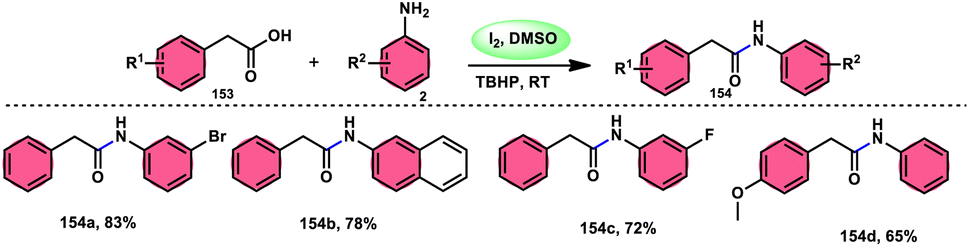
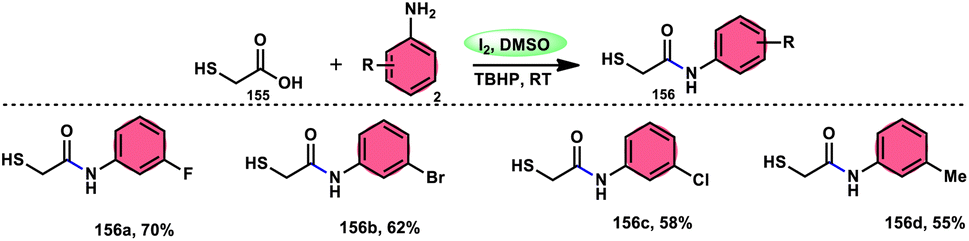
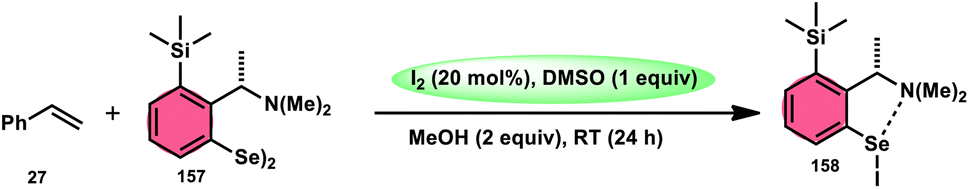
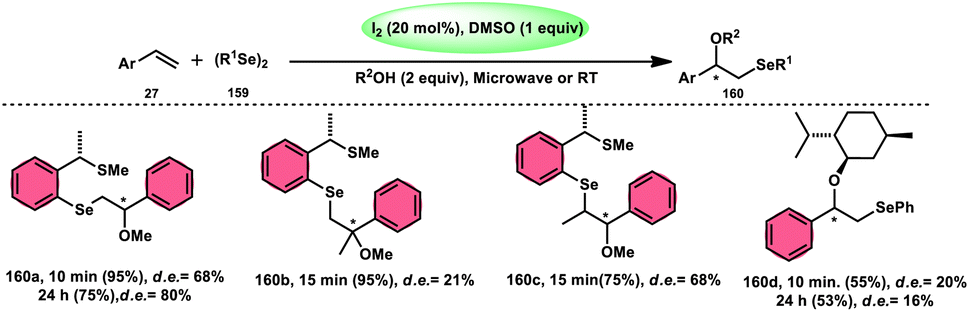
DOI:
10.1039/D3RA08685B
(Review Article)
RSC Adv., 2024,
14, 5817-5845
I2/DMSO-mediated oxidative C–C and C–heteroatom bond formation: a sustainable approach to chemical synthesis
Received
20th December 2023
, Accepted 30th January 2024
First published on 14th February 2024
Abstract
The I2/DMSO pair has emerged as a versatile, efficient, practical, and eco-friendly catalyst system, playing a significant role as a mild oxidative system, and thus employed as a good alternative to metal catalysts in synthetic chemistry. Presently, I2/DMSO is a thriving catalytic system that is used in preparing C–C and C–X (X = O/S/N/Se/Cl/Br) bonds, resulting in the formation of various bioactive molecules. Many processes utilize this system, including in situ glyoxal synthesis by diverse sp, sp2, and sp3 functionalities via iodination and subsequent Kornblum oxidation. Focusing on oxidation processes, this study examines the synergistic effect of dimethyl sulfoxide (DMSO) and molecular iodine in improving synthetic techniques. We provide a comprehensive overview of the research progress on the I2/DMSO catalytic system for the formation of C–C and C–heteroatom bonds from 2018 to the present. Additionally, the future prospects of this research field are discussed.
 Rakshanda Singhal | Ms Rakshanda Singhal obtained her Master's Degree in Organic Chemistry from the Department of Chemistry at the University of Rajasthan in 2018. Currently, she is engaged in the pursuit of her Ph.D. degree at Manipal University Jaipur, Rajasthan. Her research is being conducted under the guidance and supervision of Dr Babita Malik and Dr Meenakshi Pilania. Her area of focus includes the synthesis of biologically active azaheterocycles using N-tosylhydrazone as a coupling reagent. |
 Satya Prakash Choudhary | Mr Satya Prakash Choudhary obtained his Master's Degree in Organic Chemistry from the Department of Chemistry at the University of Rajasthan in the year 2011. Currently, he is an Assistant Professor in the College Education Department under the Government of Rajasthan. He is now engaged in the pursuit of his Ph.D. degree, under the supervision of Dr Meenakshi Pilania. This academic endeavor is being undertaken within the Department of Chemistry at Manipal University Jaipur. The primary focus of his study is the development of new and effective methodologies for the synthesis of bioactive heterocycles, using N-tosylhydrazone as a key reagent. |
 Babita Malik | Dr Babita Malik is a Professor in the field of Organic Chemistry at Manipal University Jaipur, Rajasthan. Dr Malik obtained her Master's Degree from the University of Rajasthan in 1992. In 2000, she successfully completed her doctoral studies and was awarded a Ph.D. from the University of Rajasthan. Dr Malik primary research focus covers the synthesis and physiological effects of heterocyclic compounds fused with benzene rings. |
 Meenakshi Pilania | Dr Meenakshi Pilania is an Assistant Professor of Chemistry at Manipal University Jaipur, India. Dr Pilania obtained her Master's Degree from the Department of Chemistry at the University of Rajasthan. Dr Pilania successfully completed her doctoral degree at the Birla Institute of Technology and Science Pilani, Pilani campus, Rajasthan, in 2017. She is involved in research concerning N-tosylhydrazone and hypervalent iodine chemistry, with the aim of advancing the development of innovative and effective reaction methods associated with azaheterocycles. |
1. Introduction
In the field of synthetic chemistry, researchers have consistently strived to develop reaction approaches that have the characteristics of environmental sustainability, economic viability, safety, and effectiveness. As part of this endeavor, they developed and utilized cost-effective, environmentally sustainable catalysts and solvents. In order to meet the above-mentioned requirements, the combination of I2/DMSO as a catalyst and solvent has emerged as a versatile approach.1,2 Molecular iodine (I2) has gained significant popularity as a catalyst and reagent due to its notable attributes,3–5 including its cost-effectiveness as an alternative to transition and rare earth metals, its environmentally friendly nature, non-toxicity, high stability, low sensitivity to moisture, ease of handling, and notably its exceptional performance across a wide range of reaction mixtures, including highly concentrated and solvent-free systems.6 Collectively, these characteristics make I2 a versatile and unique catalyst and reagent.7,8 Iodine is present in numerous types of cyclic and acyclic reagents in various oxidation states and cyclic and acyclic forms. Since its discovery in 1811,9–11 I2 has been shown to be a better alternative to metal catalysts due to its high efficiency as a reagent in cross-dehydrogenative coupling (CDC). Furthermore, its use dramatically reduces the number of reaction steps and produces a higher yield in multicomponent reactions (MCRs) and shortens the time for the synthesis of C–C, C–X, and C-Het bonds.12–17 Thus, molecular iodine (I2) works perfectly in MCRs, CDC, and in various tedious and hard-to-pursue reactions, providing an easy approach to the synthesis of various high-value bioactive, pharmaceutical, and industrial compounds.18 Dimethyl sulfoxide (DMSO) is a well-known aprotic polar solvent, which is less toxic than similar solvents. DMSO is used equally in chemistry and biological activities such as polymerase chain reactions (PCR), cryoprotectants, and integral parts of some drugs and medicines. In chemistry, DMSO is generally used as an inexpensive, high-boiling, and innocuous solvent. Besides, it has been utilized as a mild oxidant, methylthiolating, formylation, and cyanation agent, and as a synthon for C–S bonds.19,20 DMSO acts as a mild oxidant and plays a pivotal role in Pfitzner–Moffatt oxidation, Swern oxidation, Corey–Kim oxidation, Corey–Chaykovsky epoxidation, and Kornblum oxidation. Furthermore, it also functions as a terminal oxidant, eliminating the need for transition metals.21 Fascinatingly, the combination of these two readily accessible and benign molecules presents new avenues for simple, inexpensive, facile, eco-friendly, metal-free, and atom-economical reaction strategies, facilitating simple oxidation to chemoselective and regioselective oxidation, sulfenylation, and amination to effectively generate C–C, C–X, C–O, C–S, S–N and C–N bonds, affording various bioactive, pharmaceutical, industrial, and applied molecular structures.22–26 This review presents a detailed summary of the research advancements achieved in the I2/DMSO catalytic system for the formation of C–C and C–heteroatom bonds from 2018 to now. Furthermore, the potential future development in this research field is discussed.
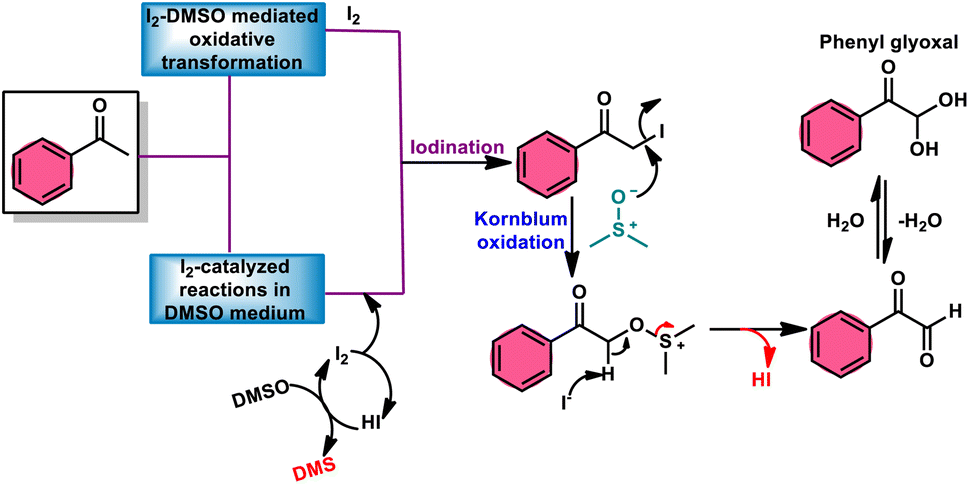
2. Kornblum oxidation
Fascinatingly, the I2/DMSO duo is very suitable for chemoselective oxidation, dehydrogenation, oxidative aromatization, protection/deprotection of various functional groups, and regioselective and stereoselective transformations.27–29 The I2/DMSO system has been extensively reported for its dual functionality, as follows: (i) the facilitation of I2-promoted activities in DMSO medium and (ii) oxidative changes via the interaction between I2 and DMSO.30–33 This method includes initial iodination of substrates including various sp, sp2, and sp3 functionalities in the presence of molecular iodine. This method proceeds via Kornblum oxidation in both instances (Fig. 1).34–36 Kornblum oxidation is a chemical process involving the transformation of a primary halide to an aldehyde, which is facilitated by the use of dimethyl sulfoxide (DMSO) as a reagent, also leading to the synthesis of dimethyl sulfide (DMS) as a byproduct.37–40 It is worth noting that this oxidation reaction is commonly conducted at elevated temperature. Besides being only a mild oxidant, I2 with K2S2O8/DMSO can be employed for the deoxygenation of N-oxides and sulfoxides,41 sulfonyation,42,43 and asymmetric alkoxy selenylation.44 Furthermore, it provides the advantage of the selective oxidation of alcohol to carbonyl or carboxylic acid. However, the application of H2O2 as an external oxidant reduces the amount of I2 reconverting iodide, whereas the addition of TBHP (tert-butyl hydroperoxide) to I2/DMSO provides opportunities to obtain various significant compounds such as benzimidazo[1,2-c]quinazolines45–47 and amidation.7,48 In general, the reaction mechanism of I2/DMSO is straightforward and unambiguous. It initiates reactions by iodinating alkenes/alkynes/carbonyls, which subsequently undergo further oxidation to form phenylglyoxal via Kornblum oxidation.49 In conclusion, the I2/DMSO duo provides beneficial and effective options to prepare various highly applicable molecules and their derivatives such as quinazoline, quinolines, isatins, thiadiazoles, triazolopyridines, trisubstituted imidazoles, benzothiazoles, benzothiadiazines and beznothiozole-fused imidazoles.50,51
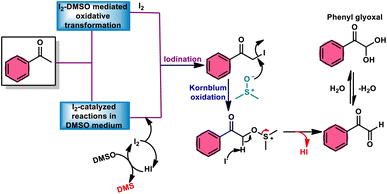 |
| | Fig. 1 Schematic illustration of Kornblum oxidation. | |
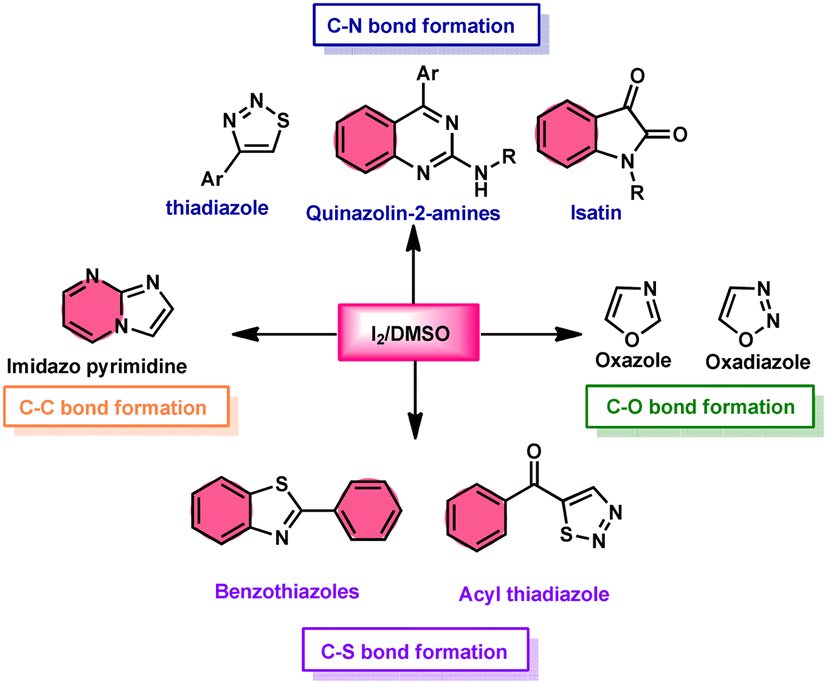
Additionally, this procedure can facilitate the basic formation of C–C, C–N, C–O, and C–S bonds using metal-free,52 atom-and-step effective, and environmentally friendly chemistry (Fig. 2). We attempt to highlight I2/DMSO-based studies reported from 2018 to 2022, showing its abundant synthetic abilities such as oxidative amination, amidation, sulfonation, sulfenylation, esterification, etherification to aryl alkyl ether, and dicarbonylation of C–H (sp3,sp2, and sp) bonds.53
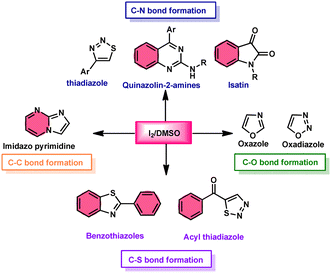 |
| | Fig. 2 I2/DMSO role in C–X bond formation. | |
3. Six-membered ring formation with one-heteroatom
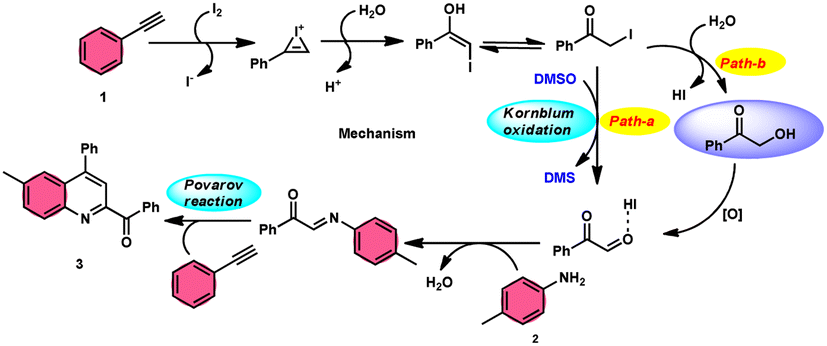
The I2/DMSO system-based synthesis of substituted quinolines 3 was demonstrated by Yan-Dong Wu, An-Xin Wu, and colleagues.54 The synthesis of 2,4-substituted quinolines 3 was achieved through a new I2-mediated Povarov reaction involving aryl acetylenes 1 and anilines 2, wherein aryl acetylene 1 initially functions as both a dienophile and diene precursor (Scheme 1). The types of suitable diene precursors have been substantially expanded by this advancement in the Povarov reaction and its substrate range has been extended to carbonyl compounds. When used in the process, aryl amines with both electron-donating and withdrawing groups produced product 3 in satisfactory yield. According to initial mechanistic research, the I2/DMSO approach achieved the oxidative carbonylation of the aryl acetylene C(sp)–H before undergoing a [4 + 2] cycloaddition process. Initially, iodine activates phenylacetylene to produce an iodonium cation and release I− species, which is subsequently attacked by water to produce an intermediate called an enol. The intermediate enol is isomerized to α-iodophenone at this point, following two paths. Path one involves the utilization of Kornblum oxidation, whereas in path two, the intermediate compound undergoes a reaction with water to produce 2-hydroxy acetophenone, which is further subjected to oxidation and the Povarov reaction (Scheme 2).
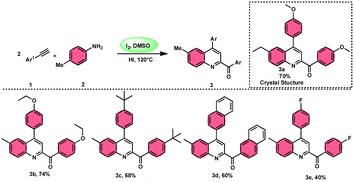 |
| | Scheme 1 Preparation of substituted quinolines 3 and its mechanism. | |
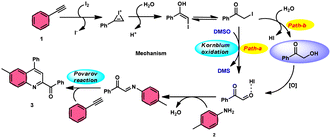 |
| | Scheme 2 Mechanism for the synthesis of substituted quinolines 3. | |
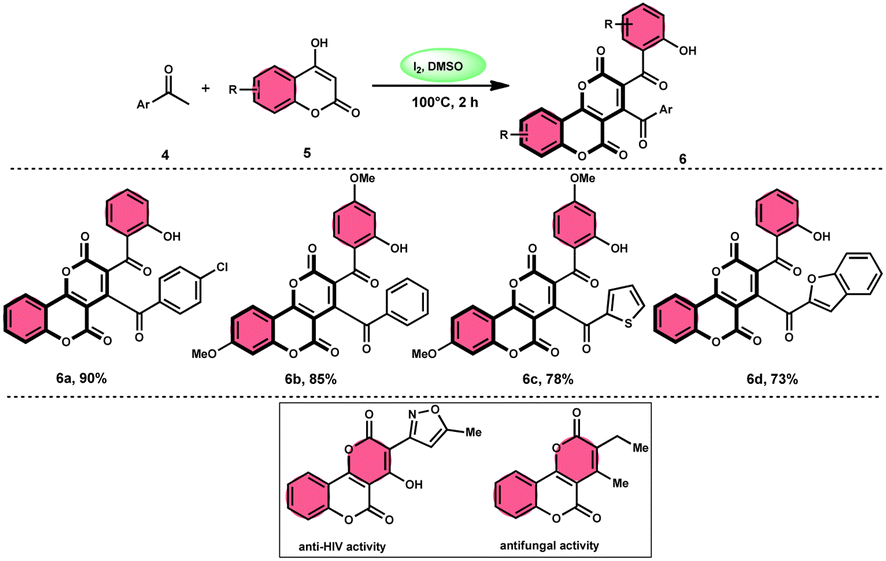
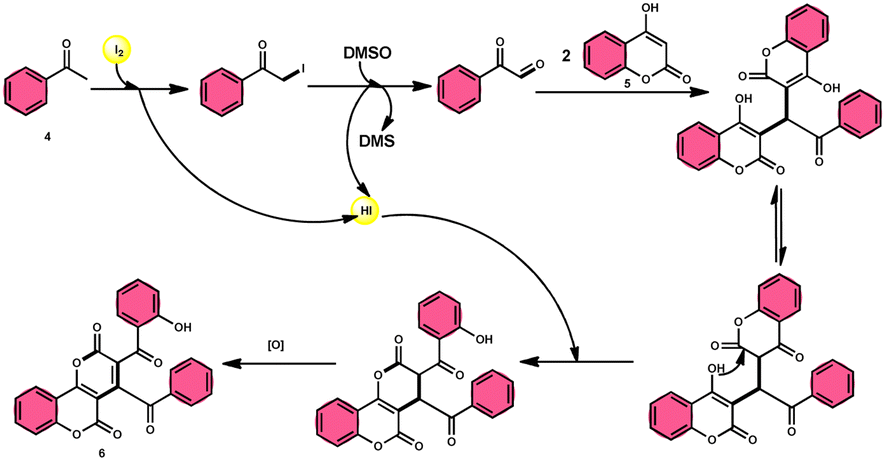
In the study by Wu et al.,55 they provided a straightforward and efficient scheme for the synthesis of several substituted pyrano[3,2-c]chromene-2,5-diones 6. This strategy involves the I2-promoted sequential cyclization of readily available aryl methyl ketones 4 and 4-hydroxycoumarins 5, as depicted in Scheme 3. Based on initial investigations into the mechanism, it was determined that the reaction follows a sequential pathway involving iodination, annulation, and Kornblum oxidation. The HI generated in the I2–DMSO system exhibited significant promotive effects, effectively enhancing the rate of the annulation process. Pyrano[3,2-c]chromene-2,5-dione 6 is a highly desirable structural motif found in heteropolycyclic complexes. Its derivatives have significant medicinal capabilities, including substantial anti-HIV activity and antifungal activity.56 It was shown that the electronic characteristics of substituted 4-hydroxycoumarins 5 had minimal effect on the yields. The aforementioned process, which serves as a viable synthetic method, provides simple and efficient access to o-heteropolycyclic scaffolds under mild conditions (Scheme 4).
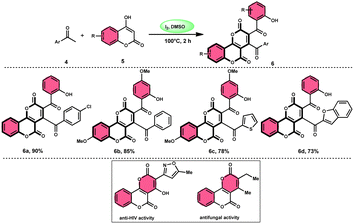 |
| | Scheme 3 Synthesis of pyranochromene-2,5-diones 6. | |
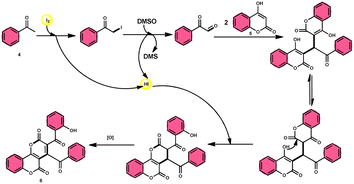 |
| | Scheme 4 Mechanism for the synthesis of pyranochromene-2,5-diones 6. | |
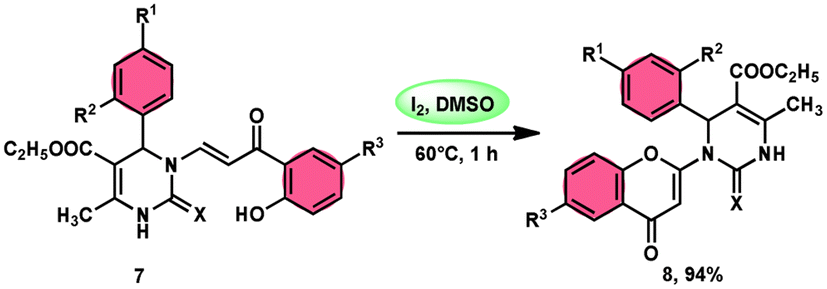
Pathan, Ali, et al.57 devised a very efficient one-pot synthesis approach using an I2/DMSO green solvent. This methodology allows the rapid synthesis of 2-substituted 4-H-1-benzopyran-4-one derivatives 8, which are considered significant pharmacophores. The notable point in this methodology is that the researchers used the molecular hybridization scheme via the path of cyclodehydration motivating the α,β-unsaturated carbonyl group, which is a very effective approach. The desired products were not observed, even in trace amounts in the absence of the green catalyst I2, although in the plausible mechanism, the investigators did not explain the role of I2. The methodology is highly feasible given that the reaction is faster based on the green medium of I2/DMSO and requires very mild conditions. The arrays of the new product 5-(ethoxycarbonyl)-4-methyl-1-(6-methyl-4-oxo-4H-chromen-2-yl)-2-oxo-6-phenyl-1,2,3,6-tetrahydropyrimidine-1-sulfide 7 work as both antibacterial and antifungal in a single compound form. The researchers performed a detailed bioactivity and pharmacological study of the products employing XRD, Petra, Osiris, Molinspiration (POS) analysis and determined that the pharmacophore sites in the compounds were some substituents on the heterocyclic and aromatic moiety (Scheme 5).
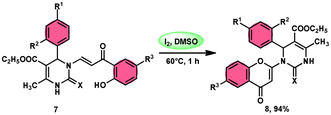 |
| | Scheme 5 Synthesis of benzopyran-4-one 8 in I2–DMSO system. | |
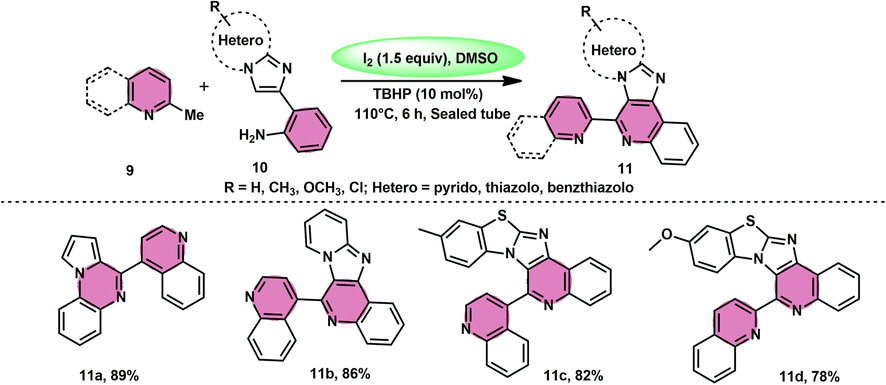
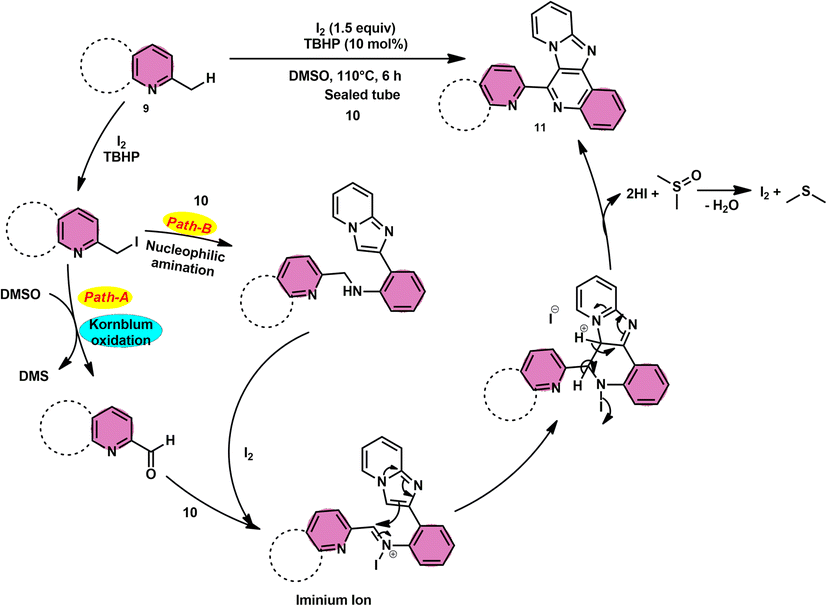
Subha Reddy and co-workers58 employed I2/DMSO and TBHP as a co-oxidant to develop a potent, one-pot, metal-free, atom-economical, simple reaction strategy to synthesize various bioactive N-fused polyheterocycle compounds 11 through the formation of C–C and C–N bonds (Scheme 6). This strategy has advantages compared to previous strategies owing to its simple path, atom economy, readily available starting materials, good yields, metal-free reagents and environmental-friendly nature. The synthesis of a range of bioactive compounds, including pyrido-/thizolo/benzthiozol-imidazo[4,5-c]quinolines and indolo-/pyrrolo-[1,2-a]quinoxalines 11a–11d, was achieved using 2-methylquinoline 9 and 2-(imidazo[1,2-a]pyridine-2yl)aniline 10, which are both readily available starting materials. The latter compound was also synthesized in the same laboratory. The role of TBHP is simply a co-oxidant, which shortens the reaction time drastically. The methodology was well tolerated by both substrates and its yields were appreciable. The following reaction mechanism starts with the iodination of the methyl group located on the 2-methylquinoline 9 compound. This is followed by the Kornblum oxidation process, resulting in the formation of the 2-carbaldehyde quinoline compound. The aldehyde functional group undergoes a reaction with the amine functional group of another substrate, leading to the development of an iminium ion. This is followed by a cyclization process via the carbon–carbon bond, ultimately yielding the expected product, as seen in Scheme 7.
