
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFacile synthesis of substituted 2-aroylbenzo[b]thiophen-3-ols to form novel triazole hybrids using click chemistry†
Asma Mukhtara,
Arif Hussaina,
Faiza Younasa,
Sammer Yousuf b and
Muhammad Saeed*a
b and
Muhammad Saeed*a
aDepartment of Chemistry and Chemical Engineering, Syed Babar Ali School of Science and Engineering, Lahore University of Management Sciences, Lahore 54792, Pakistan. E-mail: Muhammad.saeed@lums.edu.pk
bH. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi 75270, Pakistan
First published on 28th March 2024
Abstract
An efficient one-pot method is proposed for the synthesis of 2-aroylbenzo[b]thiophen-3-ols from 2-mercaptobenzoic acid and various substituted aryl bromomethyl ketones in the presence of triethylamine. The reaction is likely to proceed through SN2-type nucleophilic attack of the sulfhydryl group in thiosalicylic acid on bromomethyl ketone in the presence of a base to afford sulfanylbenzoic acid, which undergoes an intramolecular cyclization in situ to furnish 2-aroylbenzo[b]thiophen-3-ol in high yield. To investigate the utility of the synthesized benzothiophene scaffold, an alkyne moiety was introduced at the 3-hydroxy position and subsequently subjected to a click reaction to form novel benzothiophene-triazole hybrids in good yields. A simple and straightforward approach to synthesizing 2-aroylbenzo[b]thiophen-3-ols can open new avenues for discovering novel biological and pharmaceutical compounds.
1 Introduction
Heterocycles serve as fundamental components in numerous biologically relevant natural and synthetic compounds. This notion is supported by the fact that a large number of the USA Food and Drug Administration (FDA)-approved drugs contain one or several heterocyclic rings in their structures.1–3 Therefore, access to a chemical library of small molecules containing one or more “privileged” heterocyclic rings is generally considered a prerequisite for effective drug discovery endeavors.4,5 Among them, sulfur-containing heterocycles are gaining attention in organic synthesis due to their applications in materials sciences, pharmaceutical industries, and food chemistry.6,7 Benzo[b]thiophene is a versatile sulfur-containing heterocyclic scaffold, which frequently appears in pharmaceutically interesting compounds with a diverse range of biological activities.8–12 For instance, various drugs including raloxifene (1), arzoxifene (2), rintodestrant (3), and LSZ102 (4) feature the benzo[b]thiophene core and are currently clinically utilized for the treatment of estrogen-receptor-positive breast cancer (Fig. 1). Termed selective estrogen receptor modulators (SERMs), both 1 and 2 are used for the prevention and treatment of breast cancer and osteoporosis in postmenopausal women.13–20 On the other hand, 3 and 4 act as selective estrogen receptor degraders (SERDs), undergoing evaluation for the elimination of estrogen receptor-α from cancer cells in patients diagnosed with aggressive breast cancer.21–23 Along with SERM/SERD activities, several benzothiophene-based compounds show anticancer activities by targeting various cellular proteins such as tubulin, topoisomerase, and histone deacetylase.24–28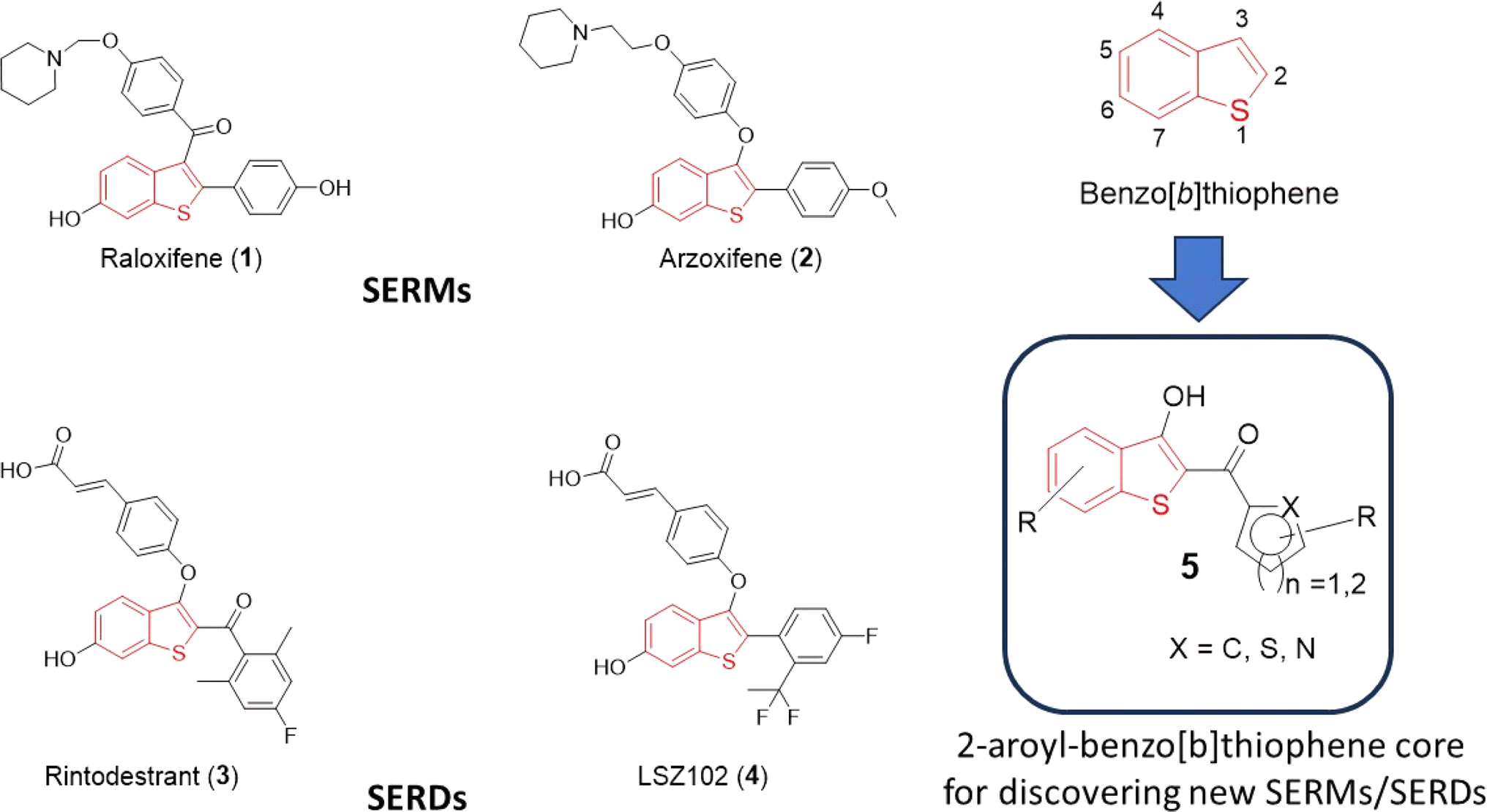 | ||
| Fig. 1 Structures of selective estrogen receptor modulators (SERMs) and selective estrogen receptor down-regulators (SERDs) containing the benzo[b]thiophene scaffold and identification of the motif 2-aroyl-benzo[b]thiophen-3-ol (5) for synthesizing new SERM/SERD hits. | ||
To exhibit specificity, substituents at the C-2 and the C-3 positions play an important role in target binding and eliciting diverse biological activities associated with benzo[b]thiophenes. Upon a cursory inspection of the structures given in Fig. 1, it seems plausible to discern that the 2-aryl and/or 2-aroyl moiety at the C-2/C-3 position of the benzo[b]thiophene core may be an essential ingredient for the SERM/SERD-like activities of 1–4. We envision that the 2-aroylbenzo[b]thiophen-3-ol core could provide access to diverse benzo[b]thiophene analogs for discovering new SERMs/SERDs. To extend our efforts on the discovery of early-phase inhibitor leads for developing antiviral and anticancer drugs,29–32 we herein report a simple, one-pot synthetic method for the preparation of substituted 2-aroyl-benzo[b]thiophen-3-ol derivatives (5a-s) by reacting 2-mercaptobenzoic acid (6) with substituted aryl bromomethyl ketones (8a-s) in the presence of triethylamine in DMF. To further extend the scope of a 2-aroyl-benzo[b]thiophen-3-ol substrate (5), we introduced an alkyne moiety at the 3-hydroxy position, conducted a click reaction, and prepared novel benzothiophene-triazole hybrids. We believe that this robust method for the direct synthesis of 5 starting from 2-mercaptobenzoic acid (6) under basic conditions will complement the previously reported method under acidic conditions.33
The core 2-aroyl-benzo[b]thiophen-3-ol (5) structure has been synthesized previously from 2-mercaptobenzoic acid (6) using a 2 or 3-step-long reaction sequence and/or using harsh conditions as shown in Scheme 1. For instance, P. Guglielmi et al.11 reported the synthesis of 5 by converting 6 to its methyl ester 7, followed by a reaction with phenacyl bromide (8a) in methanol and potassium hydroxide as a base. Several other methods have been reported where a prior conversion of 6 to 2-((2-oxo-2-phenylethyl)thio)benzoic acid (9) is required for the subsequent intramolecular cyclization reaction to 5 in an additional step, such as by using sodium acetate,12 trifluoroacetic acid,33 or zinc powder.22 In another report, 6 was first converted to thio-aspirin, then to phenacyl ester 10, and finally to 5 by using trifluoroacetic acid under reflux.33 Taking insight from these reports, we anticipated that 5 could be synthesized in a one-pot reaction using basic conditions. The reaction of the sulfhydryl (–SH) group in 6 with 8a under basic conditions is a well-known SN2-type reaction to afford sulfanyl benzoic acid 9a.34,35 Using a suitable base, the product 9a can be converted in situ to the corresponding benzo[b]thiophene (5) without its isolation.
