
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Activity against Mycobacterium tuberculosis of a new class of spirooxindolopyrrolidine embedded chromanone hybrid heterocycles†
Manal Fahad Alkalthama,
Abdulrahman I. Almansoura,
Natarajan Arumugam *a,
Siva Krishna Vagolu*bc,
Tone Tønjumbc,
Shatha Ibrahim Alaqeeld,
Saiswaroop Rajaratname and
Venketesh Sivaramakrishnane
*a,
Siva Krishna Vagolu*bc,
Tone Tønjumbc,
Shatha Ibrahim Alaqeeld,
Saiswaroop Rajaratname and
Venketesh Sivaramakrishnane
aDepartment of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia. E-mail: anatarajan@ksu.edu.sa
bDepartment of Microbiology, University of Oslo, N-0316 Oslo, Norway. E-mail: vagolu000@gmail.com
cDepartment of Microbiology, Oslo University Hospital, N-0424 Oslo, Norway
dDepartment of Chemistry, College of Science, King Saud University (034), Riyadh 11495, Saudi Arabia
eDisease Biology Lab, Department of Biosciences, Sri Sathya Sai Institute of Higher Learning, Prasanthi Nilayam, Anantapur, Andhra Pradesh, India
First published on 11th April 2024
Abstract
A new class of structurally intriguing heterocycles embedded with spiropyrrolidine, oxindole and chromanones was prepared by regio- and stereoselectively in quantitative yields using an intermolecular tandem cycloaddition protocol. The compounds synthesized were assayed for their anti-mycobacterial activity against Mycobacterium tuberculosis (Mtb) H37Rv and isoniazid-resistant (katG and inhA promoter mutations) clinical Mtb isolates. Four compounds exhibited significant antimycobacterial activity against Mtb strains tested. In particular, a compound possessing a fluorine substituted derivative displayed potent activity at 0.39 μg mL−1 against H37Rv, while it showed 0.09 μg mL−1 and 0.19 μg mL−1 activity against inhA promoter and katG mutation isolates, respectively. A molecular docking study was conducted with the potent compound, which showed results that were consistent with the in vitro experiments.
1. Introduction
Tuberculosis (TB) is one of the deadliest airborne infectious diseases, caused by various species and strains of mycobacteria. Among them, pathogenic aerobic bacteria, the bacillus Mycobacterium tuberculosis, remains the major cause of TB in humans. Mycobacterium tuberculosis usually forms an infection in the lungs of the host and is one of the leading unfavorable health problems globally. World Health Organization report in 2022 indicate that TB has replaced COVID-19 as the leading cause of death from an infectious agent on a worldwide basis.1 It is estimated that 85% of drug-susceptible TB cases can be cured with the current therapeutic treatment.2 Several first-line anti-TB medications are available, including rifampicin, isoniazid (INH), pyrazinamide, and ethambutol. These drugs have associated side effects, are ineffective in eliminating latent pathogens, and require prolonged treatment.3 Second-line drugs like bedaquiline, delamanid, or pretomanid are less effective, has a higher toxicity profile, and is more expensive than the first-line medication. Moreover, the current evolution of multidrug-resistant TB further aggravates the complications associated with TB treatment. Thus, the disease continues to be a major social health issue, particularly among low-income populations.4,5 TB in combination with HIV5 increases the overall incidence of TB by 50 times more in HIV-positive patients than HIV-negative individual.6 Therefore, it is necessary to develop structurally novel, potent, fast acting, affordable anti-TB drugs with an innovative mechanism of action and low toxicity profile that are capable of overcoming the mechanisms of resistance posed by existence of multidrug and extensively drug resistant tuberculosis (MDR-TB and XDR-TB) in order to effectively combat TB.In this context, nitrogen and oxygen comprising heterocyclic hybrid frameworks play crucial contribution in drug discovery, a significant part of which is the fact that they are the active entrants in a many number of drugs.7 In addition, molecules embedding heteroatoms possess promising drug solubility and pharmacokinetic properties. It has been argued that recent trends in the discovery of lead compounds have led to a “escape from flatland” in which planar aromatic or heteroaromatic ring systems are a rapidly replacing others with higher saturation and three-dimensionality. Such molecules are anticipated to have a better affinity for three-dimensional binding pocket in proteins and be more solubilized which is an essential property for drug development and practical applications.8,9
