
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of ciprofloxacin-linked 1,2,3-triazole conjugates as potent antibacterial agents using click chemistry: exploring their function as DNA gyrase inhibitors via in silico- and in vitro-based studies†
Upendra Kumar Patel a,
Punit Tiwarib,
Ragini Tilakb,
Gaurav Joshic,
Roshan Kumard and
Alka Agarwal*a
a,
Punit Tiwarib,
Ragini Tilakb,
Gaurav Joshic,
Roshan Kumard and
Alka Agarwal*a
aDepartment of Medicinal Chemistry, Institute of Medical Sciences, Banaras Hindu University, Varanasi, UP-221005, India. E-mail: agarwal.dralka@gmail.com
bDepartment of Microbiology, Institute of Medical Sciences, Banaras Hindu University, Varanasi, UP-221005, India
cDepartment of Pharmaceutical Sciences, Hemvati Nandan Bahuguna Garhwal University (Central University), Dist. Garhwal (Uttarakhand), Srinagar-246174, India
dDepartment of Microbiology, Central University of Punjab, Ghudda, Bathinda-151401, India
First published on 30th May 2024
Abstract
The antibacterial efficacy of some newly developed C-3 carboxylic group-containing ciprofloxacin-linked 1,2,3-triazole conjugates was studied. Twenty-one compounds from three different series of triazoles were synthesized using click chemistry and evaluated for their antibacterial activity against nine different pathogenic strains, including three Gram-positive strains, i.e. Enterococcus faecalis (ATCC29212), Staphylococcus aureus (ATCC25923), Staphylococcus epidermidis (clinical isolate), and six Gram-negative bacterial strains, i.e. Escherichia coli (ATCC25922), Pseudomonas aeruginosa (ATCC27853), Salmonella typhi (clinical isolate), Proteus mirabilis (clinical isolate), Acinetobacter baumannii (clinical isolate) and Klebsiella pneumonia (clinical isolate). Among the compounds, 10, 10a, 10b, 10c, 10d, 11a, 11f, 12c, 12e and 12f showed excellent activity with MIC values upto 12.5 μg mL−1, whereas the control ciprofloxacin showed MIC values of 0.781–25 μg mL−1 towards various strains. In addition, the low toxicity profile of the synthesized molecules revealed that they are potent antibiotics. Molecular docking and MD analysis were performed using the protein structure of E. coli DNA gyrase B, which was further corroborated with an in vitro assay to evaluate the inhibition of DNA gyrase. The analysis revealed that compound 10b was the most potent inhibitor of DNA gyrase compared to ciprofloxacin, which was employed as the positive control. Furthermore, the structure of two title compounds (11a and 12d) was characterized using single-crystal analysis.
Introduction
Fluoroquinolones are the most commonly recommended antibiotics for treating bacterial infections. They possess broad antibacterial activity, good efficacy profiles, and efficient pharmacokinetics.1 Accordingly, second-generation fluoroquinolones such as ciprofloxacin, ofloxacin, enoxacin, and norfloxacin are used to treat numerous bacterial infections. Besides, they are essential for the treatment of sexually transmitted diseases (STDs), upper and lower respiratory tract infections, and urinary tract infections (UTI) and exhibit antitubercular and antitumor activities. In addition, they can be employed to treat the infections of the skin, soft tissues, gastrointestinal system, bones, joints as well as nosocomial infections and chronic osteomyelitis.2–4 Among the second-generation fluoroquinolones, ciprofloxacin is the most popular broad-spectrum, synthetic chemotherapeutic antibiotic that has been approved for the treatment of infections, including chronic bacterial prostatitis, acute uncomplicated cystitis, shigellosis, urinary tract infections and acute sinusitis.5,6Ciprofloxacin functions by targeting bacterial DNA gyrase and topoisomerase IV (Topo IV), forming quinolone ternary complexes when it interacts with DNA and DNA gyrase or Topo IV. The antibacterial effectiveness of fluoroquinolone is due to the generation of these complexes, which hinder DNA replication and cell growth.7 However, the excessive use of ciprofloxacin has resulted in drug resistance, as discussed in the literature8,9 (Fig. 1). This developed drug resistance in bacteria can outlast other methods of prophylaxis or treatment.10 Consequently, infectious diseases carried through persistent bacteria, viruses, and fungi tend to pose a significant global problem, and if not addressed, they may result in 10 million fatalities annually by 2050.11 In this case, concomitant medication is one of the tactics that has shown some clinical effectiveness in preventing or delaying the emergence of resistance.12 It depends on the combination of two or more antibiotics, each having a unique mode of action, which reduces the chance of cells acquiring resistance.13 The pharmacokinetic characteristics of drugs in combination are considerably more likely to be changed; therefore, therapeutic results in vivo will not always coincide with in vitro outcomes.14,15 Designing hybrid antibacterial agents is another effective strategy that reduces the limitations of combination treatment, which integrates two or more active antibiotic structures with different bacterial targets in the same molecular framework via one or more chemical linkages. This approach creates a potential tool that reduces the anticipated side effects. Hybrid approaches have become popular due to their useful function in inhibiting the emergence of bacterial resistance through increased affinity and efficacy compared to the parent medications.11,16–18 They serve as novel leads with alternative actions and several pharmacological targets given that a single hybrid compound targets bacterial cells via different mechanisms of action. Novel hybrid molecules improve the ability to enhance the pharmacokinetic characteristics, toxicity profiles, and retention of drugs.19,20 A rationally designed linker between two bioactive groups may also improve the chance of both drug targets showing a lower incidence of new resistance mutations and may even be able to reduce existing drug resistance mechanisms.21
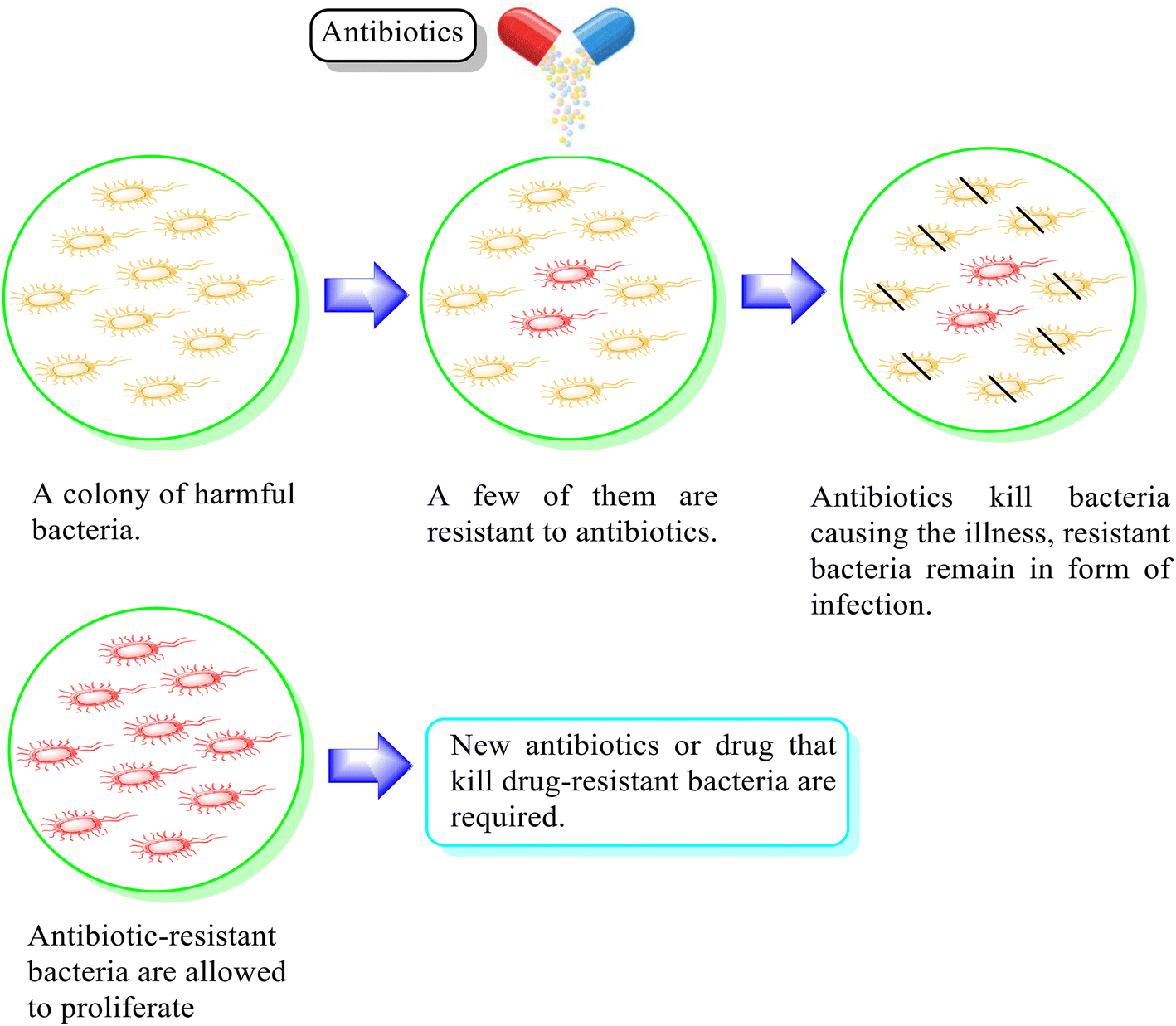 | ||
| Fig. 1 Illustration of the antimicrobial effect mediated by the antibiotic and associated mechanisms of drug resistance. | ||
To control the prevailing drug resistance, drug development to treat bacterial infections is necessary. In this case, click chemistry is a vital synthetic approach that involves a set of chemical reactions and ensures high yield with the comprehensive option of incorporating numerous chemical space parameters into a drug candidate. The versatility and efficiency click chemistry22 also ensure the outcome of the reaction, making it a convenient tool in various chemistry domains, which include drug discovery, materials science, and bioconjugation. Click chemistry also enables the fast synthesis of complex molecules and diverse heterocycles, thus paving the way for developing new pharmaceuticals with significantly improved efficacy and reduced side reactions or off-target effects. In general, click chemistry combines two similar or diverse pharmacophoric groups via a specific skeleton to yield a new analogue with synergistic biological attributes, thus ensuring that this technique is viable for lead compound discovery.23–25 The 1,2,3-triazole ring system, formed from the Huisgen 1,3-dipolar cycloaddition26 of azides and alkynes via copper-catalyzed click reaction, is a reasonably well-known pharmacophore.27–29 This moiety is a preferred linking unit because it is robust under oxidative and reductive conditions, resilient to metabolic degradation, actively binds to biomolecular targets and increases its solubility through hydrogen bonding and dipole interactions.30,31 Triazole has attracted particular interest in recent years in the search for novel antibiotics because various pharmacological molecules, i.e. cephalosporin, tazobactam, and cefatrizine, used to treat bacterial infections have a 1,2,3-triazole group.
Positions C-7 (piperazine) and C-3 (carboxylic) of ciprofloxacin, which are the most flexible sites for chemical modification and an area that considerably determines the potency, have been the focus of research on the ciprofloxacin moiety. Unfortunately, due to the insufficient information in the literature regarding ciprofloxacin analogues at the carboxylic position (C-3), they have not attracted as much attention as C-7, which has been prevalent in recent years.32 However, it has been observed that the incorporation of carboxamide (I),33 triazole (II and III)34,35 and tetrazole (IV)36 rings at the C-3 position significantly increases the antibacterial activity of synthesized hybrids in comparison to ciprofloxacin. Some C-3-modified ciprofloxacin hybrids are shown in Fig. 2. From the above discussion, there is scope for further investigation at the C-3 position of ciprofloxacin and it has often been observed that chemical transformation enhances the activity compared to a parent molecule.
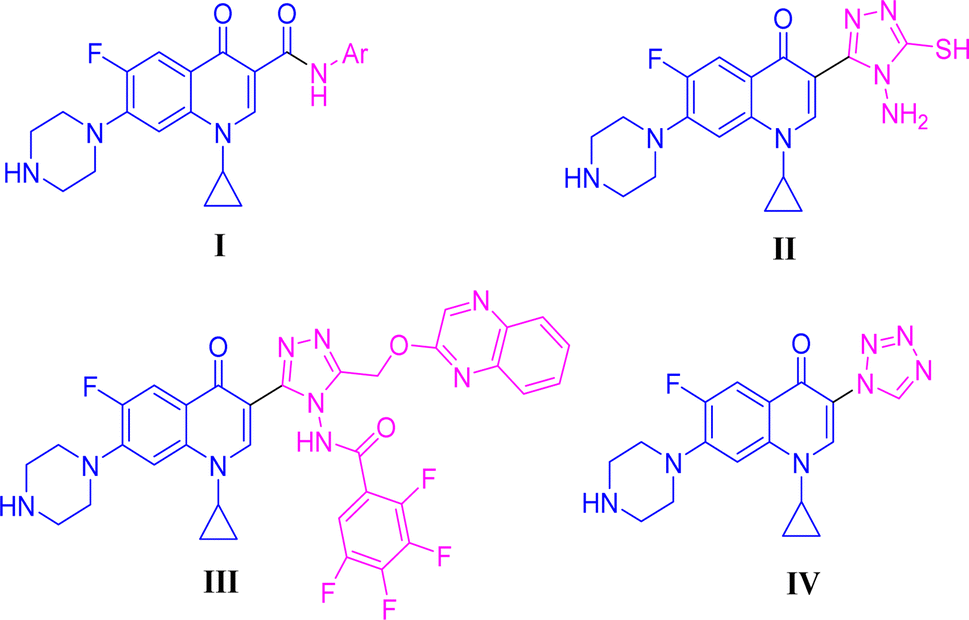 | ||
| Fig. 2 Structures of some C-3-modified ciprofloxacin hybrids. | ||
Our group has been working on the design and synthesis of various small molecules with antimicrobial activity.37,38 Encouraged by our earlier research,39,40 we aimed to develop a new and more effective series of ciprofloxacin conjugates using click chemistry for antibacterial findings (Scheme 1). Herein, we synthesized a new series of ciprofloxacin analogs with their C-3 carboxylic groups linked to various substituted 1,2,3-triazole rings and variations at the piperazine moiety. Our main goal was to improve the effectiveness of the synthesized compounds against Gram-positive and Gram-negative bacteria, while assessing their toxicity to human red blood cells. Also, they were further tested using the DNA gyrase assay as a possible target and molecular docking studies were conducted to investigate their binding mechanisms.