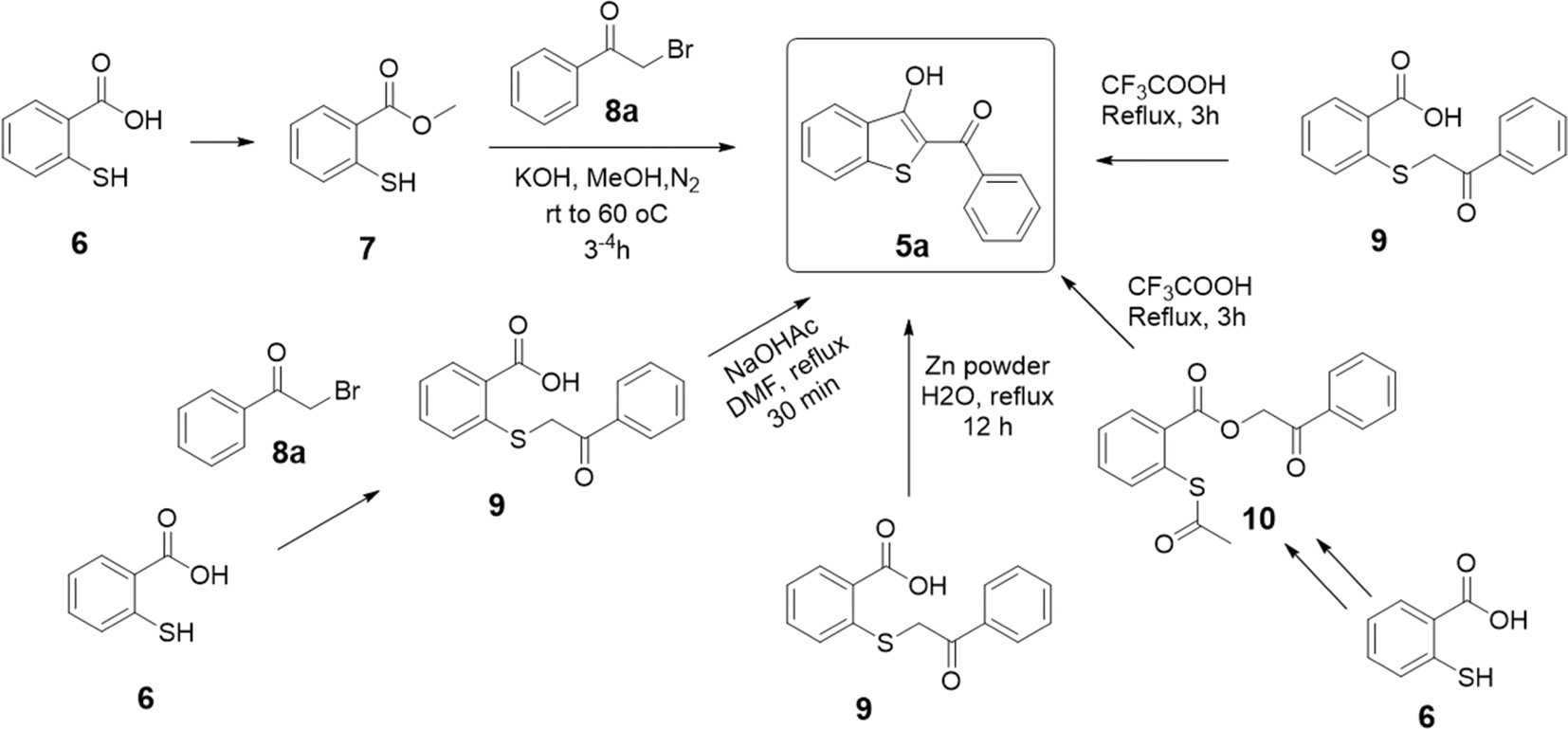 | ||
| Scheme 1 Various pathways for converting 2-mercaptobenzoic acid (6) into benzo[b]thiophene (5a). | ||
2 Results and discussion
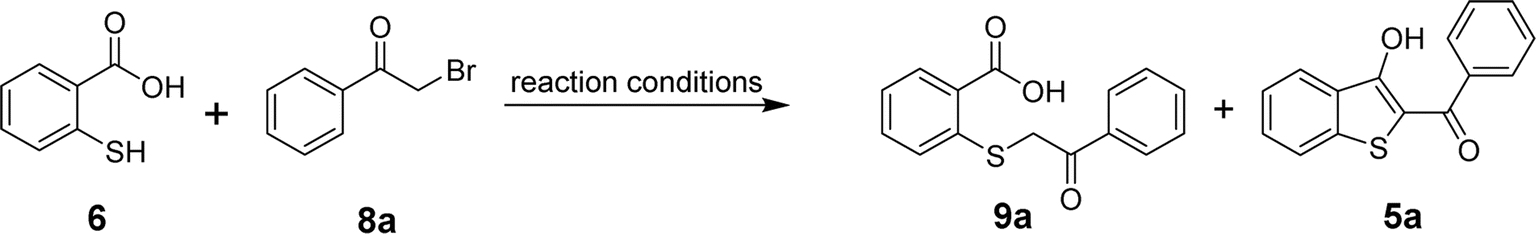
For the optimization of the reaction, we used 2-mercaptobenzoic acid (6) and phenacyl bromide (8a) as model substrates. By screening different inorganic and organic bases, reaction solvents, reaction times, and temperatures, the formation of acyclic product 9a and the targeted benzo[b]thiophenes (5a) were observed as shown in Table 1. Initially, sodium acetate was used as a base in dimethylformamide (DMF) but the in situ cyclization failed to produce 5a. The only product observed when the reaction mixture was subjected to stirring at room temperature for 8 hours (entry 1) or heated at 100 °C for 24 hours (entry 2) was acyclic 9a. Replacing the more polar solvent DMF with a less polar solvent THF was also unsuccessful in producing 5a (entries 3 and 4). When triethylamine (TEA) was used as a base in DMF, within a 30 min time frame, two new spots emerged on the TLC plate, corresponding to both acyclic 9a and the cyclized product 5a. Stirring the reaction mixture at room temperature for 3 h indicated the complete conversion of the lower (polar) spot to the desired less polar product 5a in 85% yield (entry 5). Raising the temperature to 100 °C, however, did not improve the overall yield. In fact, we observed a slightly lower yield of 5a, most probably due to the decomposition of 5a at higher temperatures (entry 6). Replacing DMF with THF did not yield 5a in the presence of TEA, and only a small amount of the acyclic intermediate 9a was obtained (entries 7 and 8). Using inorganic bases such as potassium carbonate, cesium carbonate, or sodium carbonates produced 5a in varying yields but always lower than what we could achieve using TEA (entries 9–20). Finally, the application of another organic base pyridine resulted in the formation of 5a in 65% yield when the reaction mixture was stirred in DMF at room temperature for 4–6 h (entry 21). Again, conducting the reaction in pyridine/DMF at a higher temperature reduced the yield for 5a due to its decomposition (entry 22). The reaction in the presence of pyridine also gave a poor yield of 5a when the reaction was conducted in THF at room temperature or under reflux (entries 23 and 24). Thus, among all the tested bases, triethylamine in DMF at room temperature for 3 hours yielded the maximum amounts of the desired product 5a. Finally, the formation of 5a was tested in DMSO using TEA as a base (entries 25 and 26). However, the observed yield of the target benzo[b]thiophene was lower than what we obtained using the TEA/DMF system.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Base | Solvent | Temperature | Time (h) | Yield (%) | |
| 9a | 5a | |||||
| 1 | NaOAc | DMF | Rt | 6–8 | 70 | Trace |
| 2 | NaOAc | DMF | 100 °C | 24 | 77 | Trace |
| 3 | NaOAc | THF | Rt | 6–8 | 67 | Trace |
| 4 | NaOAc | THF | Reflux | 24 | 72 | Trace |
| 5 | TEA | DMF | Rt | 3 | Trace | 85 |
| 6 | TEA | DMF | 100 °C | 2 | Trace | 60 |
| 7 | TEA | THF | Rt | 6 | 56 | Trace |
| 8 | TEA | THF | Reflux | 24 | 63 | Trace |
| 9 | K2CO3 | DMF | Rt | 4–6 | 32 | 40 |
| 10 | K2CO3 | DMF | 100 °C | 24 | 28 | 45 |
| 11 | K2CO3 | THF | Rt | 6 | 40 | 20 |
| 12 | K2CO3 | THF | Reflux | 24 | 43 | 20 |
| 13 | Cs2CO3 | DMF | Rt | 4–6 | 45 | 20 |
| 14 | Cs2CO3 | DMF | 100 °C | 24 | 57 | 10 |
| 15 | Cs2CO3 | THF | Rt | 6 | 62 | Trace |
| 16 | Cs2CO3 | THF | Reflux | 24 | 64 | Trace |
| 17 | Na2CO3 | DMF | Rt | 4–6 | 45 | 40 |
| 18 | Na2CO3 | DMF | 100 °C | 24 | 44 | 35 |
| 19 | Na2CO3 | THF | Rt | 6 | 67 | 10 |
| 20 | Na2CO3 | THF | Reflux | 24 | 52 | 10 |
| 21 | Pyridine | DMF | Rt | 4–6 | 10 | 65 |
| 22 | Pyridine | DMF | 100 °C | 24 | 22 | 55 |
| 23 | Pyridine | THF | Rt | 6 | 56 | 20 |
| 24 | Pyridine | THF | Reflux | 24 | 68 | 20 |
| 25 | TEA | DMSO | Rt | 6 | Trace | 50 |
| 26 | TEA | DMSO | Reflux | 24 | Trace | 20 |
After determining the optimized conditions for the formation of product 5a, the substrate scope of the reaction was explored. Using diverse aryl bromomethyl ketones (8a-s), we were able to obtain the target benzo[b]thiophen-3-ols (5a-s) or their keto tautomers in 45–87% yields as depicted in Scheme 2. The electronic nature of the substituent on the aromatic ring of the arylbromomethyl ketone seems to control the formation of 5 in the keto or enol form. In the case of electron-withdrawing substituents, the predominant product was obtained as the enol tautomer, while the electron-donating substituents favored the formation of the product in the 3-keto form. Further experiments to delineate the electronic factors of the reaction are under investigation in our laboratory.
