
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSolvation controlled excited-state dynamics in a donor–acceptor phenazine-imidazole derivative†
Hai-Xiong Shi*a,
Hong-Wei Baoa and
Gui-Yuan Wu *b
*b
aSchool of Chemical Engineering, Lanzhou University of Arts and Science, Lanzhou, Gansu 730000, China. E-mail: shhx2003@163.com
bAnhui Province Key Laboratory for Control and Applications of Optoelectronic Information Materials, School of Physics and Electronic Information, Anhui Normal University, Wuhu, 241002, China. E-mail: wgy@ahnu.edu.cn
First published on 28th May 2024
Abstract
In the past few decades, significant efforts have been devoted to developing phenazine derivatives in various fields such as medicine, pesticides, dyes, and conductive materials owing to their highly Stokes-shifted fluorescence and distinctive photophysical properties. The modulation of the surrounding environment can effectively influence the luminescent behavior of phenazine derivatives, prompting us to investigate the solvent effect on the excited state dynamics. Herein, we present the solvent controlled excited state dynamics of a novel triphenylamine-based phenazine-imidazole molecule (TPAIP) through steady-state spectra and femtosecond transient absorption spectra. The fluorescence emission spectrum exhibited a redshift with increasing solvent polarity, indicating the existence of a charge transfer state. Furthermore, by tracking the femtosecond transient absorption spectra of TPAIP, we found that the nature of the relaxed S1 state was strongly influenced by the solvent polarity: intersystem crossing character appears in apolar solvent, whereas intramolecular charge transfer character occurs in polar solvent because of solvation. These findings provide significant theoretical insights into the impact of solvents on the excited state dynamics within phenazine derivatives. This understanding supports diverse applications ranging from advanced biological probe design to photocatalysis and pharmaceutical research.
Introduction
Phenazine represents an important class of nitrogen-containing heterocycles, having a distinctive backbone structure with an electron-deficient π system and a lone pair of electrons on its N atoms.1–9 It has been extensively explored in the fields of medicine, pesticides, dyes, and conductive materials. Consequently, numerous phenazine derivatives have been synthesized and identified owing to their captivating performances. The functional group or ligand tuning of phenazine derivatives has been extensively documented to have a close relationship with their opto-electronic properties, which significantly impact their sensing responses. In general, the introduction of functional groups at the binding site enhances optical properties involving photoinduced electron transfer, charge transfer, excited-state intramolecular proton transfer, fluorescence resonance energy transfer, and other interaction modes.10–17 For example, Wei et al. synthesized a series of ion probes using phenazine as the framework for the selective fluorescence recognition of specific metal ions or anions.18–21 Wang et al. reported the phenazine radical cation sodium 3,3′-(phenazine-5,10-diyl)bis(propane-1-sulfonate) with a high solubility of 1.4 M and high stability in water.22 A nano-phenazine@Ketjen black anode was achieved through the in situ dissolution-precipitation method by Zhang et al., and the battery possessed a cycle life of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 times due to the stabilities and insolubilities of phenazine and its reduction products in aqueous electrolytes.23 Bode et al. reported diversity-oriented modifications of the phenazine core through two distinct BGCs in the entomopathogenic bacterium Xenorhabdus szentirmaii, which lives in symbiosis with nematodes. A previously unidentified aldehyde intermediate, which can be modified by multiple enzymatic and non-enzymatic reactions, is a common intermediate bridging the pathways encoded by these BGCs.24 Stang et al. synthesized a series of platinum(II) metallacycles via the coordination-driven self-assembly of a phenazine-cored dipyridyl donor with a 90° Pt(II) acceptor and various dicarboxylate donors. These metallacycles exhibited similar absorption profiles, but their fluorescence emission shifts to the blue as the bite angles between the carboxylate building blocks decrease. This research demonstrated the potential of coordination-driven self-assembly in creating materials with precisely tailored functionalities at the molecular level.25 Therefore, the investigation of the excited state dynamics of phenazine derivatives is crucial due to their wide range of diverse applications.26–29
