DOI:
10.1039/D4RA01674B
(Paper)
RSC Adv., 2024,
14, 16704-16712
Efficient optimization of the synthetic conditions for aerosol-assisted high-quality mesoporous CeO2 powders†
Received
4th March 2024
, Accepted 7th May 2024
First published on 22nd May 2024
Abstract
The morphology of surfactant-assisted mesoporous metal oxides was tuned to obtain high surface-area particles by utilizing the synthetic conditions for fabricating transparent thin films through an evaporation-induced self-assembly (EISA) process. For investigating their potential applications, especially for designing heterogeneous catalysts, mesoporous metal oxides should be obtained in powder forms; however, a serious limitation associated with their reproducibility persists. Herein, along with a rapid optimization approach, starting from determining and improving chemical composition for the fabrication of mesoporous metal oxide films, an advanced approach to obtain highly porous metal oxide powders is presented using a temperature-controlled spray-drying process with step-by-step but smooth optimization by combining several EISA processes, involving the utilization of a precursor solution optimized for a slow-drying process in the case of ceria (CeO2) using poly(styrene)-block-poly(ethylene oxide) (PS-b-PEO).
1. Introduction
Ordered mesoporous materials prepared using amphiphilic organic compounds such as alkyltrimethylammonium (CnTMA) halides, alkyl poly(oxy ethylene) (CnEOm), and poly(oxy ethylene)-block-poly(oxy propylene)-block-poly(oxy ethylene) (EOnPOmEOn) have potential for a wide variety of applications owing to the adjustability of mesopores, high exposure of their surfaces, and large capacity of mesopores.1–5 The morphology of ordered mesoporous materials should be designed for each application, but it is still not easy to tune them on demand in a powder form, which is generally required for the preparation of heterogeneous catalysts. For example, powder samples of mesoporous metal oxides such as alumina (Al2O3) have been mainly prepared by applying a room-temperature slow drying process of clear precursor solutions.6,7 The evaporation-induced self-assembly (EISA) process is vital for synthesizing highly ordered mesoporous metal oxide films using amphiphilic organic compounds8–10 and is also applicable for obtaining corresponding powders.11–13 However, the evaporation rate of solvents (e.g., ethanol and water) is not the same during each process of room-temperature spin-coating, temperature-controlled spray-drying, etc. Especially, the evaporation rate of such solvents during the room-temperature drying process for recovering metal oxide powders is totally different from those of other processes. The area of an air–liquid interface depends on the size of the container and is important for accelerating the evaporation of solvents, which is strongly influenced by ambient conditions, such as room temperature and humidity. This is often the main concern in the reproducibility of surfactant-assisted mesoporous metal oxides through the drying process.
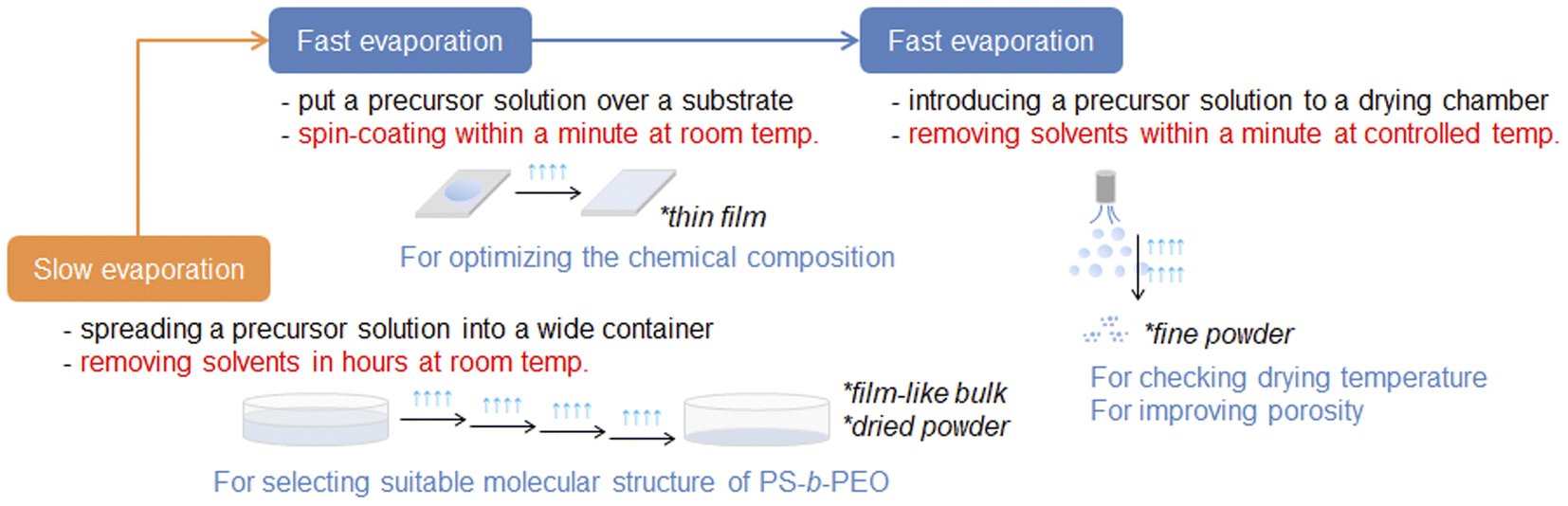
A wide variety of mesoporous metal oxide powders have thus far been prepared using surfactants by room-temperature slow drying and temperature-controlled fast spray-drying precursor solutions. Both EISA processes need long working times to investigate all the synthetic conditions, including the chemical composition of each precursor solution. From this viewpoint, our approach for obtaining high-quality mesoporous metal oxide powders is quite beneficial for the smooth optimization of synthetic conditions by combining several EISA processes. By utilizing the synthetic conditions for fabricating an ordered mesoporous metal oxide film, the chemical composition of a precursor solution can be determined very quickly via a rapid spin-coating process with fast evaporation of solvents. The same precursor solution is then applied to the fast spray-drying process and optimized further for increasing the resultant porosity. According to this realistic approach, we can save much of the working time for optimizing chemical composition, which has already been demonstrated through the synthesis of porous Al2O3 powders using poly(styrene)-block-poly(ethylene oxide) (PS-b-PEO).14–16
In this study, this synthetic approach was enhanced further as a useful choice for optimizing the chemical composition of a precursor solution very smoothly for obtaining aerosol-assisted metal oxide powders with surfactant-assisted mesopores. A precursor solution optimized for recovering mesoporous metal oxide powders by the room-temperature drying process with a slow evaporation of solvents was applied for the synthesis of the corresponding films by spin-coating. After finding the molecular structure of PS-b-PEO with the best potential for obtaining ordered and/or highly porous films, the corresponding powders were recovered by the spray-drying process (see Scheme 1). This step-by-step but smooth optimization by combining several EISA processes was proved for the synthesis of highly porous ceria (CeO2) powders in the presence of amphiphilic organic compounds, such as PS-b-PEO. This is a significant advance as a time-saving approach to obtain high-quality mesoporous metal oxide powders, which could be potentially started from the chemical compositions found in a huge number of research papers on obtaining room-temperature drying powders of mesoporous metal oxides.