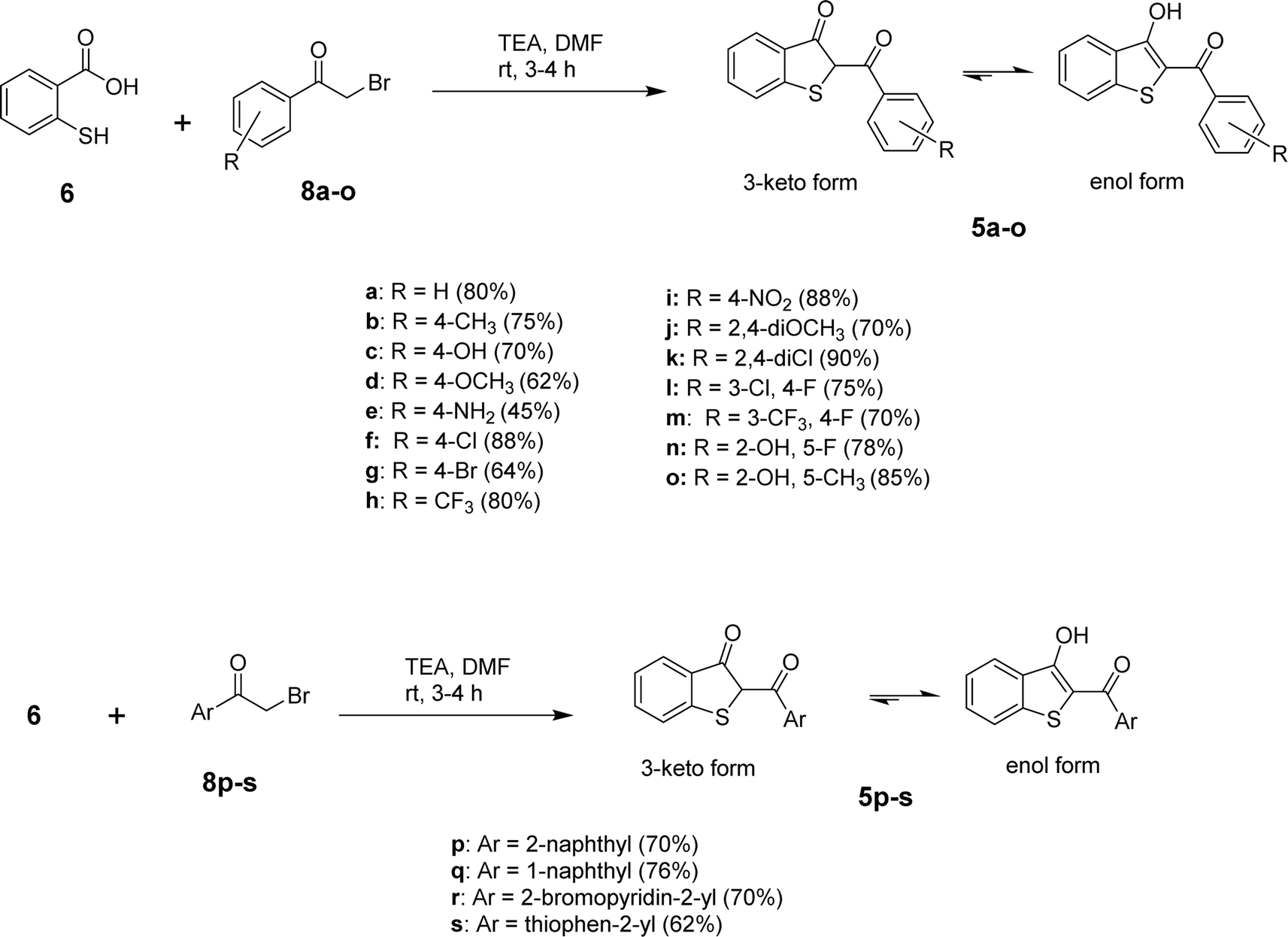 | ||
| Scheme 2 Synthesis of 2-aroylbenzo[b]thiophen-3-ols using triethylamine in DMF. | ||
All the synthesized benzo[b]thiophen-3-ol derivatives were characterized using spectroscopic techniques. We were able to obtain decent crystal of 5h and, therefore, the compound was subjected to X-ray crystallography for the corroboration of the assigned structure. Fig. 2 shows the ORTEP diagram of 5h and Table S1† reports the XRD parameters. The detailed analysis of the singel-crystal XRD data will be published eslewhere. The 1H NMR spectra exhibited a characteristic downfield signal around 13 ppm for the 3-OH moiety, indicating the formation of the enolic tautomer. The absence of this peak and the appearance of an up-field singlet around 4.5–5.5 ppm, assigned to the H-2, revealed the formation of the keto tautomer. Thus, out of the 19 benzo[b]thiophen-3-ols, most of the synthesized compounds were confirmed as enol analogs, whereas 5b, 5d, 5f and g, and 5j and k were obtained as the respective keto tautomers. The chemical shifts for aromatic protons were observed in the expected range from 6.00–8.50 ppm, whereas 13C NMR spectra exhibited the peak for the carbonyl moieties in the 187–200 ppm range.
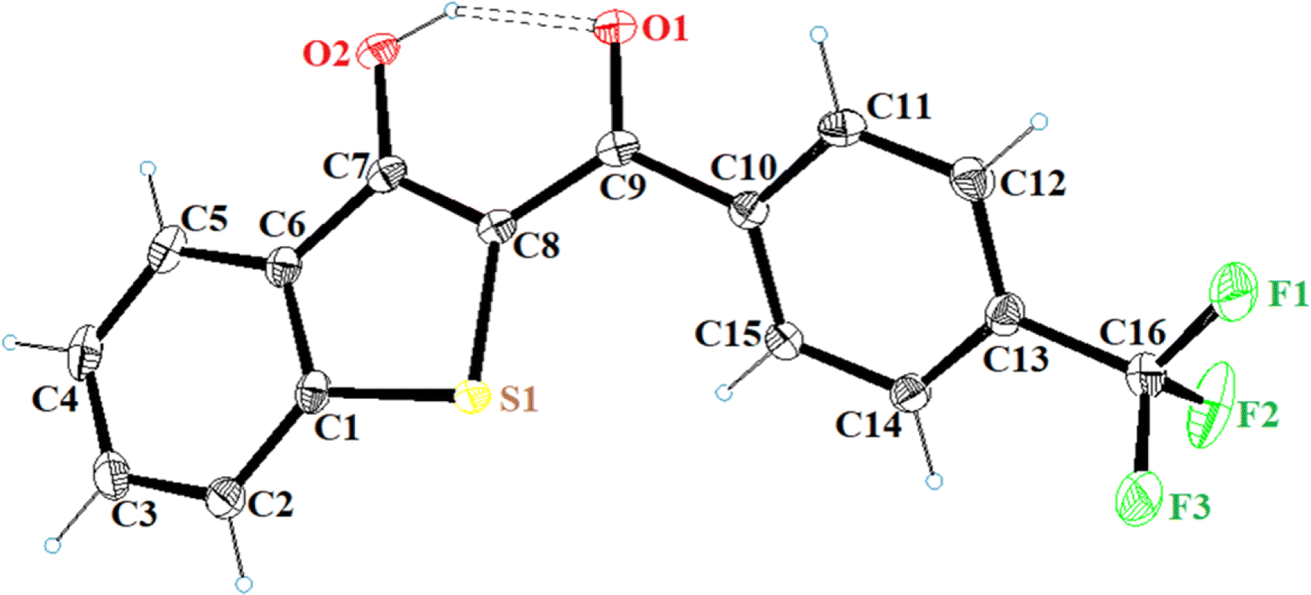 | ||
| Fig. 2 ORTEP diagram of 5h (CCDC 2297186). | ||
Based on the above results and the supporting literature,35 we propose a plausible reaction mechanism involving the initial nucleophilic attack of the sulfhydryl group of 6 on the α-carbon of the arylbromomethyl ketone (8) to eliminate the bromide ion. The SN2-type mechanism is facilitated by triethylamine, which abstracts a proton from S-atom to drive the formation of sulfanyl benzoic acid (9) and triethylammonium bromide (Et3NH+Br−). The abstraction of another proton from the α-carbon of 9 by triethylamine then leads to enolate I, which undergoes intramolecular cyclization via the attack on the carboxylic group in the presence of the triethylammonium ion to afford the intermediate II. Finally, eliminating the hydroxyl group in the form of H2O leads to the formation of benzo[b]thiophene 5 in the keto-form. The keto–enol tautomerism in the presence of acid (Et3NH+) or base (Et3N) yields 2-aroyl-benzo[b]thiophen-3-ol as the desired product, as shown in Scheme 3.