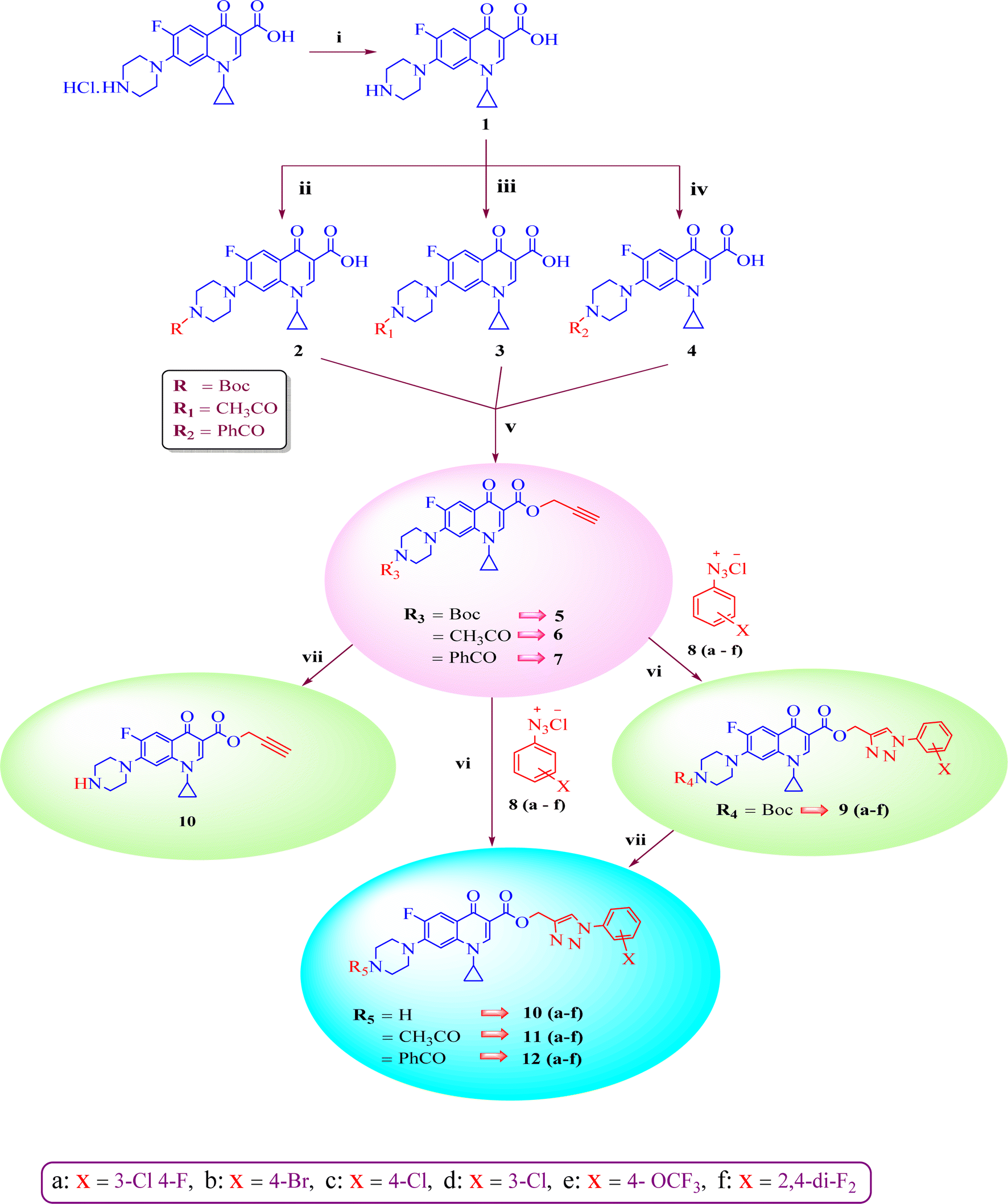 | ||
Scheme 1 Systematic strategy for the synthesis of ciprofloxacin-triazole hybrids 9–12(a–f). Reagents and conditions (i) = 5% aq. NaHCO3, (ii) = Boc2O, 1 M NaOH, THF, rt 16 h, (iii) = 2 M NaOH, CH3COC1, 1,4-dioxane, stirred, 0 °C to rt 2 h, (iv) = 2 M NaOH, PhCOC1, 1,4-dioxane, stirred, 0 °C to rt 2 h, (v) = NaHCO3, propargyl bromide, DMF, stirred 100 °C 48 h, (vi) = CuSO4·5H2O, sodium ascorbate, DMF/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), stirred 16–25 h, rt, click chemistry, (vii) = DCM(dry), TFA, rt 24 h. :1), stirred 16–25 h, rt, click chemistry, (vii) = DCM(dry), TFA, rt 24 h. | ||
Results and discussion
Chemistry
A systematic approach was used to create novel 1,2,3-triazole analogs of ciprofloxacin, as displayed in Scheme 1. Triazole derivatives 9–12(a–f) were synthesized in several steps. In the first synthetic step, free ciprofloxacin (1) was generated by solubilizing ciprofloxacin hydrochloride in a 5% aqueous solution of NaHCO3. In the next step, free ciprofloxacin was treated with Boc-anhydride, acyl chloride, and benzoyl chloride under different reaction conditions to ciprofloxacin products with a nitrogen-protected piperazine moiety (2, 3 and 4). Subsequently, the products (2, 3 and 4) were treated with propargyl bromide at 100 °C in the presence of NaHCO3 to obtain the propargylated products (5, 6 and 7, respectively). In the final step (for 11 and 12), the propargylated products (5, 6 and 7) underwent copper-catalyzed [3 + 2] cycloaddition reactions with various substituted aromatic azides 8(a–f) to afford 21 compounds 6, 7, 10 and 10–12(a–f). Compounds 5 and 9(a–f) were dissolved in a mixture of trifluoroacetic acid and dichloromethane (1:4 v/v, 5 mL) to obtain the deprotected products 10 and 10(a–f), respectively. The experimental section includes a description of the general synthesis process for each compound. The 1H NMR, 13C NMR, 19F NMR, IR and mass spectroscopy data validated the structures of all the synthesized analogues. Also, the X-ray crystallographic investigation supported the structure of compounds 11a and 12d.
Mechanism for the formation of 1,4 regioisomer
The stepwise catalytic cycle begins with the formation of a Cu–alkyne π complex (1), followed by deprotonation of the alkyne proton to form copper acetylide (2), as shown in Fig. 3. The coordination of copper increases the acidity of the acetylenic proton and facilitates deprotonation in the aqueous medium. One of the copper ions from species (2) coordinates with azide nitrogen (3) and activates it toward attack of the terminal nitrogen of the azide group on the alkyne carbon, leading to the synthesis of metallacycle (4). This metallacycle undergoes ring contraction via the transannular interaction between the lone pair of electrons present on the azide nitrogen and the carbon–copper double bond. Subsequently, Cu triazolide species (5) is formed, which undergoes protonation to generate 1,4-disubstituted triazole (6) and the Cu(I) catalyst.41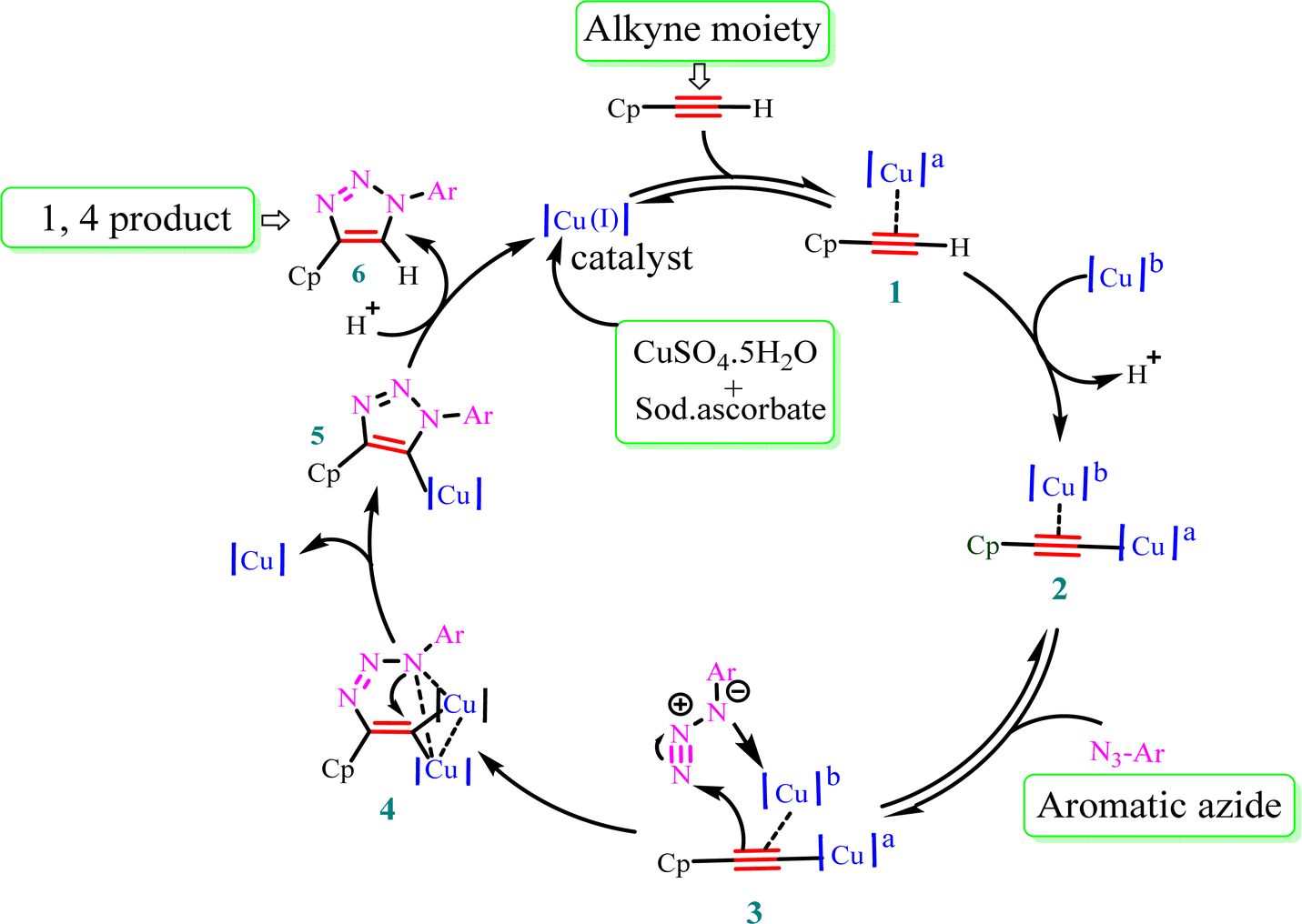 | ||
| Fig. 3 Plausible mechanism for Cu(I)-catalyzed alkyne–azide dipolar cycloaddition. Cp = ciprofloxacin and a,b = chemically equivalent copper atoms. | ||
Biological activity
 | ||
| Fig. 4 Structure–activity relationship of synthesized ciprofloxacin hybrids. | ||
Compound 10 again showed excellent activity against S. aureus (ATCC25923), and it was found to be 32-times more active with an MIC of 0.195 μg mL−1 in comparison with the standard drug (MIC 6.25 of μg mL−1), whereas against E. coli (ATCC25922), P. aeruginosa (ATCC27853) and K. pneumoniae (clinical isolate) with an MIC of 0.195 μg mL−1, it possessed very good antibacterial activity compared to the MIC of the control drug ciprofloxacin of 0.391 μg mL−1, 0.781 μg mL−1 and 0.391 μg mL−1, respectively. Further, E. faecalis (ATCC29212) and A. baumannii (clinical isolate) showed moderate to good activity with MIC of 0.781 μg mL−1 and 12.5 μg mL−1, while that of the standard drug was found to be 0.781 μg mL−1 and 25 μg mL−1, respectively. However, compound 10 was found to be ineffective against S. typhi (clinical isolate) and P. mirabilis (clinical isolate). Compound 10a containing a 3-Cl,4-F group on the phenyl-substituted triazole ring was found to be active against three strains, i.e., S. aureus (ATCC25923) and E. coli (ATCC25922) with MIC of 0.195 μg mL−1, while in A. baumannii (clinical isolate) it showed an MIC of 12.5 μg mL−1, which is half that of the standard drug. Furthermore, it showed excellent activity against S. aureus (ATCC25923), whereas it displayed moderate to weak activity against the remaining strains. Compound 10b, having a 4-Br phenyl-substituted triazole ring, was found to be 16-times more active against S. aureus (ATCC25923) with an MIC of 0.391 μg mL−1 in comparison with the standard drug ciprofloxacin (MIC 6.25 μg mL−1), while against E. coli (ATCC25922), it showed an MIC of ≤0.195 μg mL−1, which is half that of the standard drug of 0.391 μg mL−1. Compound 10c, having a 4-Cl substituent on the phenyl ring, also showed similar activity against E. coli (ATCC25922) as compound 10b. Compound 10c was found to be active against S. aureus (ATCC25923) and A. baumannii (clinical isolate) with an MIC of 1.56 μg mL−1 and 12.5 μg mL−1, respectively. Compound 10d, having a 3-Cl-substituted phenyl ring, showed excellent activity against S. aureus (ATCC25923) with an MIC of 0.391 μg mL−1 in comparison to that of the control drug of 6.25 μg mL−1. Against P. aeruginosa (ATCC27853) and A. baumannii (clinical isolate), it showed MIC values of 0.781 μg mL−1 and 12.5 μg mL−1, respectively. The control showed an MIC of 0.781 μg mL−1 and 25 μg mL−1 for these strains, respectively. The same compound was found to be ineffective against other strains. Further, compound 10e with a p-trifluoromethoxy-substituted phenyl ring showed activity against only one strain, S. aureus (ATCC25923), with an MIC value of 6.25 μg mL−1, similar to the MIC of the standard drug. Compound 10f, having a fluoro group on the ortho and para positions of the phenyl ring, did not show any activity against the tested strains. Similarly, compounds 11(a–d) did not show activity against any of the tested strains, except compound 11a, which has an acetyl group on piperazine together with a 3-Cl,4-F phenyl-substituted triazole ring, showing moderate activity against A. baumannii (clinical isolate) with an MIC of 12.5 μg mL−1. In contrast, the control drug showed an MIC of 25 μg mL−1. Similarly, compounds 11e and 11f, having 4-OCF3 and 2,4-di fluoro groups, and 12a, having a benzoyl group on piperazine together with a 3-Cl and 4-F phenyl-substituted triazole ring, showed good to moderate activity against S. aureus (ATCC25923) with MIC values of 6.25 μg mL−1, 0.391 μg mL−1, and 12.5 μg mL−1, respectively, while that of the control was 6.25 μg mL−1. Further, compounds 12b and 12d, again having a benzoyl group on piperazine at carbon 7 (C-7) with 4-Br and 3-Cl, were found to be ineffective against various strains. Compound 12c, having 4-Cl on the phenyl ring, was found to show good to moderate activity against S. aureus (ATCC25923) and S. typhi (clinical isolate) with MIC values of 1.56 μg mL−1 and 6.25 μg mL−1, respectively, while that of the control was 6.25 μg mL−1, and no activity was found in the other strains. Compound 12e, having 4-OCF3, showed excellent activity against S. aureus (ATCC25923), E. coli (ATCC25922), S. typhi (clinical isolate), and A. baumannii (clinical isolate) with MIC values of 0.391 μg mL−1, ≤0.195 μg mL−1, 1.56 μg mL−1 and 12.5 μg mL−1, while that of the control was 6.25 μg mL−1, 0.391 μg mL−1, 6.25 μg mL−1, and 25 μg mL−1 respectively. However, this compound did not show activity against E. faecalis (ATCC29212), S. epidermidis (clinical isolate), P. aeruginosa (ATCC27853), P. mirabilis (clinical isolate) and K. pneumoniae (clinical isolate). Compound 12f, having a 2,4-difluoro-substituted phenyl ring, did not show activity against any strain except E. faecalis (ATCC29212) with an MIC of 0.781 μg mL−1, which is the same as that of the control. Thus, according to Table 2 most of the compounds showed excellent activity against S. aureus with extremely good MIC values. Thus, this work can be further tuned to obtain useful results, where second-generation synthesis depending on the activity data is required and lead compounds should be used in the SAR experiments.
 | ||
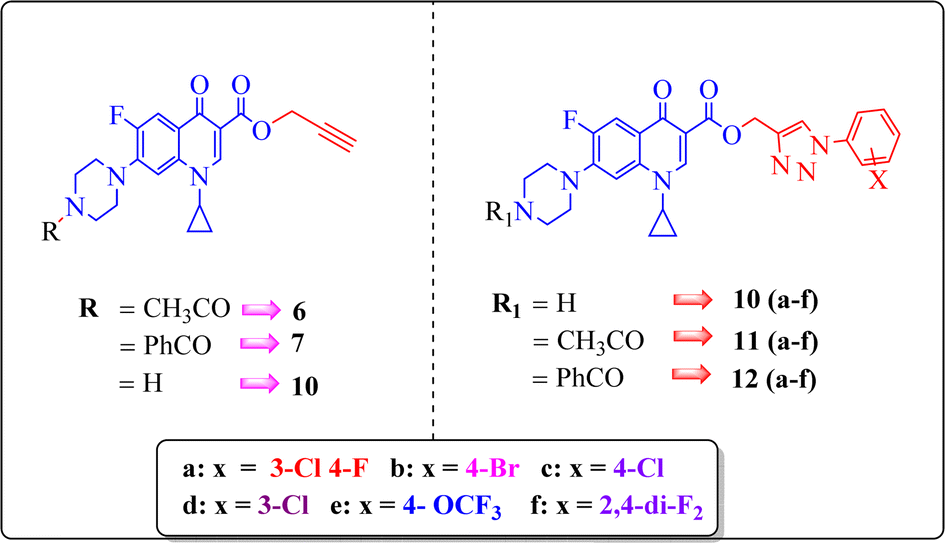
| Fig. 5 Illustration of the co-crystallized ligand (white) and redocked ligand (pink) for the calculation of RMSD (0.18 Å). | ||
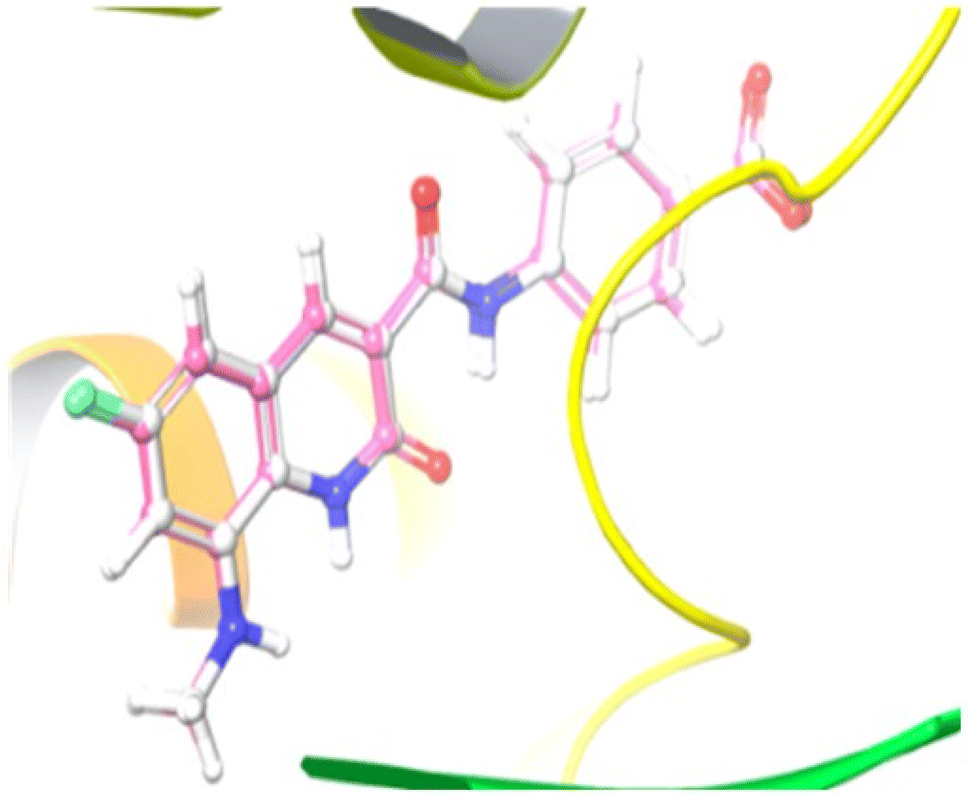
Compound 12e did not give any docking score because of its larger size and smaller grid size, and thus we excluded it. Among the compounds, compound 10b showed the highest docking score of −8.1 kcal mol−1 and the HIS83, GLU85, and GLU86 amino acids formed hydrogen bond interaction with the ligand. The other compounds showed docking scores in the range of −5.4 to −2.4 kcal mol−1. All the docked compounds with their vital amino acid interactions and docking scores are depicted in Fig. 6.
