China) Co. Ltd (Shanghai, China) were stored under N2. Methanol purchased from Shanghai Titan Technology Co. Ltd (Shanghai, China), ethanol purchased from Chengdu Jinshan Chemical Co. Ltd (Chengdu, China) and acetone purchased from Chengdu Kelon Chemicals Co. Ltd (Chengdu, China) was used as received.2.2 Polymerization
2.3 Characterization, calculation, and instrumentation
Complexation between monomer MA and Sc(OTf)3 was characterized by detecting the wavelength of carbonyl absorbance peak in the UV-vis absorption spectra recorded on a UV-2600 spectrophotometer (Shimazu Instruments (Suzhou) Co. Ltd) at a scanning speed of 400 nm min−1 in the range of 185 to 400 nm. In addition, nuclear magnetic resonance carbon spectroscopy can be used to analyze the chemical shift of carbon after the complexation of methyl acrylate with different dosages of Sc(OTf)3.NMR spectroscopies (1H NMR and 13C NMR) were used to investigate the composition of the obtained copolymer by the integration of the methoxy protons versus all the aliphatic resonances excluding hydrogen contribution of MA unit (1H NMR), and the sequence structure of the copolymer chains (13C NMR). The NMR (1H NMR, 400 MHz; 13C NMR, 400 MHz) spectra were recorded on Bruker AV II-400 MHz using deuterochloroform as solvent and tetramethylsilane as internal standard at ambient temperature. The productivity was defined and calculated as the ratio of copolymer's yield to monomer's amount. The equation for calculating the composition of the obtained copolymer is as follows.
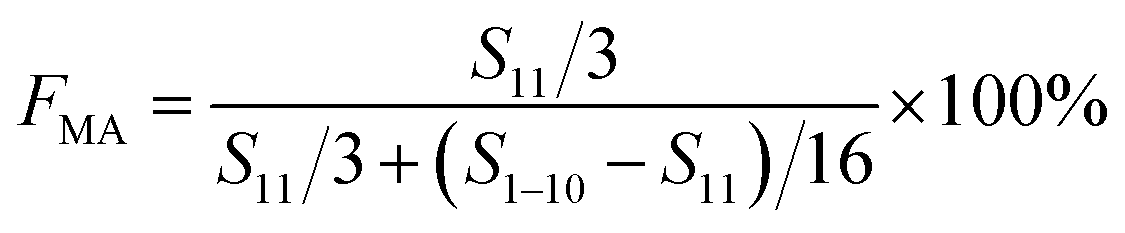 | (1) |
| F1-octene = 1 − FMA | (2) |
F MA: molar content of MA units in the copolymer, F1-octene: molar content of 1-octene units in the copolymer, S11 represents the integrated area of H on C11, and S1–10 represents the integrated area of H on C1–C10.51
The relative molecular weight (Mn) of copolymer was measured using a gel permeation chromatography (GPC, Waters 1515, America) equipped with three columns (Styragel® HR 5 THF 7.8 × 300 mm, Styragel® HR 4 THF 7.8 × 300 mm, and Styragel® HR 3 THF 7.8 × 300 mm) in series taking THF as an eluent at ambient temperature. The flow rate was set at 1 μL min−1. The polymer concentration in THF solvent was 2 mg ml−1, where the polymer was dissolved for more than 10 h in order to get a complete dissolution. Polystyrene was used as a calibration standard.
Thermo-gravimetric analyzer (Netzsch 209F1 Iris, Netzsch Instruments, Germany) was used to test and analyze the thermal weight loss process of the copolymer (temperature range: 30–600 °C, heating rate of 10 °C min−1, N2 atmosphere), in order to evaluate the heat resistance of the copolymer. Differential scanning calorimeter (TA Q250, American TA Instrument) was used to test and analyze the glass transition temperature of copolymers (temperature range: −70–140 °C, heating rate: 10 °C min−1).
The product obtained by introducing the polymerization scheme of 1,7-octadiene contains a part of acetone insoluble polymer gel. After filtering out the gel, wrap it with filter paper and put it into Soxhlet extractor. After extracting with boiling acetone solution for 2 h, vacuum dry it at 70 °C for 24 h, and the weight was recorded.
3 Results and discussions
3.1 Synthesis of alternating 1-octene–MA copolymer in the presence of Sc(OTf)3
The formation of the Sc(OTf)3 MA complex is essential for synthesizing alternating 1-octene–MA copolymer. Therefore, the interaction between Sc(OTf)3 and MA was initially analyzed using 13C NMR spectroscopy, and the results were shown in Fig. 1 and Table S1 in the ESI.† As shown in Fig. 1, in comparison with pristine MA solution, introducing far less than stoichiometric dosage of Sc(OTf)3, only about 0.1× the MA molar content, the chemical shifts of C1′ and C2′ in MA increase by 1.31 ppm and decrease by 0.39 ppm respectively, and the chemical shifts of C3′ increase by 1.09 ppm. These changes clearly suggest that the electron cloud moves from C1′ to C2′ and Sc(OTf)3 has successfully complexed with MA. As the molar ratio of Sc(OTf)3 to MA increases from 0.1 to 0.3, a larger downfield shifts in the chemical shift of C1′ (Δδ = 3.52 ppm) and C3′ (Δδ = 3.04 ppm) accompanied by a larger upfield shift in the chemical shift of C2′ (Δδ = 0.92 ppm) can be observed, suggesting that increasing Sc(OTf)3 dosage can enhance the complexation strength between Sc(OTf)3 and MA.54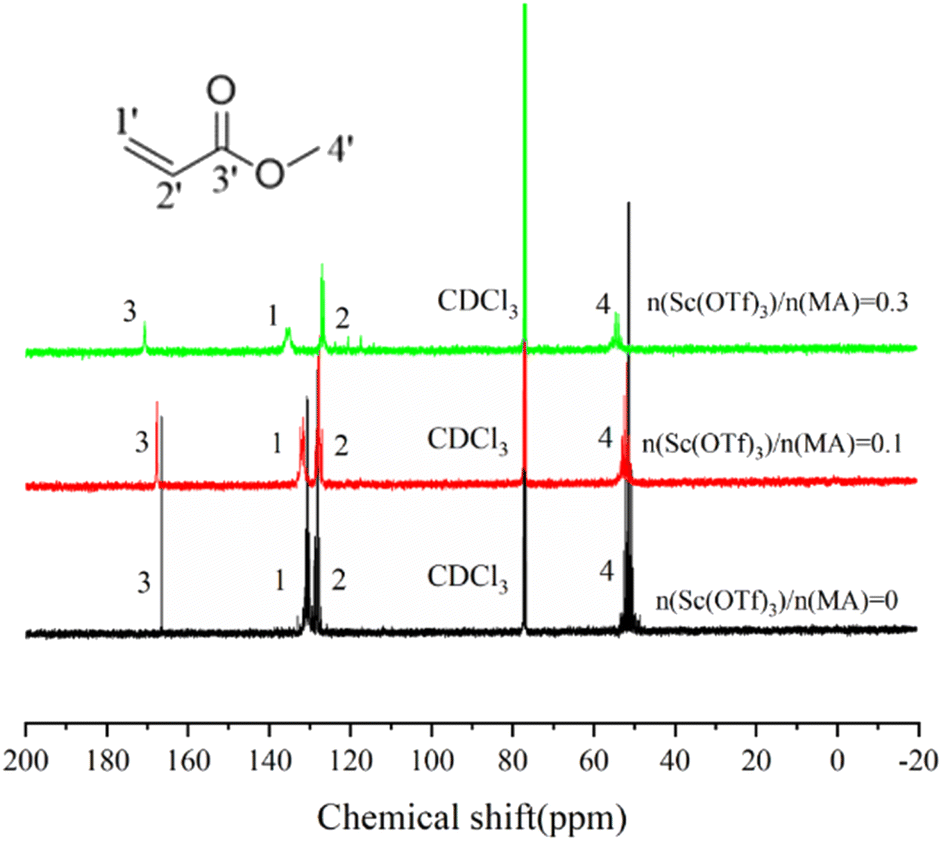 | ||
| Fig. 1 13C NMR spectra of MA complexed with Sc(OTf)3 in CDCl3. | ||
The complex between Sc(OTf)3 and MA were investigated by UV-vis absorption spectra, as shown in Fig. 2. As the molar ratio of Sc(OTf)3 to MA increases, the absorption peaks of the UV-vis absorption spectra of MA exhibit a red shift. This suggests that the addition of Sc(OTf)3 will decrease the π→π* transition energy of the lone pair electrons of carbonyl oxygen in MA molecules, increase the density of carbonyl electron clouds, and consequently decrease the density of carbon![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) carbon double bond electron clouds.55 However, the magnitude of the red shift is not positively correlated to the Sc(OTf)3/MA ratio. During the test, we found that at high Sc(OTf)3 dosage (n(Sc(OTf)3)/n(MA) ≥ 0.3), the dissolution of Sc(OTf)3 in solvent gradually deteriorates with the increase in Sc(OTf)3 dosage, and the degree of redshift of the absorption peak in the UV-visible absorption spectrum of MA is lower than that with a Sc(OTf)3/MA ratio of 0.1, and it continuously decreases with the increase of Sc(OTf)3/MA. This suggests that an excessive dosage of Sc(OTf)3 may disrupt its complexation with MA, interfering with existing complexation. Overall, when the Sc(OTf)3/MA ratio is 0.1, there is an effective complexation between Sc(OTf)3 and MA, which will benefit the alternating copolymerization of 1-octene with MA.
carbon double bond electron clouds.55 However, the magnitude of the red shift is not positively correlated to the Sc(OTf)3/MA ratio. During the test, we found that at high Sc(OTf)3 dosage (n(Sc(OTf)3)/n(MA) ≥ 0.3), the dissolution of Sc(OTf)3 in solvent gradually deteriorates with the increase in Sc(OTf)3 dosage, and the degree of redshift of the absorption peak in the UV-visible absorption spectrum of MA is lower than that with a Sc(OTf)3/MA ratio of 0.1, and it continuously decreases with the increase of Sc(OTf)3/MA. This suggests that an excessive dosage of Sc(OTf)3 may disrupt its complexation with MA, interfering with existing complexation. Overall, when the Sc(OTf)3/MA ratio is 0.1, there is an effective complexation between Sc(OTf)3 and MA, which will benefit the alternating copolymerization of 1-octene with MA.