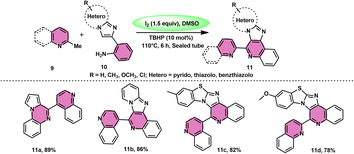 |
| | Scheme 6 I2–DMSO-based synthesis of N-fused polyheterocycles 11. | |
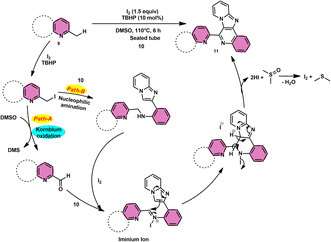 |
| | Scheme 7 Mechanism for synthesis of N-fused polyheterocycles 11. | |
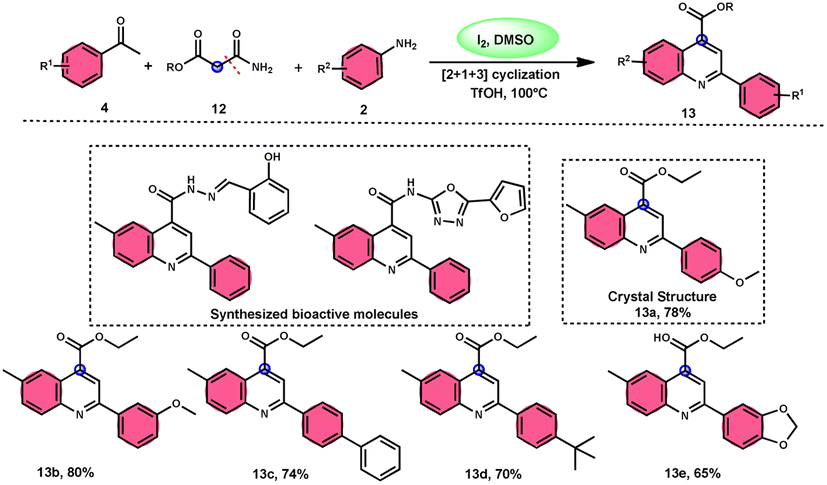
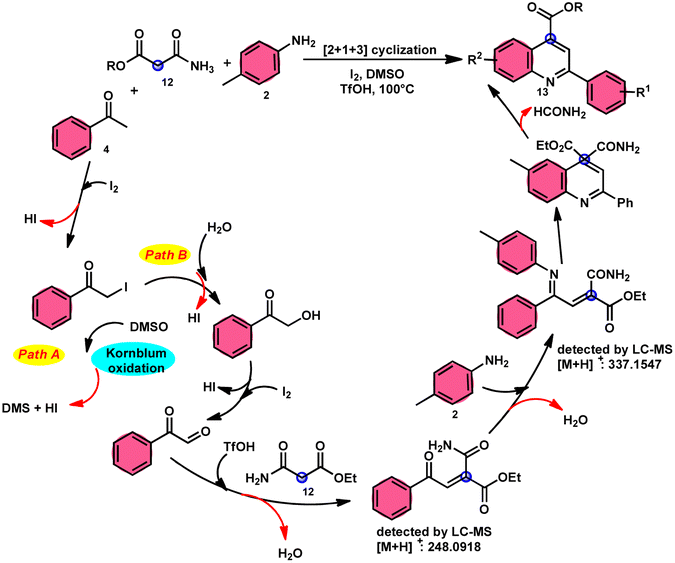
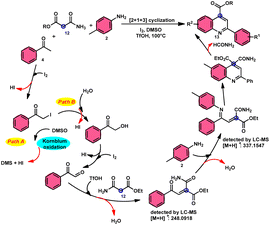
2-Arylquinoline-4-carboxylate moiety 13, which is found in many drugs, natural products, and bioactive products, was prepared by An-Xin Wu et al.59via a unique, metal-free, straightforward, and simple reaction condition-based protocol using the I2/DMSO system in an acidic medium and aryl methyl ketone, 1,3-dicarbonyl, and aryl amines (Scheme 8). The reaction was conducted in a [2 + 1 + 3] manner, including the use of aryl methyl ketone 4, 1,3-dicarbonyl 12, and arylamines 2. The C–C bond of 1,3-dicarbonyl acts as a single synthon and breaks down during the process. Three bioactive molecules were prepared using this protocol, with good tolerance of various substituents on all three substrates, where even highly steric hindered arylamines 2 and aryl methyl ketones 4 were converted into the anticipated products with good quantity, showing the efficacy of the reaction. This research team proposed a reaction mechanism in which aryl methyl ketone 4 undergoes conversion into phenylglyoxal through the use of I2/DMSO. The first step of this mechanism involves the reaction between the aldehydic carbonyl group of phenylglyoxal and 1,3-carbonyl derivatives 12. This reaction leads to the production of a carbon–carbon double bond (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C). Moreover, the ketonic carbonyl moiety present in phenylglyoxal undergoes a reaction with arylamine 2, resulting in the formation of a CN bond. Subsequently, an intramolecular cyclization process occurs, leading to the synthesis of the intended product 13 (Scheme 9).
C). Moreover, the ketonic carbonyl moiety present in phenylglyoxal undergoes a reaction with arylamine 2, resulting in the formation of a CN bond. Subsequently, an intramolecular cyclization process occurs, leading to the synthesis of the intended product 13 (Scheme 9).
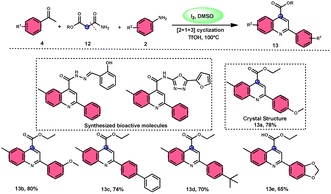 |
| | Scheme 8 I2–DMSO system-based synthesis of 2-arylquinoline-4-carboxylate 13. | |
 |
| | Scheme 9 Mechanism of 2-arylquinoline-4-carboxylate 13. | |
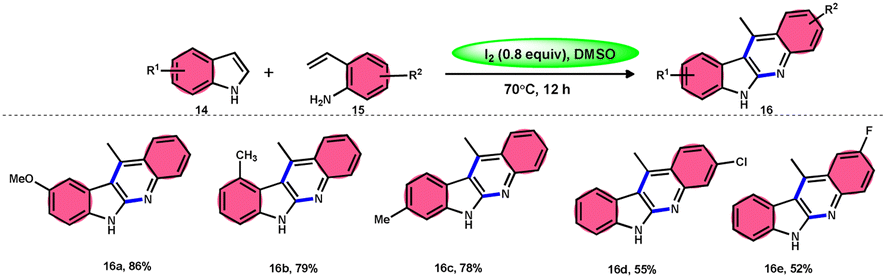
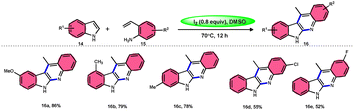
Wei, Li, and colleagues60 devised an easy one-pot method to produce a wide range of 11-methyl-6H-indolo[2,3-b]quinolines 16 (Scheme 10). This was achieved using an I2-mediated annulation process involving indoles 14 and 2-vinylanilines 15. This approach has several benefits compared with existing techniques, such as a wide range of applicable materials, gentle reaction conditions, and straightforward operation. Furthermore, this approach does not require any pre-functionalization protocols for the production of new C–C and C–N bonds and generates the necessary products in moderate to satisfactory quantities. In addition, these tetracyclic compounds were assessed for their antiviral and cytotoxicity potency against the EV71 and CVB3 viruses. The first observations indicated that some compounds showed remarkable antiviral properties against EV71 and CVB3. Additionally, these compounds efficiently suppressed virus-induced damage to cells and decreased the production of new viral particles. A potential mechanism was suggested. Initially, the reaction can only take place with 2-vinylaniline 15, which can combine with I2 to produce an iodonium ion. This is followed by 5-endo-cyclization and elimination of HI, resulting in the formation of indole 14.61 Alternatively, 2-vinylaniline 15 generates a stable benzylic carbocation C when exposed to a strong acid such as HI. This carbocation C is then targeted by indole 14, resulting in the formation of an iminium ion. Following the intramolecular cyclization of the compound, a new intermediate is formed.62 Product 16 is obtained by the oxidative aromatization of the intermediate using iodine. Throughout the procedure, iodine is continually regenerated by the oxidation of HI by DMSO.1,63,64
 |
| | Scheme 10 Synthesis of 11-methyl-6H-indolo[2,3-b] quinolines 16 and selected examples. | |
4. Six-membered ring formation with two-heteroatom
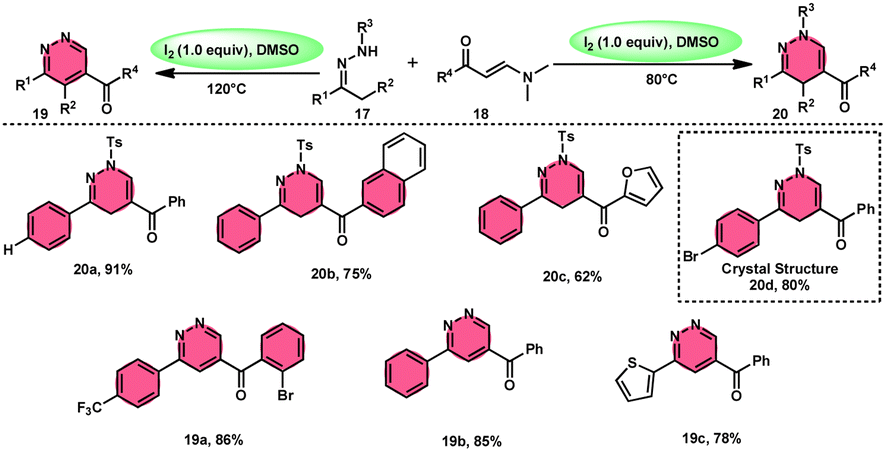
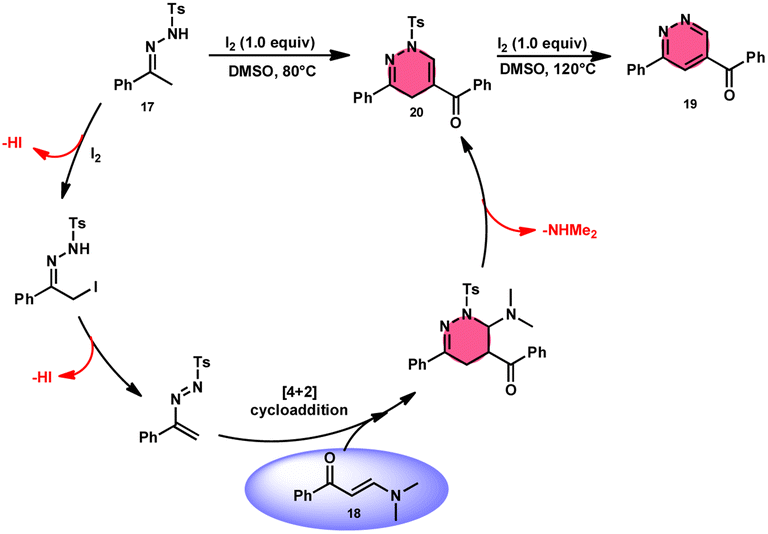
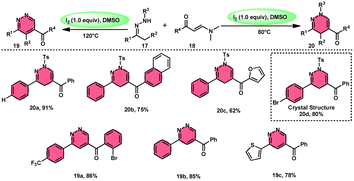
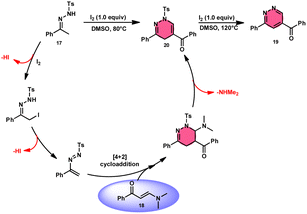
Fei Huang and colleagues developed a direct and effective method for the synthesis of 1,4-dihydropyridazines 20 and pyridazines 19. This method involves the I2-promoted [4 + 2] cycloaddition of N-tosylhydrazone 17, which is produced in situ, with enaminones 18 (Scheme 11).65 The prudent choice of the reaction temperature is responsible for the change in selectivity. The primary advantages of this work include its selective and regulated synthesis, wide functional group tolerance, good to extraordinary reaction yields, absence of metals or bases, and one-pot method adaptability. The synthesis of the pyridazine N-oxide derivative and the experiment conducted on a gram-scale demonstrate the prospective use of the developed methodology. The metal/base-free methodology has significant qualities such as regulated and selective formation, extensive functional group compatibility, and suitable for one-pot procedures. Additionally, a control experiment without I2 was conducted, which failed to produce the intended result, demonstrating the importance of I2 in this reaction. However, the effectiveness of the reaction to produce 1,4-dihydropyridazine 20 was not improved by decreasing or increasing the temperature. Remarkably, an increase in temperature to 120 °C resulted in a significant improvement in the production of pyridazine compound 19, resulting in an isolated yield of 85%. This outcome provides evidence supporting the concept that elevating the temperature facilitates the removal of the –Ts group. The reaction exhibited no progress in the presence of nitrogen. Also, this process is unaffected by the electronic properties of the substituents attached to the phenyl ring of enaminones (Scheme 12).
 |
| | Scheme 11 Synthesis of 1,4-dihydropyridazines 20, pyridazines 19 and representative examples. | |
 |
| | Scheme 12 Mechanism for the synthesis of 1,4-dihydropyridazines 20, pyridazines 19 and representative examples. | |
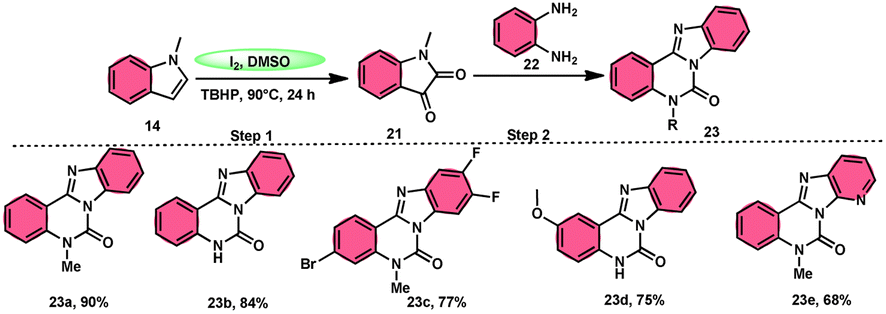
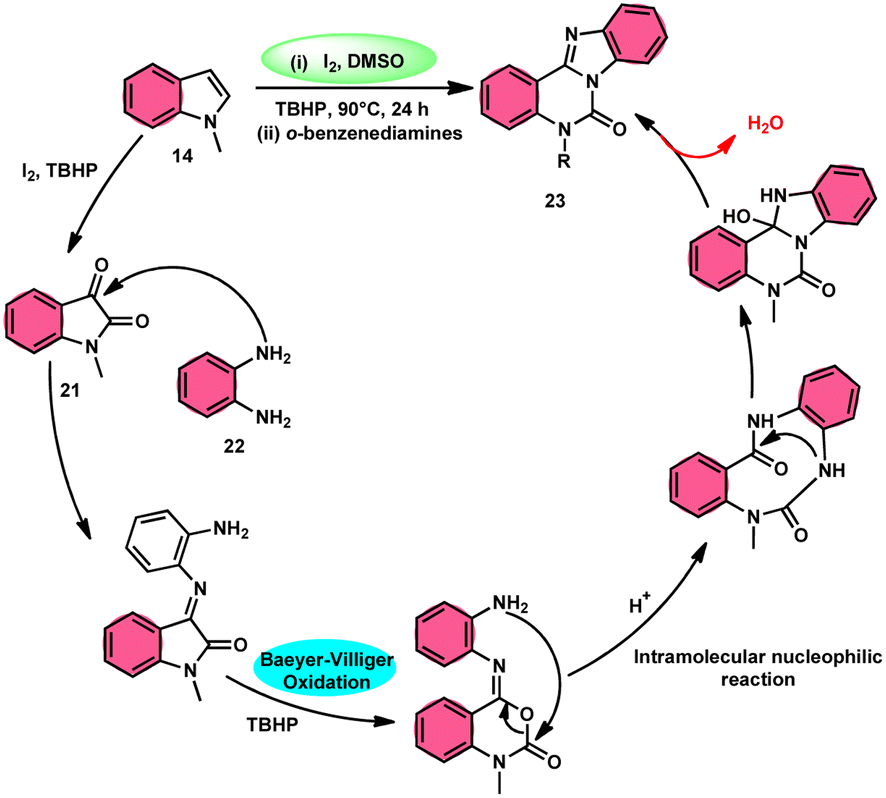
Ma et al.66 conducted a study, whereby they devised a very efficient methodology for the synthesis of benzimidazo[1,2-c]quinazolin-6-ones 23 and their derivatives (Scheme 13). This process was performed under metal-free and mild reaction conditions, utilizing I2/DMSO and TBHP (tertiary butyl hydroperoxide). This reaction is very convenient and practical because the starting reactants indole 14 and 1,2-diaminobenzene 22 are readily available, good yields are obtained, and various substituents both on indole 14 and 1,2-diaminobenzene 22 are well tolerated. The methodology proceeds in two straightforward steps, where in the first step, the indole is oxidized to isatin 21, and in the second step the addition of o-benzenediamine 22 leads to the product. However, TBHP working as an oxidant is indispensable in the reaction, especially in the second step. The investigators proposed the reaction mechanism, which was supported by spectroscopic data. In the first step, indole 14 is oxidized to isatin 21. In the second step, quinoxaline is the expected product but the reaction proceeds through a ring-expansion mechanism via Baeyer–Villiger rearrangement mediated by TBH and results in the formation of benzimidazole. 1,2-Diaminobenzene 22 as a nucleophile reacts with isatin 21, followed by Baeyer–Villiger rearrangement, and in the last step, intramolecular nucleophilic reaction results in the formation of the product (Scheme 14).
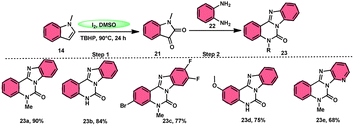 |
| | Scheme 13 Reaction of isatin 21 and representative examples. | |
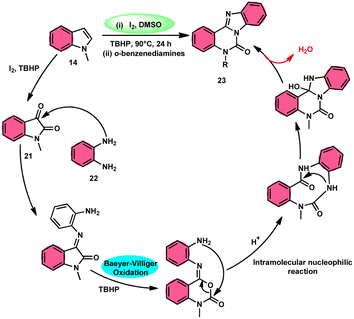 |
| | Scheme 14 Reaction mechanism and representative examples. | |
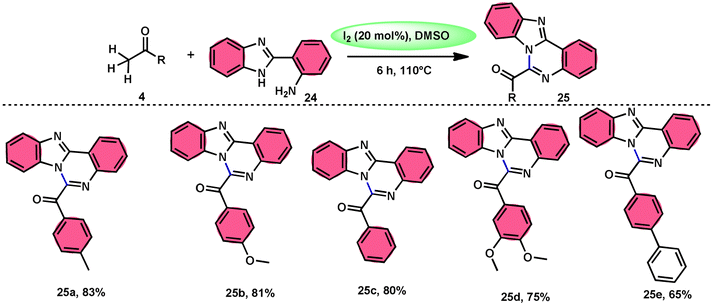
Padmini and coworkers45 prepared benzoimidazoquinazoles 25 derivates in a straightforward, one-pot, metal-free, effective new reaction methodology using the combination of I2 and DMSO. The developed technique can be considered feasible due to the fact that the I2/DMSO catalyst facilitates the oxidative amination of the C–H bond in ketones 4, resulting in favourable yields. This process demonstrated good tolerance when applied to both electron-donating groups (EDGs) and electron-withdrawing groups (EWGs) connected to aryl methyl ketones 4. Additionally, it relies on the use of conveniently accessible aryl methyl ketones 4. These ketones can be rapidly transformed into arylglyoxals by the α-iodination of the C–H (sp3) bond in the methyl group, followed by Kornblum oxidation. The proposed reaction mechanism proposed by the authors involves the use of I2 as a mediator for the α-iodination of aryl methyl ketone 4, which is followed by Kornblum oxidation. The series of chemical events described above results in the synthesis of arylglyoxal and the liberation of dimethylsulfide. The arylglyoxal compound undergoes a chemical reaction with 2-(1H-benzo[d]imidazole-2-yl) aniline 24, which acts as a nucleophilic reagent. This reaction forms an imine intermediate, which then undergoes intramolecular cycloaddition to provide a bioactive product (Scheme 15).
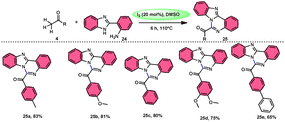 |
| | Scheme 15 I2/DMSO-catalyzed synthesis of benzoimidazo-quinazolinones 25. | |
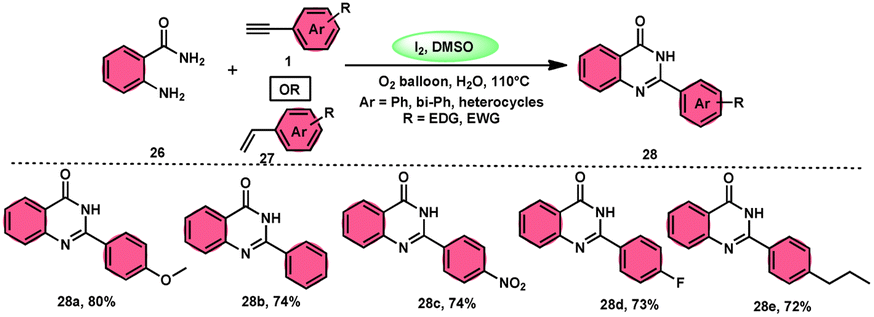
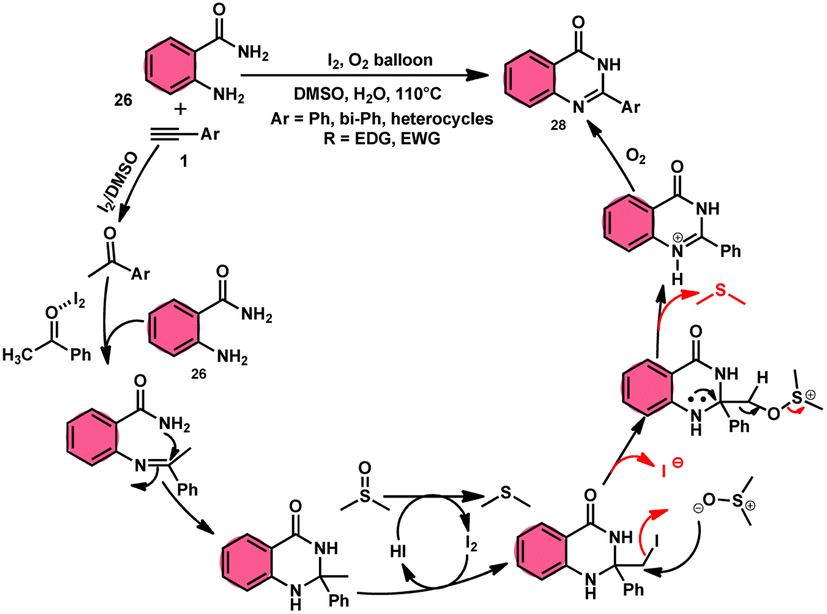
Sandip B. Bharate et al.67 synthesized a new non-sulfonyl NLRP3 inflammasome inhibitor compound 2-arylquinazolin-4(3H)-one 28 and investigated its bioactivities, interactions, binding sites, and effectiveness employing molecular docking, computations, and other methods (Scheme 16). This method employed is very elegant, economic, metal-free, and molecular O2 works as the oxidant in I2/DMSO medium, although a higher temperature (140 °C) is required but its yields are good. The condensation between the reactants, namely, 2-aminobenzoamides 26 and terminal aryl alkynes 1 or styrene 27, which are easily accessible, results in the formation of quiozoline-4(3)-ones 28. The above-mentioned conversion takes place through the process of oxidative cleavage of unsaturated carbon–carbon (C–C) bonds, subsequently leading to the formation of two carbon–nitrogen (C–N) bonds. The first carbon–nitrogen (C–N) bond is formed using a Schiff base mechanism, but the subsequent C–N bond is generated via an intramolecular cyclization process. Phenylacetylenes 1 and styrene 27 with various EDGs and EWGs are converted into the desired products with good yield; however, aliphatic alkynes and DMAD (dimethyl acetylenedicarboxylate) are not suitable to lead the reactions. The researchers revealed that quinazolin-4-(3H)-ones 28 are potent NLRP3 inflammasome inhibitors by binding with the ATP moiety via H-bonding and calibrated the inhibitory concentration value of the 5 μM on the IC50 scale, which shows its good potency (Scheme 17).
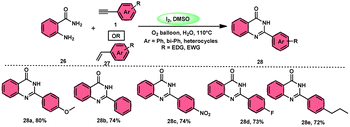 |
| | Scheme 16 Synthesis of aryl quinazolinone composition 28 and some selected examples. | |
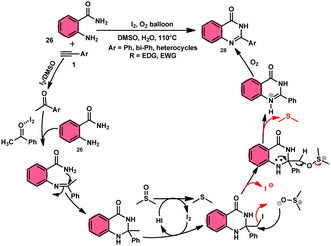 |
| | Scheme 17 Mechanism for the synthesis of aryl quinazolinone 28. | |
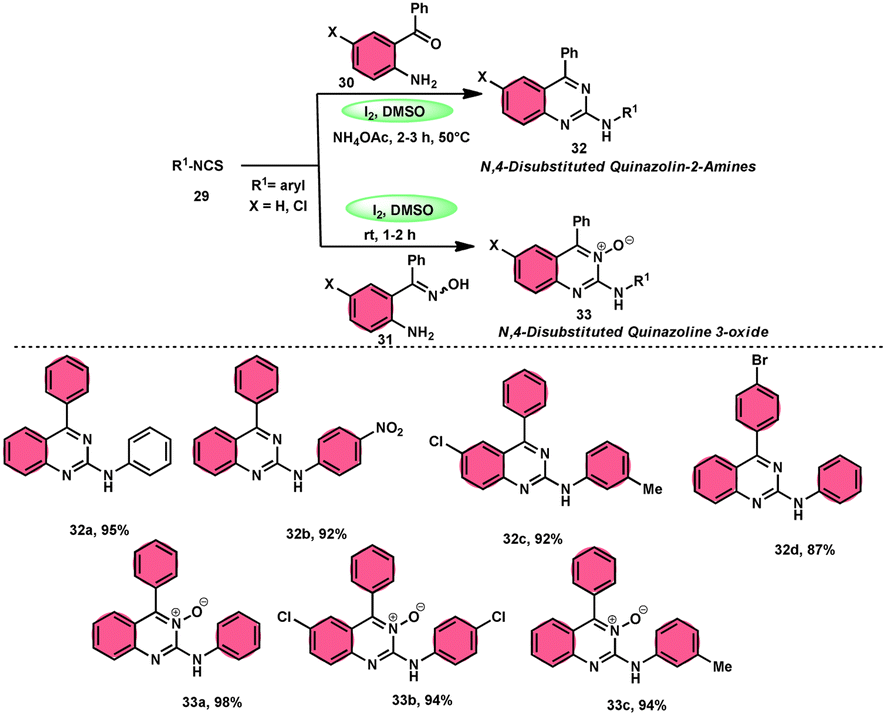
In the study by Krishna and Nagesh,68 they proposed a versatile methodology for the synthesis of N-4-disubstituted quanzoloin-2-amine 32 and 4-aryl-2-(arylamino) quinozoline-3-oxide 33. The approach used in this study involves the utilization of readily available and economically viable reagents, such as aryl isothiocyanate 29, 2-amino benzophenone 30, NH4OAc, and (2-aminophenyl)(phenyl) methanone oxime 31. The reactions are facilitated by the utilization of I2/DMSO as a promoter. The proposed methodology exhibits several advantageous characteristics, including environmentally sustainable features, cost-effectiveness, absence of transition metals, efficient time management, high production yield, and notable tolerance towards both electron-donating and electron-removing groups on phenyl isothiocyanates 29. However, it should be noted that alkyl isothiocyanates and 2-aminophenyl alkyl ketones are unable to achieve the desired outcomes within this framework. According to the plausible reaction mechanism, isocyanate moiety 29 is attacked by the amine group of the substrate, creating a C–N bond and producing a thiourea intermediate, which reacts with NH4OAc to afford the imine intermediate, and intramolecular cyclization attack by N of the imine intermediate on the carbon of the thiourea results in the formation of N-4-disubstituted quanzoloin-2-amine 32 (Scheme 18).
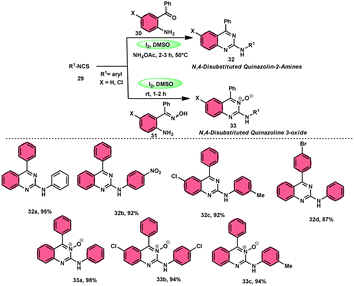 |
| | Scheme 18 Synthesis of quinazolines 32, quinazoline oxides 33 and selected examples. | |
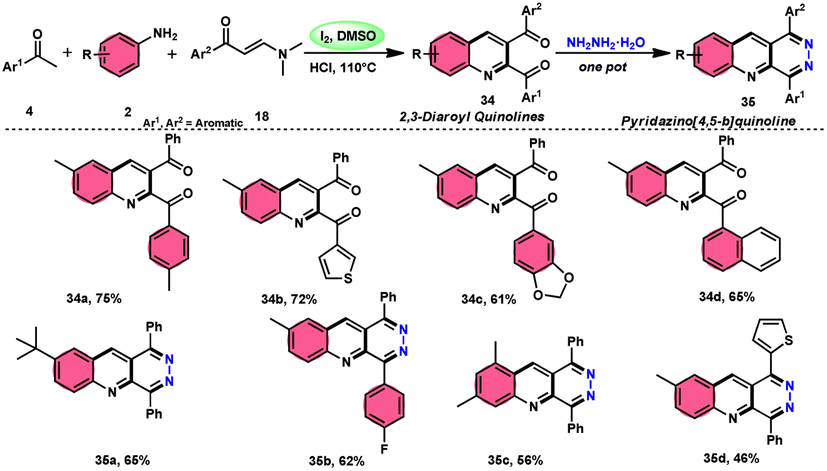
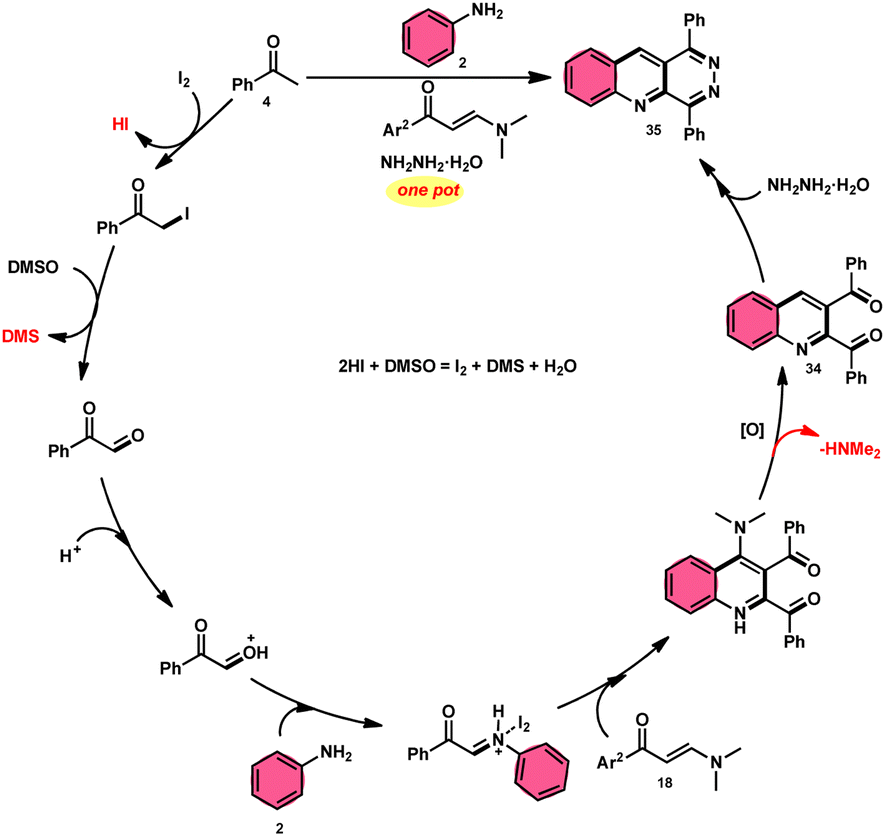
Wu et al.22 developed a successful strategy utilizing the well-known Povarov reaction to synthesize 2,3-diaroyl quinolines 34 in a direct manner (Scheme 19). I2/DMSO-controlled reaction conditions allow for the easy functionalization of these quinolines to form pyridazino[4,5-b]quinolines 35. This scheme has good potential in synthesis chemistry owing to its metal-free, one-pot, shortened, cyclocondensation in a [3 + 2 + 1] manner of three components, i.e., enaminone 18, aryl methyl ketone 4 and aryl amine 2, with mild to good yields. All three reactants having EDGs and EWGs as substituents are compatible to achieve the desired products without any significant effect on the yields; however, none of the alkyl reactants were compatible to generate the desired products. For the Kornblum oxidation, Povarov reaction, and intramolecular cyclization to occur, molecular iodine is the key ingredient. These reactions are dependent on the presence of I2, given that they cannot proceed without it (Scheme 20).