 |
| | Scheme 1 Schematic summary of the developed process to optimize the synthetic conditions for synthesizing highly porous metal oxide powders using amphiphilic organic compounds by combining several EISA processes. | |
2. Experimental
2.1 Chemicals
Ethanol (99.5%, EtOH), tetrahydrofuran (dehydrated, THF), and cerium(III) nitrate hexahydrate (Ce(NO3)3·6H2O) were obtained from FUJIFILM Wako Pure Chemical Co. A series of poly(styrene)-block-poly(ethylene oxide) (PS-b-PEO) materials were purchased from Polymer Source Inc. and utilized as received. According to the analytical data sheets, the molecular structure is expressed by using the molecular weight of PS and PEO units, like 2700-b-4000, 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000-b-19500, 10000-b-21000, 12200-b-23900, 16000-b-7500, 16400-b-39500, 18000-b-7500, 18000-b-39000, 35000-b-17000, 40000-b-25500, 40000-b-31000, 40000-b-40500, 40000-b-53000, 51000-b-28000, 53000-b-15000, 58500-b-37000, 58600-b-71000, 59000-b-31000, 59000-b-72000, 60000-b-18000, 60000-b-36000, 60000-b-61000, 60000-b-85000, 65000-b-85000, 65000-b-97000, 90000-b-45000, 100000-b-150000, 105000-b-155000, 116000-b-164000, and 125000-b-177000.
000-b-19500, 10000-b-21000, 12200-b-23900, 16000-b-7500, 16400-b-39500, 18000-b-7500, 18000-b-39000, 35000-b-17000, 40000-b-25500, 40000-b-31000, 40000-b-40500, 40000-b-53000, 51000-b-28000, 53000-b-15000, 58500-b-37000, 58600-b-71000, 59000-b-31000, 59000-b-72000, 60000-b-18000, 60000-b-36000, 60000-b-61000, 60000-b-85000, 65000-b-85000, 65000-b-97000, 90000-b-45000, 100000-b-150000, 105000-b-155000, 116000-b-164000, and 125000-b-177000.
2.2 Synthesis of PS-b-PEO templated films and powders of CeO2
According to the literature for the synthesis of a mesoporous CeO2 powder sample through a drying process in the presence of a laboratory-made PS-b-PEO,17 a PS-b-PEO containing precursor solution was prepared as follows. Ce(NO3)3·6H2O (0.30 g) was dissolved in EtOH (7.0 mL), stirred for 60 min, and then mixed with THF (7.0 mL) containing PS-b-PEO (0.10 g). The precursor solution was just spin-coated over a UV–ozone-treated clean Si wafer at room temperature (21–22 °C) with ambient humidity (∼30%). The resultant CeO2 film was heated up to 400 °C at a heating rate of 1 °C min−1 in a flow of N2, kept for 1 h under the same conditions, and then calcined for 2 h at that temperature in a flow of O2. After checking the formation of PS-b-PEO-templated mesopores, the precursor solution was spray-dried at 140 °C and the resultant powder sample was calcined at 400 °C in the same manner.
2.3 Characterization
Field-emission scanning electron microscopy (FE-SEM) images were taken using a high-resolution model HITACHI SU9000. For checking the formation of PS-b-PEO-templated large-sized mesopores, the resultant powder sample was directly observed (0.7 kV, 10 mA) using a sample folder for retarding the deposition of any conductive metals. Transmission electron microscopy (TEM) images were taken using a JEOL JEM-2010 instrument, operated at 200 kV. Nitrogen (N2) adsorption–desorption isotherms were measured using an Anton Paar Autosorb-iQ instrument at 77 K after the samples were preheated at 110 °C for 6 h under vacuum. The specific surface areas were calculated by the Brunauer–Emmett–Teller (BET) method using the adsorption data below P/P0 = 0.30. Total pore volumes were estimated using the volume of N2 at P/P0 = 0.99. X-Ray diffraction (XRD) patterns were recorded using a Rigaku RINT 2100 diffractometer with monochromated Cu Kα radiation (40 kV, 30 mA).
3. Results and discussion
The molecular structure of PS-b-PEO helpful for obtaining a highly porous spin-coated film of CeO2, as utilized with a completely equivalent chemical composition that was applied in a previous innovative paper,17 was identified by a quick screening of PS-b-PEO. In the previous paper, laboratory-made PS-b-PEO was utilized for the recovery of dried-up powders and/or a film-like bulk as porous CeO2 powders.17 Such a PS-b-PEO could not possibly be prepared elsewhere in many cases and it would be better to find commercially available ones suitable for the synthesis of porous CeO2 powders. Accordingly, the same precursor solutions containing successful PS-b-PEO compounds were spray-dried at an appropriate temperature for validating the formation of PS-b-PEO-templated pores throughout the resultant CeO2 powders after calcination. The amount of PS-b-PEO was also verified for further increasing the resultant porosity if necessary.