In this connection, spiro compounds have emerged as an attractive synthetic target in drug discovery,10–15 due to their structural complexity and rigidity, facility to reveal functionality and inherent three-dimensional structural features, which offer improved structural affinity to biological targets. Perhaps for this reason, many spiro compounds are found in biologically active alkaloids and synthetic products that have evolved to interact with target proteins of the biological system15 more readily than flat hetero aromatic ring systems. In this context, R. R. kumar et. al.16 reported an atom economic, stereoselective synthesis and antimycobacterial evaluation of spiropiperidine through cycloaddition methodology. Among these spiropiperidines, fluoro substituted compounds showed excellent activity against tested TB organism. Similarly, a library of spirooxindolopyrrolidines/pyrrolizidines/pyrrolothiazole derivatives were synthesized and assayed for their antimycobacterium tubercular activity by S. M. Rajesh et. al.17 and found that most them demonstrated excellent activity against tested TB-organism. Recently, M. Ganesh et. al.18 reported the synthesis, characterization, molecular docking simulation and antitubercular activity of new class of spiropyrrolidine oxindoles, these compounds showed significant activity against tested Mycobacterium tuberculosis H37R. In addition, to the biological precedents of the spiro compounds described above, most of the spiropyrrolidines reported in the literature demonstrated significant biological activity, including anti-TB activity,19,20 some of these spiro compounds were even more potent than reference standard.21–24 Prompted by these finding, herein we report the synthesis and anti-tubercular activity of structurally diverse spirooxindolopyrrolidine integrated chromanones using three component reactions involving 1,3-dipolar cycloaddition reaction.25–29
2. Results and discussion
2.1. Chemistry
The cycloaddition protocol initiated by the reaction of 3-arylidenechroman-4-ones 4 and in situ 1,3-dipole prepared from diketone 1 and amino acid 2 under reflux in MeOH yielded exclusively the substituted spiropyrrolidine heterocycles in quantitative yields (88–95%, Table 1) as described in Scheme 1. In the initial stage of optimization, several solvent systems were used including EtOH, MeOH, CH3CN, DMF and CH3CN![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (1:1 v/v). An excellent yield was obtained in MeOH than to other solvents, indicating that MeOH is suitable for the reaction. Thus, the mixture of arylidene chromanone 4, diketone 1, and amino acid 2 is refluxed in MeOH until the reaction is completed, as indicated by TLC and the products 5a–j are purified by crystallization. The spectroscopic analysis unambiguously assigned by the structure of compounds (vide, ESI†). Finally, X-ray diffraction analysis described in Fig. 1 confirmed the stereochemistry and regio of the structures 5a and 5g.30
:MeOH (1:1 v/v). An excellent yield was obtained in MeOH than to other solvents, indicating that MeOH is suitable for the reaction. Thus, the mixture of arylidene chromanone 4, diketone 1, and amino acid 2 is refluxed in MeOH until the reaction is completed, as indicated by TLC and the products 5a–j are purified by crystallization. The spectroscopic analysis unambiguously assigned by the structure of compounds (vide, ESI†). Finally, X-ray diffraction analysis described in Fig. 1 confirmed the stereochemistry and regio of the structures 5a and 5g.30
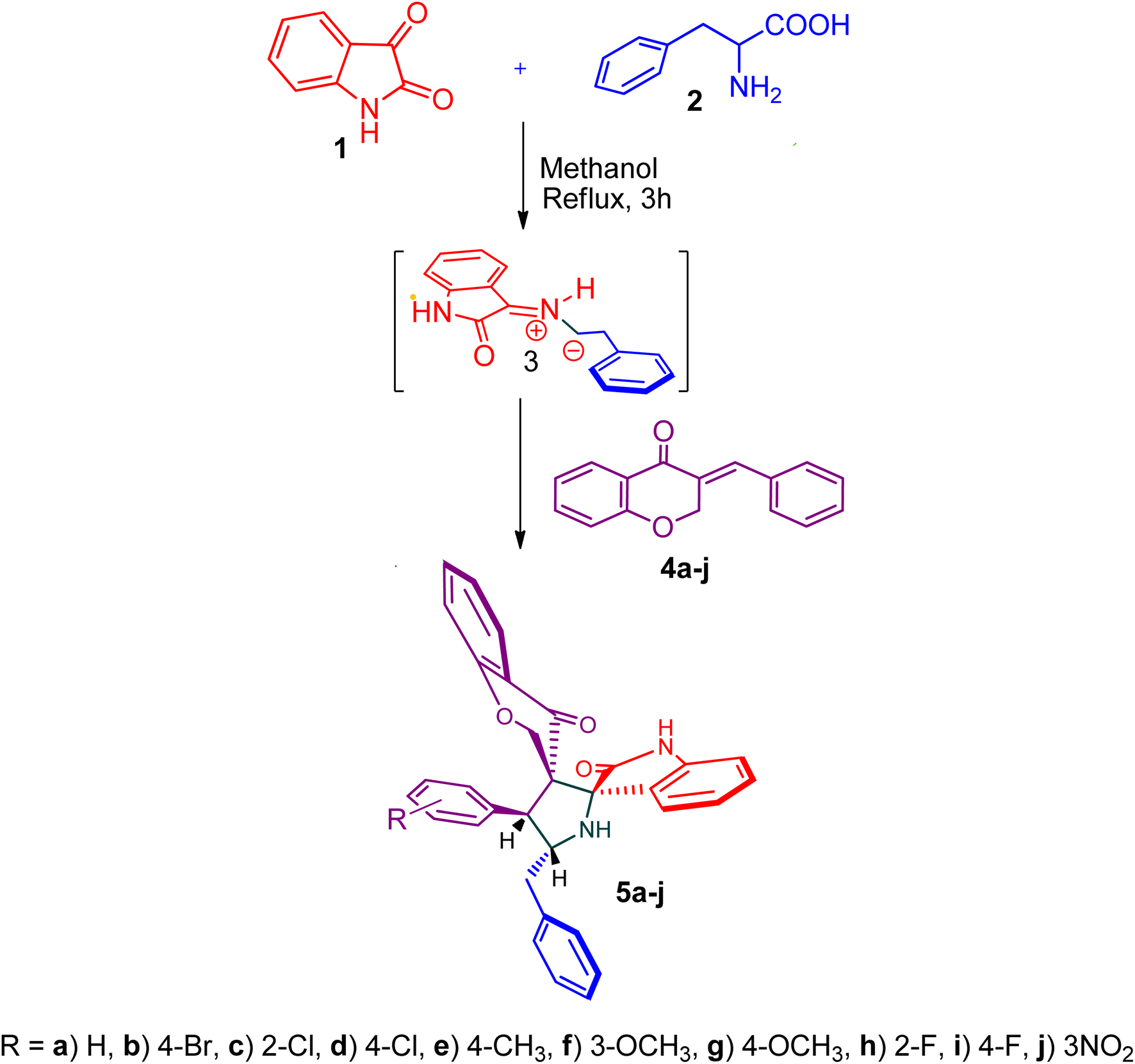 | ||
| Scheme 1 Synthesis of dispiropyrrolidine tethered chromanone hybrids, 5a–j. | ||
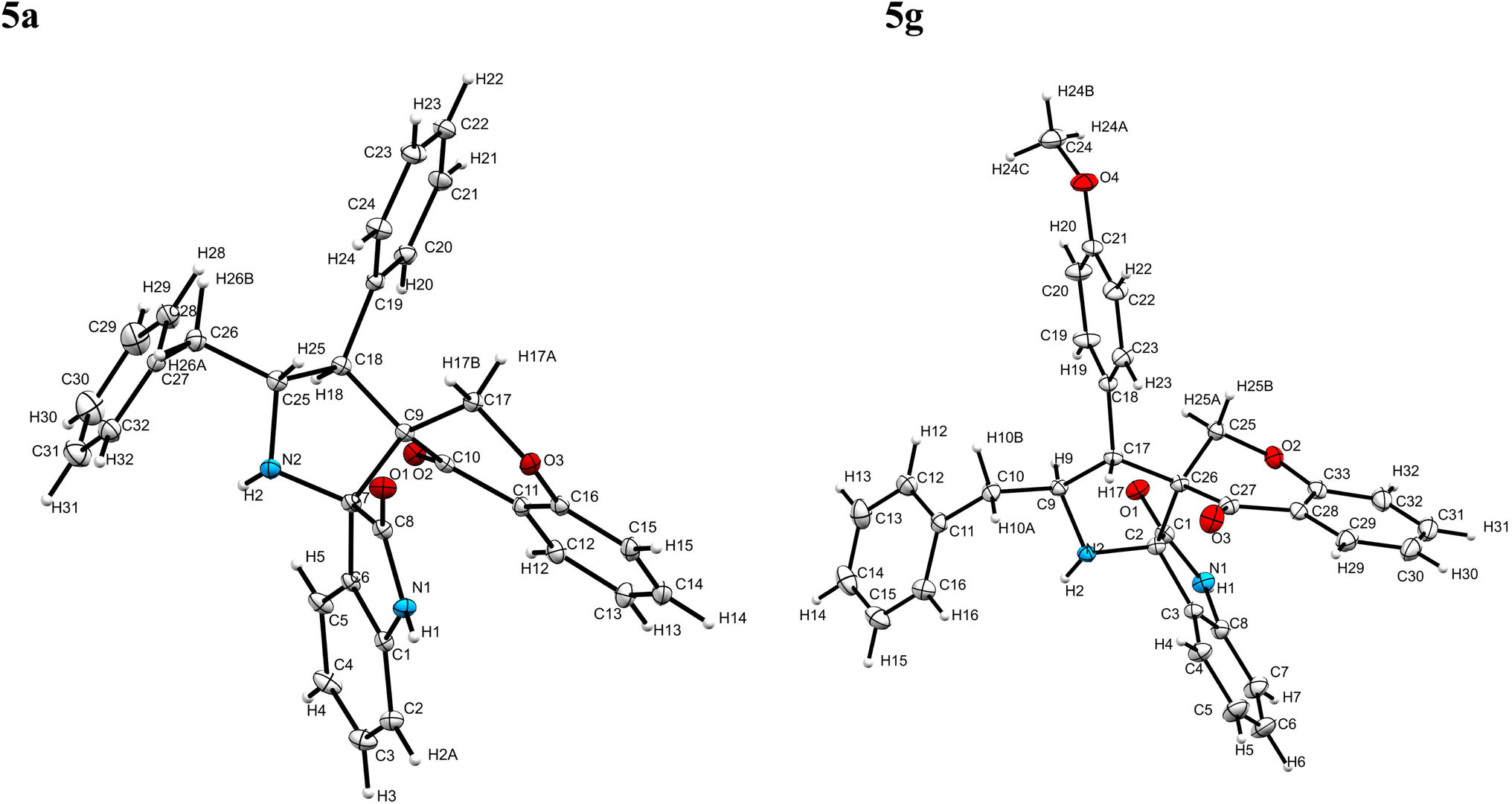 | ||
| Fig. 1 Crystal structure of compounds 5a and 5g. | ||
Scheme 2 shows a possible mechanism for the construction of spiropyrrolidine 5. In the first step, the reaction of active ketone 1 and L-phenylalanine 2 produced the non-stabilized azomethine ylide 8 via intermediates 6 and 7 under spontaneous decarboxylation reaction process. The non-stabilized ylide 8 was further reacts with benzylidene chromanone 4 yielded sole cycloadduct 5 via route A. Furthermore, the reaction proceeded regioselectively, as no traces of the possible regioisomer 9 were detected. The dipole 8 adds to the electron less carbon of the chromanone 4, producing cycloadduct 5. Four new stereogenic carbons, including two spirocarbons, were formed in this reaction process, resulting from the construction of two C–C and one C–N bonds.