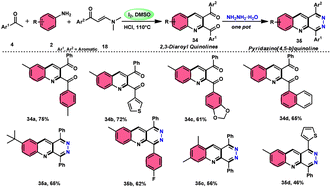 |
| | Scheme 19 Synthesis of quinolines 34, 35 and representative examples. | |
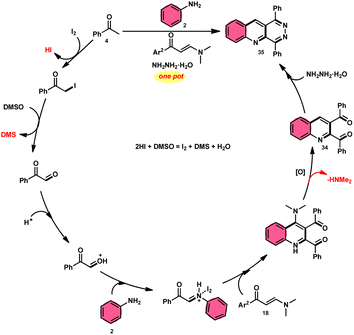 |
| | Scheme 20 Mechanism for the synthesis of quinolines 34 and 35. | |
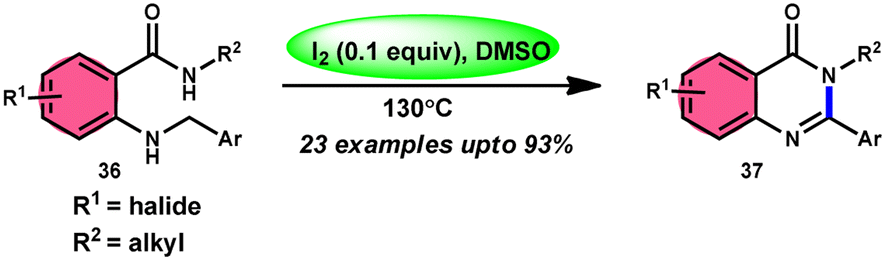
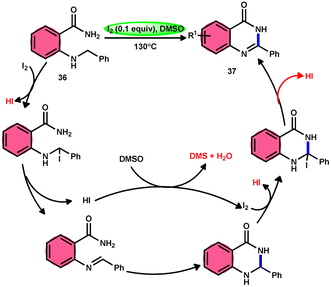
Zhou and colleagues69 presented an intramolecular C(sp3)–H/N–H oxidative cross-coupling process for the synthesis of quinazolinones 37 (Scheme 21). This approach includes the use of I2/DMSO to facilitate the intramolecular oxidative cross-coupling reaction. The reaction transforms 2-(benzylamino) benzamides 36 into arylquinazolinones 37 by forming CN bonds. This approach exhibits remarkable properties such as excellent functional group tolerance, absence of metal catalysts, straightforward procedure, practicality, and high product yields (up to 93%). According to the proposed mechanism, initially, 36 reacts with iodine, resulting in the formation of an iodine intermediate. Furthermore, the intermediate catalyzes the elimination and liberation of HI inside the molecule, resulting in the formation of imine. This imine undergoes intramolecular addition, resulting in the formation of a cyclized intermediate. This cyclized intermediate undergoes a reaction with iodine to produce another intermediate, which is iodized. The intermediate catalyzes the elimination and liberation of HI intramolecularly, resulting in the formation of the final product 37 (Scheme 22). The crucial aspect of this reaction is that HI can undergo oxidation and be restored to iodine by the action of dimethyl sulfoxide.
 |
| | Scheme 21 Intramolecular oxidative cross-coupling reaction mediated by I2/DMSO. | |
 |
| | Scheme 22 Mechanism of I2/DMSO-mediated intramolecular C(sp3)–H/N–H oxidative cross-coupling reaction. | |
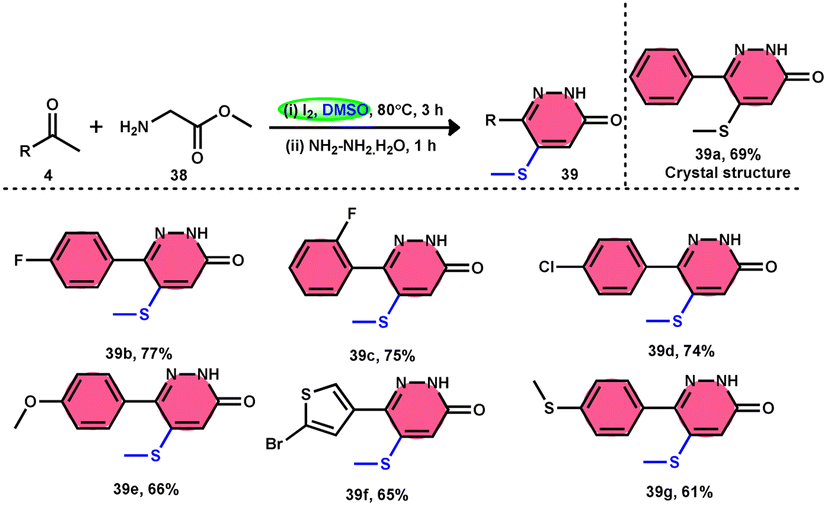
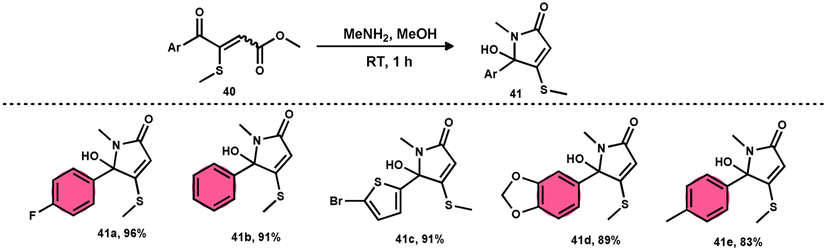
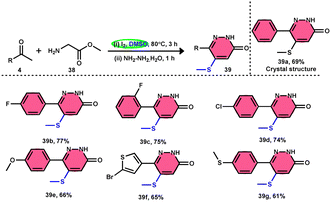
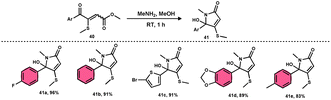
Bhat et al.70 developed a new approach for synthesizing 5-(methylthio)pyridazinone derivatives 39 (Scheme 23). This method involves the use of iodine to stimulate the deaminative coupling of glycine esters 38 with methyl ketones 4 and hydrazine hydrate in DMSO, resulting in a one-step process. In the absence of hydrazine, these modifications facilitated the production of various 3-methylthio-4-oxo-enoates 40 with high efficiency. DMSO played several roles, including an oxidant, methylthiolating reagent, and solvent. Pyridazin-3(2H)-one is a nitrogen-containing aromatic ring structure that is present in several natural products, medicines, and functional materials. The reaction is straightforward and has a wide range of substrates that can be employed, as well as being able to tolerate various functional groups. Furthermore, this technique can be readily modified to serve as an in situ generator of various alkyl-3-(methylthio)-4-oxo-enoates 40. The usefulness of 40 has been confirmed by synthesizing several new 5-hydroxy-1-methyl-4-(methylthio)-5-phenyl-1,5-dihydro-2H-pyrrol-2-ones 41 (Scheme 24).
 |
| | Scheme 23 Synthesis of 5-(methylthio)pyridazin-3(2H)-ones promoted by I2–DMSO. | |
 |
| | Scheme 24 Applications of 3-(methylthio)-4-oxo-enoates 40 and representative examples. | |
5. Six-membered ring formation with three-heteroatoms
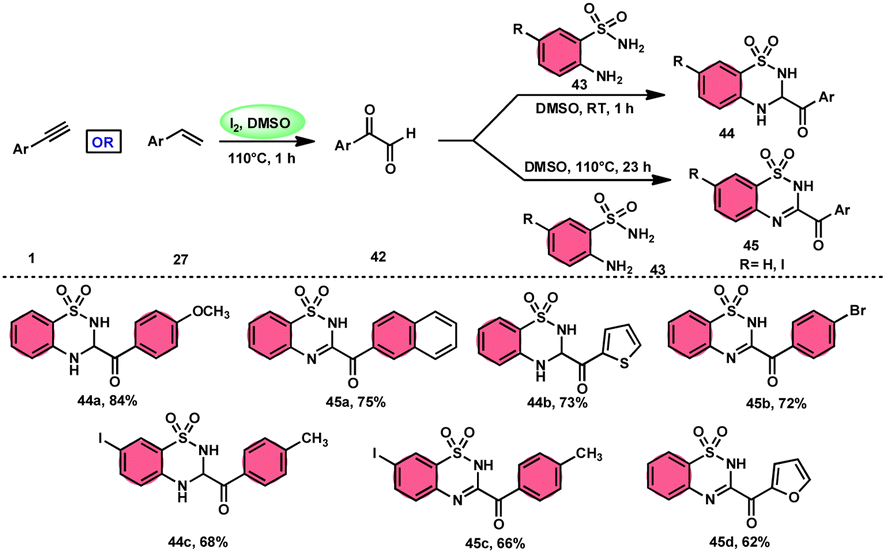
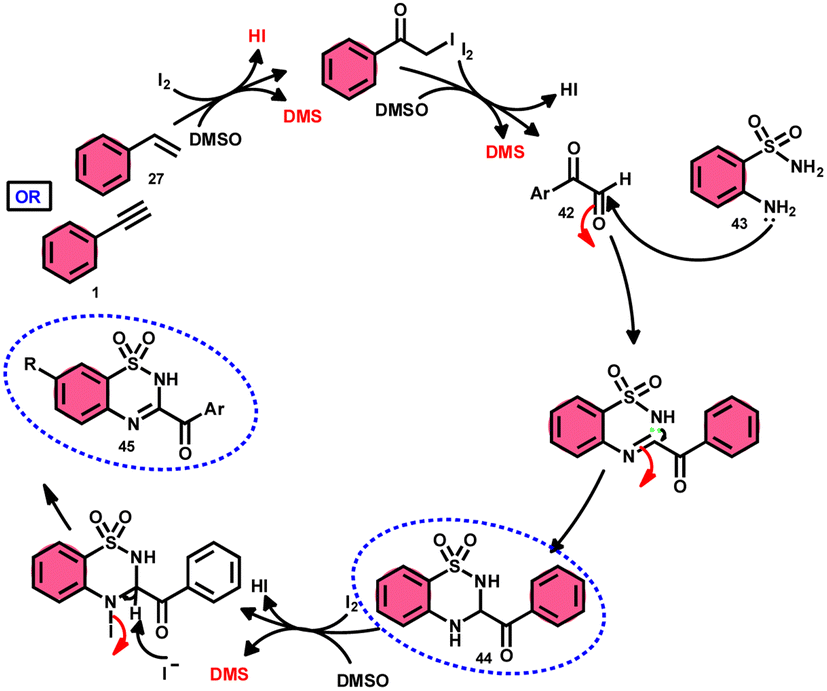
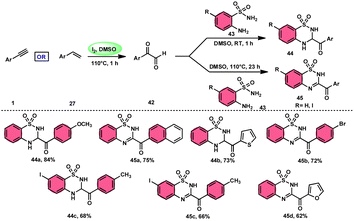
A new, metal-free, efficient, and scalable approach was devised for the production of 3-acyl-3,4-dihydro-2H-1,2,4-benzothiadiazine-1,1-dioxides 44 and 3-acyl-2H-1,2,4-benzothiadiazine-1,1-dioxide 45 (Scheme 25). In their study, Chaskar et al.71 employed affordable ethynyl arenes 1 and ethenyl arenes 27, which are readily accessible, as starting materials. These compounds consist of sp2 and sp3 carbon–hydrogen (C–H) bonds, respectively. By using Kornblum oxidation, both types of bonds were functionalized to produce phenylglyoxal 42 with the help of I2/DMSO. In the second reaction, 2-aminobenzenesulfonamides 43 are added to phenylglyoxal 42, which results in the desired product through condensation, followed by intramolecular cyclization and aromatization. However, on adding 2-amino-N-phenylbenzenesulfonamide instead of 2-aminobenzenesulfonamides, 43 does not follow the cyclization path, resulting in the formation of oxidative cross-coupling products. DMSO plays a binary role as an oxidant and solvent, whereas molecular iodine is a vital part of Kornblum oxidation, iodination, and aromatization of the product. In summary, the methodology has good value in pharmaceutical and synthetic chemistry owing to its low cost, good substrate scope, and eco-friendly nature (Scheme 26).
 |
| | Scheme 25 Synthesis of benzothiadiazine-dioxides 44 and 45 and representative examples. | |
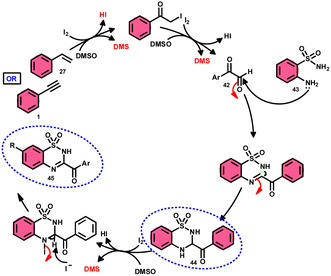 |
| | Scheme 26 Mechanism for the synthesis of benzothiadiazine-dioxides 44 and 45. | |
6. Five-membered ring formation with one-heteroatom
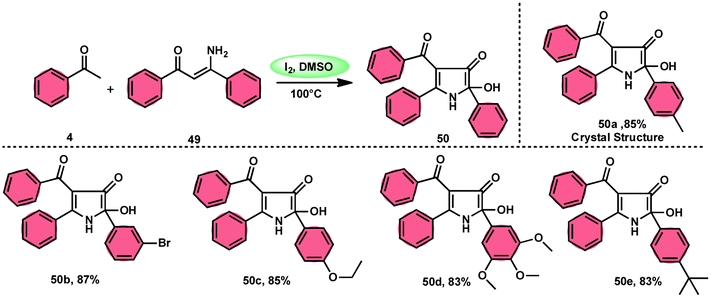
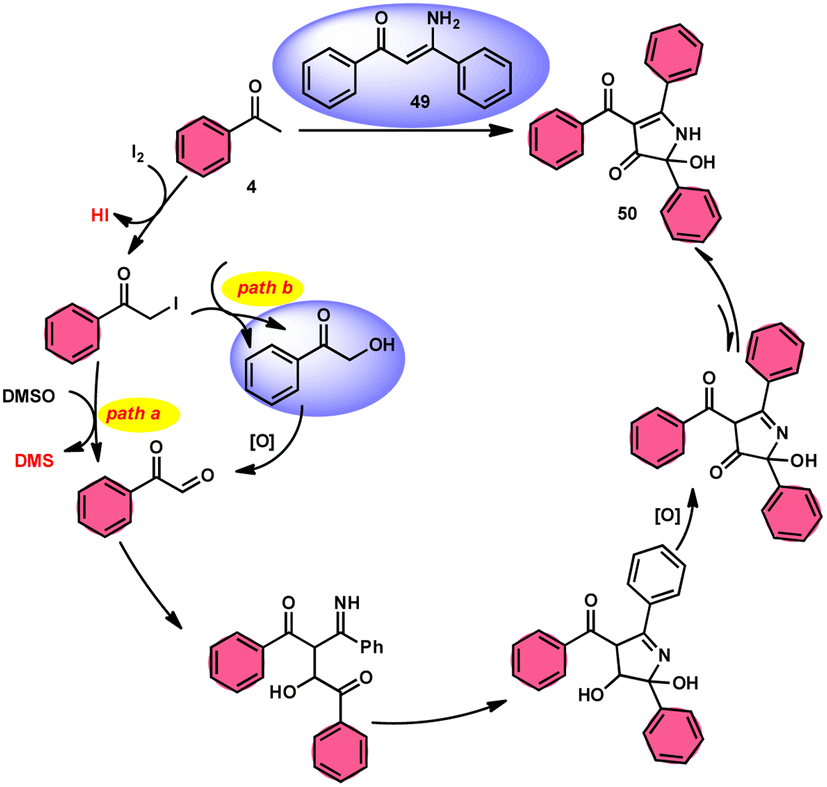
To realize the formation of 2-hydroxy-pyrrol-3(2H)-ones 39, Wu et al.72 developed a new metal-free and I2/DMSO-supported cyclization method (Scheme 27). The experimental procedure included the reaction between enaminone 49 and aryl methyl ketones 4. This approach allows for the production of 2-hydroxy-pyrrol-3(2H)-one rings 50 with diverse structural properties, resulting in high yields. Furthermore, it successfully produced quaternary alcohol. This depicts the use of group-assisted purification (GAP) chemistry, whereby the purification of the product is achieved by a simple washing process with CH2Cl2 solvent. This method avoids the need for conventional procedures such as chromatography and recrystallization. The reaction had moderate and straightforward working conditions, was very efficient, and demonstrated strong functional group compatibility. In contrast to electron-withdrawing groups, which demonstrate compatibility only with the para- or meta positions, and electron-donating groups attached to the aromatic ring of aryl methyl ketones 4 can effectively produce the desired compounds, regardless of their position in the ortho, meta, or para-positions. The suitable 2-hydroxy-pyrrol-3(2H)-ones 50 were generated with exceptional yield using halogenated aryl methyl ketones, as can be seen in Scheme 28.
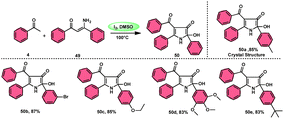 |
| | Scheme 27 Synthesis of substituted pyrrole 50. | |
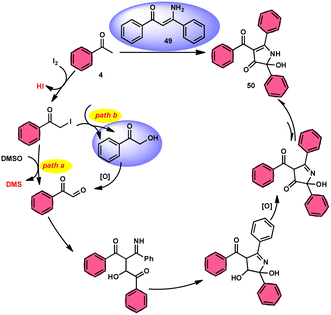 |
| | Scheme 28 Mechanism for the synthesis of substituted pyrrole 50. | |
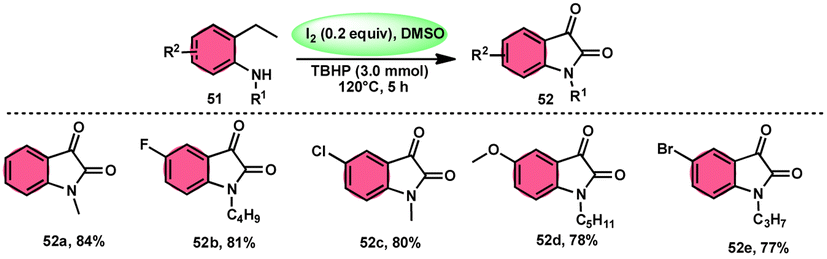
Nayaki Salvanna and coworkers73 established a novel, metal-free, and efficient methodology to prepare isatins7452 from 2-ethylanilines or 2-vinylanilines 51 through oxidative intramolecular cyclization. The I2/DMSO combination without TBHP (tertiary butyl hydroperoxide), which works as an oxidant, was unsuccessful in generating the selected product. This approach successfully functionalized both sp2 and sp3 C–H bonds. In addition, its broad substrate scope and strong functional group tolerance make it very useful. According to the researchers, TBHP initiates the reaction by generating secondary alcohol from ethylanilines 51 in a radical manner and the alcohol is easily oxidized to ketone, producing 2-aminoacetophenone by molecular iodine. 2-Aminoacetophenone produces 2-aminophenylglyoxal via Kornblum oxidation, which undergoes intramolecular cyclization, forming a C–N bond and a 2-hydroxyquinoline-3-one intermediate, which is oxidized by I2/DMSO to afford isatin 52 (Scheme 29).
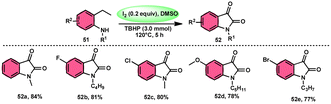 |
| | Scheme 29 Synthesis of isatin 52 catalyzed by I2–DMSO and representative examples. | |
7. Five-membered ring formation with two-heteroatoms
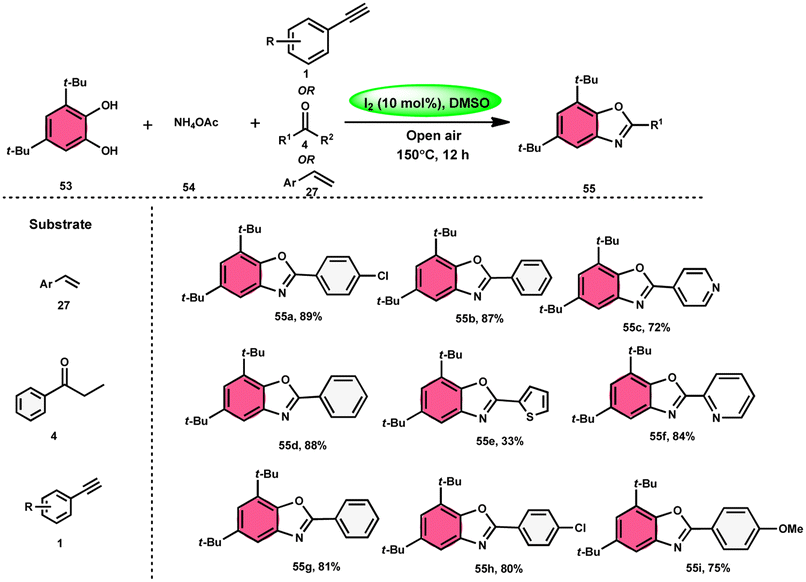
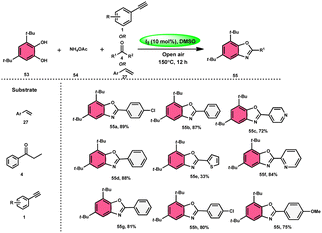
Panahi, Sharghi, and colleagues75 proposed a method to produce benzoxazoles 55 without the use of metals. This involves combining ammonium acetate 54 with catechols 53 and alkenes/alkynes/ketones. This methodology is simplified by using I2/DMSO, which is a one-pot method, suitable for large-scale procedures, and operationally efficient. When neither molecular iodine nor DMSO solvent was used, no product was detected. Besides the dimethyl sulfoxide (DMSO) solvent, only a 30% product yield was reported when using N,N-dimethylformamide (DMF). Furthermore, there was a significant decline in the yield at lower temperatures. At a temperature of 100 °C, a minute quantity of product was identified. To demonstrate the effectiveness of this process, a large-scale synthesis was conducted, resulting in the isolation of 70% of the product (Scheme 30).
 |
| | Scheme 30 Synthesis of benzoxazoles 55 from phenylacetylenes 1, styrenes 27 and different ketones 4. | |
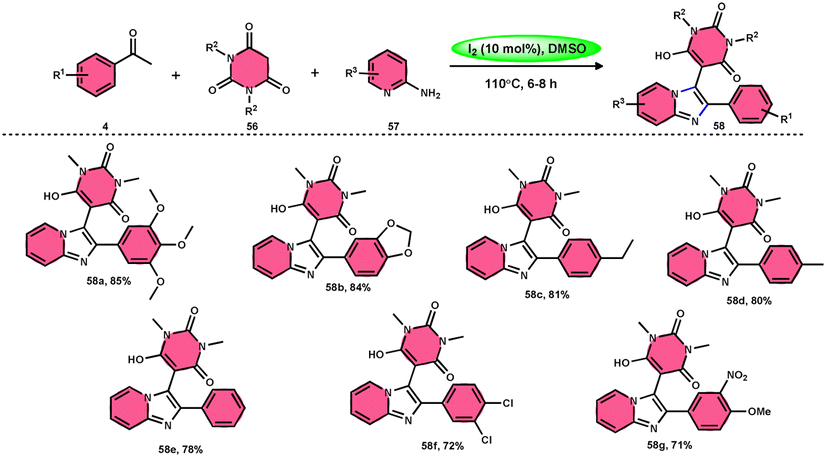
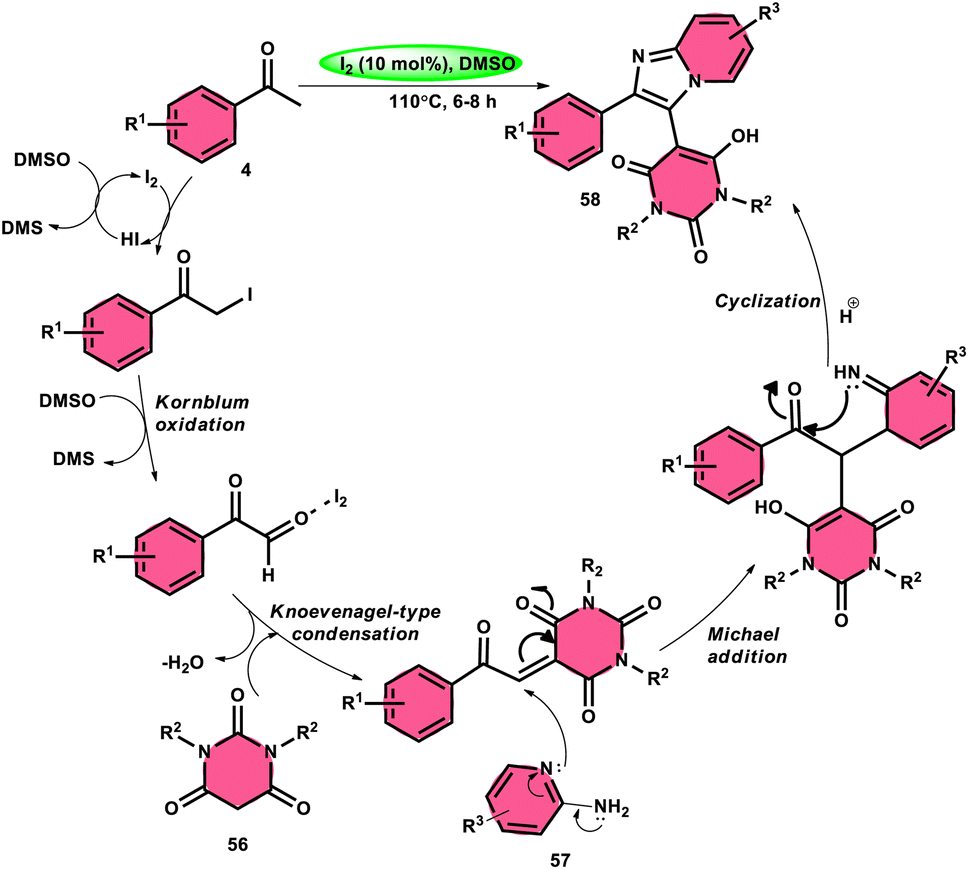
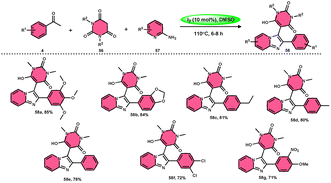
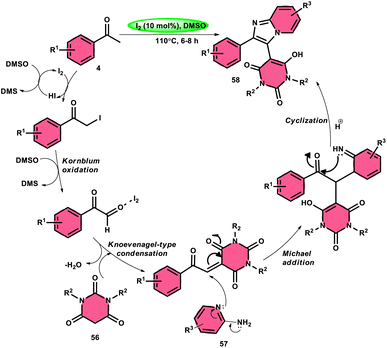
Choudhury et al.76 identified a method for synthesizing pyrimidine-linked imidazopyridine 58 (Scheme 31). This method involves the use of aryl methyl ketones 4, 2-aminopyridines 57, and barbituric acids 56 with the addition of a small quantity of molecular iodine as a catalyst. The synthesis takes place in a DMSO medium. C–H oxidation and the subsequent production of one C–C and two C–N bonds are the steps in this metal-free one-pot method. The majority of the synthesized compounds had a significant fluorescence quantum yield, ranging from very good to exceptional. The reaction process involves the first reaction of acetophenone derivative 4 with iodine, resulting in the formation of an intermediate and the byproduct HI. The presence of DMSO allows the regeneration of I2 in the following cycle. Subsequently, in the presence of DMSO, the intermediate undergoes a transformation into the equivalent phenyl glyoxal. This phenyl glyoxal then reacts with 56 by Knoevenagel condensation to get a compound. The nucleophilic addition of 56 to 57 results in the formation of an intermediate. This intermediate undergoes cyclization to form a compound. Subsequent elimination of water (H2O) from the compound leads to the formation of the intended bioactive product 58 (Scheme 32).
 |
| | Scheme 31 I2–DMSO-catalyzed synthesis of highly fluorescent pyrimidine-linked imidazopyridines 58. | |
 |
| | Scheme 32 Mechanism for the synthesis of iodine-catalyzed pyrimidine-linked imidazopyridines 58. | |
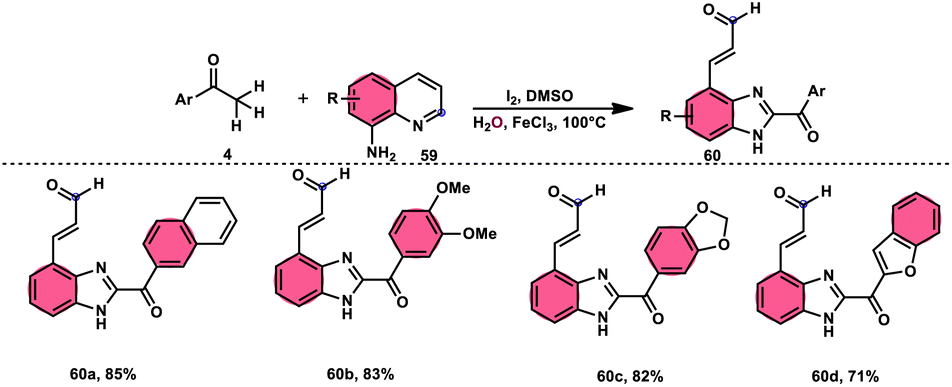
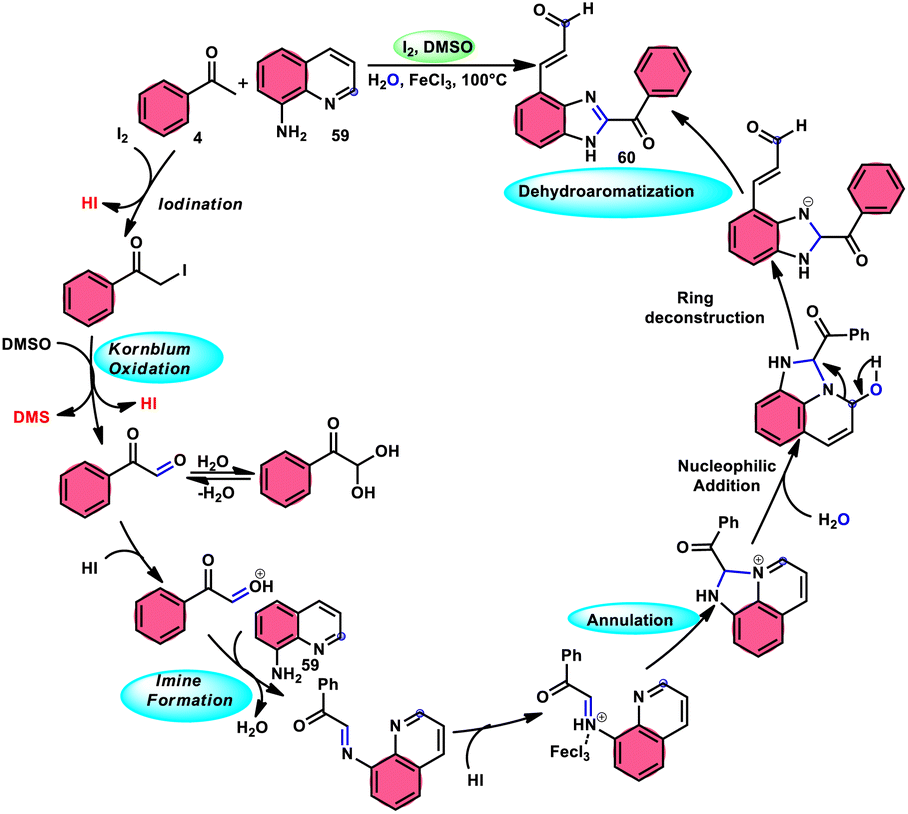
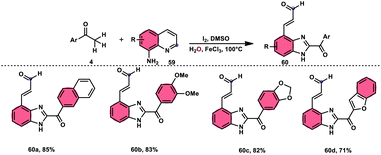
Wu et al.77 effectively demonstrated a significant cascade reaction involving aryl methyl ketones 4 and 8-aminoquinolines 59, using I2/DMSO as the catalyst (Scheme 33). The synthesis of (E)-3-(2-acyl-1H-benzo[d]imidazole-4-yl)acrylaldehydes 60 was achieved using a combination of annulation and ring deconstruction methodologies. This procedure was carried out for 8 h at 100 °C. The use of this new methodology resulted in enhanced reactivity of 8-aminoquinolines 59, therefore providing a favourable framework for the activation of unreactive N-heteroaromatic compounds through ring-opening procedures. The reaction in the absence of I2 signified the crucial functionality of molecular iodine as a prominent chemical mediator. Aryl methyl ketones 4 have a tendency to easily engage in interactions with both electron-efficient and electron-deficient groups, leading to the production of products with yields that vary from moderate to excellent. This study not only revealed the unique reactivity of 8-aminoquinolines 59, but also presented a potential structure for the initiation of unreactive N-heteroaromatic compounds by ring opening (Scheme 34).