3.1 Highly porous mesoporous CeO2 films
A wide variety of PS-b-PEO diblock copolymers were utilized to fabricate the CeO2 films by spin-coating (see Fig. S1 and S2†). As provided in Fig. 1, several PS-b-PEO molecules, especially 16000-b-7500 and 59000-b-31000, were found for obtaining high-quality porous CeO2 films (see (a) and (d) in Fig. 1). The pore diameters after calcination at 400 °C were estimated to be ∼20 nm and ∼35 nm by SEM, respectively. Besides, the molecular structures having PS > PEO (e.g., 35000-b-17000 and 40000-b-25500) seemed useful for the formation of spherical pores, but much larger pores could not be structured in resultant films thinner than the pore diameters. Scratched off powders of the CeO2 films prepared using 16000-b-7500 and 59000-b-31000 were characterized by TEM (see (b) and (e) in Fig. 1), revealing the presence of large-sized mesopores surrounded by nanoparticles of CeO2 crystallized to its cubic phase with the space group Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m.18–21 Interestingly, their particle size was almost homogeneous throughout the mesoporous CeO2 films, with the average size of around 3 nm (see (c) and (f) in Fig. 1).
m.18–21 Interestingly, their particle size was almost homogeneous throughout the mesoporous CeO2 films, with the average size of around 3 nm (see (c) and (f) in Fig. 1).
 |
| | Fig. 1 Surface SEM images of CeO2 films prepared using (a) 16000-b-7500 and (d) 59000-b-31000 by spin-coating and subsequent calcination at 400 °C, with (b and c) and (e and f) showing TEM images of their scratched films. | |
In the process of the soft-templating of mesoporous CeO2 using amphiphilic organic compounds,22 a hydrogenated poly(butadiene)-block-poly(ethylene oxide) (PHB-b-PEO, named as KLE, H[CH2CH2CH2CH(CH2CH3)]n(OCH2CH2)mOH with a molecular weight of 4400-b-3920 g mol−1) was the most successful one for obtaining an ordered mesoporous CeO2 film.18 KLE is one of the famous amphiphilic organic compounds for obtaining a wide variety of metal oxides containing large-sized spherical mesopores.23 In the case of CeO2, the formation of large-sized mesopores (14 nm and more) was confirmed by TEM and the mesoporous structure was maintained even after crystallization of the CeO2 frameworks by calcination at 550 °C.18 In the synthesis of mesoporous CeO2 films using Pluronic P123 (EO20PO70EO20), which is a commercially available EOnPOmEOn