
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotodissociation of leucine-enkephalin protonated peptide: an experimental and theoretical perspective†‡
Lara Martínez-Fernández a,
Miloš Lj. Ranković§
b,
Francis Canonc,
Laurent Nahonc,
Alexandre Giulianicd,
Aleksandar R. Milosavljević*c and
Ana Martin-Somer*e
a,
Miloš Lj. Ranković§
b,
Francis Canonc,
Laurent Nahonc,
Alexandre Giulianicd,
Aleksandar R. Milosavljević*c and
Ana Martin-Somer*e
aDepartamento de Química Física de Materiales, Instituto de Química Física de Materiales, Instituto de Química Física Blas Cabrera, CSIC, 28006, Madrid, Spain
bInstitute of Physics Belgrade, University of Belgrade, Pregrevica 118, 11080 Belgrade, Serbia
cSOLEIL, l'Orme des Merisiers, St Aubin, BP48, F-91192 Gif sur Yvette Cedex, France
dINRAE, Dpet. Transform, UAR1008, Rue de la Géraudière, BP 71627, F-44316 Nantes, France
eDepartamento de Química Física Aplicada, Universidad Autónoma de Madrid, Módulo 14, 28049, Spain. E-mail: ana.somer@uam.es
First published on 23rd May 2024
Abstract
Understanding the competing processes that govern far ultraviolet photodissociation (FUV-PD) of biopolymers such as proteins is a challenge. Here, we report a combined experimental and theoretical investigation of FUV-PD of protonated leucine-enkephalin pentapeptide ([YGGFL + H]+) in the gas-phase. Time-dependent density functional theory (TD-DFT) calculations in combination with experiments and previous results for amino acids and shorter peptides help in rationalizing the evolution of the excited states. The results confirm that fragmentation of [YGGFL + H]+ results mainly from vibrationally excited species in the ground electronic state, populated after internal conversion. We also propose fragmentation mechanisms for specific photo-fragments such as tyrosine side chain loss (with an extra hydrogen) or hydrogen loss. In general, we observe the same mechanisms as for smaller peptides or protonated Tyr and Phe, that are not quenched by the presence of other amino acids. Nevertheless, we also found some differences, as for H loss, in part due to the fact that the charge is solvated by the peptide chain and not only by the COOH terminal group.
1. Introduction
The interaction of biomolecules with ultraviolet (UV) light is of paramount interest due to the pivotal role that such interactions play in fundamental biological processes,1 ranging from DNA stability and repair2–5 to protein function,6–12 cellular signaling,13,14 photosynthesis,15,16 neurotransmission,17 immune response,18,19 or cell cycle regulation.20In addition, the interaction of peptides with UV light has significant implications in many other fields such as the study of the origin of life,21,22 photodegradation of industrially important polymeric materials23,24 or photodegradation of biopharmaceuticals that can be exposed to UV light during their manufacturing and administration process.25 Moreover, it is relevant in applications where the spectroscopic properties of the amino acid residues are applied. In this regard, the study of peptide-UV interactions becomes particularly significant in the context of mass spectrometry, where UV-induced fragmentation (UVPD, ultraviolet photodissociation) has attracted significant attention in the past years, since it is a proven and a powerful tool for structural elucidation, efficient for both small peptides and intact proteins with masses up to tens of thousands of Daltons.26,27 Investigating UV-induced processes in gas-phase peptides allows assessing their intrinsic properties without the effects due to solvent, allowing to unravel the complexities of peptide electronic and vibrational dynamics.28 It has also been suggested that gas-phase structure might be more appropriate to reproduce the biologically active geometry of peptide ligands in a hydrophobic environment than solution structures.29
For these reasons, ultraviolet and vacuum ultraviolet photodissociation as well as photoinduced dynamics of peptides have been extensively investigated in the past using both synchrotron radiation (SR) and lasers as well as from a theoretical point of view (see ref. 8–12, 28, 30–38 and references therein). In the far UV photon energy range (220–120 nm, or c.a. 5.5–10 eV) and below the ionization energy (IE), photodissociation is triggered by resonant electronic excitation of the target system, followed by competing relaxation pathways, such that UVPD can take place (i) directly from the excited state, or (ii) upon either radiative deexcitation or internal conversion (IC) into the vibrationally excited ground state (GS) where the ion subsequently fragments. Furthermore, there are two possible options prior to fragmentation in the GS: (a) the energy is redistributed statistically over many vibrational modes (intramolecular vibrational energy redistribution, IVR39) or it mostly remains in the vibrational modes of the excited bonds.40 This highlights the complexity of peptide photofragmentation that is a combination of various competing processes.
