DOI:
10.1039/D4RA00845F
(Paper)
RSC Adv., 2024,
14, 5846-5850
Highly electron-deficient 1-propyl-3,5-dinitropyridinium: evaluation of electron-accepting ability and application as an oxidative quencher for metal complexes†
Received
2nd February 2024
, Accepted 8th February 2024
First published on 14th February 2024
Abstract
Impacts of the nitro groups on the electron-accepting and oxidizing abilities of N-propylpyridinium were evaluated quantitatively. A 3,5-dinitro derivative has efficiently quenched emission from photosensitizing Ru(II) and Ir(III) complexes owing to the thermodynamically-favored electron transfer to the pyridinium whose LUMO is greatly lowered by the presence of electron-withdrawing nitro groups.
Introduction
Pyridines are typical electron-deficient heterocyclic compounds that can be seen all around us as substructures of functional materials such as pharmaceuticals, ligands, and optical and electronic devices.1 The nucleophilic ring nitrogen, furthermore, undergoes N-alkylation, resulting in pyridinium salt. Owing to their highly electron-deficient aromatic character, pyridinium skeletons have been utilized in a variety of natural/artificial systems as electron acceptors as represented by NAD+,2 methyl viologen3 and so forth.4 On the other hand, a nitro group exhibits strong electron-withdrawing ability due to both resonance and inductive effects, with the latter effect equivalent to two chloro groups.5 Therefore, a combination of the highly electron-deficient pyridinium and strong electron-withdrawing nitro group is expected to significantly increase the oxidation or electron-accepting abilities.
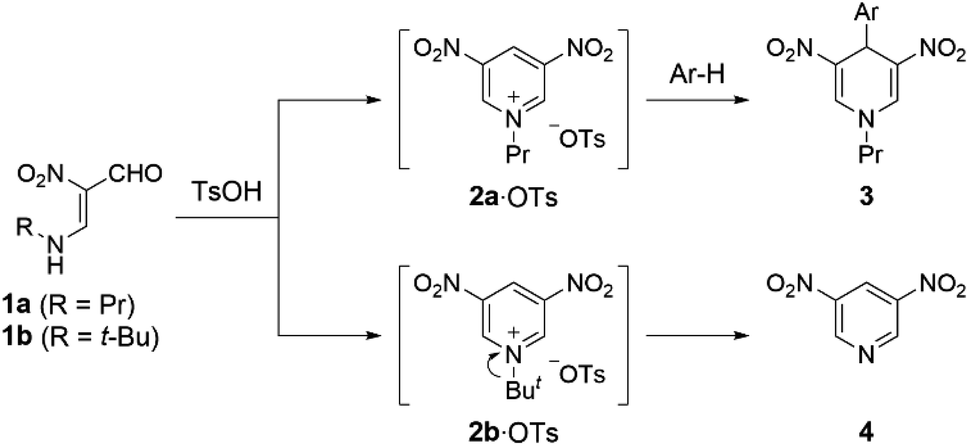
In our previous work, we have demonstrated that 1-propyl-3,5-dinitropyridinium salt 2a·OTs is formed in situ upon treatment of N-propyl-β-formyl-β-nitroenamine 1a (R = Pr) with p-toluenesulfonic acid (TsOH), and formation of the salt 2a·OTs is confirmed by trapping as 4-arylated 1,4-dihydropyridine derivatives 3 with electron-rich benzenes.6 On the other hand, 3,5-dinitropyridine 4 was obtained when N-tert-butylenamine 1b (R = t-Bu) was subjected to the same reaction, which is because a stable tert-butyl cation is readily eliminated (Scheme 1). The easy access to 3,5-dinitropyridine 4 facilitates the N-modification to afford versatile N-alkyl-3,5-dinitropyridinium salts 2. Indeed, treatment of 4 with propyl triflate (PrOTf) proceeded at room temperature to furnish 2a·OTf. This easily modifiable feature prompted us to evaluate the electron-acceptability of its N-alkylated form (2a+) by comparing with 3-nitro and unsubstituted pyridinium relatives (5+ and 6+) that are prepared from commercially available pyridines by N-propylation.