 | ||
| Fig. 6 2D and 3D interactions of compounds 10, 10a, 10b and 10c together with the co-crystallized ligand and ciprofloxacin. | ||
 | ||
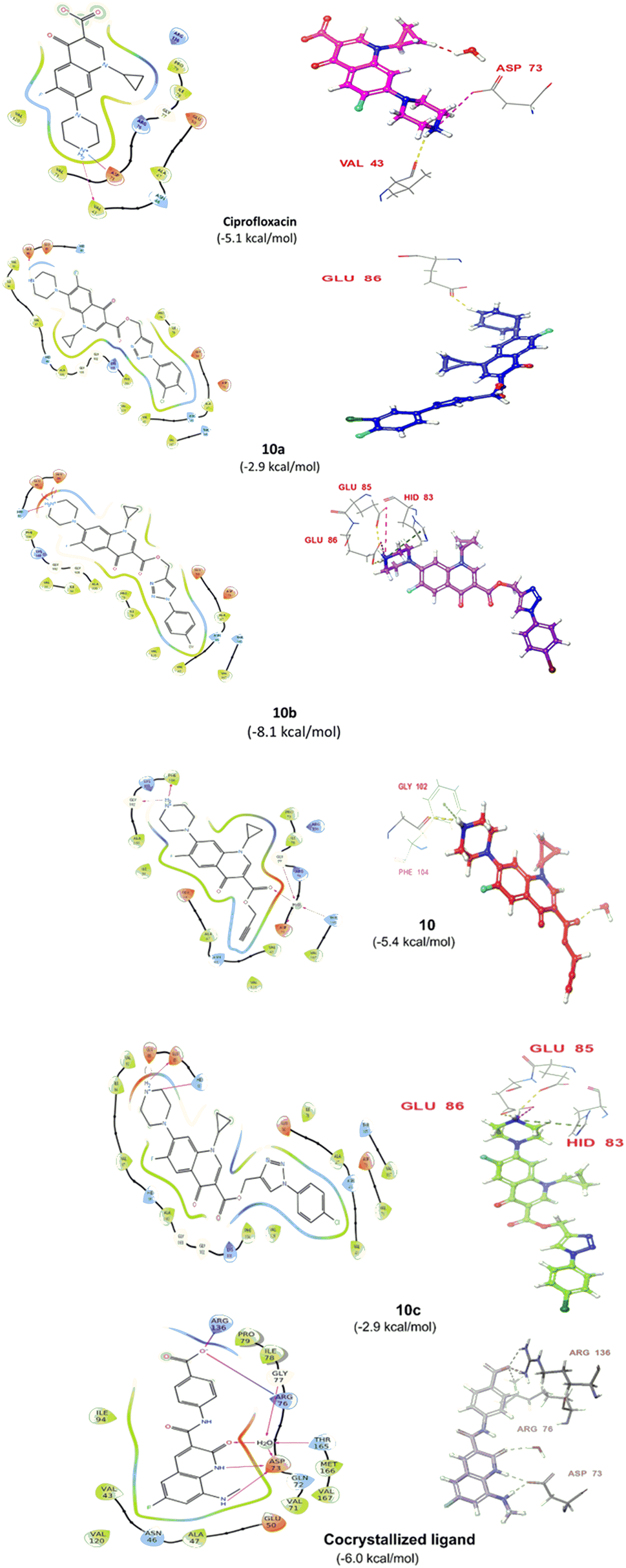
| Fig. 7 (A) PL complex RMSD of compound 10b. (B) RMSF of the protein. (C) PL-contacts of compound 10b. (D) Ligand–protein contacts of compound 10b. | ||
However, both ligand–protein complexes remain intact, which describes the ligand remaining in the protein cavity throughout the simulation. Both protein–ligand (PL) complex fluctuations were in the acceptable range, i.e. 3 Å. The root mean square fluctuation (RMSF) graph was also plotted to determine the deviations in the protein amino acid residues throughout 100 ns simulation. The RMSF graph showed that the fluctuations of the amino acid residues in the protein backbone occurred near amino acids 100–120 (3.6 Å), as shown in Fig. 7B. The PL contact histogram indicated that GLU86 formed H-bonding with the amine group of piperidine at about 57%, HIS83 was involved in the hydrophobic interaction, particularly the π-cation interaction with the amine group of piperidine, and other amino acid residues such as PRO79, ILE78 and PHE104 involved in the hydrophobic interactions provide stability to the complex. GLU85 is also engaged in hydrogen bonding for a very short time, about 40%, as depicted in Fig. 7(C and D).
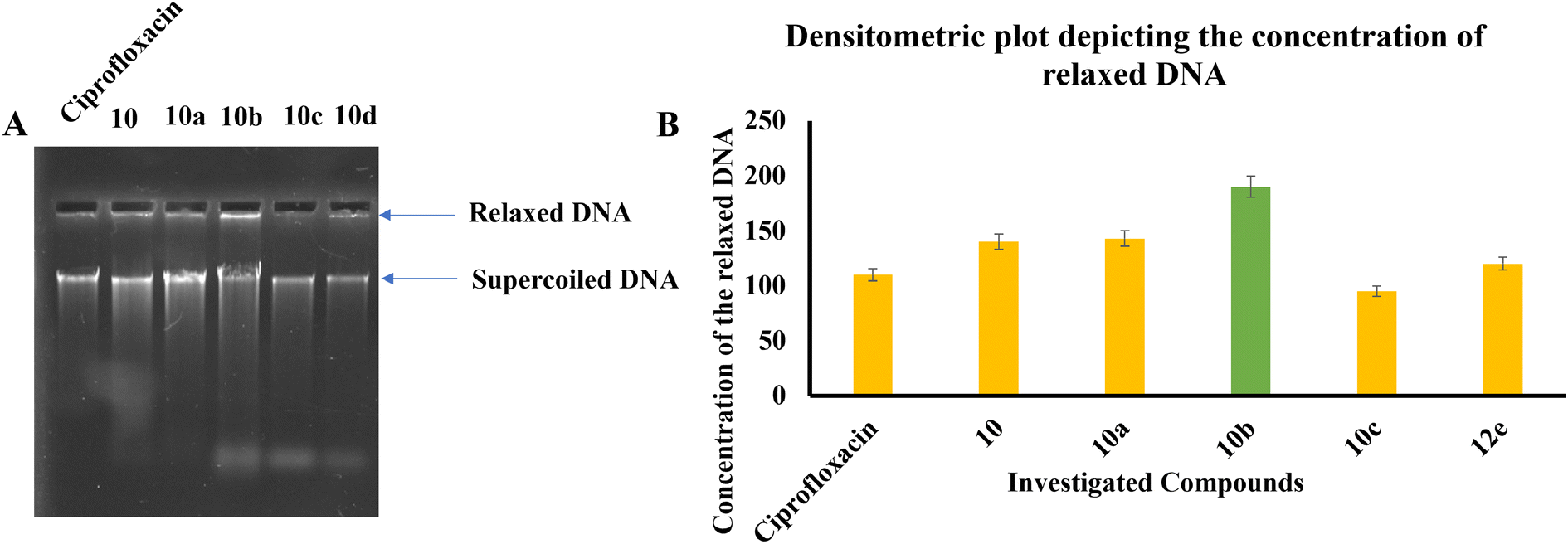 | ||
| Fig. 8 (A) Image of agarose gel depicting the movement and concentration of relaxed and supercoiled DNA upon treatment with the investigated compounds. (B) Densitometry plot illustrating the precise concentration of supercoiled DNA in the untreated sample. A decrease in supercoiled DNA suggests the inhibition of DNA gyrase. | ||
Experimental
All the chemicals and solvents used in the current study were purchased from E. Merck (India) and Sigma-Aldrich. The reactions during synthesis were monitored via thin layer chromatography (TLC) on precoated silica gel 60 F254 (mesh), and the spots were visualized using UV light. Silica gel (60–120 mesh) was employed for column chromatography. The melting points of all the synthesized compounds were determined using the open capillary method and may be uncorrected. The structural assignments of the synthesized products were based on 1H NMR, 13C NMR, 19F NMR, HRMS, IR and single-crystal XRD. NMR data were collected using a 400 MHz, JEOL JNM-ECS spectrometer in DMSO-d6 and CDCl3 using TMS as the internal standard and Delta software to process the data. In the reported spectral data, abbreviations such as s = singlet, bs = broad singlet, d = doublet, dd = doublet of doublets, t = triplet, and m = multiple are used. Mass data was produced with the use of a Bruker Compass spectrometer. X-ray analysis was performed using a Rigaku XtaLAB Synergy-i single crystal X-ray diffractometer with a CCD detector (HyPix-Bantam) using graphite monochromatized Cu Kα radiation (λ = 1.54184 Å).Regeneration of free 1-cyclopropyl-6-fluoro-4-oxo-7-piperazin-1-ylquinoline-3-carboxylic acid (1)
A clear solution was obtained by dissolving ciprofloxacin hydrochloride (5.0 g, 13.59 mmol) in water (100 mL). This solution was treated with excess 5% aqueous sodium bicarbonate solution, resulting in the formation of a white precipitate.44 The precipitate was collected by filtration to obtain hydrochloride-free ciprofloxacin 1. The free ciprofloxacin was sufficiently pure and used as the starting material for Boc protection, acetylation, and benzoylation. White solid; yield 4.5 g (96%); NMR & mp: reported45 1.Boc protection of 1-cyclopropyl-6-fluoro-4-oxo-7-piperazin-1-ylquinoline-3-carboxylic acid (2)
Compound 1 (0.5 g, 1.51 mmol) was dissolved in 1 M NaOH (5 mL) and THF (10 mL) was added, followed by the dropwise addition of Boc2O (0.36 g, 1.66 mmol), and the reaction mixture was stirred at room temperature for 16 h. The solvent was removed under reduced pressure and the resulting material was diluted with water (10 mL) and neutralized with sat. NH4Cl (aq.) solution. The precipitate was collected by vacuum filtration and washed with water to afford the product.46White solid; yield 500 mg (77%); mp: 210–212 °C; 1H NMR (400 MHz, CDCl3) δ: 14.95 (s, 1H, –COOH), 8.78 (s, 1H, Ar–H), 8.05 (d, J = 12.9 Hz, 1H, Ar–H), 7.37 (d, J = 7.1 Hz, 1H, Ar–H), 3.73–3.62 (m, 4H, 2×-NCH2), 3.53 (m, 1H, –NCH), 3.34–3.25 (m, 4H, 2×-NCH2), 1.50 (s, 9H, –C(CH3)3), 1.43–1.37 (m, 2H, –CH2), 1.24–1.17 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 177.18 (–CO), 166.99 (–CO), 154.80 (d, JC–F = 34.2 Hz), 147.58, 145.87 (d, JC–F = 10.6 Hz), 139.10, 120.17 (d, JC–F = 17.7 Hz), 112.65 (d, JC–F = 23.3 Hz), 108.29, 105.07 (d, JC–F = 2.1 Hz), 80.44, 77.41, 77.09, 76.78, 49.84, 35.37, 28.48, 8.34.
Acetylation of 1-cyclopropyl-6-fluoro-4-oxo-7-piperazin-1-ylquinoline-3-carboxylic acid (3)
To a solution of compound 1 (0.5 g, 1.3 mmol) in 1,4-dioxane (20 mL) and 2 M NaOH (5 mL) at 0 °C, acetyl chloride (0.12 g, 1.5 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 2 h and 1 M HCl (10 mL) was added to reach pH 1. The separated solid was collected by filtration and purified by column chromatography using a CH2Cl2:MeOH (98:2) mixture as the eluent.47
White solid; yield 499 mg (89%); mp: 270–272 °C; 1H NMR (400 MHz, CDCl3) δ: 14.90 (s, 1H, –COOH), 8.69 (s, 1H, Ar–H), 7.95 (d, J = 12.7 Hz, 1H, Ar–H), 7.36 (d, J = 6.9 Hz, 1H, Ar–H), 3.87 (t, J = 4.4 Hz, 2H, –NCH2), 3.70–3.73 (m, 2H, –NCH2), 3.57 (m, 1H, –NCH), 3.39–3.40 (m, 2H, –NCH2), 3.31 (t, J = 4.4 Hz, 2H, –NCH2), 2.18 (s, 3H, –CH3), 1.37–1.46 (m, 2H, –CH2), 1.22–1.29 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 176.97 (–COCH3), 169.19 (–CO), 166.83, 154.43, 152.76, 147.53, 145.45 (d, JC–F = 7.1 Hz), 139.02, 120.13 (d, JC–F = 5.1 Hz), 112.44 (d, JC–F = 15.15 Hz), 108.07, 105.18, 67.11, 50.16, 49.39, 46.10, 41.06, 35.40, 21.35, 8.29.
Benzoylation of 1-cyclopropyl-6-fluoro-4-oxo-7-piperazin-1-ylquinoline-3-carboxylic acid (4)
To a solution of compound 1 (0.5 g, 1.4 mmol) in 1,4-dioxane (20 mL) and 2 M NaOH (5 mL) at 0 °C, benzoyl chloride (0.31 g, 2.2 mmol) was added dropwise. The reaction mixture was stirred at room temperature for 2 h and 1 M HCl (10 mL) was added to reach pH 1. The separated solid was collected by filtration and purified by column chromatography using a CH2Cl2:MeOH (98:2) mixture as the eluent.47
White solid; yield 450 mg (68.5%); mp: 285–287 °C; 1H NMR (400 MHz, CDCl3:CD3OD (10% v/v)) δ: 8.83 (s, 1H, Ar–H), 8.02 (d, J = 12.7 Hz, 1H, Ar–H), 7.58 (m, 1H, Ar–H), 7.47–7.54 (m, 5H, Ar–H), 4.14–4.24 (m, 1H, –NCH), 3.65–4.04 (m, 4H, 2× –NCH2), 3.35–3.50 (m, 4H, 2×-NCH2), 1.40–1.47 (m, 2H, –CH2), 1.19–1.27 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3:CD3OD (10% v/v)) δ: 177.49 (–COAr), 171.74 (–CO), 153.87, 152.95, 148.27, 139.59 (d, JC–F = 6.1 Hz), 135.01, 133.98, 132.31, 130.62 (d, JC–F = 4.0 Hz), 128.99, 127.21, 112.46 (d, JC–F = 11.1 Hz), 107.66, 106.09, 77.97, 77.97, 77.75, 77.54, 77.54, 49.13, 48.99, 48.99, 48.85, 48.71, 48.71, 48.56, 48.42, 48.42, 48.28, 36.20, 8.22.
General procedure for the propargylation of 7-(4-(tert-butoxycarbonyl)piperazin-1-yl)/7-(4-acetylpiperazin-1-yl)/7-(4-benzoylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4dihydroquinoline-3-carboxylic acid (5), (6), (7)
Propargylation was performed using a method reported in the literature.48 To a solution of compounds 2, 3 and 4 (1 mmol) in N,N-dimethylformamide (15 mL) in separate round-bottom flasks, 25 mL of NaHCO3 solution (1.2 mmol) and (1.2 mmol) of propargyl bromide were added to each flask under vigorous stirring at room temperature. The mixture was allowed to react at 100 °C for 48 h. The progress of the reaction was monitored by TLC. After evaporating the solvent, the residue was purified by column chromatography using a CHCl3:MeOH (98:2) mixture as the eluent to obtain the desired propargylated products 5, 6 and 7.