Thus, a detailed description and understanding of the photofragmentation mechanisms offers valuable insights into the underlying photophysics of the deactivation process. However, unraveling these processes for a relatively large, isolated polypeptide remains challenging especially from a theoretical point of view28 considering the large number of degrees of freedom associated with the size and floppiness of biopolymers. Some groups have studied deactivation mechanisms in proteins employing hybrid quantum mechanics/molecular mechanics (QM/MM) approaches where only a small region is treated at TD-DFT or higher theory level.8–12 On the other hand, excited state relaxation mechanisms have been investigated in more detail for smaller systems such as N,N-dimethylformamide41,42 (smallest molecule with a peptide bond); isolated neutral and protonated amino acids43 (and references therein), related molecules such as substituted phenols,44 iodotyrosine derivatives,45 protonated adrenaline,46 and small peptides such as dipeptides31,38 tripeptides,38,47 or phosphopeptides.37 However, a comprehensive understanding of these processes for larger peptides has remained elusive.
In this context, we present a pioneering combined experimental and theoretical study on the resonant UV photodissociation of a gas-phase protonated pentapeptide: leucine-enkephalin (LeuEnk or YGGFL). LeuEnk is an endogenous opioid peptide found in the brain of animals, including humans, that acts as neurotransmitter. Among other functions, it plays a crucial role in pain modulation and nociception regulation in the body. LeuEnk has become a mass spectrometry standard,48 and there is a plethora of papers having studied its fragmentation induced by different activation techniques, including collisional induced dissociation (CID)48 and UV/VUV-induced fragmentation.33,34,49–51 LeuEnk is a large enough molecule to be a good model for biologically important peptides and at the same time small enough (MH+ = 556 Da) to be studied from a theoretical point of view.
In this article we aim to describe the intricate mechanisms involved in the deactivation processes at play at the molecular level in protonated LeuEnk after far ultraviolet (FUV) activation. To achieve this, we registered the FUV photodissociation mass spectra and we conducted a thorough TD-DFT study (CAM-B3LYP/cc-pVDZ). In particular, we have characterized the lowest-lying excited states in terms of nature, energies and intensities in the Franck–Condon (FC) region for [YGGFL + H]+ precursor ion and, subsequently, mapped their possible relaxation mechanisms.
By integrating our findings with previous results on the excited state relaxation mechanisms of neutral and protonated Tyr and Phe amino acids, as well as protonated small peptides, we propose several deactivation pathways in competition. Some of these pathways lead to specific photofragments (such as H loss and Y + H side chain loss) not observed in CID experiments. This study contributes a unique perspective to the field, shedding light on the photochemical behavior of a biologically significant peptide in the gas phase under resonant FUV conditions.
2. Methods
2.1. Experimental
Singly protonated ions of bare LeuEnk were formed from an aqueous solution of the peptide using electrospray ionization (ESI). Mass and charge selected precursor was isolated and stored in a linear ion trap. The precursor ions were then irradiated by tunable synchrotron radiation (SR) in the 5.5–8.5 eV (c.a. 225–146 nm) photon energy range,52,53 which is below the IE of protonatedLeuEnk reported at 8.9 eV.33 Resonant photoexcitation of the target led to an increased ionic fragmentation versus non-resonant photon energy domain, which was recorded by mass spectrometry as a function of photon energy, i.e. by action spectroscopy.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :25 v/v) solution with LeuEnk molecules from Sigma Aldrich at a concentration of 10 μM was prepared and introduced into nano ESI as such, without further purification. A turbo pumping vacuum stage was used to accommodate pressure differences between beamline (10−8 mbar) and LTQ spectrometer (10−5 mbar). The pressure inside the ion trap is about 10−3 mbar due to helium buffer gas that is used to cool the ions and improve trapping. A home built supporting frame for LTQ allows for fine tuning the ion trap axis with respect to the photon beam, in order to obtain high signal to noise ratio. A mechanical shutter was designed and placed in the assembly with the vacuum stage to control the irradiation time.55 Precursor cations, [YGGFL + H]+, were isolated in the ion trap and subjected to VUV monochromatic photon beam during 500 ms. The tandem mass spectra (MS2) were recorded as a function of the photon energy scanned in small steps of 0.2 eV from 5.7 eV to 8.5 eV. After the photon activation, ions were ejected and detected by two electron multiplier detectors with conversion dynodes (integral part of LTQ). Fragment ion yields, defined as the area under the peaks were extracted from recorded tandem mass spectra and normalized to the photon flux and total ionic current (or precursor ion intensity). The photon flux was measured separately under the same experimental conditions with photodiode (IRD, AXUV100) mounted in the vacuum stage assembly.