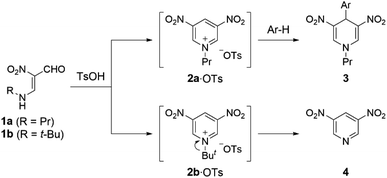 |
| | Scheme 1 Generation of 1-alkyl-3,5-dinitropyridinium salts 2·OTs in situ from N-alkyl-β-formyl-β-nitroenamines 1. | |
Experimental
Preparation of 2a·OTf
A mixture of 3,5-dinitropyridine 4 (86 mg, 0.5 mmol) and PrOTf (122 mg, 0.63 mmol) was stirred without solvent at room temperature for 2 d. Colorless precipitates were collected and washed with CH2Cl2 to afford 2a·OTf (95 mg, 0.28 mmol, 56%) as colorless powder, mp 217.0–217.8 °C. 1H NMR (400 MHz, CD3CN) δ 1.03 (t, J = 7.6 Hz, 3H), 2.12 (tq, J = 7.6, 7.6 Hz, 2H), 4.80 (t, J = 7.6 Hz, 2H), 9.78 (t, J = 2.0 Hz, 1H), 9.94 ppm (d, J = 2.0 Hz, 2H); 13C NMR (100 MHz, CD3CN) δ 10.2 (CH3), 25.3 (CH2), 66.5 (CH2), 121.8 (q, J = 318.0 Hz, CF3), 135.9 (CH), 147.3 (CH), 147.9 ppm (C); 19F NMR (376 MHz, CD3CN) δ 79.36 ppm; HRMS (ESI-TOF) calcd for C8H10N3O4 (M+): 212.0666, found: 212.0676.
Characterization of 5·OTf
mp 102.3–102.7 °C. 1H NMR (400 MHz, CD3CN) δ 1.00 (t, J = 7.2 Hz, 3H), 2.06 (tq, J = 7.2, 7.2 Hz, 2H), 4.66 (t, J = 7.2 Hz, 2H), 8.30 (dd, J = 8.4 Hz, 6.2 HZ, 1H), 9.01 (dd, J = 6.2, 2.0 Hz, 1H), 9.18 (ddd, J = 8.4, 2.0, 1.2 Hz, 1H), 9.63 ppm (d, J = 1.2 Hz 1H); 13C NMR (100 MHz, CD3CN) δ 10.3 (CH3), 25.2 (CH2), 65.2 (CH2), 125.1 (q, J = 319 Hz, CF3), 130.4 (CH), 141.0 (CH), 142.7 (CH), 147.8 (C), 150.1 ppm (CH); 19F NMR (376 MHz, CD3CN) δ 79.31 ppm; HRMS (ESI-TOF) calcd for C8H11N2O2 (M+): 167.0815, found: 167.0819.
Characterization of 6·OTf
1H NMR (400 MHz, CD3CN) δ 1.00 (t, J = 7.6 Hz, 3H), 2.06 (tq, J = 7.6, 7.6 Hz, 2H), 4.49 (t, J = 7.6 Hz, 2H), 8.03 (br, 2H), 8.51 (t, J = 7.6 Hz, 1H), 8.70 ppm (d, J = 5.6 Hz, 2H); 13C NMR (100 MHz, CD3CN) δ 9.3 (CH3), 24.0 (CH2), 62.9 (CH2), 120.9 (q, J = 318 Hz, CF3), 128.1 (CH), 144.2 (CH), 145.5 ppm (CH); 19F NMR (376 MHz, CD3CN) δ 79.30 ppm; HRMS (ESI-TOF) calcd for C8H12N (M+): 122.0964, found: 122.0966.
Other chemicals
[Ru(bpy)3](PF6)2 (bpy = 2,2′-bipyridine) is the same sample which has been used in the earlier literatures.7 [Ir(ppy)2(bpy)]PF6 (ppyH = 2-phenylpyridine) was synthesized and purified similarly to the reported procedure.8 Tetra-n-butylammonium hexafluorophosphate (TBAPF6, Wako Pure Chemical Industries) was purified by repeated recrystallizations from ethanol. Ferrocene (Wako Pure Chemical Industries) was used as supplied. Anhydrous or spectroscopic-grade CH3CN (Wako Pure Chemical Industries) was used without further purification for the electrochemical or spectroscopic measurements, respectively.
Electrochemical measurements
Cyclic voltammetry of the complexes in CH3CN at 298 K was performed by using a BAS ALS-1202A electrochemical analyzer with a three-electrode system using glassy-carbon working, Ag auxiliary, and Ag/AgNO3 reference electrodes (∼0.01 mol dm−3 (=M) in CH3CN containing ∼0.1 M TBAPF6) supplied by BAS Inc. The sample solutions containing a pyridinium salt or metal complex (∼1.0 mM) and TBAPF6 as a supporting electrolyte (∼0.1 M) in the absence or presence of ferrocene as an internal standard were deaerated by purging an argon-gas stream over 20 min prior to measurements. The potential sweep rate was 100 mV s−1.
