
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTargeting EGFR/PI3K/AKT/mTOR signaling in lung and colon cancers: synthesis, antitumor evaluation of new 1,2,4-oxdiazoles tethered 1,2,3-triazoles†
Mohammed Salah Ayoup *ab,
Islam Shawkib,
Hamida Abdel-Hamidb,
Doaa A. Ghareebcg,
Aliaa Masoudc,
Marwa F. Harrasd,
Mohamed El-Atawy*be,
Nuha Salamah Alharbif and
Magda M. F. Ismaild
*ab,
Islam Shawkib,
Hamida Abdel-Hamidb,
Doaa A. Ghareebcg,
Aliaa Masoudc,
Marwa F. Harrasd,
Mohamed El-Atawy*be,
Nuha Salamah Alharbif and
Magda M. F. Ismaild
aDepartment of Chemistry, College of Science, King Faisal University, Al-Ahsa 31982, Saudi Arabia. E-mail: mayoup@kfu.edu.sa; mohammedsalahayoup@gmail.com
bDepartment of Chemistry, Faculty of Science, Alexandria University, Alexandria, Egypt. E-mail: mohamed.elatawi@alexu.edu.eg
cBio-screening and Preclinical Trial Lab, Biochemistry Department, Faculty of Science, Alexandria University, Alexandria, Egypt
dDepartment of Pharmaceutical Medicinal Chemistry and Drug Design, Faculty of Pharmacy (Girls), Al-Azhar University, Cairo 11754, Egypt
eChemistry Department, College of Science at Yanbu, Taibah University, Yanbu 46423, Saudi Arabia
fChemistry Department, College of Sciences, Taibah University, Al-Madina 30002, Saudi Arabia
gMedical Biotechnology Department, Genetic Engineering and Biotechnology Research Institute, City of Scientific Research and Technological Applications (SRTA-City), Egypt
First published on 22nd May 2024
Abstract
The EGFR/PI3K/Akt/mTOR pathway is important for metastasis, medication resistance, apoptosis prevention, and malignant transformation. Mutations in lung and colon cancer typically change this pathway's expression. As a result, a novel class of 1,2,4-oxdiazoles that are attached to 1,2,3-triazoles, 5–11, were created as possible anticancer drugs. The produced compounds are all examined by spectroscopic and micro-analytical techniques. MTT assay results on lung (A549) colon (Caco-2) and normal lung fibroblast (WI38) revealed that compounds 6a, 6b, 8a, and 11b demonstrated strong and selective antiproliferative activities against lung (A549) and colon (Caco-2) cancer cell lines while the remaining derivatives showed moderate to low activity. qPCR data revealed that the potential hits had large fold changes in the downregulation of EGFR, mTOR, and PI3K; they upregulate the amount of p53 to support their mode of action even more. Interestingly, docking investigations validated the biological outcomes by demonstrating a strong affinity of our compounds against EGFR active regions. Computational predictions of all the synthesized compounds' pharmacokinetic profiles, physicochemical characteristics, and drug-likeness data indicated that the promising hits might be taken into consideration as drug-like prospects.
1. Introduction
Humanity is currently engaged in a struggle with a number of health-related issues, particularly those pertaining to cancer. This has fueled the desire to create strong, targeted inhibitors of tumor cells that would specifically target oncogenic proteins that play critical roles in the development and spread of cancer.1Because of its involvement in oncogenesis, EGFR is one of the receptors that has been investigated the most.2 Numerous cancer forms, including colorectal cancer,3 non-small-cell lung cancer (NSCLC),4 squamous cell carcinoma of the head and neck,5 glioblastoma,6 pancreatic cancer,7 and breast cancer,8 have been linked to the proto-oncogenic activity of EGFR. Globally, lung cancer is now the primary cause of cancer-related mortality.9 In advanced non-small cell lung cancer (NSCLC), individuals with EGFR mutations have been first-line treated with epidermal growth factor receptor tyrosine kinase inhibitors (EGFR-TKIs). However, the clinical efficacy of this treatment is significantly limited by drug resistance that develops following continuous and prolonged chemotherapy. To combat medication resistance, it is crucial to develop novel chemotherapeutic agents and treatment approaches. Likewise, EGFR is thought to be overexpressed in 60–80% of tumors in colorectal cancer (CRC), and this is linked to a poor prognosis.10 These factors have contributed to the targeting of EGFR as a therapeutic target for monoclonal antibodies and small molecule inhibitors, the former of which is used to treat metastatic illness.11
EGFR/PI3K/AKT/mTOR signaling is a key pathway in many different kinds of cancer. This route positively controls a number of cellular functions, such as metabolism, migration, survival, and proliferation. Additionally, it is essential in controlling the tumor microenvironment by attracting inflammatory cells and promoting angiogenesis.12 Furthermore, therapeutic response and metastasis are strongly influenced by the EGFR/PI3K/AKT/mTOR signaling.13,14 It should come as no surprise that the signaling pathway is frequently elevated in a range of human malignancies, given its complex roles in carcinogenesis.15,16 Combination techniques within the EGFR–PI3K–mTOR pathways may be a useful strategy to increase therapeutic efficacy when some tumor subclones develop resistance to a single treatment due to genetic alterations as well. When treating tumors connected to these changes, targeting different parts of the EGFR–PI3K–mTOR axis may prove to be an effective treatment strategy.17
In response to stress, the p53 protein acts like a conductor in a complex cellular orchestra. It controls the activity of several genes (PTEN, IGF-BP3, TSC2, and AMPKb) that act as brakes on important growth signals (Akt and mTOR). This slows down cell division and helps reverse the characteristics of cancer cells. Additionally, p53 working with LKB1 further restricts cell proliferation. But that's not all! p53 also activates a cellular recycling process called autophagy, which helps stressed cells survive.18
The literature made it evident that 1,2,3-triazole derivatives containing 1,2,4-oxadiazole and 1,3-oxazole, Ia and Ib, showed encouraging cytotoxicity against the epithelial cancer cell line A549 (lung cancer), with IC50 values of 0.79 and 0.19 μM, respectively, while etoposide, the drug used as a reference, showed IC50 value of 3.08 mM.19 When tested against A549, a library of 1,2,4-oxadiazole that included 1,2,3-triazoles, compound II20 showed strong anticancer activity (IC50 0.45 ± 0.023 μM). Furthermore, compound III was found to have a high and selective ant proliferative capability in A549 cells, with an IC50 value of 0.07 μM. It was also reported to raise the populations of early and late apoptotic/primary necrotic cells by 26.97% and 16.37%, respectively.21 The hybrid IV containing a 4-bromophenyl-thiazole moiety, exhibited potent antiproliferative activity on the same cell line, A-549, (IC50 0.40 μM). When it's EGFR suppressing effect (IC50 0.16 ± 0.05 μM) was compared to erlotinib (IC50 0.08 ± 0.04 μM), the hybrid was shown to be remarkably effective (Fig. 1).22
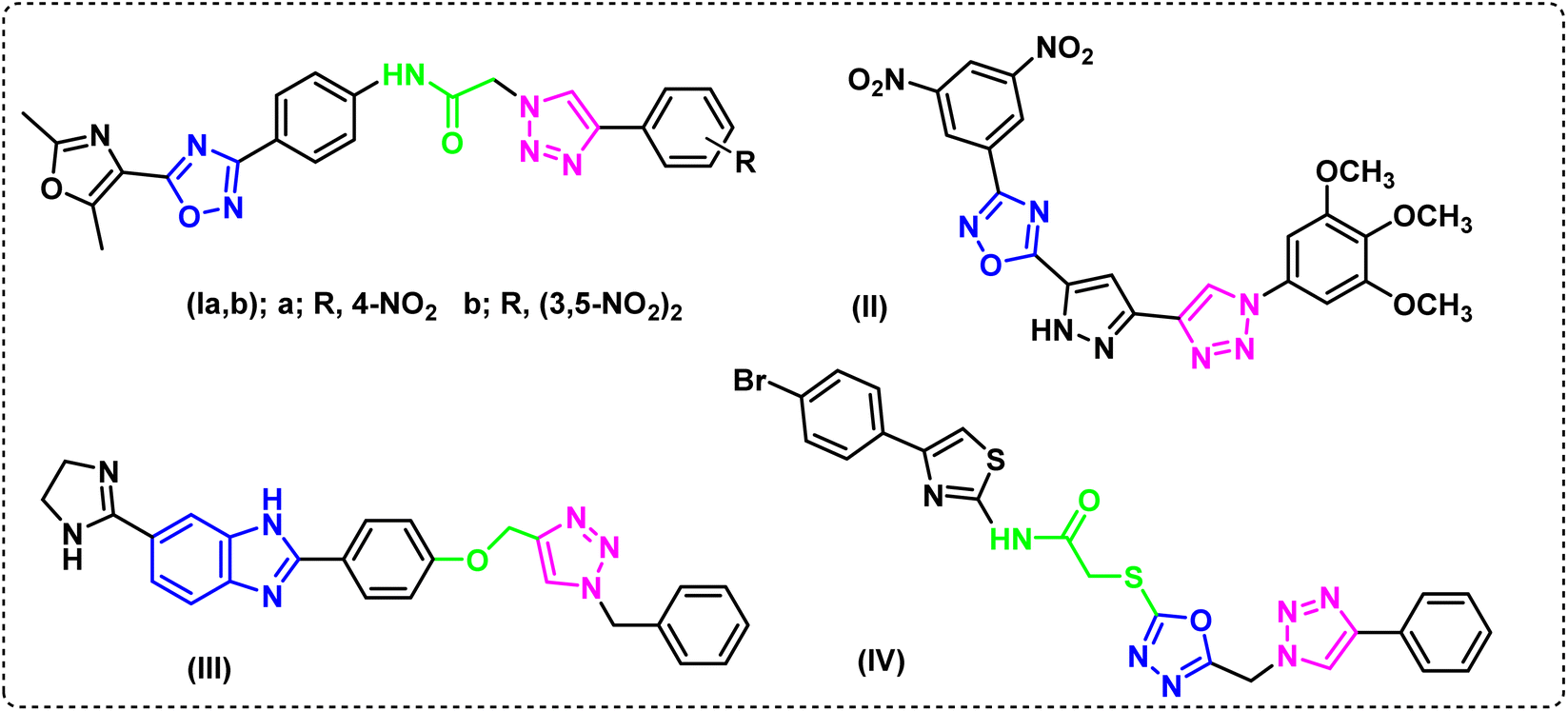 | ||
| Fig. 1 Reported anticancer compounds. | ||
As Fig. 2 shows, a few pharmacophoric properties of EGFR inhibitors are required for good affinity against the EGFR ATP binding site. The above features include: (1) a hydrophobic head that fits into a pocket (region I) and interacts with an amino acid Glu738; (2) a central flat aromatic heterocyclic moiety that sits comfortably within a pocket called adenine binding pocket allowing interactions with amino acids Met769, Leu768, Thr766, and Gln767; (3) solvent accessible area; and (4) hydrophobic pocket II. As lapatinib's analogues, two series of EGFRI were created via ligand-based drug design. As can be seen in Fig. 2, the terminal phenyl/benzyl (hydrophobic I) was planned to function as an equivalent for the chlorophenyl of lapatinib, and the middle flat aromatic heterocyclic moiety was designed to be 1,2,4-isoxazole, an isostere of pyrimidine to accommodate within the adenine binding pocket. The phenyl ring (partially structured quinazoline) will be represented by the central phenyl (hydrophobic II). Finally, lapatinib replaces furan with a side chain through the 1,2,3-triazole linked to the solvent accessible area. In order to maximise the pharmacokinetic characteristics of various aliphatic or aromatic side chains, substituent variation was also pursued (R).