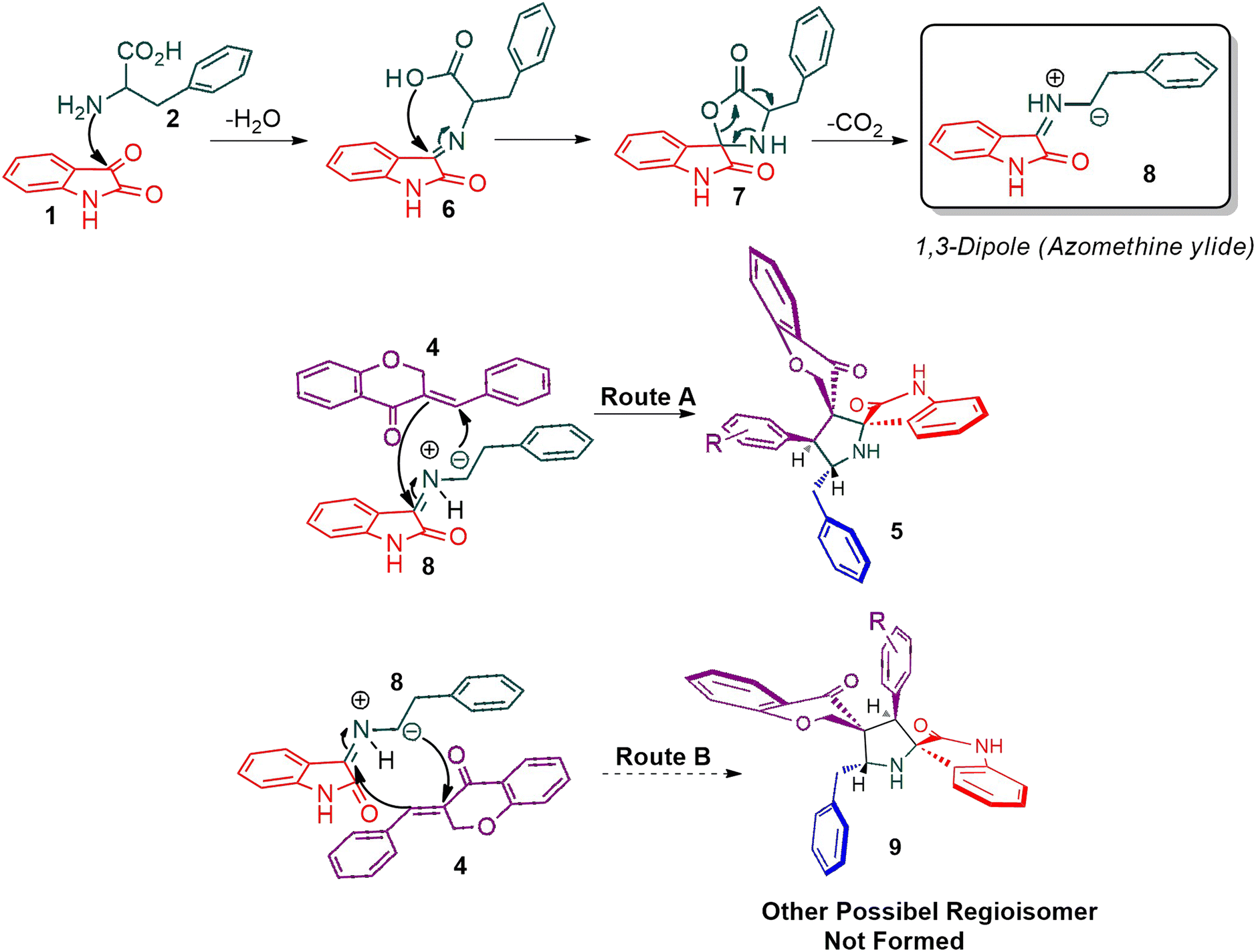 | ||
| Scheme 2 The persuasive pathway for the construction pyrrolidine, 5. | ||
2.2. Anti-Mycobacterium tuberculosis activity of spiropyrrolidine heterocyclic hybrids
The synthesized spirooxindolopyrrolidine embedded chromanones 5a–j were tested for their antimycobacterium activity against Mtb H37Rv, isoniazid-resistant (katG or inhA promoter mutation) clinical Mtb isolates using the Microplate Alamar Blue Assay (MABA) method and Middlebrook 7H9 broth + OADC (Growth supplement), with isoniazid used as the reference standard drug. Initially, we evaluated the first series of spirooxindolopyrrolidines 5a–j against Mtb H37Rv and Mtb isoniazid-resistant (katG or inhA promoter mutation). Among the compounds, four spirocompounds displayed potent antitubercular activity against the tubercular organisms tested (Table 2). Thus, the compound 5c that possessed 2-chloro substituted derivative showed excellent activity against H37Rv with MIC value 0.78 μg mL−1, which further showed potent activity against inhA promoter and katG mutation clinical isolates with MIC values 0.19 μg mL−1 and 0.39 μg mL−1, respectively. Similarly, the compound 5d bearing 4-chlorosubsitution on the aryl ring displayed good activity against H37Rv with MIC value 1.56 μg mL−1 and the compound showed excellent activity against inhA promoter and katG mutation isolates with MIC values 0.39 μg mL−1 and 1.56 μg mL−1, respectively. Compound 5h with 2-fluorosubstitution on the aryl ring showed significant activity against H37Rv with MIC value 1.56 μg mL−1, which displayed significant activity against inhA promoter and katG mutation isolates with MIC values 0.78 and 1.56 μg mL−1, respectively. Likewise, compound 5i bearing 4-fluoro