Emission quenching study
Emission spectra were recorded and emission quantum yields (Φem) were determined by the absolute method using a Hamamatsu Photonics Quantaurus-QY Plus C13534-02. Emission intensity at each wavelength was corrected for system spectral response so that the vertical axis of a spectrum corresponds to the photon number at each wavelength. Emission decay profiles of [Ir(ppy)2(bpy)]PF6 was measured by using a Hamamatsu C4334 streak camera with a C5094 polychromator by exciting at 400 nm using second harmonics of a femtosecond-pulse mode-locked Ti:sapphire laser (MKS Instruments Spectra-Physics Tsunami® 3941-M1BB and 3980 frequency doubler/pulse selector, 1 MHz) and analyzed by a single exponential decay function. Sample solutions were deaerated by purging with an argon-gas stream for over 30 min.
Free energy changes for the electron-transfer processes (−ΔG) were calculated by:9
| | | −ΔG = nF[E1/2(Q+/0) − E1/2(M*)] + ZQZMe2/Dsd = nF[E1/2(Q+/0) − E1/2(M)] + E0(M*) + ZQZMe2/Dsd | (1) |
In
eqn (1),
E1/2(Q
+/0) is the reduction potential of Q
+, and
E1/2(M) is the oxidation potential of the complex (1.32 and 1.63 V
vs. SCE for [Ru(bpy)
3]
2+ and [Ir(ppy)
2(bpy)]
+, respectively, see Fig. S1
†).
E0(M*) is the excited-state zeroth energy and has been determined to be 16
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif)
360 and 16
850 cm
−1 for [Ru(bpy)
3]
2+ and [Ir(ppy)
2(bpy)]
+, respectively, by the Franck–Condon analysis.
10ZQ and
ZM are the charges of Q
+ and complex.
d is the sum of effective radii of Q
+ and complex estimated for the optimized geometries by DFT calculations (4.7, 4.6, 4.4, 6.2 and 6.2 Å for
2a+,
5+,
6+, [Ru(bpy)
3]
2+ and [Ir(ppy)
2(bpy)]
+, respectively).
Ds,
n,
F and
e are the static dielectric constant of the solvent (relative dielectric constant of CH
3CN: 37.5), the number of electrons transferred, the Faraday constant and the formal charge, respectively. It should be noted that, in