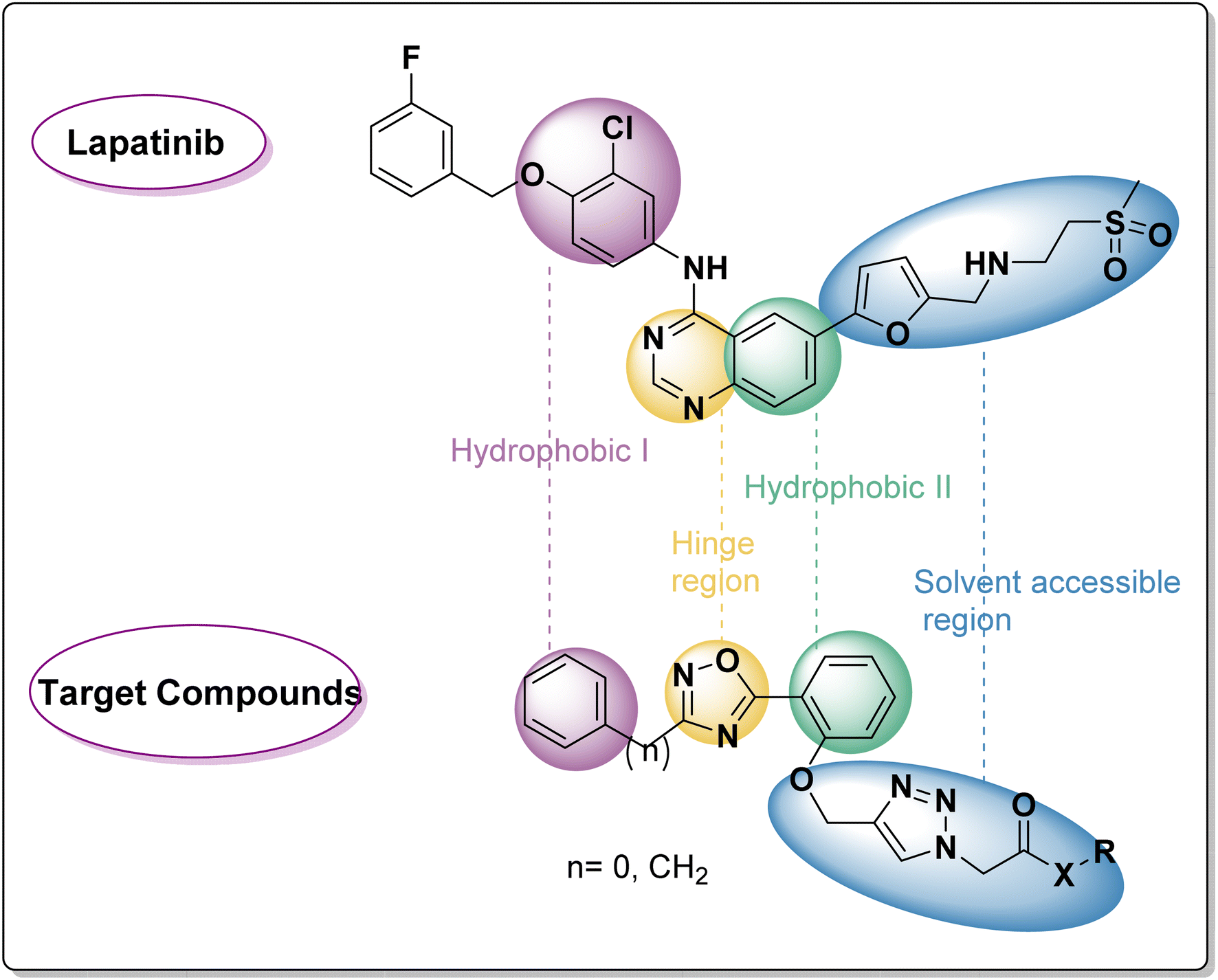 | ||
| Fig. 2 Rational of target compounds. | ||
2. Results and discussions
2.1. Chemistry
A new library of 3,5-diaryl-1,2,4-oxadiazoles23 was designed and synthesized using the 3-benzyl/phenyl derivative of 5-(2-hydroxyphenyl-1,2,4-oxadiazole) (1a and 1b) as starting materials. The primary method for creating 1,2,3-triazoles is the azide–alkyne cyclo-addition reaction.24,25 As a result, the alkylation reaction of the starting materials 1a and 1b using propargyl bromide,26 2 produced the derivatives 3a and 3b of O-alkynyl oxadiazole. Moreover, compounds 3a and 3b were used as starting materials to create a library of target compounds as quinoxaline triazole hybrids using the click reaction with ethyl 4-azidobenzoate, Cu(OAc)2, and ascorbate27 (Scheme 1). This method produced the corresponding quinoxaline-triazole esters, 5a and 5b, which were then treated with hydrochloric acid to produce their benzoic acid counterparts, 7a and 7b. Moreover, ammonolysis produced the acetamide analogues (6a and 6b) of the esters 5a and 5b upon the introduction of ammonium hydroxide. Furthermore, the reaction of 5a and 5b with hydrazine hydrate in ethanol resulted in the production of the acetohydrazides 8a and 8b (Scheme 1). FT-IR, 1H-NMR, 13C-NMR and microanalytical analysis, were employed to validate the structure of the compounds (5–11). The spectroscopy findings supported the estimated structures. IR of compounds 5–8 confirmed the successful transformation of the ester group of compounds 5 into other functionalities such as amide, carboxy or hydrazide group corresponding to compounds 6, 7, and 8 respectively. Compound 6 showed C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching near ≈1680 cm−1 rather than 1742 cm−1 that appeared for the ester functionality of 5. Moreover, IR spectrum of 6 exhibited two absorption bands at ≈3310 and ≈3185 cm−1 that can be ascribed to the typically observed absorption for N–H stretching of NH2 group. Furthermore, the carboxy group of compound 7 revealed a broad absorption peak in the region ≈3450–3300 cm−1 that is the usual OH stretching, in addition to carbonyl stretching at ≈1730 cm−1. Compound 8 displayed the carbonyl stretching absorption band at ≈1660 cm−1 and also showed three absorption bands in the region 3350–3100 cm−1 which can be recognized to stretching bands of N–H and NH2 groups.
O stretching near ≈1680 cm−1 rather than 1742 cm−1 that appeared for the ester functionality of 5. Moreover, IR spectrum of 6 exhibited two absorption bands at ≈3310 and ≈3185 cm−1 that can be ascribed to the typically observed absorption for N–H stretching of NH2 group. Furthermore, the carboxy group of compound 7 revealed a broad absorption peak in the region ≈3450–3300 cm−1 that is the usual OH stretching, in addition to carbonyl stretching at ≈1730 cm−1. Compound 8 displayed the carbonyl stretching absorption band at ≈1660 cm−1 and also showed three absorption bands in the region 3350–3100 cm−1 which can be recognized to stretching bands of N–H and NH2 groups.
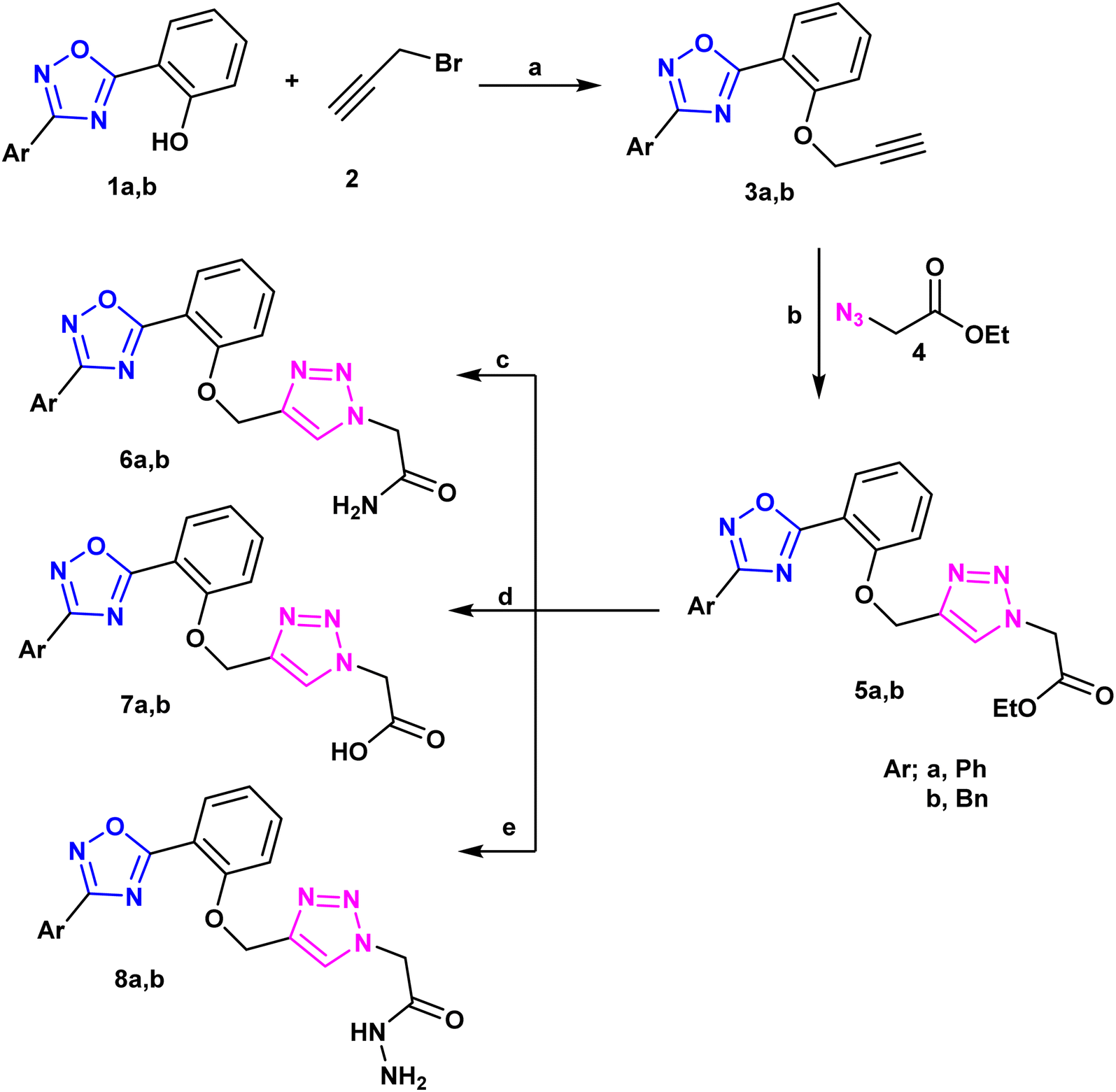 | ||
| Scheme 1 Synthesis of hybrids 1,2,4-oxadizolel1,2,4-trazole derivatives. Reagents and conditions: (a); K2CO3, acetone, reflux 8 h, (b); Cu(OAc)2, ascorbic acid, THF, r.t., reflux 16 h, (c); NH4OH, r.t., reflux 16 h, (d); THF/H2O (1:1) NaOH, pH =2; (e); EtOH/N2H2, reflux 4 h. | ||
The corresponding Schiff's bases, 9a and 9b, were obtained by condensing the acetohydrazide derivatives 8a and 8b with salicylaldehyde, as implemented in Scheme 2. Additionally, the imine derivatives 10a and 10b were made by refluxing 8a and 8b with pyridoxal-5′-phosphate monosodium salt (P5P)28 in 100% ethanol. Ultimately, compounds 11a and 11b were produced by the neucleophilic addition of phenyl isothiocyanate to the acetohydrazide derivatives 8a and 8b. Schiff bases 9, 10 have been derived from 8 via its condensation reaction with either salicylaldehyde or pyridoxal 5 phosphate (P5P). Their IR spectra showed appearance of an absorption band at ≈1600 cm−1 that matches the imine (CN) stretching. Moreover, their 1H-NMR spectra showed a highly deshielded singlet at ≈8.6 ppm that corresponds to the azomethine proton (CHN), which confirm the success of the condensation reaction. Additionally, 1H-NMR of compounds 5–11 exhibited a singlet signal in the region ≈8.14–8.40 ppm that have been ascribed to the triazolyl proton29 which validate the accomplishment of the copper-catalyzed azide–alkyne cycloaddition between compounds 3 and 4. Both 1H-NMR, 13C-NMR spectroscopy confirmed that compounds 5–11 had the expected aliphatic proton and carbons respectively. The methylene and methyl groups showed signals between 1.16 and 5.70 ppm in the proton NMR spectrum, while; in the carbon NMR spectrum, the corresponding carbons of these aliphatic groups produced signals between 14.5 and 67 ppm. Additionally, both the proton and carbon NMR spectra confirmed the presence of aromatic rings. Consequently, aromatic protons displayed downfield peaks in 1H-NMR at the region ≈7.00 to 8.10 ppm. Acidic protons (OH, NH, and NH2) of compounds 6–11 exhibited broad signals in 1H-NMR spectra and were characterized on the basis of their exchange by D2O. Furthermore, 13CNMR of compound 11 displayed a downfield signal near ≈180 ppm that has been attributed to the doubly bonded carbon of the thiocarbonyl group (CS).