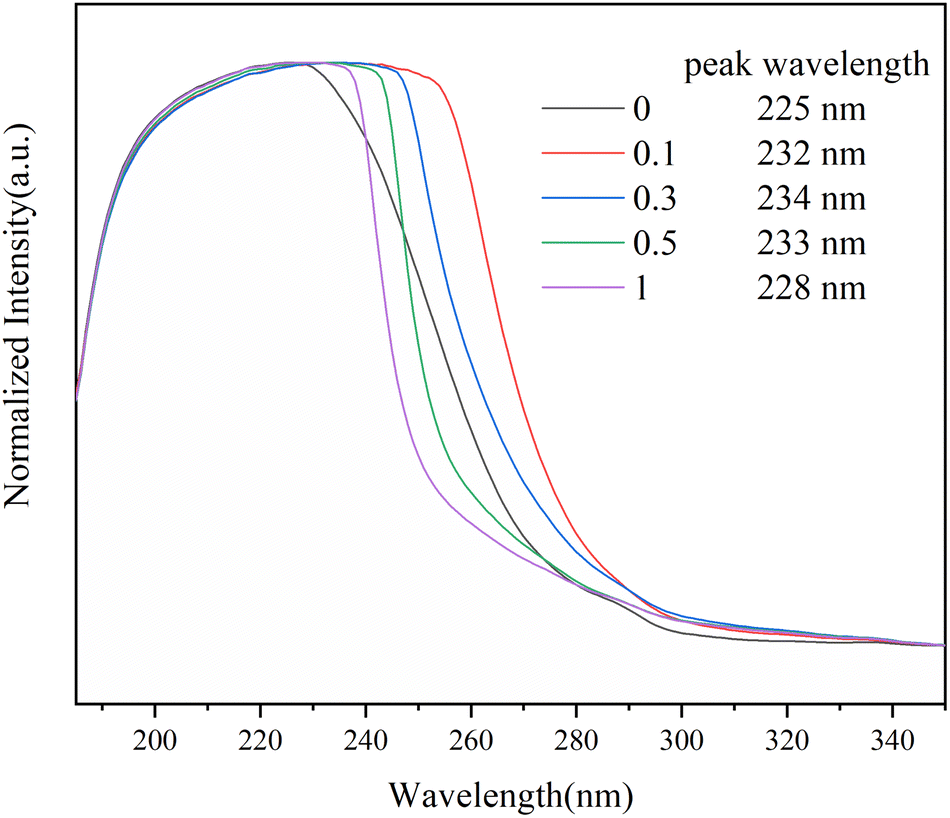 | ||
| Fig. 2 UV-vis absorption spectra of Sc(OTf)3/MA systems with different molar ratios of Sc(OTf)3 to MA in CH2Cl2. | ||
According to the above analysis, the dosage of catalyst Sc(OTf)3 for 1-octene and MA copolymerization was set to n(Sc(OTf)3)/n(MA) = 0.1. Fig. 3 exhibits the 1H NMR spectrum of the resultant copolymer. The chemical shift of the methoxy protons in MA unit can be observed at 3.58 ppm, while the chemical shifts of protons in back-bone chain and 1-octene unit present as a wide multiple peaks in the range between 0.50 ppm and 2.59 ppm. According to eqn (1) and (2), the proportion of 1-octene and MA units in the copolymer could be calculated from the 1H NMR spectrum, and the result was listed in Table 1 (entry 2). The result demonstrates that the resultant copolymer contains 45.04 mol% of 1-octene unit (F1-octene) and 54.96 mol% of MA unit, suggesting the formation of nearly 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 alternating microstructure. 13C NMR analysis were further carried out to verify the sequence structure of the copolymer, and the result is shown in Fig. 4. Neither the chemical shift of MA–MA sequence at 35 ppm nor the chemical shift of octene–octene sequence at 43 ppm were observed, clearly demonstrate the formation of the sequence structure in this case.44,51,56 We further analyzed the molecular weight and its distribution (PDI) of the resultant alternating copolymer by GPC, and the result was listed in Table 1. The Mn of the resultant 1-octene–MA copolymer is 1.79 × 104 g mol−1 with a PDI of 1.79, which is far enough to satisfy the practical requirements as a high-performance polymer.
:1 alternating microstructure. 13C NMR analysis were further carried out to verify the sequence structure of the copolymer, and the result is shown in Fig. 4. Neither the chemical shift of MA–MA sequence at 35 ppm nor the chemical shift of octene–octene sequence at 43 ppm were observed, clearly demonstrate the formation of the sequence structure in this case.44,51,56 We further analyzed the molecular weight and its distribution (PDI) of the resultant alternating copolymer by GPC, and the result was listed in Table 1. The Mn of the resultant 1-octene–MA copolymer is 1.79 × 104 g mol−1 with a PDI of 1.79, which is far enough to satisfy the practical requirements as a high-performance polymer.
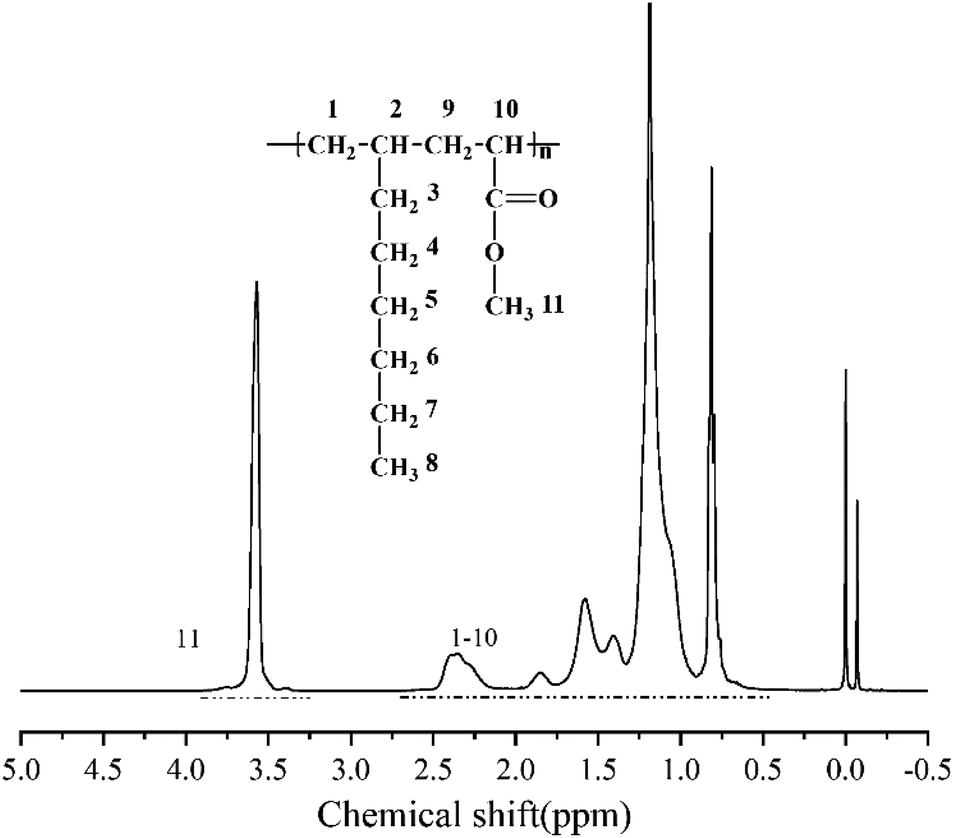 | ||
| Fig. 3 1H NMR spectrum of copolymer of MA and 1-octene in CDCl3. | ||
| #a | φ b | Integration of methoxy protons | Integration aliphatic protons | F 1-octene | F MA | M n × 10−4 (g mol−1) | M w × 10−4 (g mol−1) | PDIc |
|---|---|---|---|---|---|---|---|---|
| a Reaction conditions: n(1-octene)/n(MA) = 2.87; n(Sc(OTf)3)/n(MA) = 0.1; 45 °C. b φ value is the molar ratio of ABVN based on total amount of monomers. c Molecular weight distribution. | ||||||||
| 1 | 0.005 | 1.000 | 5.440 | 45.43 | 54.57 | 1.79 | 3.20 | 1.79 |
| 2 | 0.003 | 1.000 | 5.370 | 45.04 | 54.96 | 2.16 | 3.82 | 1.77 |
| 3 | 0.001 | 1.000 | 6.023 | 48.50 | 51.50 | 2.59 | 4.34 | 1.68 |
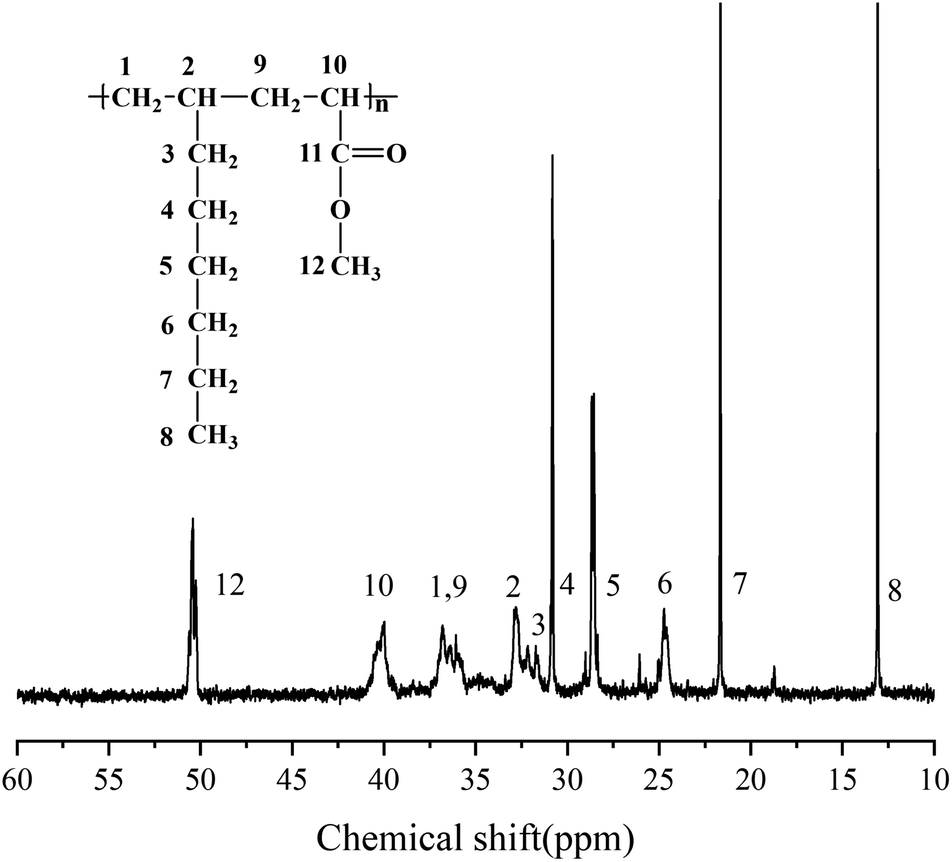 | ||
| Fig. 4 13C NMR spectrum of copolymer of MA and 1-octene in CDCl3. | ||
Following the successful synthesis of the alternating 1-octene–MA copolymer, we further attempted to increase its molecular weight by reducing the dosage of initiator, and the results are shown in Table 1. As the initiator dosage decreases from 0.5 mol% to 0.1 mol%, the molecular weight (Mn) of the copolymer increases from 1.79 × 104 g mol−1 to 2.59 × 104 g mol−1, while the molecular weight distribution is slightly decreased from 1.79 to 1.68. It is noteworthy that nearly 1:1 alternating copolymers have been consistently achieved in all cases, as indicated by ratio of 1-octene unit (F1-octene) to MA unit (FMA) in