type triblock copolymer, the aging time of the CeO2 sol, the relative humidity during dip-coating, and the calcination temperature were very important for tuning the size of the CeO2 nanocrystals that should be accommodated inside the limited space of the thin CeO2 frameworks around the EOnPOmEOn micelles.24 Compared to the size of the resultant micelles of the asymmetric KLE and symmetric Pluronic P123, KLE seemed preferable for accommodating large-sized mesopores with the formation of thick CeO2 frameworks. In this study, the high-molecular-weight PS-b-PEO type amphiphilic organic compounds were asymmetric and thus useful for obtaining large-sized mesoporous CeO2, as in the case of other metal oxides.18,23,25–28
3.2 Highly porous mesoporous CeO2 powders
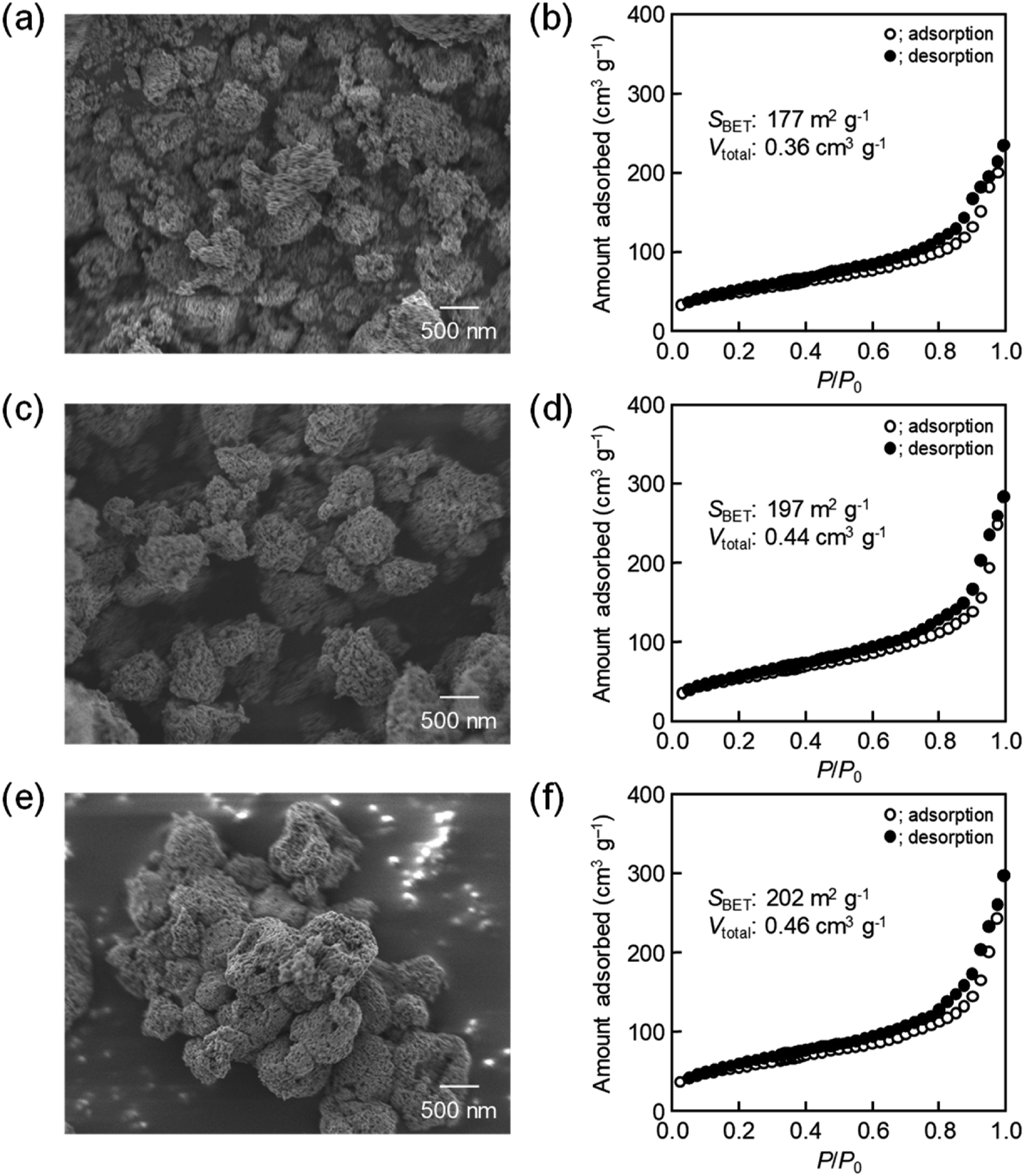
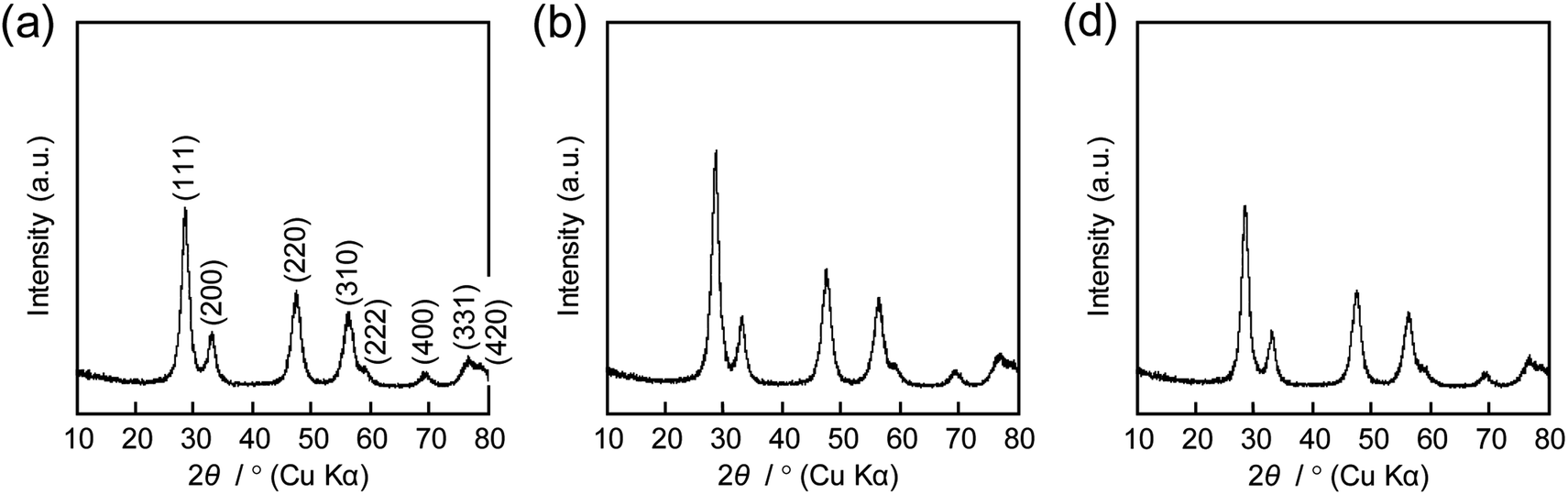
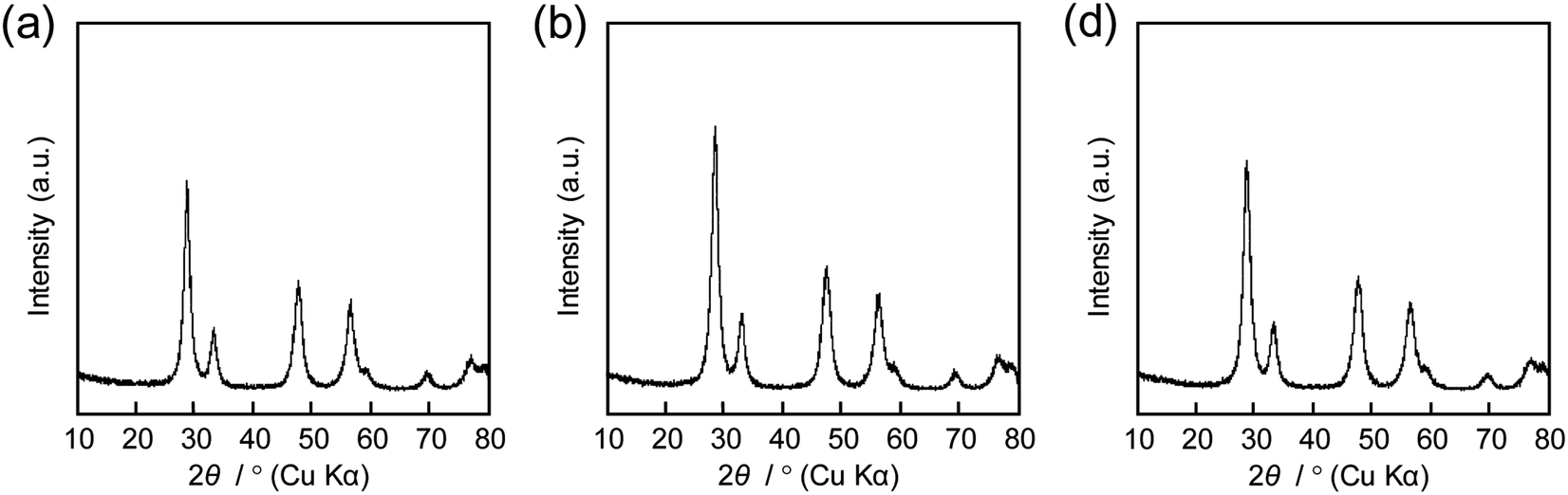
The same precursor solutions were spray-dried at an inlet temperature at 140 °C and the recovered powders were heated up to 400 °C in a flow of N2, kept for 1 h under the same conditions, and then calcined at that temperature in a flow of O2 for 2 h. The SEM image of the calcined sample prepared using 16000-b-7500 (0.10 g) clearly showed the opening of PS-b-PEO-templated large-sized mesopores throughout the CeO2 particles after calcination at 400 °C (see (a) in Fig. 2). The morphology seemed different from that observed for mesoporous metal oxide particles, which were typically aggregated as spherical ones, prepared by an aerosol-assisted synthesis using amphiphilic organic molecules.14–16,27 This was likely be due to the strongly ethanolic synthesis without water showing a high surface tension. The TEM images revealed that the PS-b-PEO-templated mesopores were surrounded by fine CeO2 nanoparticles (around 3 nm) (see (a) and (b) in Fig. 3). The N2 adsorption–desorption isotherm was almost type V (see (b) in Fig. 2) as indicated by the presence of large-sized mesopores.16,25–27 The BET surface area and the total pore volume were 160 m2 g−1 and 0.35 cm3 g−1, respectively.31 These extremely large values come from the presence of intercrystalline spaces of the resultant fine nanoparticles of CeO2 in addition to the surfaces of the PS-b-PEO-templated mesopores. Some diffraction peaks that could be assigned to the typical cubic Fmm phase of CeO2 were clearly detected in the wide-angle XRD pattern (see (a) in Fig. 4).