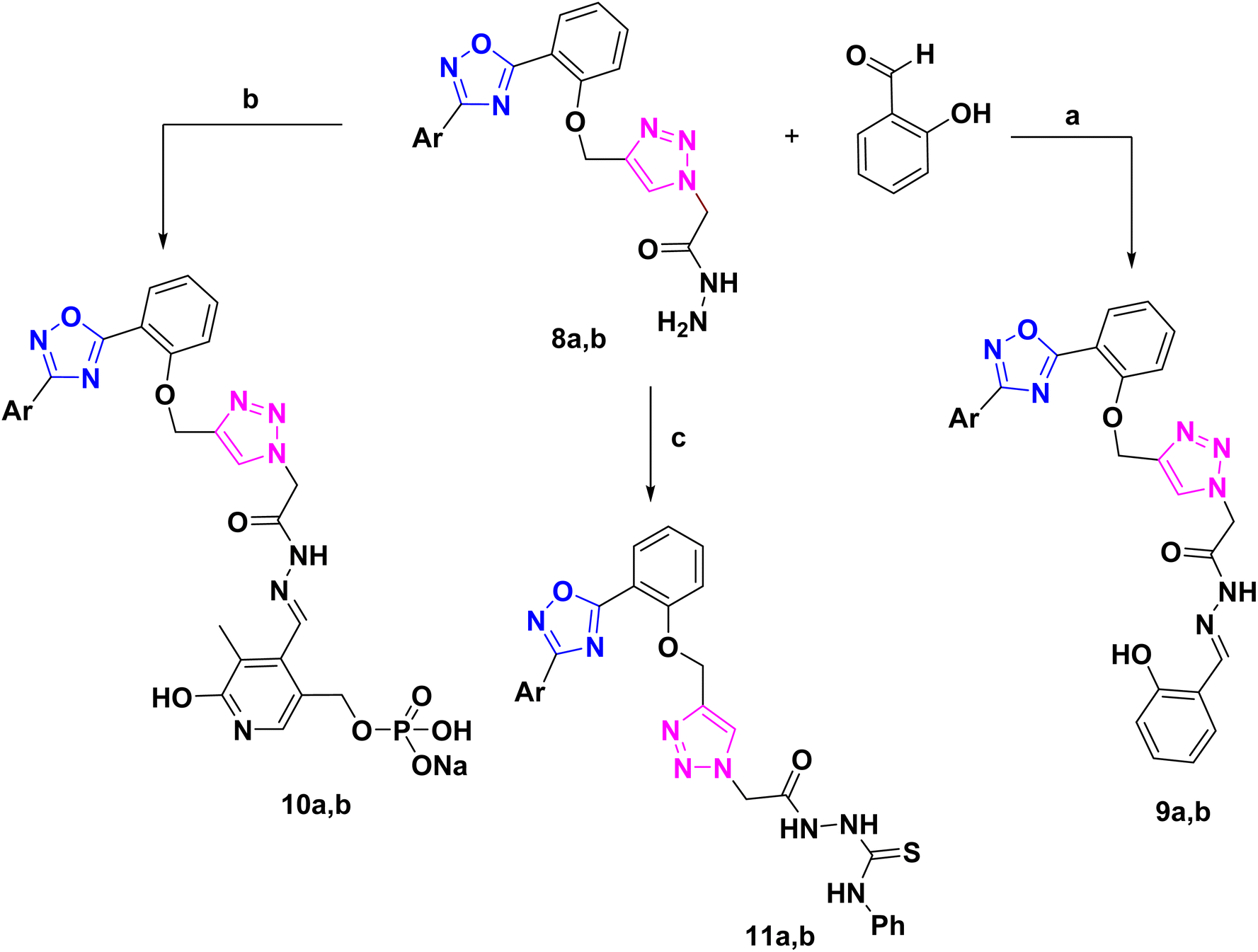 | ||
| Scheme 2 Synthsis of Schiff's bases and thiourea derivatives based 1,2,4-oxadiazole. Reagents and conditions: (a), EtOH reflux 8 h, (b); P5P, EtOH, reflux 8h, (c); PhNCS, EtOH, reflux 8 h. | ||
2.2. Biological evaluation
| Compd no. | WI38 | A549 | SI | CaCo-2 | SI |
|---|---|---|---|---|---|
| 5a | 14.27 | 7.40 | 1.93 | 26.31 | 0.54 |
| 5b | 19.54 | 10.47 | 1.87 | 47.69 | 0.41 |
| 6a | 29.78 | 12.73 | 2.34 | 14.09 | 2.11 |
| 6b | 15.89 | 3.56 | 4.46 | 11.53 | 1.38 |
| 7a | 22.38 | 210.2 | 0.11 | 98.11 | 0.23 |
| 7b | 83.83 | 86.03 | 0.97 | 173.32 | 0.48 |
| 8a | 30.63 | 13.09 | 2.34 | 12.89 | 2.38 |
| 8b | 3.60 | 4.27 | 0.84 | 4.44 | 0.81 |
| 9a | 6.10 | 6.52 | 0.93 | 3.37 | 1.81 |
| 9b | 2.12 | 2.51 | 0.84 | 2.89 | 0.73 |
| 10a | 9.02 | 29.50 | 0.31 | 26.71 | 0.34 |
| 10b | 32.80 | 37.31 | 0.88 | 44.50 | 0.74 |
| 11a | 21.59 | 12.37 | 1.75 | 32.18 | 0.67 |
| 11b | 34.02 | 10.33 | 3.29 | 16.38 | 2.08 |
| Doxorubicin | 4.32 | 1.0 | 4.32 | 1.3 | 3.32 |
Noticeably, when comparing the cytotoxicity of the following pairs of compounds (6a, 6b), (8a, 8b), (9a, 9b) and (11a, 11b), their benzyl derivatives 6b, 8b, 9b and 11b were superior in activity than their phenyl analogs 6a, 8a, 9a and 11a which may be attributed to +I effect of the benzyl substituents.
| Compd no. | A549 | Caco-2 | ||||||
|---|---|---|---|---|---|---|---|---|
| PI3K | mTOR | EGFR | P53 | PI3K | mTOR | EGFR | p53 | |
| Cancer | 1 ± 0.0003 | 1 ± 0.004 | 1 ± 0.003 | 1 ± 0.007 | 1 ± 0.008 | 1 ± 0.001 | 1 ± 0.005 | 1 ± 0.01 |
| 6a | 0.28 ± 0.002 | 0.27 ± 0.01 | 0.25 ± 0.007 | 6.5 ± 0.05 | 0.45 ± 0.001 | 0.35 ± 0.005 | 0.37 ± 0.08 | 4.75 ± 0.03 |
| 6b | 0.24 ± 0.03 | 0.25 ± 0.0008 | 0.23 ± 0.004 | 5.9 ± 0.04 | ND | ND | ND | ND |
| 8a | 0.3 ± 0.004 | 0.28 ± 0.009 | 0.26 ± 0.03 | 5.5 ± 0.003 | 0.43 ± 0.02 | 0.32 ± 0.09 | 0.35 ± 0.03 | 5.13 ± 0.02 |
| 11b | 0.36 ± 0.005 | 0.31 ± 0.04 | 0.29 ± 0.08 | 5.4 ± 0.01 | 0.53 ± 0.0004 | 0.365 ± 0.01 | 0.423 ± 0.005 | 4.36 ± 0.05 |
| Doxorubicin | 0.1 ± 0.008 | 0.18 ± 0.07 | 0.16 ± 0.02 | 8.3 ± 0.02 | 0.19 ± 0.003 | 0.16 ± 0.002 | 0.15 ± 0.03 | 7.9 ± 0.03 |
A correlation between downregulation of PI3K, mTOR and EGFR and upregulation of p53 expression was evident, thus the activity order of our hits towards the tested oncogenic mediators was 6b > 6a > 8a > 11b (Table 2 and Fig. 3–5).