 |
| | Scheme 33 Synthesis of benzoimidazolylacrylaldehyde 60 and selected examples. | |
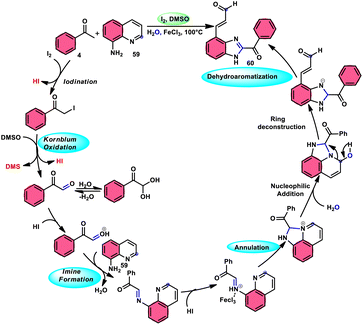 |
| | Scheme 34 Mechanism for the synthesis of benzoimidazolylacrylaldehydes 60. | |
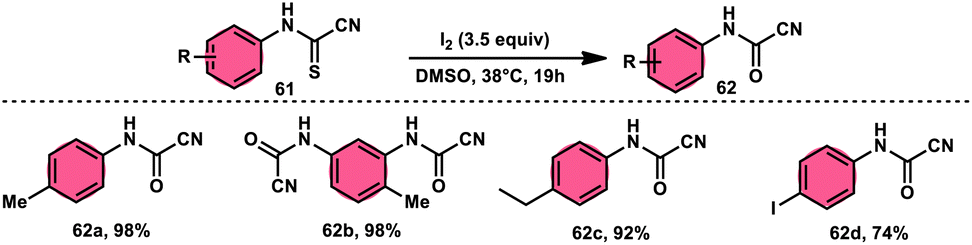
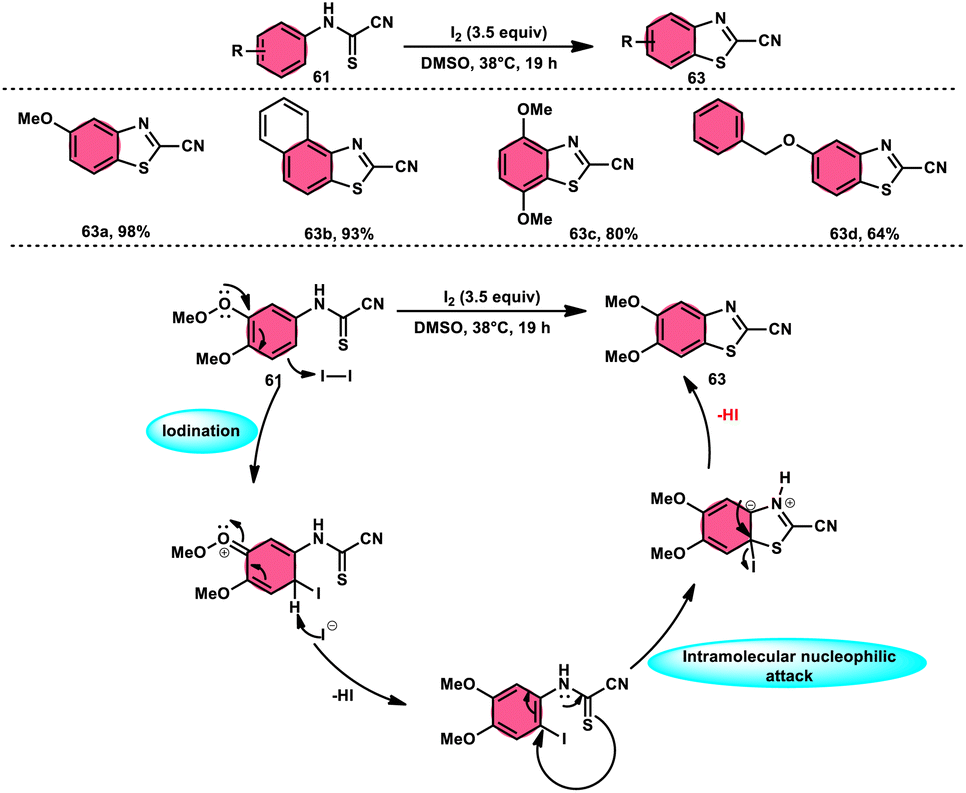
Ziad Moussa et al.78 provided information regarding the viability of using the I2–DMSO oxidative system for the production of N-arylcyanoformamides 62 at 38 °C using N-arylcyanothioformamides 61 (Scheme 35). The synthesis of important intermediates and bioactive compounds frequently involves the use of cyanoformamides as useful building blocks. This synthetic approach employed a diverse array of substrates, operated under gentle conditions, and exhibited exceptional reaction efficiency. Furthermore, it presents a novel and unconventional means to obtain 2-cyanobenzothiazoles 63 (as seen in Scheme 36). These compounds serve as valuable substrates for the identification of distinct luciferin analogues. The reaction exhibited tolerance towards a diverse array of functional groups, including various alkoxides, halides, esters, nitro, thiomethyl, cyano, and trifluoromethyl functionalities, resulting in the formation of a broad spectrum of products. Because there was no apparent improvement in conversion, KI was not an acceptable alternative for iodine. The biological actions of the benzothiazole nucleus are extremely diverse. For example, improve breast cancer diagnosis and therapy, 2-cyanobenzothiazole 63 was recently integrated in gold nanoparticles.79
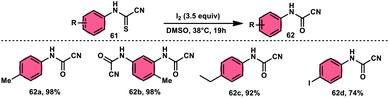 |
| | Scheme 35 Synthesis of N-arylcyanoformamide 62 and representative examples. | |
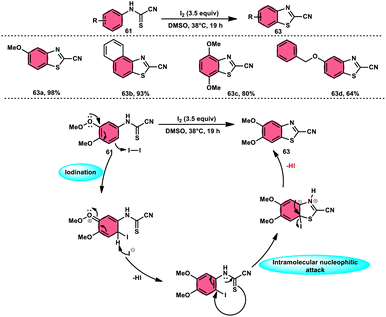 |
| | Scheme 36 Synthesis and mechanism of 2-cyanobenzothiazoles 63. | |
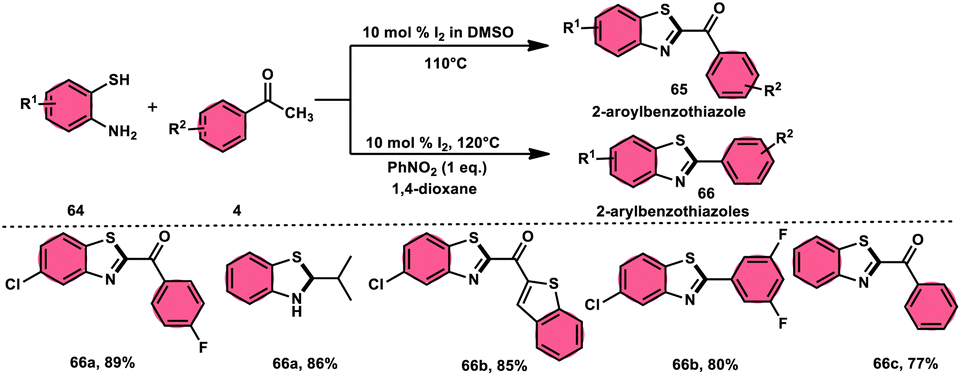
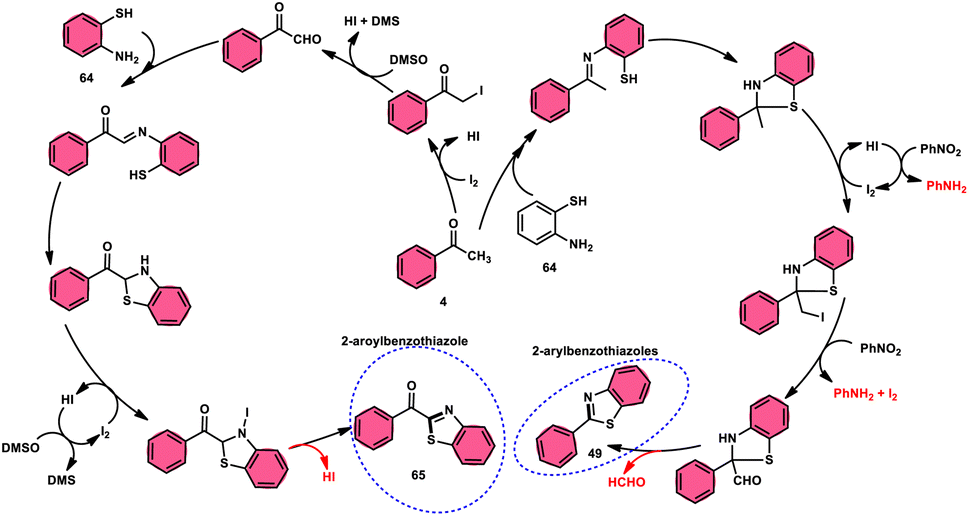
Ma et al.80 developed a unique, domino solvent-based selective methodology to prepare 2-aryl benzothiazole 66 and 2-aroylbenzothioazole 65 mediated by I2/DMSO or nitrobenzene/1,4-dioxane, respectively, starting with substrates aryl methyl ketone 4 and 2-aminobenzene thiol 64 (Scheme 37). Both EWG and EDG substituents on both substrates were well tolerated and the yields remained unaltered owing to presence of substituents. The generation of different products is due to the oxidation of aryl methyl ketone 4 to phenylglyoxal in the DMSO/I2-mediated reaction, where the aldehydic group of the phenyl glyoxal reacts with amino moiety of the substrate, leading to the formation of 2-aroylbenzothiazole 65, whereas in PhNO2/dioxane, the ketonic group without any oxidation step directly reacts with the amine moiety of aminobenzene thiol 64 and results in the formation of 2-aryl benzothiazole 66. The formation of the imine intermediate is initiated through the reaction between the aldehydic or ketonic group and the amine of aminobenzene thiol 64. This is followed by an intramolecular cyclization process, facilitated by the thiol group of the latter substrate, resulting in the formation of a C–S bond. In this reaction scheme, PhNO2 acts as an oxidant, oxidizing the methyl group of the aryl benzothiazole intermediate to an aldehydic group. Subsequently, the aldehydic group undergoes elimination, producing formaldehyde as a byproduct. Ultimately, this series of reactions leads to the formation of 2-aryl benzothiazole 66 (Scheme 38).
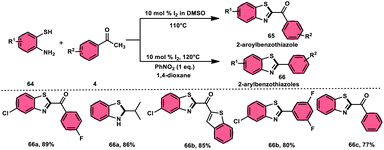 |
| | Scheme 37 Synthesis of aroylbenzothiazole 65 and arylbenzothiazoles 66 in I2–DMSO system. | |
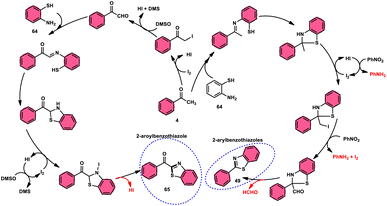 |
| | Scheme 38 Mechanism for the synthesis of aroylbenzothiazole 65 and arylbenzothiazoles 66 using I2–DMSO system. | |
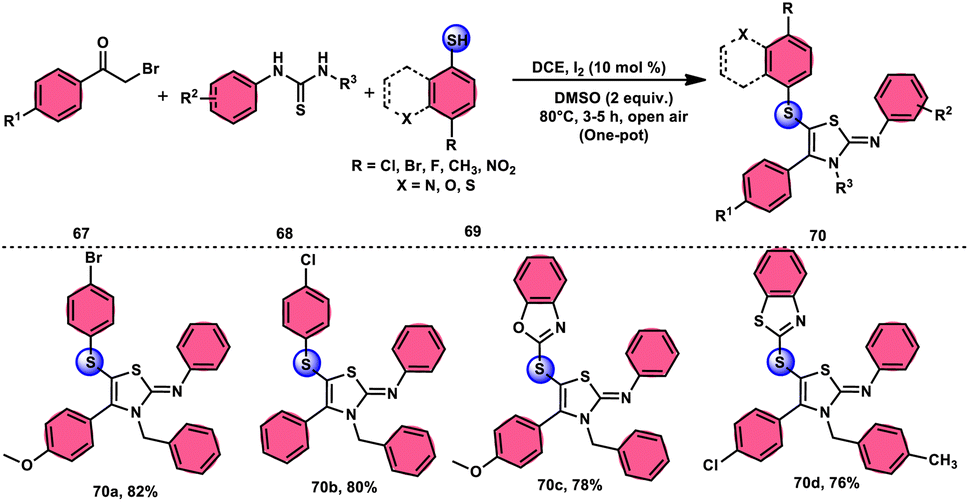
Animesh Pramanik and Bodhak81 proposed a metal-free, open-air, one-pot method for the regioselective sulfenylation of 2-iminothiazoline 70 at the C-5 position. This approach utilizes the I2/DMSO combination as a catalytic oxidant. The reaction takes place in the C2H4Cl2 (DCE) solvent and involves the interaction between phenacyl bromide 67 and thiourea 68. This interaction leads to the formation of the 2-iminothiazoline intermediate. The C–H bond in this intermediate becomes reactive due to the presence of I2 as a catalyst. Subsequently, the oxidant DMSO facilitates the regioselective sulfenylation at the C-5 position. This series of reactions ultimately yields the desired product, which is 5-sulfenyl-2-iminothiazoline derivative 70. The applicability of this methodology increases when the chemoselective bridged S-atom of the desired product is oxidized into sulfoxide, and the reaction also proceeds smoothly whether all three substrates have EDGs or EWGs, pyridyls or heterocyclic thiols and gives good yields in all cases, showing its good functional tolerance. Interestingly, 2-iminothiozoline intermediate 70 achieved through the condensation between phenacyl bromide 67 and thiourea 68 does not require I2/DMSO; however, the diaryl disulfide intermediate, which forms in situ from aromatic thiols 69 and nucleophilic attack on the 2-iminothiozolines intermediate on the S atom of the former intermediate through C–H bond activation leads to the desired product, although I2/DMSO is indispensable in both steps (Scheme 39).
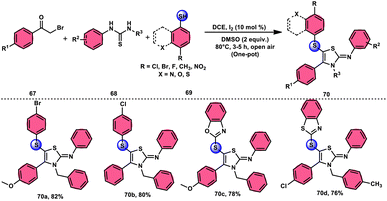 |
| | Scheme 39 I2–DMSO-based preparation of 5-sulfenyl-2-iminothiazolines 70 and representative examples. | |
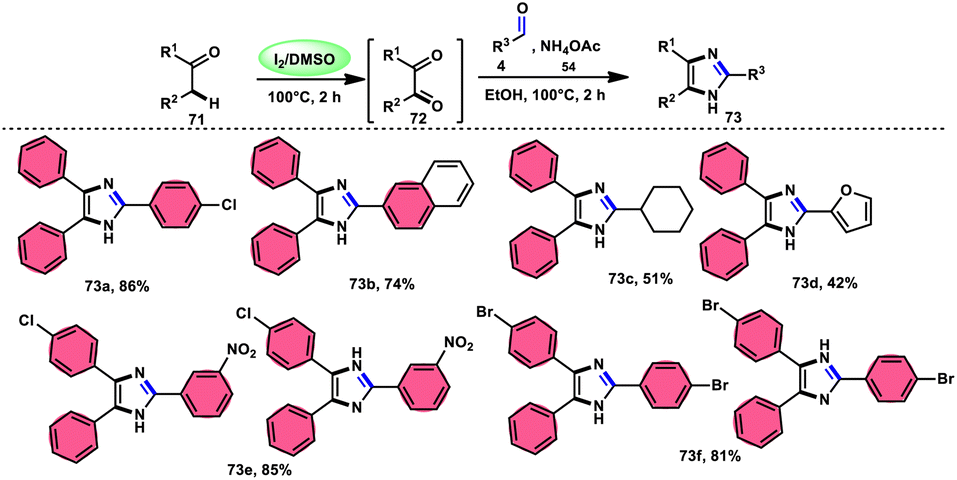
In the study by Jeena and Jayram,82 they proposed an enhanced and environmentally sustainable approach for the construction of 2,4,5-trisubstituted imidazole 73 and its bioactive scaffold. This technique eliminates the need for acid/base and transition metal catalysts, resulting in a more time-efficient and cost-effective process. The authors used I2/DMSO as a promoter in the oxidative cyclization of the C–H bond, leading to increased yields and reduced environmental impact. The reactants α-methylene ketone 71, aldehydes and NH4OAc 54, which is used as a nitrogen source, are readily accessible and inexpensive. Furthermore, the broad substrate scope of this methodology makes its applicable in the synthesis of various derivates of the target product, which can be used as pharmaceutical scaffolds. The use of benzyl alcohol instead of benzyl aldehyde is feasible given that benzyl alcohol and benzyl phenyl ketone are concurrently oxidized in benzene aldehyde and benzil, respectively, leading to the target product in moderate yields through the domino convergent reaction path approach. According to the plausible reaction mechanism, molecular iodine converts methylene ketone 71 into diketones 72via the radical path, and both carbonyl reactants further react with the in situ-generated NH3 from NH4OAC 54, affording the imine intermediate. Finally, the condensation between these imine intermediates results in the desired products 73 (Scheme 40).
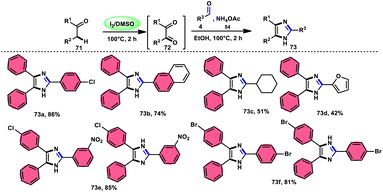 |
| | Scheme 40 I2/DMSO-catalyzed synthesis of trisubstituted-imidazoles 73 and representative examples. | |
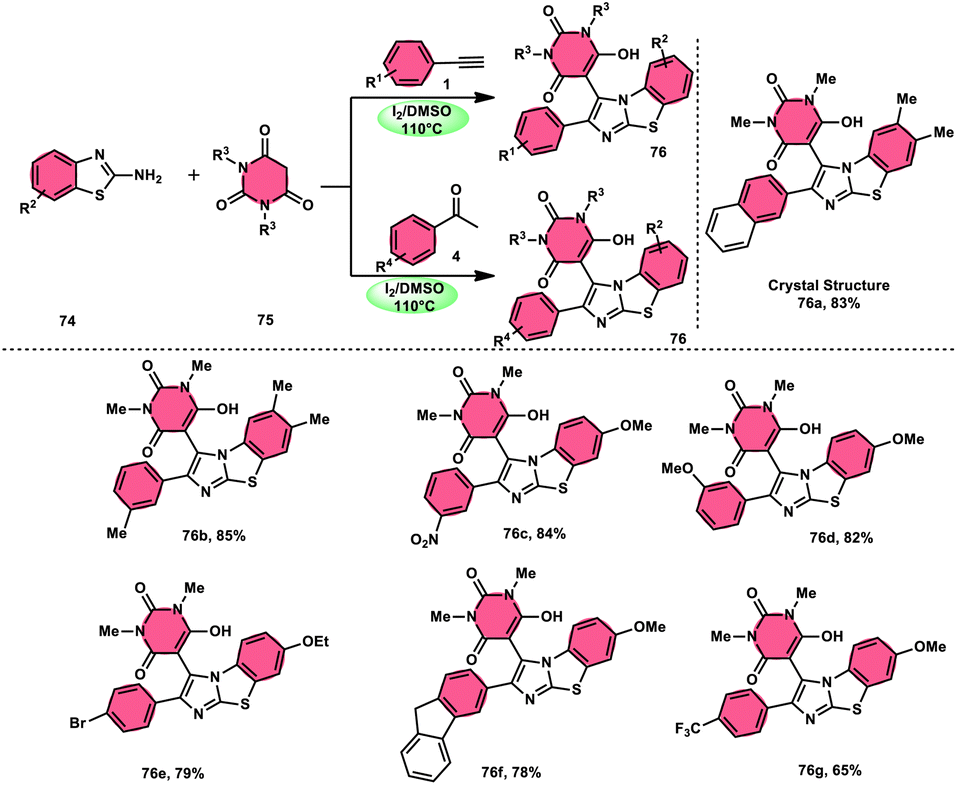
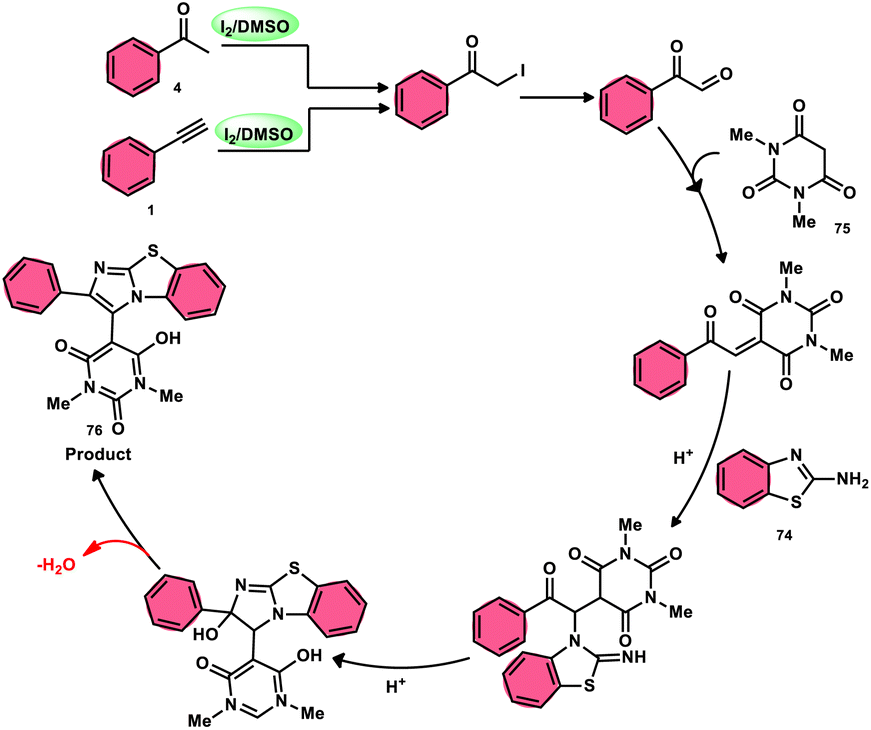
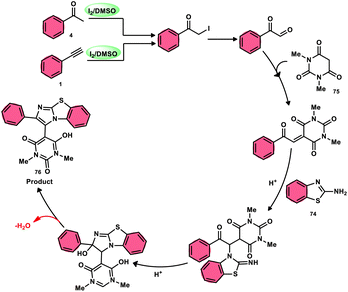
The research group led by Lokman H. Choudhury devised a method, as shown in Scheme 41,50 for the synthesis of 2-arylbenzo[d]imidazo[2,1-b]thiazoles 76 derived from barbituric acid 75. This method utilizes an I2/DMSO-based reaction system, which is both effective and environmentally benign. Furthermore, this metal-free, one-pot, three-component reaction (MCR) offers an efficient approach for the synthesis of the desired compounds. The methodology has practical value owing to the fact that its product and its arrays have medicinal applications, its good substrate scope, competence under traditional or microwave heating condition, and utilized reactants, i.e., barbituric acid 75, 2-aminobenzothizole 74 and aryl methyl ketone 4 or aryl acetylene 1. The reaction is easily accessible, inexpensive and its yields are good to excellent. The methodology proceeds via Kornblum oxidation, converting aryl methyl ketone 4 into aryl glyoxal, which undergoes Knoevenagel condensation, forming a C–C bond with barbituric acid, aza-Michael addition by 2-aminobenzothiazole and intramolecular cyclization of the intermediate, affording two C–N bonds, which results in the targeted products, and molecular iodine plays an important role in the oxidation and cyclization (Scheme 42).
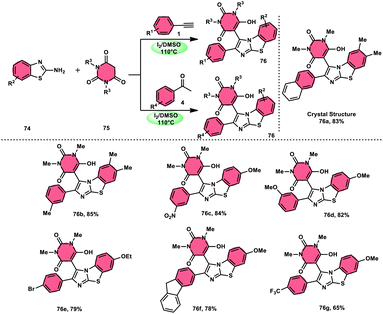 |
| | Scheme 41 Synthesis of 2-arylbenzo[d]imidazo[2,1-b]thiazole 76 catalyzed by I2/DMSO. | |
 |
| | Scheme 42 Mechanism for the synthesis of 2-arylbenzo[d]imidazo[2,1-b]thiazole 76 catalyzed by I2/DMSO. | |
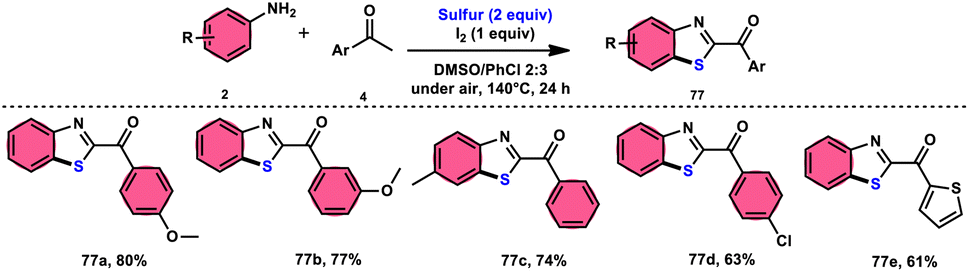
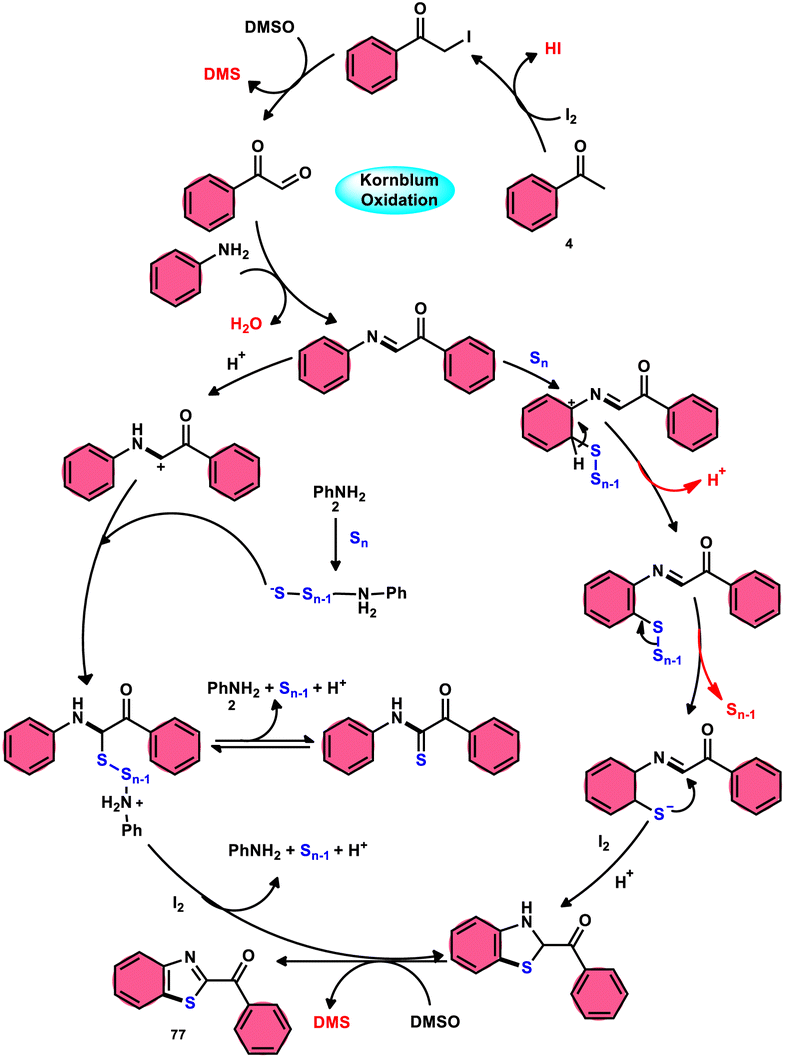
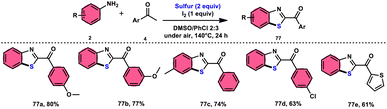
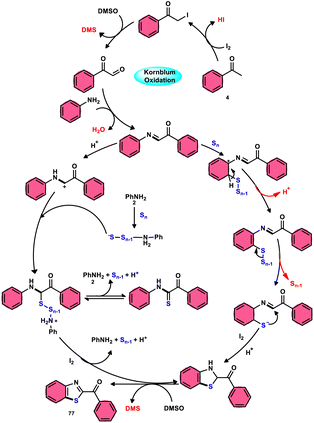
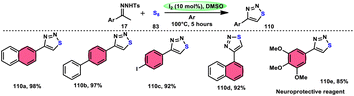
Phan et al.83 proposed a new, straightforward, environmentally friendly, transition metal-free, three-component reaction. This reaction utilizes readily available and cost-effective starting materials including elemental sulfur (S8), acetophenones 4, and anilines 2 to produce 2-aroylbenzothiazoles 77 (Scheme 43). The molecular iodine-promoted reaction gave the best yields in the solvent combination of DMSO/PhCl in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio, whereas DMSO as an oxidant leads to Kornblum oxidation and aromatization. This strategy exhibits economical value due to the non-toxic nature and commercial availability of the substrates, as well as the favourable functional group tolerance of the substituted acetophenones 4 and anilines 2. According to the proposed reaction mechanism, the combination of I2 and DMSO facilitates the Kornblum oxidation, resulting in the conversion of acetophenone 4 to phenylglyoxal. Subsequently, the condensation of an amine ketone forms an imine intermediate. The imine intermediate then undergoes electrophilic attack by S8 at the ortho position of benzene, followed by intramolecular cyclization to yield the desired product (Scheme 44).