000 times due to the stabilities and insolubilities of phenazine and its reduction products in aqueous electrolytes.23 Bode et al. reported diversity-oriented modifications of the phenazine core through two distinct BGCs in the entomopathogenic bacterium Xenorhabdus szentirmaii, which lives in symbiosis with nematodes. A previously unidentified aldehyde intermediate, which can be modified by multiple enzymatic and non-enzymatic reactions, is a common intermediate bridging the pathways encoded by these BGCs.24 Stang et al. synthesized a series of platinum(II) metallacycles via the coordination-driven self-assembly of a phenazine-cored dipyridyl donor with a 90° Pt(II) acceptor and various dicarboxylate donors. These metallacycles exhibited similar absorption profiles, but their fluorescence emission shifts to the blue as the bite angles between the carboxylate building blocks decrease. This research demonstrated the potential of coordination-driven self-assembly in creating materials with precisely tailored functionalities at the molecular level.25 Therefore, the investigation of the excited state dynamics of phenazine derivatives is crucial due to their wide range of diverse applications.26–29
Upon photoexcitation, D–A chromophores can generate a locally excited state and/or an intramolecular charge transfer state.30–32 The presence of these states is influenced not only by the strength of the donor–acceptor interaction but also by the surrounding environment, such as solvent polarity. The locally excited state, characterized by an electronic transition within the same moiety, typically exhibits a small change in its permanent dipole moment upon excitation.33–35 Conversely, the intramolecular charge transfer state displays a solvatochromic effect due to a significant dipole moment change. The potential energy level of the intramolecular charge transfer state can be easily adjusted by varying both the strength of the D–A interaction and the solvent polarity. As systems with intramolecular charge transfer state character exhibit a strong interaction between solvent and solute, increasing solvent polarity reduces the energy level of this state while minimally affecting the energy level of the LE state due to its small dipole moment change. Furthermore, transitioning from a locally excited to an intramolecular charge transfer state by modulating solvent properties induces changes in both electronic structure and geometry, resulting in reduced overlap between the highest occupied molecular orbital and lowest unoccupied molecular orbital. This alteration, combined with spin–orbit coupling changes, significantly impacts the intersystem crossing process.36–38
Herein, the novel D–A molecule TPAIP, which contains the electron-donating group triphenylamine (TPA) and the electron-withdrawing phenazine, was designed and synthesized via the cyclization reaction of 2,3-diamino-phenazine with 4-(N,N-diphenylamino)benzaldehyde. Femtosecond transient absorption spectroscopy confirmed that the TPAIP excited state transitions from the initial localized excited state to the solvent-induced charge transfer excited state exhibited dipole characteristics in polar solvents. The time constants of the excited state deactivation process was extracted from the femtosecond transient absorption experimental data. However, in non-polar solvents, molecular excited states directly relax from local excited states to triple excited states through intersystem conversion. This implies that solvent polarity primarily influences the redistribution of charges in TPAIP's excited state.
Results and discussion
Steady-state spectroscopy
The spectral characteristics of TPAIP were investigated using UV-vis and fluorescence experiments across a range of solvents, including dimethyl sulfoxide, acetonitrile, dimethylformamide, methanol, ethanol, acetone, tetrahydrofuran, ethyl acetate, dichloromethane, chloroform, and toluene. In the UV-vis absorption spectrum, TPAIP displayed distinct absorption peaks at 250–350 nm and 375–500 nm corresponding to the UV absorptions of the triphenylamine and phenazine units, respectively. Simultaneously, solvent polarity-induced solvatochromism led to an approximately 27 nm redshift when transitioning from a low polarity solvent to a high polarity solvent. Furthermore, fluorescence measurements for TPAIP were conducted in various organic solvents (λex = 435 nm). The fluorescence emission intensity of TPAIP not only manifested solvation effects, with the maximum emission peak shifting to longer wavelengths with increasing solvent polarity, but also exhibited significantly lower fluorescence emission intensities in highly polar solvents (dimethyl sulfoxide, dimethylformamide, acetonitrile, methanol, ethanol) compared to other solvents, leading to fluorescence quenching. For instance, the maximum fluorescent emission wavelengths of TPAIP in toluene, tetrahydrofuran, chloroform and acetone solvents were 510, 540, 570, and 600 nm, respectively. In methanol and acetonitrile solvents, the fluorescence emission was nearly quenched due to the aggregation-caused quenching effect. The photophysical properties, including the quantum yield (Φf) and fluorescent lifetime of TPAIP in various organic solvents, are summarized in Table 1. The fluorescence emission of TPAIP exhibited a redshift with increasing solvent polarity, indicating its characteristic intramolecular charge transfer state behavior (Fig. 1b).| Solvent | λabs/nm | λem/nm | τF/ns | Φ/% |