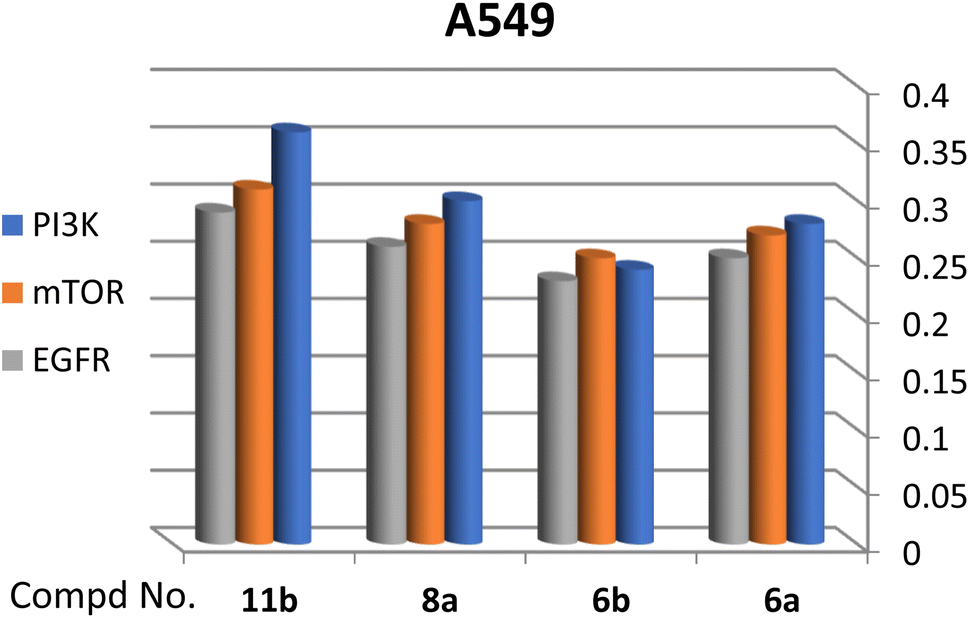 | ||
| Fig. 3 Effect of test compounds on the fold change of PI3K, mTOR, EGFR in A549 cell line. | ||
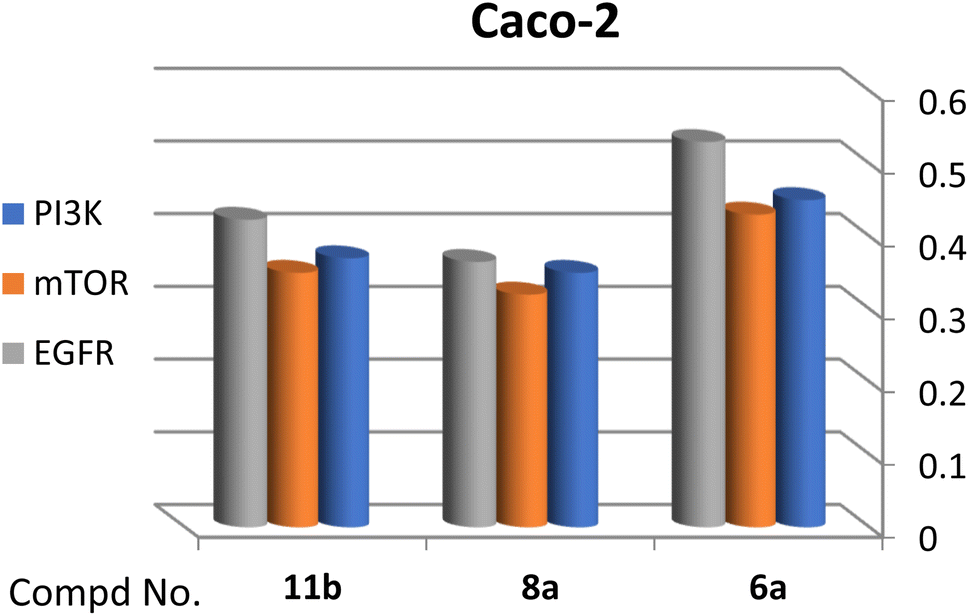 | ||
| Fig. 4 Effect of test compounds on the fold change of PI3K, mTOR, EGFR in Caco-2 cell line. | ||
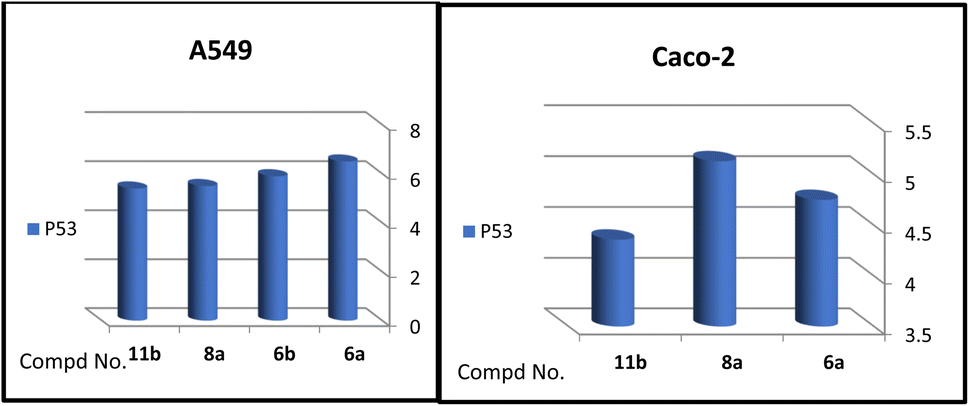 | ||
| Fig. 5 Effect of test compounds on the fold change of p53 in A549 and Caco-2 cell lines. | ||
| Compd no. | HBD | HBA | M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP logP |
M. Wt | No. of rot. bonds | Lipinski's violations | Veber's violations |
|---|---|---|---|---|---|---|---|
| 5a | 0 | 8 | 2.18 | 421.45 | 9 | 0 | 0 |
| 5b | 0 | 8 | 2.12 | 435.48 | 10 | 0 | 0 |
| 6a | 1 | 7 | 1.10 | 376.37 | 7 | 0 | 0 |
| 6b | 1 | 7 | 1.06 | 390.40 | 8 | 0 | 0 |
| 7a | 1 | 8 | 1.51 | 377.35 | 7 | 0 | 0 |
| 7b | 1 | 8 | 1.47 | 391.38 | 8 | 0 | 0 |
| 8a | 2 | 8 | 1.14 | 391.38 | 8 | 0 | 0 |
| 8b | 2 | 8 | 1.10 | 405.41 | 9 | 0 | 0 |
| 9a | 2 | 9 | 2.08 | 511.53 | 10 | 1 | 1 |
| 9b | 2 | 9 | 2.02 | 525.56 | 11 | 1 | 1 |
| 10a | 4 | 14 | 1.96 | 620.51 | 13 | 2 | 2 |
| 10b | 4 | 14 | 0.26 | 634.54 | 14 | 2 | 2 |
| 11a | 4 | 8 | 1.53 | 544.58 | 12 | 1 | 2 |
| 11b | 4 | 8 | 1.47 | 558.61 | 13 | 1 | 2 |
| Erlotinib | 1 | 6 | 1.89 | 393.44 | 10 | 0 | 0 |
As adopted in Table 4, it is well established that the topological polar surface area (TPSA)38 is a trustworthy indicator of intestinal medication absorption (TPSA less than 140 Å2) and drug penetration through the blood–brain barrier (TPSA less than 60 Å2). All compounds, with the exception of 9–11, satisfy Veber's rule since their computed TPSA values fall within the range that allows them to pass through cell membranes. Additionally, absorption (% ABS) was computed using the formula % ABS = 109 − (0.345 × TPSA). The results showed that the calculated % ABS of all these hits varied from 60.51% to 72.72%, suggesting that these synthetic derivatives—aside from compounds 10 and 11—may have the required bioavailability and cell membrane permeability (Table 4).
| Cpd. no. | TPSA | % ABS |
|---|---|---|
| 5a | 105.16 | 72.72 |
| 5b | 105.16 | 72.72 |
| 6a | 121.95 | 66.93 |
| 6b | 121.95 | 66.93 |
| 7a | 116.16 | 68.92 |
| 7b | 116.16 | 68.92 |
| 8a | 133.98 | 62.78 |
| 8b | 133.98 | 62.78 |
| 9a | 140.55 | 60.51 |
| 9b | 140.55 | 60.51 |
| 10a | 230.01 | 29.65 |
| 10b | 230.01 | 29.65 |
| 11a | 173.34 | 49.20 |
| 11b | 173.34 | 49.20 |
| Erlotinib | 74.73 | 83.22 |
Additionally, the test compounds' medicinal chemistry properties and drug-likeness were predicted using the SwissADME software (Table 5). With the exception of compounds 9–11, all compounds have high GI absorption and high bioavailability ratings (0.55). For the majority of substances, the SwissADME also forecasted no alerts. Furthermore, according to a study by SwissADME, all of the analogues had synthetic accessibility scores ranging from 3.43 to 4.74, meaning that they could all be readily synthesised on a large scale and were on par with the reference drug, erlotinib (3.19).
| Cpd. no. | GI absorption | BBB permeation | P-gp substrate | Bioavailability score | Pains alerts | Synthetic accessibility |
|---|---|---|---|---|---|---|
| 5a | High | No | Yes | 0.55 | 0 | 3.86 |
| 5b | High | No | Yes | 0.55 | 0 | 3.95 |
| 6a | High | No | Yes | 0.55 | 0 | 3.43 |
| 6b | High | No | Yes | 0.55 | 0 | 3.53 |
| 7a | High | No | No | 0.56 | 0 | 3.43 |
| 7b | High | No | No | 0.56 | 0 | 3.52 |
| 8a | High | No | Yes | 0.55 | 0 | 3.57 |
| 8b | High | No | Yes | 0.55 | 0 | 3.67 |
| 9a | Low | No | Yes | 0.17 | 1 | 4.37 |
| 9b | Low | No | Yes | 0.17 | 1 | 4.46 |
| 10a | Low | No | Yes | 0.11 | 0 | 4.63 |
| 10b | Low | No | Yes | 0.11 | 0 | 4.74 |
| 11a | Low | No | Yes | 0.17 | 0 | 4.25 |
| 11b | Low | No | Yes | 0.17 | 0 | 4.35 |
| Erlotinib | High | Yes | No | 0.55 | 0 | 3.19 |
It's interesting to note that our hits, which are systemically targeted compounds, will have little to no central nervous system side effects because they do not penetrate the blood–brain barrier, an advantage over erlotinib. With the exception of compounds 7a and 7b, the majority of our novel compounds are P-gp substrates. All things considered, it was possible to determine that our hits had drug-likeness values and medicinal chemistry criteria that were appropriate, indicating that they might be considered as potential drugs.
The investigation of the binding mode between gefitinib and the EGFR active site showed the crucial hydrogen bond interaction with the important residue Met793 in the hinge region. The moiety of 3-chloro-4-fluoroaniline expanded into the hydrophobic pocket I located at the back of the ATP-binding site forming hydrophobic interactions with the Val726, Lys745, and Leu788 residues. In addition, the quinazoline and the methoxy substituent were in hydrophobic contacts with Leu718, Ala743, and Pro794 residues. On the other hand, the propylmorpholine moiety was extended in the solvent region forming electrostatic and carbon–hydrogen bond interactions with Asp800 (Fig. 6).
