
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePd/NHC catalyzed reduction and coupling of nitroaromatics for the synthesis of diarylamines†
Wei Chen *a,
Kai Chenb,
Xuejie Wangb,
Linjie Yangb and
Wanzhi Chen*b
*a,
Kai Chenb,
Xuejie Wangb,
Linjie Yangb and
Wanzhi Chen*b
aSchool of Biological and Chemical Engineering, Zhejiang University of Science and Technology, Hangzhou, China. E-mail: 123069@zust.edu.cn
bDepartment of Chemistry, Zhejiang University, Hangzhou, China. E-mail: chenwzz@zju.edu.cn
First published on 22nd May 2024
Abstract
Herein, we report a one-pot approach to diarylamines through the reductive homocoupling of nitroaromatics, employing triethylsilane as the reducing agent and Pd/NHC as the catalyst. This method enables nitroaromatics to serve both as electrophilic reagents and as precursors of nucleophilic reagents, allowing for the direct preparation of diarylamines without the need to isolate aromatic primary amines.
Introduction
Diarylamines and their derivatives are important structural constituents of many pharmaceuticals, pesticides, dyes and pigments. Numerous synthetic methodologies have been developed by using readily available starting materials. Amongst the plethora of methodologies for the synthesis of diarylamines, Buchwald–Hartwig coupling1 and Ullmann coupling2 of haloarenes and aromatic amines have proved to be the most valuable synthetic approaches. The success of these C–N approaches pivots on the choice of metal catalysts and appropriate ligands. Various phosphines,3 amides,2 and NHC4,5 ligands were employed in the C–N coupling reactions showing satisfactory efficiency.Pertinent statistics elucidate that aromatic amines are predominantly derived from the reduction of nitroaromatic hydrocarbons.6 Transition metal catalyzed direct reductive C–N coupling of nitroarenes with an electrophilic partner would be an ideal approach for diarylamines, which diminishes the necessity for an aromatic amine separation step.
Recently, Xue and coworkers described the C–N coupling of aryl halides with nitroarenes by nickel-bipyridine catalysis under light irradiation and mild basic conditions.7 Some readily available nitroarenes undergo coupling with a variety of aryl halides and proceed via the addition of an aryl radical, generated from a NiI/NiIII cycle, to a nitrosoarene intermediate (Fig. 1a). Weix and coworkers reported the reductive arylation of nitroarenes with chloroarene to form diarylamines using manganese as the reducing agent (Fig. 1b).8 Under reducing conditions, palladium–phosphine complexes catalyze the dual N-arylation of typically inert azoarenes generated via the in situ reduction of nitroarenes. Kazemnejadi and coworkers described a one-pot synthetic approach to diarylamines through bimetallic nickel–palladium catalyzed sequential reduction of nitroarenes to aromatic amines and further C–N coupling with haloarenes (Fig. 1c).9 The reductive coupling reaction is well performed by the bimetallic catalyst in the presence of 2.0 equiv. of NaBH4.