|---|---|---|---|---|
| DMSO | 450 | 640 | 0.33 | 0.32 |
| ACN | 437.5 | 640 | 0.29 | 0.32 |
| DMF | 446 | 625 | 0.43 | 0.62 |
| MeOH | 450 | — | — | — |
| EtOH | 449 | 550 | — | — |
| Acetone | 438 | 600 | 0.73 | 3.76 |
| THF | 439 | 540 | 2.21 | 45.13 |
| EA | 431 | 530 | 1.99 | 38.08 |
| DCM | 452 | 600 | 3.1 | 50.68 |
| CHCl3 | 448 | 570 | 2.87 | 61.72 |
| Tol | 439 | 510 | 1.28 | 19.58 |
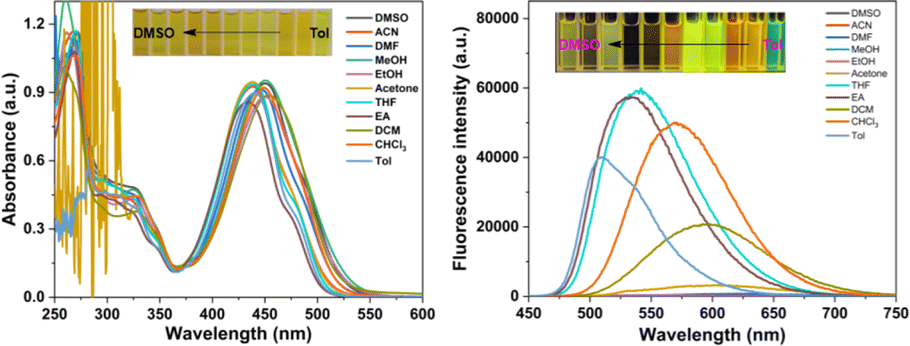 | ||
| Fig. 1 The UV-vis absorption and fluorescence spectra of TPAIP (20 μM) in various solvents, including dimethyl sulfoxide (DMSO), acetonitrile (ACN), dimethylformamide (DMF), methanol (MeOH), ethanol (EtOH), acetone, tetrahydrofuran (THF), ethyl acetate (EA), dichloromethane (DCM), chloroform (CHCl3), and toluene (Tol). | ||
To explore the aggregation-induced quenching effects of TPAIP, the UV absorption and fluorescence emission spectra of TPAIP were examined at various concentrations in different solvents including toluene, acetone, methanol, and dimethyl sulfoxide. The absorbance in the UV absorption spectra increased with an elevation in TPAIP concentration (Fig. S1†). In the fluorescence emission spectra (Fig. S2†), the fluorescence initially increased and then decreased as the concentration of TPAIP rose in toluene, acetone, and dimethyl sulfoxide, indicating a pronounced aggregation-induced quenching effect. However, in methanol solvent, although there was also an initial increase followed by a decrease in fluorescence intensity, it remained consistently weak across all concentrations due to phenazine's propensity for easy aggregation resulting in fluorescence quenching. Overall, the highest fluorescence emission intensity was observed at a TPAIP concentration of 20 μM, which indicated a dispersed state. Therefore, this specific concentration was utilized to examine subsequent transient absorption spectra.
UV-vis absorption and fluorescence spectra were employed to investigate the aggregation-caused quenching effect of TPAIP in mixed solutions of dimethyl sulfoxide/H2O with varying degrees of aggregation. The UV absorption spectrum of TPAIP exhibited a decrease as the water ratio increased, indicating molecular aggregation (Fig. S3†). In the fluorescence spectrum (Fig. S3†), the intensity of fluorescent emission from TPAIP significantly decreased when the H2O concentration reached 10%. Within the range of 20–100% H2O content, the fluorescent emission intensity continued to decrease until it was completely quenched, suggesting that molecular aggregation caused a rapid decline in emission intensity due to an increase in H2O content. To the best of our knowledge, the aggregation-caused quenching effect can be induced by the aggregation of phenazine groups. Thus, we verified whether the fluorescence quenching of TPAIP in methanol was caused by molecular aggregation. The effectiveness of measuring TPAIP aggregation in solution was demonstrated through dynamic light scattering experiments. TPAIP (20 μM) was subjected to dynamic light scattering in various solvents (toluene, dimethyl sulfoxide, acetone, ethanol, and methanol), revealing substantial aggregation sizes in methanol. However, the aggregate size of TPAIP in other solvents fell below the instrument's limit, precluding determination of the aggregate size. At the consistent concentration of 20 μM, the average aggregation size of TPAIP in methanol was measured at 212 nm (Fig. S4†). These data unequivocally established the formation of aggregates by TPAIP in methanol. Molecular aggregation relies on intermolecular interactions, such as π–π stacking, hydrogen bonding, hydrophobic or electrostatic interactions, and van der Waals forces. These interactions govern the degree of molecular aggregation and exert influence over the photophysical behaviors of the molecule.
In order to gain a deeper understanding of the molecular structure in its solid form, TPAIP crystals were cultivated for X-ray analysis through the gradual evaporation of their methanol solution. The crystal structure illustrated in Fig. 2 revealed that the phenazine-imidazole units of TPAIP exhibit a well-defined planar configuration. Furthermore, numerous weak interactions between molecules, such as C–H⋯π and π–π stacking, can be observed within the crystalline state (Fig. 2). When viewed from a side perspective, a single crystal is formed by stacking two TPAIP molecular dimers that are not completely face-to-face stacked. These dimers display intermolecular C–H⋯π and π–π packing interactions at distances of 2.77 Å and 3.34 Å, respectively. Additionally, these dimers stack with two outer reversed dimer heads to form columns through strong C–H⋯π interactions (at a distance of 2.71 Å), among other interactions. Moreover, one molecule within each dimer engages in π–π stacking interaction with another phenazine unit at a distance of 3.3 Å. From this perspective, three types of C–H⋯π interactions (at distances of 2.93 Å, 3.15 Å, and 3.07 Å) can be identified between benzene and other molecules within the TPA unit.