on the phenyl ring exhibited potent activity against H37Rv with MIC value 0.39 μg mL−1, which showed 0.09 μg mL−1 and 0.19 μg mL−1 against inhA promoter and katG mutation isolates, respectively. The above mentioned antitubercular results revealed that the spirooxindolopyrrolidine integrated chromanones with halogen substituted derivative showed significant activity against the tubercular organisms tested. In particular, the compound possessing a fluoro substitution on the aryl ring showed more potent activity which was comparable against the standard drug isoniazid (INH).| Compounds | H37Rv (MIC μg mL−1) | inhA promoter mutation (MIC μg mL−1) | katG mutation (MIC μg mL−1) |
|---|---|---|---|
| a Compounds with promising activity. | |||
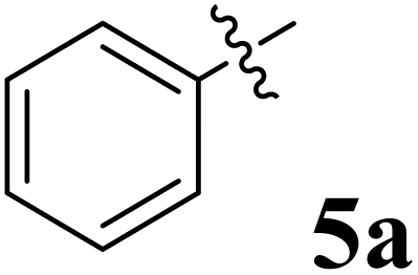 |
>50 | >50 | >50 |
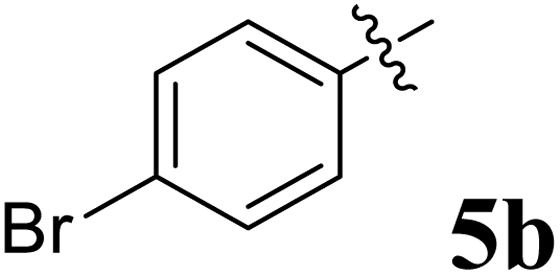 |
>50 | >50 | >50 |
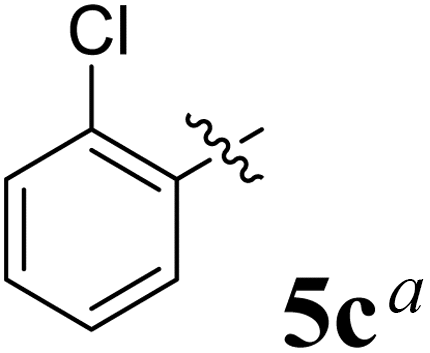 |
0.78 | 0.19 | 0.39 |
 |
1.56 | 0.39 | 1.56 |
 |
>50 | >50 | >50 |
 |
>50 | >50 | >50 |
 |
>50 | >50 | >50 |
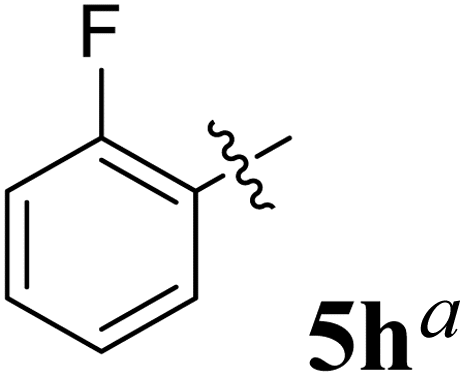 |
1.56 | 0.78 | 1.56 |
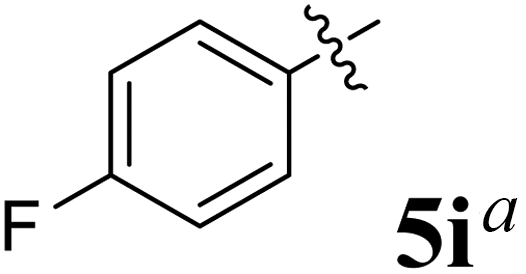 |
0.39 | 0.09 | 0.19 |
 |
>50 | >50 | >50 |
| Isoniacid (INH) | 0.19 μM | 1.56 μM | 12.5 μM |
2.3. Molecular docking study
Molecular docking studies showed that the ligand had a strong binding affinity with the active sites of Type II dehydroquinase (−9.59) and decaprenyl phosphoryl-beta-D-ribose oxidase from Mycobacterium tuberculosis (−10.81). Further, it showed a stronger binding affinity compared to known natural ligands. Hydrophobic interactions were formed between the amino acids PRO11, ASN12, LEU13, LEU16, TYR24, ASN75, GLY77, GLY78 of Type II dehydroquinase (1H0R) active sites and the ligand. In the case of