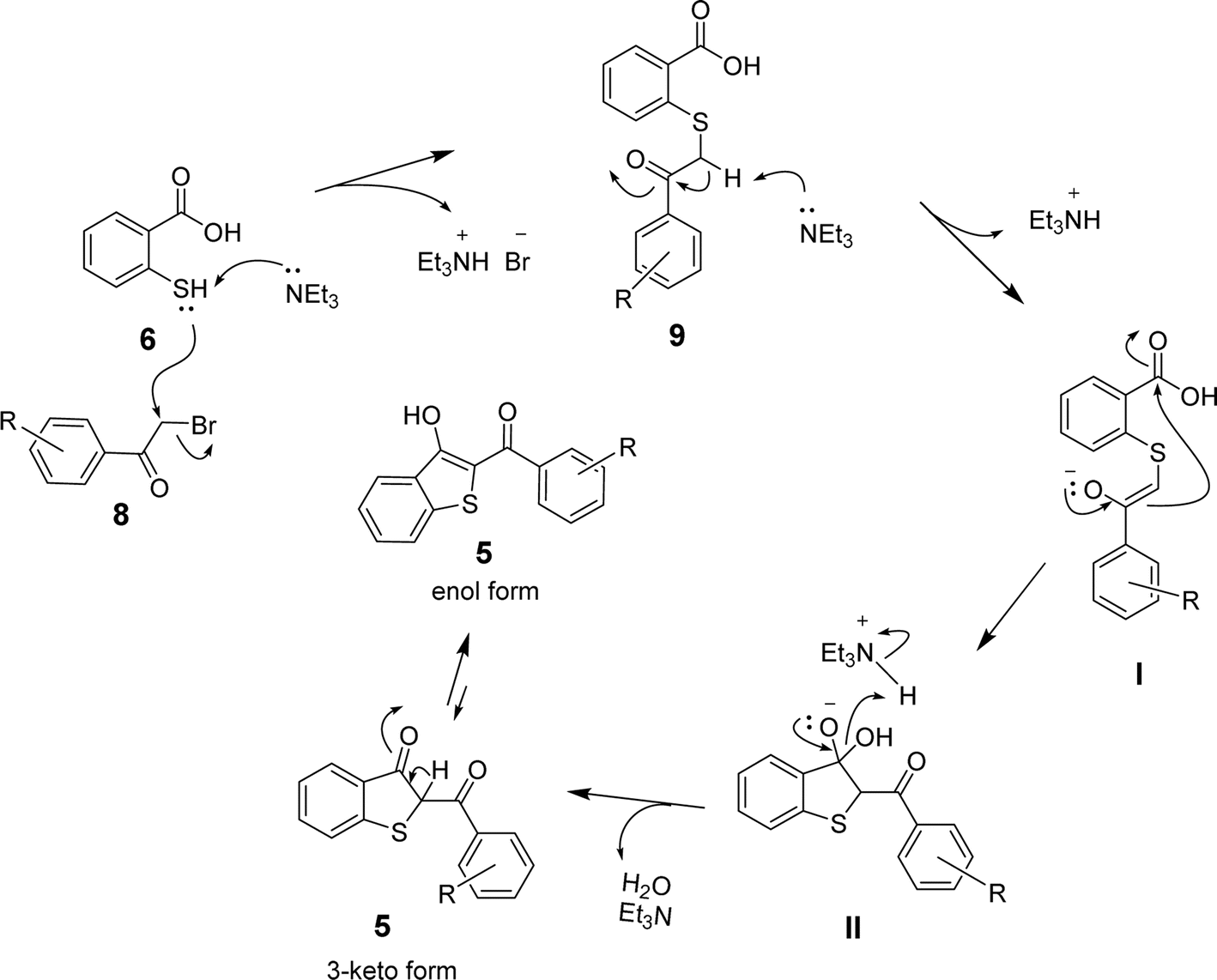 | ||
| Scheme 3 The proposed mechanism for the formation of 2-aroyl-benzo[b]-thiophen-3-ol (5) from 2-mercaptobenzoic acid (6) and arylbromomethyl ketone (8). | ||
After the successful construction of the 2-aroyl-benzo[b]thiophen-3-ol scaffold, the strategic utilization of the 3-hydroxyl group was examined for the introduction of an alkyne moiety. Various bases were attempted, and we found sodium hydride (NaH) to be a suitable base for converting selected benzothiophenes, 5a, 5h, and 5i to the corresponding benzothiophen-3-olates, which were individually reacted with propargyl bromide to afford the alkyne derivatives 11a, 11b, and 11c, respectively in 45–50% yields. Alkynes 11a–c were then reacted with benzyl azide 13, prepared in parallel by reacting benzyl bromide with sodium azide (NaN3) in the same flask, to accomplish the copper-catalyzed azide–alkyne cycloaddition (CUAAC) reaction in the presence of copper sulfate (CuSO4) and ascorbic acid. Thus, the target triazols 14a–c, novel hybrid pharmacophores containing benzo[b]thiophene and triazole together, were prepared in 50%, 42%, and 50% yields, respectively as shown in Scheme 4.
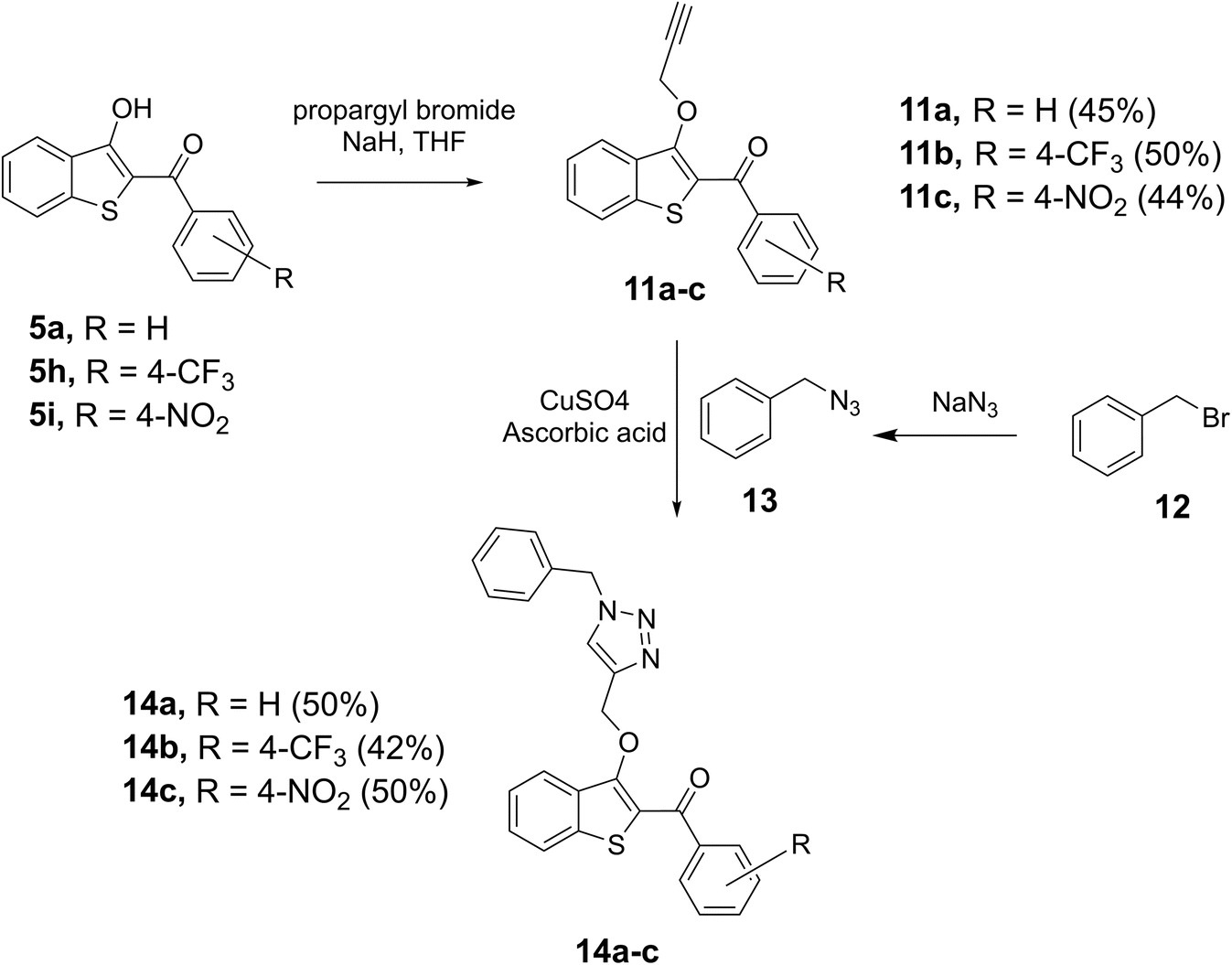 | ||
| Scheme 4 Synthesis of benzo[b]thiophene-triazole hybrids. | ||
3 Experimental
3.1. Materials
All the chemicals and reagents were purchased from different vendors (Merck, Sigma-Aldrich & TCI) and were used without further purification. Solvents for syntheses and chromatographic work were distilled and properly dried before use (if required). The progress of the reactions was monitored by TLC plates (aluminium sheets pre-coated with silica gel) by visualization under a UV lamp at 254 nm wavelength. The reaction mixtures were purified using silica gel column chromatography (pore size 60 Å, 230–400 mesh size, 40–63 μm). FTIR spectra were recorded using a Bruker Alpha Platinum AT by applying the ATR sampling technique through neat samples. NMR spectra were recorded in deuterated solvent using a Bruker 600 MHz NMR instrument (Avance Neo Bruker). GCMS data were obtained using a 1300 GC-MS (Thermo Scientific). Melting points were measured using the STUART SMP3 apparatus and have been reported uncorrected.3.2. General procedure for the synthesis of 2-acyl-benzo[b]thiophen-3-ols
To a stirred solution of 2-mercaptobenzoic acid (1 mmol) and arylbromomethyl ketone (1.2 mmol) in 5 mL of DMF, triethylamine (1 mL) was added, and the reaction mixture was stirred for 2–4 h at room temperature. The progress of the reaction was monitored through TLC analysis. After completion of the reaction, the mixture was poured into 30 mL of ice-cold water and the pH was adjusted to approximately 7 using a 2 N solution of hydrochloric acid. The precipitate of benzo[b]thiophen-3-ol was filtered via a Buchner funnel and washed with cold water. The ppt was recrystallized using hot methanol to afford the target.(3-Hydroxybenzo[b]thiophen-2-yl)(phenyl)methanone 5a. Yellow powder (205 mg, 80%), m.p. = 103–107 °C. 1H NMR (600 MHz, chloroform-d): δ (ppm) = 13.45 (s, 1H, OH), 8.08–8.05 (m, 3H, C6H4), 7.74 (d, J = 8.2 Hz, 1H, C6H4), 7.62 (t, J = 7.4 Hz, 1H, C6H5), 7.57–7.54 (m, 3H, C6H5), 7.44 (t, J = 7.6 Hz, 1H, C6H5). 13C NMR (150 MHz, chloroform-d) δ (ppm) = 191.9(CO), 165.5 (C-3), 140.9, 138.3, 132.7, 130.3, 130.2, 128.8, 128.5, 124.9, 124.1, 123.1 (C6H5 and C6H4) 109.7 (C-2). IR (cm−1): 3473 (broad OH); 1682 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O); 1590 (CC). EI-MS (m s−1): Calcd. for C15H10O2S [M]+: 254.30. GC-MS (EI) m/z (%relative intensity) 254.04 (M+, 100%), 197.05 (10%), 176.04 (95%).