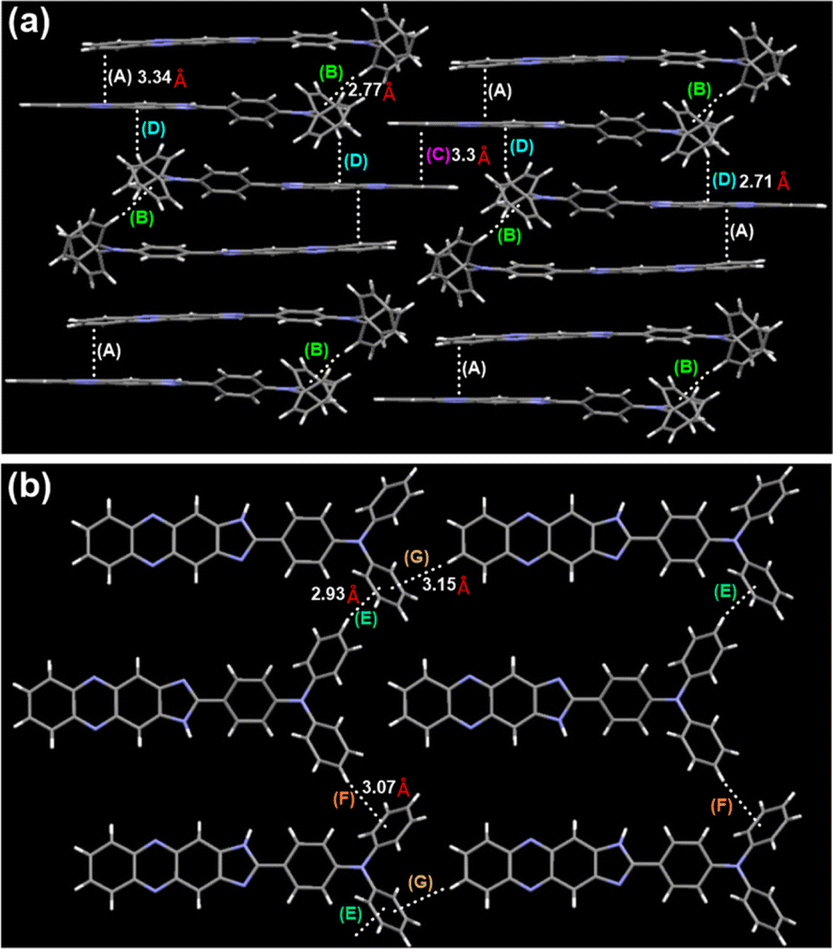 | ||
| Fig. 2 TPAIP crystal analysis. The molecular conformation in (a) side and (b) top views. (A), (C), (D), π–π stacking; (B), (E), (F), (G) C–H⋯π interactions. | ||
Femtosecond transient absorption spectroscopy
Transient absorption measurements have the capability to identify the pathways and rates of transitions between energy levels of excited states in samples.39 In order to investigate the impact of TPAIP on excited state dynamics, we conducted femtosecond transient absorption spectroscopy on TPAIP (20 μM) using different solvents such as toluene, acetone, ethanol, methanol, and dimethyl sulfoxide at 380 nm pulse excitation (Fig. 3). Interestingly, TPAIP exhibited a distinct transient absorption spectrum in toluene compared to those in acetone, ethanol, methanol, and dimethyl sulfoxide. The initial transient absorption spectra displayed a negative ground-state bleaching signal along with excited-state absorption bands within specific wavelength ranges: 400–455 nm and 455–650 nm, respectively (Fig. 3). Notably, in toluene solvent (Fig. 3a and b), the absorption intensity of TPAIP's excited state was weaker between 455 and 530 nm compared to that between 530 and 650 nm due to the presence of overlapping stimulated emission and excited-state absorption signals. Additionally, slight blue-shifts were observed in the excited-state absorption bands, indicating solvent–solute interaction or solvation effects. This can be attributed to excitation occurring on the red side of ground-state absorption leading to a transition from the initially populated higher vibrational state of lowest singlet excited state towards a relaxed S1 state.40 As the time delay increased to 300 ps, the stimulated emission signal became completely obscured by the excited-state absorption band and disappeared entirely. Simultaneously, the ground-state bleaching signal started to surpass zero due to the overlap between the excited-state absorption and the ground-state bleaching bands. Furthermore, a blue-shifted excited-state absorption signal was observed and reached its maximum intensity, indicating that triplet species formation might be responsible for these phenomena. Subsequently, throughout the entire duration of our experiment (7 ns), the signal did not fully diminish. In the acetone solution of TPAIP, we observed distinct blue-shifted excited state absorption bands along with a newly formed excited-state absorption band in the range of 465–575 nm. These additional absorption bands were attributed to intramolecular charge transfer from TPA moieties to phenazine units (Fig. 4). After 3000 ps had elapsed, both ground-state bleaching and excited-state absorption signals returned to their baseline levels, suggesting an absence of