the copolymer. From the above results it can rationally conclude that: (1) in comparison to traditional LA catalysts, which requires a stoichiometric amount of LA for alternating copolymerization, catalytic amount of Sc(OTf)3 (n(Sc(OTf)3)/n(MA) = 0.1) is adequate for catalyzing the alternating copolymerization of 1-octene with MA in our case;51 (2) the molecular weight of the 1-octene–MA copolymer could be significantly improved by reducing the initiation dosage, however, further reducing initiator dosage may lead to an extremely low polymerization rate and unsatisfactory productivity. Hence, improving the molecular weight of 1-octene–MA copolymer in an efficient way remains a challenge.
3.2 The effect of the difunctional comonomer 1,7-octadiene on the molecular weight and its distribution
To increase the molecular weight of the copolymer, the difunctional comonomer 1,7-octadiene was introduced to copolymer with methyl acrylate and 1-octene in the presence of Sc(OTf)3. Since both of the ene-groups can participate in the polymerization, 1,7-octadiene, the difunctional comonomer, can be served as “chain extender” to get a significant increase in molecular weight of the resulting copolymer. The polymerization parameters and conditions for Sc(OTf)3 catalyzed copolymerization in the presence of different dosage of 1,7-octadiene were listed in Table 2. Under different polymerization conditions, the introduction of 1,7-octadiene can increase the molecular weight (Mn) of the copolymer from 2.59 × 104 g mol−1 to 4.48 × 104 g mol−1, and the molecular weight distribution is increased from 1.68 to 2.14. The results indicate that the incorporation of difunctional comonomer leads to a significant increase in the molecular weight (Mn) and its distribution of the copolymers synthesized.| #b | n(1,7-octadiene)/n(MA) | n(AIBN)/n(Monomer) (%) | F 1-octene (%) | F MA (%) | M n × 10−4 (g mol−1) | M w × 10−4 (g mol−1) | PDIc |
|---|---|---|---|---|---|---|---|
| a The equations for calculating the composition of the obtained copolymer is eqn S1 in the ESI. b Reaction conditions: n(1-octene)/n(MA) = 2.87; n(Sc(OTf)3)/n(MA) = 0.1; temperature 60 °C. c Molecular weight distribution. | |||||||
| 1 | 0.01 | 0.2 | 40.10 | 59.26 | 3.24 | 7.37 | 2.28 |
| 2 | 0.03 | 0.1 | 40.18 | 59.60 | 4.48 | 9.60 | 2.14 |
| 3 | 0.03 | 0.5 | 39.80 | 60.06 | 3.86 | 8.65 | 2.24 |
| 4 | 0.04 | 0.5 | 38.15 | 60.31 | 2.93 | 8.37 | 2.86 |
| 5 | 0.05 | 0.5 | 38.83 | 60.14 | 3.56 | 8.82 | 2.48 |
In addition, with the increase of the proportion of 1,7-octadiene in the monomer, the PDI of the copolymer increases, and the molecular weight distribution curve shows a widening phenomenon (Fig. 5). This suggests that the molecular chains in the polymer undergo branching, resulting in an increase in molecular weight. The results of the obtained copolymer GPC confirm that the 1,7-octadiene unit in the copolymerization system are involved in the copolymerization.52,57
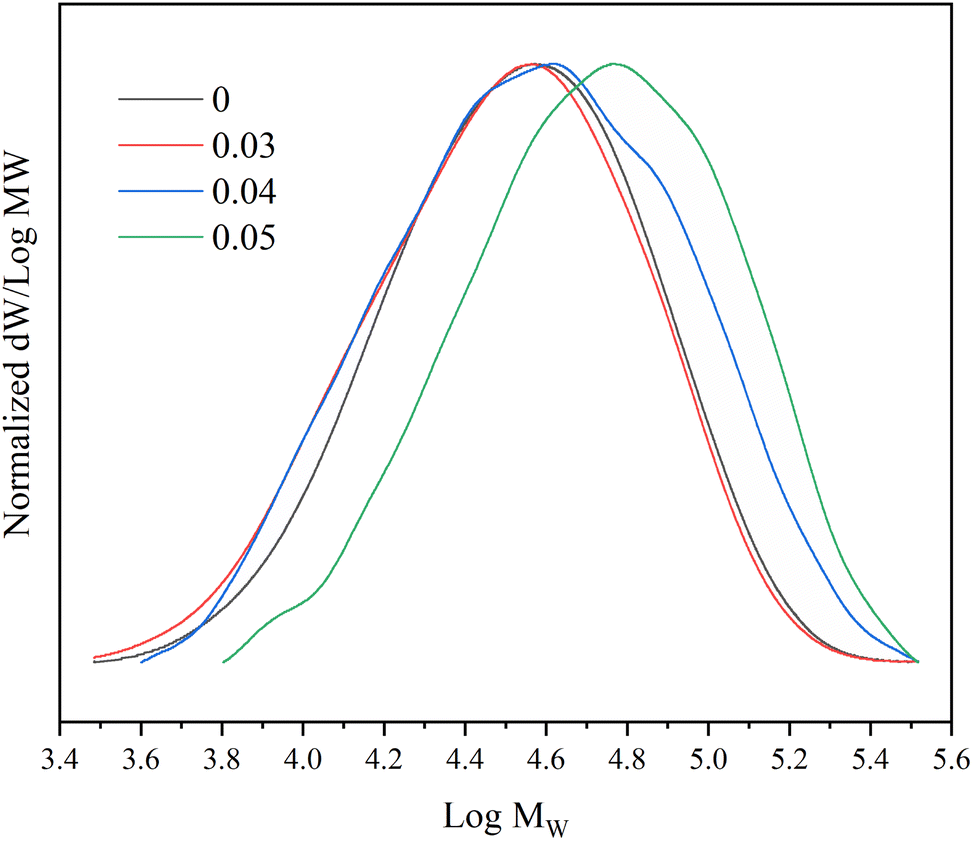 | ||
| Fig. 5 Molecular weight distribution curves of copolymers with different 1,7-octadiene/MA molar ratio. Reaction conditions: n(Sc(OTf)3)/n(MA) = 0.1; n(1-octene)/n(MA) = 2.87; n(ABVN)/n(Monomer) = 0.001; temperature 45 °C. | ||
The impact of 1,7-octadiene on the sequence structures of the resultant copolymers was examined. Compared the proportion of 1-octene (F1-octene) of copolymers synthesized in the absence of 1,7-octadiene (as shown in Table 1) and in the presence of 1,7-octadiene (as shown in Table 2), it can be found that F1-octene slightly decreased from 40–50% to 35–45% as 1,7-octadiene were introduced. This can be rationally attributed to the reaction activity of 1,7-octadiene, which is stronger than that of 1-octene.58,59 Thus, 1,7-octadiene will compete with 1-octene to copolymerize with MA-Sc(OTf)3 complex.