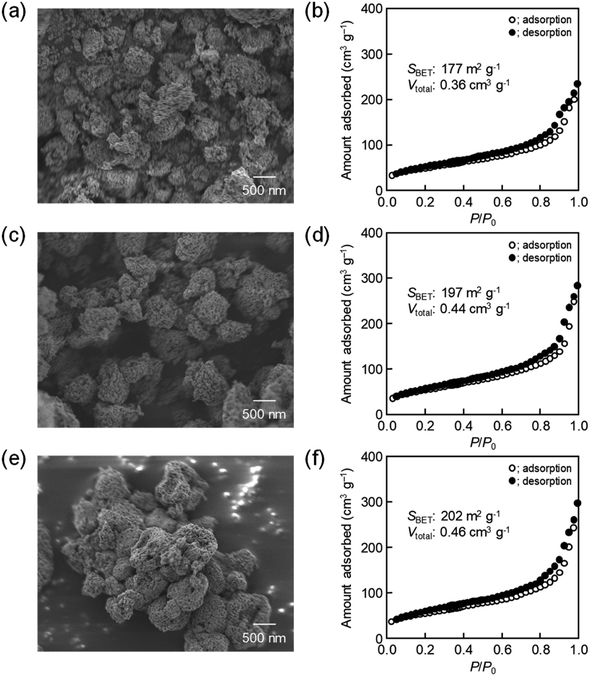 |
| | Fig. 2 SEM images of CeO2 powders prepared using (a) 0.10 g, (c) 0.12 g, and (e) 0.14 g of 16000-b-7500 via spray-drying and calcination at 400 °C, with (b, d and f) corresponding N2 adsorption–desorption isotherms. | |
 |
| | Fig. 3 TEM images of CeO2 powders prepared using (a and b) 0.10 g, (c and d) 0.12 g, and (e and f) 0.14 g of 16000-b-7500 via spray-drying and calcination at 400 °C. | |
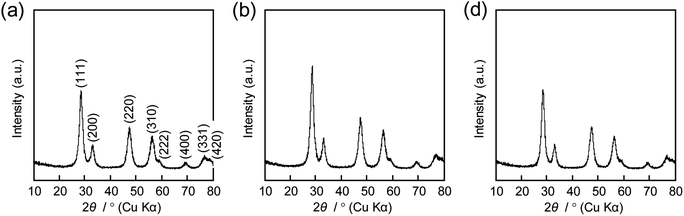 |
| | Fig. 4 XRD patterns of CeO2 powders prepared using (a) 0.10 g, (b) 0.12 g, and (c) 0.14 g of 16000-b-7500 via spray-drying and calcination at 400 °C. | |
In the early papers on the synthesis of mesoporous CeO2 samples using Pluronic P123, it was rather difficult to structure ordered mesopores by a drying process.29,30 Still, the mesoporous structure was stable after calcination below 500 °C (120–150 m2 g−1 after calcination at 400 °C and 80–110 m2 g−1 after calcination at 500 °C) even when the CeO2 frameworks were crystallized to the cubic Fmm phase.19–21 The mesopores though were deformed and finally collapsed by further crystallization at around 600 °C and the grain growth of CeO2 nanocrystals at higher temperature.30 Large-sized mesopores that are surrounded by thick frameworks allow abundant crystallization and the extra phase transformation of metal oxide species.14–16 The use of preformed CeO2 nanocrystals (∼3 nm) is a suitable approach for obtaining mesoporous CeO2 powders using EOnPOmEOn smaller than KLE and PS-b-PEO (for example, 160 m2 g−1 and 0.35 cm3 g−1 after calcination at 500 °C).19,21,31 In summary, according to the added benefits of using PS-b-PEO, this study was very successful at obtaining highly porous powders structured by highly crystallized CeO2 nanocrystals.