triplet states during the entire excited state evolution process. Similarly, when using solvents with higher polarities, such as ethanol, methanol and dimethyl sulfoxide, clear features of intramolecular charge transfer states were observed in the transient spectra (Fig. 3c–h and S5†). However, these intramolecular charge transfer signals decayed more rapidly compared to those in acetone solution. Additionally, the stimulated emission signal was barely discernible, which aligns with the low fluorescence quantum yields indicated in Table 1.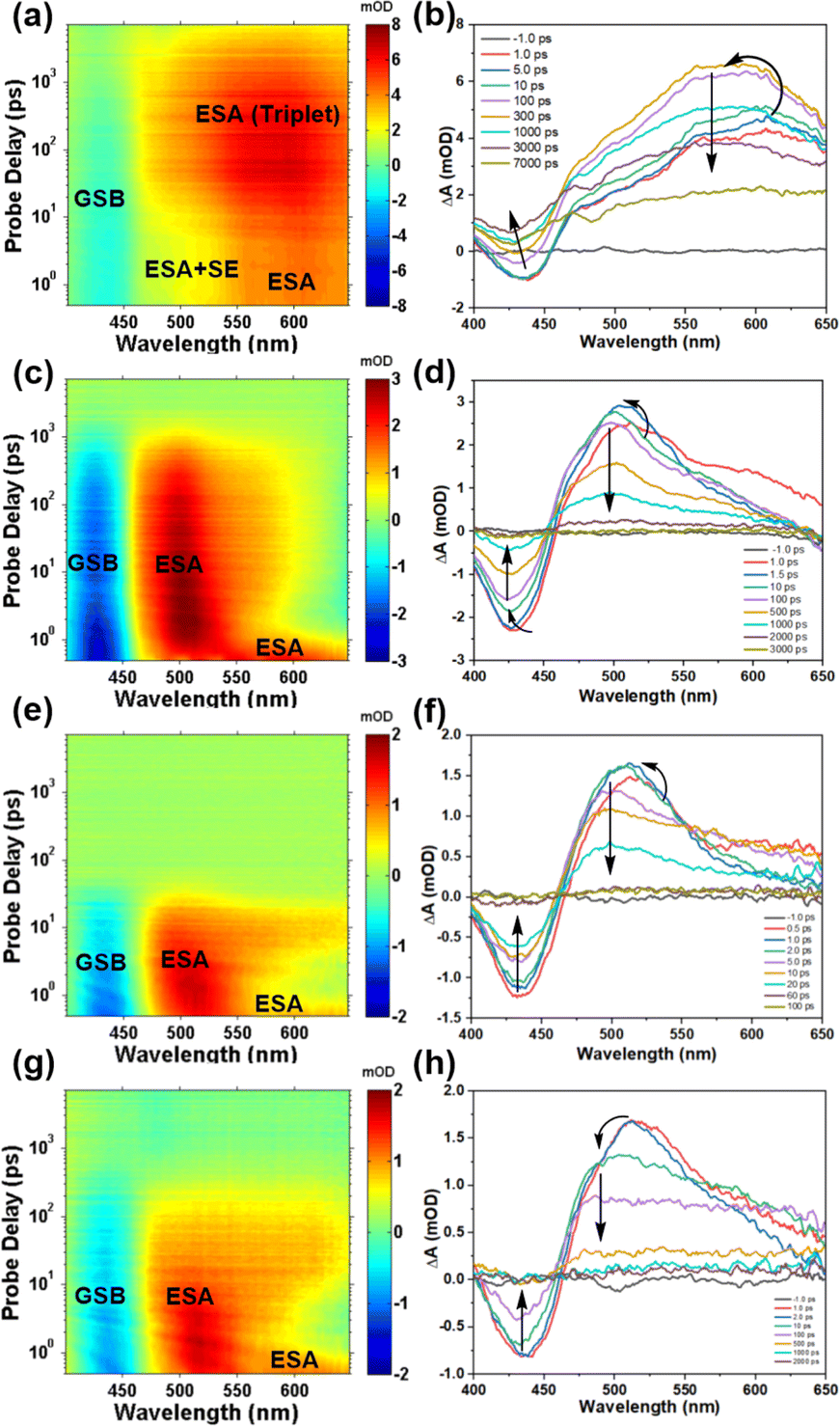 | ||
| Fig. 3 Femtosecond broadband transient absorption spectra of the TAS data matrix after 380 nm excitation (left column: toluene (a), acetone (c), methanol (e) and dimethyl sulfoxide (g)) and time evolution of the transient absorption spectra (right column: toluene (b), acetone (d), methanol (f) and dimethyl sulfoxide (h)) of TPAIP (20 μM). | ||
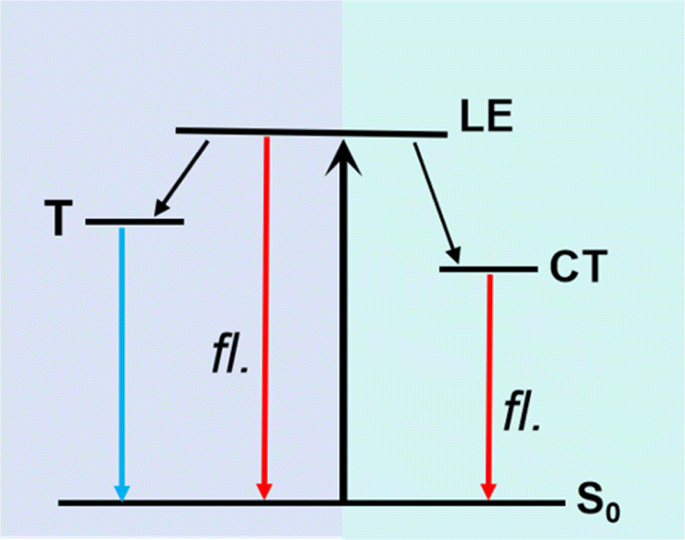 | ||
| Fig. 4 Solvent-polarity dependent excited-state charge redistribution mechanism of TPAIP. LE: locally excited state, CT: charge transfer state, T: triplet. | ||
Solvent-dependent excited-state dynamics mechanism of TPAIP
Global fitting of transient absorption data was conducted to comprehensively understand the process of these spectral changes, and the time constants of the corresponding excited state processes are presented in Table 2. The target evolution models and species-associated difference spectra (SADS) can be found in Fig. S6,† while Fig. S7† illustrates the temporal concentrations of related species and kinetics traces at various selected wavelengths. For TPAIP in toluene, the initial species-associated spectra (SADS-1) displayed a ground-state bleaching signal at 430 nm and an excited-state absorption signal at 490 nm. The relaxation process associated with solvation-stabilization had a time constant of 20.2 ps. This timescale suggested that solvent reorganization facilitated by conformational relaxation occurs within the locally excited state. Subsequently, one portion of the locally excited state relaxes back to the S0 state, while another portion relaxes to the triplet state through intersystem conversion. Nanosecond transient absorption spectroscopy was utilized to investigate the characteristics of the triplet state in toluene under both deaerated and aerobic conditions (Fig. S8†). The lifetime of TPAIP monomers in the triplet state was found to be twice as long under deaerated conditions (6 μs) compared to that in aerated conditions (200 ns) in toluene. This observation indicates that TPAIP was responsible for generating the triplet state, as oxygen can quench it. However, for TPAIP in acetone, ethanol, methanol, and dimethyl sulfoxide solvents, only two