:3 ratio, whereas DMSO as an oxidant leads to Kornblum oxidation and aromatization. This strategy exhibits economical value due to the non-toxic nature and commercial availability of the substrates, as well as the favourable functional group tolerance of the substituted acetophenones 4 and anilines 2. According to the proposed reaction mechanism, the combination of I2 and DMSO facilitates the Kornblum oxidation, resulting in the conversion of acetophenone 4 to phenylglyoxal. Subsequently, the condensation of an amine ketone forms an imine intermediate. The imine intermediate then undergoes electrophilic attack by S8 at the ortho position of benzene, followed by intramolecular cyclization to yield the desired product (Scheme 44).
 |
| | Scheme 43 Synthesis of 2-aroylbenzothiazoles 77 catalyzed by I2/DMSO. | |
 |
| | Scheme 44 Mechanism for the synthesis of 2-aroylbenzothiazoles 77 catalyzed by I2/DMSO. | |
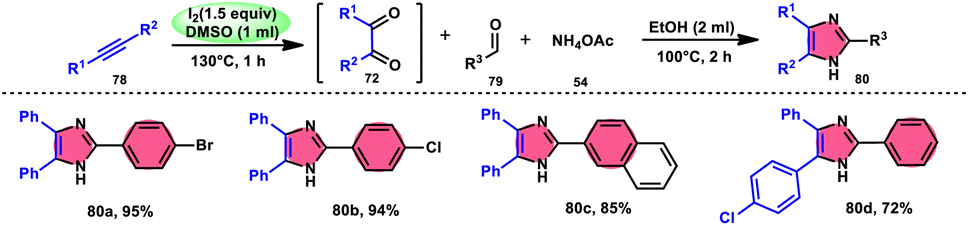
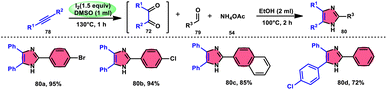
Vineet Jeena and Shivani Naidoo84 developed a new, practical, economical, metal-free, acid-free, and environmental-benign I2/DMSO-mediated approach to prepare trisubstituted imidazoles 80 using the cheap reactants internal alkyne 78, aldehyde 79, and ammonium acetate 54 (Scheme 45). The one-pot oxidative cyclization procedure is concluded in two steps, where in the first step, alkynes 78 are transformed into α-diketone, which further reacts with aldehyde and NH4OAc 54via cyclic condensation, affording the desired product 80. Benzaldehydes substituted with various EDGs and EWGs are compatible to achieve the target products; however, aliphatic aldehydes result in unsatisfactory yields. The utilization of molecular iodine as a catalyst and DMSO as an oxidant plays significant roles in the described transformation (Scheme 46).
 |
| | Scheme 45 I2–DMSO-catalyzed oxidation of internal alkynes 78 and selected examples. | |
 |
| | Scheme 46 Mechanism for the I2–DMSO-catalyzed of oxidation of internal alkynes 78. | |
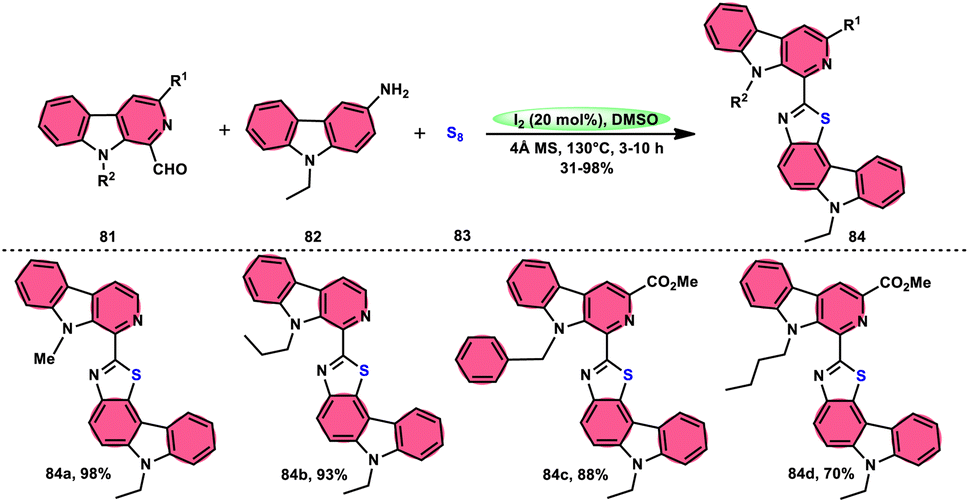
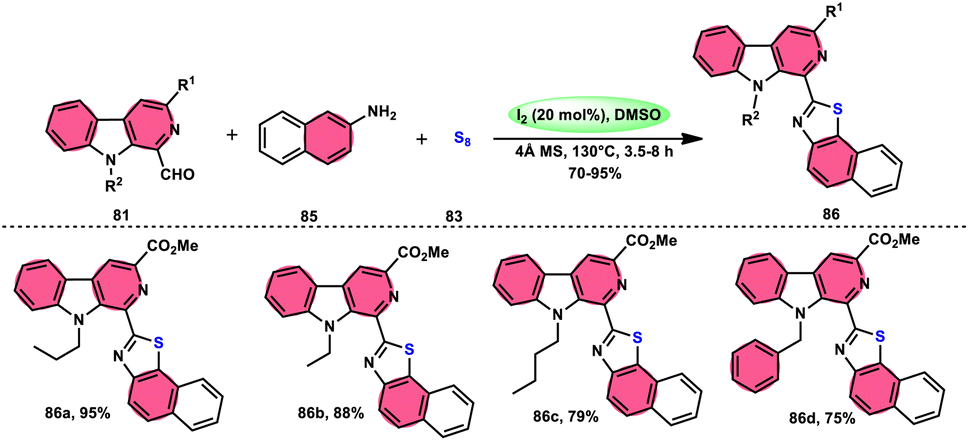
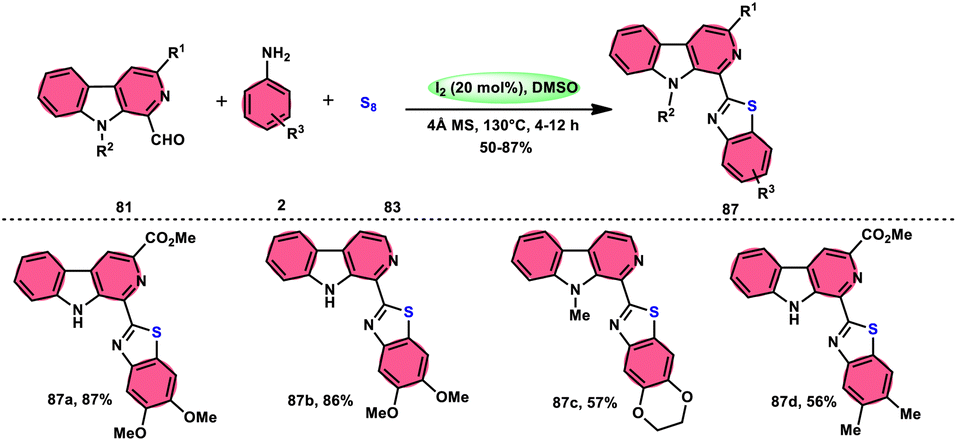
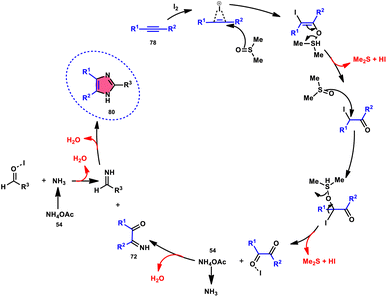
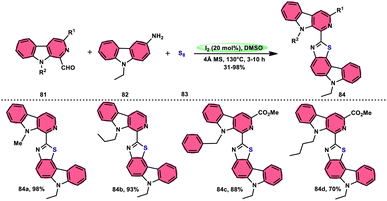
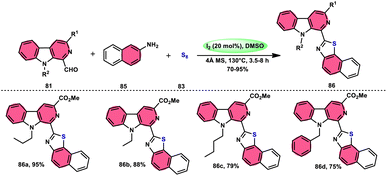
Singh et al.85 developed an innovative and effective method, free of metal catalysts, for synthesizing highly fluorescent compounds. This method involves the preparation of β-carboline C-1(3) linked thiazolo[4,5-c]carbozoles 84 (Scheme 47), naphtho[2,1-d]thioazoles 86 (Scheme 48), and benzothiazole 87 (Scheme 49) using I2/KI as a mediator in DMSO solvent. All three variant products were highly fluorescent, and also prepared on a gram scale with outstanding yield of above 90%, making this technique industrially applicable. Both acetal and aldehyde derivatives of Kumujian C (1-formyl-9H-β-carbolines) 81, which serve as the base model, exhibit high efficiency in their reaction with elemental sulfur (S8) 83, as well as 3-aminocarbazole 82, anilines 2, 2-aminopyridine, and naphthylamine 85. These reactions yield the desired products derived from the corresponding aryl amine derivatives. This methodology has good functional group tolerance on β-carboline (Kumujian) 81 and aniline 2; however, aniline having EWG groups failed to generate the desired products. This strategy involves the formation of two C–S bonds and one C–N bond in a single step through the utilization of an imine intermediate. This reaction is considered to be highly atom economic. Additionally, both potassium iodide (KI) and iodine (I2) exhibit equal efficiency in catalyzing these reactions. The research team also deeply investigated the luminous nature of derivates of all three products. All the derivates had good fluorescence with a fluorescence quantum (ΦF) yield of up to 92%, although thiazolo[4,5-c]carbazole 84 had the highest fluorescence properties, which can be used in medical science.
 |
| | Scheme 47 Synthesis of β-carboline C-1-tethered thiazolo[4,5-c]carbazole 84 and selected examples. | |
 |
| | Scheme 48 Synthesis of β-carboline C-1-substituted naphtho[2,1-d]thiazole 86 and selected examples. | |
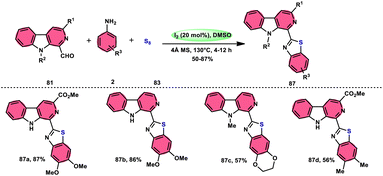 |
| | Scheme 49 Synthesis of β-carboline C-1-substituted benzothiazole 87 and selected examples. | |
8. Five-membered ring formation with three-heteroatoms
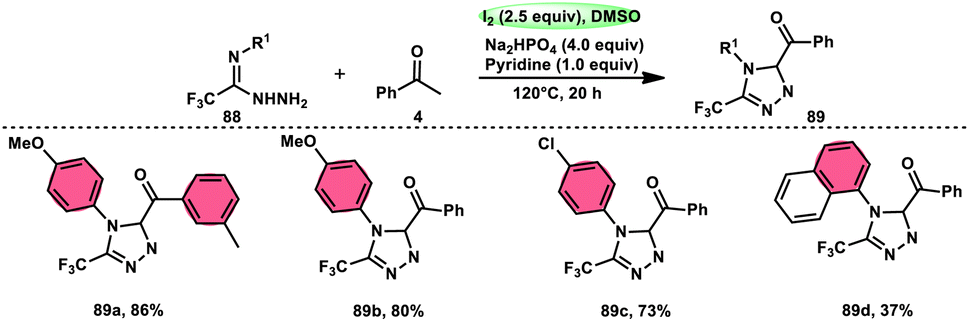
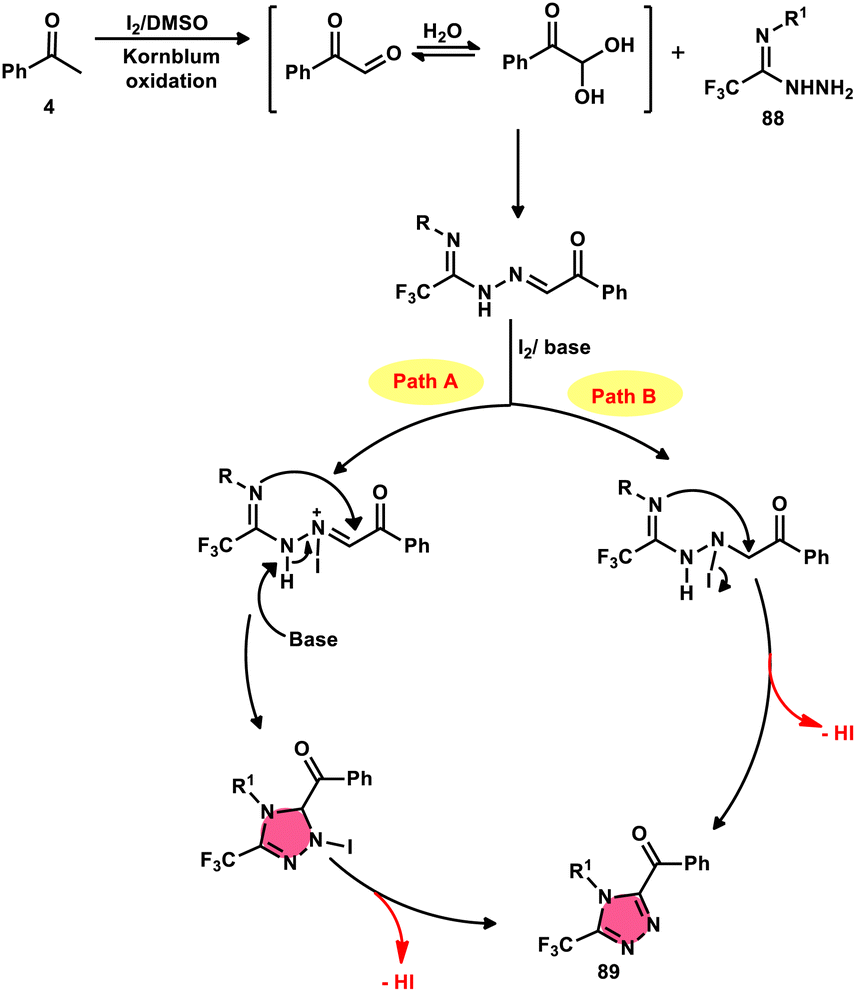
The formation of 5-trifluoromethyl-1,2,4-triazoles 89 was achieved by Zhengkai Chen, Xiao-Feng Wu, and colleagues using a metal-free methodology.86 This method involves the I2-mediated [4 + 1] annulation of readily accessible trifluoroacetimidohydrazides 88 and methyl ketones 4, as illustrated in Scheme 50. The procedure involves iodination/Kornblum oxidation, intermolecular dehydration condensation, and an intramolecular cyclization–aromatization sequence facilitated by iodine. The utilization of the synthesis method presents a viable alternative for obtaining functionalized 1,2,4-triazole derivatives87 possessing therapeutic features. The results indicate that the electron factors and steric hindrance of the trifluoroacetimidohydrazides have a limited effect on the outcome of the reaction, as evidenced by the observed comparable efficiency (Scheme 51).
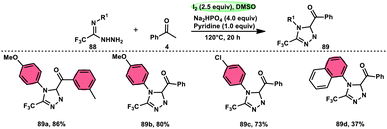 |
| | Scheme 50 Synthesis of substituted triazoles 74 and selected examples. | |
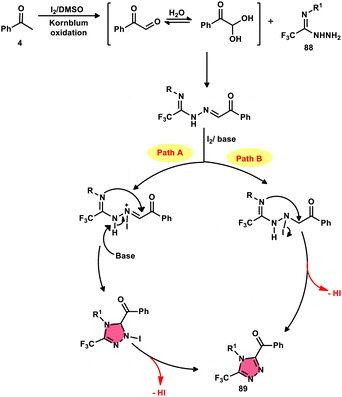 |
| | Scheme 51 Mechanism for the synthesis of substituted triazoles 89. | |
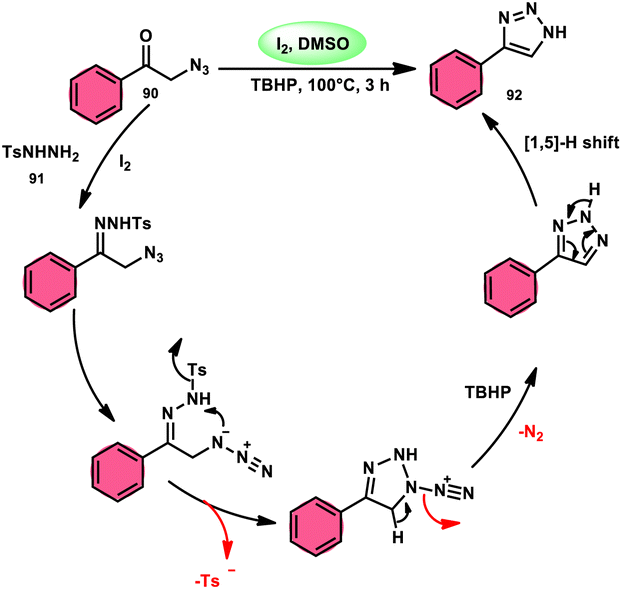
NH-1,2,3-Triazoles 92, together with their derivatives, represent a crucial class of heterocyclic compounds that possess diverse and significant pharmacological and biological properties. These properties include the inhibition of HER2, hMetAP2, IDO, VIM-2, as well as anticancer activity.88–91 Hence, by employing readily available α-azido acetophenones 90 and p-toluene sulfonyl hydrazide 91 in the presence of DMSO solvent, Wen-Ming Shu et al. successfully devised a condensation/cyclization methodology mediated by molecular iodine for the production of 4-aryl-NH-1,2,3-triazoles 92 (Scheme 52).92 Under ideal circumstances, this reaction presents a method that does not require the use of metals to sequentially develop C–N and N–N bonds. The compounds that correspond to α-azido ketones and possess either an electron-donating or withdrawing group can be synthesized in high yields. Substrates with halo-substituted groups (4-F, 3,4-2Cl, 3-Cl, 4-Cl, 3-Br, and 4-Br) also exhibited a favourable performance, resulting in the production of the desired products in significant quantities. The utilization of cyclopropyl α-azido ketone in the annulation reaction was prohibited, resulting in the failure to attain the intended outcome (Scheme 53).
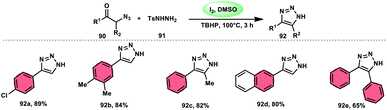 |
| | Scheme 52 Synthesis of triazoles 92 and selected examples. | |
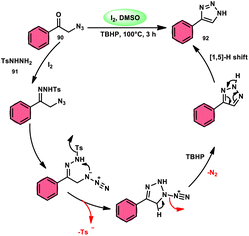 |
| | Scheme 53 Mechanism involving the synthesis of triazoles 92. | |
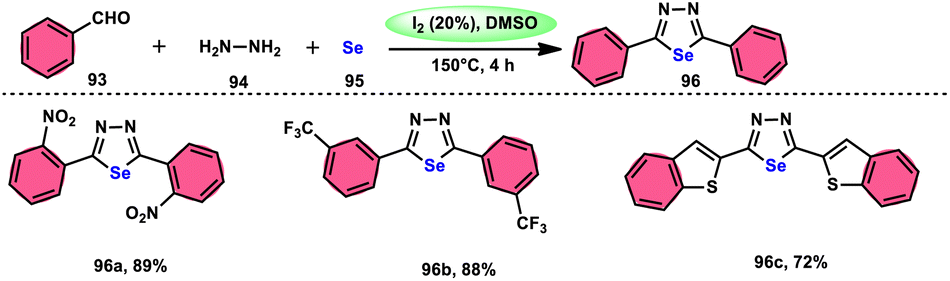
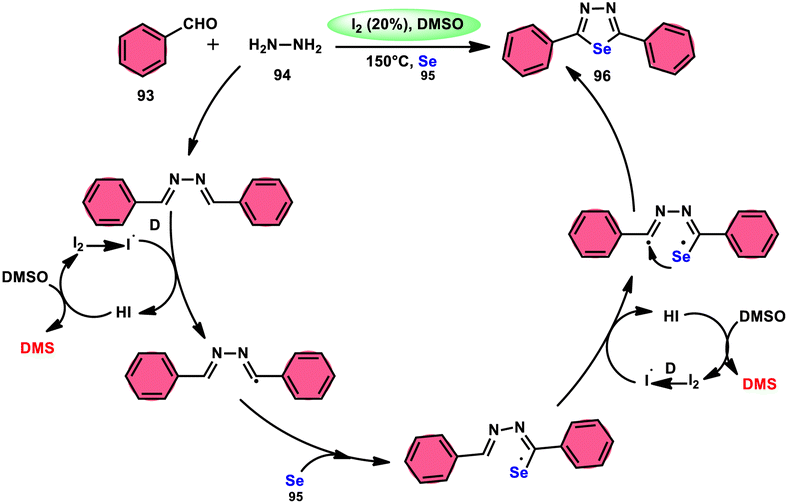
The synthesis of 1,3,4-selenadiazoles 96 was explained by Bavanthula and colleagues in 2021 utilizing a three-component process involving arylaldehydes 93, hydrazine 94, and elemental selenium 95 in the presence of the I2/DMSO system (Scheme 54).93 This methodology demonstrates the ability to produce the desired products in moderate to favorable yields. It is characterized by its operational simplicity and effectiveness across a range of functional groups. The reaction tolerating a radical operation is predicted by the postulated mechanism. 1,3,4-Selenadiazoles 96 have demonstrated a wide range of biological actions, ranging from pesticides, fungicides, analgesics, anticancer, anticonvulsants, and anti-inflammatory medications.94–96 At 150 °C, the reaction was finished in 4 h. In the control experiment, dibutylhydroxytoluene (BHT) and (tetramethylpiperidin-1-yl)oxidanyl were used as radical inhibitors to stop the reaction. The advantages of the current approach are its basic operation and avoidance of metal. Under the optimal reaction conditions, benzaldehyde with substituents at the ortho (o), meta (m), and para (p) positions of the aromatic ring that are neutral, electron-donating, or electron-withdrawing exhibited efficient reactivity, resulting in the formation of the desired product in significant yields (Scheme 55).
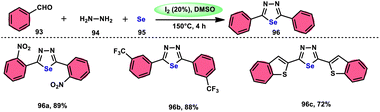 |
| | Scheme 54 Synthesis of 1,3,4-selenadiazoles 96 and selected examples. | |
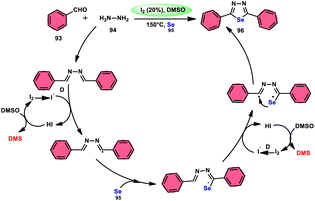 |
| | Scheme 55 Mechanism involving the synthesis of 1,3,4-selenadiazoles 96. | |
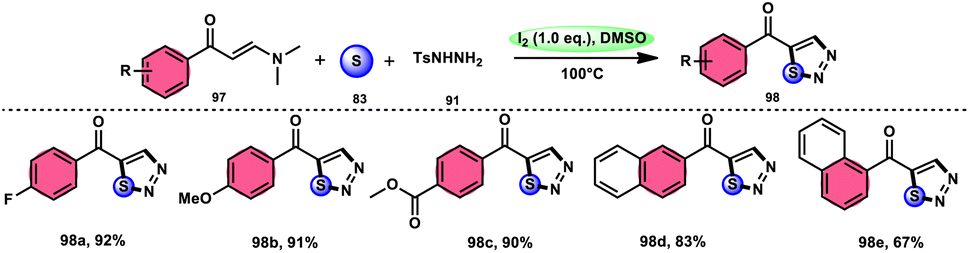
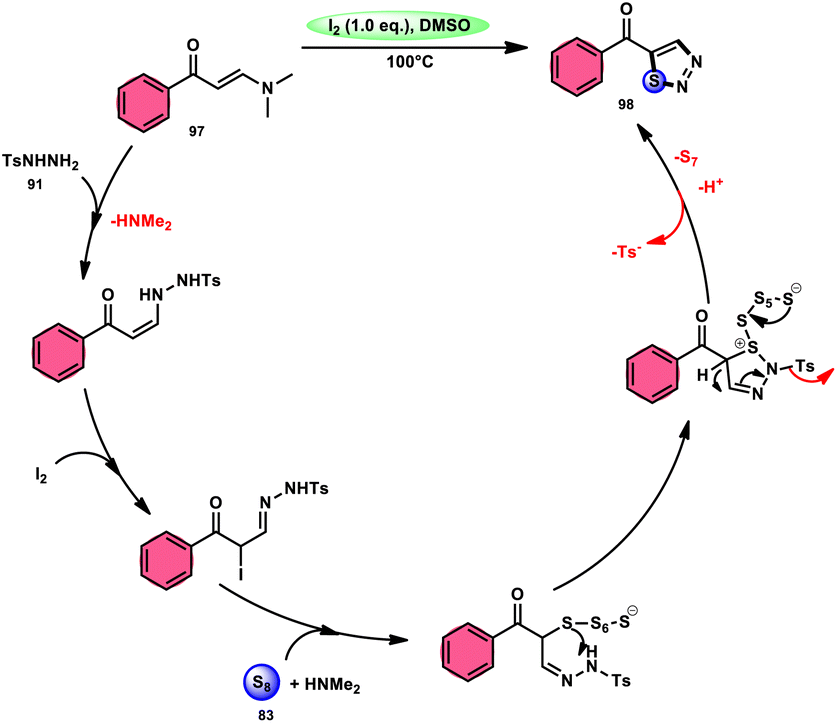
In the study by Zhou et al.,97 they developed a highly efficient method involving three components, without the use of transition metals, to synthesize 5-acyl-1,2,3-thiozole 98. This protocol involves the reaction of enaminones 97, tosylhydrazine (p-toluenesulfonyl hydrazide) 91, and elemental sulfur 83, promoted by I2/DMSO (as shown in Scheme 56). This strategy facilitates the formation of three significant C–S, C–S, and S–N bonds in a direct manner, resulting in yields ranging from moderate to outstanding. Additionally, the utilization of affordable and easily accessible reactants enhances the practicality of this methodology.98,99 The functional group tolerance on aromatic enaminones is very good, whereas alkyl enaminones are incompatible to achieve the targeted product (Scheme 57).
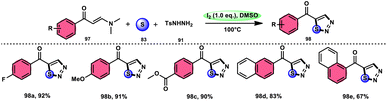 |
| | Scheme 56 I2/DMSO-based synthesis of thiadiazols 98 and selected examples. | |
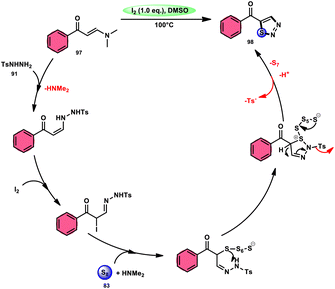 |
| | Scheme 57 Mechanism for the synthesis of thiadiazols 98 catalyzed by I2/DMSO. | |
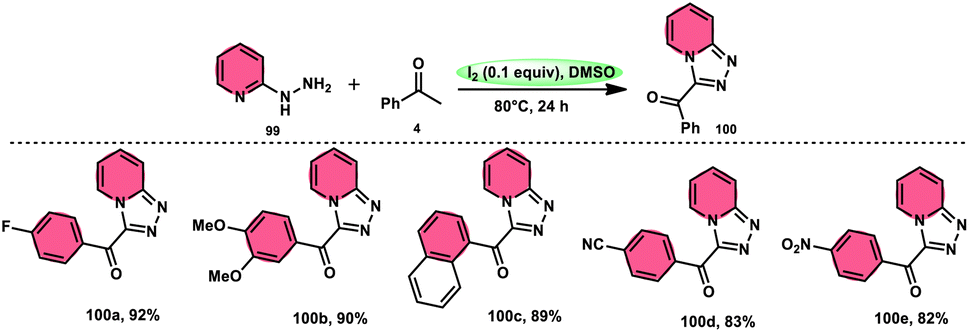
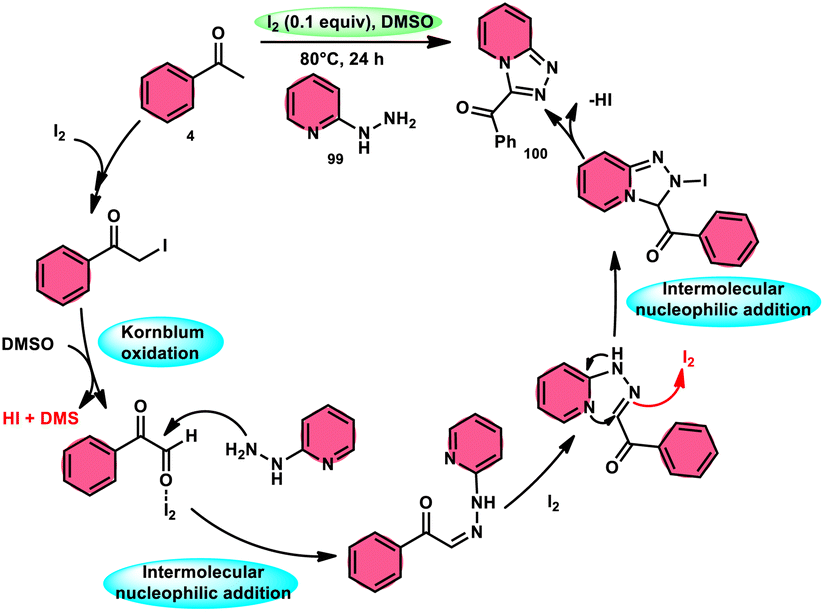
B. V. Subba Reddy and fellows100 developed an environmentally friendly, one-pot, metal-free, conducive and highly effective protocol to prepare the bioactive scaffold 3-aryl[1,2,4]triazolo[4,3-a]pyridines 100 promoted by I2 and DMSO (Scheme 58). This methodology has a broad scope of reactants including acetophenone 4, phenylacetylene, ethyl benzoylacetate, styrene and alcohol with 2-hydrazinopyridine 99; however, styrene and alcohol require 2 equiv. of IBX (2-iodo benzoic acid) besides the general conditions, where IBX converts these reactants to phenylacyl iodide. Further, this strategy is scalable and has a high functional group tolerance on all the reactants and its yields are good to excellent. Molecular iodine plays pivotal role in the oxidative cyclization via Kornblum oxidation, producing phenylglyoxal, working as a Lewis acid to induce the intramolecular nucleophilic addition of 2-hydrazinopyridine and intramolecular cycloaddition, which results in the target product (Scheme 59).
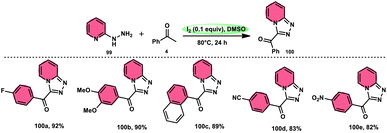 |
| | Scheme 58 Metal-free I2–DMSO-based reaction of 2-hydrazinylpyridines 99 and selected examples. | |
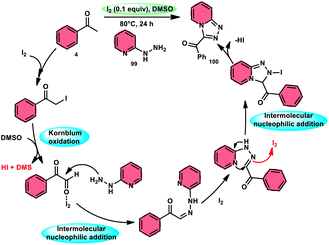 |
| | Scheme 59 Mechanism for reaction of 2-hydrazinylpyridines 99. | |
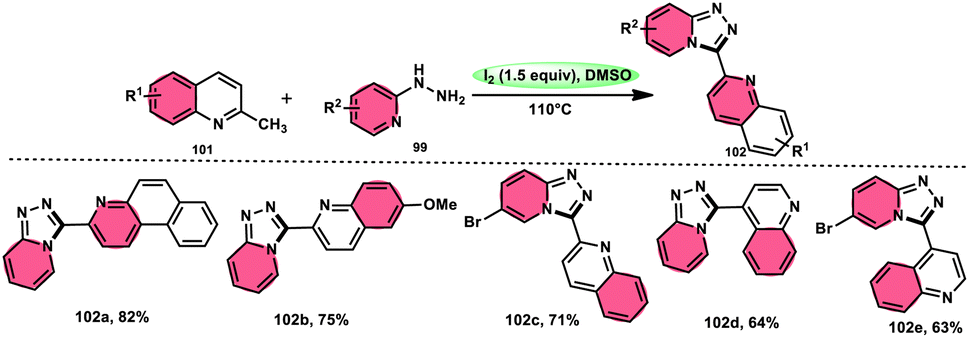
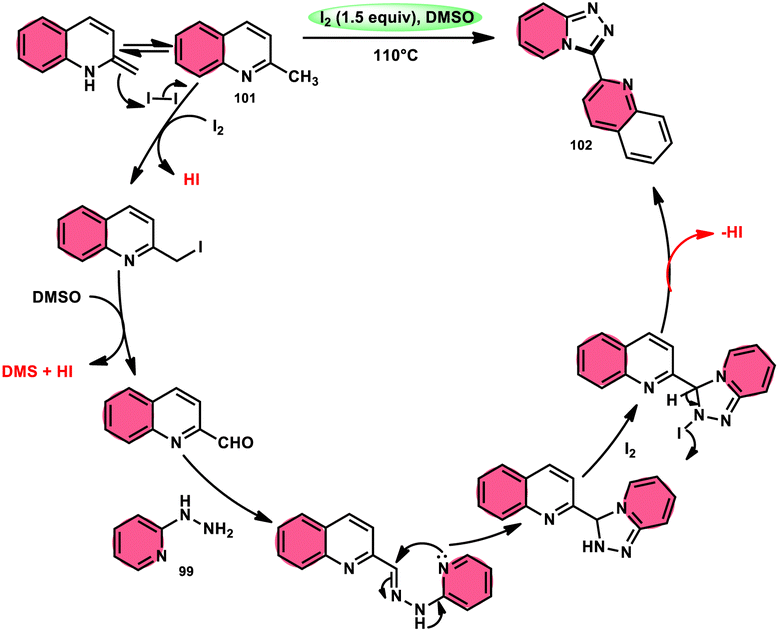
Wu et al.101 developed a new reaction method that does not involve the use of metals or acid/base catalysts. This one-pot, environmentally friendly approach enables the synthesis of diheterocycles containing 1,2,4-traizolo[4,3-a] pyridine and quinoline moieties 102. The reaction utilizes 2-methyl quinoline derivatives 101 and 2-hydrazinepyridine derivatives 99 as starting materials and is facilitated by the I2/DMSO system (Scheme 60). In this protocol, the oxidative activation of the C–H (sp3) bond reacting with 2-hydrazinepyridine 99via (1 + 4 ring forming procedure) affords 1,2,4-traizolo[4,3-a] pyridine 102 in good yield. The compatibility of methyl quinoline 101 and 2-hydrazinepyridine 99, together with their derivatives substituted with diverse functional groups allows for their conversion into the desired products. According to the proposed reaction mechanism, molecular iodine is crucial in the transformation, which starts the reaction via iodination of methyl quinoline 101 followed by Kornblum oxidation, which results in the formation of quinoline-2-carbaldehyde, and it further reacts with 2-hydrazinepyridine 99via condensation, followed by the annulation to afford the target products (Scheme 61).