O); 1590 (CC). EI-MS (m s−1): Calcd. for C15H10O2S [M]+: 254.30. GC-MS (EI) m/z (%relative intensity) 254.04 (M+, 100%), 197.05 (10%), 176.04 (95%).
(3-Hydroxybenzo[b]thiophen-2-yl)(p-tolyl)methanone 5b. Yellow amorphous solid (134 mg, 50%), m.p. = 99–101 °C. 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 8.05–8.03 (dd, J = 7.62 Hz, 1H), 7.92 (d, J = 8.16 Hz, 2H), 7.70 (m, 1H), 7.66 (m, 1H), 7.63 (m, 1H), 7.40 (d, J = 7.98 Hz, 2H), 5.69 (s, 1H), 2.42 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ (ppm) = 193.15, 191.39, 165.63, 144.95, 136.57, 132.27, 131.50, 129.60, 129.41, 127.94, 66.60, 21.79. EI-MS: Calcd. for C16H12O2S [M]+: 268.33. GC-MS (EI) m/z (%relative intensity) 268.06 (M+, 70%), 254.01(15%), 176.04 (100%).
(3-Hydroxybenzo[b]thiophen-2-yl)(4-hydroxyphenyl)methanone 5c. Light yellow amorphous solid (189 mg, 70%). 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 10.47 (bs, 1H), 7.96 (d, J = 8.4 Hz, 2H), 7.89–7.88 (d, J = 7.8 Hz, J = 1.2 Hz, 1H), 7.49–7.47 (m, 1H), 7.44–7.43 (m, 1H), 7.21 (t, J = 7.8 Hz, 1H), 6.88 (d, J = 9.0 Hz, 2H).13C NMR (150 MHz, DMSO-d6) δ (ppm) = 193.23, 168.00, 162.96, 140.89, 132.75, 131.73, 131.36, 127.53, 126.46, 124.51, 115.77. EI-MS: Calcd. for C15H10O3S [M]+: 270.30. GC-MS (EI) m/z (%relative intensity) 270.04 (M+, 100%), 254.04 (40%), 176.04 (90%).
2-(4-Methoxybenzoyl)benzo[b]thiophen-3(2H)-one 5d. Yellow amorphous solid (176 mg, 62%), m.p. = 182–183 °C. 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 8.05–7.99 (m, 4H), 7.71–7.62 (m, 2H), 7.16–7.10 (d, J = 7.8 Hz, 2H), 5.66 (s, 1H), 3.87 (s, 3H). 13C NMR (150 MHz, DMSO-d6): δ (ppm) = 193.09, 191.23, 165.64, 164.21, 137.25, 134.21, 132.94, 131.34, 130.77, 130.31, 128.29, 127.17, 123.96, 114.66, 67.53, 56.12. EI-MS: Calcd. for C16H12O3S [M]+: 284.33. GC-MS (EI) m/z (%relative intensity) 284.05 (M+, 70%), 254.04 (30%), 176.04 (100%).
(4-Aminophenyl)(3-hydroxybenzo[b]thiophen-2-yl)methanone 5e. Light Yellow powder (40 mg, 45%); mp 164–166 °C. 1H NMR (600 MHz, DMSO): δ (ppm) = 11.10 (brs, 1H), 7.90 (d, J = 7.2 Hz, 2H), 7.47 (d, J = 8.4 Hz, 2H), 7.21 (t, J = 7.2 Hz, 2H), 7.09 (t, J = 7.2 Hz, 2H), 4.40 (brs, 2H–NH2). 13C NMR (150 MHz, DMSO-d6): δ (ppm) = 189.08, 164.56, 139.58, 130.05, 129.99, 129.20, 123.76, 123.03, 121.89, 114.92, 114.77. EI-MS: Calcd. for C15H11NO2S [M]+: 269.32 GC-MS (EI) m/z (%relative intensity) 269.05 (M+, 30%), 254.04 (70%), 176.04 (100%).
(4-Chlorophenyl)(3-hydroxybenzo[b]thiophen-2-yl)methanone 5f. Yellow powder (240 mg, 88%), m.p. = 164–166 °C. 1H NMR (600 MHz, chloroform-d) δ (ppm) = 8.17 (d, J = 7.8 Hz, 1H), 7.85 (d, J = 7.8 Hz, 2H), 7.58 (m, 2H), 7.51 (m, 1H), 7.30 (d, J = 7.8 Hz, 2H), 5.53 (s, 1H). 13C NMR (150 MHz, chloroform-d) δ (ppm) = 198.28, 191.52, 165.75, 145.07, 136.70, 132.40, 131.62, 129.75, 129.72, 129.53, 128.10, 128.06. EI-MS: Calcd. for C15H9ClO2S [M + H]+: 288.75. GC-MS (EI) m/z (%relative intensity) 288.0 (M+, 70%), 176.04 (100%), 112.01 (40%).
(4-Bromophenyl)(3-hydroxybenzo[b]thiophen-2-yl)methanone 5g. Yellow amorphous solid (180 mg, 64%), m.p. = 168–170 °C. 1H NMR (600 MHz, chloroform-d): δ (ppm) = 8.15 (m, 2H), 7.87 (m, 2H), 7.40 (m, 4H), 7.20 (t, J = 7.3 Hz, 1H), 4.46 (s, 1H). 13C NMR (150 MHz, chloroform-d): δ (ppm) = 193.91, 168.10, 166.50, 164.88, 140.19, 132.78, 132.50, 132.18, 132.12, 131.31, 126.44, 124.64, 116.34, 116.20. EI-MS: Calcd. for C15H9BrO2S [M]+: 333.20. GC-MS (EI) m/z (%relative intensity) 331.95 (M + 1, 97.8%), 333.95 (M+, 100%), 176.04 (70%).
(3-Hydroxybenzo[b]thiophen-2-yl)[4-(trifluoromethyl)phenyl]methanone 5h. Yellow amorphous solid (258 mg, 80%), m.p. = 150–152 °C. 1H NMR (600 MHz, DMSO-d6) δ (ppm) = 11.96 (s, 1H), 8.08 (d, J = 8.0 Hz, 1H), 8.04 (d, J = 8.0 Hz, 2H), 7.98 (d, J = 8.16 Hz, 1H), 7.93 (d, J = 8.1 Hz, 2H), 7.62 (t, J = 7.3 Hz, 1H), 7.49 (t, J = 7.3 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ (ppm) = 206.95, 158.04, 142.89, 139.93, 131.85, 130.20 (q, 2JCF = 32.07 Hz), 129.50, 125.80, 125.78 (q, 3JCF = 3.85 Hz), 125.38, 124.11 (q, 1JCF = 272.98 Hz), 124.07, 123.466, 114.01. EI-MS: Calcd. for C16H9F3O2S [M]+: 322.30. GC-MS (EI) m/z (%relative intensity) 322.03 (M+, 80%), 176.04 (100%), 146.03 (20%).
(3-Hydroxybenzo[b]thiophen-2-yl)(4-nitrophenyl)methanone 5i. Orange powder (230 mg, 88%), m.p. = 202–204 °C. 1H NMR (600 MHz, DMSO-d6): δ (ppm) = 12.01 (s, 1H), 8.37 (d, J = 8.7 Hz, 2H), 8.36 (m, 3H), 7.98 (d, J = 8.4 Hz, 1H), 7.62 (t, J = 1H), 7.49 (t, 1H). 13C NMR (150 MHz, DMSO-d6) δ (ppm) = 193.09, 164.22, 137.25, 134.21, 132.95, 131.34, 131.16, 130.75, 130.71, 130.31, 128.30, 127.18, 123.96. EI-MS: Calcd. for C15H9NO4S [M]+: 299.30. GC-MS (EI) m/z (%relative intensity) 299.03 (M+, 70%), 176.04 (100%), 123.03 (20%).