distinct processes were observed: a locally excited state and an intramolecular charge transfer state (Fig. 4). The lifetimes of SADS-1 (locally excited state) were measured as 1.2 ps, 2.5 ps, 6.9 ps, and 18.2 ps for TPAIP in acetone, ethanol, methanol, and dimethyl sulfoxide, respectively. Therefore, SADS-2 represented a geometrically relaxed intramolecular charge transfer state with radiative pathway time constants of 118.4 ps for acetone, 18.1 ps for ethanol, 1.0 ns for methanol, and 324.9 ps (τ2) for dimethyl sulfoxide.41 Due to the longer radiative pathway time constant observed in acetone solvent compared to the others mentioned above, the fluorescence emission intensity from TPAIP was relatively weaker in ethanol, methanol, and dimethyl sulfoxide solvents than that observed in acetone.| Solvent | τ1 (ps) | τ2 | τ3 (ns) |
|---|---|---|---|
| Toluene | 20.2 ± 0.1 | 1.1 ± 0.1 ns | Long-lived |
| Acetone | 1.2 ± 0.1 | 1.0 ± 0.1 ns | — |
| Ethanol | 2.5 ± 0.1 | 118.4 ± 0.2 ps | — |
| Methanol | 7.0 ± 0.1 | 61.2 ± 1.0 ps | — |
| Dimethyl sulfoxide | 18.2 ± 0.1 | 324.9 ± 0.9 ps | — |
Conclusions
In conclusion, this study delved into the excited-state charge redistribution dynamics of the D–A molecule TPAIP upon excitation. Utilizing steady-state and femtosecond transient absorption spectroscopy, we found that in non-polar solvents, the molecule underwent intersystem crossing from its local excited state to generate a triplet state. In polar solvents, a population conversion from the local excited state to the charge transfer state was detected. Moreover, with increasing solvent polarity, this conversion slowed down, while the decay of the charge transfer state accelerated. These findings indicated that solvent fluctuations play a crucial role in the charge redistribution process. The investigation presented here highlights how femtosecond transient absorption can be an effective technique for tracking photoinduced delocalization/localization processes in phenazine derivatives such as TPAIP. Understanding the underlying mechanism governing TPAIP's response to different solvent polarities is invaluable for guiding the design of novel fluorescent molecular chem- or biosensors with numerous potential applications.Author contributions
H.-X Shi, G.-Y Wu: conceived the project, performed most of the experiments, analyzed the data, acquired funding, writing – review & editing. Hong-Wei Bao: formal analysis, data curation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 22301079), the Natural Science Foundation of Gansu Province (No. 21JR7RA199) and Natural Science Foundation of Anhui Province (No. 2108085QB79).References
- K. X. Zhao, F. Yu, W. B. Liu, Y. J. Huang, A. A. Said, Y. Li and Q. C. Zhang, J. Org. Chem., 2020, 85, 101–107 CrossRef CAS PubMed.