eqn (1), an electrostatic work term for the electron-transfer products was omitted since the reduced pyridiniums are charge-neutral.
Theoretical calculations
Theoretical calculations for the compounds were conducted with Gaussian 09W software (Revision C.01).11 The ground-state geometries of the pyridinium cations were optimized by using density functional theory (DFT) using the restricted B3LYP functional with 6-31+G(d,p) basis set. All the optimized geometries did not gave any negative frequencies under identical methodologies. Lowest-energy unoccupied molecular orbitals were plotted using GaussView 5.12 All the calculations were carried out as in acetonitrile by using a polarizable continuum model (PCM).
Results and discussion
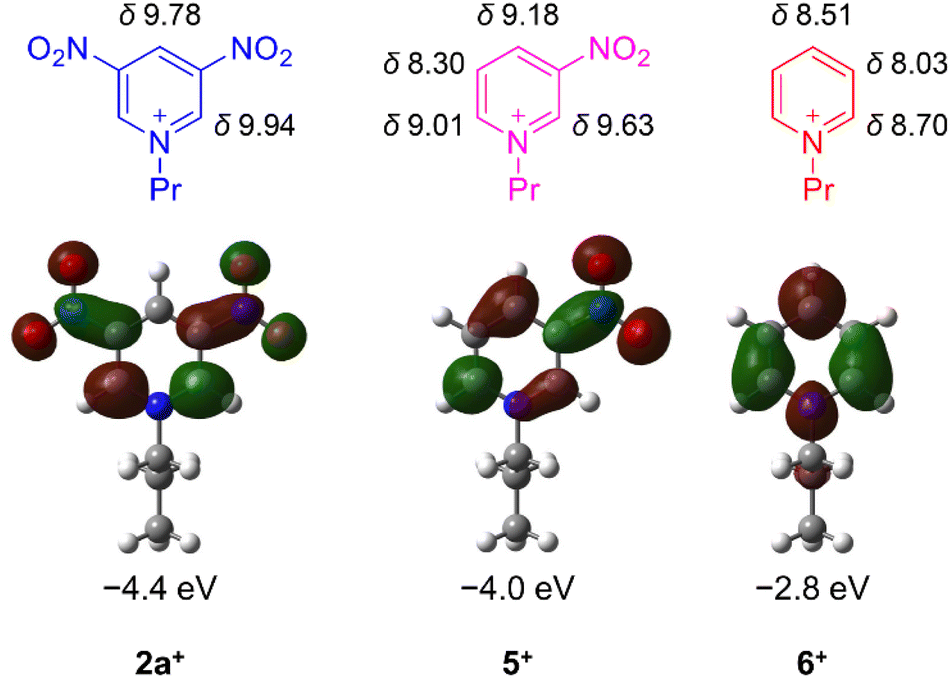
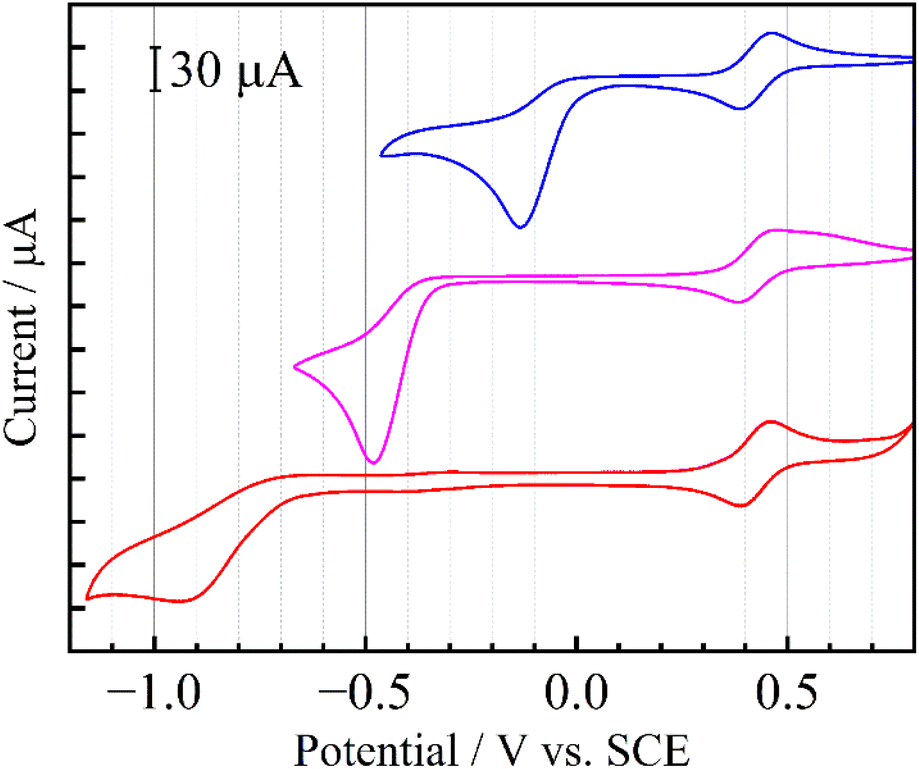
Down-field shifts of the ring protons in the 1H NMR spectra in CD3CN were observed as the number of nitro groups increased (Fig. 1), indicating a decrease in the electron density of the pyridine ring. All the pyridiniums 2a+, 5+ and 6+ in CH3CN exhibited an irreversible reduction wave as shown in Fig. 2. Half reduction potential (E1/2) was shifted to a positive potential region with increasing the nitro group (E1/2 = −0.061 (2a+), −0.41 (5+) and −0.80 V (6+) vs. saturated calomel electrode (SCE)). These tendencies were supported by DFT calculations, as the LUMO energy of 2a+ was lowered to −4.4 eV (see Fig. 1). It is, furthermore, worth emphasizing that the E1/2 value of 2a+ is surprisingly positive even by comparing with that of methyl viologen (−0.44 V vs. sodium saturated calomel electrode (SSCE)13). In contrast to fully reversible redox behavior of methyl viologen and resulting applications as a redox shuttle,3 the highly-positive reduction potential of 2+ is advantageously utilizable as a sacrificial electron acceptor in the various photochemical systems.