The influence of Sc(OTf)3 dosage on the sequence structure and the molecular weight of 1-octene–MA copolymer synthesized in the presence of varying dosages of 1,7-octadiene were presented in Fig. 6. As the ratio of n(Sc(OTf)3)/n(MA) increased to 0.2, the proportion of 1-octene units in copolymers with different dosages of 1,7-octadiene remained consistent within the range of 40% to 50%. The copolymer obtained exhibits an alternating microstructure close to 1:1, similar to the copolymer synthesized under conditions of n(Sc(OTf)3)/n(MA) = 0.1 (refer to Fig. S1 in the ESI†). This observation indicates that the presence of 1,7-octadiene does not impede the catalytic activity of Sc(OTf)3. In comparison to the copolymer synthesized in the absence of 1,7-octadiene, introducing 0.03–0.05 stoichiometric amount of 1,7-octadiene (based on MA) could significantly increase the molecular weight of the resultant copolymers. The maximum Mn of 4.12 × 104 g mol−1 and Mw of 8.40 × 104 g mol−1 were achieved under the conditions of 42.34%. Meanwhile, it is evident from Fig. 6 that the dosage of Sc(OTf)3 has a minimal effect on the molecular weight. Considering that increasing Sc(OTf)3 dosage did not result in a substantial improvement in the proportion of 1-octene unit in copolymers or the molecular weight of copolymers, the Sc(OTf)3 dosage was set at n(Sc(OTf)3)/n(MA) = 0.1 for the subsequent studies.
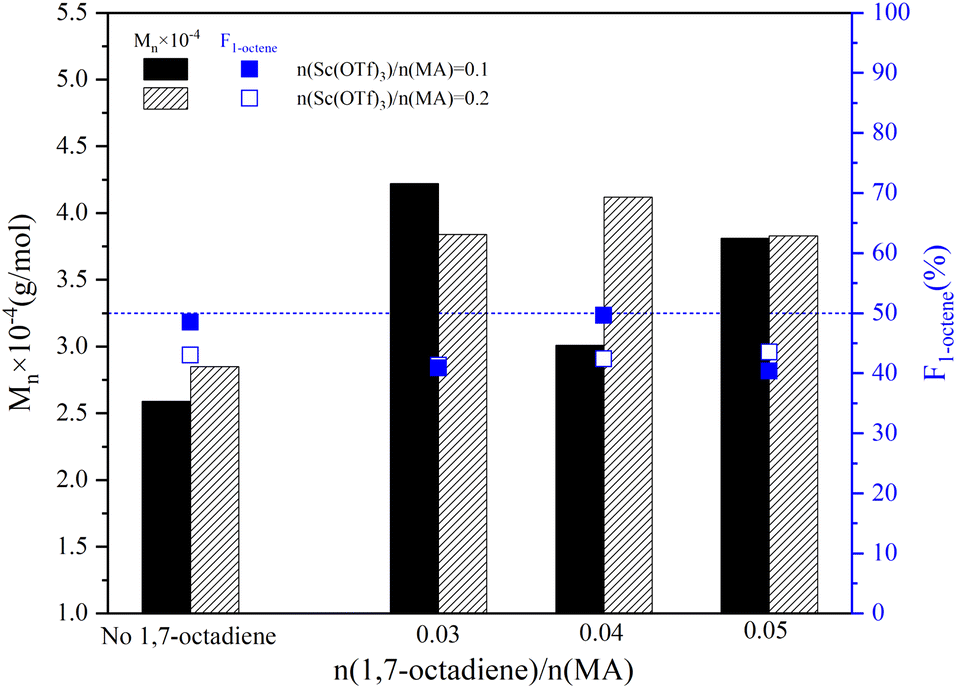 | ||
| Fig. 6 The effect of the 1,7-octadiene on the molecular weight and its distribution. Reaction conditions: n(1-octene)/n(MA) = 2.87; n(ABVN)/n(monomer) = 0.001; temperature 45 °C. | ||
Fig. 7 illustrates the influence of 1,7-octadiene dosage on Mn and PDI of the resultant copolymers. The Mn and PDI presented in Fig. 7 are derived from 28 sets of copolymerization experiments conducted under different 1,7-octadiene dosages, with polymerization parameters and conditions are listed in Table S2,† and each data point in the figure corresponds to each entry in the table. Compared to copolymers synthesized in the absence of 1,7-octadiene, introducing 1 mol% of 1,7-octadiene (based on the amount of MA) to the copolymerization system could increase the molecular weight by 2 times, as shown in Fig. 7a. However, if the dosage of 1,7-octadiene exceeds 1 mol%, the molecular weight of the resultant copolymer decreases, accompanied by a broadening of PDI (Fig. 7b). Meanwhile, gels were generated during the polymerization, and a negative relationship between the gel content and the molecular weight was observed, as shown in Fig. 8 and Table S3.† The phenomenon clearly indicates the dual functions of 1,7-octadiene: serving as the branching site to extend the kinetic chain length and as the crosslinking agent causing irreversible gels. The optimal balance between chain extension and crosslinking is achieved at n(1,7-octadiene)/n(MA) ratio of 0.01. Under the optimal conditions, a copolymer with the highest Mn of ca. 6.77 × 104 g mol−1 and Mw of ca. 13.45 × 104 g mol−1 was obtained, while also maintaining a high degree of the alternating 1-octene–MA sequence (F1-octene = 41%).
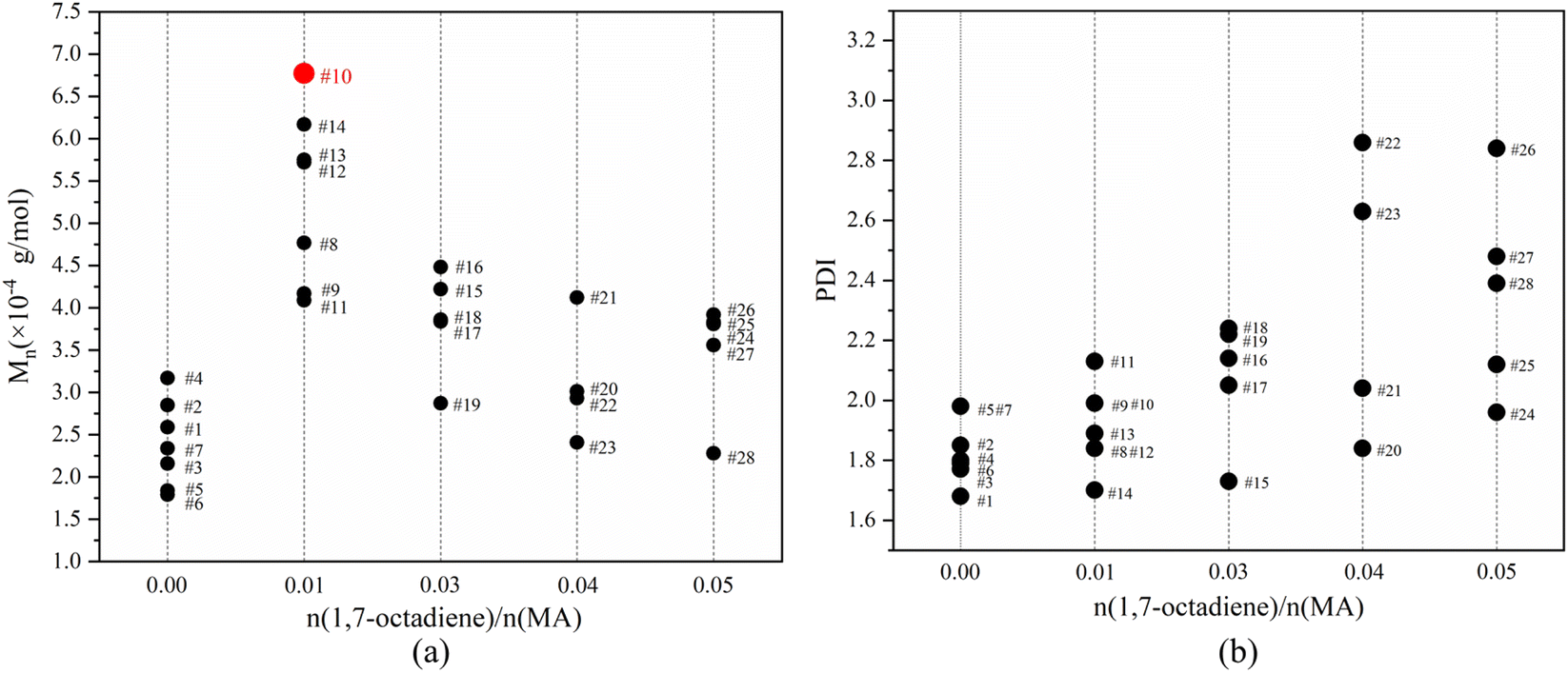 | ||
| Fig. 7 The influence of 1,7-octadiene/MA molar ratio on the molecular weight (a) and PDI (b) of copolymers. The data points were labeled with entry numbers that align with the corresponding experiments listed in Table S2 in the ESI.† | ||
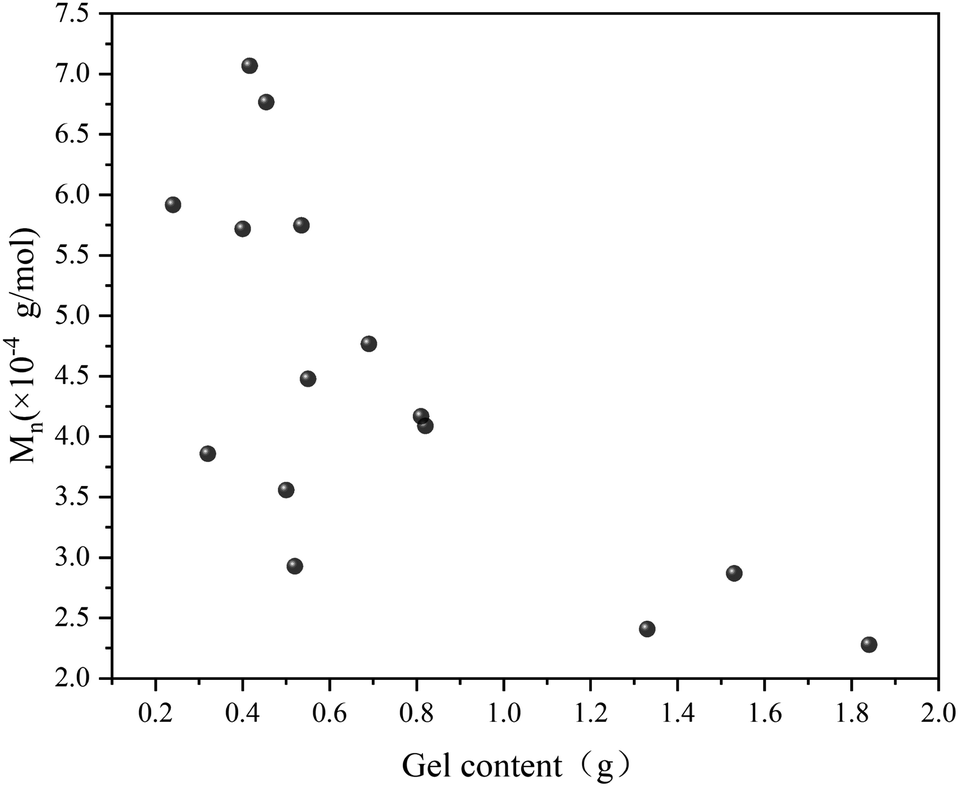 | ||
| Fig. 8 Correlation between molecular weight and gel content in Sc(OTf)3-catalyzed 1-octene–MA copolymerization system in the presence of 1,7-octadiene. Reaction conditions are in Table S3 in the ESI.† | ||
Compared to the copolymers obtained from coordination polymerization and traditional Lewis acid catalytic systems, Sc(OTf)3-catalyzed polymerization of MA and 1-octene can achieve an increase in molecular weight to above 10 × 104 g mol−1 while maintaining alternating structures.19,60