:25 v/v) solution with LeuEnk molecules from Sigma Aldrich at a concentration of 10 μM was prepared and introduced into nano ESI as such, without further purification. A turbo pumping vacuum stage was used to accommodate pressure differences between beamline (10−8 mbar) and LTQ spectrometer (10−5 mbar). The pressure inside the ion trap is about 10−3 mbar due to helium buffer gas that is used to cool the ions and improve trapping. A home built supporting frame for LTQ allows for fine tuning the ion trap axis with respect to the photon beam, in order to obtain high signal to noise ratio. A mechanical shutter was designed and placed in the assembly with the vacuum stage to control the irradiation time.55 Precursor cations, [YGGFL + H]+, were isolated in the ion trap and subjected to VUV monochromatic photon beam during 500 ms. The tandem mass spectra (MS2) were recorded as a function of the photon energy scanned in small steps of 0.2 eV from 5.7 eV to 8.5 eV. After the photon activation, ions were ejected and detected by two electron multiplier detectors with conversion dynodes (integral part of LTQ). Fragment ion yields, defined as the area under the peaks were extracted from recorded tandem mass spectra and normalized to the photon flux and total ionic current (or precursor ion intensity). The photon flux was measured separately under the same experimental conditions with photodiode (IRD, AXUV100) mounted in the vacuum stage assembly.Collision induced dissociation (CID) experiments were conducted using the LTQ methods on the precursor ion. The nature of the fragment ion was assessed by the comparison of the m/z with theoretical values and by checking the isotopic ratio of the ions, from which the charge could be determined.
2.2. Computational details
Density functional theory (DFT) and its Time-Dependent version (TD-DFT) together with cc-pVDZ basis set were used to optimize the ground state (GS) minimum of LeuEnk, characterize the excited state (ES) at this geometry and their potential energy surfaces (PES), respectively. We assessed the performance of different functionals and basis set (see Fig. S2 and S3‡). In particular, the CAM-B3LYP56 functional was selected, due to its good performance in treating such kind of extended π systems.12,57,58To simulate the vertical absorption spectrum, we computed the vertical absorption energies (VAEs) and oscillator strengths (f) for the 100 lowest ES. For an easier comparison with the experimental spectra, each VAE was shifted by −0.35 eV and a phenomenological broadening was used by adding a Gaussian with half width half maximum of 0.2 eV. This shift accounts for all different sources of approximations in our calculations (functional, incomplete basis set, lack of vibronic and thermal effects).59 TD-DFT absorption energies are typically blue-shifted compared to experimental absorption spectra. For small or medium-size molecules allowing the use of large basis set, the error is ∼0.2–0.3 eV,58 although it can be larger when the basis set is reduced.
The characterization of the ES nature in such complex systems is not straightforward since, usually, they are a mixture of different states and furthermore the molecular orbitals are delocalized along the molecule. Excited-ground state density difference provides a good alternative because it shows from where the density moves when populating a given excited state, being, then, very useful. Throughout this manuscript the color code is that the electron density moves from the blue region (negative density difference) of the molecule to the green areas (positive density difference). All the above calculations were performed with the Gaussian 16 suite of programs.60
We also performed the analysis of the non-covalent interactions (NCI) plots, using NCI code.61,62 NCI plots allow to visualize the different non-covalent interaction regions using a color code to rank those interactions. Red is used for destabilizing interactions, blue for stabilizing interactions and green for delocalized weak interactions. The intensity of these colors is associated with the interaction strength.