decaprenyl phosphoryl-beta-D-ribose oxidase, interactions with both the hydrogen bond (CYS129) and hydrophobic amino acids (ILE131, ALA417, ARG58, GLY125, VAL121, SER59, THR118, TYR60 and TYR415) were observed. This could probably contribute to stronger binding affinity of the ligand with the decaprenyl phosphoryl-beta-D-ribose oxidase compared to Type II dehydroquinase.2.4. ADME studies
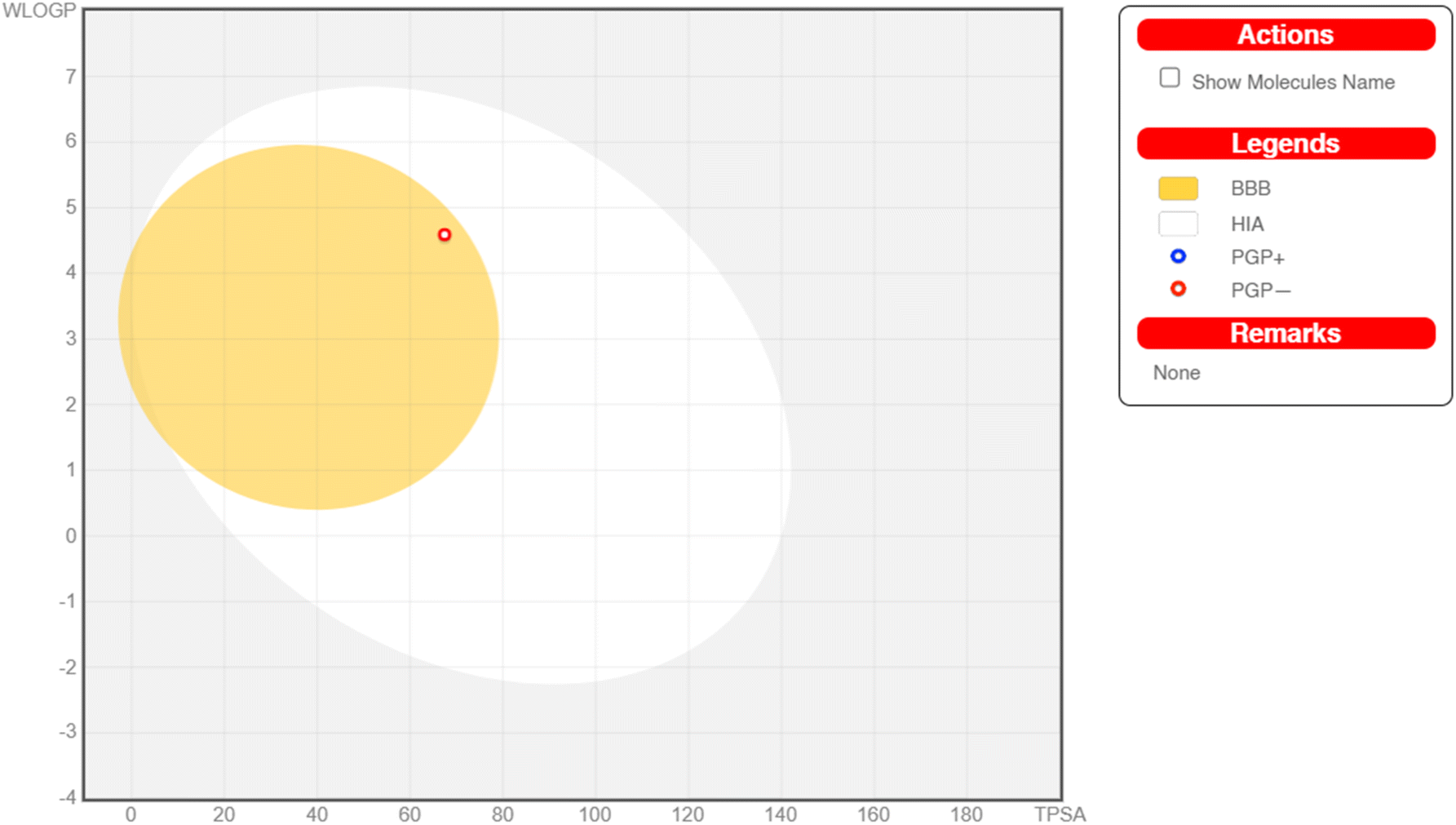
Absorption, distribution, metabolism, and excretion (ADME) properties of the ligand were assessed using lipophilicity, pharmacokinetics, water solubility, physio-chemical properties and drug-likeness. The ligand lacked flexibility and polar interactions. This was reflected in the interactions observed in the docking studies. However, in general, the ligand exhibited good druggability properties. Swiss Target Prediction was used to bioinformatically forecast additional possible targets to understand the cross-reactivity of the drug molecule. Swiss Target Prediction uses molecular similarity and Shape to forecast prospective targets. Our study demonstrated that the ligand has a 46.7% chance of binding to proteases, 26.7% with G-protein coupled receptors and 13.3% with hydrolases. Overall, the molecule shows good properties in order to be used as a possible drug for TB.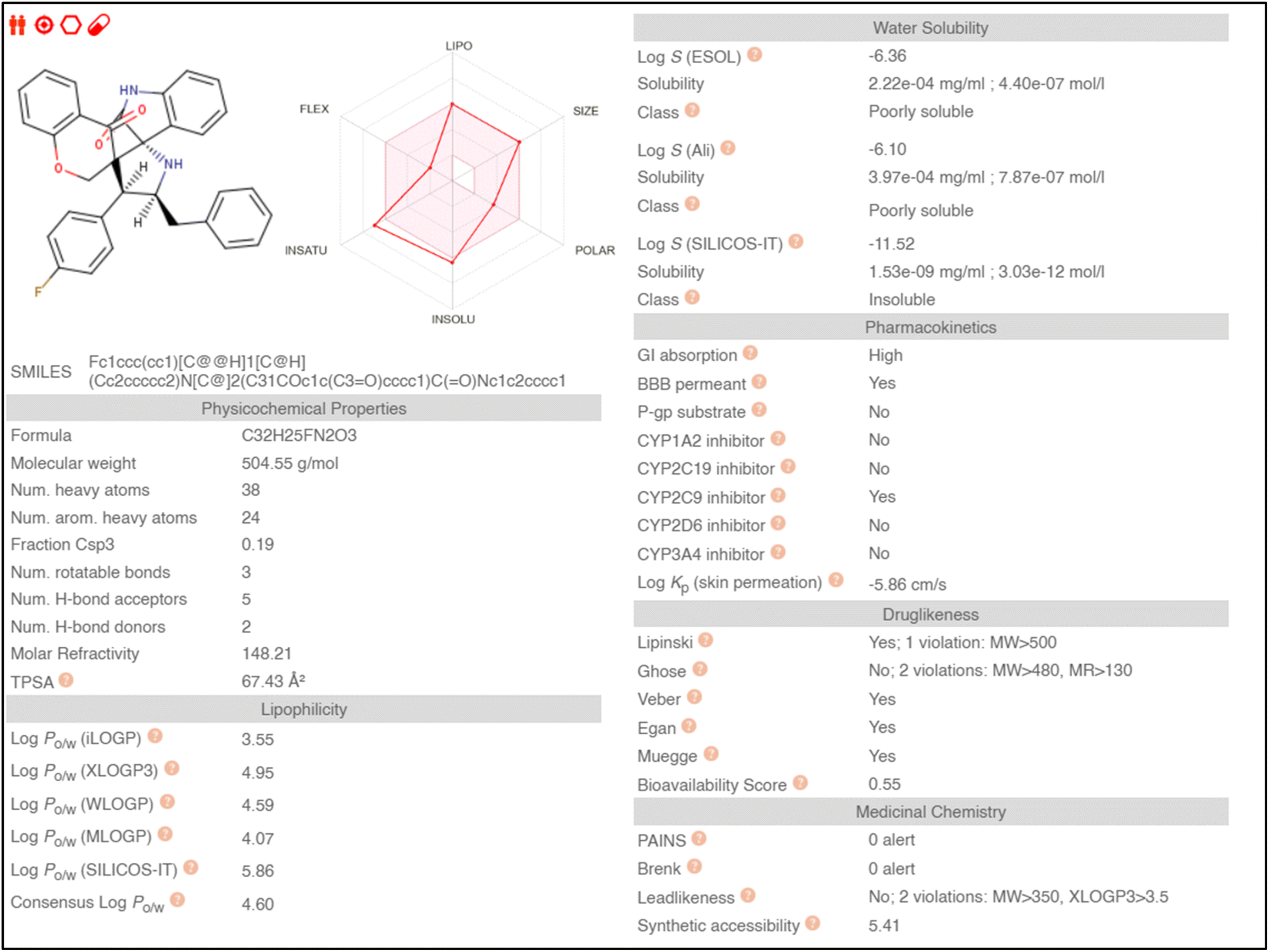
Binding energy of 5i with the active site of 1H0R (decaprenyl phosphoryl-beta-D-ribose oxidase from Mtb): −10.81.
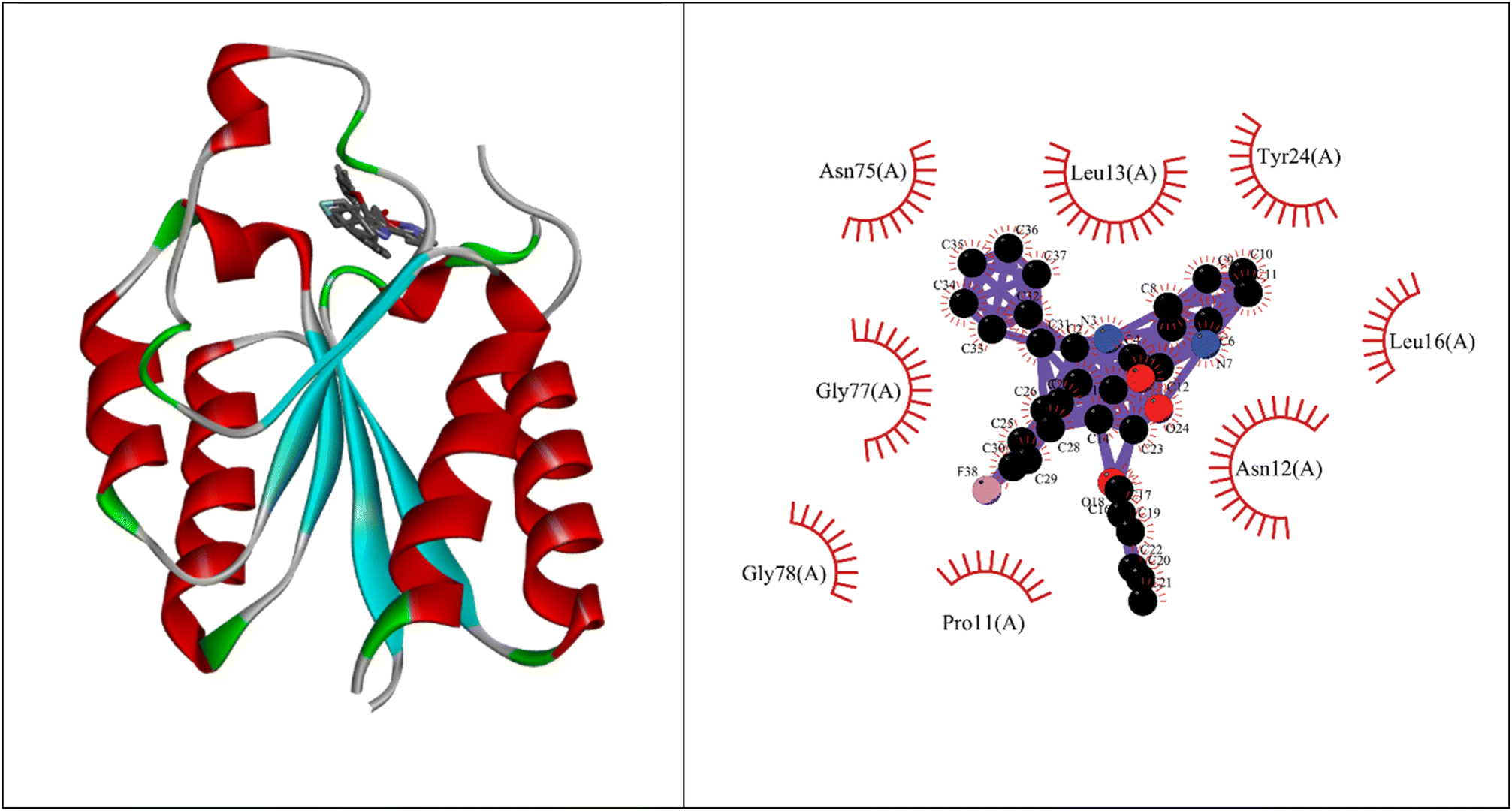
Binding energy of known ligand (FAD) with the active site of 1H0R (decaprenyl phosphoryl-beta-D-ribose oxidase from Mtb): −7.05.
Binding energy of 5i with the active site of 1H0R (decaprenyl phosphoryl-beta-D-ribose oxidase from Mtb): −10.81.