The chemical structure of the copolymer synthesized in the presence of 1,7-octadiene was characterized using 1H NMR spectroscopy. The 1H NMR spectra (Fig. 9) demonstrate the identification of chemical shifts from 1-octene, MA, and 1,7-octadiene units, confirming the successful copolymerization of 1,7-octadiene with 1-octene and MA. Furthermore, it is noteworthy to observe the presence of residual double bonds in 1-octene–MA–1,7-octadiene copolymers. The chemical shifts of 4.8 ppm and 5.7 ppm correspond to the H connected to CC, suggesting that a portion of 1,7-octadiene contributes only one ene-group for copolymerization with 1-octene and MA, while another ene-group remains available as a crosslinkable site.61–63
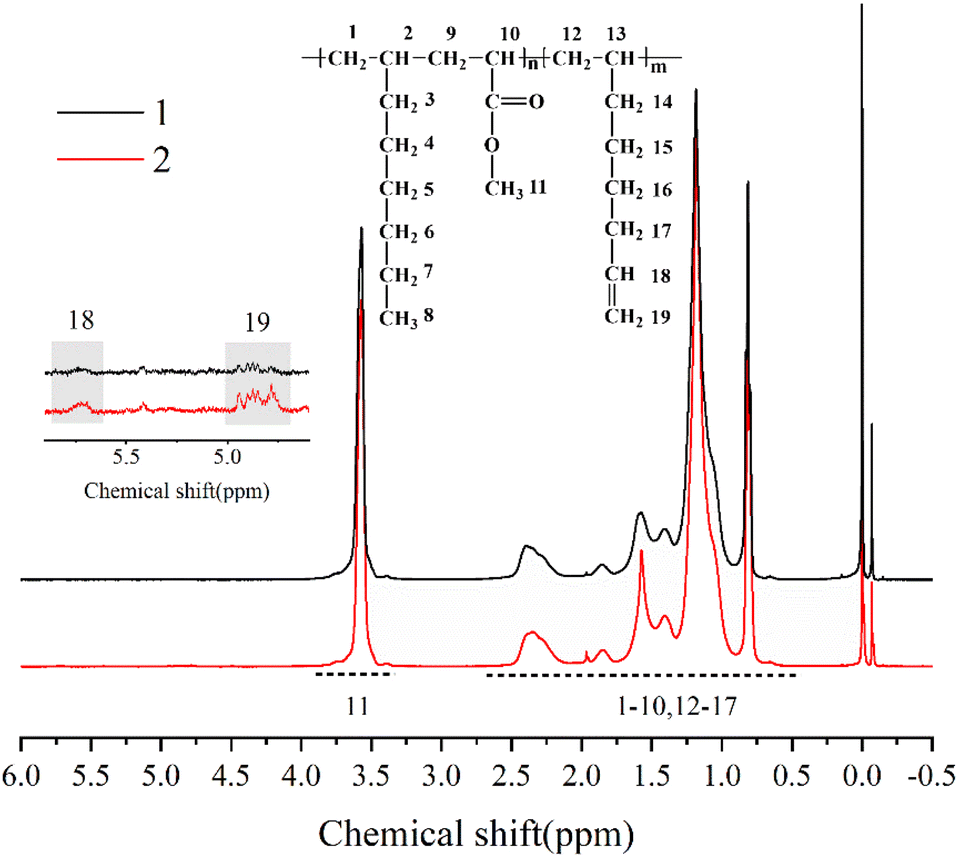 | ||
| Fig. 9 1H NMR spectrum of copolymer of MA, 1-octene and 1,7-octadiene in CDCl3. Reaction conditions: n(1-octene)/n(MA) = 2.87; n(Sc(OTf)3)/n(MA) = 0.1; temperature 60 °C; curve 1: n(1,7-octadiene)/n(MA) = 0.01, AIBN 0.2 mol%, curve 2: n(1,7-octadiene)/n(MA) = 0.03, AIBN 0.1 mol%. | ||
3.3 Influence of polymerization parameters on Sc(OTf)3-Catalyzed 1-octene/MA copolymerization
In order to gain a deeper understanding of the Sc(OTf)3-catalyzed 1-octene/MA copolymerization, a systematic investigation was conducted to examine the impact of polymerization parameters such as solvent, monomer concentration, and temperature. Moreover, copolymerization of α-olefins with different chain lengths including 1-hexene, 1-decene and 1-tetradecane was also carried out to confirm the universality of this strategy.Chlorobenzene and 1,4-dioxane, which are good solvent of Sc(OTf)3, were employed as solvents to carry out the Sc(OTf)3-catalyzed 1-octene/MA copolymerization. As shown in Table 3, Sc(OTf)3-catalyzed 1-octene/MA copolymerization has good suitability toward various solvents. However, it is noteworthy that the proportion of 1-octene units (F1-octene) slightly decreased in comparison to copolymer synthesized in DCM (F1-octene = 41%). This phenomenon may be attributed to the cation–π interaction between Sc3+ and the benzene ring in PhCl, or the coordination between Sc(OTf)3 and the ether group in 1,4-dioxane. Such solvent effect could potentially weaken the MA-Sc(OTf)3 complex, thereby resulting in an increase in the formation of MA–MA units in the resultant copolymers, and consequently causing the deviation of FMA/F1-octene from 1:1.
| #a | Solvent | F 1-octene (%) | F MA (%) | M n × 10−4 (g mol−1) | M w × 10−4 (g mol−1) | PDIb |
|---|---|---|---|---|---|---|
| a Reaction conditions: n(1-octene)/n(MA) = 2.87; n(Sc(OTf)3)/n(MA) = 0.1; Solvent: 30 ml; temperature 40 °C; n(Initiator)/n(Monomer) = 0.001; n(1,7-octadiene)/n(MA) = 0.01. b Molecular weight distribution. | ||||||
| 1 | PhCl | 38.14 | 61.86 | 5.25 | 11.84 | 2.25 |
| 2 | 1,4-Dioxane | 35.43 | 64.57 | 6.17 | 10.50 | 1.70 |
The impact of monomer concentration on the copolymerization was shown in Table 4. When the monomer concentration increased, the molecular weight of the copolymer increased and the molecular weight distribution remained relatively stable. The proportion of 1-octene in the copolymer composition remains relatively stable without obvious change. When the polymerization temperature is increased, the same law also applies. Higher monomer concentration can increase the collision probability between comonomer molecules, which is beneficial for chain growth and thus increases the molecular weight of the copolymer.64,65 The good complexation between Sc(OTf)3 and MA can satisfy the transfer of Sc(OTf)3 from MA units in copolymer chain to MA monomer, facilitating the formation of alternating octene–MA structure and inhibiting the formation of homopolymer units.