2-(2,4-Dimethoxybenzoyl)benzo[b]thiophen-3(2H)-one 5j. Light yellow amorphous solid (220 mg, 70%) m.p. = 200–204 °C. 1H NMR (600 MHz, DMSO-d6) δ (ppm) = 8.19–8.18 (dd, J = 7.8 Hz, J = 1.2 Hz, 1H), 8.00 (d, J = 8.8 Hz, 1H), 7.59 (m, 1H), 7.55 (m, 1H), 7.50 (m, 1H), 6.58 (dd, J = 8.4 Hz, J = 1.4 Hz, 1H), 6.47 (d, J = 2.4 Hz, 1H), 5.41 (s, 1H), 3.94 (s, 3H), 3.88 (s, 3H). 13C NMR (150 MHz, DMSO-d6) δ (ppm) = 193.35, 190.73, 165.87, 165.49, 161.52, 136.48, 133.79, 133.24, 132.01, 131.48, 129.35, 128.79, 117.20, 105.92, 98.05, 55.66, 55.60. EI-MS: Calcd. for C17H14O4S [M]+: 314.36. GC-MS (EI) m/z (%relative intensity) 314.06 (M+, 80%), 254.04 (70%), 176.04 (100%).
(2,4-Dichlorophenyl)(3-hydroxybenzo[b]thiophen-2-yl)methanone 5k. Yellow amorphous solid (290 mg, 90%), m.p. = 149–152 °C. 1H NMR (600 MHz, chloroform-d) δ (ppm) = 8.08 (dd, J = 7.8 Hz, 1H), 7.50 (t, J = 8.4 Hz, 1H), 7.44 (d, J = 2.4 Hz, 1H), 7.37 (m, 3H), 7.28 (m, 1H), 7.25 (m, 1H), 4.37 (s, 1H).13C NMR (150 MHz, chloroform-d) δ (ppm) = 196.78, 170.51, 140.40, 138.21, 135.73, 133.51, 132.55, 132.18, 131.03, 130.44, 127.52, 126.96, 126.73, 125.26. EI-MS: Calcd. for C15H8Cl2O2S [M]+: 323.19. GC-MS (EI) m/z (%relative intensity) 321.96 (M+, 80%), 176.04 (100%), 145.97 (30%).
(3-Chloro-4-fluorophenyl)(3-hydroxybenzo[b]thiophen-2-yl)methanone 5l. Yellow amorphous solid (230 mg, 75%) m.p. = 167–169 °C. 1H NMR (600 MHz, chloroform-d): δ (ppm) = 11.94 (s, 1H), 8.08 (m, 2H), 7.98 (d, J = 8.16 Hz, 1H), 7.90 (m. 1H), 7.62 (m, 2H), 7.48 (m, 1H). 13C NMR (150 MHz, chloroform-d) δ (ppm) = 195.18, 167.93, 140.56, 136.01, 134.11, 132.81 (d, 1JCF = 245.26 Hz), 131.41, 129.25 (d, 3JCF = 8.74 Hz), 129.06, 126.50 (d, 2JCF = 23.15 Hz), 124.63. EI-MS: Calcd. for C15H8ClFO2S [M]+: 306.74. GC-MS (EI) m/z (%relative intensity) 307.99 (M + 1, 36.6%), 305.99 (M+, 100%), 176.04 (90%).
(4-Fluoro-3-(trifluoromethyl)phenyl)(3-hydroxybenzo[b]thiophen-2-yl)methanone 5m. Yellow amorphous solid (238 mg, 70%), m.p. = 160–162 °C. 1H NMR (600 MHz, DMSO-d6) δ (ppm) = 11.91 (s, 1H), 8.22 (m, 2H), 7.99 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 7.8 Hz, 1H), 7.65 (t, J = 8.8 Hz, 1H), 7.55 (t, J = 7.2 Hz, 1H), 7.40 (t, J = 7.2 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ (ppm) = 190.00, 162.00, 140.19 (d, 1JCF = 245.26 Hz), 136.53, 136.26, 129.84(m, 2JCF = 32.93 Hz), 128.36, 124.79 (m, 3JCF = 3.98 Hz), 124.31 (q, 1JCF = 273.49 Hz), 123.90(d, 3JCF = 8.74 Hz), 117.56 (d, 2JCF = 23.15 Hz), 117.42. EI-MS: Calcd. for C16H8F4O2S [M]+: 340.29. GC-MS (EI) m/z (%relative intensity) 341.02 (M+, 18.3%), [M]+: 340.02 (100%), 176.04 (100%).
(5-Fluoro-2-hydroxyphenyl)(3-hydroxybenzo[b]thiophen-2-yl)methanone 5n. White amorphous solid (224 mg, 78%) m.p. = 166–169 °C. 1H NMR (600 MHz, DMSO-d6) δ (ppm) = 13.48 (bs, 1H), 8.04–8.02 (dd, J = 7.8 Hz, J = 1.8 Hz, 1H), 7.93–7.91 (dd, J = 7.8 Hz, J = 1.2 Hz, 1H), 7.62 (m, 1H), 7.58–7.55 (m, 1H), 7.52–7.51 (d, J = 8.4 Hz, 1H), 7.38–7.34 (m, 1H), 7.19 (t, J = 7.8 Hz, 1H), 3.37 (bs, 1H). 13C NMR (150 MHz, DMSO-d6) δ (ppm) = 200.46, 168.09, 139.40, 133.76, 132.83, 132.06 (d, 1JCF = 245.26 Hz), 131.83, 131.44, 128.54 (d, 3JCF = 8.66 Hz), 126.98, 126.47 (d, 2JCF = 24.11 Hz), 125.47, 124.90. EI-MS: Calcd. for C15H9FO3S [M]+: 288.29. GC-MS (EI) m/z (%relative intensity) 289.03 (M + 1, 17.3%), 288.03 (M+, 100%), 176.04 (70%)
(2-Hydroxy-5-methylphenyl)(3-hydroxybenzo[b]thiophen-2-yl)methanone 5o. Yellow solid (227 mg, 85%) m.p. = 130–132 °C. 1H NMR (600 MHz, DMSO-d6) δ (ppm) = 12.87 (s, 1H), 12.16 (s, 1H), 7.85 (m, 2H), 7.56 (m, 3H), 7.54 (dd, J = 7.8 Hz, J = 1.2 Hz, 1H), 6.90 (d, J = 7.8 Hz, 2H). 13C NMR (150 MHz, DMSO-d6) δ (ppm) = 195.18, 167.93, 140.56, 136.01, 134.11, 132.81, 131.41, 129.25, 129.06, 126.50, 124.63, 31.17. EI-MS: Calcd. for C16H12O3S [M]+: 284.33. GC-MS (EI) m/z (%relative intensity) 284.03 (M+, 100%), 254.04 (30%), 176.04 (70%)
(3-Hydroxybenzo[b]thiophen-2-yl)(naphthalen-2-yl)methanone 5p. Yellow amorphous solid (210 mg, 70%) m.p. = 126–128 °C. 1H NMR (600 MHz, DMSO-d6) δ (ppm) = 12.85 (s, 1H), 7.96 (m, 2H), 7.70 (m, 3H), 7.54 (m, 1H), 7.47 (m, 3H), 7.06 (d, J = 8.4 Hz, 1H), 6.98 (t, J = 7.8 Hz, 1H). 13C NMR (150 MHz, DMSO-d6) δ (ppm) = 193.90, 163.61, 145.53, 136.47, 134.60, 130.98, 130.56, 129.69, 129.00, 128.71, 128.44, 120.02, 118.90, 118.79. EI-MS: Calcd. for C19H12O2S [M]+: 304.36 GC-MS (EI) m/z (%relative intensity) 304.06 (M+, 60%), 176.04 (100%), 128.06 (30%).
(3-Hydroxybenzo[b]thiophen-2-yl)(naphthalen-1-yl)methanone 5q. Yellow amorphous solid (231 mg, 76%) m.p. = 162–166 °C. 1H NMR (600 MHz, DMSO-d6) δ (ppm) = 13.18 (s, 1H), 8.92 (m, 1H), 8.20 (m, 2H), 7.96 (m, 2H), 7.66 (m, 1H), 7.50 (m, 1H), 7.72 (m, 3H), 7.57 (m, 1H). 13C NMR (150 MHz, DMSO-d6) δ (ppm) = 195.10, 167.91, 140.73, 135.47, 134.60, 130.98, 130.56, 129.69, 129.01, 128.71, 128.44, 124.35, 124.03. EI-MS: Calcd. for C19H12O2S [M]+: 304.36. GC-MS (EI) m/z (%relative intensity) 304.06 (M+, 60%), 176.04 (100%), 128.06 (30%).
(6-Bromopyridin-2-yl)(3-hydroxybenzo[b]thiophen-2-yl)methanone 5r. Light Yellow amorphous solid (233 mg, 70%), m.p. = 138–142 °C. 1H NMR (600 MHz, DMSO-d6) δ (ppm) = 13.17 (s, 1H), 8.02 (s, 1H), 7.99 (m, 2H), 7.90 (dd, J = 7.8 Hz, J = 1.2 Hz, 1H), 7.50 (t, J = 7.2 Hz, 1H), 7.45 (d, J = 7.8 Hz, 1H), 7.22 (t, J = 7.2 Hz, 1H).13C NMR (150 MHz, DMSO-d6) δ (ppm) = 194.10, 167.96, 153.32, 141.52, 141.17, 140.30, 133.08, 132.92, 131.47, 128.46, 126.31, 124.77, 122.14. EI-MS: Calcd. for C14H8BrNO2S [M]+: 334.19. GC-MS (EI) m/z (%relative intensity) 332.95 (M + 1, 98.3%), 334.94 (M+, 100%), 176.04 (70%)
(3-Hydroxybenzo[b]thiophen-2-yl)(thiophen-2-yl)methanone 5s. Yellow amorphous solid, (161 mg, 62%), m.p. = 182–184 °C. 1H NMR (600 MHz, chloroform-d) δ (ppm) = 13.24 (s, 1H),8.04 (m, 1H), 7.88 (m, 1H), 7.77 (m, 1H), 7.60 (m, 1H), 7.47 (m, 1H), 7.20 (m, 1H). 13C NMR (150 MHz, chloroform-d) δ (ppm) = 181.13, 166.07, 144.59, 140.22, 134.43, 132.97, 131.79, 130.97, 130.63, 125.29, 124.23, 124.08, 123.51, 123.27, 123.20. EI-MS: Calcd. for C13H8O2S2 [M]+: 260.33. GC-MS (EI) m/z (%relative intensity) 260.00 (M+, 60%), 176.04 (100%), 84.06 (30%).