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), 1.43 (s, 9H, –C(CH3)3), 1.17–1.28 (m, 2H, –CH2), 1.08–1.10 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 171.64 (–CO), 163.36 (–CO), 153.35 (d, JC–F = 27.3 Hz), 151.24, 147.41, 143.34 (d, JC–F = 8.1 Hz), 136.76, 121.91 (d, JC–F = 6.1 Hz), 112.11 (d, JC–F = 19.2 Hz), 108.11, 103.99, 79.10, 77.00, 76.18, 75.93, 75.67, 73.66, 50.93, 48.77, 33.55, 28.55, 27.29, 7.03; HRMS (ESI): anal. calcd. For C25H28FN3O5, 469.5134 [M]+; found 470.2075 [M + H]+.CH), 2.17 (s, 3H, –CH3), 1.31–1.36 (m, 2H, –CH2), 1.14–1.18 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 172.81 (–COCH3), 169.15 (–CO), 164.58, 154.62, 152.15, 148.67, 144.14 (d, JC–F = 10.6 Hz), 137.93, 123.41 (d, JC–F = 7.1 Hz), 113.47 (d, JC–F = 23.1 Hz), 109.40, 105.26, 78.13, 77.41, 77.09, 76.77, 74.86, 52.16, 50.46, 49.57, 46.25, 41.18, 34.71, 21.39, 8.23; 19F NMR (565 MHz CDCl3) δ: −123.65; HRMS (ESI): anal. calcd. For C22H22FN3O4,411.4334 [M]+; found 412.1651 [M + H]+.CH), 1.25–1.35 (m, 2H, –CH2), 1.12–1.21 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 175.61 (–COAr), 172.84 (–CO), 170.66 (–CO), 164.44, 154.19, 152.54, 148.64, 144.14 (d, JC–F = 7.1 Hz), 137.93, 135.17, 130.14, 128.67, 127.20, 123.27 (d, JC–F = 5.1 Hz), 113.37 (d, JC–F = 15.2 Hz), 109.32, 105.34, 78.11, 77.30, 77.09, 76.88, 74.86, 52.13, 50.27, 34.77, 20.81, 8.22; 19F NMR (565 MHz CDCl3) δ: −123.62; HRMS (ESI): anal. calcd. For C27H24FN3O4, 473.5044 [M]+; found 474.1840 [M + H]+.
CH), 1.43 (s, 9H, –C(CH3)3), 1.17–1.28 (m, 2H, –CH2), 1.08–1.10 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 171.64 (–CO), 163.36 (–CO), 153.35 (d, JC–F = 27.3 Hz), 151.24, 147.41, 143.34 (d, JC–F = 8.1 Hz), 136.76, 121.91 (d, JC–F = 6.1 Hz), 112.11 (d, JC–F = 19.2 Hz), 108.11, 103.99, 79.10, 77.00, 76.18, 75.93, 75.67, 73.66, 50.93, 48.77, 33.55, 28.55, 27.29, 7.03; HRMS (ESI): anal. calcd. For C25H28FN3O5, 469.5134 [M]+; found 470.2075 [M + H]+.CH), 2.17 (s, 3H, –CH3), 1.31–1.36 (m, 2H, –CH2), 1.14–1.18 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 172.81 (–COCH3), 169.15 (–CO), 164.58, 154.62, 152.15, 148.67, 144.14 (d, JC–F = 10.6 Hz), 137.93, 123.41 (d, JC–F = 7.1 Hz), 113.47 (d, JC–F = 23.1 Hz), 109.40, 105.26, 78.13, 77.41, 77.09, 76.77, 74.86, 52.16, 50.46, 49.57, 46.25, 41.18, 34.71, 21.39, 8.23; 19F NMR (565 MHz CDCl3) δ: −123.65; HRMS (ESI): anal. calcd. For C22H22FN3O4,411.4334 [M]+; found 412.1651 [M + H]+.CH), 1.25–1.35 (m, 2H, –CH2), 1.12–1.21 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 175.61 (–COAr), 172.84 (–CO), 170.66 (–CO), 164.44, 154.19, 152.54, 148.64, 144.14 (d, JC–F = 7.1 Hz), 137.93, 135.17, 130.14, 128.67, 127.20, 123.27 (d, JC–F = 5.1 Hz), 113.37 (d, JC–F = 15.2 Hz), 109.32, 105.34, 78.11, 77.30, 77.09, 76.88, 74.86, 52.13, 50.27, 34.77, 20.81, 8.22; 19F NMR (565 MHz CDCl3) δ: −123.62; HRMS (ESI): anal. calcd. For C27H24FN3O4, 473.5044 [M]+; found 474.1840 [M + H]+.General procedure for the synthesis of azides 8(a–f)
Azides were synthesized according to the established method.49 Briefly, aniline (1 eq.) was dissolved in 6 N HCl solution (10 mL mmol−1 of aniline) at room temperature and cooled to 0 °C, and then NaNO2 (1.2 eq.) solution added under stirring. After 10 min, sodium azide (1.2 eq.) was added to the reaction mixture at the same temperature under stirring. Again, this mixture was stirred at room temperature for 2–3 h. Subsequently, the mixture was extracted with ethyl acetate. The organic layer was washed with brine solution and dried over Na2SO4. After evaporation of the solvent, crude products 8(a–f) were sufficiently pure for further reactions.General procedure for the synthesis of 1,2,3-triazole scaffolds 9–12(a–f)
Triazoles were synthesized according to the literature-reported method.50 In brief, compounds 5, 6 and 7 (1 eq.) and substituted aromatic azides (a–f, 1.2 eq.) were suspended in a mixture of DMF and water (4:1, 25 mL mmol−1 of alkyne). Then, a solution of sodium ascorbate (0.4 eq. in minimum water) was added, followed by copper(II) sulfate pentahydrate solution (0.2 eq. in minimum water). At room temperature, the heterogeneous mixture was rapidly stirred until the consumption of alkyne and the progress of the reaction was monitored by TLC. After completion of the reaction, the reaction mixture was poured into ice water to get the precipitate, which was collected by filtration. The required products were purified by column chromatography using a CHCl3:MeOH (98:3) mixture as the eluent.
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH of triazole), 8.25 (d, J = 4.0 Hz, 1H, Ar–H), 7.98 (d, J = 8.7 Hz, 1H, Ar–H), 7.75 (d, J = 13.1 Hz, 1H, Ar–H), 7.68 (t, J = 8.9 Hz, 1H, Ar–H), 7.46 (d, J = 7.3 Hz, 1H, Ar–H), 5.38 (s, 2H, –OCH2), 3.64–3.81 (m, 1H, –NCH), 3.54 (s, 4H, 2×-NCH2), 3.20 (s, 4H, 2×-NCH2), 1.43 (s, 9H, –C(CH3)3), 1.23–1.28 (m, 2H, –CH2), 1.09–1.18 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 172.60 (–CO), 164.08 (–CO), 161.55, 156.21 (d, JC–F = 14 Hz), 148.58, 143.78 (d, JC–F = 5 Hz), 138.57 (d, JC–F = 5.1 Hz), 135.68, 132.87, 123.12, 122.18 (d, JC–F = 7.1 Hz), 121.50, 111.71, 108.52, 106.81, 103.65, 79.20, 56.87, 49.45, 40.00, 39.86, 39.72, 39.58, 39.44, 39.31, 39.17, 28.11, 7.64; HRMS (ESI): anal. calcd. For C31H31ClF2N6O5,640.2013 [M]+; found 641.2063 [M + H]+.CH of triazole), 8.04 (d, J = 13.1 Hz, 1H, Ar–H), 7.82 (d, J = 8.7 Hz, 2H, Ar–H), 7.38 (d, J = 8.7 Hz, 2H, Ar–H), 7.27 (d, J = 6.2 Hz, 2H, Ar–H), 5.54 (s, 2H, –OCH2), 3.65–3.73 (m, 4H, 2×-NCH2), 3.42 (s, 1H, –NCH), 3.22 (s, 4H, 2×-NCH2), 1.49 (s, 9H, –C(CH3)3), 1.23–1.34 (m, 2H, –CH2), 1.14–1.18 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 171.86 (–CO), 164.37 (–CO), 163.81, 154.09 (d, JC–F = 9.1 Hz), 148.93 (d, JC–F = 13.3), 138.45, 135.66, 133.52, 130.23, 123.46, 122.19, 112.16 (d, JC–F = 34.3 Hz), 108.91, 107.19, 79.38, 57.97, 49.82, 40.28, 40.14, 40.00, 39.86, 39.72, 39.58, 39.31, 39.44, 28.39, 7.91; HRMS (ESI): anal. calcd. For C31H32BrFN6O5, 666.1602 [M]+; found 667.1660 [M + H]+.CH of triazole), 8.04 (d, J = 13.1 Hz, 1H, Ar–H), 7.82 (d, J = 8.7 Hz, 2H, Ar–H), 7.38 (d, J = 8.7 Hz, 2H, Ar–H), 7.27 (d, J = 6.2 Hz, 2H, Ar–H), 5.54 (s, 2H, –OCH2), 3.65–3.73 (m, 4H, 2×-NCH2), 3.42 (s, 1H, –NCH), 3.22 (s, 4H, 2×-NCH2), 1.49 (s, 9H, –C(CH3)3), 1.23–1.34 (m, 2H, –CH2), 1.14–1.18 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 171.86 (–CO), 164.37 (–CO), 163.81, 154.10 (d, JC–F = 9.1 Hz), 148.92 (d, JC–F = 13.1 Hz), 138.45, 135.66, 133.52, 130.23, 123.46, 122.19, 112.16 (d, JC–F = 34.3 Hz), 111.99, 108.91, 107.19, 79.38, 75.97, 57.19, 49.82, 40.28, 40.14, 40.00, 39.86, 39.72, 39.58, 39.44, 35.20, 28.39, 7.91; HRMS (ESI): anal. calcd. For C31H32ClFN6O5, 622.2107 [M]+; found 623.2218 [M + H]+.CH of triazole), 8.07 (s, 1H, Ar–H), 7.91–7.95 (m, 1H, Ar–H), 7.76 (d, J = 13.4 Hz, 1H, Ar–H), 7.64 (t, J = 8.2 Hz, 1H, Ar–H), 7.58 (d, J = 7.6 Hz, 1H, Ar–H), 7.44–7.47 (m, 1H, Ar–H), 5.39 (s, 2H, –OCH2), 3.65–3.82 (m, 1H, –NCH), 3.54 (s, 4H, 2×-NCH2), 3.21 (s, 4H, 2×-NCH2), 1.43 (s, 9H, –C(CH3)3), 1.19–1.28 (m, 2H, –CH2), 1.09–1.19 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 171.91 (–CO), 164.42 (–CO), 154.16 (d, JC–F = 5.1 Hz), 148.94, 144.13 (d, JC–F = 4 Hz), 138.42, 137.97, 134.64, 132.07, 129.02, 123.64, 122.50, 120.37, 119.17, 111.98 (d, JC–F = 15.2 Hz), 108.95, 107.16 (d, JC–F = 3 Hz), 79.56, 57.20, 49.90, 49.90, 40.36, 40.22, 40.08, 39.94, 39.80, 39.66, 39.53, 35.30, 28.47, 8.00; HRMS (ESI): anal. calcd. For C31H32ClFN6O5, 622.2145 [M]+; found 623.2218 [M + H]+.CH of triazole), 8.04 (d, J = 13.1 Hz, 1H, Ar–H), 7.82 (d, J = 8.7 Hz, 2H, Ar–H), 7.38 (d, J = 8.7 Hz, 2H, Ar–H), 7.27 (d, J = 6.2 Hz, 2H, Ar–H), 5.54 (s, 2H, –OCH2), 3.65–3.73 (m, 4H, 2×-NCH2), 3.42 (s, 1H, –NCH), 3.22 (s, 4H, 2×-NCH2), 1.49 (s, 9H, –C(CH3)3), 1.23–1.34 (m, 2H, –CH2), 1.14–1.18 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 171.95 (–CO), 166.33 (–CO), 164.01 (d, JC–F = 4 Hz), 154.18 (d, JC–F = 8.1 Hz), 152.46, 149.09, 148.48, 144.18 (d, JC–F = 10.1 Hz), 139.55, 138.43, 135.82, 123.84, 123.05, 122.61, 112.07, 108.93, 108.51, 107.18, 79.57, 77.88, 57.45, 56.82, 51.85, 49.83 (d, JC–F = 18.2 Hz), 40.37, 40.23, 40.09, 39.95, 39.81, 39.67, 39.53, 36.63, 35.35, 28.48, 8.00; HRMS (ESI): anal. calcd. For C32H32F4N6O6, 672.6376 [M]+; found 673.2434 [M + H]+.CH of triazole), 7.88–7.96 (m, 1H, Ar–H), 7.68–7.76 (m, 2H, Ar–H), 7.41–7.47 (m, 1H, Ar–H), 7.38 (dd, J = 16.8, 9.0 Hz, 1H, Ar–H), 5.40 (s, 2H, –OCH2), 3.64 (s, 1H, –NCH), 3.54 (m, 4H, 2×-NCH2), 3.20 (m, 4H, 2×-NCH2), 1.43 (s, 9H, –C(CH3)3), 1.23–1.30 (m, 2H, –CH2), 1.09–1.19 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 171.96 (–CO), 164.52 (–CO), 154.21 (d, JC–F = 12.5 Hz), 148.97, 144.20 (d, JC–F = 10.1 Hz), 143.48, 138.43, 128.11 (d, JC–F = 10.5 Hz), 126.75 (d, JC–F = 9.8 Hz), 113.21, 113.00, 112.00 (d, JC–F = 21.1 Hz), 108.95, 107.14, 106.17, 79.57, 57.25, 49.92, 40.58, 40.37, 40.16, 39.95, 39.75, 39.54, 39.33, 35.31, 29.44, 28.48, 8.00; HRMS (ESI): anal. calcd. For C31H31F3N6O5, 624.6212 [M]+; found 625.2357 [M + H]+.