| # | [Monomers] (mmol ml−1) | Temp. (°C) | F 1-octene (%) | F MA (%) | M n × 10−4 (g mol−1) | M w × 10−4 (g mol−1) | PDIb |
|---|---|---|---|---|---|---|---|
| a Reaction conditions: n(1-octene)/n(MA) = 2.87; n(Sc(OTf)3)/n(MA) = 0.1; n(Initiator)/n(Monomer) = 0.001; Initiator: AIBN; n(1,7-octadiene)/n(MA) = 0.01. b Molecular weight distribution. | |||||||
| 1 | 1.651 | 60 | 38.47 | 61.53 | 4.09 | 8.72 | 2.13 |
| 2 | 2.384 | 60 | 38.53 | 61.47 | 4.17 | 8.32 | 1.99 |
| 3 | 1.651 | 50 | 37.46 | 62.54 | 5.72 | 10.53 | 1.84 |
| 4 | 2.384 | 50 | 38.34 | 61.66 | 7.07 | 14.25 | 2.02 |
The impact of polymerization temperature on the Sc(OTf)3-catalyzed 1-octene/MA copolymerization behaviors is shown Fig. 10 and 11. From Fig. 10, it can be seen that the molecular weight of the copolymer increased sharply with decreasing polymerization temperature, while the PDI remains relatively stable within the range of 1.60 to 1.95. The average molecular weight of the copolymer at a polymerization temperature of 40 °C (6.17 × 104 g mol−1) exceeds twice that of copolymer synthesized at a polymerization temperature of 60 °C (2.55 × 104 g mol−1). This result can be rationally explained as follows: The low temperature during the polymerization process helps to reduce the concentration of free radicals, which inhibits chain termination and transfer processes, thereby increasing the lifespan of free radicals.66–68
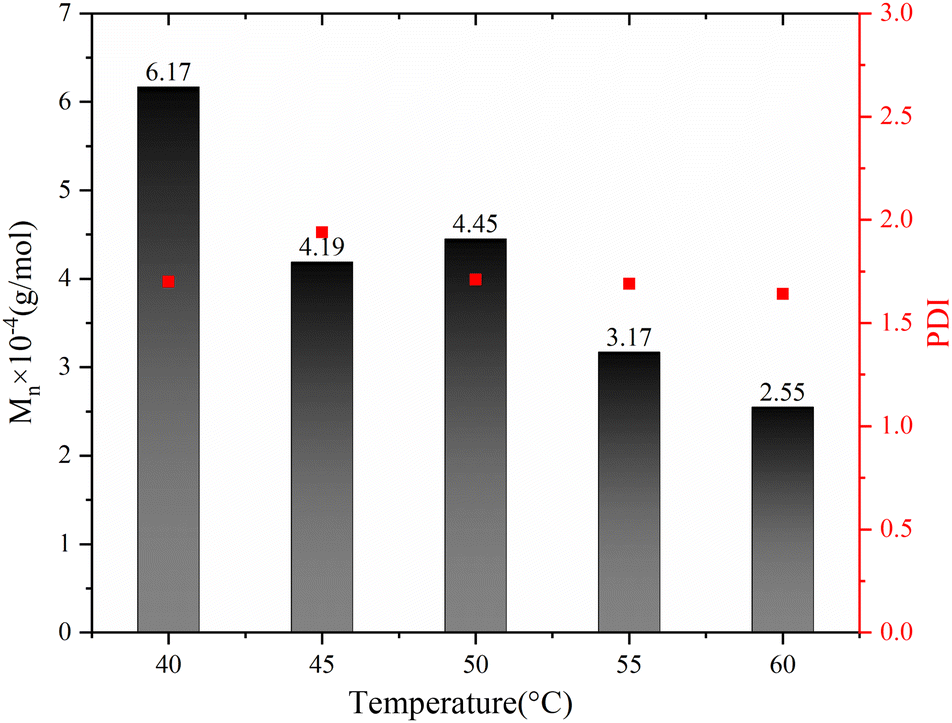 | ||
| Fig. 10 The molecular weights and their distribution of the copolymers with polymerization temperature. Reaction conditions: n(1-octene)/n(MA) = 2.87; n(Sc(OTf)3)/n(MA) = 0.1; n(Initiator)/n(Monomer) = 0.001; n(1,7-octadiene)/n(MA) = 0.01. | ||
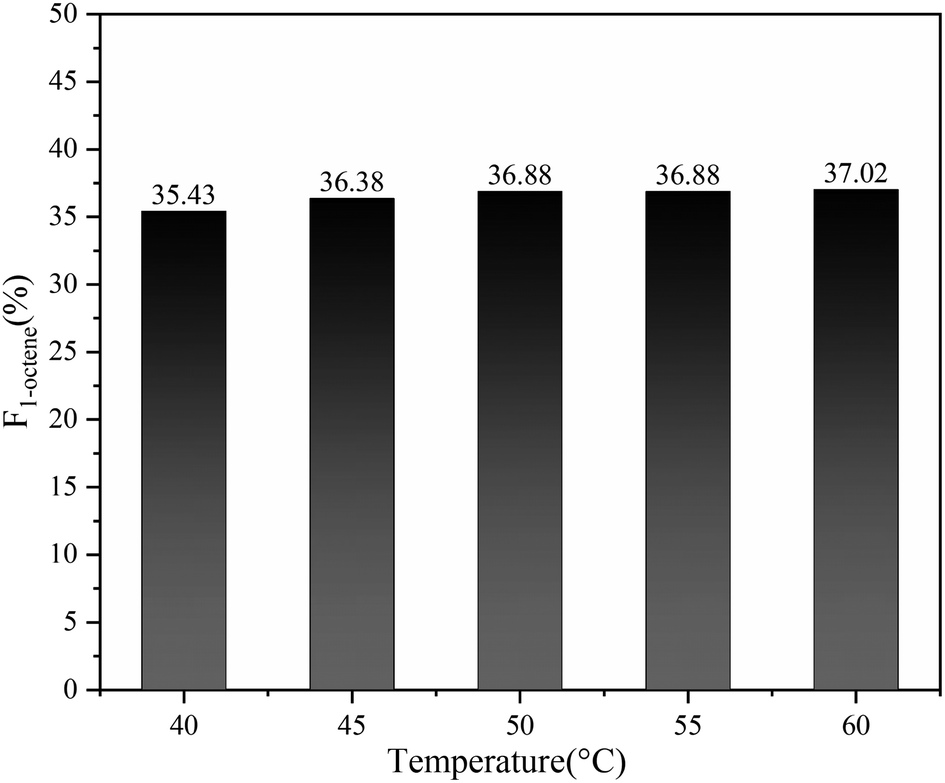 | ||
| Fig. 11 The mole fractions of 1-octene unit (F1-octene) of copolymers with polymerization temperature. Reaction conditions: n(1-octene)/n(MA) = 2.87; n(Sc(OTf)3)/n(MA) = 0.1; n(Initiator)/n(Monomer) = 0.001; n(1,7-octadiene)/n(MA) = 0.01. | ||
In addition, benefiting from the excellent capability of Sc(OTf)3 in controlling the alternating sequence, 1-octene–MA copolymers synthesized at temperatures between 40–60 °C display a notably high proportion of 1-octene units, all exceeding 35%, as shown in Fig. 11.69,70
Based on the success in the synthesis of alternating 1-octene–MA copolymer, α-olefins with different chain lengths such as 1-hexene, 1-decene and 1-tetradecane were also employed to copolymer with MA in the presence of Sc(OTf)3. The 1H NMR spectrums of copolymer and equations for calculating the composition of the obtained copolymer are shown in Fig. S2–S4† and eqn (2) and S4 in the ESI.† The results are shown in Fig. 12, the proportion of α-olefins in the copolymer is above 30%, and the molecular weight reaches over 5 × 104 g mol−1. This suggests that MA after complexing with Sc(OTf)3 can copolymerize with different olefin monomers and maintain an alternating sequence structure of the copolymer. The strategy of increasing the molecular weight of copolymers mentioned earlier has universality for different long chain olefins.
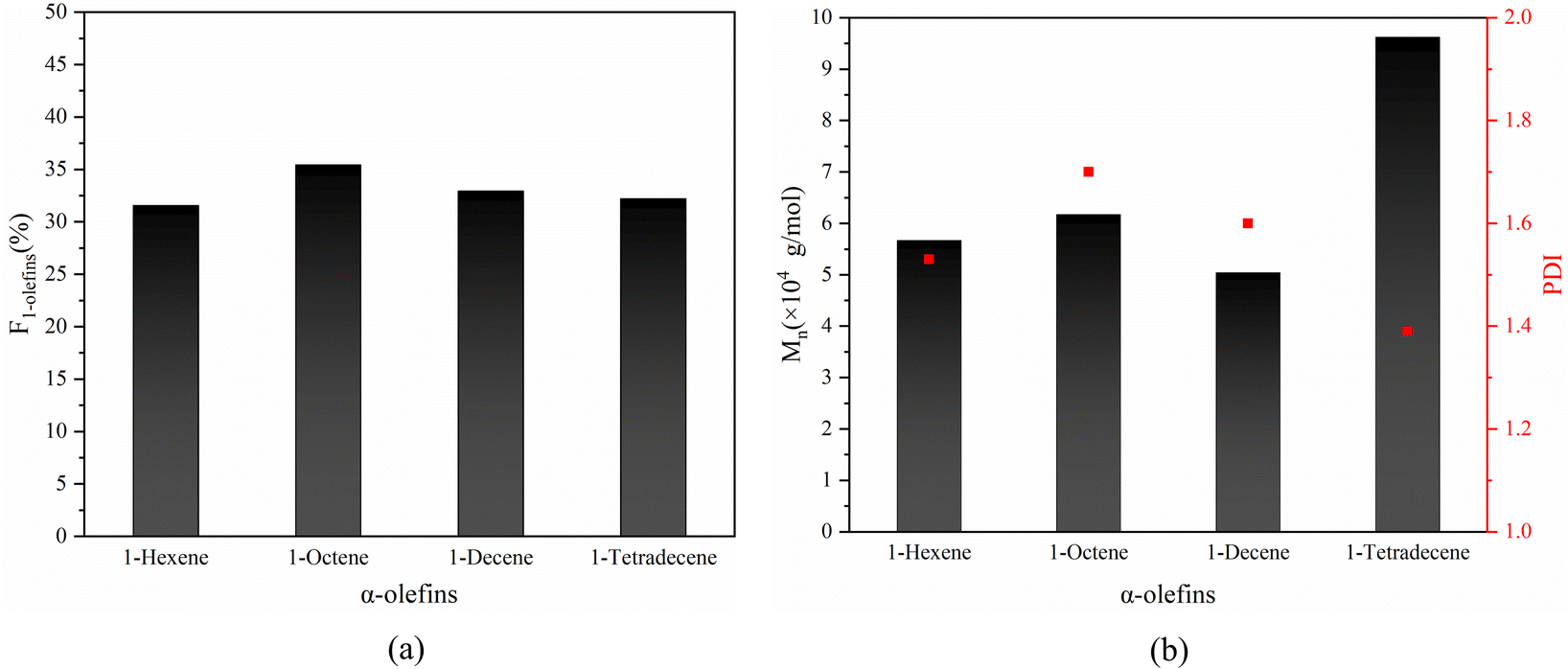 | ||
| Fig. 12 The influence of different types α-olefins on the mole fractions of α-olefins unit (F1-olefins) (a) and the molecular weight/PDI (b) of copolymers. Reaction conditions: n(Sc(OTf)3)/n(MA) = 0.1; n(Initiator)/n(Monomer) = 0.001; n(1,7-octadiene)/n(MA) = 0.01; temperature 40 °C. | ||
Based on the successful synthesis of alternating 1-octene–MA copolymer, α-olefins with varying chain lengths such as 1-hexene, 1-decene, and 1-tetradecane were also employed to copolymerize with MA in the presence of Sc(OTf)3. As depicted in Fig. 12, the proportion of α-olefins in the copolymer exceeds 30%, and the molecular weight exceeds 5 × 104 g mol−1. This indicates that MA, upon complexing with Sc(OTf)3, has the ability to copolymerize with various olefin monomers while preserving an alternating sequence structure in the resulting copolymer. The strategy of increasing the molecular weight of copolymers, as discussed previously, exhibits universality towards various α-olefins.