- P. Y. Gu, F. Zhou, J. K. Gao, G. Li, C. Y. Wang, Q. F. Xu, Q. C. Zhang and J. M. Lu, J. Am. Chem. Soc., 2013, 135, 14086–14089 CrossRef CAS PubMed.
- J. B. Laursen and J. Nielsen, Chem. Rev., 2004, 104, 1663–1686 CrossRef CAS PubMed.
- G. Li, Y. C. Wu, J. K. Gao, C. Y. Wang, J. B. Li, H. C. Zhang, Y. Zhao and Q. C. Zhang, J. Am. Chem. Soc., 2012, 134, 20298–20301 CrossRef CAS PubMed.
- F. Zhu, W. Guo and Y. Fu, Chem. Soc. Rev., 2023, 52, 8410–8446 RSC.
- L. Wang, F. Chen, P.-C. Qian and J. Cheng, Tetrahedron Lett., 2022, 88, 153550–153553 CrossRef CAS.
- T. Shigehiro, S. Yagi, T. Maeda, H. Nakazumi, H. Fujiwara and Y. Sakurai, Tetrahedron Lett., 2014, 55, 5195–5198 CrossRef CAS.
- G. Q. Wei, Y. C. Tao, J. J. Wu, Z. Z. Li, M. P. Zhuo, X. D. Wang and L. S. Liao, J. Phys. Chem. Lett., 2019, 10, 679–684 CrossRef CAS PubMed.
- E. R. Clifford, R. W. Bradley, L. T. Wey, J. M. Lawrence, X. L. Chen, C. J. Howe and J. Z. Zhang, Chem. Sci., 2021, 12, 3328–3338 RSC.
- X. M. Wu, X. R. Sun, Z. Q. Guo, J. B. Tang, Y. Q. Shen, T. D. James, H. Tian and W. H. Zhu, J. Am. Chem. Soc., 2014, 136, 3579–3588 CrossRef CAS PubMed.
- G. Qian, X. Z. Li and Z. Y. Wang, J. Mater. Chem., 2009, 19, 522–530 RSC.
- X. Chen, S. W. Nam, G. H. Kim, N. Song, Y. Jeong, I. Shin, S. K. Kim, J. Kim, S. Park and J. Yoon, Chem. Commun., 2010, 46, 8953–8955 RSC.
- X. H. Cheng, R. L. Tang, H. Z. Jia, J. Feng, J. G. Qin and Z. Li, ACS Appl. Mater. Interfaces, 2012, 4, 4387–4392 CrossRef CAS PubMed.
- S. Kothavale and N. Sekar, Dyes Pigm., 2017, 136, 31–45 CrossRef CAS.
- X. Zhou, F. Su, H. Lu, P. Senechal-Willis, Y. Tian, R. H. Johnson and D. R. Meldrum, Biomaterials, 2012, 33, 171–180 CrossRef CAS PubMed.
- M. Maniyazagan, R. Mariadasse, M. Nachiappan, J. Jeyakanthan, N. Lokanath, S. Naveen, G. Sivaraman, P. Muthuraja, P. Manisankar and T. Stalin, Sens. Actuators, B, 2018, 254, 795–804 CrossRef CAS.
- X. Zhang, Y. Xiao and X. Qian, Angew. Chem., Int. Ed., 2008, 47, 8025–8029 CrossRef CAS PubMed.
- T. B. Wei, G. Y. Wu, B. B. Shi, Q. Lin, H. Yao and Y. M. Zhang, Chin. J. Chem., 2014, 32, 1238–1244 CrossRef CAS.
- G. Y. Gao, W. Y. Qu, B. B. Shi, Q. Lin, H. Yao, Y. M. Zhang, J. Chang, Y. Cai and T. B. Wei, Sens. Actuators, B, 2015, 213, 501–507 CrossRef CAS.