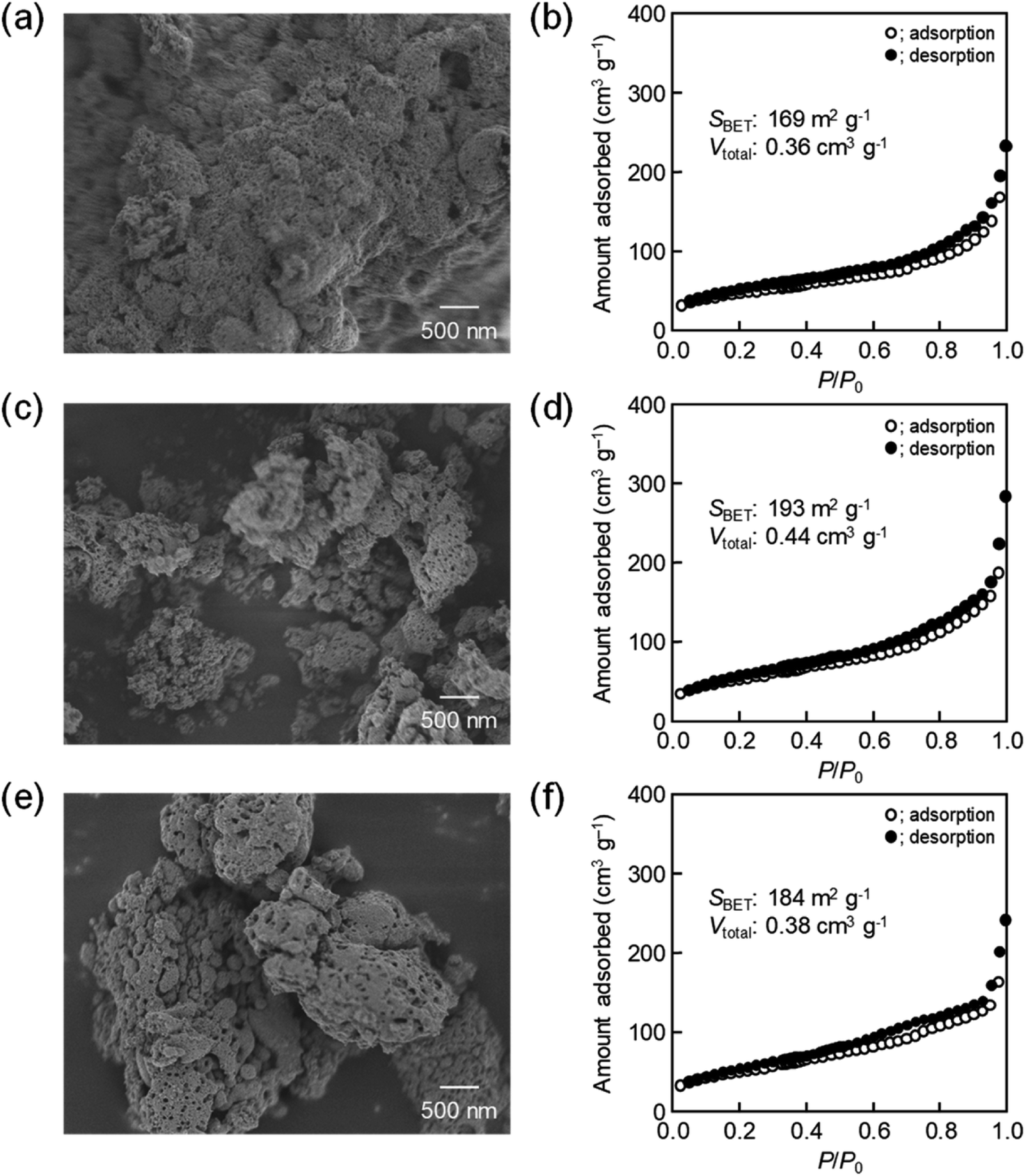
The amount of 16000-b-7500 was, besides, increased to 0.12 g and 0.14 g for checking the possibility to increase the porosity. The specific surface area and the total pore volume are summarized in Table 1, including those prepared using 0.10 g of 16000-b-7500. The analytical data, such as the SEM images with the N2 adsorption–desorption isotherms, the TEM images, and the XRD patterns, are presented in Fig. 2–4, being almost analogous to those observed for the highly porous CeO2 particles prepared using 0.10 g of 16000-b-7500. By increasing the amount of 16000-b-7500, the BET surface area and the total pore volume gradually increased up to around 200 m2 g−1 and around 0.46 cm3 g−1, respectively. Likewise, the powder samples were synthesized by spray-drying precursor solutions containing different amounts of 59000-b-31000 (0.10 g, 0.12 g, and 0.14 g) at 140 °C and then calcined at 400 °C. The SEM images with the N2 adsorption–desorption isotherms, the TEM images, and the XRD patters are shown in Fig. 5–7, respectively. The resultant porosity (e.g., specific surface area and total pore volume) are listed in Table 1. Although the analytical data seemed similar to those observed for the CeO2 particles prepared using 16000-b-7500, the BET surface area and the total pore volume were maximized to around 190 m2 g−1 and around 0.44 cm3 g−1 in the case of using 0.12 g of 59000-b-31000.
Table 1 Porosities of PS-b-PEO-templated CeO2 powders
| PS-b-PEO |
|
BET surface area/m2 g−1 |
Total pore volume/cm3 g−1 |
| 16000-b-7500 |
0.10 g |
177 |
0.36 |
| 0.12 g |
197 |
0.44 |
| 0.14 g |
202 |
0.46 |
| 59000-b-31000 |
0.10 g |
169 |
0.36 |
| 0.12 g |
193 |
0.44 |
| 0.14 g |
184 |
0.28 |
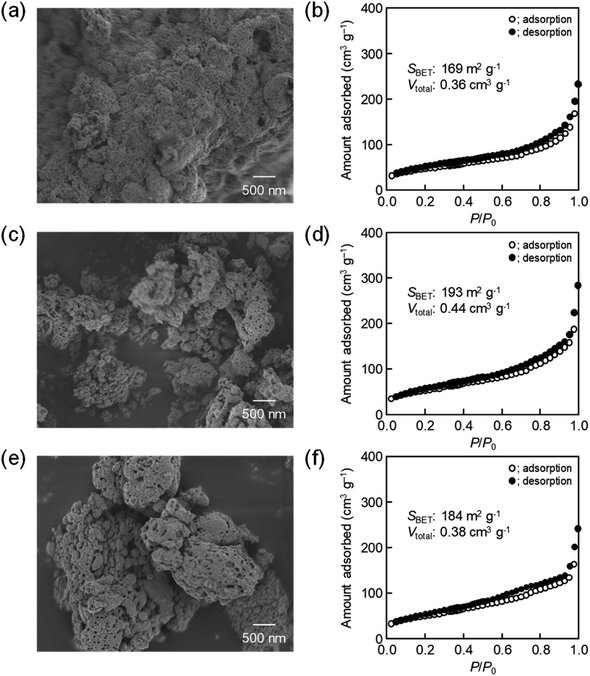 |
| | Fig. 5 SEM images of CeO2 powders prepared using (a) 0.10 g, (c) 0.12 g, and (e) 0.14 g of 59000-b-31000 via spray-drying and calcination at 400 °C with (b, d and f) corresponding N2 adsorption–desorption isotherms. | |
 |
| | Fig. 6 TEM images of CeO2 powders prepared using (a and b) 0.10 g, (c and d) 0.12 g, and (e and f) 0.14 g of 59000-b-31000 via spray-drying and calcination at 400 °C. | |
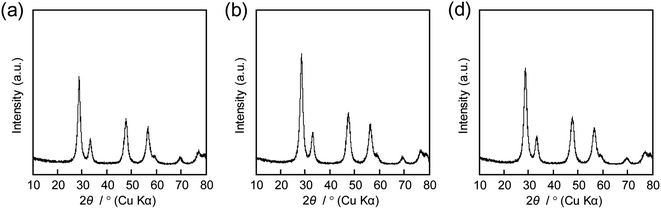 |
| | Fig. 7 XRD patterns of CeO2 powders prepared using (a) 0.10 g, (b) 0.12 g, and (c) 0.14 g of 16000-b-7500 via spray-drying and calcination at 400 °C. | |
Highly porous CeO2 powders (e.g., specific surface area of 200 m2 g−1 and total pore volume of 0.45 cm3 g−1, prepared using 16000-b-7500 after calcination at 400 °C) were constructed by fine nanocrystals of CeO2 (around 3 nm) through a thermal calcination, being much higher than those synthesized using different amphiphilic organic compounds (pore size of 5.5 nm, specific surface area of 125 m2 g−1, and total pore volume of 0.20 cm3 g−1, prepared using EOnPOmEOn after calcination at 500 °C; and pore size of 12 nm, specific surface area of 87 m2 g−1, and total pore volume of 0.12 cm3 g−1, prepared using KLE after calcination at 500 °C).20,21 Even in the synthesis of pure CeO2 using the laboratory-made 17000-b-5000, the specific surface area and total pore volume were limited to 107 m2 g−1 and 0.32 cm3 g−1 with the formation of ∼14 nm pores.17 Such nano-sized CeO2 crystallites are powerful materials for oxygen and charge storage32,33 and useful for the design of metal-supported catalysts that depend on strong interaction with the surfaces of crystallized CeO2 particles.34–36 For example, a strong interaction of Pt to nanostructured CeO2 surfaces is helpful for accelerating oxygen transfer from CeO2 to Pt, leading to the formation of Pt–O species.34 The size of CeO2 nanocrystals (several nm order) is important for controlling the catalytic performance of atomically dispersed palladium (Pd).36 The spray-pyrolysis technique is also very interesting for the design of single-atom catalysts over mesoporous metal oxides that are useful for low-temperature CO oxidation.37–40 The resultant CeO2 powders also possibly have potential for the design of metal-supported catalysts with an oxygen-storage capacity (OSC) for the purification of exhaust gases from automobiles.