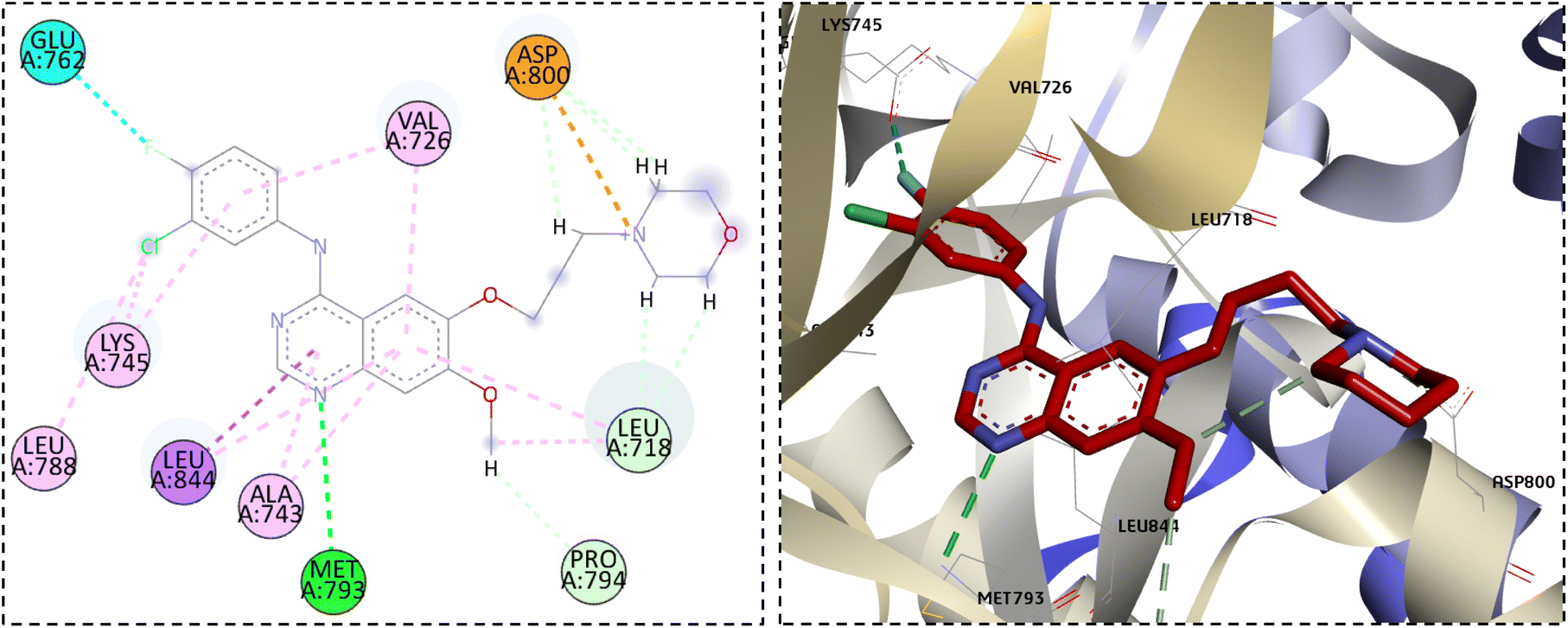 | ||
| Fig. 6 The docked pose of gefitinib in the EGFR active site. | ||
Interestingly, when the new compounds (6a, 6b, 8a, and 11b) docked to EGFR, they produced a favourable binding through a binding pattern close to that of gefitinib, yielding docking scores of −6.85, −7.04, −7.49, and −7.91 kcal mol−1, respectively. The oxadiazole ring of the docked derivatives was oriented in the hinge region, with its oxygen atom acting as a hydrogen bond acceptor for the key amino acid Met793, in addition to hydrophobic interactions with Val726, Ala743, and Leu844 residues. The terminal phenyl group of these compounds was positioned in the back hydrophobic pocket, forming non polar interactions with the Val726 and Lys745 residues, while the phenyl ring at the 5 position of the oxadiazole ring occupied the hydrophobic pocket II, interacting with Leu718 through a Pi-alkyl or Pi-sigma interaction. Finally, the substituted triazolyl moiety expanded near the solvent area (Fig. 7). The triazolylacetamide motif of compound 6a was found to be rotated towards the hydrophobic pocket I, forming carbon–hydrogen bond with Gly719 and a hydrophobic interaction with Val726. As well, the acetohydrazide group of 8a and the N-phenylhydrazine-1-carbothioamide moiety of 11b were rotated close to Asp855 side chain, forming an electrostatic interaction. However, the acetamide group of compound 6b showed a different orientation in which it was extended near Cys797 and Asp800 at the edge of the active site, forming H-bonds with them (Fig. 7). Overlay of the reference drug gefitinib and the new target compounds 6a, 6b, 8a, and 11b in the EGFR active site is shown in Fig. 8.
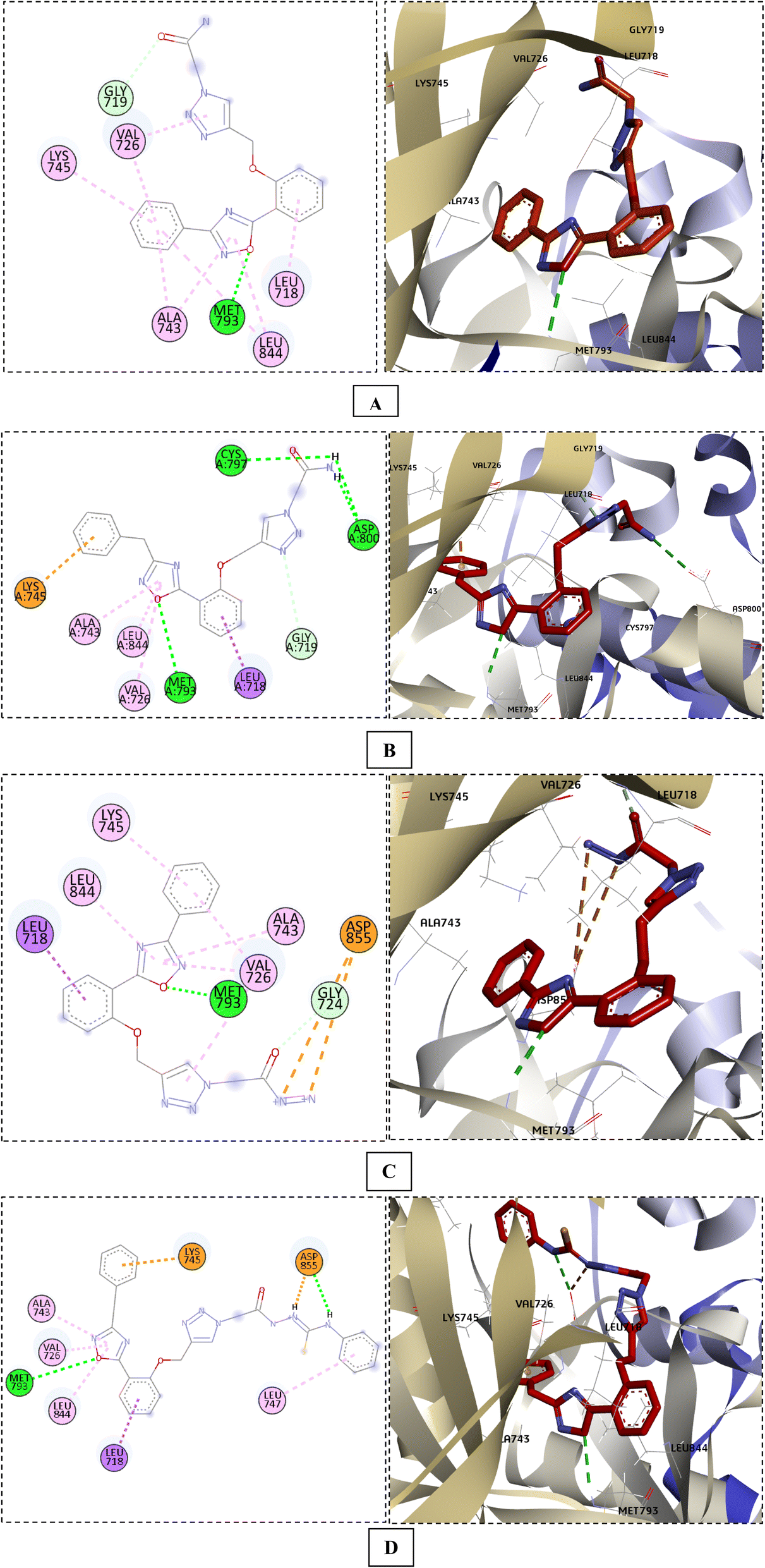 | ||
| Fig. 7 The docking poses of compounds 6a (A), 6b (B), 8a (C), and 11b (D) in the EGFR active site. | ||
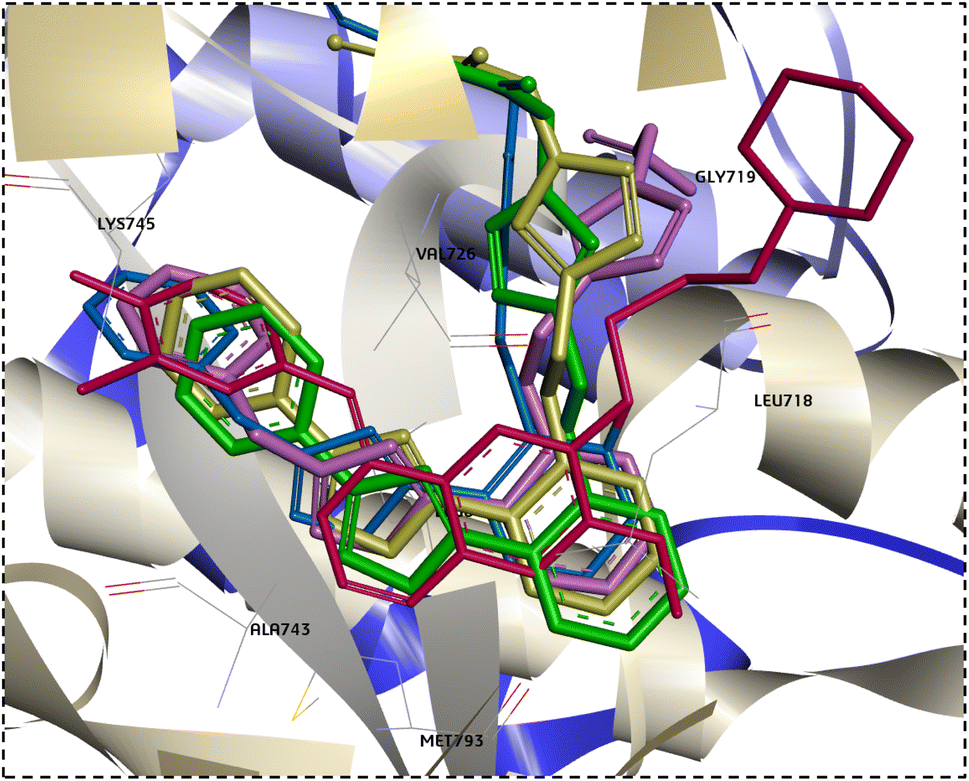 | ||
| Fig. 8 Overlay docking alignment of gefitinib (red), 6a (green), 6b (purple), 8a (yellow), and 11b (blue) in the active site of EGFR. | ||
3. Conclusions
We synthesized and examined the anticancer properties of fourteen 1,2,4-oxadiazole tethered 1,2,3-triazole derivatives. The MTT assay was used to assess the cytotoxicity of these drugs against A549, Caco-2, and WI38 cell lines, with doxorubicin serving as the reference standard. Potent and specific cytotoxicity against lung cancer (A549) and colon cancer (Caco-2) was demonstrated by four compounds: 6a, 6b, 8a, and 11b. It was looked into how they affected the PI3K/mTOR/EGFR pathway. The promising hits showed significant fold changes in the downregulation of EGFR, mTOR, and PI3K, according to qPCR data. Moreover, they upregulate the amount of p53, supporting their mechanism of action. Furthermore, our compounds may be considered safe drug-like candidates based on their physicochemical and drug-likeness findings.It is feasible to conclude from the findings of this investigation that the freshly synthesized compounds have the potential to be a novel class of anticancer drugs for lung and colon tumors, and this warrants further exploration based on the study's findings.