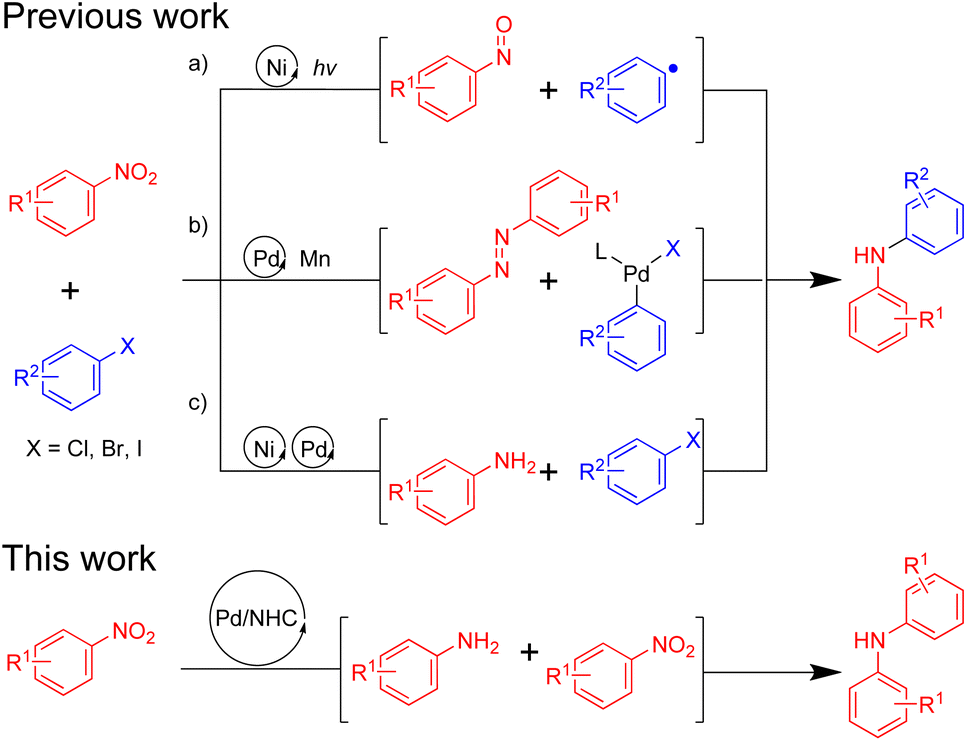 | ||
| Fig. 1 Transition metal-catalyzed reductive coupling of nitroarenes and haloarenes. | ||
In addition to serving as precursors for nucleophilic reagents, nitroaromatics also possess unique advantages as electrophilic reagents, drawing attention.10–17 Employing nitroaromatics instead of halogenated aromatic can circumvent halogenated waste contamination and certain halogenated aromatic compounds are synthesized through multistep reactions, using nitroaromatics as starting materials.18
In light of the dual roles of nitroaromatics in cross-coupling reactions, we contemplated the introduction of a reducing agent into the Pd/NHC catalytic system previously reported by our group.11,19,20 This would allow nitroaromatics to act both as an electrophilic reagent and as a nitrogen source for the synthesis of diarylamines, thereby broadening the application scope of nitroaromatics and facilitating a novel mode of nitroarene reductive cross-coupling. Although this strategy appears promising, there remain a few challenges to address: (1) achieving the complete reduction of nitroaromatic hydrocarbons; (2) ensuring compatibility between the reduction and the palladium catalytic processes.
Result and discussion
We herein delineate a streamlined synthetic methodology for the construction of diarylamines in a singular step, utilizing nitroaro-matic compounds as the sole aromatic source. This one-step pro-tocol for the synthesis of diarylamines from nitroaromatics cir-cumvents the necessity for amine separation, thereby reducing the overall complexity and number of synthetic steps. To optimize this reaction system, we employed Pd/NHC catalyzed reduction and coupling of 4-nitrotoluene as a model reaction for our inves-tigation.Initially, we employed 4-nitrotoluene as the archetype substrate. A range of reducing agents such as B2Pin2, KBH4, Et3SiH, and Ph2SiH2 were subjected to a rigorous screening process, with the resulting reactions documented in Fig. 2. B2Pin2, B2(OH)2, and KBH4 are frequently observed in the study of nitroaromatic compound reduction for the synthesis of aromatic amines.9,21 However, these reagents have proven to be less than ideal within this catalytic system (Fig. 2, entries 1–3), possibly due to their predilection for exhibiting optimal performance in polar solvents, making them unsuitable for deployment in weakly polar environments.22 Methanol can serve as a hydrogen source, effectuating the reduction of nitroaromatic compounds through hydrogen transfer.23 It was unfortunately incompatible with this specific reaction environment (Fig. 2, entries 4 and 5). Silanes demonstrate an advantageous ability for the reduction of nitroaromatic compounds invarious polar or weakly polar solvents.24,25 The amount of triethylsilane has a great influence on the reaction. Utilizing 2.0 equivalents of triethylsilane as the reducing agent results in a yield of 55% for the subsequent product. Alterations in the quantity of the reducing agent, whether an increase or decrease, result in corresponding decreased yields (Fig. 2, entries 7–9).