4. Conclusion
A step-by-step but smooth optimization of the synthetic conditions was demonstrated as a rational approach for obtaining highly porous metal oxide powders through a spray-drying EISA process. As a typical example, a precursor solution, disclosed in the formative paper on the synthesis of a dried-up film-like bulk of CeO2 with the formation of PS-b-PEO-templated mesopores during a slow evaporation of solvent(s) such as EtOH,17 was utilized for screening the molecular structure of PS-b-PEO. After finding the molecular structure of PS-b-PEO that was most suitable for fabricating highly porous CeO2 films by quick spin-coating (e.g., 16000-b-7500 and 59000-b-31000), the same precursor solution was spray-dried at an appropriate temperature (e.g., 140 °C) for recovering high-quality porous CeO2 powders, and the porosity was checked and/or increased by changing the amount of PS-b-PEO. In addition to our previous approach starting from the synthetic conditions optimized by a fast spin-coating process,14 this extended approach to combine several EISA processes is promising for a smooth optimization of the synthetic conditions (e.g., the chemical composition of the starting mixture and the molecular structure of the amphophilic organic compound) for obtaining highly porous metal oxides. Numerous papers concerning the dried-up powders and spin-coated films of metal oxides would be helpful for reproducing highly porous metal oxide powders by this temperature-controlled spray-drying process in the presence of a wide variety of amphiphilic organic compounds.41,42
Conflicts of interest
The author declares no competing financial interest.
Acknowledgements
This work has been mainly supported as one of the research projects (JPNP18016, commissioned by the New Energy and Industrial Technology Development Organization, NEDO) in the “Moonshot R&D program (Moonshot Goal 4: Realization of sustainable resource circulation to recover the global environment by 2050)”.
References
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature, 1992, 359, 710–712 CrossRef CAS.
- Q. Huo, R. Leon, P. M. Petroff and G. D. Stucky, Science, 1995, 268, 1324–1327 CrossRef CAS PubMed.
- D. Zhao, Q. Huo, J. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- D. Zhao, J. Feng, Q. Huo, N. Melosh, G. H. Fredrickson, B. F. Chmelka and G. D. Stucky, Science, 1998, 279, 548–552 CrossRef CAS.
- K.-W. Kim, B. Park, J. Kim, C. Jo and J. K. Kim, J. Mater. Chem. A, 2023, 11, 7358–7386 RSC.
- Q. Yuan, A.-X. Yin, C. Luo, L.-D. Sun, Y.-W. Zhang, W.-T. Duan, H.-C. Liu and C.-H. Yan, J. Am. Chem. Soc., 2008, 130, 3465–3472 CrossRef CAS PubMed.
- X. Xu, S. K. Megarajan, Y. Zhang and H. Jiang, Chem. Mater., 2020, 32, 3–26 CrossRef CAS.
- Y. Lu, R. Ganguli, C. A. Drewien, M. T. Anderson, C. J. Brinker, W. Gong, Y. Guo, H. Soyez, B. Dunn, M. H. Huang and J. I. Zink, Nature, 1997, 389, 364–368 CrossRef CAS.
- D. Grosso, F. Cagnol, G. J. de A. A. Soler-Illia, E. L. Crepaldi, H. Amenitsch, A. Brunet-Bruneau, A. Bourgeois and C. Sanchez, Adv. Funct. Mater., 2004, 14, 309–322 CrossRef CAS.