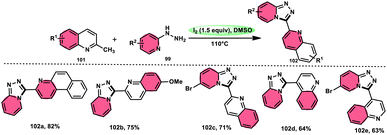 |
| | Scheme 60 Synthesis of triazolo-pyridines 102 and representative examples. | |
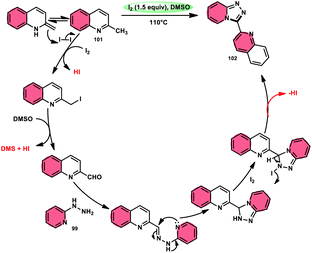 |
| | Scheme 61 Mechanism for the synthesis of triazolo-pyridines catalyzed by I2/DMSO. | |
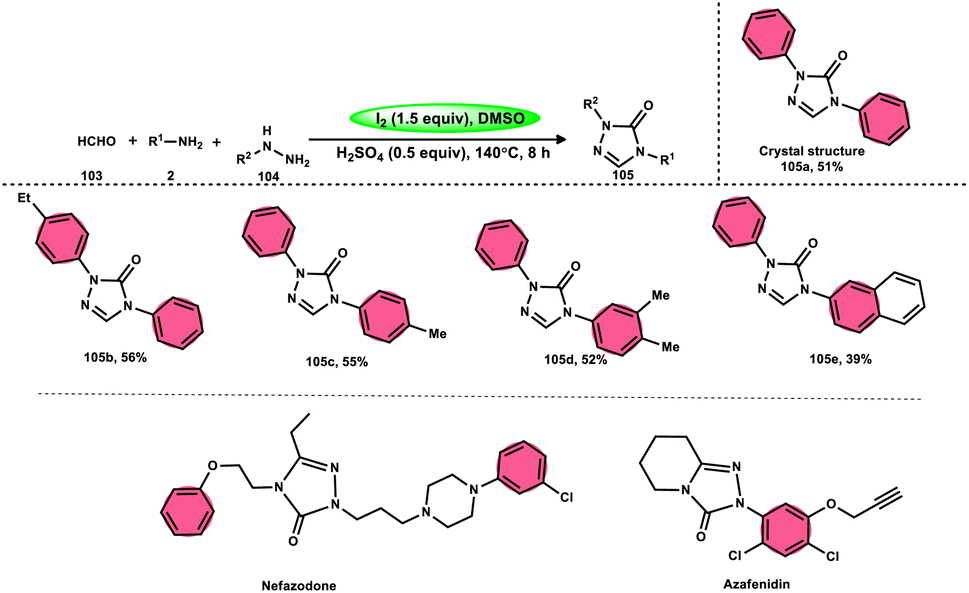
Zhu et al.102 introduced a method to produce 2,4-disubstituted 1,2,4-triazole-3-ones 105 by a three-component reaction involving formaldehyde 103, amines 2, and hydrazines 104, which is facilitated by iodine and DMSO. 1,2,4-Triazole-3-ones 105 are significant heterocyclic compounds that exhibit a diverse range of biological functions.103–105 However, the reaction does not occur at a temperature of 100 °C and only produces a yield of 28%. Further, absence of I2 resulted in minimal product formation, highlighting the vital importance of I2 in this reaction. The desired product could not be obtained when anilines with strong electron-withdrawing substituents, such as nitro groups, were used as the reactants. Also, the desired product could not be obtained if the temperature is lower than 100 °C. Both I2 and DMSO are essential given that the lack of either resulted in a small or insufficient quantity of the desired product. Also, I2 must be introduced after all other reactants are added; otherwise, complicated products will be produced throughout the process. Alternatively, an atmosphere of either O2 or N2 did not affect the yield of the product (Scheme 62).
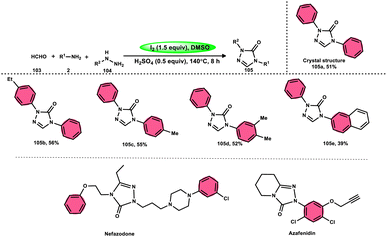 |
| | Scheme 62 Synthesis of 2,4-disubstituted-1,2,4-triazole-3-ones 105, representative examples and their biologically active compounds. | |
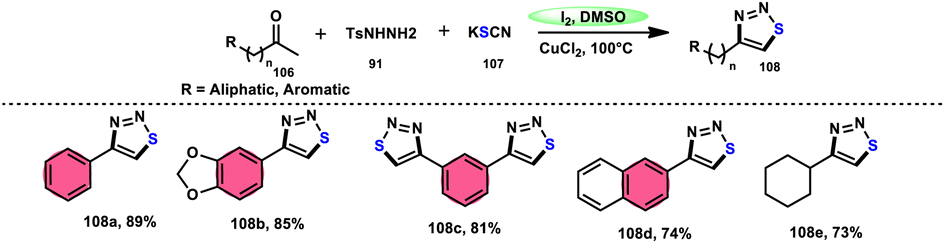
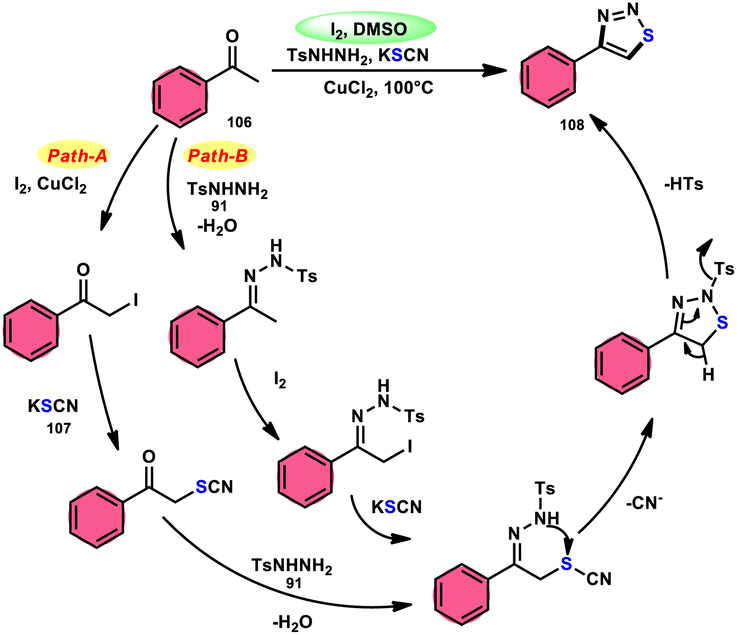
Wu et al.106 developed a concise and efficient method for the synthesis of 1,2,3-thiadiazoles 108 using a one-pot, three-component protocol. The reaction utilizes readily accessible and cost-effective reactants, including TsNHNH291, KSCN 107, and aliphatic or aromatic methyl ketones 106. The reaction is facilitated by the use of I2/CuCl2 as a catalyst in DMSO solvent, as depicted in Scheme 63. The methodology exhibits a broad substrate scope, including aryl/heteroaryl/chained-aliphatic/cyclic aliphatic methyl ketones 106, which are capable of producing the desired compounds. Additionally, the methodology demonstrates good compatibility with various substituents on the aryl moiety of the aryl methyl ketones, resulting in high yields in the majority of cases. The conventional Kornblum oxidation of ketones to glyoxal derivatives was found to be incompatible with the proposed reaction mechanism. Instead, an alternative pathway was observed. Initially, an intermediate α-iodo ketone is formed, followed by substitution of the iodine atom with the nucleophile SCN−. Then, a condensation reaction between ketone 106 and TsNHNH291 occurs, resulting in the formation of a C–N bond. Next, intramolecular cyclization leads to the desired product 108. Molecular iodine plays a key role in the methodology; however, the yield without the catalyst CuCl2 is lower, and thus CuCl2 increases the yield to an outstanding level (Scheme 64).
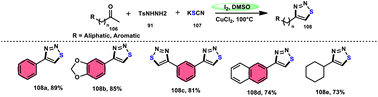 |
| | Scheme 63 Iodine–DMSO-catalyzed synthesis of 1,2,3-thiadiazoles 108 and representative examples. | |
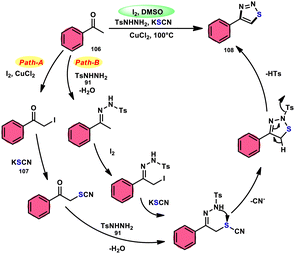 |
| | Scheme 64 Iodine–DMSO-catalyzed mechanism for the synthesis of 1,2,3-thiadiazoles 108. | |
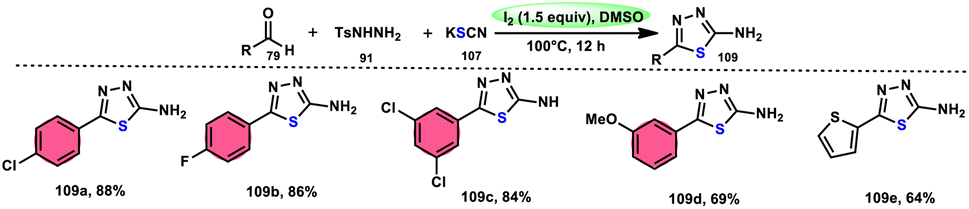
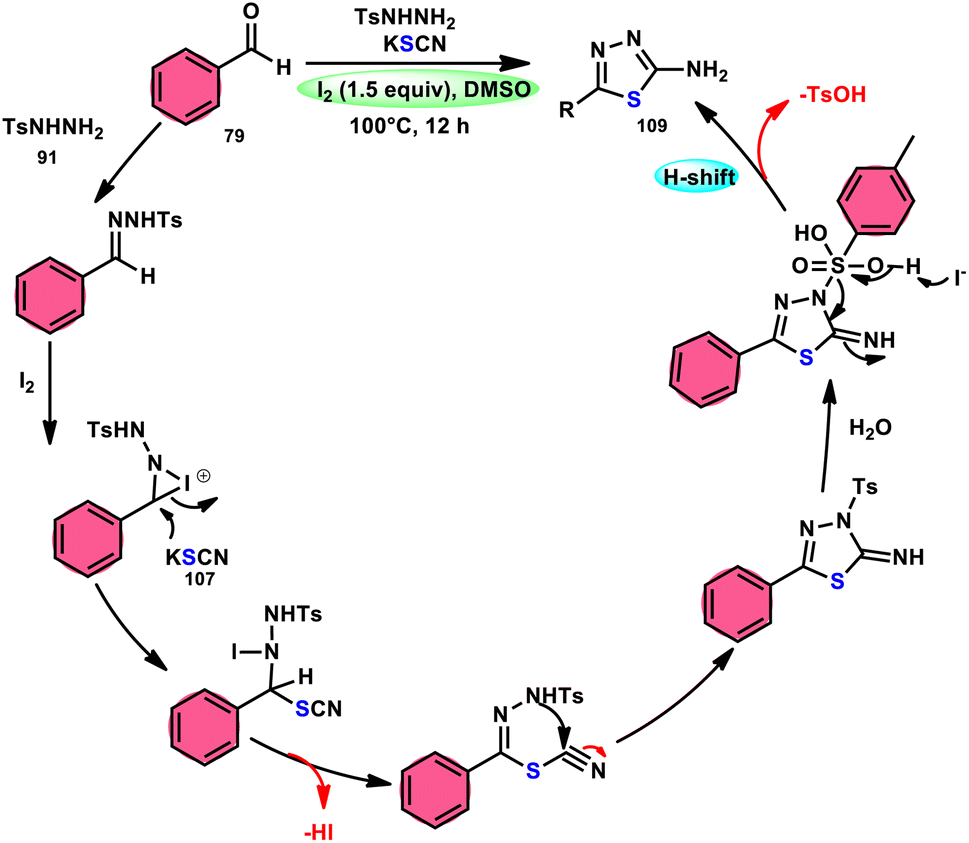
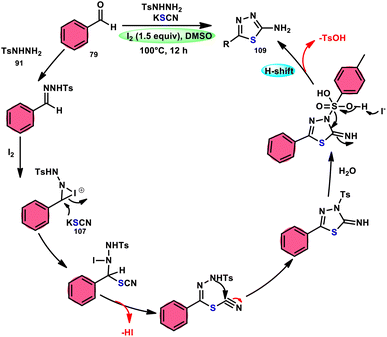
Sen Lin et al.107 designed a unique protocol to synthesize 2-aminothiadiazole 109 employing aldehyde 79, NH2NHTs (p-toluenesulfonyl hydrazide) 91, and KSCN 107, which works as an odourless, effective source of S without emitting toxic cyanide byproduct (Scheme 65). The three-component, metal-free, I2/DMSO-mediated procedure proceeds in a one-pot fashion, producing the target product in satisfactory yields and on a gram scale. Benzaldehydes having EDGs or EWGs, multi-substituents or heteroaryl aldehydes are competent to afford the desired products; however, aliphatic substituents cannot be converted to the target product. According to the plausible reaction mechanism, aldehyde 79 reacts with TsNHNH291 to afford N-tosylhydrazone, which reacts with iodine, resulting in an iodonium salt intermediate. Subsequently, −SCN attacks this intermediate, and in the last step intramolecular cyclization results in the formation of an N–C bond, generating the desired product 109 (Scheme 66).
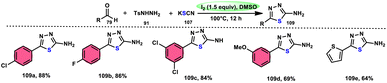 |
| | Scheme 65 Synthesis of aminothiadiazoles 109 catalyzed by I2–DMSO and selected examples. | |
 |
| | Scheme 66 Mechanism for the synthesis of aminothiadiazoles 109 catalyzed by I2–DMSO. | |
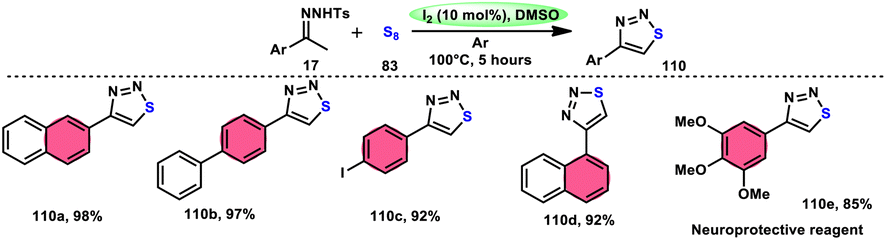
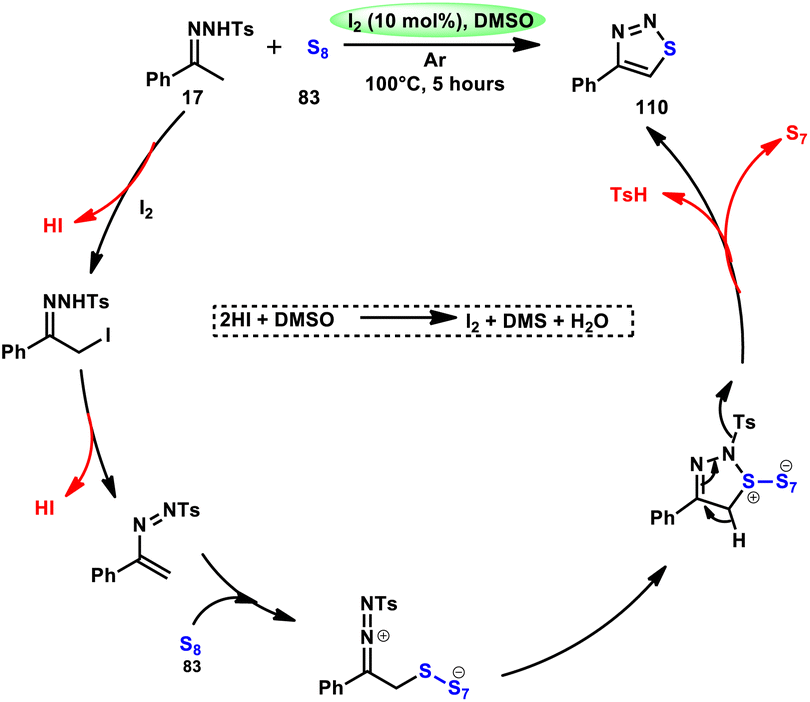
Liu, Gu, and colleagues108 discussed the synthesis of 4-aryl-1,2,3-thiadiazoles 110. They described a selective cyclization process using iodine/DMSO as the catalyst, which enabled the cyclization of N-tosylhydrazones 17 with sulphur 83 without the need for an additional oxidant (Scheme 67). The crucial aspect of this procedure involves the oxidation of HI using DMSO as both the oxidizing agent and solvent. This enabled the retrieval of elemental iodine, thereby confirming the achievement of the synthesis. The distinguishing features of this protocol include its user-friendly nature, efficient utilization of steps (one-pot approach), wide range of applicable substrates, and potential for scalability. This methodology can be employed for the synthesis of compounds on a gram scale. A one-pot synthesis method was also developed, enabling the direct utilization of ketone as a precursor without the need for isolating N-tosylhydrazone intermediate 17. The efficacy of this method was also demonstrated in the production of neuroprotective drug 110e. The utilization of this approach offers a notable advantage given that it eliminates the need for the utilization of external oxidizing agents (Scheme 68).
 |
| | Scheme 67 Synthesis of thiadiazoles 110 using I2/DMSO system. | |
 |
| | Scheme 68 Mechanism for the synthesis of thiadiazoles 110 using I2/DMSO system. | |
9. Other reactions
9.1 Sulfenylation
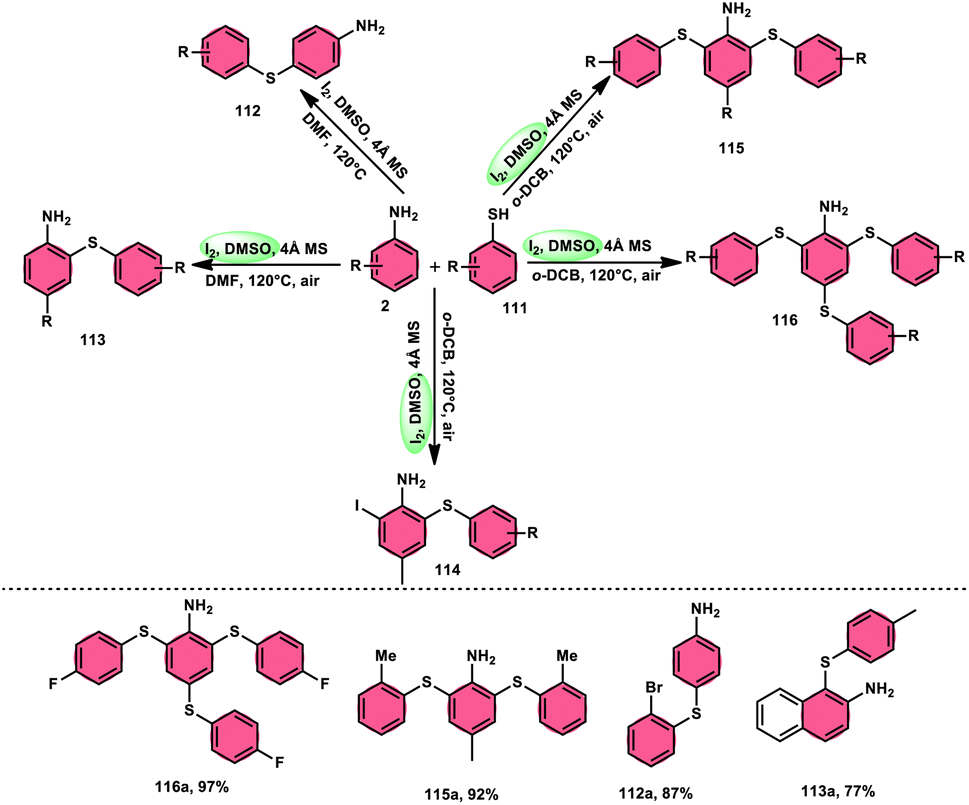
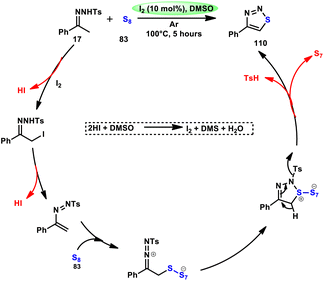
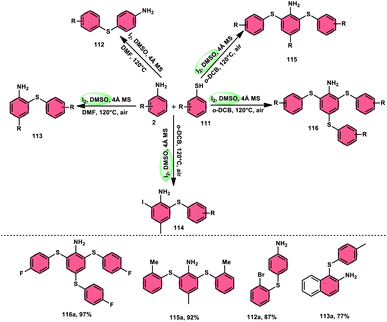
The synthesis of sulfide anilines 112–116 was developed by Zhang, Deng, and research team. This method involves the iodine-promoted divergent thiolation of unprotected anilines 2 using thiols 111.109 The utilization of a mixture comprising I2 and DMSO played a significant role in facilitating this particular reaction, avoiding the use for a metal catalyst or potent oxidizing agents. To produce mono-112–113, bis-115, and trisubstituted diaryl sulphide derivatives 116, the reaction selectivity was carefully regulated. Significantly, the simultaneous iodination and sulfenylation processes can yield very useful iodoaniline molecules that possess several functional groups. Under the conditions of mild reactions, this methodology offers a successful procedure for synthesizing C–S and C–I bonds by the activation of C–H bonds. The oxidative reaction conditions utilizing DMSO as a solvent demonstrated favorable outcomes when applied to synthetically valuable functional groups, such as halogens. Given that DMSO is crucial in this thiolation reaction, the coupling reaction in its absence did not produce any products. The desired compounds were successfully synthesized in high yields through the reaction of aniline 2 with different thiol derivatives 111 possessing methyl and halogen substituents (–F, –Cl, and –Br) on the aromatic ring (Scheme 69).
 |
| | Scheme 69 Reaction of aniline 2 with thiols 111 in a I2/DMSO environment. | |
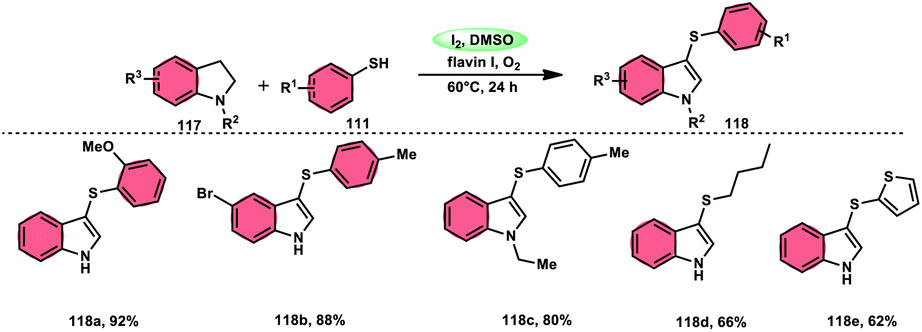
Chuanming Yu and colleagues110 discovered a method for the aerobic oxidative sulfenylation of aryl-fused cyclic amines, using a range of thiols 111 and flavin/I2 as catalysts. The conversion facilitated by flavin II resulted in the replacement of the para-position on the aryl ring with a sulfenyl group, leading to the synthesis of 6-sulfenylquinolines. In contrast, flavin I was shown to act as a catalyst in the sulfenylation process of indolines, resulting in the synthesis of 3-sulfenylindoles 118. The investigation focused on the benefits of using ambient oxygen as the ultimate oxidizing agent in this metal-free oxidative C–S coupling method, which was conducted under ecologically sustainable conditions. The results demonstrated the significant atom efficiency and high degree of compatibility with various functional groups. This study represents the initial occurrence of a sequential process including the dehydrogenation and sulfenylation of indolines, employing thiophenols 111. The procedure presented in this study offers a metal-free approach for the production of environmentally sustainable water given that it is the only by-product, using molecular oxygen as the final oxidizing agent. The intended compounds were successfully synthesized in high yields by the incorporation of several electron-donating groups (–Me, –OMe, and –Ph) and electron-withdrawing groups (–F, –Cl, –Br, and –CF3) at different positions of thiophenol, as well as at the C-2, C-4, C-5, and C-6 positions of the indoline ring (Scheme 70).
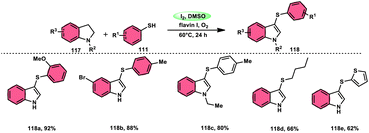 |
| | Scheme 70 Formation of sulfenylindoles 118 using I2/DMSO framework. | |
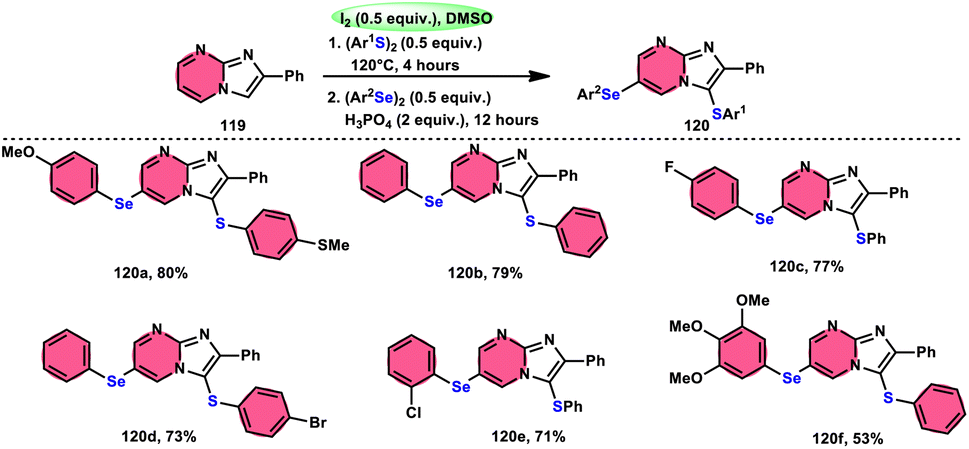
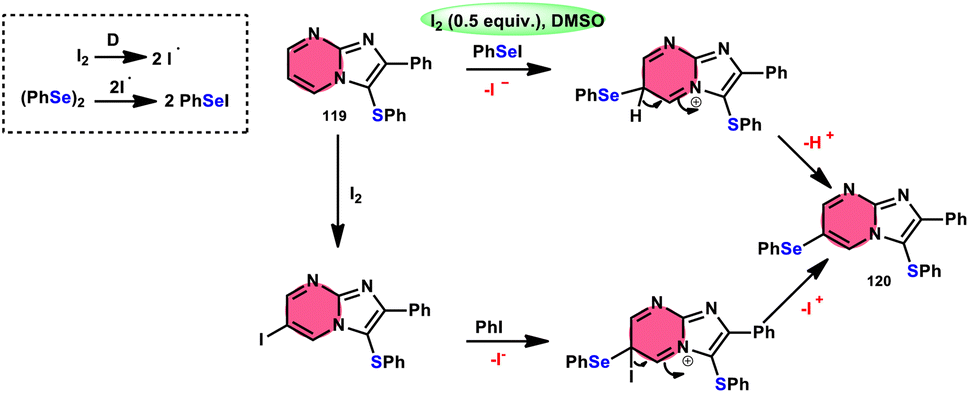
Vanelle, Redon, and colleagues111 documented the process of dichalcogenation of imidazoheterocycles 119, leading to the successful functionalization of the C6-position of the imidazo[1,2-a]pyrimidine moiety 119 (Scheme 71). The process of iodine/DMSO treatment of diaryldichalcogenides commenced with C3-chalcogenation, and then by C6-selanylation of 120, which was facilitated in acidic conditions using phosphoric acid. The addition of stronger acids proved the beneficial effect on the reaction efficiency. This unique stepwise dichalcogenation technique performed well in terms of regioselectivity and yield. However, the yield was considerably degraded (5%) under an inert atmosphere (N2), highlighting the importance of O2 in the process. The use of the C6-halogenated intermediate was shown to provide benefits in promoting cross-coupling events, which resulted in the formation of C–C bonds. The mechanistic studies suggested that C6-selanylation of 120 can be related to electrophilic aromatic substitution (Scheme 72).
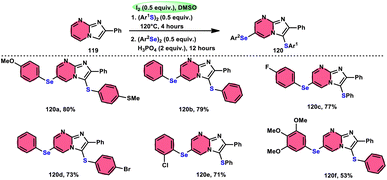 |
| | Scheme 71 I2/DMSO-based dichalcogenation of imidazo[1,2-a]pyrimidine 120. | |
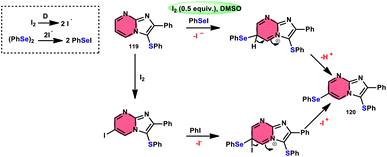 |
| | Scheme 72 I2/DMSO-based dichalcogenation mechanism of imidazo[1,2-a]pyrimidine 120. | |
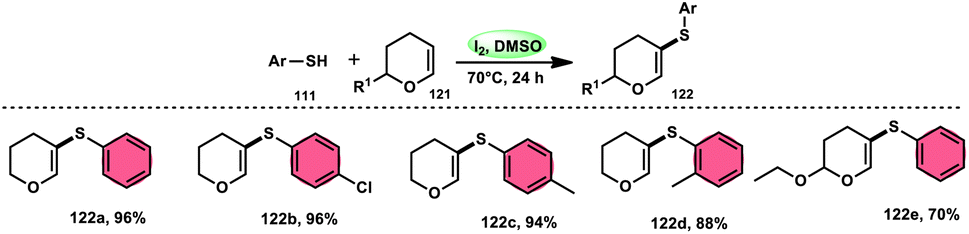
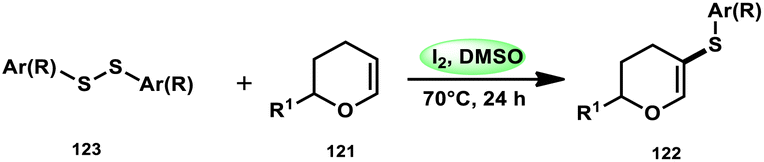
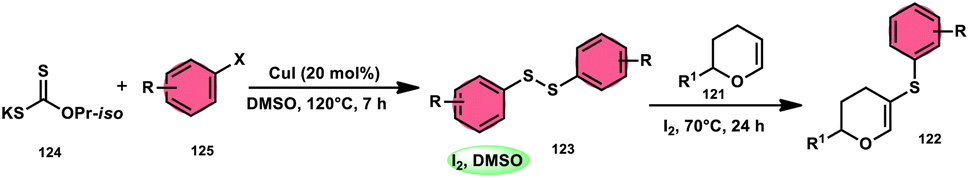
Nejmoh Nowrouzi et al.112 devised a series approach for the sulfenylation of the C(sp2)–H bond of dihydropyrans employing thiols 111, disulfides 123, and aryl halides 125 as co-reagents. The first methodology was carried out in one-pot, straightforward fashion, and without any metal-catalyst using dihydropyrans as the substrate and aromatic thiols or disulfides reactants promoted by the I2/DMSO combination (Schemes 73 and 74). In another repugnant-free methodology, aryl halide 125 was deployed instead of thiols or disulfides and CuI, potassium isopropyl, and molecular I2 worked as catalysts to achieve sulfonated dihydropyrans (Scheme 75). Under all these conditions, the yield was excellent, although aliphatic thiols and aryl chlorides were not potent to afford the desired products 122, and in addition, dihydropyrans bearing EWGs gave lower yields. I2 plays a central role in all these reactions, such as oxidizing thiols first to disulfides, and further converting them to ArSI, making it susceptible to nucleophilic attack by dihydropyran working as a good leaving group.