 | ||
| Fig. 2 Optimization of reaction conditions. | ||
Among the bases scrutinized, K3PO4·3H2O emerged as the most efficacious for this reaction. Transitioning from K3PO4·3H2O to anhydrous K3PO4 resulted in diminished product formation (Table S1,† entries 1 and 2). The employment of organic bases fails to yield the desired product (Table S1,† entry 6). Following a solvent screening, we found that the reaction struggles to proceed in highly polar solvents (Table S1,† entry 8), whereas it exhibits modest efficiency in less polar and nonpolar solvents. Amongst these, the nonpolar solvent n-octane emerged as the most optimal reaction medium (Table S1,† entry 12). Ultimately, we selected n-octane as the reaction solvent. An appraisal of palladium sources revealed Pd(acac)2 as the superior catalyst (Fig. 2, entry 11). Pd2(dba)3, also exhibited catalytic capability, while some palladium(II) catalysts scarcely produced the desired product (Fig. 2, entries 13 and 14). The experiments showed that upon employing [Pd(allyl)Cl]2 or Pd(OAc)2 as a catalyst, bubbles appeared immediately after the reducing agent's introduction, and the solution exhibited a black and turbid appearance. This observation suggested that [Pd(allyl)Cl]2 and Pd(OAc)2 underwent rapid reduction and inactivation. Upon examining the steric hindrance of ligands, we found that both smaller and larger steric hindrances negatively impact the reaction. Among the ligands investigated, L3·HCl was identified as the superior choice. After we determined the optimal reaction conditions, we explored the substrate scope for this catalytic system. Upon screening various substrates, we discovered that nitroarenes featuring various alkyl groups could give the coupling products with satisfactory yields (2a–2e). The reductive coupling nitrobenzene bearing an ortho-methoxy afforded the corresponding product in 67% yield (2f). However, the same reaction of nitrobenzene having a para-methoxy exhibited a reduced yield, with the primary side reaction involving the reduction of para-methoxy nitrobenzene to 4-methoxyaniline in 81% yield. Generally, nitroarenes bearing electron-donating groups at para-positions often presented an abundance of reduction products with minimal coupling products (Fig. 3).
 | ||
| Fig. 3 Substrate scope for the palladium-catalyzed reductive arylation of nitroarenes. | ||
Fluorine and trifluoromethyl-containing nitrobenzenes could react smoothly giving the target products in satisfactory yields (2h–2k). Nitrobenzenes with ortho or meta phenyl substituents gave their corresponding products in 71% and 73% yields, respectively (2l and 2m). Nitrobiphenyls bearing electron-withdrawing groups were moderately efficient coupling partners, and the corresponding di(biphenyl)amines were attained in lower yields (2n and 2o). Naphthalene derivatives were also suitable for the reductive coupling reaction, and di(naphthalen-1-yl)amine was obtained in 79% yield (2p). More steric 2-methyl nitronaphthalene demonstrated a diminished reaction yield, probably due to steric hindrance effects (2q). The homocoupling reaction of 2-methoxy-3-nitropyridine yielded product 2 s in a 69% yield. Moreover, using an analog of the drug molecule Celecoxib26 as the starting material, the reductive coupling product 2t could be obtained in a yield of 78%.