- Q. Lin, F. Zheng, T. T. Lua, J. Liu, H. Lia, T. B. Wei, H. Yao and Y. M. Zhang, Sens. Actuators, B, 2017, 251, 250–255 CrossRef CAS.
- H. L. Zhang, W. T. Li, W. J. Qu, T. B. Wei, Q. Lin, Y. M. Zhang and H. Yao, RSC Adv., 2017, 7, 47547–47551 RSC.
- L. Li, Y. Su, Y. Ji and P. Wang, A Long-Lived Water-Soluble Phenazine Radical Cation, J. Am. Chem. Soc., 2023, 145, 5778–5785 CrossRef CAS PubMed.
- L. Li, L. Chen, Y. Wen, T. Xiong, H. Xu, W. Zhang, G. Cao, Y. Yang, L. Mai and H. Zhang, J. Mater. Chem. A, 2020, 8, 26013–26022 RSC.
- Y.-M. Shi, A. O. Brachmann, M. A. Westphalen, N. Neubacher, N. J. Tobias and H. B. Bode, Nat. Chem. Biol., 2019, 15, 331–339 CrossRef CAS PubMed.
- Z. Zhou, D.-G. Chen, M. L. Saha, H. Wang, X. Li, P.-T. Chou and P. J. Stang, J. Am. Chem. Soc., 2019, 141, 5535–5543 CrossRef CAS PubMed.
- W. Chen, C.-L. Chen, Z. Zhang, Y.-A. Chen, W.-C. Chao, J. Su, H. Tian and P.-T. Chou, J. Am. Chem. Soc., 2017, 139, 1636–1644 CrossRef CAS PubMed.
- J. E. Barnsley, G. E. Shillito, C. B. Larsen, H. v. d. Salm, R. Horvath, X. Z. Sun, X. Wu, M. W. George, N. T. Lucas and K. C. Gordon, Inorg. Chem., 2019, 58, 9785–9795 CrossRef CAS PubMed.
- L. M. Loftus, E. C. Olson, D. J. Stewart, A. T. Phillips, K. Arumugam, T. M. Cooper, J. E. Haley and T. A. Grusenmeyer, Inorg. Chem., 2021, 60, 16570–16583 CrossRef CAS PubMed.
- P. Ulukan, E. E. Bas, R. B. Ozek, C. D. Kaynak, A. Monari, V. Aviyente and S. Catak, Phys. Chem. Chem. Phys., 2022, 24, 16167–16182 RSC.
- X. Niu, Z. Kuang, M. Planells, Y. Guo, N. Robertson and A. Xia, Phys. Chem. Chem. Phys., 2020, 22, 15743–15750 RSC.
- Z. Szakács, M. Tasior, D. T. Gryko and E. Vauthey, ChemPhysChem, 2020, 21, 1718–1730 CrossRef PubMed.
- B. Dereka, D. Svechkarev, A. Rosspeintner, A. Aster, M. Lunzer, R. Liska, A. M. Mohs and E. Vauthey, Nat. Commun., 2020, 11, 1925–1935 CrossRef CAS PubMed.
- F. Philippi, K. Goloviznina, Z. Gong, S. Gehrke, B. Kirchner, A. A. H. Pádua and P. A. Hunt, Phys. Chem. Chem. Phys., 2022, 24, 3144–3162 RSC.
- F. Chen, W. Zhang, Z. Liu, L. Meng, B. Bai, H. Wang and M. Li, RSC Adv., 2019, 9, 1–10 RSC.
- V. L. Nadurata and C. Boskovic, Inorg. Chem. Front., 2021, 8, 1840–1864 RSC.
- T. J. Frankcombe, Phys. Chem. Chem. Phys., 2015, 17, 3295–3302 RSC.
- W. Zhang, J. Kong, D. Hu, M. Tao, X. Niu, S. Vdovic, D. Aumiler, Y. Ma and A. Xia, J. Phys. Chem. C, 2020, 124, 5574–5582 CrossRef CAS.
- J. Kong, W. Zhang, Y. Guo, X. Niu, T. Yamao, K. Yamashita and A. Xia, J. Phys. Chem. C, 2020, 124, 18946–18955 CrossRef CAS.
- W. Zhang, W. Xu, G. Zhang, J. Kong, X. Niu, J. M. W. Chan, W. Liu and A. Xia, J. Phys. Chem. B, 2021, 125, 4456–4464 CrossRef CAS PubMed.
- J. Kong, W. Zhang, G. Li, D. Huo, Y. Guo, X. Niu, Y. Wan, B. Tang and A. Xia, J. Phys. Chem. Lett., 2020, 11, 10329–10339 CrossRef CAS PubMed.
- Z. Kuang, Q. Guo, X. Wang, H. Song, M. Maroncelli and A. Xia, J. Phys. Chem. Lett., 2018, 9, 4174–4181 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2323974. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02417f |
| This journal is © The Royal Society of Chemistry 2024 |