CH of triazole), 8.25 (d, J = 4.0 Hz, 1H, Ar–H), 7.98 (d, J = 8.7 Hz, 1H, Ar–H), 7.75 (d, J = 13.1 Hz, 1H, Ar–H), 7.68 (t, J = 8.9 Hz, 1H, Ar–H), 7.46 (d, J = 7.3 Hz, 1H, Ar–H), 5.38 (s, 2H, –OCH2), 3.64–3.81 (m, 1H, –NCH), 3.54 (s, 4H, 2×-NCH2), 3.20 (s, 4H, 2×-NCH2), 1.43 (s, 9H, –C(CH3)3), 1.23–1.28 (m, 2H, –CH2), 1.09–1.18 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 172.60 (–CO), 164.08 (–CO), 161.55, 156.21 (d, JC–F = 14 Hz), 148.58, 143.78 (d, JC–F = 5 Hz), 138.57 (d, JC–F = 5.1 Hz), 135.68, 132.87, 123.12, 122.18 (d, JC–F = 7.1 Hz), 121.50, 111.71, 108.52, 106.81, 103.65, 79.20, 56.87, 49.45, 40.00, 39.86, 39.72, 39.58, 39.44, 39.31, 39.17, 28.11, 7.64; HRMS (ESI): anal. calcd. For C31H31ClF2N6O5,640.2013 [M]+; found 641.2063 [M + H]+.CH of triazole), 8.04 (d, J = 13.1 Hz, 1H, Ar–H), 7.82 (d, J = 8.7 Hz, 2H, Ar–H), 7.38 (d, J = 8.7 Hz, 2H, Ar–H), 7.27 (d, J = 6.2 Hz, 2H, Ar–H), 5.54 (s, 2H, –OCH2), 3.65–3.73 (m, 4H, 2×-NCH2), 3.42 (s, 1H, –NCH), 3.22 (s, 4H, 2×-NCH2), 1.49 (s, 9H, –C(CH3)3), 1.23–1.34 (m, 2H, –CH2), 1.14–1.18 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 171.86 (–CO), 164.37 (–CO), 163.81, 154.09 (d, JC–F = 9.1 Hz), 148.93 (d, JC–F = 13.3), 138.45, 135.66, 133.52, 130.23, 123.46, 122.19, 112.16 (d, JC–F = 34.3 Hz), 108.91, 107.19, 79.38, 57.97, 49.82, 40.28, 40.14, 40.00, 39.86, 39.72, 39.58, 39.31, 39.44, 28.39, 7.91; HRMS (ESI): anal. calcd. For C31H32BrFN6O5, 666.1602 [M]+; found 667.1660 [M + H]+.CH of triazole), 8.04 (d, J = 13.1 Hz, 1H, Ar–H), 7.82 (d, J = 8.7 Hz, 2H, Ar–H), 7.38 (d, J = 8.7 Hz, 2H, Ar–H), 7.27 (d, J = 6.2 Hz, 2H, Ar–H), 5.54 (s, 2H, –OCH2), 3.65–3.73 (m, 4H, 2×-NCH2), 3.42 (s, 1H, –NCH), 3.22 (s, 4H, 2×-NCH2), 1.49 (s, 9H, –C(CH3)3), 1.23–1.34 (m, 2H, –CH2), 1.14–1.18 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 171.86 (–CO), 164.37 (–CO), 163.81, 154.10 (d, JC–F = 9.1 Hz), 148.92 (d, JC–F = 13.1 Hz), 138.45, 135.66, 133.52, 130.23, 123.46, 122.19, 112.16 (d, JC–F = 34.3 Hz), 111.99, 108.91, 107.19, 79.38, 75.97, 57.19, 49.82, 40.28, 40.14, 40.00, 39.86, 39.72, 39.58, 39.44, 35.20, 28.39, 7.91; HRMS (ESI): anal. calcd. For C31H32ClFN6O5, 622.2107 [M]+; found 623.2218 [M + H]+.CH of triazole), 8.07 (s, 1H, Ar–H), 7.91–7.95 (m, 1H, Ar–H), 7.76 (d, J = 13.4 Hz, 1H, Ar–H), 7.64 (t, J = 8.2 Hz, 1H, Ar–H), 7.58 (d, J = 7.6 Hz, 1H, Ar–H), 7.44–7.47 (m, 1H, Ar–H), 5.39 (s, 2H, –OCH2), 3.65–3.82 (m, 1H, –NCH), 3.54 (s, 4H, 2×-NCH2), 3.21 (s, 4H, 2×-NCH2), 1.43 (s, 9H, –C(CH3)3), 1.19–1.28 (m, 2H, –CH2), 1.09–1.19 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 171.91 (–CO), 164.42 (–CO), 154.16 (d, JC–F = 5.1 Hz), 148.94, 144.13 (d, JC–F = 4 Hz), 138.42, 137.97, 134.64, 132.07, 129.02, 123.64, 122.50, 120.37, 119.17, 111.98 (d, JC–F = 15.2 Hz), 108.95, 107.16 (d, JC–F = 3 Hz), 79.56, 57.20, 49.90, 49.90, 40.36, 40.22, 40.08, 39.94, 39.80, 39.66, 39.53, 35.30, 28.47, 8.00; HRMS (ESI): anal. calcd. For C31H32ClFN6O5, 622.2145 [M]+; found 623.2218 [M + H]+.CH of triazole), 8.04 (d, J = 13.1 Hz, 1H, Ar–H), 7.82 (d, J = 8.7 Hz, 2H, Ar–H), 7.38 (d, J = 8.7 Hz, 2H, Ar–H), 7.27 (d, J = 6.2 Hz, 2H, Ar–H), 5.54 (s, 2H, –OCH2), 3.65–3.73 (m, 4H, 2×-NCH2), 3.42 (s, 1H, –NCH), 3.22 (s, 4H, 2×-NCH2), 1.49 (s, 9H, –C(CH3)3), 1.23–1.34 (m, 2H, –CH2), 1.14–1.18 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 171.95 (–CO), 166.33 (–CO), 164.01 (d, JC–F = 4 Hz), 154.18 (d, JC–F = 8.1 Hz), 152.46, 149.09, 148.48, 144.18 (d, JC–F = 10.1 Hz), 139.55, 138.43, 135.82, 123.84, 123.05, 122.61, 112.07, 108.93, 108.51, 107.18, 79.57, 77.88, 57.45, 56.82, 51.85, 49.83 (d, JC–F = 18.2 Hz), 40.37, 40.23, 40.09, 39.95, 39.81, 39.67, 39.53, 36.63, 35.35, 28.48, 8.00; HRMS (ESI): anal. calcd. For C32H32F4N6O6, 672.6376 [M]+; found 673.2434 [M + H]+.CH of triazole), 7.88–7.96 (m, 1H, Ar–H), 7.68–7.76 (m, 2H, Ar–H), 7.41–7.47 (m, 1H, Ar–H), 7.38 (dd, J = 16.8, 9.0 Hz, 1H, Ar–H), 5.40 (s, 2H, –OCH2), 3.64 (s, 1H, –NCH), 3.54 (m, 4H, 2×-NCH2), 3.20 (m, 4H, 2×-NCH2), 1.43 (s, 9H, –C(CH3)3), 1.23–1.30 (m, 2H, –CH2), 1.09–1.19 (m, 2H, –CH2); 13C NMR (101 MHz, DMSO-d6) δ: 171.96 (–CO), 164.52 (–CO), 154.21 (d, JC–F = 12.5 Hz), 148.97, 144.20 (d, JC–F = 10.1 Hz), 143.48, 138.43, 128.11 (d, JC–F = 10.5 Hz), 126.75 (d, JC–F = 9.8 Hz), 113.21, 113.00, 112.00 (d, JC–F = 21.1 Hz), 108.95, 107.14, 106.17, 79.57, 57.25, 49.92, 40.58, 40.37, 40.16, 39.95, 39.75, 39.54, 39.33, 35.31, 29.44, 28.48, 8.00; HRMS (ESI): anal. calcd. For C31H31F3N6O5, 624.6212 [M]+; found 625.2357 [M + H]+.General procedure for the removal of di-tert-butyl dicarbonate (Boc) 10, 10(a–f)
Removal of Boc group was carried out according to the method reported in the literature.51 Briefly compounds 5 and 9(a–f) (0.156 mmol) were dissolved in a mixture of trifluoroacetic acid and dichloromethane (1:4 v/v, 5 mL) and stirred at room temperature for 24 h, respectively. The reaction mixture was diluted with dichloromethane and washed with saturated aqueous NaHCO3 to remove the acid. The organic phase was dried over Na2SO4, filtered, and evaporated in vacuo to obtain the deprotected 1,2,3-triazole scaffolds 10 and 10(a–f) as a powder, respectively.
CH), 1.29–1.41 (m, 2H, –CH2), 1.12–1.20 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 172.89 (–CO), 164.62 (–CO), 154.29, 152.64, 148.46, 145.04 (d, JC–F = 7.1 Hz), 137.96, 122.78 (d, JC–F = 4 Hz), 113.23 (d, JC–F = 16.2 Hz), 109.22, 104.80, 78.16, 77.25, 77.04, 76.83, 74.75, 52.07, 51.08, 45.93, 34.64, 8.16; 19F NMR (565 MHz CDCl3) δ: −123.38; IR (KBr) cm−1: 3192, 2975, 2864, 1729, 1701, 1621, 1584, 1482, 1423, 1243, 1159, 1078, 1032, 998, 889, 754, 618,549; HRMS (ESI): anal. calcd. For C20H21FN3O3, 369.3964 [M]+; found 370.1583 [M + H]+.
(1-(3-Chloro-4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate (10a). Pale yellow solid; yield 72 mg (85%); mp: 205–207 °C; 1H NMR (400 MHz, CDCl3) δ: 8.48 (s, 1H, Ar–H), 8.28 (s, 1H, –C
CH of triazole), 7.83–7.92 (m, 2H, Ar–H), 7.60 (dd, J = 5.2, 3.1 Hz, 1H, Ar–H), 7.19–7.24 (m, 4H, Ar–H), 5.44 (s, 2H, –OCH2), 3.31–3.37 (m, 1H, –NCH), 3.20 (m, 4H, 2×-NCH2), 3.05 (m, 4H, 2×-NCH2), 1.26 (m, 2H, –CH2), 1.15–1.20 (m, 2H, –CH2), 1.07 (m, 1H, –NH(CH2)2); 13C NMR (101 MHz, CDCl3) δ: 172.32 (–CO), 163.69 (–CO), 157.92, 155.92, 153.48, 151.51, 147.42, 143.96, 143.53, 137.00, 132.51, 122.09, 121.83, 121.55, 121.40, 119.31 (d, JC–F = 6.1 Hz), 116.59 (d, JC–F = 19.2 Hz), 112.20 (d, JC–F = 19.2 Hz), 108.15, 103.88, 76.28, 76.03, 75.77, 56.76, 44.61, 33.71, 30.96, 28.67, 28.34, 21.68, 13.27, 7.20; 19F NMR (565 MHz CDCl3) δ: −123.27, −114.24; IR (KBr) cm−1: 3154, 2972, 2919, 2825, 1723, 1697, 1620, 1469, 1401, 1340, 1246, 1160, 1015, 904, 829, 773, 712, 462; HRMS (ESI): anal. calcd. For C26H23ClF2N6O3, 540.9558 [M]+; found 541.1583 [M + H]+.
(1-(4-Bromophenyl)-1H-1,2,3-triazol-4-yl)methyl1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate (10b). Green solid; yield 70 mg (82%); mp: 218–220 °C; 1H NMR (400 MHz, CDCl3) δ: 8.52 (s, 1H, Ar–H), 8.31 (s, 1H, –C
CH of triazole), 7.92–8.00 (m, 1H, Ar–H), 7.60–7.65 (m, 4H, Ar–H), 7.24–7.39 (m, 1H, Ar–H), 5.53 (s, 2H, –OCH2), 3.42–3.48 (m, 1H, –NCH), 3.21 (s, 4H, 2×-NCH2), 3.06 (s, 4H, 2×-NCH2), 1.21–1.41 (m, 3H, –CH2, and –NH(CH2)2), 1.05–1.14 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 173.12 (–CO), 164.67 (CO), 152.53, 148.50, 144.65, 137.96, 136.26, 132.84, 122.60 (d, JC–F = 14.9 Hz), 121.97, 113.25 (d, JC–F = 20.0 Hz), 109.18 (d, JC–F = 23.23 Hz), 104.71, 77.53, 57.41, 50.74, 45.84, 40.53, 39.90, 39.10, 34.29, 8.18; IR (KBr) cm−1: 3147, 2977, 2360, 1724, 1697, 1619, 1489, 1422, 1389, 1341, 1246, 1159, 1077, 1019, 904, 831, 774, 617, 544; HRMS (ESI): anal. calcd. For calculated for C26H24BrFN6O3, 566.1077 [M]+; found 567.1171 [M + H]+.
(1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate (10c). Light brown solid; yield 80 mg (95%); mp: 215–217 °C; 1H NMR (400 MHz, CDCl3) δ: 8.55 (s, 1H, Ar–H), 8.33 (s, 1H, –C
CH of triazole), 7.99 (d, J = 13.3 Hz, 1H, Ar–H), 7.72 (d, J = 8.8 Hz, 2H, Ar–H), 7.49 (d, J = 8.8 Hz, 2H, Ar–H), 7.26 (d, J = 8.2 Hz, 1H, Ar–H), 5.52 (s, 2H, –OCH2), 3.43–3.57 (m, 1H, –NCH), 3.25–3.31 (m, 4H, 2×-NCH2), 3.11 (s, 4H, 2×-NCH2), 1.22–1.41 (m, 3H, –CH2, and –NH(CH2)2), 1.14–1.22 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 173.35 (–CO), 164.64 (–CO), 148.44, 145.16 (d, JC–F = 7.5 Hz), 138.07, 135.53, 129.97, 122.73 (d, JC–F = 8.2 Hz), 113.29 (d, JC–F = 23.5 Hz), 109.31, 104.87, 77.43, 77.12, 76.80, 57.88, 51.13, 45.98, 34.73, 29.77, 8.27; IR (KBr) cm−1: 3074, 2973, 1694, 1619, 1476, 1421, 1333, 1245, 1161, 1122, 1028, 892, 831, 800, 775, 703, 703, 623, 544, 464; HRMS (ESI): anal. calcd. For C26H24ClFN6O3, 522.9654 [M]+; found 523.1685 [M + H]+.
(1-(3-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate (10d). White solid; yield 65 mg (77%); mp: 190–192 °C; 1H NMR (400 MHz, CDCl3) δ: 8.55 (s, 1H, Ar–H), 8.35 (s, 1H, –C
CH of triazole), 7.98 (d, J = 13.3 Hz, 1H, Ar–H), 7.86 (d, J = 14.8 Hz, 1H, Ar–H), 7.63–7.68 (m, 1H, Ar–H), 7.39–7.52 (m, 2H, Ar–H), 7.25–7.28 (m, 1H, Ar–H), 5.52 (s, 2H, –OCH2), 3.44 (s, 1H, –NCH), 3.25–3.32 (m, 4H, 2×-NCH2), 3.11 (s, 4H, 2×-NCH2), 1.25–1.40 (m, 3H, –CH2, and –NH(CH2)2), 1.14–1.22 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 172.60 (–CO), 164.75 (–CO), 152.38, 149.26, 144.52 (d, JC–F = 4.5 Hz), 138.59 (d, JC–F = 39.3 Hz), 135.19, 132.14, 129.19, 123.81 (d, JC–F = 1.8 Hz), 122.28, 120.29, 119.25, 112.49 (d, JC–F = 9.0 Hz), 109.01, 106.54, 79.66, 57.61, 51.51, 46.00, 41.24, 40.23, 35.70, 29.81, 8.44; HRMS (ESI): anal. calcd. For C26H24ClFN6O3, 522.9654 [M]+; found 523.1679 [M + H]+.
(1-(4-(Trifluoromethoxy)phenyl)-1H-1,2,3-triazol-4-yl)methyl1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate (10e). White solid; yield 70 mg (80%); mp: 205–207 °C; 1H NMR (400 MHz, CDCl3) δ: 8.57 (s, 1H, Ar–H), 8.32 (s, 1H, –C
CH of triazole), 8.04 (d, J = 13.1 Hz, 1H, Ar–H), 7.82 (d, J = 8.7 Hz, 2H, Ar–H), 7.38 (d, J = 8.7 Hz, 2H, Ar–H), 7.27 (d, J = 6.2 Hz, 1H, Ar–H), 5.54 (s, 2H, –OCH2), 3.65–3.73 (m, 4H, 2×-NCH2), 3.42 (m, 1H, –NCH), 3.22 (m, 4H, 2×-NCH2), 1.23–1.34 (m, 3H, –NH(CH2)2), 1.14–1.18 (m, 2H, –CH2); 13CNMR (101 MHz, CDCl3) δ: 172.43 (–CO), 164.58 (–CO), 152.22, 149.10, 144.35 (d, JC–F = 4.5 Hz), 138.42 (d, JC–F = 39.3 Hz), 135.03, 131.98, 129.03, 123.64 (d, JC–F = 1.8 Hz), 122.12, 120.12, 119.09, 112.33 (d, JC–F = 9.0 Hz), 109.31, 104.87, 106.38, 77.43, 77.12, 76.80, 57.88, 51.19 (d, JC–F = 29.9 Hz), 45.98, 34.73, 8.27; 19F NMR (565 MHz CDCl3) δ: −123.56, −57.98; HRMS (ESI): anal. calcd. For C27H24F4N6O3, 572.5206 [M]+; found 573.1847 [M + H]+.
(1-(2,4-Difluorophenyl)-1H-1,2,3-triazol-4-yl)methyl1-cyclopropyl-6-fluoro-4-oxo-7-(piperazin-1-yl)-1,4-dihydroquinoline-3-carboxylate (10f). Brown solid; yield 79 mg (94%); mp: 197–199 °C; 1H NMR (400 MHz, CDCl3) δ: 8.56 (s, 1H, Ar–H), 8.31 (s, 1H, –C
CH of triazole), 7.97 (d, J = 13.1 Hz, 1H, Ar–H), 7.86 (dd, J = 14.0, 8.2 Hz, 1H, Ar–H), 7.28 (d, J = 3.6 Hz, 1H, Ar–H), 7.06 (t, J = 8.4 Hz, 2H, Ar–H), 5.53 (s, 2H, –OCH2), 3.44–3.50 (m, 1H, –NCH), 3.24–3.35 (m, 4H, 2×-NCH2), 3.11–3.24 (m, 4H, 2×-NCH2), 1.22–1.34 (m, 3H, –NH(CH2)2), 1.14–1.23 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 173.33 (–CO), 164.62 (–CO), 154.30, 152.65, 148.39, 144.96, 143.81, 138, 126.44 (d, JC–F = 7.1 Hz), 125.67 (d, JC–F = 3 Hz), 122.74, 121.80, 113.18 (d, JC–F = 16.2 Hz), 112.51 (d, JC–F = 15.2 Hz), 109.16, 105.49 (d, JC–F = 13 Hz), 105.23, 104.92, 77.25, 77.04, 76.83, 65.92, 58.22, 57.63, 50.47, 45.55, 34.71, 29.64, 18.43, 15.27, 8.21; 19F NMR (565 MHz CDCl3) δ: −123.51, −121.23, −114.33; HRMS (ESI): anal. calcd. For C26H23F3N6O3, 524.5042 [M]+; found 525.1843 [M + H]+.