4. Experimental
4.1. Chemistry
:1).
4.1.1.1 Ethyl 2-(4-((2-(3-phenyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetate (5a). To afford 5a as a white solid (72% yield): m.p = 102–104 °C; Rf = 0.53 (EtOAc: n-hexane, 1
:2); IR (KBr) νmax/(cm−1): 1749 (CO, ester). 1H NMR (500 MHz, DMSO-d6) δH: 8.26 (s, 1H, Triazole–H), 8.08–8.02 (m, 3H, Ar–H), 7.67 (t, J = 8.5 Hz, 1H, Ar–H), 7.59–7.51 (m, 4H, Ar–H), 7.18 (t, J = 7.5 Hz, 1H, Ar–H), 5.4 (d, J = 4.5 Hz, 4H, O–CH2, N–CH2), 4.13 (q, 2H, CH2), 1.16 (t, J = 7.0 Hz, 3H, CH3), 13C NMR (125 MHz, DMSO-d6) δC: 175.5, 168.1, 167.7, 157.4, 143.0, 135.3, 132.1, 131.9, 129.8, 127.6, 126.8, 126.6, 121.9, 114.9, 113.2, 62.7, 62.0, 51.0, 14.5. Anal. calcd for C21H19N5O4 (M. wt: 405.41): C, 62.22; H, 4.72; N, 17.27; found: C, 62.43; H, 4.89; N, 17.51.
4.1.1.2 Ethyl 2-(4-((2-(3-benzyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetate (5b). To afford 5b as a white solid (75% yield) m.p = 114–116 °C; Rf = 0.53 (EtOAc: n-hexane, 1
:2); IR (KBr) νmax/(cm−1): 1742 (CO, ester). 1H NMR (500 MHz, DMSO-d6) δH: 8.19 (s, 1H, Triazole–H), 7.92 (d, J = 9.5 Hz, 1H, Ar–H), 7.61 (t, J = 7.5 Hz, 1H, Ar–H), 7.47 (d, J = 8.0 Hz, 1H, Ar–H), 7.33–7.20 (m, 5H, Ar–H), 7.11 (t, J = 7.5 Hz, 1H, Ar–H), 5.37 (d, J = 16 Hz, 4H, N–CH2, O–CH2), 4.13 (q, 4H, CH3–CH2, Ph–CH2), 1.16 (t, 7 Hz, 3H, CH2–CH3), 13C NMR (125 MHz, DMSO-d6) δC: 175.1, 169.7, 167.7, 157.3, 143.0, 136.4, 135.1, 131.8, 129.5, 129.1, 127.4, 126.5, 121.8, 114.8, 113.2, 62.6, 62.0, 50.9, 31.9, 14.5. Anal. calcd for C22H21N5O4 (M.wt: 419.43): C, 63.00; H, 5.05; N, 16.70; found: C, 63.24; H, 5.21; N, 16.93.
4.1.2.1 2-(4-((2-(3-Phenyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (6a). To afford compound 6a as grey solid (76% yield); m.p = 148–150 °C; Rf = 0.32 (ethyl acetate: n-hexane: MeOH, 1
:2: 0.5); IR (KBr): νmax/(cm−1): 3303, 3273 (NH2), 1684 (OCN). 1H NMR (500 MHz, DMSO-d6) δH: 8.19 (s, 1H, Triazole–H), 8.08–8.03 (m, 3H, Ar–H), 7.75 (s, 1H, Ar–H), 7.67 (t, J = 7.5 Hz, 1H, Ar–H), 7.56–7.53 (m, 4H, Ar–H, NH2), 7.39 (s, 1H, Ar–H), 7.18 (t, J = 7.5 Hz, 1H, Ar–H), 5.38 (s, 2H, O–CH2), 5.08 (s, 2H, N–CH2). 13C NMR (125 MHz, DMSO-d6) δC: 175.4, 168.1, 167.8, 157.5, 142.6, 135.3, 132.1, 131.9, 129.8, 127.6, 126.8, 126.7, 121.9, 114.9, 113.2, 62.8, 52.0. Anal. Calcd for C19H16N6O3 (M. wt: 376.38): C, 60.63; H, 4.29; N, 22.33; found: C, 60.91; H, 4.45; N, 22.60.
4.1.2.2 2-(4-((2-(3-Benzyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (6b). To afford 6b as grey solid (60% yield): m.p = 220–222 °C; Rf = 0.28 (ethyl acetate: n-hexane: methanol, 1
:2: 0.5); IR (KBr) νmax/(cm−1): 3318, 3184 (NH2), 1683 (OCN). 1H NMR (500 MHz, DMSO-d6) δH: 8.13 (s, 1H, Triazole-H), 7.92 (d, J = 6.5 Hz, 1H, Ar–H), 7.74 (s, 1H, Ar–H), 7.61 (t, J = 7.5 Hz, 1H, Ar–H), 7.48 (d, J = 9.0 Hz, 1H, Ar–H), 7.39 (s, 1H, Ar–H), 7.33–7.27 (m, 4H, Ar–H, NH2), 7.23–7.20 (m, 1H, Ar–H), 7.10 (t, J = 7.5 Hz, 1H, Ar–H), 5.33 (s, 2H, N–CH2), 5.06 (s, 2H, O–CH2), 4.11 (S, 2H, Ph–CH2). 13C NMR (125 MHz, DMSO-d6) δC: 175.1, 169.7, 167.8, 157.3, 142.6, 136.4, 135.1, 131.8, 129.5, 129.1, 127.4, 126.7, 121.8, 114.9, 113.2, 62.7, 52.0, 31.9. Anal. Calcd for C20H18N6O3 (M.wt: 390.40): C, 61.53; H, 4.65; N, 21.53; found: C, 61.80; H, 4.73; N, 21.68.
4.1.3.1 2-(4-((2-(3-Phenyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetic acid (7a). As off-white powder (85% yield); m.p= 198–200 °C Rf = 0.53 (EtOAc: n-hexane, 1
:2); IR (KBr) νmax/(cm−1): 3031 (OH, carboxyl), 1723 (CO, carboxyl). 1H NMR (500 MHz, DMSO-d6) δH: 13.42 (bs, 1H, OH), 8.24 (s, 1H, Triazole–H), 8.08–8.02 (m, 3H, Ar–H), 7.66 (t, J = 8.0 Hz, 1H, Ar–H), 7.57–7.51 (m, 4H, Ar–H), 7.17 (t, J = 7.5 Hz, 1H, Ar–H), 5.39 (s, 2H, O–CH2), 5.29 (s, 2H, N–CH2) 13C NMR (125 MHz, DMSO-d6) δC: 175.4, 169.2, 168.1, 157.4, 142.9, 135.3, 132.1, 131.9, 129.8, 127.6, 126.8, 126.5, 121.9, 114.9, 113.2, 62.8, 51.1. Anal. Calcd for C19H15N5O4 (M. wt: 377.35): C, 60.47; H, 4.01; N, 18.56; found: C, 60.70; H, 4.25; N, 18.82.
4.1.3.2 2-(4-((2-(3-Benzyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetic acid (7b). As off-white powder (80% yield); m.p = 175–177 °C; Rf = 0.53 (EtOAc: n-hexane, 1
:2); IR (KBr) νmax/(cm−1): 3456 (OH, carboxyl), 1735 (CO, carboxyl). 1H NMR (500 MHz, DMSO-d6) δH: 13.42 (bs, 1H, OH), 8.17 (s, 1H, Triazole–H), 7.92 (d, J = 9.5 Hz, 1H, Ar–H), 7.61 (t, J = 7.0 Hz, 1H, Ar–H), 7.48 (d, J = 8.0 Hz, 1H, Ar–H), 7.30–7.27 (m, 4H, Ar–H), 7.23–7.20 (m, 1H, Ar–H), 7.11 (t, J = 7.5 Hz, 1H, Ar–H), 5.34 (s, 2H, O–CH2), 5.27 (s, 2H, N–CH2), 4.11 (s, 2H, Ph–CH2), 13C NMR (125 MHz, DMSO-d6) δC: 175.1, 169.7, 169.1, 157.3, 142.9, 136.4, 135.1, 131.8, 129.5, 129.1, 127.4, 126.5, 121.8, 114.9, 113.2, 62.7, 51.1, 31.9. Anal. Calcd for C20H17N5O4 (M. wt: 391.38): C, 61.38; H, 4.38; N, 17.89; found: C, 61.21; H, 4.49; N, 18.03.
4.1.4.1 2-(4-((2-(3-Phenyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetohydrazide (8a). To give 8a off-white solid (64% yield): m.p = 194–196 °C; Rf = 0.53 (ethyl acetate: n-hexane, 3
:1); IR (KBr) νmax/(cm−1): 3330, 3307 (NH2), 3248 (NH), 1653 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 9.52 (bs, 1H, NH), 8.22 (s, 1H, Triazole–H), 8.08–8.03 (m, 3H, Ar–H), 7.67 (t, J = 7.5 Hz, 1H, Ar–H), 7.56–7.52 (m, 4H, Ar–H), 7.17 (t, J = 8.0 Hz, 1H, Ar–H), 5.38 (s, 2H, N–CH2), 5.04 (s, 2H, O–CH2), 4.56 (s, 1H, NH), 4.34 (bs, 1H, NH). 13C NMR (125 MHz, DMSO-d6) δC: 175.5, 168.1, 157.5, 142.7, 135.3, 132.1, 131.9, 129.8, 127.6, 126.9, 126.8, 126.6, 121.8, 114.9, 113.2, 62.7, 51.0. Anal. Calcd for C19H17N7O3 (M. wt: 391.39): C, 58.31; H, 4.38; N, 25.05; found: C, 58.47; H, 4.50; N, 24.97.