Moreover, the impact of molecular weight (Mw) and the proportion of 1-octene units (F1-octene) in poly(1-octene-alt-MA) copolymers on their thermal properties, specifically heat resistance and glass transition temperature (Tg), was examined. The investigation involved analyzing the thermal decomposition performance of copolymers with varying Mw and F1-octene. The results were shown in Fig. 13a, which indicate that the initial thermal decomposition temperature (temperature at 5% weight loss, T95%) of poly(1-octene-alt-MA) is more responsive to changes in Mw than F1-octene. An increase in Mw was observed to result in a noticeable rise in T95%, from 348.4 °C (4.19 × 104 g mol−1, curve 3 in Fig. 13a) to 355.2 °C (6.77 × 104 g mol−1, curve 1 in Fig. 13a) or 354.0 °C (6.17 × 104 g mol−1, curve 2 in Fig. 13). Furthermore, the influence of Mw and F1-octene on the Tg of poly(1-octene-alt-MA) was investigated using DSC measurement. The findings revealed that Tg is more sensitive to changes in F1-octene than Mw. Notably, an increase in the proportion of 1-octene unit led to a decrease in Tg, aligning with the expectation that a higher proportion of 1-octene unit enhances the flexibility of the molecular chain, thereby reducing Tg.71,72 In conclusion, maintaining a high proportion of 1-octene and increasing the molecular weight of the copolymer are beneficial for expanding the potential temperature range for the practical application of poly(1-octene-alt-MA).
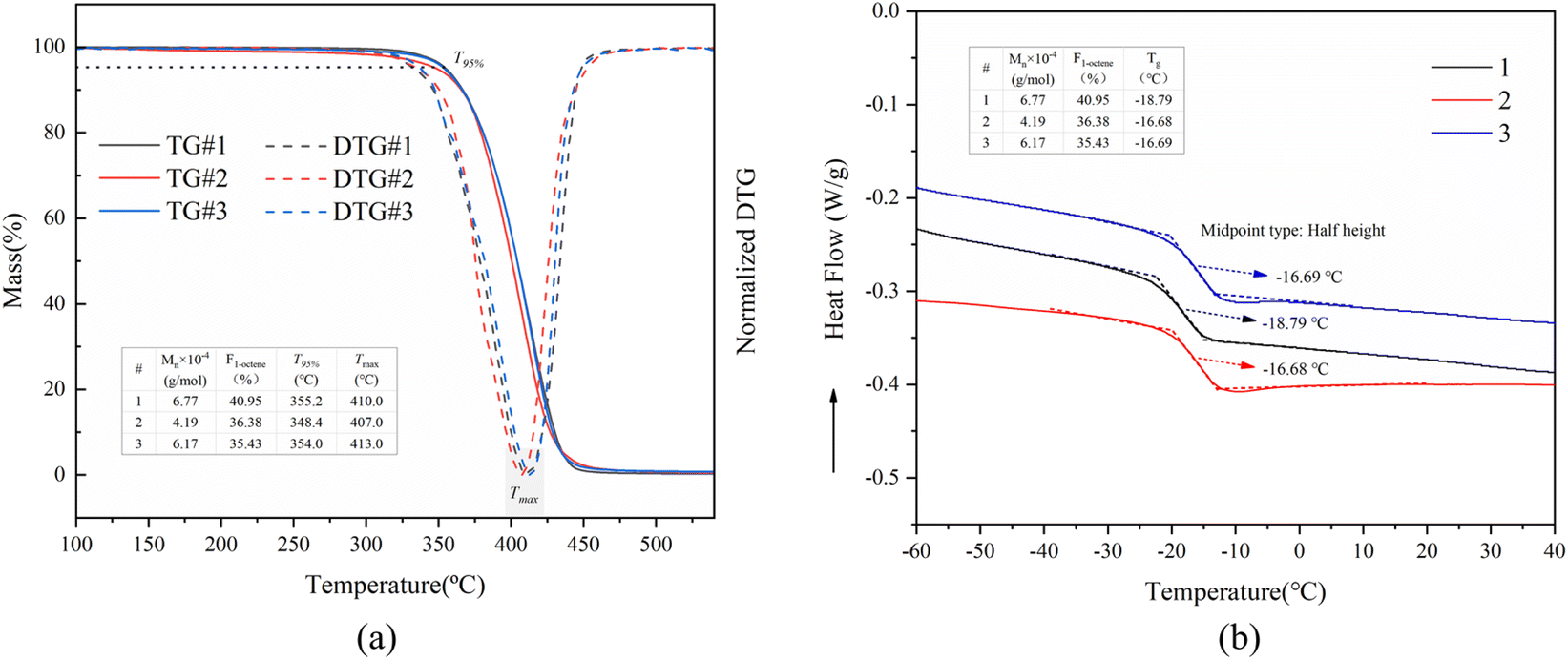 | ||
| Fig. 13 TGA and DTG curves (a) and DSC curves (b) for poly(1-octene-alt-MA) copolymers with different mole fractions of 1-octene unit and molecular weight. | ||
4 Conclusions
With the goal of developing a practical method to convert low-value α-olefins, by-products of ethylene cracking process, into high-performance polymers, we successfully conducted alternating radical copolymerization of olefins with methyl acrylate using 1-octene as the representative of olefins. The control of the alternating sequence and molecular weight has been achieved by optimizing the polymerization parameters, such as monomer concentration and initiator dosage, n(Sc(OTf)3)/n(MA) ratio, temperature, and copolymerizing with a difunctional comonomer. The results indicate that:(1) A catalytic dosage Sc(OTf)3 (n(Sc(OTf)3)/n(MA) = 0.1) is sufficient to catalyze the alternating radical copolymerization of 1-octene and MA.
(2) Introducing 1,7-octadiene as “chain extender” to copolymerization with 1-octene and MA is an efficient way to significant increase the molecular weight, although its loading dosage needs to be finely controlled to avoid gelation. In addition, the carboncarbon double bonds in 1,7-octadiene units not participating in the copolymerization can serve as crosslinking sites for further vulcanization.
(3) As the 1,4-dioxane is used as a solvent, the copolymer can completely dissolve while ensuring high molecular weight and almost alternating sequence structure, making it a more favorable solvent for subsequent material development. The molecular weight of the copolymer increases with the increase of monomer concentration and polymerization temperature.
(4) The strategy of maintaining the alternating sequence structure of copolymers and increasing the molecular weight of copolymers is universal for different long chain α-olefins such as 1-hexene and 1-decene.
(5) The initial decomposition temperature of the copolymer shows an upward trend with the increase of molecular weight. And the glass transition temperature of copolymers is generally below −16 °C, which decreases with the increase of the proportion of 1-octene in the copolymer.
This study has the potential to create new opportunities for producing high-performance polymers from α-olefins, by-products of ethylene cracking process. This could help reduce the depletion of non-renewable petrochemical resources and enable their efficient and valuable utilization.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge the financial support from the Natural Science Foundation of Sichuan Province (No. 2023NSFSC0307).References
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- C. W. S. Yeung, J. Y. Q. Teo, X. J. Loh and J. Y. C. Lim, ACS Mater. Lett., 2021, 3, 1660–1676 CrossRef CAS.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y.-X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS.
- G. Zanchin and G. Leone, Prog. Polym. Sci., 2021, 113, 101342 CrossRef CAS.
- Y. Xiao, P. Liu, W.-J. Wang and B.-G. Li, Macromolecules, 2021, 54, 10381–10387 CrossRef CAS.
- L. Chen and Z. Lin, Polyethylene: Properties, Production and Applications, in 2021 3rd IEEE International Academic Exchange Conference on Science and Technology Innovation, 2021, pp. 1191–1196 Search PubMed.
- D. Jubinville, E. Esmizadeh, S. Saikrishnan, C. Tzoganakis and T. Mekonnen, Sustainable Mater. Technol., 2020, 25, e00188 CrossRef CAS.
- M. Alabdullah, A. Rodriguez-Gomez, T. Shoinkhorova, A. Dikhtiarenko, A. D. Chowdhury, I. Hita, S. R. Kulkarni, J. Vittenet, S. M. Sarathy, P. Castaño, A. Bendjeriou-Sedjerari, E. Abou-Hamad, W. Zhang, O. S. Ali, I. Morales-Osorio, W. Xu and J. Gascon, Nat. Catal., 2021, 4, 233–241 CrossRef CAS.
- T. Ren, S. Liu, H. Li, A. Zhang, X. Li, W. Yuan and X. Gao, Sep. Purif. Technol., 2022, 282, 120012 CrossRef CAS.
- L. Guo, W. Chen, W. Wang, W. An, S. Gao, Y. Zhao, M. Luo, G. He and T. Liu, J. Catal., 2022, 413, 8–19 CrossRef CAS.