In order to gain deeper knowledge on the N-heterocyclic carbene ligands with an imidazo[1,5-a]pyridine core in palladium-catalyzed C–N coupling of nitroarenes, we prepared [Pd(L3)(acac)Cl] metal complexes through reaction of L3·HCl and Pd(acac)2 at 80 °C in 1,4-dioxane (Scheme S1†). The structure of [Pd(L3)(acac)Cl] was undoubtedly established using spectroscopic and analytical methods, and [Pd(L3)(acac)Cl] was crystallographically characterized (Fig. 4). It can be seen from the single crystal structure that the interaction between the triisopropylphenyl group and the palladium center causes a slight distortion of the structure. X-ray crystallography indicates that the Pd–C bond length in the complex falls within the typical range for Pd/NHC complexes.27–29 The distance between the Pd1 and C4 measures 3.135(2) Å. The dihedral angle of Pd1–C1–C3–C4 is 26.073°, suggesting a noticeable offset between the palladium atom and the triisopropylphenyl group, possibly due to weak interactions between them.
 | ||
| Fig. 4 X-ray crystal structure of [Pd(L3)(acac)Cl]. Thermal ellipsoids are given at 50% probability, H atoms are omitted. Selected bond lengths (Å): Pd1–C1 = 1.962(2), Pd1–O1 = 2.012(1), Pd1–O2 = 2.031(1), Pd1–Cl1 = 2.300(1). | ||
To gain an understanding of the reaction mechanism, a series of controlled experiments were carried out. We found that in the absence of NHC ligands Pd(acac)2 itself was not efficient, in such case only naphthalen-1-amine was formed, which suggests that the NHC ligand plays an important role in the denitrative homocoupling (Fig. 5a). The same reduction reaction could be achieved using the classical Pd/C catalyst (Fig. 5b). When [Pd(L3)(acac)Cl] was used as catalysts, the reaction could proceed effectively (Fig. 5c). However, mercury test has shown that when Pd(acac)2 or Pd/NHC complexes were used as the catalysts, the reduction of nitroarenes could not be carried out (Fig. 5d and e). The reduction of nitroarenes may be partially catalyzed by acetylacetone palladium via the generation of palladium black. Nevertheless, the role of NHC–Pd in cataly tic reduction remains ambiguous.30,31
 | ||
| Fig. 5 Mechanism studies. | ||
Conclusions
In summary, we have developed a one-pot synthetic approach for the preparation of diarylamine compounds, utilizing nitroaromatics as both the electrophilic reagent and nitrogen source and triethylsilane as a reducing agent. This method streamlines the reaction steps and enhances overall efficiency. We successfully isolated an Pd/NHC precatalyst and conducted preliminary investigations into the reaction mechanism. Further research in this area is currently underway.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (22071214).Thanks the financial support from Zhejiang University of Science and Technology (F701103N08) and Wenzhou Huanuo Technology Co., Ltd.References
- P. A. Forero-Cortés and A. M. Haydl, The 25th anniversary of the Buchwald–Hartwig amination: development, applications, and outlook, Org. Process Res. Dev., 2019, 23, 1478–1483 CrossRef.
- Q. Yang, Y. Zhao and D. Ma, Cu-mediated Ullmann-type cross-coupling and industrial applications in route design, process development, and scale-up of pharmaceutical and agrochemical processes, Org. Process Res. Dev., 2022, 26, 1690–1750 CrossRef CAS.
- D. van der Westhuizen, A. C. Castro, N. Hazari and A. Gevorgyan, Bulky, electron-rich, renewable: analogues of Beller's phosphine for cross-couplings, Catal. Sci. Technol., 2023, 13, 6733–6742 RSC.
- M. M. Rahman, Q. Zhao, G. Meng, R. Lalancette, R. Szostak and M. Szostak, [IPr#-PEPPSI] : A Well-Defined, Highly Hindered and Broadly Applicable Pd (II)–NHC (NHC = N-Heterocyclic Carbene) Precatalyst for Cross-Coupling Reactions, Molecules, 2023, 28, 5833 CrossRef CAS PubMed.