4.1.4.2 2-(4-((2-(3-Benzyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetohydrazide (8b). To give 8b white solid (72% yield): m.p = 223–225 °C; Rf = 0.53 (EtOAc: n-hexane, 1
:2); IR (KBr) νmax/(cm−1): 3338, 3285 (NH2), 3139 (NH), 1653 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 9.53 (bs, 1H, NH), 8.17 (s, 1H, Triazole–H), 7.92 (s, 1H, Ar–H), 7.61 (s, 1H, Ar–H), 7.48 (s, 1H, Ar–H), 7.40–7.20 (m, 5H, Ar–H), 7.10 (s, 1H, Ar–H), 5.33 (s, 2H, N–CH2), 5.03 (s, 2H, O–CH2), 4.55 (s, 1H, NH), 4.35 (s, 1H, NH), 4.11 (s, 2H, Ph–CH2). 13C NMR (125 MHz, DMSO-d6) δC: 175.1, 169.7, 157.3, 142.7, 136.4, 135.1, 131.8, 129.5, 129.1, 127.4, 126.5, 121.8, 114.8, 113.2, 62.6, 51.0, 50.7, 31.9. Anal. Calcd for C20H19N7O3 (M. wt: 405.42): C, 59.25; H, 4.72; N, 24.18; found: C, 59.53; H, 4.91; N, 24.43.
4.1.5.1 N′-(2-Hydroxybenzylidene)-2-(4-((2-(3-phenyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetohydrazide (9a). Recrystallized from ethanol to form white solid (78% yield); m.p = 212–214 °C; Rf = 0.53 (Ethyl Acetate: n-hexane, 3
:1); IR (KBr) νmax/(cm−1): 3550 (OH), 3480 (NH), 1684 (OCN), 1604 (CN). 1H NMR (500 MHz, DMSO-d6) δH: 11.76 (s, 1H, NH), 10.12 (s, 1H, OH), 8.44–8.21 (m, 2H, NCH, Ar–H), 8.09–8.03 (m, 3H, Triazole–H, Ar–H), 7.75 (d, J = 7.5 Hz, 1H, Ar–H), 7.67 (t, J = 7.5 Hz, 1H, Ar–H), 7.55 (s, 4H, Ar–H), 7.27–7.17 (m, 2H, Ar–H), 6.88–6.81 (m, 2H, Ar–H), 5.70 (s, 1H, N–CH), 5.50–5.29 (m, 3H, N–CH, O–CH2). 13C NMR (125 MHz, DMSO-d6) δC: 175.4, 168.1, 167.6, 157.5, 157.0, 142.1, 135.3, 132.2, 132.0, 131.9, 129.8, 127.6, 126.9, 126.8, 126.7, 121.9, 120.5, 119.9, 116.9, 116.7, 114.9, 113.2, 62.9, 51.2. Anal. Calcd for C26H21N7O4 (M. wt: 495.49): C, 63.02; H, 4.27; N, 19.79; found: C, 62.96; H, 4.41; N, 20.05.
4.1.5.2 2-(4-((2-(3-Benzyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N'-(2-hydroxybenzylidene)acetohydrazide (9b). Recrystallized from ethanol to form white solid (73% yield); m.p = 178–180 °C; Rf = 0.53 (ethyl acetate: n-hexane, 3
:1); IR (KBr) νmax/(cm−1): 3560 (OH), 3493 (NH), 1682 (OCN), 1599 (CN). 1H NMR (500 MHz, DMSO-d6) δH: 11.76 (s, 1H, NH), 10.10 (s, 1H, OH), 8.43–8.14 (m, 2H, Triazole–H, NCH), 7.92 (d, J = 7.0 Hz, 1H, Ar–H), 7.74 (d, J = 7.5 Hz, 1H, Ar–H), 7.62 (t, J = 7.5 Hz, 1H, Ar–H), 7.55–7.49 (m, 1H, Ar–H), 7.30–7.21 (m, 6H, Ar–H), 7.11 (t, J = 10.0 Hz, 1H, Ar–H), 6.88–6.81 (m, 2H, Ar–H), 5.67 (s, 1H, O–CH), 5.48–5.27 (m, 3H, O–CH, N–CH2), 4.11 (s, 2H, Ph–CH2). 13C NMR (125 MHz, DMSO-d6) δC: 175.1, 169.7, 167.6, 157.8, 157.3, 156.9, 142.3, 136.4, 135.2, 132.0, 131.8, 129.4, 129.1, 127.4, 121.8, 120.5, 120.0, 116.9, 116.7, 113.2, 62.7, 62.6, 51.3, 51.1, 31.9. Anal. Calcd for C27H23N7O4 (M. wt: 509.52): C, 63.65; H, 4.55; N, 19.24; found: C, 63.89; H, 4.67; N, 19.48.
4.1.5.3 Sodium-(6-hydroxy-5-methyl-4-((2-(2-(4-((2-(3-phenyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetyl)hydrazonol)methyl)pyridin-3-yl)methyl hydrogen phosphate (10a). To give compound 10a as brown solid (87% yield), m.p = 218–220 °C; Rf = 0.53 (EtOAc: n-hexane, 1
:2); IR (KBr) νmax/(cm−1): 3410 (OH), 1705 (OCN). 1H NMR (500 MHz, DMSO-d6)δH: 14.35 (bs, 1H, Pyridine–OH), 12.15 (bs, 1H, CO–NH), 8.91 (s, 1H, NC–H), 8.35 (s, 1H, Triazole–H), 8.08–7.84 (m, 4H, Ar–H), 7.67 (t, J = 7.0 Hz, 1H, Ar–H), 7.53 (s, 4H, Ar–H), 7.17 (t, J = 7.5 Hz, 1H, Ar–H), 5.48 (s, 2H, N–CH2), 5.39 (s, 2H, PO–CH2), 4.83 (s, 2H, O–CH2), 2.46 (s, 3H, Pyridine–CH3). 13C NMR (125 MHz, DMSO-d6) δC: 175.3, 168.1, 167.2, 163.7, 163.6, 157.5, 157.4, 148.8, 148.6, 147.4, 142.8, 135.3, 132.0, 131.8, 129.8, 127.6, 126.8, 121.8, 121.0, 114.9, 113.2, 62.8, 62.2, 51.3, 19.4. Anal. Calcd for C27H24N8NaO8P (M. wt: 642.49): C, 50.47; H, 3.77; N, 17.44; found: C, 50.54; H, 3.96; N, 17.72.
4.1.5.4 Sodium-(4-((2-(2-(4-((2-(3-benzyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetyl)hydrazineylidene)methyl)-6-hydroxy-5-methylpyridin-3-yl)methyl hydrogen phosphate (10b). To give compound 10b as brown solid (61% yield), m.p = 210–212 °C; Rf = 0.53 (EtOAc: n-hexane, 1
:2); IR (KBr) νmax/(cm−1): 3411 (OH), 1707 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 14.35 (bs, 1H, Pyridine–OH), 12.16 (bs, 1H, CO–NH), 8.91 (s, 1H, NC–H), 8.28 (s, 1H, Triazole–H), 7.91 (d, J = 7.5 Hz, 2H, Ar–H), 7.61 (t, J = 7.0 Hz, 1H, Ar–H), 7.48 (d, J = 8.5 Hz, 1H, Ar–H), 7.29–7.19 (m, 5H, Ar–H), 7.10 (t, J = 8.0 Hz, 1H, Ar–H), 5.47 (s, 2H, N–CH2), 5.34 (s, 2H, OP–CH2), 4.83 (s, 2H, O–CH2), 4.11 (s, 1H, OP–OH), 2.46 (s, 3H, pyridine–CH3), 2.35 (s, 2H, Ph–CH2). 13C NMR (125 MHz, DMSO-d6) δC: 175.0, 172.9, 169.7, 163.6, 157.3, 148.9, 147.4, 142.8, 141.4, 139.7, 139.3, 136.4, 135.1, 131.7, 129.5, 129.1, 127.3, 126.8, 121.7, 121.0, 115.1, 114.9, 113.3, 62.8, 51.3, 31.9. Anal. Calcd for C28H26N8NaO8P (M. wt: 656.52): C, 51.23; H, 3.99; N, 17.07; found: C, 51.52; H, 4.15; N, 17.34.
4.1.6.1 N-Phenyl-2-(2-(4-((2-(3-phenyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetyl)hydrazine-1-carbothioamide (11a). 11a white solid (80% yield); m.p = 115–117 °C; Rf = 0.53 (EtOAc: n-hexane, 1
:2); IR (KBr) νmax/(cm−1): 3216 (NH), 1688 (OCN), 744 (CS); 1H NMR (500 MHz, DMSO-d6) δH: 11.04 (s, 1H, N–H), 9.80 (s, 1H, NH), 8.30 (s, 1H, Triazole–H), 8.08–8.03 (m, 2H, Ar–H), 7.69–7.53 (m, 4H, CO–NH, Ar–H), 7.37–7.30 (m, 6H, Ar–H), 7.19–7.10 (m, 3H, Ar–H), 5.40, 5.24 (2s, 2H, N–CH2), 4.84 (s, 2H, O–CH2).13C NMR (125 MHz, DMSO-d6) δC: 175.5, 168.2, 168.1, 157.5, 142.8, 139.1, 139.0, 135.3, 132.1, 131.9, 129.9, 129.4, 129.0, 128.7, 127.6, 126.8, 122.2, 121.9, 114.9, 113.2, 67.8, 65.7, 62.7, 51.1. Anal. Calcd for C26H22N8O3S (M. wt: 526.57): C, 59.31; H, 4.21; N, 21.28; found: C, 59.52; H, 4.13; N, 21.43.
4.1.6.2 2-(2-(4-((2-(3-Benzyl-1,2,4-oxadiazol-5-yl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetyl)-N-phenylhydrazine-1-carbothioamide (11b). 11b (76% yield); m.p = 95–97 °C; Rf = 0.53 (EtOAc: n-hexane, 1
:2); IR (KBr) νmax/(cm−1): 3216 (NH), 1596 (OCN), 741 (CS). 1H NMR (500 MHz, DMSO-d6) δH: 11.0 (s, 3H, 3NH), 7.60 (bs, 2H, Triazole–H, Ar–H), 7.30 (s, 10H, Ar–H), 7.09 (bs, 3H, Ar–H), 4.48 (s, 6H, Ph–CH2, N–CH2, O–CH2). 13C NMR (125 MHz, DMSO-d6) δC: 188.4, 187.7, 139.2, 139.1, 139.0, 138.9, 138.6, 138.4, 138.3, 129.4, 129.0, 125.6, 125.1, 123.6, 122.2, 67.8, 65.7, 14.5. Anal. Calcd for C27H24N8O3S (M. wt: 540.60): C, 59.99; H, 4.47; N, 20.73; S, 5.93; found: C, 60.12; H, 4.45; N, 20.44.
4.2. Biological activity
| PI3K | Forward: 5′-CTG CCTGCG ACAGATGAG TG-3 |
| Reverse: 5′-TCCGAT TACCAAGTG CTC TTTC-3 | |
| mTOR | Forward: 5′-ACA ACT TTG GTATCG TGG AAGG-3′ |
| Reverse: 5′-GCC ATC ACG CCACAG TTTC-3′ | |
| EGFR | Forward: 5′-TGGAGC TACGGG GTGACCGT-3 |
| Reverse: 5′-GGT TCAGAG GCT GAT TGT GAT-3 | |
| p53 | Forward: CCTCAGCATCTTATCCGAGTGG |
| Reverse: TGGATGGTGGTACAGTCAGAGC | |
| B-actin | Forward: 5′-TCA AGA AGG TGG TGA AGC AGG-3′ |
| Reverse: 5′-AGCGTC AAAGGTGGAGGAGTG-3′ |
Conflicts of interest
The authors declare that none of their relationships or conflicting financial interests could have influenced the findings of this study.Acknowledgements
“MSA acknowledge the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia for financial support under the annual funding track [Project No. GrantA316]”.References
- S. Jin, Y. Sun, X. Liang, X. Gu, J. Ning, Y. Xu, S. Chen and L. Pan, Emerging new therapeutic antibody derivatives for cancer treatment, Signal Transduction Targeted Ther., 2022, 7, 39 CrossRef CAS PubMed.