- T. Kimura, Chem. Rec., 2016, 16, 445–457 CrossRef CAS PubMed.
- Y. Fu, H. Fan, A. Stump, T. L. Ward, T. Rieker and C. J. Brinker, Nature, 1999, 398, 223–226 CrossRef.
- C. Boissiere, L. Nicole, C. Gervais, F. Babonneau, M. Antonietti, H. Amenitsch, C. Sanchez and D. Grosso, Chem. Mater., 2006, 18, 5238–5243 CrossRef CAS.
- C.-K. Tsung, J. Fan, N. Zheng, Q. Shi, A. J. Forman, J. Wang and G. D. Stucky, Angew. Chem., Int. Ed., 2008, 47, 8682–8686 CrossRef CAS PubMed.
- H. Maruoka and T. Kimura, Bull. Chem. Soc. Jpn., 2019, 92, 1859–1866 CrossRef CAS.
- T. Kimura and H. Maruoka, Chem. Commun., 2019, 55, 10003–10006 RSC.
- Md. I. Saidul, R. Wakabayashi and T. Kimura, Dalton Trans., 2021, 50, 7191–7197 RSC.
- J. Ge, X. Yang, J. Luo, J. Ma, Y. Zou, J. Li, W. Luo, X. Cheng and Y. Deng, Appl. Mater. Today, 2019, 15, 482–493 CrossRef.
- T. Brezesinski, C. Erpen, K. Iimura and B. Smarsly, Chem. Mater., 2005, 17, 1683–1690 CrossRef CAS.
- K. Suzuki and A. K. Sinha, J. Mater. Chem., 2007, 17, 2547–2551 RSC.
- J.-Y. Chane-Ching, F. Cobo, D. Aubert, H. G. Harvey, M. Airiau and A. Corma, Chem.–Eur. J., 2005, 11, 979–987 CrossRef CAS PubMed.
- A. S. Deshpande, N. Pinna, B. Smarsly, M. Antonietti and M. Niederberger, Small, 2005, 1, 313–316 CrossRef CAS PubMed.
- M. Dubey, S. Wadhwa, A. Mathur and R. Kumar, Appl. Surf. Sci. Adv., 2022, 12, 100340 CrossRef.
- C. Reitz, J. Haetge, C. Suchomski and T. Brezesinski, Chem. Mater., 2013, 25, 4633–4642 CrossRef CAS.
- Z. Lu, D. Rébiscoul, J. Causse, X. le Goff, N. Mollard and X. Deschanels, J. Sol-Gel Sci. Technol., 2020, 94, 174–185 CrossRef CAS.
- X. Yang, X. Cheng, H. Song, J. Ma, P. Pan, A. A. Elzatahry, J. Su and Y. Deng, Adv. Healthcare Mater., 2018, 7, 1800149 CrossRef.
- X. Yang, X. Cheng, J. Ma, Y. Zou, W. Luo and Y. Deng, Small, 2019, 15, 1903058 CrossRef.
- Y. Zhang, R. Wakabayashi and T. Kimura, Dalton Trans., 2023, 52, 1543–1550 RSC.
- Y. Deng, T. Yu, Y. Wan, Y. Shi, Y. Meng, D. Gu, L. Zhang, Y. Huang, C. Liu, X. Wu and D. Zhao, J. Am. Chem. Soc., 2007, 129, 1690–1697 CrossRef CAS.
- M. Lundberg, B. Skårman, F. Cesar and L. R. Wallenberg, Microporous Mesoporous Mater., 2002, 54, 97–103 CrossRef CAS.
- M. Lundberg, B. Skårman and L. R. Wallenberg, Microporous Mesoporous Mater., 2004, 69, 187–195 CrossRef CAS.
- A. Corma, P. Atienzar, H. García and J.-Y. Chane-Ching, Nat. Mater., 2004, 3, 394–397 CrossRef CAS.
- T. Brezesinski, J. Wang, R. Senter, K. Brezesinski, B. Dunn and S. H. Tolbert, ACS Nano, 2010, 4, 967–977 CrossRef CAS PubMed.
- P. Hartmann, T. Brezesinski, J. Sann, A. Lotnyk, J.-P. Eufinger, L. Kienle and J. Janek, ACS Nano, 2013, 7, 2999–3013 CrossRef CAS PubMed.
- J. A. Farmer and C. T. Campbell, Science, 2010, 329, 933–936 CrossRef CAS PubMed.
- G. N. Vayssilov, Y. Lykhach, A. Migani, T. Staudt, G. P. Petrova, N. Tsud, T. Skála, A. Bruix, F. Illas, K. C. Prince, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2011, 10, 310–315 CrossRef CAS PubMed.
- V. Muravev, A. Parastaev, Y. van den Bosch, B. Ligt, N. Claes, S. Bals, N. Kosinov and E. J. M. Hensen, Science, 2023, 380, 1174–1178 CrossRef CAS PubMed.
- R. Li, Y. Yang, N. Sun and L. Kuai, Chem.–Eur. J., 2019, 25, 15586–15593 CrossRef CAS PubMed.
- Q. Tao, J. Song, N. Sun, Y. Ren, L. Xiang, S. Liu and L. Kuai, Inorg. Chem., 2022, 61, 11932–11938 CrossRef CAS PubMed.
- N. Sun, L. Xiang, B. Zhuge, E. Kan, N. Yu, L. Li and L. Kuai, Inorg. Chem., 2023, 62, 782–791 CrossRef CAS PubMed.
- L. Xiang, S. Wang and L. Kuai, Microporous Mesoporous Mater., 2024, 363, 112809 CrossRef CAS.
- J. Wei, Z. Sun, W. Luo, Y. Li, A. A. Elzatahry, A. M. Al-Enizi, Y. Deng and D. Zhao, J. Am. Chem. Soc., 2017, 139, 1706–1713 CrossRef CAS PubMed.
- Y. Zou, X. Zhou, J. Ma, X. Yang and Y. Deng, Chem. Soc. Rev., 2020, 49, 1173–1208 RSC.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*