3.3. General procedure for the synthesis of phenyl(3-(prop-2-yn-1-yloxy)benzo[b]thiophen-2-yl)methanone
In a 50 mL round-bottom flask, 1 mmol of a benzothiophene derivative (compound 5) and 4 mmol of NaH were dissolved in 8 mL of DMF. To this stirred solution, 10 mmol of propargyl bromide was added. The stirring was continued at room temperature for approximately 1 hour. After this time, the reaction progress was assessed using TLC, revealing the presence of some benzothiophene derivative remaining in its protonated form. Consequently, 4 mmol of NaH and 5 mmol of propargyl bromide were added. Following overnight stirring, the reaction mixture was transferred to a separating funnel and dissolved in a solution consisting of 5 mL of ice-cold deionized water, 5 mL of brine, and 5 mL of ethyl acetate. The excess ethyl acetate was removed using a rotary evaporator, and the organic layer was collected in a clean vial. A brownish-yellow semi-liquid was obtained, which was ultimately purified through column chromatography to yield a brownish-yellow solid.Phenyl(3-(prop-2-yn-1-yloxy)benzo[b]thiophen-2-yl)methanone 11a. Compound 11a was synthesized from 5a following the general procedure. Brownish-yellow solid, (45%). 1H NMR (600 MHz, chloroform-d) δ (ppm): 8.050–8.080 (m, 3H, Ar), 7.775–7.761 (m, 1H, benzothiphene), 7.765–7.761 (m, 1H, benzothiphene), 7.510–7.490 (m, 3H, Ar), 7.463–7.445 (m, 1H, benzothiophene), 4.678–4.567 (m, 2H, CH2), 2.226–2.222 (m, 1H, CH). 13C NMR (150 MHz, chloroform-d) δ (ppm) = 189.27, 152.17, 138.68, 138.33, 133.98, 132.88, 129.63, 128.26, 128.08, 124.83, 124.73, 123.46, 122.90, 78.03, 76.89, 62.26. EI-MS (m s−1): Calcd. For C18H12O2S [M]+: 292.35. GC-MS (EI) m/z (%relative intensity) 260.00 (M+, 60%), 188.04 (100%), 84.06 (30%).
(3-(Prop-2-yn-1-yloxy)benzo[b]thiophen-2-yl)(4-(trifluoromethyl)phenyl)methanone 11b. Compound 11b was synthesized from 5h by following the general procedure. Brown solid, (50%). 1H NMR (600 MHz, chloroform-d) δ (ppm): 8.07 (d, J = 8.0 Hz, 1H), 8.01 (d, J = 8.0 Hz, 2H), 7.98 (d, J = 8.16 Hz, 1H), 7.92 (d, J = 8.1 Hz, 2H), 7.62 (t, J = 7.3 Hz, 1H), 7.53 (t, J = 7.3 Hz, 1H), 4.71 (s, 2H), 3.53 (s, 1H). 13C NMR (150 MHz, DMSO-d6) δ (ppm) = 206.95, 152.70, 142.06, 138.83, 133.44, 130.26 (q, 2JCF = 32.07 Hz), 129.25, 126.03, 125.87 (q, 3JCF = 3.85 Hz), 125.73, 125.71, 124.14 (q, 1JCF = 272.98 Hz), 123.92, 80.75, 62.64. EI-MS (m s−1): Calcd. For C19H11F3O2S [M]+: 360.35. GC-MS (EI) m/z (%relative intensity) 360.04 (M+, 60%), 188.03 (100%), 174.03 (30%).
(4-Nitrophenyl)(3-(prop-2-yn-1-yloxy)benzo[b]thiophen-2-yl)methanone 11c. Compound 11c was synthesized from 5i by following the general procedure. Brown solid, (44%). 1H NMR (600 MHz, chloroform-d) δ (ppm): 7.90 (m, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.72 (m, 2H), 7.55 (d, J = 8.0 Hz, 2H), 7.35 (dd, J = 7.8 Hz, J = 1.2 Hz, 1H), 7.25 (D, 1H), 4.84 (s, 1H), 4.24 (s, 2H). 13C NMR (150 MHz, DMSO-d6) δ (ppm) = 194.34, 167.79, 144.51, 132.48, 132.44, 131.55, 131.30, 131.10, 130.90, 129.35, 128.87, 128.81, 126.95, 80.95, 68.16, 52.75. EI-MS (m s−1): Calcd. For C19H11F3O2S [M]+: 337.35 GC-MS (EI) m/z (%relative intensity) 337.04 (M+, 60%), 188.03 (100%), 151.03 (30%).
3.4. General procedure for the synthesis of (3-((1-benzyl-1H-1,2,3-triazol-4-yl)methoxy)benzo[b]thiophen-2-yl)(phenyl)methanone
In a 25 mL round-bottom flask, a solution was prepared by dissolving 1.5 mmol of benzyl bromide and 1 mmol of the acetylene derivative. To this stirred solution, 1 mmol of sodium azide was added, along with a 2 mL mixture of tBuOH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (1 mL:1 mL), and the reaction mixture was stirred at room temperature for approximately 5 minutes. Subsequently, 0.1 mmol of sodium ascorbate and 0.64 mmol of Cu(II) sulfate pentahydrate were added. The reaction mixture was stirred at 60 °C for 2–3 hours. The completion of the reaction was confirmed by TLC, which indicated the consumption of the starting material and the formation of the desired product. After overnight stirring, the reaction mixture was dissolved in 5 mL of ice-cold deionized water, followed by the dropwise addition of 2 mL of aqueous ammonia. The mixture was stirred for 5 minutes. Finally, the reaction mixture was extracted with 12 mL of ethyl acetate. A yellow liquid was obtained, which was ultimately purified through column chromatography to yield a yellowish liquid.
:H2O (1 mL:1 mL), and the reaction mixture was stirred at room temperature for approximately 5 minutes. Subsequently, 0.1 mmol of sodium ascorbate and 0.64 mmol of Cu(II) sulfate pentahydrate were added. The reaction mixture was stirred at 60 °C for 2–3 hours. The completion of the reaction was confirmed by TLC, which indicated the consumption of the starting material and the formation of the desired product. After overnight stirring, the reaction mixture was dissolved in 5 mL of ice-cold deionized water, followed by the dropwise addition of 2 mL of aqueous ammonia. The mixture was stirred for 5 minutes. Finally, the reaction mixture was extracted with 12 mL of ethyl acetate. A yellow liquid was obtained, which was ultimately purified through column chromatography to yield a yellowish liquid.
(3-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)benzo[b]thiophen-2-yl)(phenyl)methanone 14a. Compound 14a was synthesized from 11a by following the general procedure. Brown solid (50%). 1H NMR (600 MHz, chloroform-d): δ (ppm) = 7.89 (m, 3H), 7.80 (m, 1H), 7.52 (m, 2H), 7.40 (m, 6H), 7.20 (m, 2H), 6.97 (s, 1H), 5.44 (s, 2H), 5.04 (s, 2H). 13C NMR (150 MHz, chloroform-d) δ (ppm) = 189.32, 152.85, 143.36, 138.93, 138.31, 134.31, 133.71, 132.68, 129.58, 129.14, 128.84, 128.17, 128.10, 124.90, 123.38, 123.22, 123.02, 68.56, 54.10. EI-MS (m s−1): Calcd. For C25H19N3O2S [M]+: 425.51 GC-MS (EI) m/z (%relative intensity) 425.12 (M + 1, 29%), 426.12 (M+, 100%).
(3-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)benzo[b]thiophen-2-yl)(4-(trifluoromethyl)phenyl)methanone 14b. Compound 14b was synthesized from 11b by following the general procedure. Brownish Yellow solid (42%). 1H NMR (600 MHz, chloroform-d): δ (ppm) = 7.75 (m, 2H), 7.60 (s, 1H), 7.38 (m, 6H), 7.32 (m, 4H), 6.49 (s, 1H), 5.56 (s, 2H), 5.34 (s, 2H). 13C NMR (150 MHz, chloroform-d) δ (ppm) = 206.10, 150.00, 144.30, 137.64, 134.40, 132.05 (q, 2JCF = 32.07 Hz), 129.19, 128.86, 128.13, 125.25 (q, 3JCF = 3.85 Hz), 123.73 (q, 1JCF = 272.98 Hz), 122.80, 120.94, 97.51, 63.90, 54.30. EI-MS (m s−1): Calcd. For C26H18F3N3O2S [M]+: 493.50 GC-MS (EI) m/z (%relative intensity) 494.11 (M + 1, 29%), 493.11 (M+, 100%).