- S. Sigismund, D. Avanzato and L. Lanzetti, Emerging functions of the EGFR in cancer, Mol. Oncol., 2018, 12, 3–20 CrossRef PubMed.
- J.-P. Spano, C. Lagorce, D. Atlan, G. Milano, J. Domont, R. Benamouzig, A. Attar, J. Benichou, A. Martin and J.-F. Morere, Impact of EGFR expression on colorectal cancer patient prognosis and survival, Ann. Oncol., 2005, 16, 102–108 CrossRef.
- W. Pao and J. Chmielecki, Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer, Nat. Rev. Cancer, 2010, 10, 760–774 CrossRef CAS PubMed.
- S. J. Rogers, K. J. Harrington, P. Rhys-Evans, P. O-Charoenrat and S. A. Eccles, Biological significance of c-erb B family oncogenes in head and neck cancer, Cancer Metastasis Rev., 2005, 24, 47–69 CrossRef CAS PubMed.
- J. C. Lee, I. Vivanco, R. Beroukhim, J. H. Y. Huang, W. L. Feng, R. M. DeBiasi, K. Yoshimoto, J. C. King, P. Nghiemphu and Y. Yuza, Epidermal growth factor receptor activation in glioblastoma through novel missense mutations in the extracellular domain, PLoS Med., 2006, 3, e485 CrossRef PubMed.
- M. Oliveira-Cunha, W. G. Newman and A. K. Siriwardena, Epidermal growth factor receptor in pancreatic cancer, Cancers, 2011, 3, 1513–1526 CrossRef CAS PubMed.
- H.-W. Lo, S.-C. Hsu and M.-C. Hung, EGFR signaling pathway in breast cancers: from traditional signal transduction to direct nuclear translocalization, Breast Cancer Res. Treat., 2006, 95, 211–218 CrossRef CAS PubMed.
- S. Yan, H. Wu, Y. Zhu, S. Shen, K. Zhang, W. Wu and N. Liu, FGFC1 selectively inhibits erlotinib-resistant non-small cell lung cancer via elevation of ROS mediated by the EGFR/PI3K/Akt/mTOR pathway, Front. Pharmacol, 2022, 12, 764699 CrossRef PubMed.
- R. B. Cohen, Epidermal growth factor receptor as a therapeutic target in colorectal cancer, Clin. Colorectal Cancer, 2003, 2, 246–251 CrossRef CAS.
- B. Pabla, M. Bissonnette and V. J. Konda, Colon cancer and the epidermal growth factor receptor: Current treatment paradigms, the importance of diet, and the role of chemoprevention, World J. Clin. Oncol., 2015, 6, 133 CrossRef.
- Y. He, M. M. Sun, G. G. Zhang, J. Yang, K. S. Chen, W. W. Xu and B. Li, Targeting PI3K/Akt signal transduction for cancer therapy, Signal Transduct. Targeted Ther., 2021, 6, 425 CrossRef CAS.
- R. Liu, Y. Chen, G. Liu, C. Li, Y. Song, Z. Cao, W. Li, J. Hu, C. Lu and Y. Liu, PI3K/AKT pathway as a key link modulates the multidrug resistance of cancers, Cell Death Dis., 2020, 11, 797 CrossRef CAS.
- Y. Peng, Y. Wang, C. Zhou, W. Mei and C. Zeng, PI3K/Akt/mTOR pathway and its role in cancer therapeutics: are we making headway?, Front. Oncol., 2022, 12, 819128 CrossRef CAS PubMed.
- L. Yu; J. Wei and P. Liu Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer, in Proceedings of the Seminars in Cancer Biology, 2022, pp. 69–94 Search PubMed.
- J. Bang, M. Jun, S. Lee, H. Moon and S. W. Ro, Targeting EGFR/PI3K/AKT/mTOR signaling in hepatocellular carcinoma, Pharmaceutics, 2023, 15, 2130 CrossRef CAS PubMed.
- M. Colardo, M. Segatto and S. Di Bartolomeo, Targeting RTK-PI3K-mTOR axis in gliomas: An update, Int. J. Mol. Sci., 2021, 22, 4899 CrossRef CAS PubMed.
- A. J. Levine and A. M. Puzio-Kuter, The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes, Science, 2010, 330, 1340–1344 CrossRef CAS.
- M. M. Alam, 1, 2, 3-Triazole hybrids as anticancer agents: a review, Arch. Pharm., 2022, 355, 2100158 CrossRef CAS PubMed.
- G. Mohan, G. Sridhar, E. Laxminarayana and M. T. Chary, Synthesis and biological evaluation of 1, 2, 4-oxadiazole incorporated 1, 2, 3-triazole-pyrazole derivatives as anticancer agents, Chem. Data Collect., 2021, 34, 100735 CrossRef.
- K. Bozorov, J. Zhao and H. A. Aisa, 1, 2, 3-Triazole-containing hybrids as leads in medicinal chemistry: A recent overview, Bioorg. Med. Chem., 2019, 27, 3511–3531 CrossRef CAS.
- M. A. Mahmoud, A. F. Mohammed, O. I. Salem, H. A. Gomaa and B. G. Youssif, New 1, 3, 4-oxadiazoles linked with the 1, 2, 3-triazole moiety as antiproliferative agents targeting the EGFR tyrosine kinase, Arch. Pharm., 2022, 355, 2200009 CrossRef CAS.
- S. K. Verma, R. Verma, K. S. S. Kumar, L. Banjare, A. B. Shaik, R. R. Bhandare, K. P. Rakesh and K. S. Rangappa, A key review on oxadiazole analogs as potential methicillin-resistant Staphylococcus aureus (MRSA) activity: Structure-activity relationship studies, Eur. J. Med. Chem., 2021, 219, 113442 CrossRef CAS PubMed.
- S. K. Verma, R. Verma, Y. R. Girish, S. Verma, K. Pramoda, Y. Vaishnav, J. Saji and K. S. S. Kumar, Two-dimensional Ti3C2Tx MXenes as a catalyst support for the synthesis of 1, 4-disubstituted-1, 2, 3-triazoles via azide-nitroalkene oxidative cycloaddition, J. Mol. Struct., 2023, 1281, 135145 CrossRef CAS.
- Y. Hu, Z. Liu, G. Zha, S. Long, M. Sridhara, K. S. S. Kumar and K. P. Rakesh, Triazole derivatives as potential antifungal agents: A structure-activity relationship (SAR) studies, Process Biochem., 2023, 102–118 CrossRef CAS.
- S. Mandal, S. Mandal, S. Biswas, S. Banerjee and B. Saha, Synthesis of 2-(ethynyloxy) naphthaene-1-carbaldehyde using 2-hydroxy benzyl alcohol and propargyl bromide in aqueous micellar media, Res. Chem. Intermed., 2018, 44, 2169–2177 CrossRef CAS.
- M. S. Singh, S. Chowdhury and S. Koley, Advances of azide-alkyne cycloaddition-click chemistry over the recent decade, Tetrahedron, 2016, 72, 5257–5283 CrossRef CAS.
- A. Amadasi, M. Bertoldi, R. Contestabile, S. Bettati, B. Cellini, M. Luigi di Salvo, C. Borri-Voltattorni, F. Bossa and A. Mozzarelli, Pyridoxal 5'-phosphate enzymes as targets for therapeutic agents, Curr. Med. Chem., 2007, 14, 1291–1324 CrossRef CAS PubMed.
- A. Bolje, D. Urankar and J. Košmrlj, Synthesis and NMR Analysis of 1, 4-Disubstituted 1, 2, 3-Triazoles Tethered to Pyridine, Pyrimidine, and Pyrazine Rings, Eur. J. Org Chem., 2014, 2014, 8167–8181 CrossRef CAS.
- F. M. Freimoser, C. A. Jakob, M. Aebi and U. Tuor, The MTT [3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide] assay is a fast and reliable method for colorimetric determination of fungal cell densities, Appl. Environ. Microbiol., 1999, 65, 3727–3729 CrossRef CAS.
- M. El-Zeftawy, D. Ghareeb, E. R. ElBealy, R. Saad, S. Mahmoud, N. Elguindy, A. F. El-kott and M. El-Sayed, Berberine chloride ameliorated PI3K/Akt-p/SIRT-1/PTEN signaling pathway in insulin resistance syndrome induced in rats, J. Food Biochem., 2019, 43, e13049 CrossRef PubMed.
- D. A. Guertin and D. M. Sabatini, Defining the role of mTOR in cancer, Cancer Cell, 2007, 12, 9–22 CrossRef CAS.
- G. Lurje and H.-J. Lenz, EGFR signaling and drug discovery, Oncology, 2010, 77, 400–410 CrossRef.
- J. T. Zilfou and S. W. Lowe, Tumor suppressive functions of p53, Cold Spring Harbor Perspect. Biol., 2009, 1, a001883 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules, Sci. Rep., 2017, 7, 42717 CrossRef PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings, Adv. Drug Delivery Rev., 1997, 23, 3–25 CrossRef CAS.
- D. F. Veber, S. R. Johnson, H.-Y. Cheng, B. R. Smith, K. W. Ward and K. D. Kopple, Molecular properties that influence the oral bioavailability of drug candidates, J. Med. Chem., 2002, 45, 2615–2623 CrossRef CAS PubMed.
- M. Maximo da Silva, M. Comin, T. Santos Duarte, M. A. Foglio, J. E. De Carvalho, M. do Carmo Vieira and F. A. S. Nazari, Synthesis, antiproliferative activity and molecular properties predictions of galloyl derivatives, Molecules, 2015, 20, 5360–5373 CrossRef CAS PubMed.
- E. Ostertagová and O. Ostertag, Methodology and Application of One-way ANOVA, Am. J. Mech. Eng., 2013, 1, 256–261 Search PubMed.
- T. Mosmann, Rapid colorimetric assay for cellular growth and survival: application to proliferation and cytotoxicity assays, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS.
- C. Scholz, S. Knorr, K. Hamacher and B. Schmidt, DOCKTITE A Highly Versatile Step-by-Step Workflow for Covalent Docking and Virtual Screening in the Molecular Operating Environment, J. Chem. Inf. Model., 2015, 55, 398–406 CrossRef CAS PubMed.
- S. Vilar, G. Cozza and S. Moro, Medicinal chemistry and the molecular operating environment (MOE): application of QSAR and molecular docking to drug discovery, Curr. Top. Med. Chem., 2008, 8, 1555–1572 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02222j |
| This journal is © The Royal Society of Chemistry 2024 |