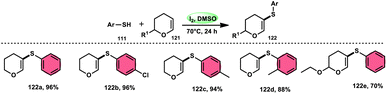 |
| | Scheme 73 Thioarylation of dihydropyrans with different thiols 111. | |
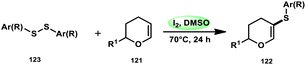 |
| | Scheme 74 Thioarylation of dihydropyrans with disulfides 123. | |
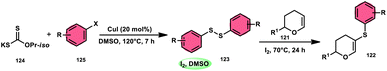 |
| | Scheme 75 Thioarylation of dihydropyrans with aryl halides 125. | |
9.2 Decarbonylation
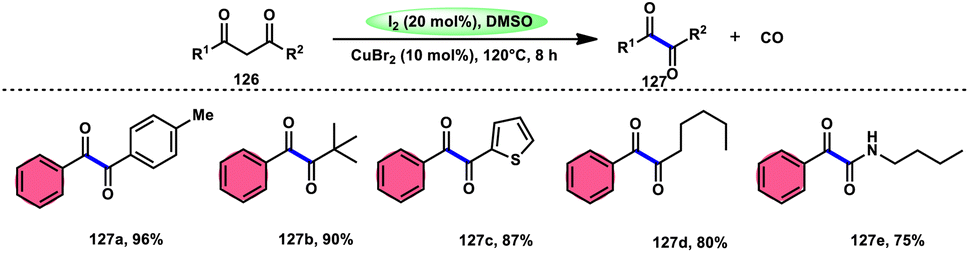
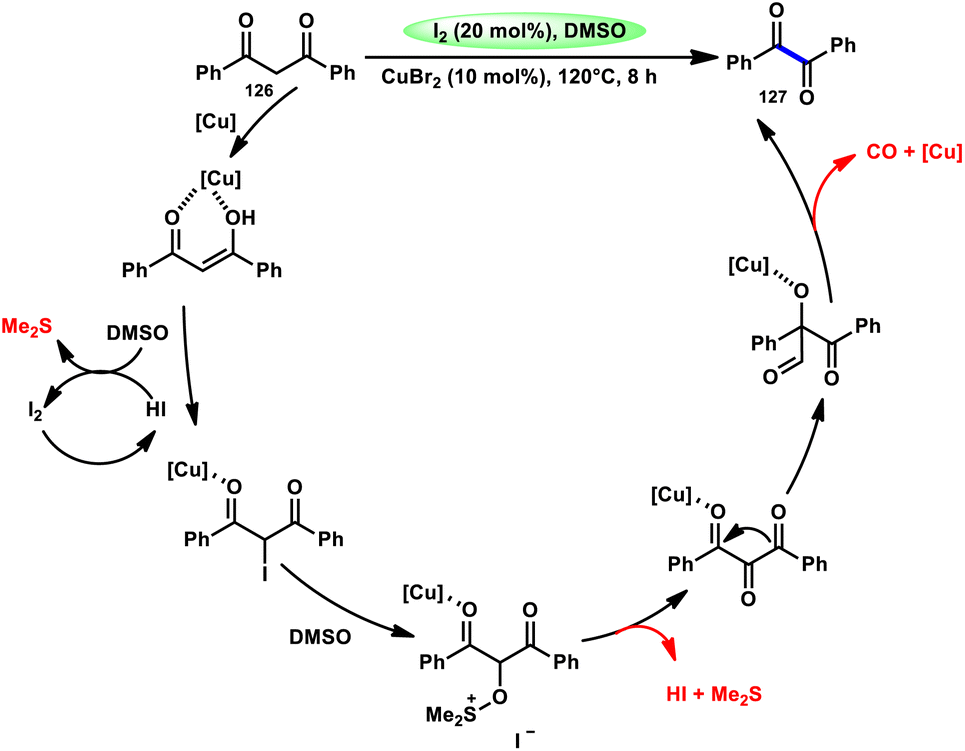
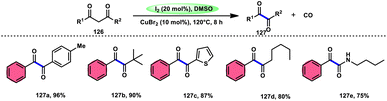
For the oxidative C–C bond breakage of 1,3-dicarbonyl compounds 126, Chen and colleagues113 developed a coordinated catalytic system including CuBr2 and I2 (Scheme 76). The oxidant used in this process was DMSO, resulting in the formation of 1,2-dicarbonyl compounds 127, which hold considerable synthetic value. Significant selectivity was achieved in the breaking of the C–C bonds during the process of carbon monoxide (CO) release when 1,2-dicarbonyl molecules 127 were considered. Moreover, this specific methodology can be used for a wide range of 1,3-dicarbonyl compounds, involving 1,3-diketones, 1,3-keto esters, and 1,3-keto amides. The use of a copper catalyst to improve the reactivity of iodine affords a prospect for the investigation and progression of a novel theoretical approach for cleaving C–C bonds. The observed reaction exhibited a dependence on temperature, whereby a decrease in temperature from 120 °C to either 100 °C or 80 °C resulted in a reduction in the product yields. A variety of substituents, including –Me, –MeO, –Cl, –CO2Me, –F, and –CF3, was shown to be compatible with 1,3-dicarbonyl compounds. The reaction exhibited some sensitivity to both electronic and steric hindrance factors (Scheme 77).
 |
| | Scheme 76 Synthesis of 1,2-dicarbonyl compounds 126 and selected examples. | |
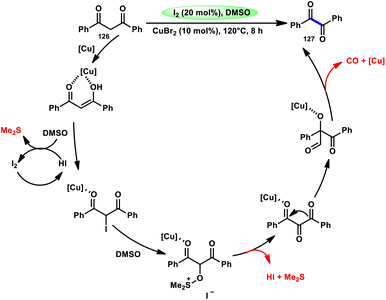 |
| | Scheme 77 Mechanism for the synthesis of 1,2-dicarbonyl compounds 126. | |
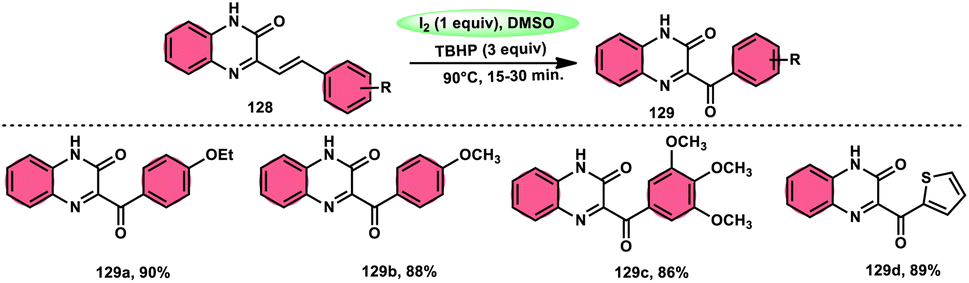
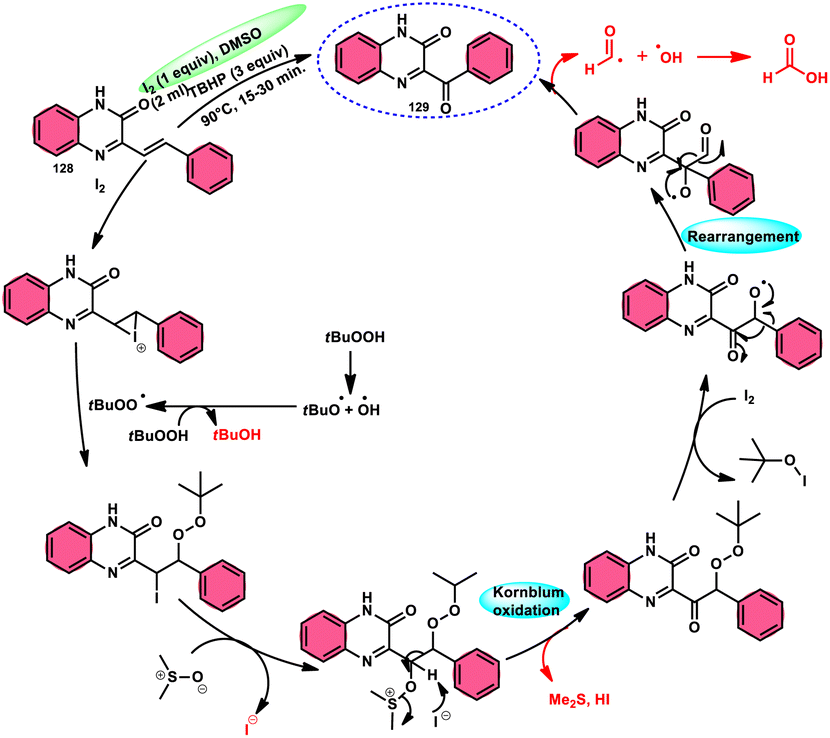
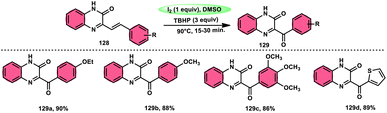
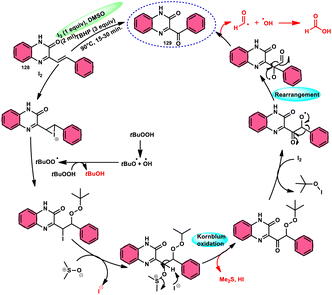
A new approach was developed by Kamal K. Kapoor et al.114 to produce 3-aroylquinoxaline-2(1H)-ones 112 (Scheme 78), which is both efficient and ecologically sustainable, while also being free of any metal components. The researchers used the combination of I2 and DMSO, together with tert-butyl hydroperoxide (TBHP) as a co-oxidant. This particular combination of I2/DMSO/TBHP is widely recognized for its effectiveness in facilitating the oxidative rearrangement of α,β-unsaturated ketones 111. The reaction protocol exhibits favorable tolerance towards a range of styrylquinoxalin-2(1H)-one substrates, resulting in satisfactory yields. It is worth mentioning that substrates including electron-donating groups exhibit greater yields in comparison to that incorporating electron-withdrawing groups. This discovery implies that the reaction initiates with the involvement of electron-rich olefinic groups as nucleophiles. According to the plausible reaction mechanism, the substrate initiates the reactions by attacking I2, generating the iodonium intermediate. The reaction further leads via Kornblum oxidation followed by a radical rearrangement. TBHP (tertiary butyl hydroperoxide) generates the radicals and the reaction continues in a radical manner, resulting in the desired product 112 through a carbon degrading reaction (Scheme 79).
 |
| | Scheme 78 I2/DMSO-mediated metal-free reaction. | |
 |
| | Scheme 79 Mechanistic pathway for I2/DMSO-mediated metal-free reaction mechanism. | |
9.3 α-Oxygenation and deoxygenation
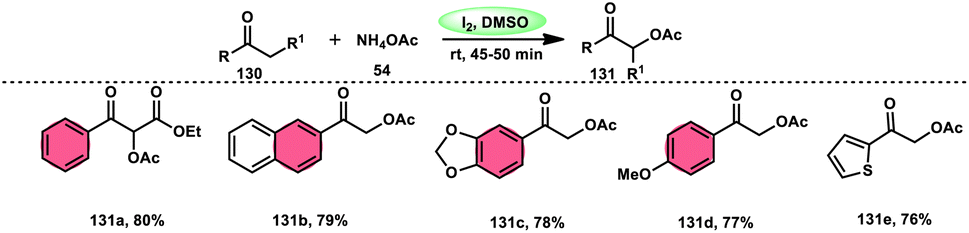
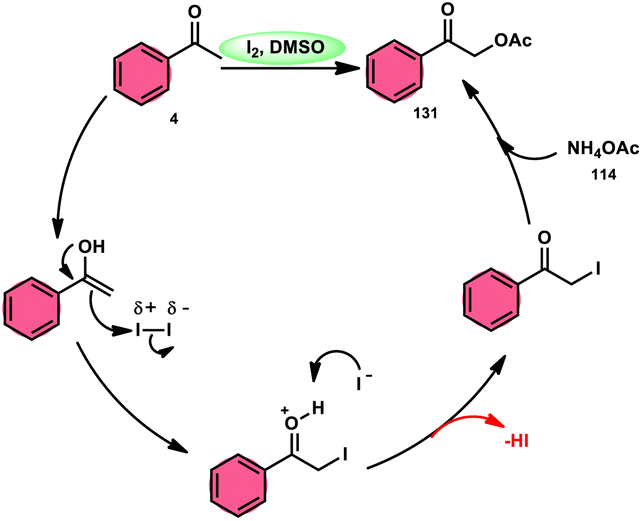
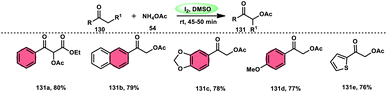
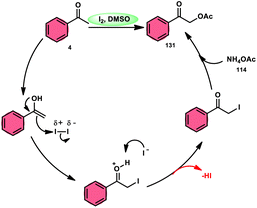
Prapurna et al. presented a procedure for the iodine-mediated oxidative α-functionalization of ketones 130 using NH4OAC 54. This method presents a direct method for the production of α-acetoxy and α-alkoxy ketones 131 using readily available chemicals (Scheme 80).115 The reaction begins with iodination, and then moves to nucleophilic substitution with acetate. The product was produced within one hour of the reaction and completed at room temperature. Under the specified reaction conditions, acetophenone, which has electronic substituents on its phenyl ring, exhibited favourable reactivity, leading to the synthesis of α-keto-acetals in modest yields. Nevertheless, the attempted reactions using cyclohexanone and ethylmethyl ketone did not provide the anticipated products. Consequently, the proposed reaction is a helpful alternative to conventional methods for α-oxygenation due to its simple synthesis method, broad substrate range, and quicker reaction times (Scheme 81).
 |
| | Scheme 80 I2-mediated direct α-functionalization of ketones 113. | |
 |
| | Scheme 81 I2–DMSO-mediated mechanism of direct α-functionalization of ketones 130. | |
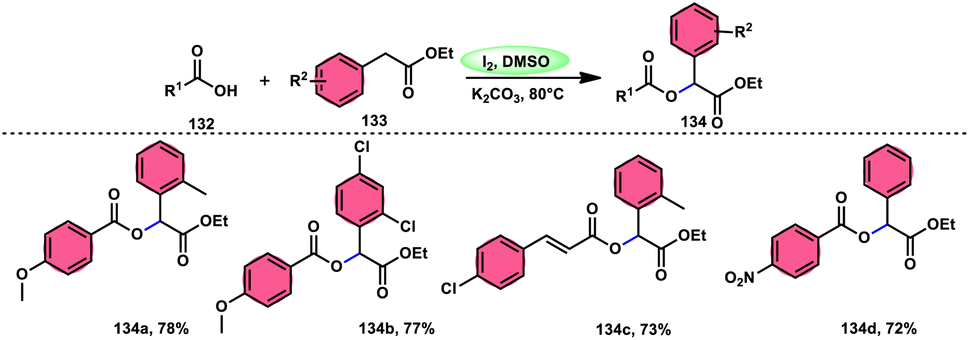
A practical, economic, simple, transition-metal free method of cross dehydrogenative coupling (CDC) promoted by I2/DMSO was developed by the team of Krishan Nand Singh.116 The researchers synthesized α-acyloxy esters derivates 134 from the inexpensive easily accessible reactants aryl carboxylic acid 132 and ethyl arylacetates 133 in basic medium of K2CO3 in the appropriate yield. This methodology has good functional tolerance on both reactants without any effect from EDG and EWG groups. as Also, α,β-unsaturated aryl carboxylic acids were equally compatible; however, aliphatic esters could not produce the desired products. According to the reaction mechanism proposed by the investigators, the ester is converted into an α-iodo ester intermediate, and further a carboxylate ion (originates from the carboxylic acid by K2CO3) participates in nucleophilic substitution attack on the ester intermediate, leading to the desired product (Scheme 82).
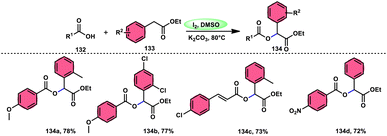 |
| | Scheme 82 Synthesis of α-acyloxy esters 134 and representative examples. | |
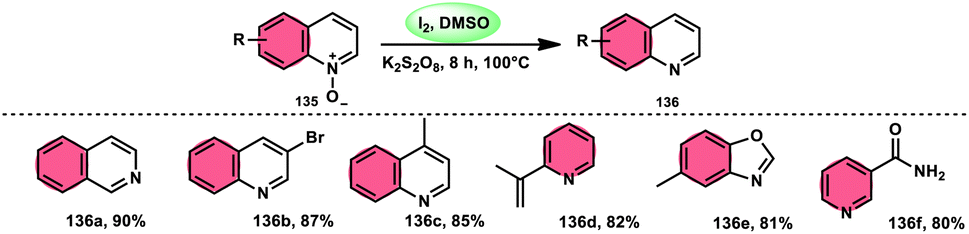
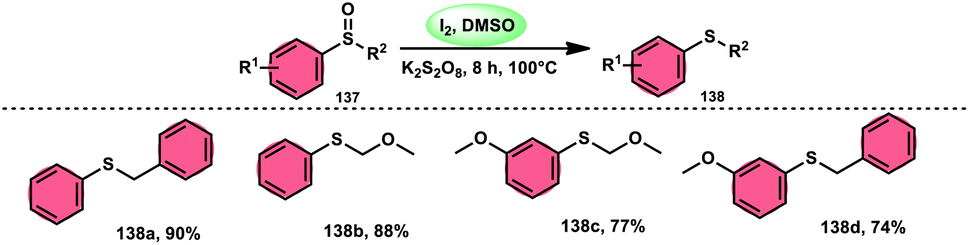
The I2/DMSO combo in presence of K2S2O8 was applied in a different approach to generate amine 136 and sulfides 138 from amine N-oxides 135 (Scheme 83) and sulfoxides (Scheme 84), respectively, by the team of Dushyant Singh Raghuvanshi.117 The metal-free, straightforward, deoxygenative technique has practical value owing to the use of the inexpensive and green I2/K2S2O8 catalyst, its excellent yields, good functional group tolerance, and broad substrate scope including pyridine N-oxides, secondary/tertiary amine N-oxides and aryl sulfoxides. However, the nitro group positioned at C-8 in quinoline was not reduced to the desired products and aliphatic sulfoxides were not compatible with this scheme. The molecular iodine plays a pivotal role in reacting such as an electrophile initiating the reaction, whereas homolysis activities occur with K2S2O8.
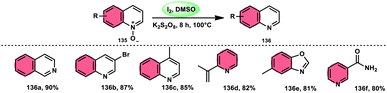 |
| | Scheme 83 Deoxygenation of N-oxides 135 and representative examples. | |
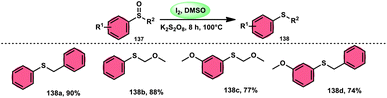 |
| | Scheme 84 Deoxygenation of sulfoxides 137 and representative examples. | |
9.4 Oxidation
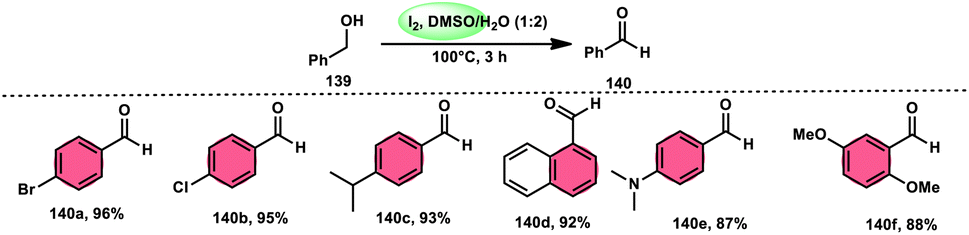
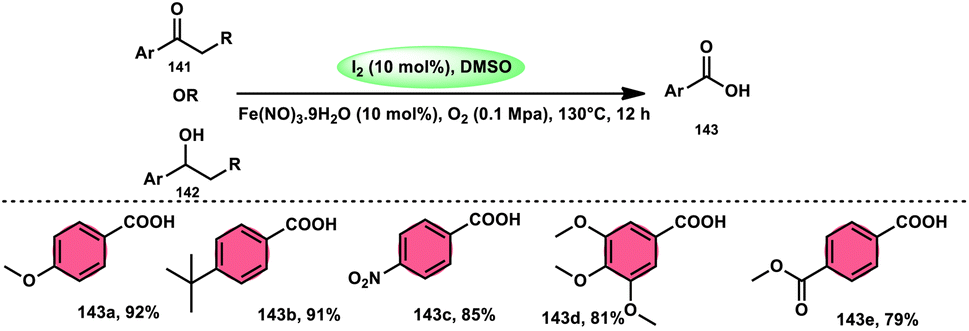
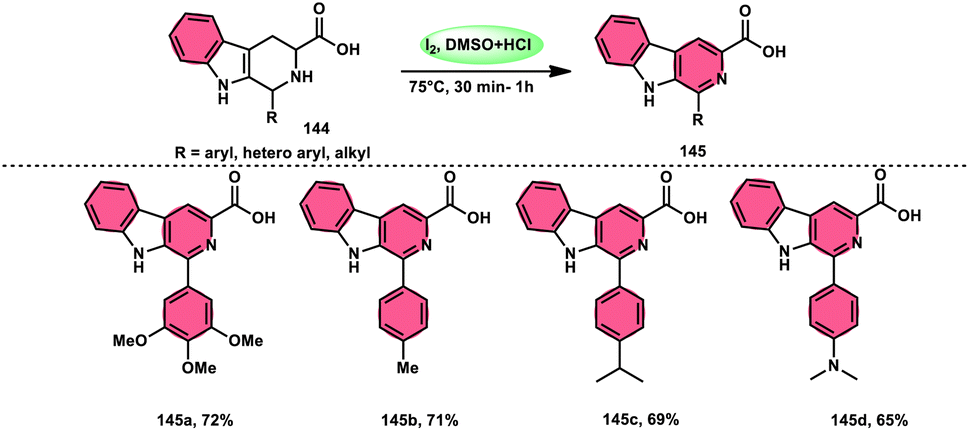
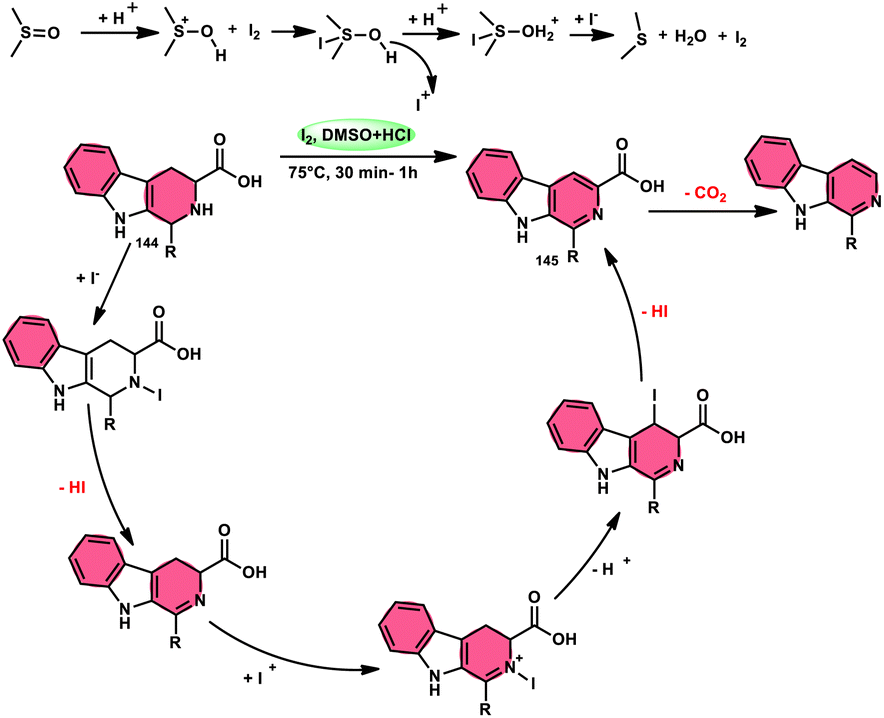
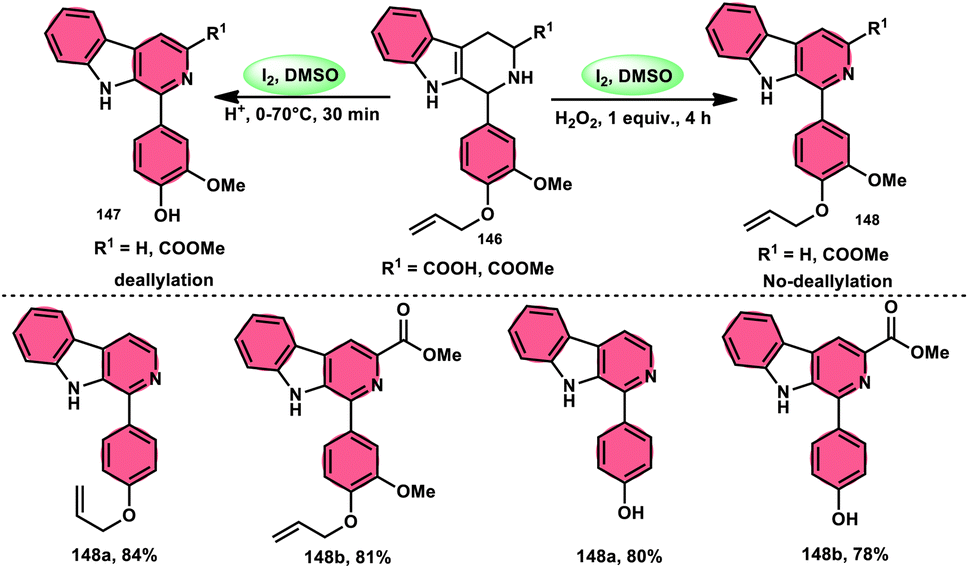
A new, easy, low-cost, environmental-friendly and highly chemoselective and proficient protocol to access benzaldehyde 140 from benzyl alcohol 139 was designed by Ehsan Sheikhi and coworkers.118 Although many known methodologies are applied in the oxidation of the benzylic alcohol 139 to aromatic aldehyde 140, this tactic selectively oxidizes only benzylic alcohol 139, resulting in the formation of the desired product. This strategy has several advantages compared to previous methodologies such as no further purification of the product, no by-product, fascinating yields of up to 96%, no co-catalyst such as N2H4, K2CO3, MeCN and KI are required, where only the readily available DMSO dissolved in water (DMSO:H2O) in a ratio of 1:2 and molecular iodine are required (Scheme 85).
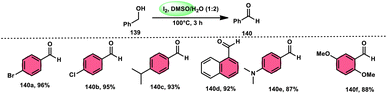 |
| | Scheme 85 I2–DMSO-based chemoselective oxidation of benzylic alcohols 139. | |
Li et al.119 devised an economically viable, uncomplicated, ecologically sustainable, and feasible methodology for synthesizing carboxylic acids 143. This method employs I2/Fe(NO3)3·9H2O as the catalysts and DMSO/O2 as the oxidants. This method utilized aryl alkyl ketones 141 or sec-benzylic alcohols 142 as feedstocks. It has good substrate scope, covering various substituted aryl alkyl ketones to heteroaryl alkyl ketones; however, to avoid polymerization reactions, the temperature must not be above 110 °C when heteroaryl is employed as the substrate. The approach used in this study demonstrates favourable to outstanding yields. However, it was noted that substrates containing electron-donating groups (EDGs) exhibit greater yields compared to substrates containing electron-withdrawing groups (EWGs). According to the precise analysis of the reaction mechanism by the researchers, initially, both types of substrates are converted into phenylglyoxal via Kornblum oxidation, whereas Fe3+ smoothly cleaves the C–C bond of phenylglyoxal, resulting in the formation of HCOOH as the by-product and benzaldehyde, which is oxidized by O2 to carboxylic acids. Interestingly, the use of N2 instead of O2 resulted in the disproportion of the ratio of desired products and by-products (Scheme 86).
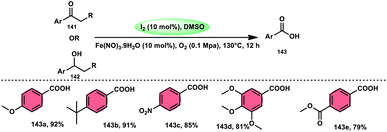 |
| | Scheme 86 Oxidation of aryl methyl ketones 141 by I2/Fe(NO3)3·9H2O. | |
9.5 Aromatization
Bhosale, Lokhande, and colleagues120 developed a new catalytic method utilizing I2/DMSO for the selective aromatization of tetrahydro-β-carboline-3-carboxylic acids (THβC-carboxylic acids 144), aiming to produce β-carboline-3-carboxylic acid 145 (Scheme 87). This approach has the advantage of utilizing easily accessible starting materials. This study presents a metal-free aromatization methodology that includes a controlled decarboxylation phase. The developed approach offers several advantages, including mild reaction temperatures, convenient handling, and the utilization of readily accessible chemicals. In comparison to conventional approaches, using I2/DMSO to produce HI and I+ is a more environmentally friendly process. Additionally, this technique can be improved to aromatize new hybrid β-carboline compounds 145 and THβC methyl esters with great yield. Density functional theory (DFT) calculations were employed to investigate the process in a cost-effective manner. The chemo selectivity of β-carboline-3-carboxylic acid 145 was further validated using computational analysis. This research revealed that the carboxylic acid group was retained, leading to the significant production of β-carboline-3-carboxylic acid as the primary product 145 (Scheme 88).
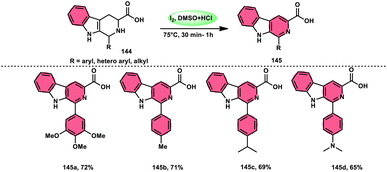 |
| | Scheme 87 I2/DMSO-based chemoselective mechanism. | |
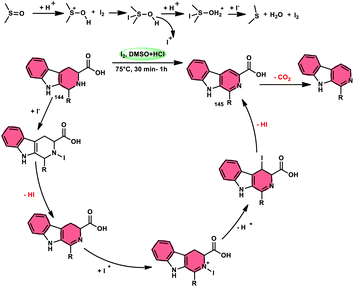 |
| | Scheme 88 I2/DMSO-based chemoselective reaction and representative examples. | |
Sunil V. Gaikwad and others27 designed a metal-free, green, economical, fast, and low temperature-based approach for the chemoselective aromatization tetrahydro-β-carboline (THβC) 147, as well as providing the option of obtaining deallylated or non-deallylated 148 of THβC. Catalyst I2 at 100% mol, DMSO as an oxidant, and H2O2 as an external oxidant all contributed to the high yield of the desired products. This methodology was employed to successfully synthesize flavones and has high functional tolerance, demonstrating its usefulness. The deallylation work-up occurred by adding just a drop of HCl to the reaction mixture. Overall, this strategy was proven to be superior to the conventional methods owing to its eco-friendly nature, low cost, shorter reaction time, and practical value (Scheme 89).
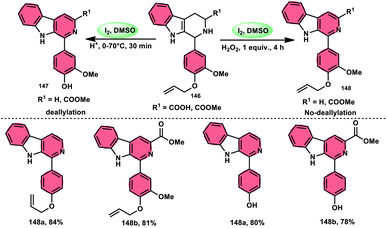 |
| | Scheme 89 Deallylation of O-allyl tetrahydro-β-carboline 146. | |
The synthesis of aryl alkyl ethers 150 through a new, metal-free, simple, and economical protocol was possible by employing commercially available nonaromatic cyclohexenones 149 and alcohols 139 (Scheme 90). In this study Jiao and colleagues121 used an approach using I2/DMSO, whereby molecular iodine serves as the catalyst and DMSO acts as the oxidant, facilitating the regeneration of I2 from I−. The broad substrate scope of alcohols 139 and compatibility of cyclohexenones 149 bearing various substituents to afford the desired products show the excellent substrate and functional group tolerance of this methodology. Besides, meta-substituted aromatic ethers are smoothly prepared using this strategy, which is tedious by conventional methods. According to the proposed reaction mechanism, iodine enhances the electrophilicity of the carbonyl group, leading to the aromatization of cyclohexenone 149via iodination; however, the reaction mechanism requires further investigation (Scheme 91).