(1-(3-Chloro-4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-acetylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (11a). Pale pink solid; yield 94 mg (95%); mp: 169–171 °C; 1H NMR (400 MHz, CDCl3) δ: 8.57 (s, 1H, Ar–H), 8.32 (s, 1H, –C
CH of triazole), 7.96–8.03 (m, 1H, Ar–H), 7.90 (dd, J = 6.2, 2.6 Hz, 1H, Ar–H), 7.65–7.68 (m, 1H, Ar–H), 7.27–7.33 (m, 3H, Ar–H), 5.52 (s, 2H, –OCH2), 3.69–3.91 (m, 4H, 2×-NCH2), 3.42–3.46 (m, 1H, –NCH), 3.25 (m, 4H, 2×-NCH2), 2.17 (s, 3H, –CH3), 1.25–1.37 (m, 2H, –CH2), 1.13–1.21 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 173.30 (–CO), 169.07 (–COCH3), 164.59 (–CO), 159.36, 156.6 (d, JC–F = 26.3 Hz), 148.48, 144.11 (d, JC–F = 26.4 Hz), 137.89, 133.65, 122.89 (d, JC–F = 31.8 Hz), 120.22, 117.57 (d, J = 23.2 Hz), 113, 109.75, 105.17, 77.33, 77.00, 76.68, 57.72, 49.92, 46.14, 41.08, 34.65, 21.27, 8.18; 19F NMR (565 MHz CDCl3) δ: −123.54, −114.39; HRMS (ESI): anal. calcd. For C28H25 ClF2N6O4, 582.9928 [M]+; found 583.1649 [M + H]+.
(1-(4-Bromophenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-acetylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (11b). Brick red solid; yield 68 mg (66%); mp: 201–203 °C; 1H NMR (400 MHz, CDCl3) δ: 8.55 (s, 1H, Ar–H), 8.29 (s, 1H, –C
CH of triazole), 8.00 (d, J = 13.1 Hz, 1H, Ar–H), 7.62–7.73 (m, 4H, Ar–H), 7.27 (d, J = 8.1 Hz, 1H, Ar–H), 5.52 (s, 2H, –OCH2), 3.68–3.91 (m, 4H, 2×-NCH2), 3.41–3.45 (m, 1H, –NCH), 3.22–3.29 (m, 4H, 2×-NCH2), 2.17 (s, 3H, –CH3), 1.25–1.34 (m, 2H, –CH2), 1.12–1.19 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 173.27 (–CO), 169.04 (–COCH3), 164.65 (–CO), 154.29, 151.71, 148.43, 144.21 (d, JC–F = 18.0 Hz), 137.85, 135.71 (d, JC–F = 21.7 Hz), 132.81, 122.14 (d, JC–F = 50.0 Hz), 121.89, 113.32 (d, JC–F = 23.0 Hz), 109.29, 105.16, 77.32, 77.00, 76.67, 57.70, 50.35, 49.44, 46.12, 41.06, 34.61, 29.63, 21.28, 8.16; HRMS (ESI): anal. calcd. For C28H26BrFN6O4, 609.4564 [M]+; found 610.1307 [M + H]+.
(1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-acetylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (11c). Light brick red solid; yield 69 mg (72%); mp: 197–199 °C; 1H NMR (400 MHz, CDCl3) δ: 8.55 (s, 1H, Ar–H), 8.30 (s, 1H, –C
CH of triazole), 8.01 (d, J = 12.2 Hz, 1H, Ar–H), 7.72 (d, J = 6.5 Hz, 2H, Ar–H), 7.50 (d, J = 6.7 Hz, 2H, Ar–H), 7.28 (s, 1H, Ar–H), 5.52 (s, 2H, –OCH2), 3.69–3.84 (m, 4H, 2×-NCH2), 3.37–3.43 (m, 1H, –NCH), 3.25 (m, 4H, 2×-NCH2), 2.17 (s, 3H, –CH3), 1.26–1.40 (m, 2H, –CH2), 1.03–1.20 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 173.06 (–CO), 169.04 (–COCH3), 164.52 (–CO), 155.83, 152.07, 148.44, 144.25 (d, JC–F = 9.1 Hz), 137.85, 135.35, 134.51, 129.85, 122.39 (d, JC–F = 32.3 Hz), 121.67, 113.35 (d, JC–F = 23.4 Hz), 109.43, 105.16, 77.32, 77.00, 76.68, 57.72, 50.37, 49.44, 46.12, 41.06, 34.61, 21.28, 8.17; HRMS (ESI): anal. calcd. For C28H26ClFN6O4, 565.0024 [M]+; found 566.1810 [M + H]+.
(1-(3-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-acetylpiperazin-1-yl)-1cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (11d). Light pink solid; yield 70 mg (73%); mp: 205–207 °C; 1H NMR (400 MHz, CDCl3) δ: 8.56 (s, 1H, Ar–H), 8.32 (s, 1H, –C
CH of triazole), 8.01 (d, J = 13.1 Hz, 1H, Ar–H), 7.83 (s, 1H, Ar–H), 7.66 (d, J = 7.8 Hz, 1H, Ar–H), 7.40–7.48 (m, 2H, Ar–H), 7.27 (d, J = 6.3 Hz, 1H, Ar–H), 5.52 (s, 2H, –OCH2), 3.68–3.86 (m, 4H, 2×-NCH2), 3.41–3.45 (m, 1H, –NCH), 3.22–3.29 (m, 4H, 2×-NCH2), 2.17 (s, 3H, –CH3), 1.29–1.36 (m, 2H, –CH2), 1.14–1.21 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 173.05 (–CO), 169.04 (–COCH3), 164.54 (–CO), 154.66, 151.92, 148.44, 144.17 (d, JC–F = 29.7 Hz), 137.77 (d, JC–F = 16.7 Hz), 135.48, 130.76, 128.80, 122.55 (d, JC–F = 18.2 Hz), 120.75, 118.44, 113.32 (d, JC–F = 23.2 Hz), 109.27, 105.16, 77.32, 77.00, 76.68, 57.70, 50.35, 49.46, 46.12, 41.06, 34.63, 29.63, 21.27, 8.16; HRMS (ESI): anal. calcd. For C28H26ClFN6O4, 565.0024 [M]+; observed 566.1810 [M + H]+.
(1-(4-(Trifluoromethoxy)phenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-acetylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (11e). Radish brown solid; yield 61 mg (58%); mp: 220–222 °C; 1H NMR (400 MHz, CDCl3) δ: 8.55 (s, 1H, Ar–H), 8.30 (s, 1H, –C
CH of triazole), 8.01 (d, J = 12.2 Hz, 1H, Ar–H), 7.72 (d, J = 6.5 Hz, 2H, Ar–H), 7.50 (d, J = 6.7 Hz, 2H, Ar–H), 7.28 (s, 1H, Ar–H), 5.52 (s, 2H, –OCH2), 3.69–3.84 (m, 4H, 2×-NCH2), 3.37–3.43 (m, 1H, –NCH), 3.25 (m, 4H, 2×-NCH2), 2.17 (s, 3H, –CH3), 1.26–1.40 (m, 2H, –CH2), 1.03–1.20 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 169.09 (–CO), 166.35 (–COCH3), 164.62 (–CO), 148.62, 144.14 (d, JC–F = 31 Hz), 137.91, 123.19, 113.51 (d, JC–F = 56.6 Hz), 109.48, 105.14, 78.06, 77.33, 77.01, 76.69, 74.78, 52.13 (d, JC–F = 22.2 Hz), 50.97, 49.50, 46.20, 41.13, 34.62, 29.68, 21.31, 8.18; 19F NMR (565 MHz CDCl3) δ: −123.56, −57.98 HRMS (ESI): anal. calcd. For C29H26F4N6O5, 614.5576 [M]+; found 615.1995 [M + H]+.
(1-(2,4-Difluorophenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-acetylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (11f). Creamy white solid; yield 34 mg (35%); mp: 219–221 °C; 1H NMR (400 MHz, CDCl3) δ: 8.54 (s, 1H, Ar–H), 8.33 (s, 1H, –C
CH of triazole), 8.00–8.07 (m, 1H, Ar–H), 7.81–7.89 (m, 2H, Ar–H), 7.38 (d, J = 8.5 Hz, 2H, Ar–H), 7.27 (d, J = 4.8 Hz, 1H, Ar–H), 5.53 (s, 2H, –OCH2), 3.68–3.97 (m, 4H, 2× –NCH2), 3.43 (s, 1H, –NCH), 3.21–3.29 (m, 4H, 2×-NCH2), 2.17 (s, 3H, –CH3), 1.29–1.41 (m, 2H, –CH2), 1.14–1.20 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 173.02 (–CO), 169.05 (–COCH3), 164.58 (–CO), 154.98, 152.09, 148.71 (d, JC–F = 50.7 Hz), 144.16 (d, JC–F = 21.4 Hz), 137.88, 135.43 (d, JC–F = 36.4 Hz), 122.68, 122.19, 121.97, 118.93, 113.39 (d, JC–F = 22.8 Hz), 109.35, 105.16, 77.32, 77.00, 76.68, 57.75, 50.34, 49.90, 46.13, 41.06, 34.62, 21.29, 8.18; 19F NMR (565 MHz CDCl3) δ: −123.69, −121.30, −118.55; HRMS (ESI): anal. calcd. For C28H25F3N6O4, 566.5412 [M]+; found 567.1976 [M + H]+.
(1-(3-Chloro-4-fluorophenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-benzoylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (12a). Light green solid; yield 105 mg (96%); mp: 210–212 °C; 1H NMR (400 MHz, CDCl3) δ: 8.56 (s, 1H, Ar–H), 8.30 (s, 1H, –C
CH of triazole), 8.00 (dd, J = 12.9, 8.5 Hz, 1H, Ar–H), 7.89 (dd, J = 6.2, 2.5 Hz, 1H, Ar–H), 7.63–7.67 (m, 1H, Ar–H), 7.45 (s, 5H, Ar–H), 7.27–7.32 (m, 3H, Ar–H), 5.52 (s, 2H, –OCH2), 3.70–4.01 (m, 4H, 2×-NCH2), 3.23–3.43 (m, 5H, –NCH and 2×-NCH2), 1.33–1.40 (m, 2H, –CH2), 1.15–1.22 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 173.01 (–CO), 170.50 (–COAr), 164.59 (–CO), 148.49, 144.11 (d, JC–F = 31.3 Hz), 143.95, 138.17, 137.89, 135.13, 130.07, 128.61, 127.11, 123.07, 122.90 (d, JC–F = 33.0 Hz), 120.30, 117.57 (d, JC–F = 22.8 Hz), 113.53, 109.45, 105.22, 77.32, 77.00, 76.68, 57.75, 51.51, 50.54, 49.78, 35.27, 34.65, 8.21; 19F NMR (565 MHz CDCl3) δ: −123.52, −114.37; HRMS (ESI): anal. calcd. For C33H27ClF2N6O4, 645.0638 [M]+; found 646.1860 [M + H]+.
(1-(4-Bromophenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-benzoylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (12b). Light green solid; yield 74 mg (65.5%); mp: 172–174 °C; 1H NMR (400 MHz, CDCl3) δ: 8.56 (s, 1H, Ar–H), 8.31 (s, 1H, –C
CH of triazole), 7.98 (d, J = 12.8 Hz, 1H, Ar–H), 7.65–7.73 (m, 4H, Ar–H), 7.45–7.53 (m, 5H, Ar–H), 7.27–7.33 (m, 2H, Ar–H), 5.55 (s, 2H, –OCH2), 3.68–4.13 (m, 4H, 2×-NCH2), 3.22–3.43 (m, 5H, –NCH and 2×-NCH2), 1.28–1.34 (m, 2H, –CH2), 1.14 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 172.29 (–CO), 170.48 (–COAr), 164.54 (–CO), 148.45, 144.58 (d, JC–F = 52.8 Hz), 135.48 (d, JC–F = 70.7 Hz), 132.81, 130.04, 128.58, 127.09, 122.48, 121.90, 113.25, 109.27, 105.22, 77.32, 77.00, 76.67, 57.73, 50.52, 49.46, 34.62, 8.18; HRMS (ESI): anal. calcd. For C33H28BrFN6O4, 671.5274 [M]+; found 672.1319 [M + H]+.
(1-(4-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-benzoylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (12c). Light mint solid; yield 75 mg (71%); mp: 168–170 °C; 1H NMR (400 MHz, CDCl3) δ: 8.57 (s, 1H, Ar–H), 8.30 (s, 1H, –C
CH of triazole), 8.03 (d, J = 12.9 Hz, 1H, Ar–H), 7.71 (d, J = 8.7 Hz, 2H, Ar–H), 7.45–7.53 (m, 7H, Ar–H), 7.28 (d, J = 7.8 Hz, 2H, Ar–H), 5.52 (s, 2H, –OCH2), 3.69–4.10 (m, 4H, 2×-NCH2), 3.42–3.32 (m, 5H, –NCH and 2×-NCH2), 1.23–1.36 (m, 2H, –CH2), 1.10–1.19 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 173.10 (–CO), 170.50 (–COAr), 164.57 (–CO), 155.92, 154.74, 148.45, 144.34, 141.72, 137.88, 136.55, 135.16, 134.34, 129.96 (d, JC–F = 19.2 Hz), 128.61, 127.12, 123.36 (d, JC–F = 6.1 Hz), 122.55, 121.71, 113.45 (d, JC–F = 23.2 Hz), 109.40, 105.22, 77.32, 77.00, 76.68, 57.78, 41.73, 34.62, 32.31, 8.20; HRMS (ESI): anal. calcd. For C33H28ClFN6O4, 627.0734 [M]+; found 628.1961 [M + H]+.
(1-(3-Chlorophenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-benzoylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (12d). Light orange solid; yield 98 mg (92%); mp: 189–191 °C; 1H NMR (400 MHz, CDCl3) δ: 8.58 (s, 1H, Ar–H), 8.35 (s, 1H, –C
CH of triazole), 8.03 (d, J = 13.1 Hz, 1H, Ar–H), 7.85 (s, 1H, Ar–H), 7.67–7.70 (m, 1H, Ar–H), 7.40–7.50 (m, 7H, Ar–H), 7.29–7.31 (m, 1H, Ar–H), 5.55 (s, 2H, –OCH2), 3.71–4.12 (m, 4H, 2×-NCH2), 3.43–3.35 (m, 5H, –NCH and 2×-NCH2), 1.28–1.38 (m, 2H, –CH2), 1.14–1.23 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 173.00 (–CO), 170.51 (–COAr), 164.57 (–CO), 154.75, 152.40, 148.49, 144.33, 137.79 (d, JC–F = 17.5 Hz), 135.32 (d, JC–F = 36.9 Hz), 131.10, 130.38, 129.16, 128.72 (d, JC–F = 22.1 Hz), 127.43, 122.92, 121.11, 118.79, 113.38 (d, JC–F = 23.0 Hz), 109.32, 105.25, 77.34, 77.02, 76.71, 57.73, 50.60, 49.72, 41.65, 34.65, 29.66, 8.20; HRMS (ESI): anal. calcd. For C33H28ClFN6O4, 627.0734 [M]+; found 628.1966 [M + H]+.
(1-(4-(Trifluoromethoxy)phenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-benzoylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (12e). Light peach solid; yield 99 mg (87%); mp: 195–197 °C; 1H NMR (400 MHz, CDCl3) δ: 8.58 (s, 1H, Ar–H), 8.32 (s, 1H, –C
CH of triazole), 7.99–8.07 (m, 1H, Ar–H), 7.81 (d, J = 8.9 Hz, 2H, Ar–H), 7.37–7.53 (m, 7H, Ar–H), 7.27–7.29 (m, 2H, Ar–H), 5.54 (s, 2H, –OCH2), 3.69–4.16 (m, 4H, 2×-NCH2), 3.45–3.22 (m, 5H, –NCH and 2×-NCH2), 1.29–1.38 (m, 2H, –CH2), 1.14–1.29 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 170.68 (–CO), 164.63 (–CO), 159.41, 149.12 (d, JC–F = 21.7 Hz), 148.36, 144.29, 137.92, 134.86 (d, JC–F = 29.2 Hz), 130.08, 128.62, 127.13, 122.11 (d, JC–F = 21.1 Hz), 113.65 (d, JC–F = 20.9 Hz), 104.99 (d, JC–F = 26.3), 77.31, 77.00, 76.68, 57.49 (d, JC–F = 13.6 Hz).49.76, 40.71, 34.62, 8.22; 19F NMR (565 MHz CDCl3) δ −123.21, −57.86; IR (KBr) cm−1: 3145, 2975, 2360, 1725, 1619, 1493, 1424, 1388, 1340, 1314, 1252, 1213, 1157, 1077, 1009, 906, 832, 802, 777, 702, 618, 544; HRMS (ESI): anal. calcd. For C34H28F4N6O5, 676.6286 [M]+; found 677.2151 [M + H]+.