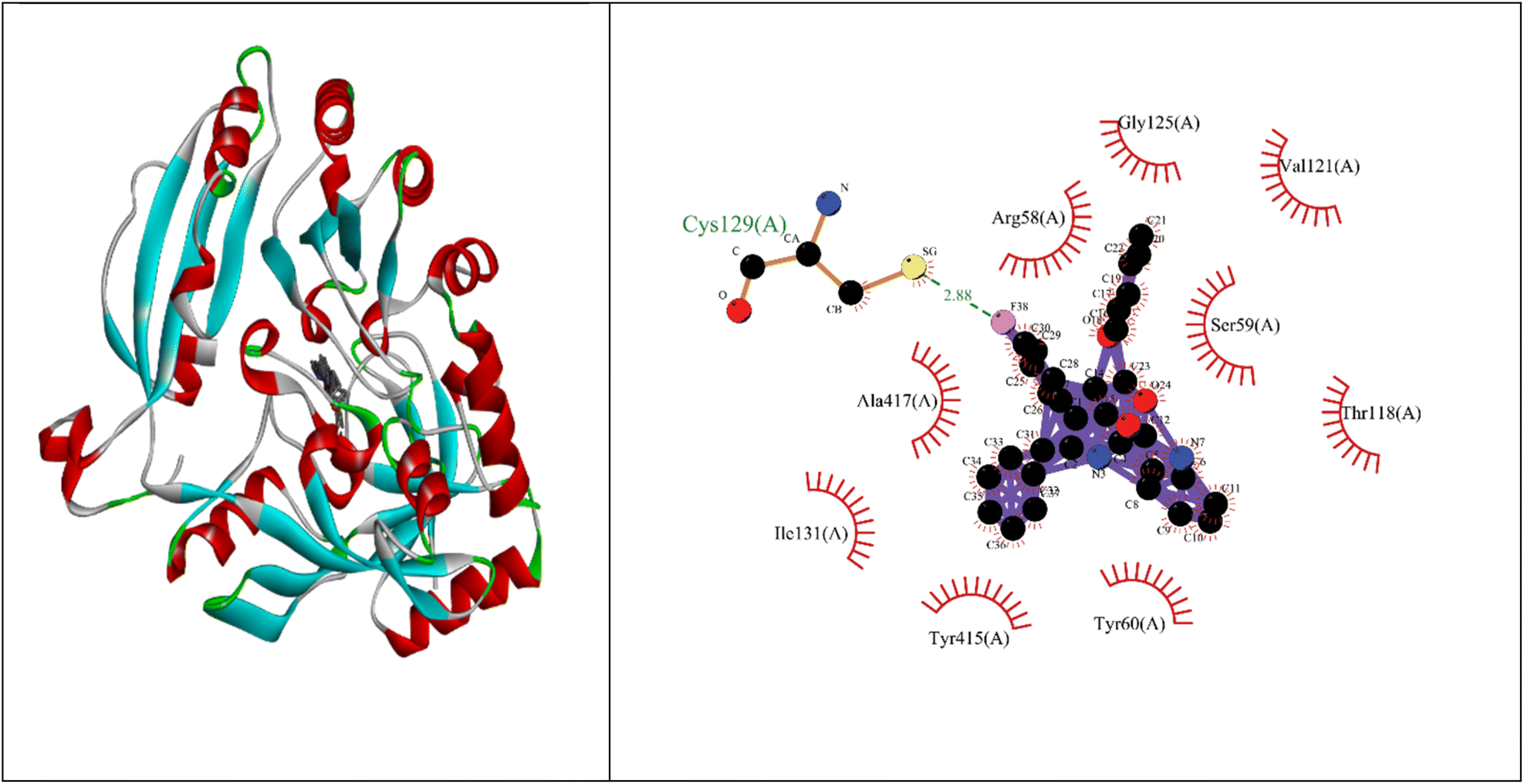
Binding energy of known ligand (FAD) with the active site of 1H0R (decaprenyl phosphoryl-beta-D-ribose oxidase from Mtb): −7.05.
2.5. Swiss target prediction
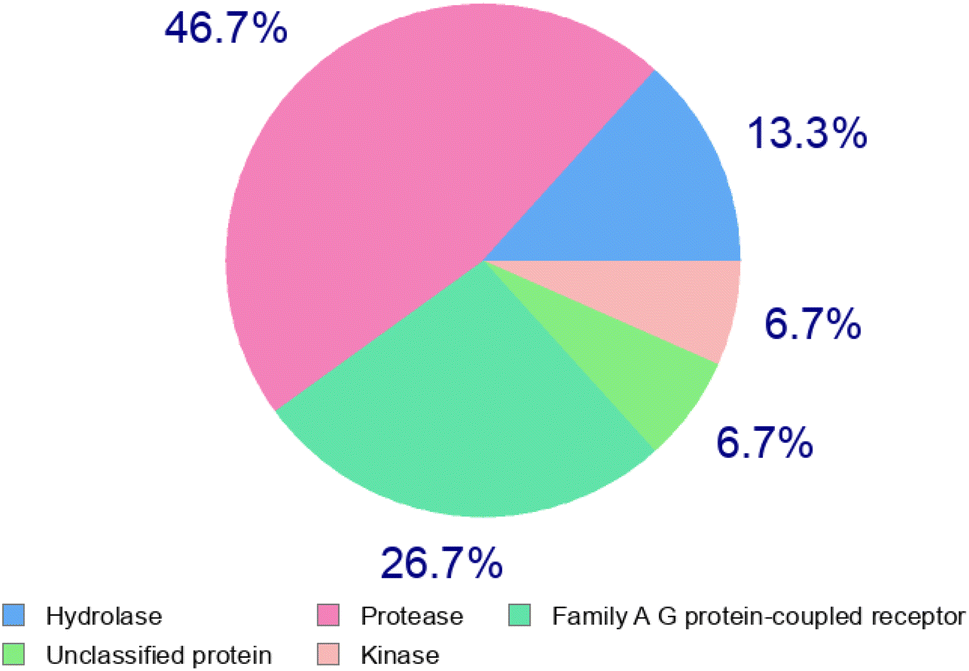
| Target | Common name | Target class | Probability |
|---|---|---|---|
| Acetylcholinesterase | ACHE | Hydrolase | 0.121906255 |
| Thrombin | F2 | Protease | 0.121906255 |
| Urotensin II receptor | UTS2R | Family A G protein-coupled receptor | 0.121906255 |
| ADAMTS5 | ADAMTS5 | Protease | 0.121906255 |
| Phospholipase D1 | PLD1 | Hydrolase | 0.121906255 |
| Dipeptidyl peptidase II | DPP7 | Protease | 0.121906255 |
| Dipeptidyl peptidase VIII | DPP8 | Protease | 0.121906255 |
| Dipeptidyl peptidase IX | DPP9 | Protease | 0.121906255 |
| Menin | MEN1 | Unclassified protein | 0.121906255 |
| Gonadotropin-releasing hormone receptor | GNRHR | Family A G protein-coupled receptor | 0.121906255 |
3. Conclusion
A series of structurally new class of spirooxindolopyrrolidine tethered chromanones were synthesized in quantitative yields using cycloaddition methodology. The spiro compounds were evaluated for their antitubercular activity against Mycobacterium tuberculosis H37Rv, isoniazid-resistant (katG and inhA promoter mutation) clinical Mycobacterium tuberculosis isolates. The spirooxindolopyrrolidine integrated chromanones that possessed an electron withdrawing group substituted derivative displayed significant activity against tested Mycobacterium tuberculosis H37Rv, as well as isoniazid-resistant Mycobacterium tuberculosis strains. Among them, the compound bearing fluorine derivative displayed significant activity against tested antimycobacterial pathogens which were comparable to the activity of the reference standard drug isoniazid. Molecular docking simulation was also performed with the most active compounds, and the docking results in relation with ADME and receptor specificity studies confirmed the possible role of the molecule as a potential drug for treating TB efficiently.4. Experimental section
4.1. Preparation of spiropyrrolidines, 5a–j
The reaction consisted of a mixture of L-phenylalanine 2 (1.1 mmol), diketone 1 and dipolarophile 4a–j (1 mmol) was heated to reflux in MeOH at 75 °C for 2 h. TLC was used to verify the completeness of the reaction, and filtration was used to obtain the pure products.Compound 5c: 1H NMR: δH 2.68–2.72 (1H, dd, J = 13.5, 5.0 Hz), 2.88–2.92 (1H, m), 3.28 (1H, d, J = 12.0 Hz), 4.34 (1H, J = 12.0 Hz), 4.64–4.68 (1H, m), 4.73 (1H, d, J = 9.5 Hz), 6.44 (1H, ArH, t, J = 8.0 Hz), 6.49 (1H, ArH, d, J = 8.0 Hz), 6.54 (1H, ArH, d, J = 7.0 Hz), 6.71 (1H, ArH, d, J = 7.5 Hz), 6.80–6.84 (2H, ArH, m), 6.99 (1H, ArH, t, J = 7.5 Hz), 7.07–7.16 (6H, ArH, m), 7.27 (2H, ArH, t, J = 8.0 Hz), 7.71–7.73 (1H, ArH, dd, J = 8.0, 1.5 Hz), 7.88 (1H, ArH, d, J = 8.0 Hz), 10.52 (1H, s, 1H); 13C NMR: δC 39.7, 50.5, 59.8, 63.9, 72.4, 72.7, 109.5, 117.1, 121.1, 121.3, 121.9, 126.2, 126.3, 127.0, 127.6, 127.9, 128.4, 129.1, 129.3, 129.7, 131.1, 135.3, 135.4, 135.9, 139.6, 142.5, 161.4, 178.7, 192.1; mass m/z: 521 (M+).