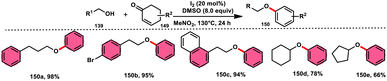 |
| | Scheme 90 Coupling of cyclohexenones 149 and alcohols 139 catalyzed by I2–DMSO. | |
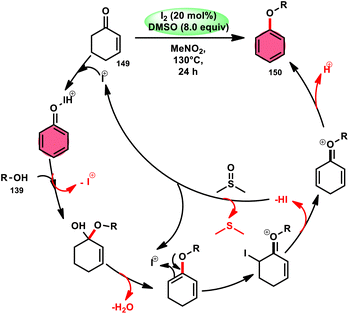 |
| | Scheme 91 I2–DMSO-catalyzed mechanism of the reaction. | |
9.6 Amidation
The oxidative amidation of the methyl ketone to obtain α-ketoamides 152 in a unique, effective, atom-economical, and metal-free manner was developed by An-Xin Wu and coworkers (Scheme 92).122 This methodology promoted by I2/DMSO employs methyl ketones 4 and anthranils 151 as feedstock where anthranils 151 works as a masked N-nucleophile, with the addition of trifluoromethanesulfonic acid (TfOH) enhance the yield to a satisfactory level. Iodine plays a vital role in transforming the methyl ketone to phenylglyoxal via Kornblum oxidation and in situ generating 2-amino-4-iodobenzaldehyde to form anthranils via reductive ring opening besides iodination of this intermediate at 4-position. This process exhibits favourable functional group tolerance and a wide range of substrates in relation to anthranils and methyl ketones. However, it was observed that methyl ketones with electron-donating groups (EDGs) yielded higher quantities compared to ketones with electron-withdrawing groups (EWGs) on aryl rings. Regrettably, aliphatic ketones are not viable for producing the required products. This methodology provides further scope for the transformation of good target products bearing iodo and formyl groups. The iodo and formyl groups of the desired product N-(2-formyl-4-iodophenyl)-2-oxo-2-phenylacetamide further extend the scope of various important transformation (Scheme 93).
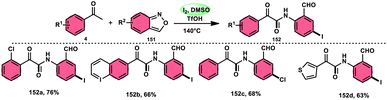 |
| | Scheme 92 I2/DMSO-catalyzed synthesis of α-ketoamides 152 and representative examples. | |
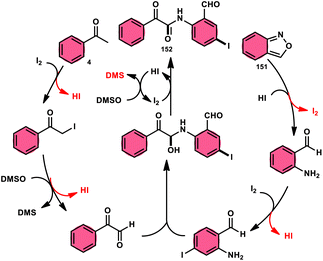 |
| | Scheme 93 I2/DMSO-catalyzed mechanism of α-ketoamides 152. | |
Sakram Boda et al.123 devised a facile, metal-free, environmental-benign technique to afford amide employing I2/TBHP as a catalyst in DMSO solvent. The reaction proceeds under mild reaction conditions utilizing carboxylic acid 153 and amines 2 as feedstock. Additionally, this methodology has a broad substrate scope covering aliphatic, aryl, bulky carboxylic acids and amines substituted with various functional groups (Scheme 94), and even thioglycolic acids 155 and phenoxy acetic acids were well tolerated in the reaction (Scheme 95). The feasible reaction starts by the reaction of TBHP and I2, forming the tertiary-butoxy radical, which abstracts H from acid and generates a carboxy radical, further attacking I2 to generate acetic hypoiodous anhydrides (ArCOOI). In conclusion, amine nucleophilic attacking acetic hypoiodous anhydrides generate amide 154 and HOI 156 as the final products.
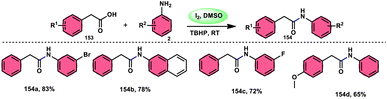 |
| | Scheme 94 I2–DMSO-catalyzed synthesis of amides 154 and representative examples. | |
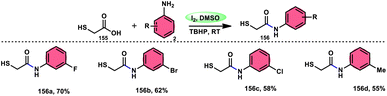 |
| | Scheme 95 Synthesis of amides 156 using thioglycolic acids 155. | |
9.7 Selenization
The emerging catalytic combo I2/DMSO was applied by Julian B. Azeredo and coworkers124 to pursue the asymmetric alkoxy-selenization of styrene 27 and its derivatives in a metal-free, solvent-free, and environmental-benign methodology. These researchers detected that [(1S)-1-(methylthio)ethyl]phenyl diselenide 157 was the most effective non-racemic chiral diselenide reagent, and methanol works as the best alcohol, although menthol behaving as an alcohol source also shows feasibility of using natural products. The investigating team revealed that the high yield and diastereoisomeric excess resulting from [(1S)-1-(methylthio)ethyl]phenyl diselenide were attributed to the interaction between the S and Se atom, which brings the stereogenic center closer to the reaction center to form R–Se–I complex. It was further proven by ORTEP analysis that employing the diselenide reagent bearing an N-atom results in the formation of the N–Se–I complex (Scheme 96). The Se atom behaving as an electrophilic center directs the protocol in a Markovnikov regiochemistry manner, producing asymmetric alkoxy-selenized 160. The targeted products could be achieved using either microwave irradiation or heat as the energy source, providing equal results (Scheme 97).
 |
| | Scheme 96 N–Se–I interactions detected by ORTEP analysis. | |
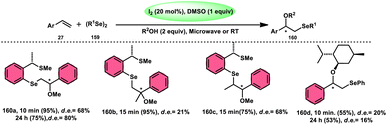 |
| | Scheme 97 I2–DMSO-catalyzed alkoxy-selenization of styrenes 27. | |
In the research by Zhu, Sun, and colleagues,125 they developed a new approach employing an iodine-promoted oxidative domino annulation and carbonylation methodology. The use of this approach facilitates the production of physiologically significant aza-arene-substituted bis pyrazolo pyridines (BPPs) 163, o-amino diheteroaryl ketones 164, and diuracilpyridines (Scheme 98). Using readily available 5-aminouracils 161, modified 5-aminopyrazoles 162, and methyl aza-arenes 101, the domino process was carried out. This basic approach does not require the use of metals and it works well with numerous different substrates and functional groups. Additionally, this process can be exploited to synthesize gram-scale dipyrazolo/diuracil-fused pyridines 163 and 164. Substituted methyl quinolines incorporating both electron-donating and electron-removing groups exhibit a remarkable degree of tolerance.
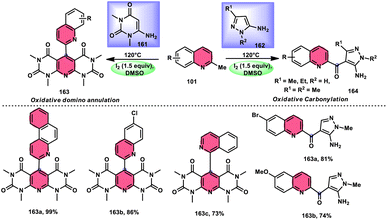 |
| | Scheme 98 Synthesis of substituted pyridines 163 and diheteroaryl ketones 164. | |
Zheng Fang, Kai Guo and coworkers126 developed a practical, environmentally friendly, fast, metal- and amine catalyst-free, two-step continuous flow reaction strategy to prepare C-3 dicarbonyl indole derivates 166 from phenylacetaldehyde 165 and N-methyl indole 14 promoted by I2/DMSO. This approach also offers potential for synthesizing α-ketoamides127167 and α-ketoesters 168 simultaneously under the same reaction conditions, utilizing secondary amines and alcohols, respectively, which demonstrates its broad application. The reaction occurs in a two-step process within two microreactors to achieve a maximum yield of up to 91%. In the first step, phenylacetaldehyde 165 undergoes oxidation to form phenylglyoxal through the Kornblum oxidation method. In the subsequent step, indole is continuously introduced via a syringe, where it reacts with the iodine-activated aldehyde of phenylglyoxal to form a benzoin intermediate. This intermediate is then oxidized to yield the desired product. The research team employed a DMSO18-labelled experiment to establish the reaction mechanism. The methodology has good functional group tolerance for the substrates, where EDGs and EWGs are equally tolerated on both substrates; however, primary amine and aniline could not be converted into α-ketoamide 167 (Scheme 99).
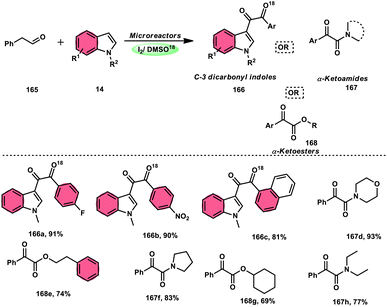 |
| | Scheme 99 Selective oxidative coupling reactions facilitated by I2/DMSO. | |
10. Conclusion
This review presented the broad applicability of I2/DMSO working not merely as green oxidative agent but also a versatile catalyst–solvent combination having good compatibility for the formation of various important and diverse bonds, providing more beneficial paths to generate various important compounds. This review also highlighted the recent trends of employing transition metals, Fe/Cu, etc. as co-catalysts, which enables more proficient methodologies in achieving various significant compounds. Besides, I2/DMSO working as a solvent-free catalytic system is equally effective in enabling the formation of C–Se bonds under metal-free reaction conditions. Overall, this review will help readers understand the tendencies, reaction mechanism, and applicability of I2/DMSO without any metal catalyst or in the presence of metal catalysts. In this work, the synthetic progress made possible by the combination of DMSO and molecular iodine was highlighted. The majority of the biologically active compounds was produced synthetically, and these developments have had a significant impact on their preparation. Thus, the I2/DMSO catalytic system is anticipated to be used for the synthesis of significant structures and frameworks with biological activities in the future. This will enhance the application value of heteroatomic molecules in medicine, agricultural chemicals, materials, and fine chemicals. Nevertheless, the I2/DMSO catalytic system typically requires an elevated reaction temperature and its reaction activity is subject to certain limitations. Accordingly, further research is required to enhance these synthetic approaches for the production of new molecules with distinct pharmacological properties. For instance, the appropriate incorporation of certain additives may induce a reaction at standard ambient temperature.
Also, additional thorough studies are required to further enhance the reaction system and develop more effective catalytic reactions. In the future, it is anticipated that the use of I2/DMSO, an environmentally friendly catalytic system, will be expanded in organic synthesis to create a variety of beneficial heterocyclic compounds with biological properties.
Conflicts of interest
There are no conflicts to declare.
References
- A. Monga, S. Bagchi and A. Sharma, New J. Chem., 2018, 42, 1551–1576 RSC.
- J.-Q. Wang, Z.-Y. Zuo and W. He, Catalysts, 2022, 12, 821 CrossRef.
- H. Zhang, H. Wang, Y. Jiang, F. Cao, W. Gao, L. Zhu, Y. Yang, X. Wang, Y. Wang, J. Chen, Y. Feng, X. Deng, Y. Lu, X. Hu, X. Li, J. Zhang, T. Shi and Z. Wang, Chem.–Eur. J., 2020, 26, 17289–17317 CrossRef CAS PubMed.
- Monika, Chander, S. Ram and P. K. Sharma, Asian J. Org. Chem., 2023, 12, e202200616 CrossRef CAS.
- N. Chatterjee and A. Goswami, Tetrahedron Lett., 2015, 56, 1524–1527 CrossRef CAS.
- J. V. Matlock, T. D. Svejstrup, P. Songara, S. Overington, E. M. McGarrigle and V. K. Aggarwal, Org. Lett., 2015, 17, 5044–5047 CrossRef CAS.
- P. N. Reddy, B. V. S. Reddy and P. Padmaja, Curr. Org. Synth., 2018, 15, 815–838 CrossRef CAS.
- Y.-F. Liang, K. Wu, S. Song, X. Li, X. Huang and N. Jiao, Org. Lett., 2015, 17, 876–879 CrossRef CAS.
- L. Rosenfeld, J. Chem. Educ., 2000, 77, 984 CrossRef CAS.
- R. Singhal, S. P. Choudhary, B. Malik and M. Pilania, Org. Biomol. Chem., 2023, 21, 4358–4378 RSC.
- R. Singhal, B. Malik and M. Pilania, AIP Conf. Proc., 2023, 2723 Search PubMed.
- P. T. Parvatkar, R. Manetsch and B. K. Banik, Chem.–Asian J., 2019, 14, 6–30 CrossRef CAS.
- P. Chauhan, S. Mahajan and D. Enders, Chem. Rev., 2014, 114, 8807–8864 CrossRef CAS PubMed.
- C. Shen, P. Zhang, Q. Sun, S. Bai, T. A. Hor and X. Liu, Chem. Soc. Rev., 2015, 44, 291–314 RSC.
- M. S. Yusubov and V. V. Zhdankin, Resour.-Effic. Technol., 2015, 1, 49–67 Search PubMed.
- F. C. Küpper, M. C. Feiters, B. Olofsson, T. Kaiho, S. Yanagida, M. B. Zimmermann, L. J. Carpenter, G. W. Luther III, Z. Lu and M. Jonsson, Angew. Chem., Int. Ed., 2011, 50, 11598–11620 CrossRef.
- E. A. Merritt, V. M. Carneiro, L. F. Silva Jr and B. Olofsson, J. Org. Chem., 2010, 75, 7416–7419 CrossRef CAS.
- X. Wang, F. Yan and Q. Wang, Synth. Commun., 2021, 51, 1763–1781 CrossRef CAS.
- J. Zhao and X. Jiang, Chin. Chem. Lett., 2018, 29, 1079–1087 CrossRef CAS.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, J. Med. Chem., 2014, 57, 2832–2842 CrossRef CAS.
- X. Li, X. Wang, Y. Li, J. Xiao and Y. Du, Org. Biomol. Chem., 2022, 20, 4471–4495 RSC.
- P. Zhao, X. Wu, Y. Zhou, X. Geng, C. Wang, Y.-d. Wu and A.-X. Wu, Org. Lett., 2019, 21, 2708–2711 CrossRef CAS.
- C. Jacob, Nat. Prod. Rep., 2006, 23, 851–863 RSC.
- M. E. Cinar and T. Ozturk, Chem. Rev., 2015, 115, 3036–3140 CrossRef CAS PubMed.
- M. Feng, B. Tang, S. H Liang and X. Jiang, Curr. Top. Med. Chem., 2016, 16, 1200–1216 CrossRef CAS.
- J. V. Matlock, S. P. Fritz, S. A. Harrison, D. M. Coe, E. M. McGarrigle and V. K. Aggarwal, J. Org. Chem., 2014, 79, 10226–10239 CrossRef CAS.
- S. V. Gaikwad, M. V. Gaikwad and P. D. Lokhande, J. Heterocycl. Chem., 2021, 58, 1408–1414 CrossRef CAS.
- B. Deka, G. K. Rastogi, M. L. Deb and P. K. Baruah, Top. Curr. Chem., 2022, 380, 1–38 CrossRef CAS.
- Y.-F. Liang, X. Li, X. Wang, M. Zou, C. Tang, Y. Liang, S. Song and N. Jiao, J. Am. Chem. Soc., 2016, 138, 12271–12277 CrossRef CAS.
- S. A. Rather, A. Kumar and Q. N. Ahmed, Chem. Commun., 2019, 55, 4511–4514 RSC.
- J. Jayram, B. A. Xulu and V. Jeena, Tetrahedron, 2019, 75, 130617 CrossRef.
- C. Xie, Z. Zhang, B. Yang, G. Song, H. Gao, L. Wen and C. Ma, Tetrahedron, 2015, 71, 1831–1837 CrossRef CAS.
- Y.-F. Liang, S. Song, L. Ai, X. Li and N. Jiao, Green Chem., 2016, 18, 6462–6467 RSC.
- G. Xu, J. P. Wu, X. M. Ai and L. R. Yang, Chin. Chem. Lett., 2007, 18, 643–646 CrossRef CAS.
- A. A. Malik, N. A. Dangroo and T. Ara, ChemistrySelect, 2020, 5, 12965–12970 CrossRef CAS.
- N. Kornblum, J. W. Powers, G. J. Anderson, W. J. Jones, H. O. Larson, O. Levand and W. M. Weaver, J. Am. Chem. Soc., 1957, 79, 6562 CrossRef CAS.
- C. F. Brayton, Cornell Vet., 1986, 76, 61–90 CAS.
- B. Swanson, Rev. Clin. Basic Pharmacol., 1985, 5, 1–33 CAS.
- J. Galvao, B. Davis, M. Tilley, E. Normando, M. R. Duchen and M. F. Cordeiro, FASEB J., 2014, 28, 1317–1330 CrossRef CAS.
- K. V. Balakin, N. P. Savchuk and I. V. Tetko, Curr. Med. Chem., 2006, 13, 223–241 CrossRef CAS.
- D. S. Raghuvanshi, N. Verma and A. Gupta, ChemistrySelect, 2019, 4, 2075–2078 CrossRef CAS.
- S. Liang, K. Hofman, M. Friedrich, J. Keller and G. Manolikakes, ChemSusChem, 2021, 14, 4878–4902 CrossRef CAS PubMed.
- O. M. Mulina, A. I. Ilovaisky, V. D. Parshin and A. O. Terent'ev, Adv. Synth. Catal., 2020, 362, 4579–4654 CrossRef CAS.
-
J. B. Azeredo, A. L. Braga and C. Santi, 1st International Electronic Conference on Catalysis Sciences, 2020, vol. 10, p. 30 Search PubMed.
- S. Ambethkar, M. Kalaiselvi, J. Ramamoorthy and V. Padmini, ACS Omega, 2018, 3, 5021–5028 CrossRef CAS PubMed.
- K. Donthiboina, G. S. Mani, N. Shankaraiah and A. Kamal, ChemistrySelect, 2020, 5, 3923–3928 CrossRef CAS.
- C. Cimarelli, M. Di Nicola, S. Diomedi, R. Giovannini, D. Hamprecht, R. Properzi, F. Sorana and E. Marcantoni, Org. Biomol. Chem., 2015, 13, 11687–11695 RSC.
- R. Deshidi, S. Devari and B. A. Shah, Eur. J. Org Chem., 2015, 2015, 1428–1432 CrossRef CAS.
- D. Liu and A. Lei, Chem.–Asian J., 2015, 10, 806–823 CrossRef CAS PubMed.
- A. Jana, P. Bhaumick, A. K. Panday, R. Mishra and L. H. Choudhury, Org. Biomol. Chem., 2019, 17, 5316–5330 RSC.
- Y.-M. Ren, C. Cai and R.-C. Yang, RSC Adv., 2013, 3, 7182–7204 RSC.
- M. Saroha, J. Sindhu, S. Kumar, K. K. Bhasin, J. M. Khurana, R. S. Varma and D. Tomar, ChemistrySelect, 2021, 6, 13077–13208 CrossRef CAS.
- S. Ambethkar, M. Kalaiselvi, J. Ramamoorthy and V. Padmini, ACS Omega, 2018, 3, 5021–5028 CrossRef CAS PubMed.
- X.-X. Yu, P. Zhao, Y. Zhou, C. Huang, L.-S. Wang, Y.-D. Wu and A.-X. Wu, J. Org. Chem., 2021, 86, 8381–8388 CrossRef CAS PubMed.
- Q. Cai, S. Zhuang, M. Yang, N. Peng, Y. Liu and A. Wu, Tetrahedron, 2019, 75, 130756 CrossRef.
- R.-R. Zhang, J. Liu, Y. Zhang, M.-Q. Hou, M.-Z. Zhang, F. Zhou and W.-H. Zhang, Eur. J. Med. Chem., 2016, 116, 76–83 CrossRef CAS.
- N. Pathan, P. Ali, A. Rahatgaonkar and K. Al-Mousa, J. Heterocycl. Chem., 2021, 58, 1675–1689 CrossRef CAS.
- B. Sridevi, S. Reddy Kandimalla and B. Subba Reddy, Eur. J. Org Chem., 2019, 2019, 6800–6806 CrossRef CAS.
- Y. Zhou, P. Zhao, L.-S. Wang, X.-X. Yu, C. Huang, Y.-D. Wu and A.-X. Wu, Org. Lett., 2021, 23, 6461–6465 CrossRef CAS PubMed.
- J. Yan, Z. Fang, J. Su, Q. He, N. Al-Maharik, Q. Zhang, Y. Wei and D. Li, Org. Chem. Front., 2023, 10, 6192–6199 RSC.
- K. Hu, Y. Zhang, Z. Zhou, Y. Yang, Z. Zha and Z. Wang, Org. Lett., 2020, 22, 5773–5777 CrossRef CAS PubMed.
- S. Badigenchala, V. Rajeshkumar and G. Sekar, Org. Biomol. Chem., 2016, 14, 2297–2305 RSC.
- S. K. Samanta and M. K. Bera, Org. Biomol. Chem., 2019, 17, 6441–6449 RSC.
- S. Bhukta, R. Chatterjee and R. Dandela, Synthesis, 2023, 55, 846–856 CrossRef CAS.
- J. Feng, T. He, Y. Xie, Y. Yu, J. B. Baell and F. Huang, Org. Biomol. Chem., 2020, 18, 9483–9493 RSC.
- Z. Dai, S. Li, Y. Li, L. Feng and C. Ma, Tetrahedron, 2019, 75, 2012–2017 CrossRef CAS.
- M. Abdullaha, S. Mohammed, M. Ali, A. Kumar, R. A. Vishwakarma and S. B. Bharate, J. Org. Chem., 2019, 84, 5129–5140 CrossRef CAS PubMed.
- N. Jatangi and R. K. Palakodety, Org. Biomol. Chem., 2019, 17, 3714–3717 RSC.
- S. Wen, Y. Du, Y. Liu, X. Cui, Q. Liu and H. Zhou, Eur. J. Org Chem., 2022, 2022, e202101187 CrossRef CAS.
- S. A. Bhat, M. Y. Bhat, S. A. Rather, S. Jameel, K. A. Bhat and Q. N. Ahmed, Org. Lett., 2023, 25, 2382–2387 CrossRef CAS PubMed.
- P. A. Sathe, K. S. Vadagaonkar, M. V. Vhatkar, L. Melone and A. C. Chaskar, ChemistrySelect, 2018, 3, 277–283 CrossRef CAS.
- X.-X. Yu, P. Zhao, Y. Zhou, C. Huang, L.-S. Wang, Y.-D. Wu and A.-X. Wu, J. Org. Chem., 2021, 86, 12141–12147 CrossRef CAS PubMed.
- N. Salvanna, L. M. Reddy, R. A. Kumar and B. Das, ChemistrySelect, 2018, 3, 8019–8022 CrossRef CAS.
- V. Rajeshkumar, S. Chandrasekar and G. Sekar, Org. Biomol. Chem., 2014, 12, 8512–8518 RSC.
- J. Aboonajmi, F. Panahi, M. A. Hosseini, M. Aberi and H. Sharghi, RSC Adv., 2022, 12, 20968–20972 RSC.
- S. S. Acharya, P. Bhaumick, R. Kumar and L. H. Choudhury, ACS Omega, 2022, 7, 18660–18670 CrossRef CAS PubMed.
- C. Xu, G. Yin, F.-C. Jia, Y.-D. Wu and A.-X. Wu, Org. Lett., 2021, 23, 2559–2564 CrossRef CAS PubMed.
- Z. Moussa, Z. M. Judeh, A. Alzamly, S. A. Ahmed, H. T. Al-Masri, B. Al-Hindawi, F. Rasool and S. Saada, RSC Adv., 2022, 12, 6133–6148 RSC.
- Y. Yang, Q. Chen, S. Li, W. Ma, G. Yao, F. Ren, Z. Cai, P. Zhao, G. Liao and J. Xiong, J. Biomed. Nanotechnol., 2018, 14, 1396–1408 CrossRef CAS PubMed.
- R. Ma, Y. Ding, R. Chen, Z. Wang, L. Wang and Y. Ma, J. Org. Chem., 2020, 86, 310–321 CrossRef PubMed.
- C. Bodhak and A. Pramanik, J. Org. Chem., 2019, 84, 7265–7278 Search PubMed.
- J. Jayram and V. Jeena, RSC Adv., 2018, 8, 37557–37563 RSC.
- T. V. Huynh, K. V. Doan, N. T. Luong, D. T. Nguyen, S. H. Doan, T. T. Nguyen and N. T. Phan, RSC Adv., 2020, 10, 18423–18433 Search PubMed.
- S. Naidoo and V. Jeena, Eur. J. Org Chem., 2019, 2019, 1107–1113 CrossRef CAS.
- M. Singh, P. Awasthi and V. Singh, Eur. J. Org Chem., 2020, 2020, 1023–1041 Search PubMed.
- J. Zhang, J. Tang, Z. Chen and X. F. Wu, Adv. Synth. Catal., 2021, 363, 3060–3069 CrossRef CAS.
- D. Kumar, V. B. Reddy and R. S. Varma, Tetrahedron Lett., 2009, 50, 2065–2068 Search PubMed.
- K. Dabak, Ö. Sezer, A. Akar and O. Anaç, Eur. J. Med. Chem., 2003, 38, 215–218 CrossRef CAS PubMed.
- L. S. Kallander, Q. Lu, W. Chen, T. Tomaszek, G. Yang, D. Tew, T. D. Meek, G. A. Hofmann, C. K. Schulz-Pritchard and W. W. Smith, J. Med. Chem., 2005, 48, 5644–5647 CrossRef CAS PubMed.
- H. Duan, S. Sengupta, J. L. Petersen and X. Shi, Organometallics, 2009, 28, 2352–2355 CrossRef CAS.
- T. Weide, S. A. Saldanha, D. Minond, T. P. Spicer, J. R. Fotsing, M. Spaargaren, J.-M. Frère, C. Bebrone, K. B. Sharpless and P. S. Hodder, ACS Med. Chem. Lett., 2010, 1, 150–154 CrossRef CAS PubMed.
- M. T. Ren, M. Li, A. J. Wang, J. Gao, X. X. Zhang and W. M. Shu, Eur. J. Org Chem., 2020, 2020, 2233–2236 CrossRef CAS.
- S. K. Bowroju and R. Bavanthula, RSC Adv., 2021, 11, 5724–5728 RSC.
- H. N. Dogan, A. Duran, S. Rollas, G. Sener, M. K. Uysal and D. Gülen, Bioorg. Med. Chem., 2002, 10, 2893–2898 CrossRef CAS PubMed.
- S. Schenone, O. Bruno, A. Ranise, F. Bondavalli, W. Filippelli, G. Falcone, L. Giordano and M. R. Vitelli, Bioorg. Med. Chem., 2001, 9, 2149–2153 CrossRef CAS PubMed.
- J.-Y. Chou, S.-Y. Lai, S.-L. Pan, G.-M. Jow, J.-W. Chern and J.-H. Guh, Biochem. Pharmacol., 2003, 66, 115–124 CrossRef CAS PubMed.
- Z. Yang, Y. Liang, A. Li, K. Liu, L. Li, T. Yang and C. Zhou, J. Org. Chem., 2019, 84, 16262–16267 CrossRef CAS PubMed.
- R. Singhal, S. P. Choudhary, B. Malik and M. Pilania, ChemistrySelect, 2022, 7, e202200134 CrossRef CAS.
- V. Vaishya, R. Singhal, T. Kriplani and M. Pilania, Synthesis, 2022, 54, 3941–3961 CrossRef CAS.
- L. M. Reddy, V. V. Reddy, P. S. Prathima, C. K. Reddy and B. S. Reddy, Adv. Synth. Catal., 2018, 360, 3069–3073 CrossRef CAS.
- W.-Z. Weng, Y.-H. Gao, X. Zhang, Y.-H. Liu, Y.-J. Shen, Y.-P. Zhu, Y.-Y. Sun, Q.-G. Meng and A.-X. Wu, Org. Biomol. Chem., 2019, 17, 2087–2091 RSC.
- H. Liao, Z. Huang and Q. Zhu, Org. Biomol. Chem., 2022, 20, 3721–3725 RSC.
- A. Demirbas, D. Sahin, N. Demirbas and S. A. Karaoglu, Eur. J. Med. Chem., 2009, 44, 2896–2903 CrossRef CAS PubMed.
- J. M. Kane, B. M. Baron, M. W. Dudley, S. M. Sorensen, M. A. Staeger and F. P. Miller, J. Med. Chem., 1990, 33, 2772–2777 CrossRef CAS PubMed.
- S. Han, F.-F. Zhang, X. Xie and J.-Z. Chen, Eur. J. Med. Chem., 2014, 74, 73–84 CrossRef CAS PubMed.
- C. Wang, X. Geng, P. Zhao, Y. Zhou, Y.-D. Wu, Y.-F. Cui and A.-X. Wu, Chem. Commun., 2019, 55, 8134–8137 RSC.
- F. Zhu, Z. Yan, C. Ai, Y. Wang and S. Lin, Eur. J. Org Chem., 2019, 2019, 6561–6565 CrossRef CAS.
- W. Li, J. Zhang, J. He, L. Xu, L. Vaccaro, P. Liu and Y. Gu, Front. Chem., 2020, 8, 466 CrossRef CAS PubMed.
- W. Zhao, F. Zhang and G.-J. Deng, J. Org. Chem., 2020, 86, 291–301 CrossRef PubMed.
- X. Jiang, Z. Zhao, Z. Shen, K. Chen, L. Fang and C. Yu, Eur. J. Org Chem., 2020, 2020, 3889–3895 CrossRef CAS.
- A. R. Obah Kosso, Y. Kabri, J. Broggi, S. b. Redon and P. Vanelle, J. Org. Chem., 2020, 85, 3071–3081 CrossRef CAS PubMed.
- E. Safari, N. Nowrouzi, M. Abbasi and A. Hasaninejad, ChemistrySelect, 2019, 4, 8771–8775 CrossRef CAS.
- L. S. Chen, L. B. Zhang, Y. Tian, J. H. Li and Y. Q. Liu, Eur. J. Org Chem., 2020, 2020, 5523–5526 CrossRef CAS.
- N. Slathia, A. Gupta and K. K. Kapoor, Tetrahedron Lett., 2021, 78, 153268 CrossRef CAS.
- N. Pogaku, P. R. Krishna and Y. L. Prapurna, ChemistrySelect, 2019, 4, 12333–12336 CrossRef CAS.
- A. K. Sharma, P. Kumar, R. K. Vishwakarma and K. N. Singh, Asian J. Org. Chem., 2018, 7, 688–691 CrossRef CAS.
- D. S. Raghuvanshi, N. Verma and A. Gupta, ChemistrySelect, 2019, 4, 2075–2078 CrossRef CAS.
- E. Sheikhi, M. Adib and M. A. Karajabad, Org. Prep. Proced. Int., 2020, 52, 120–126 CrossRef CAS.
- L. Xu, Y. Chen, Z. Shen, Y. Wang and M. Li, Tetrahedron Lett., 2018, 59, 4349–4354 CrossRef CAS.
- S. V. Gaikwad, D. N. Nadimetla, M. A. Kobaisi, M. Devkate, R. Joshi, R. G. Shinde, M. V. Gaikwad, M. D. Nikalje, S. V. Bhosale and P. D. Lokhande, ChemistrySelect, 2019, 4, 10054–10059 Search PubMed.
- Y. F. Liang, Y. Yuan, T. Shen, S. Song and N. Jiao, Chin. J. Chem., 2018, 36, 233–240 CrossRef CAS.
- S.-Y. Zhuang, Y.-X. Tang, X.-L. Chen, Y.-D. Wu and A.-X. Wu, Org. Biomol. Chem., 2021, 19, 4258–4262 RSC.
- R. Sirgamalla, A. Kommakula, S. Banoth, R. Dharavath, K. Adem and S. Boda, ChemistrySelect, 2018, 3, 1062–1065 CrossRef CAS.
-
J. B. Azeredo, A. L. Braga and C. Santi, Presented at the 1st International Electronic Conference on Catalysis Sciences, 2020, vol. 10, p. 30 Search PubMed.
- X. K. Zhang, X. Y. Miao, H. R. Jiang, F. Ge, J. C. Sun, R. Y. Zhang, Q. Ouyang, W. Y. Fan, Y. P. Zhu and Y. Y. Sun, Adv. Synth. Catal., 2021, 363, 4632–4638 CrossRef CAS.
- S. Guo, J. Hua, Z. Dai, Z. Yang, Z. Fang and K. Guo, ACS Sustain. Chem. Eng., 2018, 6, 7979–7988 CrossRef CAS.
- A. Muthukumar, S. Sangeetha and G. Sekar, Org. Biomol. Chem., 2018, 16, 7068–7083 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *
*