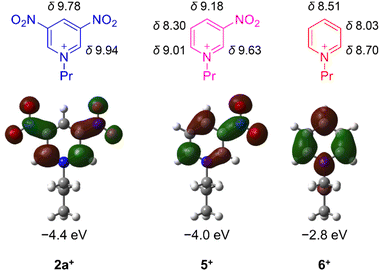 |
| | Fig. 1 Chemical shifts of 1H NMR in CD3CN (given in ppm) and LUMO distributions/energies of pyridiniums 2a+, 5+ and 6+. | |
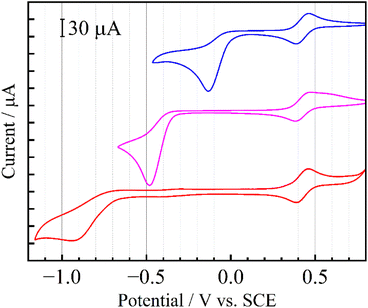 |
| | Fig. 2 Cyclic voltammograms of 2a·OTf (blue), 5·OTf (pink) and 6·OTf (red) in deaerated CH3CN containing 0.1 M TBAPF6. Reversible waves at around +0.43 V represent redox couples of ferrocene as an internal standard. | |
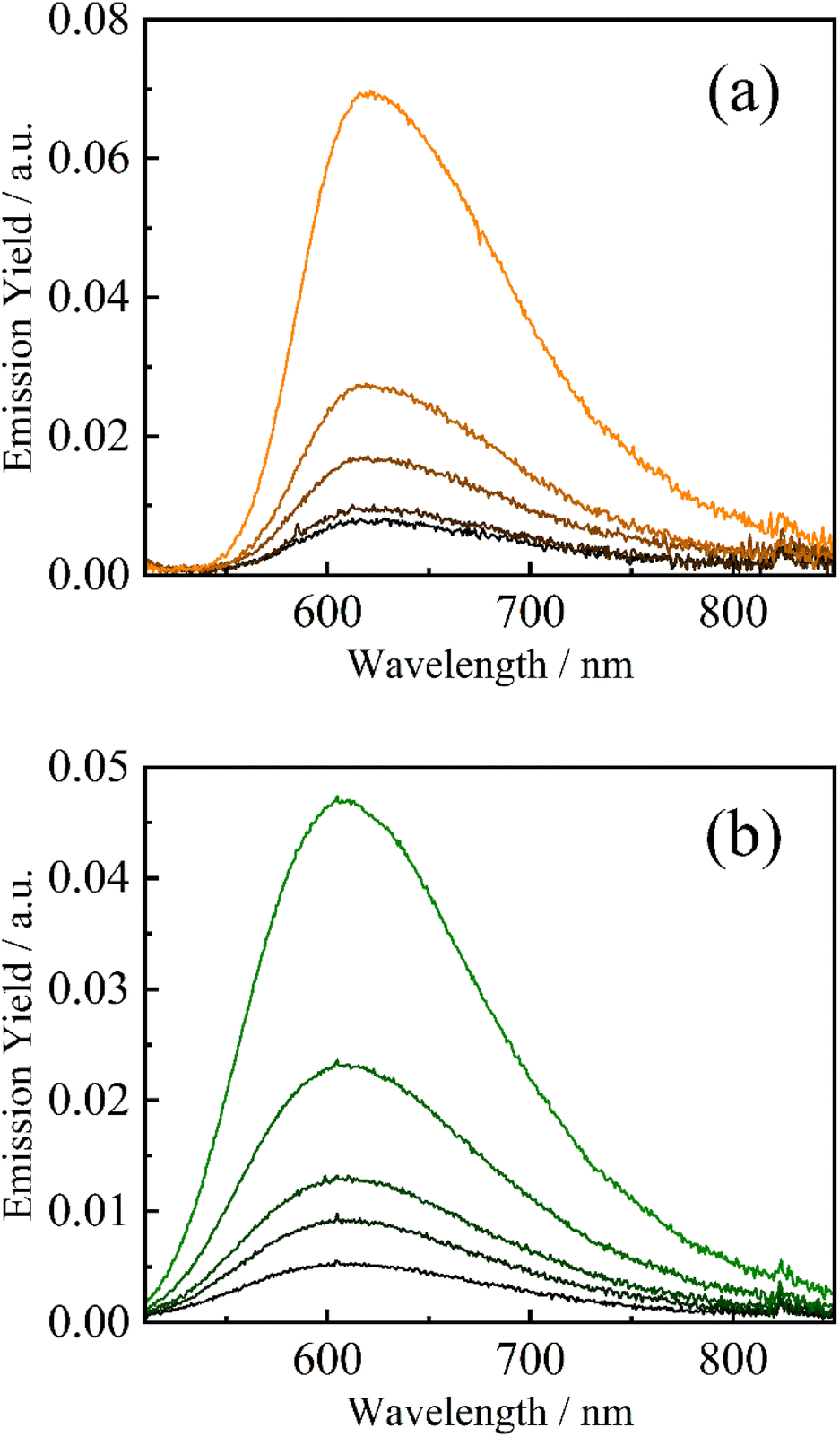
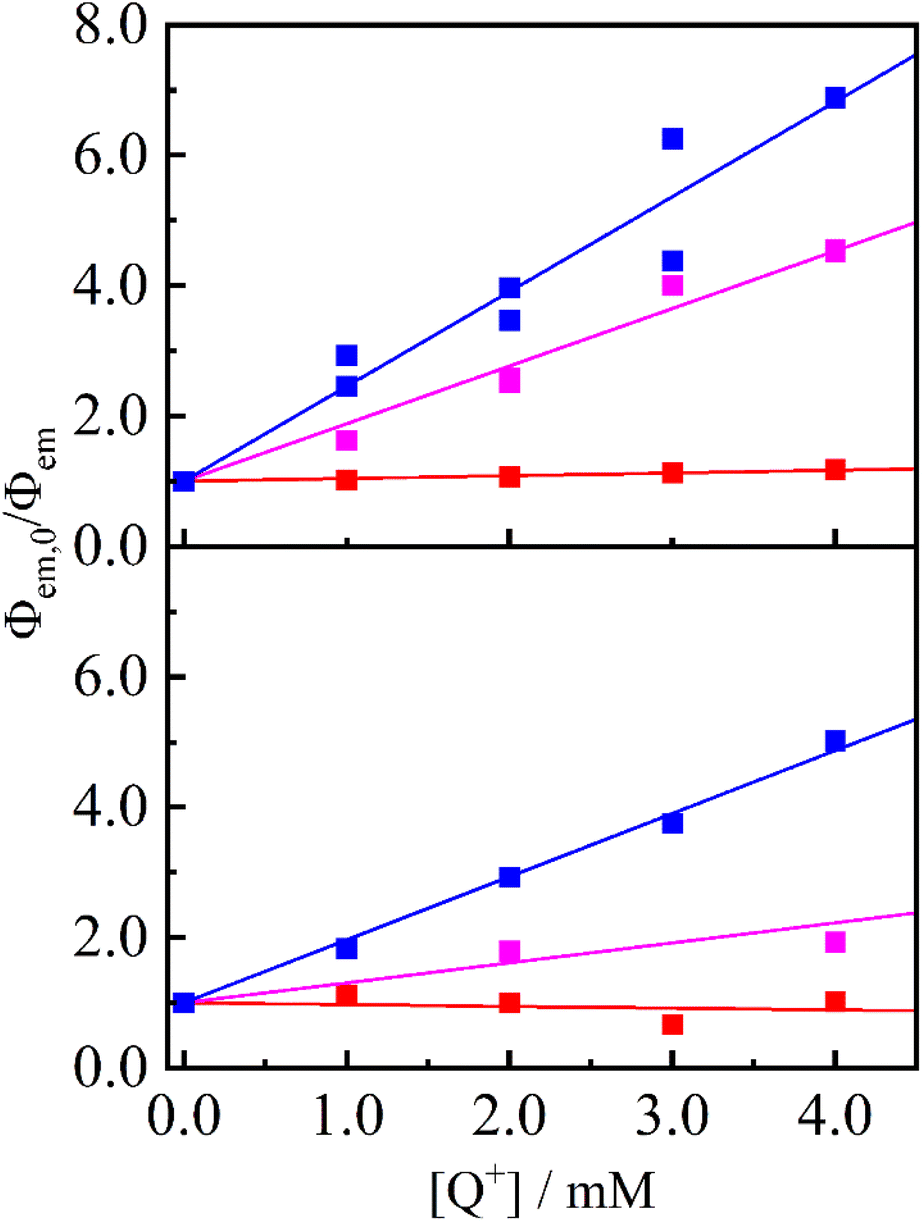
The strong electron-accepting ability of 2a+ is utilizable as an oxidative quencher in photoinduced electron-transfer reactions. As shown in Fig. 3(a), emission from a famous photosensitizer [Ru(bpy)3]2+ (ref. 14) in CH3CN (3.8 × 10−5 M) was reduced upon addition of 2a+ ((0.0–4.0) × 10−3 M), and emission quantum yield (Φem) of [Ru(bpy)3]2+ was decreased from 0.096 to 0.014 in the presence of 2a+ (4.0 × 10−3 M). Emission from a cyclometalated iridium(III) complex [Ir(ppy)2(bpy)]+ in CH3CN (3.8 × 10−4 M) was also quenched when 2a+ coexisted in a solution (Φem = 0.085 and 0.017 in the absence and presence (4.0 × 10−3 M) of 2a+, respectively) as shown in Fig. 3(b). Stern–Volmer plots for emission quenching of the complexes by 2a+ are shown in Fig. 4, together with those by 5+ and 6+ (emission spectra are shown in Fig. S2–S5†). The plots exhibited good linear dependences, irrespective of the complex and pyridinium, as expressed by the Stern–Volmer equation: Φem,0/Φem = 1 + kqτ0[Q+] with Φem,0 and Φem the emission quantum yields in the absence and presence of the quencher (i.e., 2a+, 5+ or 6+), respectively, kq the quenching rate constant, τ0 the excited-state lifetime of the complex in the absence of the quencher (890 and 300 ns for [Ru(bpy)3]2+15 and [Ir(ppy)2(bpy)]+, respectively), and [Q+] the quencher concentration. As clearly seen in Fig. 4 and Table 1, emission quenching by 2a+ (kq = 1.6 × 109 and 3.2 × 109 M−1 s−1 for [Ru(bpy)3]2+ and [Ir(ppy)2(bpy)]+, respectively) was more efficient than those by 5+ and 6+ (kq ≤ 1.0 × 109 M−1 s−1), indicating that the more nitro group, the stronger the quenching ability.