Compound 5d: 1H NMR: δH 2.75 (2H, d, J = 6.0 Hz), 3.29 (1H, d, J = 12.0 Hz), 4.25 (1H, d, J = 11.0 Hz), 4.43–4.48 (1H, m), 4.73 (1H, d, J = 12.0 Hz), 6.40 (1H, ArH, d, J = 8.0 Hz), 6.51–6.57 (2H, m, ArH), 6.68–6.73 (2H, m, ArH), 6.80 (1H, t, J = 8.0 Hz, ArH), 7.06–7.17 (7H, m, ArH), 7.31–7.35 (ArH, m, 3H), 7.41 (1H, ArH, d, J = 8.0 Hz), 10.34 (1H, s, NH); 13C NMR: δC 39.8, 52.9, 61.1, 62.3, 70.6, 71.8, 109.2, 117.0, 120.9, 121.1, 121.4, 126.2, 127.6, 127.7, 128.1, 128.4, 129.1, 129.2, 129.5, 132.4, 135.8, 139.7, 142.5, 161.0, 179.5, 191.9; mass m/z: 521 (M+).
Compound: 5h: 1H NMR: δH 2.84–2.88 (2H, m), 3.27–3.40 (1H, m), 4.60–4.68 (3H, m), 6.51–6.57 (3H, m, ArH), 6.72–6.83 (3H, m, ArH), 7.03–7.34 (9H, m, ArH), 7.63–7.68 (2H, m, ArH), 10.48 (1H, s, NH); 13C NMR: δC 40.0, 59.9, 62.6, 67.9, 72.0, 72.3, 109.4, 117.1, 118.5, 121.1, 121.7, 121.9, 122.6, 124.9, 126.3, 127.4, 127.9, 128.5, 129.3, 129.4, 129.9, 135.9, 136.9, 139.7, 142.6, 160.5, 161.4, 162.5, 178.9, 191.9; mass m/z: 504 (M+).
Compound 5i: 1H NMR: δH 2.77–2.86 (2H, m), 3.18 (1H, d, J = 8.8 Hz), 4.29 (1H, d, J = 10.0 Hz), 4.40–4.51 (1H, m), 4.76 (1H, d, J = 11.6 Hz), 6.43 (1H, d, J = 7.2 Hz, ArH), 6.57–6.62 (2H, m, ArH), 6.74–6.77 (2H, m, ArH), 6.80–6.88 (1H, m, ArH), 7.11–7.21 (8H, m, ArH), 7.44 (3H, d, J = 8.0 Hz, ArH), 10.34 (1H, s, NH); 13C NMR: δH 39.3, 52.4, 54.1, 60.6, 64.9, 70.1, 108.6, 115.6, 116.4, 120.5, 120.6, 125.9, 127.7, 127.8, 128.1, 128.6, 128.9, 129.1, 132.4, 135.2, 135.5, 139.3, 141.9, 160.1, 160.5, 162.5, 179.0, 191.6; mass m/z: 504 (M+).
4.2. Anti-tubercular screening
The strains H37Rv of Mycobacterium tuberculosis, clinical isolates with inhA promoter mutation and katG mutation was streaked onto 7H10+OADC agar plates and incubated at 37 °C. Pure colonies grown on OADC-enriched liquid 7H9 medium were grown to mid-log phase. A cultural growth was followed by inoculation into 7H9 medium on 96-well plates at progressively higher concentrations of the testing chemicals, using approximately 4 × 105 CFU mL−1 in 200 μL of culture per well. Plates were incubated for one week at 37 °C before receiving 32.5 μL of a Resazurin-tween mixture (8:5 ratio of 0.6 mM Resazurin in PBS to 20% Tween 80). Resorufin produced by fluorescent resorufin is used for determining the minimum inhibitory concentration (MIC) of compounds.31
4.3. Methodology
Molecular docking studies were performed using Autodock (MGL-Tools) and Cygwin. Type II dehydroquinase (1H0R) and decaprenyl phosphoryl-beta-D-ribose oxidase from Mtb (4FDN) were used as target proteins. The methodology and the active sites followed for docking studies were obtained from literature.32,33 Docking studies were also carried out with the natural ligands and the binding efficiency of the ligands were assessed. The ligand interaction plots were made using Discovery Studio and LigPlus softwares. Subsequently, ADME properties (absorption, distribution, metabolism and excretion) and other potential targets for the ligand was assessed using Swiss ADME online portals (http://www.swissadme.ch/) and Swiss Target Prediction (https://www.swisstargetprediction.ch/). The interaction image was made using Liplot.34Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was funded by Researchers Supporting Project number (RSP2024R143), King Saud University, Riyadh, Saudi Arabia. Tone Tønjum acknowledges financial support by the Research Council of Norway (RCN project numbers #234506, #261669, and #309592) and EU JPIAMR (RCN project #298410).References
- World Health Organization, Global Tuberculosis Report, 2022, https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022 Search PubMed.
- WHO Global Tuberculosis Report, World Health Organization, Geneva, 2018, Available at https://apps.who.int/medicinedocs/en/m/abstract/Js23553en/ Search PubMed.
- A. Somoskovi, L. M. Parson and M. Salfinger, The Molecular Basis of Resistance to Isoniazid, Rifampin, and Pyrazinamide in Mycobacterium tuberculosis, Respir. Res., 2001, 2, 164–168 CrossRef CAS PubMed.
- J. C. Palomino, D. F. Ramos, P. Almeida and D. Silva, New antituberculosis drugs: strategies, sources and new molecules, Curr. Med. Chem., 2009, 16, 1898 CrossRef CAS PubMed.
- M. A. Arbex, M. de C. L. Varella, H. R. de Siqueira and F. A. F. de Mello, Antituberculosis drugs: drug interactions, adverse effects, and use in special situations. Part 1: first-line drugs, J. Bras. Pneumol., 2010, 36, 626–640 CrossRef PubMed.
- A. Koul, E. Arnoult, N. Lounis, J. Guillemont and K. Andries, The challenge of new drug discovery for tuberculosis, Nature, 2011, 469, 483–490 CrossRef CAS PubMed.
- S. Samanta, S. Kumar, E. K. Aratikatla, S. R. Ghorpade and V. Singh, Recent developments of imidazo[1,2-a]pyridine analogues as antituberculosis agents, RSC Med. Chem., 2023, 14, 644–657 RSC.
- S. Prashant, A. G. George and A. T. Paul, Recent advances in the pharmacological diversification of quinazoline/quinazolinone hybrids, RSC Adv., 2020, 10, 41353–41392 RSC.
- R. Yadav, D. Meena, K. Singh, R. Tyagi, Y. Yadav and R. Sagar, Recent advances in the synthesis of new benzothiazole based anti-tubercular compounds, RSC Adv., 2023, 13, 21890–21925 RSC.
- S. Haddad, S. Boudriga, T. N. Akhaja, J. P. Raval, F. Porzio, A. Soldera, M. Askri, M. Knorr, Y. Rousselin, M. M. Kubicki and D. Rajani, A strategic approach to the synthesis of functionalized spirooxindole pyrrolidine derivatives: in vitro antibacterial, antifungal, antimalarial and antitubercular studies, New J. Chem., 2015, 39, 520–528 RSC.