(1-(2,4-Difluorophenyl)-1H-1,2,3-triazol-4-yl)methyl7-(4-benzoylpiperazin-1-yl)-1-cyclopropyl-6-fluoro-4-oxo-1,4-dihydroquinoline-3-carboxylate (12f). Creamy white solid; yield 70.5 mg (66.5%); mp: 203–205 °C; 1H NMR (400 MHz, CDCl3) δ: 8.57 (s, 1H, Ar–H), 8.26 (s, 1H, –C
CH of triazole), 8.01–8.06 (m, 1H, Ar–H), 7.82–7.89 (m, 1H, Ar–H), 7.47 (s, 5H, Ar–H), 7.28 (d, J = 7.7 Hz, 1H, Ar–H), 7.04–7.09 (m, 2H, Ar–H), 5.54 (s, 2H, –OCH2), 3.66–4.10 (m, 4H, 2×-NCH2), 3.22–3.44 (m, 5H, –NCH and 2×-NCH2), 1.25–1.43 (m, 2H, –CH2), 1.12–1.23 (m, 2H, –CH2); 13C NMR (101 MHz, CDCl3) δ: 172.94 (–CO), 170.46 (–COAr), 164.56 (–CO), 154.41, 148.41, 145.96, 143.44 (d, JC–F = 14.14 Hz), 138.04, 135.20 (d, JC–F = 18.2 Hz), 130.02, 128.56, 127.08, 126.38, 125.91 (d, JC–F = 73.2 Hz), 123.45, 113.32, 112.43 (d, JC–F = 15.9 Hz), 109.24, 105.12 (d, JC–F = 5.4 Hz), 77.27, 76.95, 76.63, 57.63, 34.56, 8.16; 19F NMR (565 MHz CDCl3) δ: −123.51, −121.23, −114.33; HRMS (ESI): anal. calcd. For C33H27F3N6O4, 628.6122 [M]+; found 629.214 [M + H]+.
X-ray crystallographic analysis
The single-crystal X-ray analysis further verified the structure of the synthesized triazole derivatives 11a and 12d. Briefly, the crystals were formed via the slow evaporation solution technique with ethyl alcohol. Graphite monochromatized Cu Kα radiation (λ = 1.54184 Å) was used to measure the X-ray diffraction intensity data at 293 K using the X-ray scan method on a Rigaku XtaLAB Synergy-i single-crystal X-ray diffractometer with a CCD-detector (HyPix-Bantam). The structure of compounds 11a and 12d was established by the direct method using the Olex2-1.5 software.52 Then, it was refined using the full-matrix least-squares method on F2 by SHELXL.53 Fig. 9 and 10 show the thermal ellipsoid plot prepared using ORTEP III54 of compounds 11a and 12d, which were crystallized in an orthorhombic and triclinic system with the Pbcn and![[P with combining macron]](https://www.rsc.org/images/entities/i_char_0050_0304.gif) 1 space group, respectively. Table 1 provides information about the single crystal X-ray crystallographic structures of compounds 11a and 12d.
1 space group, respectively. Table 1 provides information about the single crystal X-ray crystallographic structures of compounds 11a and 12d.
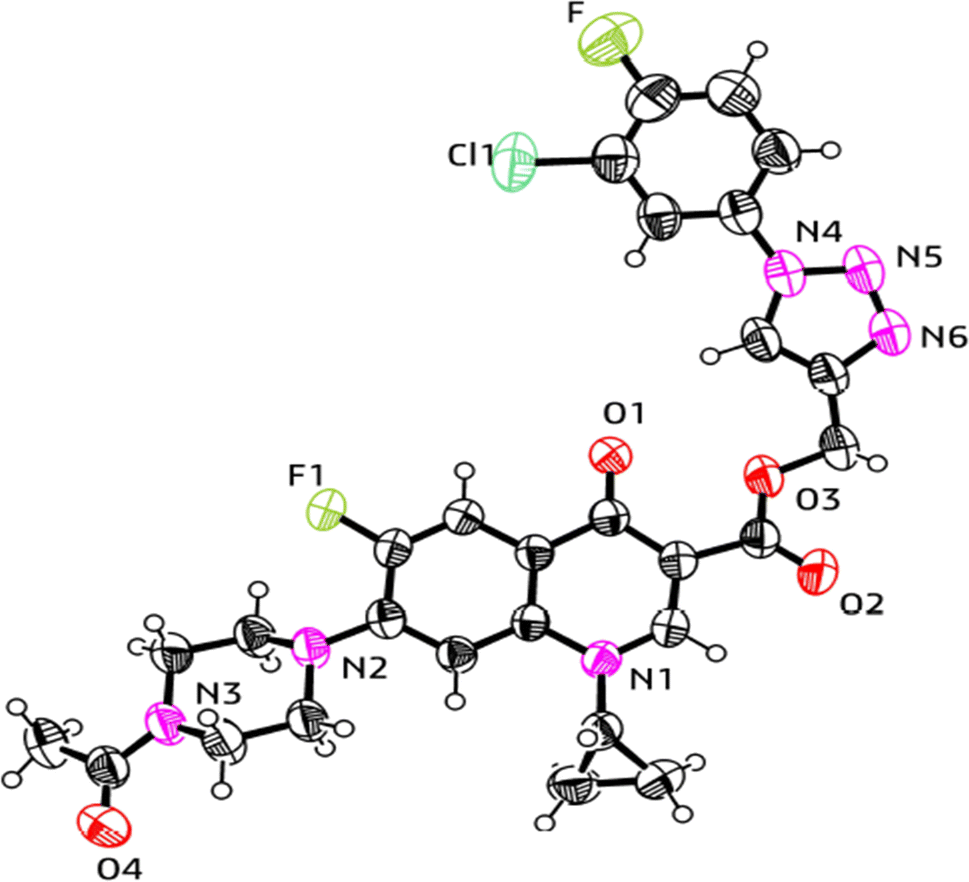 | ||
| Fig. 9 X-ray structure of the 1,2,3-triazole-linked carboxylic group of ciprofloxacin conjugate 11a. | ||
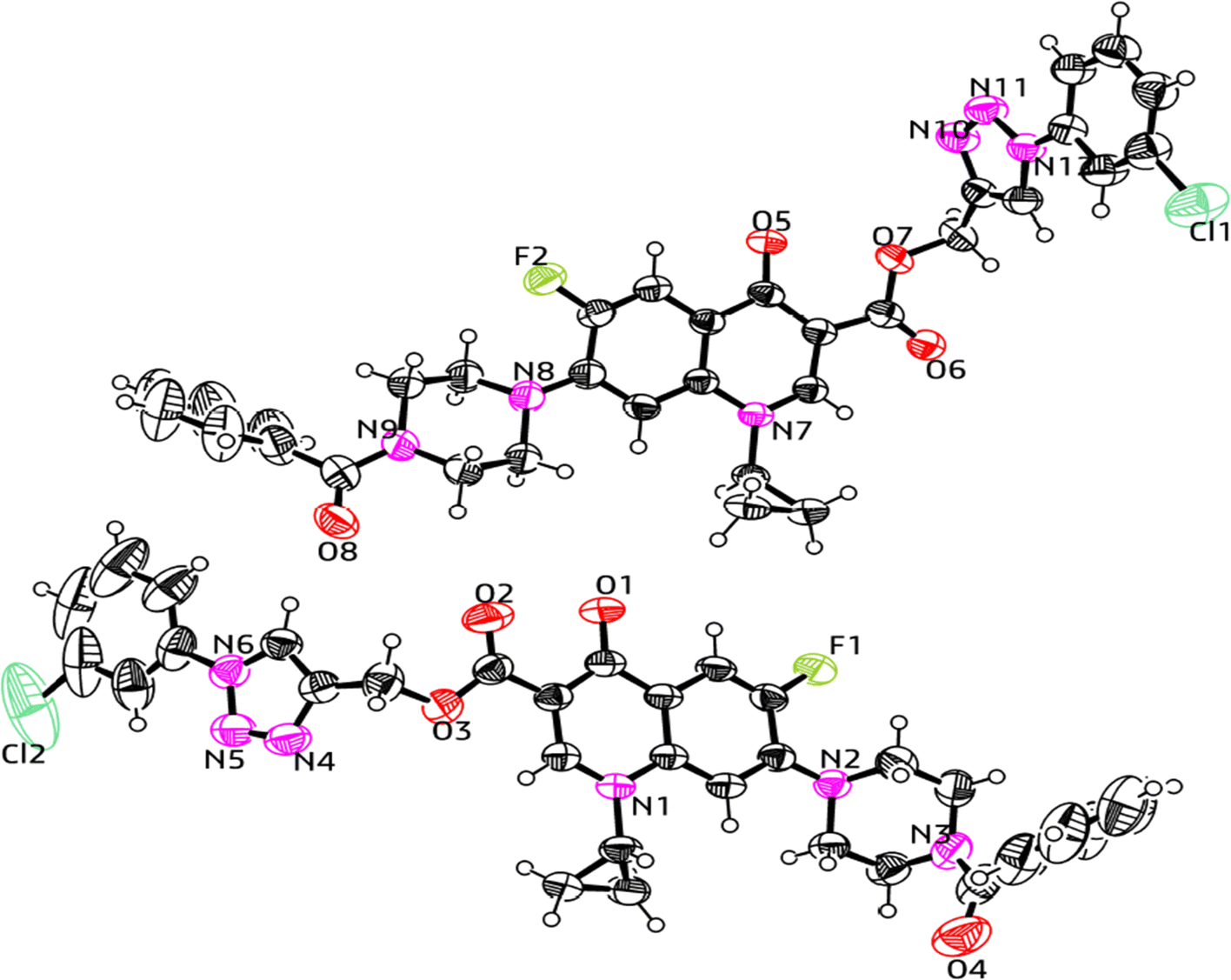 | ||
| Fig. 10 X-ray structure of the 1,2,3-triazole-linked carboxylic group of ciprofloxacin conjugate 12d. | ||
| Compound no. | 11a | 12d |
| CCDC no. | 2268965 | 2268988 |
| Empirical formula | C28H25ClF2N6O4 | C33H28ClFN6O4 |
| Formula weight | 582.99 | 627.06 |
| Temperature | 293(2) K | 293(2) K |
| Wavelength | 1.54184 Å | 1.54184 Å |
| Crystal system | Orthorhombic | Triclinic |
| Space group | Pbcn | 1 |
| Hall group | −P2n2ab | −P1 |
| Unit cell dimensions | a = 22.1700(4) Å, α = 90 | a = 11.7334(2) Å, α = 97.2620(10) |
| b = 7.54141(12) Å, β = 90 | b = 12.3293(2) Å, β = 98.652(2) | |
| c = 34.4865(4) Å, γ = 90 | c = 22.1027(4) Å, γ = 102.2660(10) | |
| Volume | 5765.92(16) | 3047.14(9) |
| Z | 8 | 4 |
| Density | 1.343 g cm−3 | 1.367 g cm−3 |
| Absorption coefficient | 1.668 mm−1 | 1.572 mm−1 |
| F(000) | 2416.0 | 1304.0 |
| Theta range for data collection | 5.124 to 136.332 | 7.438 to 144.282 |
| Index ranges | −25 ≤ h ≤ 26 | −14 ≤ h ≤ 14 |
| −8 ≤ k ≤ 9 | −15 ≤ k ≤ 14 | |
| −41 ≤ l ≤ 41 | −27 ≤ l ≤ 27 | |
| Reflections collected | 34455 |
58235 |
| Completeness to theta | 99.9% | 99.9% |
| Absorption correction | Multi-scan | Multi-scan |
| Refinement method | Full-matrix least-squares on F2 | Full-matrix least-squares on F2 |
| Goodness-of-fit on F2 | 1.063 | 1.069 |
| Final R indices [I > 2sigma(I)] | R1 = 0.0539, wR2 = 0.1532 | R1 = 0.0655, wR2 = 0.1963 |
| S. No. | Compound | Gram +ve strains | Gram −ve strains | % Hemolysis | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| E. faecalis (ATCC 29212) | S. aureus (ATCC 25923) | S. epidermidis (clinical) | E. coli (ATCC 25922) | P. aeruginosa (ATCC 27853) | S. typhi (clinical) | P. mirabilis (clinical) | A. baumannii (clinical) | K. pneumoniae (clinical) | |||
| 6 |  |
50 | 6.25 | 100 | 0.391 | 12.5 | 100 | 50 | 100 | 12.5 | 10.20 |
| 7 | 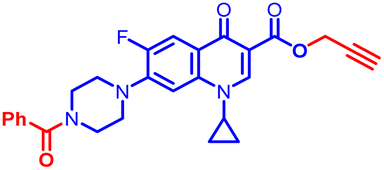 |
100 | 100 | ≥50 | 3.15 | 100 | 50 | 100 | 12.5 | 50 | 6.15 |
| 10 |  |
0.781 | 0.195 | 6.25 | 0.195 | 0.195 | 12.5 | 12.5 | 12.5 | 0.195 | 3.43 |
| 10a | 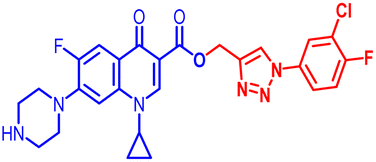 |
6.25 | 0.195 | 12.5 | ≤0.195 | 6.25 | 25 | 50 | 12.5 | 6.25 | 6.58 |
| 10b | 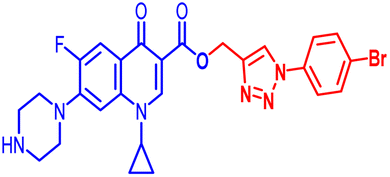 |
6.25 | 0.391 | 50 | ≤0.195 | 3.12 | 25 | 50 | 12.5 | 3.12 | 4.06 |
| 10c | 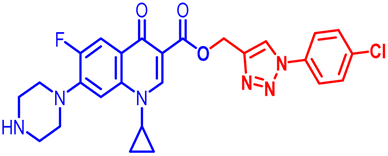 |
6.25 | 1.56 | 25 | ≤0.195 | 6.25 | 25 | 50 | 12.5 | 6.25 | 1.68 |
| 10d | 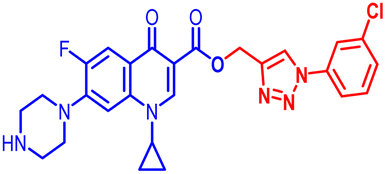 |
12.5 | 0.391 | 50 | 100 | 0.781 | 25 | 50 | 12.5 | 1.56 | 1.17 |
| 10e | 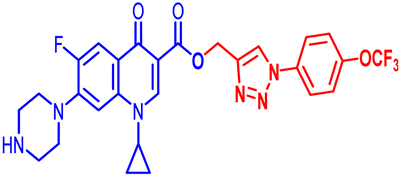 |
3.12 | 6.25 | 50 | 12.5 | 100 | 50 | 25 | 100 | 100 | 7.65 |
| 10f | 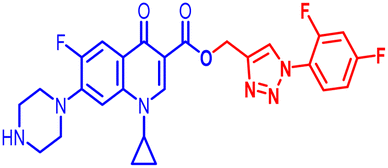 |
100 | 12.5 | 100 | 25 | 50 | 25 | 25 | 100 | 25 | 7.00 |
| 11a | 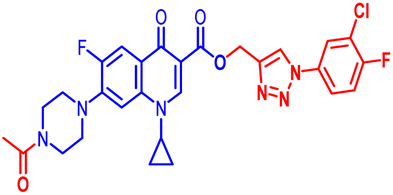 |
25 | 100 | 50 | 100 | 100 | ≥100 | 100 | 12.5 | 12.5 | 5.74 |
| 11b | 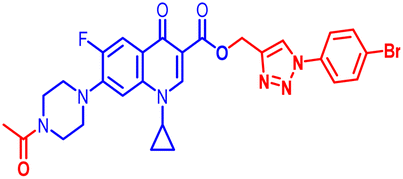 |
100 | 100 | ≥100 | 100 | 100 | 25 | 100 | 100 | 50 | 2.99 |
| 11c | 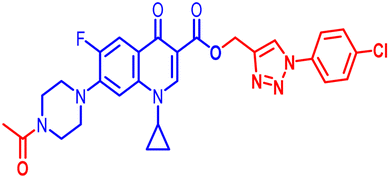 |
100 | 100 | ≥100 | 100 | 100 | 100 | 100 | 100 | 50 | 7.62 |
| 11d | 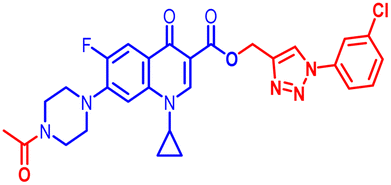 |
100 | 100 | 50 | 25 | 50 | 50 | 50 | 50 | 25 | 24.50 |
| 11e |  |
50 | 6.25 | ≥100 | 25 | 50 | 25 | 25 | 50 | 50 | 7.09 |
| 11f |  |
25 | 0.391 | 50 | 25 | 50 | 50 | 25 | 50 | 50 | 1.42 |
| 12a |  |
100 | 12.5 | 50 | 100 | 50 | 25 | 100 | 50 | ≥ 50 | 7.23 |
| 12b |  |
100 | 25 | 100 | 100 | 100 | 12.5 | 100 | 50 | ≥ 50 | 8.06 |
| 12c | 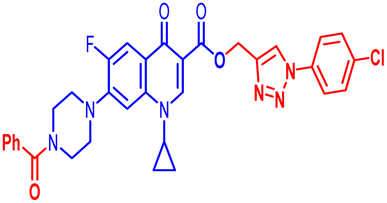 |
100 | 1.56 | 100 | 100 | 100 | 6.25 | 50 | 100 | 50 | 6.24 |
| 12d | 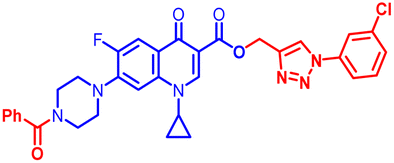 |
3.12 | 100 | >100 | 12.5 | 100 | 50 | 12.5 | 100 | 50 | 4.80 |
| 12e | 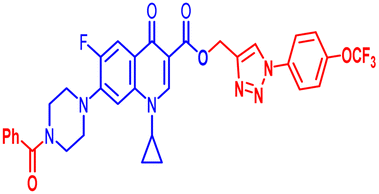 |
100 | 0.391 | 50 | ≤0.195 | 1.56 | 1.56 | 50 | 12.5 | 1.56 | 8.41 |
| 12f | 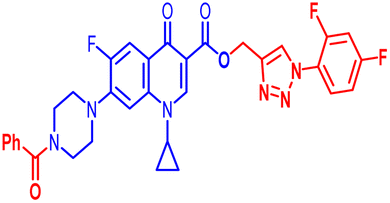 |
0.781 | 100 | ≥100 | 100 | 100 | 25 | 100 | 100 | 50 | 24.29 |
| Standard | Ciprofloxacin | 0.781 | 6.25 | 0.391 | 0.391 | 0.781 | 6.25 | 1.56 | 25 | 0.391 | 5.23 |
Biological assays
000 rpm for 10 min. The DNeasy Tissue Kit (Qiagen, Cat. No. 69504) was used according to the manufacturer's protocol to recover DNA efficiently. The extracted DNA preparation was quantified by measuring the absorbance at 260 nm. The value of one absorbance at A260 is equivalent to 50 μg mL−1 for standard DNA. The integrity of the isolated genomic DNA was determined by 0.8% agarose (Ameresco, USA) gel electrophoresis, which was carried out for 1 h at 75 V against a 1 kb molecular weight marker (Fermentas, USA). The ratio of absorbance at 260 nm and 280 nm was calculated to check the purity of DNA. The genomic DNA was obtained from all treatments using a commercially available kit method and resolved in agarose gel electrophoresis. The results showed that the DNA was intact, and the concentration of all samples was nearly uniform.Conclusions
In total, 21 new synthetic ciprofloxacin analogues were tested against nine different antibacterial strains. Five compounds from a pool of a small library, i.e., 10, 10a, 10b, 10c and 12e, showed excellent activity against E. coli (ATCC 25922) strain with MIC of ≤0.195 μg mL−1, while compound 10 showed excellent activity against S. aureus (ATCC 25923), E. coli (ATCC 25922), P. aeruginosa (ATCC27853) and K. pneumonia (clinical isolate) with an MIC of 0.195 μg mL−1, respectively. Moreover, four compounds, i.e., 10b, 10d, 11f and 12e, showed excellent activity with an MIC of 0.391 μg mL−1 against S. aureus (ATCC25923), whereas the control drug, ciprofloxacin, showed an MIC of 6.25 μg mL−1. Further, the electron-withdrawing groups such as OCF3, F, Cl, and Br in the benzene ring at the para-position play a significant role in defining the antibacterial activity. The hemotoxicity testing results showed that all substances had a very low toxicity profile. To create second-generation compounds for antibacterial investigations, it is necessary to analyze the structure–activity relationship (SAR) of potential lead molecules and lower their further effective doses, while increasing their antibacterial action. Subsequently, the lead molecules from the second-generation compounds are coupled with current clinical medications to create multifunctional hybrids. This approach will undoubtedly help reduce the problem of bacterial drug resistance to a certain extent. Moreover, in silico studies were performed on the most potent compounds and it was found that compound 10b showed the highest docking score of −8.1 kcal mol−1. Thus, due to its highest docking score, it was further subjected to MD analysis with E. coli. DNA gyrase B complex protein for 100 ns. Also, the in vitro assay indicated that 10b was a more potent inhibitor of DNA gyrase compared to ciprofloxacin employed as the positive control. The single-crystal X-ray analysis of the compounds further confirmed their structure and design criteria. Extensive studies involving a large number of compounds are required to reach any meaningful conclusion in the future.Author contributions