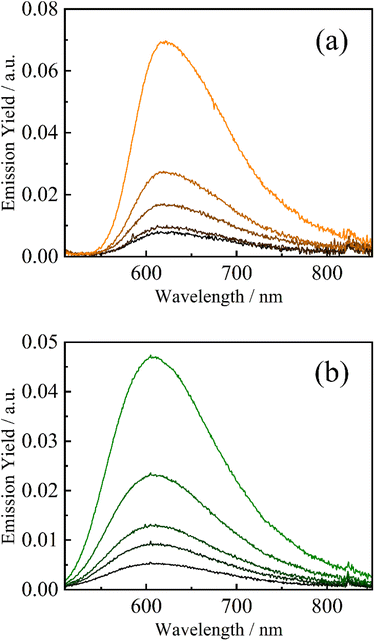 |
| | Fig. 3 Emission spectra of [Ru(bpy)3](PF6)2 (a, 3.8 × 10−5 M, λex = 500 nm) and [Ir(ppy)2(bpy)]PF6 (b, 3.8 × 10−4 M, λex = 470 nm) in the absence and presence of dinitropyridinium salt 2a·OTf ((0.0–4.0) × 10−3 M: orange/green → black) in deaerated CH3CN. | |
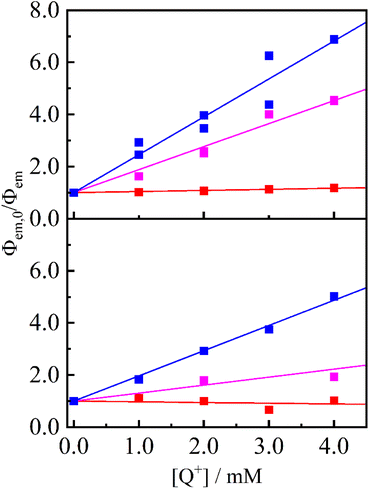 |
| | Fig. 4 Stem–Volmer plots for [Ru(bpy)3]2+ (top panel) and [Ir(ppy)2(bpy)]+ (bottom panel) quenching by 2a+ (blue), 5+ (pink) and 6+ (red) in deaerated CH3CN. Solid lines represent linear regressions with the intercept fixed at 1. | |
Table 1 Reduction potentials, driving forces for electron transfer and quenching rate constants of pyridiniums 2a+, 5+ and 6+
| Q+ |
E
1/2/V |
[Ru(bpy)3]2+ |
[Ir(ppy)2(bpy)]+ |
| −ΔG/eV |
k
q/109 M−1 s−1 |
−ΔG/eV |
k
q/109 M−1 s−1 |
|
2a+
|
−0.061 |
+0.72 |
1.6 |
+0.43 |
3.2 |
|
5+
|
−0.41 |
+0.37 |
0.99 |
+0.084 |
1.0 |
|
6+
|
−0.80 |
+0.016 |
0.048 |
−0.30 |
<0.01 |
The efficient emission quenching by 2a+ can be discussed in terms of a driving force of the electron-transfer process (−ΔG, Table 1), which is calculated from the reduction potentials of the pyridinium, the oxidation potentials of the excited-state complexes and so forth. The kq value correlates well with the −ΔG value, suggesting that the observed emission quenching originates in the electron transfer from the metal complex (i.e., [Ru(bpy)3]2+ or [Ir(ppy)2(bpy)]+) in the excited state to pyridinium (i.e., 2a+, 5+ or 6+). It is worth noting that the electron transfer between [Ir(ppy)2(bpy)]+* to 6+ is highly an endergonic process (−ΔG = −0.30 eV) and, therefore, no emission quenching has been observed. Thus, an introduction of a nitro group(s) into a pyridinium skeleton improves the electron-accepting ability.
Conclusions
A combination of an electron-deficient pyridinium and electron-withdrawing nitro group(s) enhanced electron-accepting and oxidizing abilities. Each nitro-group introduction lowered the LUMO by several tenths of an electron volt, and dinitropyridinium 2a+ especially served as an excellent oxidative quencher in photoinduced electron-transfer reactions. Since pyridinium derivatives have attracted increasing interest and utilized in a variety of photochemical systems such as natural/artificial photosynthesis, these nitropyridiniums are possible candidates as a new class of electron acceptors.