- M. A. M. Knorr, C. Strohmann and L. Brieger, Synthesis, antidiabetic activity and molecular docking study of Rhodanine-substituted spirooxindole pyrrolidine derivatives as novel α-amylase inhibitors, Bioorg. Chem., 2021, 106, 104507 CrossRef PubMed.
- N. Arumugam, A. I. Almansour, R. S. Kumar, S. I. Alaqeel, V. Siva Krishna and D. Sriram, Anti-tubercular activity of novel class of spiropyrrolidine tethered indenoquinoxaline heterocyclic hybrids, Bioorg. Chem., 2020, 99, 103799 CrossRef CAS PubMed.
- N. Arumugam, A. I. Almansour, R. S. Kumar, M. Altaf, R. Padmanaban, P. Sureshbabu, G. Angamuthu, D. Kotresha, T. S. Manohar and S. Venketesh, Spiropyrrolidine/spiroindolizino[6,7-b]indole heterocyclic hybrids: Stereoselective synthesis, cholinesterase inhibitory activity and their molecular docking study, Bioorg. Chem., 2018, 79, 64–71 CrossRef CAS PubMed.
- H. M. Hügel, N. H. de Silva, A. Siddiqui, E. Blanch and A. Lingham, Natural spirocyclic alkaloids and polyphenols as multi target dementia leads, Bioorg. Med. Chem., 2021, 43, 116270 CrossRef PubMed.
- L. Hao, Y. Ma, L. Zhao, Y. Zhang, X. Zhang, Y. Ma, R. H. Dodd, H. Sun and P. Yu, Synthesis of tetracyclic oxindoles and evaluation of their α-glucosidase inhibitory and glucose consumption-promoting activity, Bioorg. Med. Chem. Lett., 2020, 15, 127264 CrossRef PubMed.
- R. R. Kumar, S. Perumal, P. Senthil kumar, P. Yogeeswari and D. Sriram, Discovery of antimycobacterial spiro-piperidin-4-ones: an atom economic, stereoselective synthesis, and biological intervention, J. Med. Chem., 2008, 51, 5731–5735 CrossRef CAS PubMed.
- S. M. Rajesh, S. Perumal, J. C. Menéndez, P. Yogeeswari and D. Sriram, Antimycobacterial activity of spirooxindolo-pyrrolidine, pyrrolizine and pyrrolothiazole hybrids obtained by a three-component regio- and stereoselective 1,3-dipolar cycloaddition, Med. Chem. Commun., 2011, 2, 626–630 RSC.
- M. P. Rao, A. Chauhan, L. pintilie, S. K. Singh and M. Ganesh, Concise route to stereoselective chlorobenzene-based spiropyrrolidine oxindoles for pursuit as antitubercular agents, J. Chem. Sci., 2023, 135, 20 CrossRef CAS.
- K. K. Alluri, R. S. Reshma, R. Suraparaju, S. Gottapu and D. Sriram, Synthesis and evaluation of 4',5'-dihydrospiro[piperidine-4,7'-thieno[2,3-c]pyran] analogues against both active and dormant Mycobacterium tuberculosis, Bioorg. Med. Chem., 2018, 26, 1462–1469 CrossRef CAS PubMed.
- R. S. Kumar, A. I. Almansour, N. Arumugam, F. Mohammad, W. S. Alshahrani, D. Kotresha, M. Altaf, M. Azam and J. C. Menéndez, Highly functionalized pyrrolidine analogues: stereoselective synthesis and caspase-dependent apoptotic activity, RSC Adv., 2018, 8, 41226–41236 RSC.
- N. Arumugam, A. I. Almansour, R. S. Kumar, D. Kotresha, R. Saiswaroop and S. Venketesh, Dispiropyrrolidinyl-piperidone embedded indeno[1,2-b]quinoxaline heterocyclic hybrids: Synthesis, cholinesterase inhibitory activity and their molecular docking simulation, Bioorg. Med. Chem., 2019, 15, 2621–2628 CrossRef PubMed.
- A. I. Almansour, N. Arumugam, R. S. Kumar, D. Kotresha, T. S. Manohar and S. Venketesh, Design, synthesis and cholinesterase inhibitory activity of novel spiropyrrolidine tethered imidazole heterocyclic hybrids, Bioorg. Med. Chem. Lett., 2020, 15, 126789 CrossRef PubMed.
- N. Arumugam, A. I. Almansour, R. Suresh Kumar, A. J. M. Ali Al-Aizari, S. I. Alaqeel, S. Kansız, V. Siva Krishna, D. Sriram and N. Dege, Regio- and diastereoselective synthesis of spiropyrroloquinoxaline grafted indole heterocyclic hybrids and evaluation of their anti-Mycobacterium tuberculosis activity, RSC Adv., 2020, 10, 23522–23531 RSC.
- N. Arumugam, A. I. Almansour, R. S. Kumar, V. Siva Krishna, D. Sriram and N. Dege, Stereoselective synthesis and discovery of novel spirooxindolopyrrolidine engrafted indandione heterocyclic hybrids as antimycobacterial agents, Bioorg. Chem., 2021, 110, 104798 CrossRef CAS PubMed.
- K. E. O. Ylijoki and J. M. Stryker, [5 + 2] Cycloaddition Reactions in Organic and Natural Product Synthesis, Chem. Rev., 2013, 113, 2244–2266 CrossRef CAS PubMed.
- C. G. Lima, A. Ali, S. S. Van Berkel, B. Westermann and M. W. Paixão, Emerging approaches for the synthesis of triazoles: beyond metal-catalyzed and strain-promoted azide-alkyne cycloaddition, Chem. Commun., 2015, 51, 10784–10796 RSC.
- V. P. Ananikov and E. G. Gordeeva, Novel [4 + 2] cycloaddition reactions of alkyne and enyne key-units: Direct access to bicyclic aromatic and heteroaromatic products, A theoretical mechanistic study, Chem. Sci., 2011, 2, 2332–2341 RSC.
- P. R. Mali, N. B. Khomane, B. Sridhar, H. M. Meshram and P. R. Likhar, Synthesis of new spiro pyrrole/pyrrolizine/thiazole derivatives via (3+2) cycloaddition reactions, New J. Chem., 2018, 42, 13819–13827 RSC.
- A. Ali, A. G. Corrêa, D. Alves, J. Zukerman-Schpector, B. Westermann, M. A. B. Ferreiraa and M. W. Paixão, An efficient one-pot strategy for the highly regioselective metal-free synthesis of 1,4-disubstituted-1,2,3-triazoles, Chem. Commun., 2014, 50, 11926–11929 RSC.
- N. Arumugam, M. Fahad Alkaltham, V. Viswanathan, A. I. Almansour, R. S. Kumar, S. I. Alaqeel, S. Yeswant Kumar, R. Padmanaban, S. M. Mahalingam and N. Dege, Design, synthesis, structural characterization and computational studies of new class of dispirooxindolopyrrolidine embedded chromanone hybrid heterocycles, J. Mol. Struct., 2023, 1286, 135539 CrossRef CAS.
- E. K. Aslan, V. S. Krishna, V. S. Armaković, S. J. Armaković, S. Şahin, O. Tønjum and T. M. G. Gündüz, Linking azoles to isoniazid via hydrazone bridge: Synthesis, crystal structure determination, antitubercular evaluation and computational studies, J. Mol. Liq., 2022, 354, 118873 CrossRef.
- S. M. Rizvi, S. Shakil and M. Haneef, A simple click by click protocol to perform docking: AutoDock 4.2 made easy for non-bioinformaticians, EXCLI J., 2013, 23(12), 831–857 Search PubMed.
- M. Z. Hassan, M. Ashraf Ali, H. Osman, R. S. Kumar and N. Arumugam, Design, Synthesis and Antimycobacterial Activity of Dispiropyrrolidines Derivatives, Med. Chem., 2016, 6, 486–491 Search PubMed.
- R. A. Laskowski and M. B. Swindells, LigPlot+: multiple ligand-protein interaction diagrams for drug discovery, J. Chem. Inf. Model., 2011, 51, 2778–2786 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2126482 (5a) and 2126502 (5g). For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra01501k |
| This journal is © The Royal Society of Chemistry 2024 |