Upendra Kumar Patel did the conceptualization, methodology, software, visualization, experimental work and writing the main text. Punit Tiwari did the antibacterial and hemolytic activity under the supervision of Ragini Tilak. Gaurav Joshi and Roshan Kumar did molecular docking studies, molecular dynamic (MD) analysis, DNA gyrase expression assay, software and visualization. Alka Agarwal helped with supervision, reviewing, and editing the original draft. All authors reviewed the manuscript.Conflicts of interest
The authors have no conflict of Interest for manuscript publication.Acknowledgements
Upendra Kumar Patel is thankful to the Council of Scientific & Industrial Research (CSIR) HRDG New Delhi, India (Grant no. 09/013(0933)/2020-EMR-I) for the Junior Research Fellowship and Senior Research Fellowship. Alka Agarwal is thankful to Banaras Hindu University and Institute of Eminence (IoE no. Dev scheme no. 6031), Varanasi, India, for the financial support.References
- H. H. H. Mohammed, E. S. M. N. Abdelhafez, S. H. Abbas, G. A. I. Moustafa, G. Hauk, J. M. Berger, S. Mitarai, M. Arai, R. M. Abd E-Baky and G. E. D. A. Abuo-Rahma, Bioorg. Chem., 2019, 88, 102952 CrossRef CAS PubMed.
- E. N. Esfahani, M. Mohammadi-Khanaposhtani, Z. Rezaei, Y. Valizadeh, R. Rajabnia, M. Hassankalhori, F. Bandarian, M. A. Faramarzi, N. Samadi, M. R. Amini, M. Mahdavi and B. Larijani, Res. Chem. Intermed., 2019, 45, 223–236 CrossRef CAS.
- Z. Xu, S. J. Zhao, Z. S. Lv, F. Gao, Y. Wang, F. Zhang, L. Bai and J. L. Deng, Eur. J. Med. Chem., 2019, 162, 396–406 CrossRef CAS PubMed.
- N. Suresh, H. N. Nagesh, J. Renuka, V. Rajput, R. Sharma, I. A. Khan and C. S. K. V. Gowri, Eur. J. Med. Chem., 2014, 71, 324–332 CrossRef CAS PubMed.
- G. L. Patrick, An Introduction to Medicinal Chemistry, Oxford University Press, Oxford, 3rd edn, 2005 Search PubMed.
- R. Kant, V. Singh, G. Nath, S. K. Awasthi and A. Agarwal, Eur. J. Med. Chem., 2016, 124, 218–228 CrossRef CAS PubMed.
- R. J. Reece, A. Maxwell and J. C. Wang, J. Biochem. Mol. Biol., 1991, 26, 335–375 CrossRef CAS.
- T. Plech, B. Kaproń, A. Paneth, U. Kosikowska, A. Malm, A. Strzelczyk, P. Stączek, Ł. Świątek, B. Rajtar and M. Polz-Dacewicz, Molecules, 2015, 20, 6254–6272 CrossRef CAS PubMed.
- E. H. Yee, S. S. Cheng, G. A. Knappe and C. A. Moomau, MIT Sloan Manag. Rev., 2020, 1, 10–17 Search PubMed.
- B. Aslam, W. Wang, M. I. Arshad, M. Khurshid, S. Muzammil, M. H. Rasool, M. A. Nisar, R. F. Alvi, M. A. Aslam, M. U. Qamar, M. K. F. Salamat and Z. Baloch, Infect. Drug Resist., 2018, 11, 1645–1658 CrossRef CAS PubMed.
- A. Mermer, O. Faiz, A. Demirbas, N. Demirbas, M. Alagumuthu and S. Arumugam, Bioorg. Chem., 2019, 85, 308–318 CrossRef CAS PubMed.
- T. F. Schäberle and I. M. Hack, Trends Microbiol., 2014, 22, 165–167 CrossRef PubMed.
- J. B. Michel, P. J. Yeh, R. Chait, R. C. Moellering and R. Kishony, Proc. Natl. Acad. Sci. U.S.A., 2008, 105, 14918–14923 CrossRef CAS PubMed.
- S. Loewe, Arzneim. Forsch., 1953, 3, 285–290 CAS.
- M. Shavit, V. Pokrovskaya, V. Belakhov and T. Baasov, Bioorg. Med. Chem., 2017, 25, 2917–2925 CrossRef CAS PubMed.
- S. B. Ozdemir, N. Demirbas, A. Demirbas and N. Colak, ChemistrySelect, 2018, 3, 2144–2151 CrossRef CAS.
- D. Sarkar, V. M. Khedkar, F. A. K. Khan and N. Jaiprakash, Eur. J. Med. Chem., 2016, 5, 385–399 Search PubMed.
- M. J. Mitton-fry, S. J. Brickner, J. C. Hamel, R. Barham, L. Brennan, J. M. Casavant, X. Ding, S. Finegan, J. Hardink, T. Hoang, M. D. Huband, M. Maloney, A. Marfat, S. P. Mccurdy, D. Mcleod, C. Subramanyam, M. Plotkin, J. Schafer, G. G. Stone, D. P. Uccello, T. Wisialowski, K. Yoon, R. Zaniewski and C. Zook, Bioorg. Med. Chem. Lett., 2017, 27, 3353–3358 CrossRef CAS PubMed.
- G. T. Robertson, E. J. Bonventre, T. B. Doyle, Q. Du, L. Duncan, T. W. Morris, E. D. Roche, D. Yan and A. S. Lynch, Antimicrob. Agents Chemother., 2008, 52, 2313–2323 CrossRef CAS PubMed.
- J. B. Bremner, Pure Appl. Chem., 2007, 79, 2143–2153 CrossRef CAS.
- V. Pokrovskaya and T. Baasov, Expert Opin. Drug Discovery, 2010, 5, 883–902 CrossRef CAS PubMed.
- A. Keivanloo, S. Sepehri, M. Bakherad and M. Eskandari, ChemistrySelect, 2020, 5, 4091–4098 CrossRef CAS.
- B. Negi, D. Kumar, W. Kumbukgolla, S. Jayaweera, P. Ponnan, R. Singh, S. Agarwal and D. S. Rawat, Eur. J. Med. Chem., 2016, 115, 426–437 CrossRef CAS PubMed.
- H. M. Savanur, K. N. Naik, S. M. Ganapathi, K. M. Kim and R. G. Kalkhambkar, ChemistrySelect, 2018, 3, 296–5303 Search PubMed.
- D. K. R. Vennam, R. K. Thatipamula, S. B. Haridasyam and S. K. Koppula, Chem. Heterocycl. Compd., 2018, 54, 630–637 CrossRef CAS.
- R. Huisgen, G. Szeimies and L. Mobius, Chem. Ber., 1967, 100, 2494–2507 CrossRef CAS.
- A. Massarotti, S. Aprile, V. Mercalli, E. D. Grosso, G. Grosa, G. Sorba and G. C. Tron, ChemMedChem, 2014, 9, 2497–2508 CrossRef CAS PubMed.
- S. G. Agalave, S. R. Maujan and V. S. Pore, Chem.–Asian J., 2011, 6, 2696–2718 CrossRef CAS PubMed.
- D. Gonzalez-Calderon, M. G. Mejía-Dionicio, M. A. Morales-Reza, A. Ramírez-Villalva, M. Morales-Rodríguez, B. Jauregui-Rodríguez, E. Díaz-Torres, C. Gonzalez-Romero and A. Fuentes-Benítes, Eur. J. Med. Chem., 2016, 112, 60–65 CrossRef CAS PubMed.
- H. C. Kolb and K. B. Sharpless, Drug Discovery Today, 2003, 8, 1128–1137 CrossRef CAS PubMed.
- K. Bezouska, Rev. Mol. Biotechnol., 2002, 90, 269–290 CrossRef CAS PubMed.
- E. N. Esfahani, M. Mohammadi-Khanaposhtani, Z. Rezaei, Y. Valizadeh, R. Rajabnia, M. Hassankalhori, F. Bandarian, M. A. Faramarzi, N. Samadi, M. R. Amini, M. Mahdavi and B. Larijani, Res. Chem. Intermed., 2019, 45, 223–236 CrossRef CAS.
- N. Sultana, M. S. Arayne, S. B. S. Rizvi and U. Haroon, Bull. Korean Chem. Soc., 2011, 32, 483–488 CrossRef CAS.
- S. Jubie, R. Kalirajan and P. Yadav, Eur. J. Chem., 2012, 9, 980–987 CAS.
- N. B. Patel, A. R. Shaikh, H. Soni, R. B. Parmar and J. A. Patel, Chem. Biol. Interface, 2018, 8, 184–193 CAS.
- C. S. Azad and A. K. Narula, RSC Adv., 2016, 6, 19052–19059 RSC.
- R. Kant, D. Kumar, D. Agarwal, R. D. Gupta, R. Tilak, S. K. Awasthi and A. Agarwal, Eur. J. Med. Chem., 2016, 113, 34–49 CrossRef CAS PubMed.
- M. K. Singh, R. Tilak, G. Nath, S. K. Awasthi and A. Agarwal, Eur. J. Med. Chem., 2013, 63, 635–644 CrossRef CAS PubMed.
- R. Kant, V. Singh, G. Nath, S. K. Awasthi and A. Agarwal, Eur. J. Med. Chem., 2016, 124, 218–228 CrossRef CAS PubMed.
- A. Agarwal, P. Singh, A. Maurya, U. K. Patel, A. Singh and G. Nath, ACS Omega, 2022, 7, 2725–2736 CrossRef CAS PubMed.
- B. T. Worrell, J. A. Malik and V. V. Fokin, Science, 2013, 340, 457 CrossRef CAS PubMed.
- S. F. Nielsen, M. Larsen, T. Boesen, K. Schønning and H. Kromann, J. Med. Chem., 2005, 48, 2667–2677 CrossRef CAS PubMed.
- F. Ushiyama, H. Amada, Y. Mihara, T. Takeuchi, N. Tanaka-Yamamoto, M. Mima, M. Kamitani, R. Wada, Y. Tamura, M. Endo, A. Masuko, I. Takata, K. Hitaka, H. Sugiyama and N. Ohtake, Bioorg. Med. Chem., 2020, 28, 115776 CrossRef CAS PubMed.
- Y. M. Thakre and M. D. Choudhary, J. Chem. Pharm. Res., 2012, 4, 1048–1051 CAS.
- R. Kant, V. Singh, G. Nath, S. K. Awasthi and A. Agarwal, Eur. J. Med. Chem., 2016, 124, 218–228 CrossRef CAS PubMed.
- L. E. Evans, A. Krishna, Y. Ma, T. E. Webb, D. C. Marshall, C. L. Tooke, J. Spencer, T. B. C. A. Armstrong and A. M. Edwards, J. Med. Chem., 2019, 62, 4411–4425 CrossRef CAS PubMed.
- B. Marquez, V. Pourcelle, C. M. Vallet, M. P. Mingeot-Leclercq, P. M. Tulkens, J. Marchand-Bruynaert and F. V. Bambeke, Pharm. Res., 2014, 31, 1290–1301 CrossRef CAS PubMed.
- J. C. Mc Pherson III, R. Runner, T. B. Buxton, J. F. Hartmann, D. Farcasiu, I. Bereczki, E. Roth, S. Tollas, E. Ostorházi, F. Rozgonyi and P. Herczegh, Eur. J. Med. Chem., 2012, 47, 615–618 CrossRef PubMed.
- M. Hu, J. Li and S.-Q. Yao, Org. Lett., 2008, 10, 5529–5531 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- S. W. Svenningsen, R. F. Frederiksen, C. Counil, M. Ficker, J. J. Leisner and J. B. Christensen, Molecules, 2020, 25, 1389 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- S. F. Nielsen, M. Larsen, T. Boesen, K. Schønning and H. Kromann, J. Med. Chem., 2005, 48, 2667–2677 CrossRef CAS PubMed.
- I. Wiegand, K. Hilpert and R. E. W. Hancock, Nat. Protoc., 2008, 3, 163–175 CrossRef CAS PubMed.
- G. M. Shaikh, M. Murahari, S. Thakur, M. S. Kumar and Y. C. Mayur, Modelling, J. Mol. Graphics Modell., 2022, 112, 108–114 CrossRef PubMed.
- R. Kataria and A. Khatkar, Curr. Pharm. Biotechnol., 2019, 20, 410–421 CAS.
- M. Sharma, S. Thakur, H. R. Jadhav and S. B. Bharate, ChemistrySelect, 2023, 8, 202301879 CrossRef.
- P. Vishwakarma, N. F. Siddiqui, S. Thakur and H. Jadhav, J. Biomol. Struct. Dyn., 2023, 1–18, DOI:10.1080/07391102.2023.2276315.
- S. A. Hollingsworth and R. O. Dror, J. Neuron., 2018, 99, 1129–1143 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2268965 and 2268988. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra01332h |
| This journal is © The Royal Society of Chemistry 2024 |