- F. Mazars, K. S. Etsè, G. Zaragoza and L. Delaude, Pd-PEPPSI catalysts bearing N-heterocyclic carbene ligands derived from caffeine and theophylline for Mizoroki–Heck and C(sp2)–H arylation reactions, J. Organomet. Chem., 2024, 1003, 122928 CrossRef.
- M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Recent developments in the reduction of aromatic and aliphatic nitro compounds to amines, Org. Process Res. Dev., 2016, 22, 430–445 CrossRef.
- G. Li, L. Yang, J.-J. Liu, W. Zhang, R. Cao, C. Wang, Z. Zhang, J. Xiao and D. Xue, Light-Promoted C–N Coupling of Aryl Halides with Nitroarenes, Angew. Chem., Int. Ed., 2021, 60, 5230–5234 CrossRef CAS PubMed.
- B. D. Akana-Schneider and D. J. Weix, Reductive Arylation of Nitroarenes with Chloroarenes: Reducing Conditions Enable New Reactivity from Palladium Catalysts, J. Am. Chem. Soc., 2023, 145, 16150–16159 CrossRef CAS PubMed.
- M. Kazemnejadi, R. O. Ahmed and B. Mahmoudi, Ni/Pd-catalyzed Suzuki–Miyaura Cross-Coupling of Alcohols and Aldehydes and C–N Cross-Coupling of Nitro and Amines via Domino Redox Reactions: Base-Free, Hydride Acceptor-Free, RSC Adv., 2020, 10, 43962–43974 RSC.
- F. Inoue, M. Kashihara, M. R. Yadav and Y. Nakao, Buchwald-Hartwig Amination of Nitroarenes, Angew. Chem., Int. Ed., 2017, 56, 13307–13309 CrossRef CAS PubMed.
- W. Chen, K. Chen, W. Chen, M. Liu and H. Wu, Well-Designed N-Heterocyclic Carbene Ligands for Palladium-Catalyzed Denitrative C–N Coupling of Nitroarenes with Amines, ACS Catal., 2019, 9, 8110–8115 CrossRef CAS.
- K. Muto, T. Okita and J. Yamaguchi, Transition-Metal-Catalyzed Denitrative Coupling of Nitroarenes, ACS Catal., 2020, 10, 9856–9871 CrossRef CAS.
- M. Kashihara and Y. Nakao, Cross-Coupling Reactions of Nitroarenes, Acc. Chem. Res., 2021, 54, 2928–2935 CrossRef CAS PubMed.
- J. Yao, L. Yu, W. Duan and C.-J. Li, Palladium-Catalyzed C–Si Bond Formation via Denitrative Cross-Coupling of Nitroarenes with Hexamethyldisilane, Org. Chem. Front., 2023, 10, 524–530 RSC.
- K. K. Asahara, K. Muto and J. Yamaguchi, Pd-Catalyzed Denitriative Intramolecular Mizoroki–Heck Reaction, Chem. Lett., 2023, 52, 299–302 CrossRef CAS.
- M. Kashihara, R.-L. Zhong, K. Semba, S. Sakaki and Y. Nakao, Pd/NHC-catalyzed Cross-Coupling Reactions of Nitroarenes, Chem. Commun., 2019, 55, 9291–9294 RSC.
- T. Zhou, P. Gao, E. Bisz, B. Dziuk, R. Lalancette, R. Szostak and M. Szostak, Well-defined, air-and moisture-stable palladium–imidazo [1,5-a] pyridin-3-ylidene complexes: a versatile catalyst platform for cross-coupling reactions by L-shaped NHC ligands, Catal. Sci. Technol., 2022, 12, 6581–6589 RSC.
- A. Makhloutah, D. Hatych, T. Chartier, L. Rocard, A. Goujon, F.-X. Felpin and P. Hudhomme, An Investigation of Palladium-Catalyzed Stille-type Cross-Coupling of Nitroarenes in Perylenediimide Series, Org. Biomol. Chem., 2022, 20, 362–365 RSC.