Author contributions
A. Ito: conceptulization, data curation, writing – original draft, and supervision. Y. Kuroda, K. Iwai and S. Yokoyama: investigation. N. Nishiwaki: supervision and writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Notes and references
-
J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley-Blackwell, New York, 5th edn, 2010 Search PubMed
 .
.
- A. Fulton and L. E. Lyons, Aust. J. Chem., 1967, 20, 2267 CrossRef CAS ; G. Unden and J. Bongaerts, Biochim. Biophys. Acta, 1997, 1320, 217 CrossRef PubMed ; J. Barber, Chem. Phys. Rev., 2009, 38, 185 Search PubMed .
- K. Kalyanasundaram, J. Kiwi and M. Grätzel, Helv. Chim. Acta, 1978, 61, 2720 CrossRef CAS ; H. Misawa, H. Sakuragi, Y. Usui and K. Tokumaru, Chem. Lett., 1983, 12, 1021 CrossRef ; A. Bose, P. He, C. Liu, B. D. Ellman, R. J. Twieg and S. D. Huang, J. Am. Chem. Soc., 2002, 124, 4 CrossRef ; A. Ito, D. J. Stewart, Z. Fang, M. K. Brennaman and T. J. Meyer, Proc. Natl. Acad. Sci. U.S.A., 2012, 109, 15132 CrossRef ; H. Yamamoto, M. Taomoto, A. Ito and D. Kosumi, J. Photochem. Photobiol., A, 2020, 401, 112771 CrossRef .
- D. Mauzerall and F. H. Westheimer, J. Am. Chem. Soc., 1955, 77, 2261 CrossRef CAS ; S. Fukuzumi, S. Koumitsu, K. Hironaka and T. Tanaka, J. Am. Chem. Soc., 1987, 109, 305 CrossRef ; A. Kobayashi, H. Konno, K. Sakamoto, A. Sekine, Y. Ohashi, M. Iida and O. Ishitani, Chem.–Eur. J., 2005, 11, 4219 CrossRef PubMed ; S. Ikeyama and Y. Amao, ChemCatChem, 2017, 9, 833 CrossRef .
- N. Nishiwaki and M. Ariga, Top. Heterocycl. Chem., 2007, 8, 43 CAS .
- H. Asahara, M. Hamada, Y. Nakaike and N. Nishiwaki, RSC Adv., 2015, 5, 90778 RSC ; Y. Nakaike, N. Nishiwaki, M. Ariga and Y. Tobe, J. Org. Chem., 2014, 79, 2163 CrossRef CAS PubMed .
- A. Ito, D. J. Stewart, Z. Fang, M. K. Brennaman and T. J. Meyer, Proc. Natl. Acad. Sci. USA, 2012, 109, 15132 CrossRef CAS PubMed ; A. Ito, D. J. Stewart, T. E. Knight, Z. Fang, M. K. Brennaman and T. J. Meyer, J. Phys. Chem. B, 2013, 117, 3428 CrossRef ; A. Ito, Z. Fang, M. K. Brennaman and T. J. Meyer, Phys. Chem. Chem. Phys., 2014, 16, 4880 RSC .
- J. Sun, F. Zhong, X. Yi and J. Zhao, Inorg. Chem., 2013, 52, 6299 CrossRef CAS PubMed .
-
Gibbs energy of photoinduced electron transfer, in IUPAC Compendium of Chemical Terminology, International Union of Pure and Applied Chemistry (IUPAC), 3rd edn, 2019, DOI:10.1351/goldbook.GT07388.
- A. Ito and T. J. Meyer, Phys. Chem. Chem. Phys., 2012, 14, 13731 RSC .
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 (Revision C.01), Gaussian, Inc., Wallingford, CT, 2009 Search PubMed .
-
R. Dennington, T. Keith and J. Millam, GaussView Version 5, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed .
- J. N. Younathan, W. E. Jones and T. J. Meyer, J. Phys. Chem., 1991, 95, 488 CrossRef CAS .
- D. W. Thompson, A. Ito and T. J. Meyer, Pure Appl. Chem., 2013, 85, 1257 CrossRef CAS .
- A. Nakagawa, E. Sakuda, A. Ito and N. Kitamura, Inorg. Chem., 2015, 54, 10287 CrossRef CAS PubMed .
Footnotes |
| † Electronic supplementary information (ESI) available: Cyclic voltammograms of the metal complexes and emission quenching data by 5+ and 6+. See DOI: https://doi.org/10.1039/d4ra00845f |
| ‡ Present address: The Institute of Scientific and Industrial Research (SANKEN), Osaka University, Ibaraki, Osaka 567-0047, Japan. |
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Yasuyuki
Kuroda
a,
Kento
Iwai
*ab,
Yasuyuki
Kuroda
a,
Kento
Iwai