- G. Leone, M. Mauri, I. Pierro, G. Ricci, M. Canetti and F. Bertini, Polymer, 2016, 100, 37–44 CrossRef CAS.
- A. Gollwitzer, T. Dietel, W. P. Kretschmer and R. Kempe, Nat. Commun., 2017, 8, 1226 CrossRef.
- N. Patel, V. Valodkar and G. Tembe, Polym. Chem., 2023, 14, 2542–2571 RSC.
- X.-Y. Wang, Y. Gao and Y. Tang, Prog. Polym. Sci., 2023, 143, 101713 CrossRef CAS.
- B. Young, T. R. Hawkins, C. Chiquelin, P. Sun, U. R. Gracida-Alvarez and A. Elgowainy, J. Cleaner Prod., 2022, 359, 131884 CrossRef CAS.
- J. Merna, P. Vlcek, V. Volkis and J. Michl, Chem. Rev., 2016, 116, 771–785 CrossRef CAS PubMed.
- C. Tan, C. Zou and C. Chen, Macromolecules, 2022, 55, 1910–1922 CrossRef CAS.
- A. Schöbel, M. Winkenstette, T. M. J. Anselment and B. Rieger, Copolymerization of Alkenes and Polar Monomers by Early and Late Transition Metal Catalysts, 2012, vol. 3, pp. 779–823 Search PubMed.
- H. Zheng, Z. Qiu, D. Li, L. Pei and H. Gao, J. Polym. Sci., 2023, 61, 2987–3021 CrossRef CAS.
- C. Tan, M. Chen and C. Chen, Trends Chem., 2023, 5, 147–159 CrossRef CAS.
- Y. Zhang and Z. Jian, Polym. Chem., 2022, 13, 4966–4972 RSC.
- S. Kawauchi, A. Akatsuka, Y. Hayashi, H. Furuya and T. Takata, Polym. Chem., 2022, 13, 1116–1129 RSC.
- Z. Wu, Z. Wang, B.-W. Wang, C.-H. Peng and X. Fu, Macromolecules, 2020, 53, 212–222 CrossRef CAS.
- M. Li, X. Wang, Y. Luo and C. Chen, Angew. Chem., Int. Ed., 2017, 56, 11604–11609 CrossRef CAS PubMed.
- T. Rünzi, D. Fröhlich and S. Mecking, J. Am. Chem. Soc., 2010, 132, 17690–17691 CrossRef.
- Y.-N. Zhou, J.-J. Li, T.-T. Wang, Y.-Y. Wu and Z.-H. Luo, Prog. Polym. Sci., 2022, 130, 101555 CrossRef CAS.
- N. Corrigan, K. Jung, G. Moad, C. J. Hawker, K. Matyjaszewski and C. Boyer, Prog. Polym. Sci., 2020, 111, 101311 CrossRef CAS.
- X. Wang and H. Gao, Polymers, 2017, 9, 188 CrossRef PubMed.
- F. Li, F. Zhou, D. Romano and S. Rastogi, Macromolecules, 2023, 56, 1995–2008 CrossRef CAS.
- A. Destephen, L. Lezama and N. Ballard, Polym. Chem., 2020, 11, 5757–5766 RSC.
- J. Jiang, W.-J. Wang, B.-G. Li and S. Zhu, Ind. Eng. Chem. Res., 2019, 58, 18997–19008 CrossRef CAS.
- H. Lai, C. Jin, J. Park, R. Ikura, Y. Takashima and M. Ouchi, Angew. Chem., Int. Ed., 2023, 62, e202218597 CrossRef CAS PubMed.
- Z. Huang, Q. Shi, J. Guo, F. Meng, Y. Zhang, Y. Lu, Z. Qian, X. Li, N. Zhou and Z. Zhang, Nat. Commun., 2019, 10, 1918 CrossRef PubMed.
- K. G. Goswami, S. Mete, S. S. Chaudhury, P. Sar, E. Ksendzov, C. D. Mukhopadhyay, S. V. Kostjuk and P. De, ACS Appl. Polym. Mater., 2020, 2, 2035–2045 CrossRef CAS.
- S. Han, P. Wen, H. Wang, Y. Zhou, Y. Gu, L. Zhang, Y. Shao-Horn, X. Lin and M. Chen, Nat. Mater., 2023, 1–8 Search PubMed.
- S. Ida, M. Ouchi and M. Sawamoto, J. Am. Chem. Soc., 2010, 132, 14748–14750 CrossRef CAS PubMed.
- Y. Hibi, M. Ouchi and M. Sawamoto, Nat. Commun., 2016, 7, 11064 CrossRef CAS PubMed.
- M. Ouchi, Polym. J., 2020, 53, 239–248 CrossRef.
- C. Yang, K. B. Wu, Y. Deng, J. Yuan and J. Niu, ACS Macro Lett., 2021, 10, 243–257 CrossRef CAS PubMed.
- X. Xia, R. Suzuki, T. Gao, T. Isono and T. Satoh, Nat. Commun., 2022, 13, 163 CrossRef CAS.
- J.-F. Lutz, M. Ouchi, D. R. Liu and M. Sawamoto, Science, 2013, 341, 6146 CrossRef.
- A. L. Logothetis and J. M. McKenna, J. Polym. Sci., Polym. Chem. Ed., 2003, 15, 1431–1439 CrossRef.
- A. L. Logothetis and J. M. McKenna, J. Polym. Sci., Polym. Chem. Ed., 2003, 15, 1441–1455 CrossRef.
- Y. Chen and A. Sen, Macromolecules, 2009, 42, 3951–3957 CrossRef CAS.
- A. L. Logothetis and J. M. McKenna, J. polym. sci., Polym. lett. ed., 2003, 12, 131–137 CrossRef.
- M. Hirooka, H. Yabuuchi, S. Morita, S. Kawasumi and K. Nakaguchi, J. Polym. Sci., Part B: Polym. Lett., 2003, 5, 47–55 CrossRef.
- M. Imoto, T. Otsu, B. Yamada and A. Shimizu, Macromol. Chem. Phys., 1965, 82, 277–280 CrossRef CAS.
- M. Nagel, D. Poli and A. Sen, Macromolecules, 2005, 38, 7262–7265 CrossRef CAS.
- S. Akita and K. Nozaki, Polym. J., 2021, 53, 1057–1060 CrossRef CAS.
- H. Fan, Y. Liao and S. Dai, Polymer, 2022, 254, 125076 CrossRef CAS.
- C. Xiao, Y. Yu, L. Jiang and Y. Dan, Ind. Eng. Chem. Res., 2018, 57, 16604–16614 CrossRef CAS.
- C. Xiao, L. Jiang and Y. Dan, Ind. Eng. Chem. Res., 2019, 58, 7857–7865 CrossRef CAS.
- R. Luo and A. Sen, Macromolecules, 2007, 40, 154–156 CrossRef CAS.
- J. Otera, Modern Carbonyl Chemistry, John Wiley & Sons, 2008 Search PubMed.
- J. E. Gautrot, P. Hodge, D. Cupertino and M. Helliwell, New J. Chem., 2006, 30, 1801–1807 RSC.
- S. Kaur, G. Singh and V. K. Gupta, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 2156–2162 CrossRef CAS.
- T. Zhao, Y. Zheng, J. Poly and W. Wang, Nat. Commun., 2013, 4, 1873 CrossRef PubMed.
- R. Chanerika, M. L. Shozi, M. Prato and H. B. Friedrich, Catalysts, 2023, 13, 746 CrossRef CAS.
- A. Ali, M. K. Tufail, M. I. Jamil, W. Yaseen, N. Iqbal, M. Hussain, A. Ali, T. Aziz, Z. Fan and L. Guo, Molecules, 2021, 26, 2037 CrossRef CAS PubMed.
- Y. Wang, R. Gao, Q. Gou, J. Lai, R. Zhang, X. Li and Z. Guo, Eur. Polym. J., 2022, 181, 111693 CrossRef CAS.
- Y. Liu, Y. Qin and J.-Y. Dong, Ind. Eng. Chem. Res., 2020, 59, 12038–12047 CrossRef CAS.
- H. Chen and J. Kong, Polym. Chem., 2016, 7, 3643–3663 RSC.
- H. Li, G. Rojas and K. B. Wagener, ACS Macro Lett., 2015, 4, 1225–1228 CrossRef CAS PubMed.
- R. Han, S. Lu, Y. Wang, X. Zhang, Q. Wu and T. He, Electrochim. Acta, 2015, 173, 796–803 CrossRef CAS.
- R. Ye, X. Sun, X. Mao, F. S. Alfonso, S. Baral, C. Liu, G. W. Coates and P. Chen, Nat. Chem., 2024, 16, 210–217 CrossRef CAS PubMed.
- L. Sollka and K. Lienkamp, Macromol. Rapid Commun., 2020, 42, 2000546 CrossRef.
- J. M. Kaiser and B. K. Long, Coordin. Chem. Rev., 2018, 372, 141–152 CrossRef CAS.
- T. Pirman, M. Ocepek and B. Likozar, Ind. Eng. Chem. Res., 2021, 60, 9347–9367 CrossRef CAS.
- L. Zhong, H. Zheng, C. Du, W. Du, G. Liao, C. S. Cheung and H. Gao, J. Catal., 2020, 384, 208–217 CrossRef CAS.
- R. Kalepu and S. Mishra, Chem. Sci. J., 2020, 132, 48 CrossRef CAS.
- Y. Kametani and M. Ouchi, ACS Polym. Au, 2021, 1, 10–15 CrossRef CAS PubMed.
- C.-C. Huang, M.-X. Du, B.-Q. Zhang and C.-Y. Liu, Macromolecules, 2022, 55, 3189–3200 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra00165f |
| This journal is © The Royal Society of Chemistry 2024 |