- K. Chen, W. Chen, X. Yi, W. Chen, M. Liu and H. Wu, Sterically Hindered N-heterocyclic Carbene/Palladium(II) Catalyzed Suzuki–Miyaura Coupling of Nitrobenzenes, Chem. Commun., 2019, 55, 9287–9290 RSC.
- W. Chen, W. Chen, M. Liu and H. Wu, Construction of heterobiaryl skeletons through Pd-catalyzed cross-coupling of nitroarenes and heterocyclic arylborononate esters with a sterically demanding NHC ligand, Org. Lett., 2022, 24, 6983–6987 CrossRef CAS PubMed.
- H.-C. Du, N. Simmons, J. C. Faver, Z. Yu, M. Palaniappan, K. Riehle and M. M. Matzuk, A Mild, DNA-Compatible Nitro Reduction Using B2(OH)4, Org. Lett., 2019, 21, 2194–2199 CrossRef CAS PubMed.
- M. Jang, T. Lim, B. Y. Park and M. S. Han, Metal-Free, Rapid, and Highly Chemoselective Reduction of Aromatic Nitro Compounds at Room Temperature, J. Org. Chem., 2022, 87, 910–919 CrossRef CAS PubMed.
- Y. Liu, W. Miao, W. Tang, D. Xue, J. Xiao, C. Wang and C. Li, Rhodium-terpyridine Catalyzed Transfer Hydrogenation of Aromatic Nitro Compounds in Water, Chem.–Asian J., 2021, 16, 1725–1729 CrossRef CAS PubMed.
- R. J. Rahaim and R. E. Maleczka, Pd-Catalyzed Silicon Hydride Reductions of Aromatic and Aliphatic Nitro Groups, Org. Lett., 2005, 7, 5087–5090 CrossRef CAS PubMed.
- N. S. Shaikh, Sustainable Amine Synthesis: Iron Catalyzed Reactions of Hydrosilanes with Imines, Amides, Nitroarenes and Nitriles, ChemistrySelect, 2019, 4, 6753–6777 CrossRef CAS.
- R. J. Flower, The development of COX2 inhibitors, Nat. Rev. Drug Discovery, 2003, 2, 179–191 CrossRef CAS PubMed.
- O. Navarro, N. Marion, N. M. Scott, J. González, D. Amoroso, A. Bell and S. P. Nolan, Synthesis of Novel (NHC)Pd(Acac)Cl Complexes (Acac=acetylacetonate) and Their Activity in Cross-Coupling Reactions, Tetrahedron, 2005, 61, 9716–9722 CrossRef CAS.
- S. Meiries, A. Chartoire, A. M. Z. Slawin and S. P. Nolan, [Pd(IPr*)(Acac)Cl] : An Easily Synthesized, Bulky Precatalyst for C–N Bond Formation, Organometallics, 2012, 31, 3402–3409 CrossRef CAS.
- S. Meiries, K. Speck, D. B. Cordes, A. M. Z. Slawin and S. P. Nolan, [Pd(IPr*OMe)(Acac)Cl] : Tuning the N-Heterocyclic Carbene in Catalytic C–N Bond Formation, Organometallics, 2013, 32, 330–339 CrossRef CAS.
- V. M. Chernyshev, A. V. Astakhov, I. E. Chikunov, R. V. Tyurin, D. B. Eremin, G. S. Ranny, V. N. Khrustalev and V. P. Ananikov, Pd and Pt catalyst poisoning in the study of reaction mechanisms: what does the mercury test mean for catalysis?, ACS Catal., 2019, 9, 2984–2995 CrossRef CAS.
- I. C. Chagunda, T. Fisher, M. Schierling and J. S. McIndoe, Poisonous Truth about the Mercury Drop Test: The Effect of Elemental Mercury on Pd (0) and Pd (II) ArX Intermediates, Organometallics, 2023, 42, 2938–2945 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2328226. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra00921e |
| This journal is © The Royal Society of Chemistry 2024 |