(3-((1-Benzyl-1H-1,2,3-triazol-4-yl)methoxy)benzo[b]thiophen-2-yl)(3-nitrophenyl)methanone 14c. Compound 14c was synthesized from 11c by following the general procedure. Brown solid (50%). 1H NMR (600 MHz, chloroform-d): δ (ppm) = 8.84 (t, J = 7.8 Hz, 1H), 8.40 (m, 1H), 8.37 (m, 1H), 7.65 (m, 3H), 7.38 (m, 5H), 7.33 (m, 3H), 5.56 (s, 2H), 5.50 (s, 2H). 13C NMR (150 MHz, chloroform-d) δ (ppm) = 200.01, 164.34, 148.23, 142.52, 135.42, 134.22, 131.48, 129.69, 129.22, 128.95, 128.22, 127.62, 124.76, 124.05, 58.73, 54.34. EI-MS (m s−1): Calcd. for C25H18N4O4S [M]+: 470.50. GC-MS (EI) m/z (%relative intensity) 471.11 (M + 1, 29%) [M]+: 470.10 (M+, 100%).
4 Conclusion
In conclusion, an expeditious one-pot method has been developed to synthesize 2-aroyl-benzo[b]thiophen-3-ols. The method is applicable to a wide range of arylbromomethyl ketones, demonstrating its substrate tolerability. The synthesis of benzothiophene was extended further toward introducing the triazole moiety, exemplifying the efficiency and selectivity inherent in click chemistry methodologies. The outlined approach not only underscores the versatility of click chemistry but also contributes to the expanding repertoire of synthetic methods for constructing complex molecular architectures with potential applications in medicinal and materials science.Author contributions
Asma Mukhtar: syntheses of benzo[b]thiophenes and writing the manuscript. Arif Hussain and Faiza Younus: assistance in the synthesis of benzo[b]thiophenes. Sammer Yousuf: X-ray data acquisition and curation, Muhammad Saeed: supervision, experimental planning project administration, writing – review & editing, funding acquisition.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by an internal funding from Lahore University of Management Sciences (LUMS) under the catoegory of the faculty initiative funds (FIF # 800/850-CH) awarded to MS.Notes and references
- M. C. Egbujor, P. Tucci, U. C. Onyeije, C. N. Emeruwa and L. Saso, Molecules, 2023, 28, 2751 CrossRef CAS PubMed.
- C. Rizzo, S. Amata, I. Pibiri, A. Pace, S. Buscemi and A. Palumbo Piccionello, Int. J. Mol. Sci., 2023, 24, 7728 CrossRef CAS PubMed.
- E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274 CrossRef CAS PubMed.
- M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247–44311 RSC.
- J. Shearer, J. L. Castro, A. D. G. Lawson, M. MacCoss and R. D. Taylor, J. Med. Chem., 2022, 65, 8699–8712 CrossRef CAS PubMed.
- S. Sharma, C. C. Malakar and V. Singh, Asian J. Org. Chem., 2020, 9, 1857–1868 CrossRef CAS.
- S. Sharma, D. Singh, S. Kumar, Vaishali, R. Jamra, N. Banyal, Deepika, C. C. Malakar and V. Singh, Beilstein J. Org. Chem., 2023, 19, 231–244 CrossRef CAS PubMed.
- R. S. Keri, K. Chand, S. Budagumpi, S. Balappa Somappa, S. A. Patil and B. M. Nagaraja, Eur. J. Med. Chem., 2017, 138, 1002–1033 CrossRef CAS PubMed.
- T. Al-Warhi, N. M. Rashad, H. Almahli, M. M. Abdel-Aziz, Z. M. Elsayed, M. I. Shahin and W. M. Eldehna, Arch. Pharm., 2024, 357, e2300529 CrossRef PubMed.
- T. Barbier, A. Barbry, J. Magand, C. Badiou, F. Davy, A. Baudouin, Y. Queneau, O. Dumitrescu, G. Lina and L. Soulere, Biomolecules, 2022, 12, 131 CrossRef CAS PubMed.
- P. Guglielmi, D. Secci, A. Petzer, D. Bagetta, P. Chimenti, G. Rotondi, C. Ferrante, L. Recinella, S. Leone, S. Alcaro, G. Zengin, J. P. Petzer, F. Ortuso and S. Carradori, J. Enzyme Inhib. Med. Chem., 2019, 34, 1511–1525 CrossRef CAS PubMed.
- M. Gerard, L. Caroline and K. Micheline, French Pat., FR2862646, 2005 Search PubMed.
- T. W. Burke and C. L. Walker, Gynecol. Oncol., 2003, 90, S40–S46 CrossRef CAS PubMed.
- E. Esposito, A. Iacono, G. M. Raso, M. Pacilio, A. Coppola, R. Di Carlo and R. Meli, Endocrinology, 2005, 146, 3301–3308 CrossRef CAS PubMed.
- S. Gizzo, C. Saccardi, T. S. Patrelli, R. Berretta, G. Capobianco, S. Di Gangi, A. Vacilotto, A. Bertocco, M. Noventa, E. Ancona, D. D'Antona and G. B. Nardelli, Obstet. Gynecol. Surv., 2013, 68, 467–481 CrossRef PubMed.
- I. Y. Kim, B. C. Kim, D. H. Seong, D. K. Lee, J. M. Seo, Y. J. Hong, H. T. Kim, R. A. Morton and S. J. Kim, Cancer Res., 2002, 62, 5365–5369 CAS.
- S. S. Ko and V. C. Jordan, Expert Opin. Pharmacother., 2011, 12, 657–674 CrossRef CAS PubMed.
- C. Marchisano-Karpman and M. W. DeGregorio, IDrugs, 2003, 6, 880–885 CAS.
- P. N. Munster, Expert Opin. Invest. Drugs, 2006, 15, 317–326 CrossRef CAS PubMed.
- M. B. Sporn, Clin. Cancer Res., 2004, 10, 5313–5315 CrossRef CAS PubMed.
- R. Iqbal, M. Yaqub, D. E. Oprea-Lager, Y. Liu, A. M. Luik, A. P. Beelen, R. C. Schuit, A. D. Windhorst, R. Boellaard and C. W. Menke-van der Houven van Oordt, J. Nucl. Med., 2022, 63, 694–699 CAS.
- K. Jhaveri, D. Juric, Y. S. Yap, S. Cresta, R. M. Layman, F. P. Duhoux, C. Terret, S. Takahashi, J. Huober, N. Kundamal, Q. Sheng, A. Balbin, Y. Ji, W. He, A. Crystal, S. De Vita and G. Curigliano, Clin. Cancer Res., 2021, 27, 5760–5770 CrossRef CAS PubMed.
- G. S. Tria, T. Abrams, J. Baird, H. E. Burks, B. Firestone, L. A. Gaither, L. G. Hamann, G. He, C. A. Kirby, S. Kim, F. Lombardo, K. J. Macchi, D. P. McDonnell, Y. Mishina, J. D. Norris, J. Nunez, C. Springer, Y. Sun, N. M. Thomsen, C. Wang, J. Wang, B. Yu, C. L. Tiong-Yip and S. Peukert, J. Med. Chem., 2018, 61, 2837–2864 CrossRef CAS PubMed.
- J. Ceramella, A. Caruso, M. A. Occhiuzzi, D. Iacopetta, A. Barbarossa, B. Rizzuti, P. Dallemagne, S. Rault, H. El-Kashef, C. Saturnino, F. Grande and M. S. Sinicropi, Eur. J. Med. Chem., 2019, 181, 111583 CrossRef PubMed.
- N. R. Penthala, V. N. Sonar, J. Horn, M. Leggas, J. S. Yadlapalli and P. A. Crooks, Medchemcomm, 2013, 4, 1073–1078 RSC.
- C. V. Do, A. Faouzi, C. Barette, A. Farce, M. O. Fauvarque, E. Colomb, L. Catry, O. Berthier-Vergnes, M. Haftek, R. Barret and T. Lomberget, Bioorg. Med. Chem. Lett., 2016, 26, 174–180 CrossRef CAS PubMed.
- R. Romagnoli, D. Preti, E. Hamel, R. Bortolozzi, G. Viola, A. Brancale, S. Ferla, G. Morciano and P. Pinton, Bioorg. Chem., 2021, 112, 104919 CrossRef CAS PubMed.
- D. J. Witter, S. Belvedere, L. Chen, J. P. Secrist, R. T. Mosley and T. A. Miller, Bioorg. Med. Chem. Lett., 2007, 17, 4562–4567 CrossRef CAS PubMed.
- F. Batool, E. U. Mughal, K. Zia, A. Sadiq, N. Naeem, A. Javid, Z. Ul-Haq and M. Saeed, J. Biomol. Struct. Dyn., 2022, 40, 3777–3788 CrossRef CAS PubMed.
- F. Batool, M. Saeed, H. N. Saleem, L. Kirschner and J. Bodem, Pathogens, 2021, 10, 464 CrossRef CAS PubMed.
- H. N. Saleem, S. Kousar, A. H. Jiskani, I. Sohail, A. Faisal and M. Saeed, Arch. Pharm., 2023, 356, e2300292 CrossRef PubMed.
- H. A. Younus, M. Saeed, A. Mahmood, M. S. K. Jadoon, A. Hameed, A. Asari, H. Mohamad, J. Pelletier, J. Sevigny, J. Iqbal and M. Al-Rashida, Bioorg. Chem., 2023, 134, 106450 CrossRef CAS PubMed.
- P. Trapani, L. Kvapil, P. Hradil and M. Soural, Synlett, 2018, 29, 810–814 CrossRef CAS.
- D. W. Plank and J. B. Howard, J. Biol. Chem., 1988, 263, 8184–8189 CrossRef CAS PubMed.
- G. A.-N. Gohar, J. Phys. Org. Chem., 2012, 25, 343–350 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2297186. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra01146e |
| This journal is © The Royal Society of Chemistry 2024 |