3. Results and discussion
3.1. Protonated leucine-enkephalin structure
The sequence for protonated LeuEnk is shown in Fig. 1 together with the backbone fragments named following Biemann's nomenclature.63 The 3D structure for GS [YGGFL + H]+ was elucidated by Burke et al.64 combining IR-UV double resonance spectroscopy of cryogenically cooled protonated LeuEnk with DFT calculations (M052X/6-31+G(d)). We re-optimized that geometry at CAM-B3LYP/cc-pVDZ level of theory used to describe the photodissociation of protonated LeuEnk. The optimized structure is shown in Fig. 1 together with the NCI isosurface.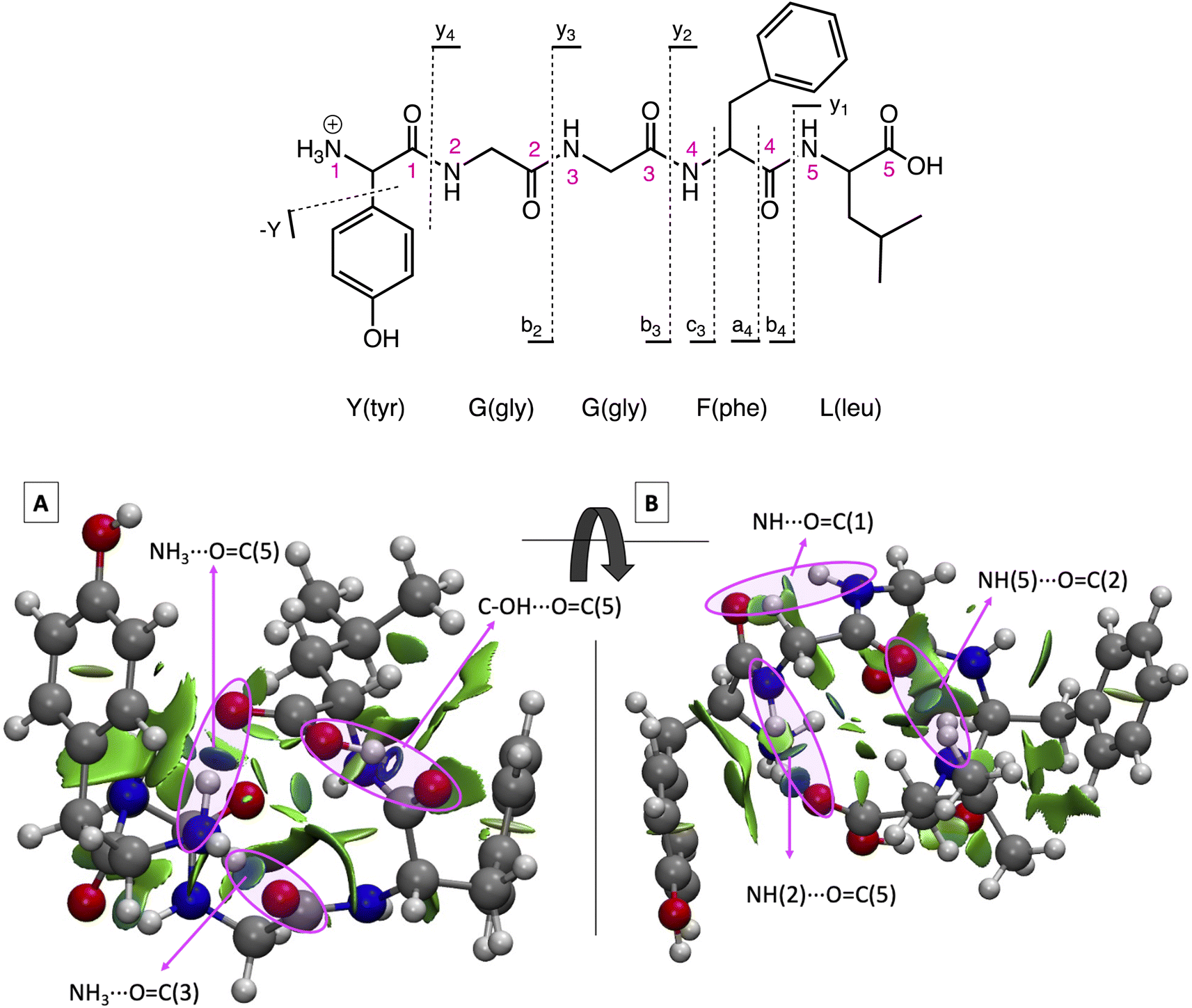 | ||
| Fig. 1 (Top) Protonated LeuEnk, [YGGFL + H]+, schematic structure together with the backbone fragments named following Biemann's nomenclature.63 (Bottom) Optimized 3D structure showing the NCI plots for the ground state minimum, min0. (A) and (B) are two different views of the same structure. Pink arrows point the NCI surfaces corresponding to the H-bonds. | ||
As we can see in Fig. 1, the proton is located on the N-terminal amino group, which is the most basic site. There is a net of H-bonds keeping the peptide in a hairpin like structure. These bonds are revealed by the blue (strong attractive interaction) flat circular regions of the NCI plots (marked with pink arrows in Fig. 1). In the 3D structure we can observe that the positive charge in the nitrogen is solvated by H bonds formed with (i) the carbonyl groups of the 3rd peptide backbone and (ii) the C-terminal COOH.
3.2. Photofragmentation spectra
Fig. 2 shows tandem mass spectra (MS2) of protonated LeuEnk, at the resonant photon activation energy of 6.7 eV together with the CID spectra for comparison.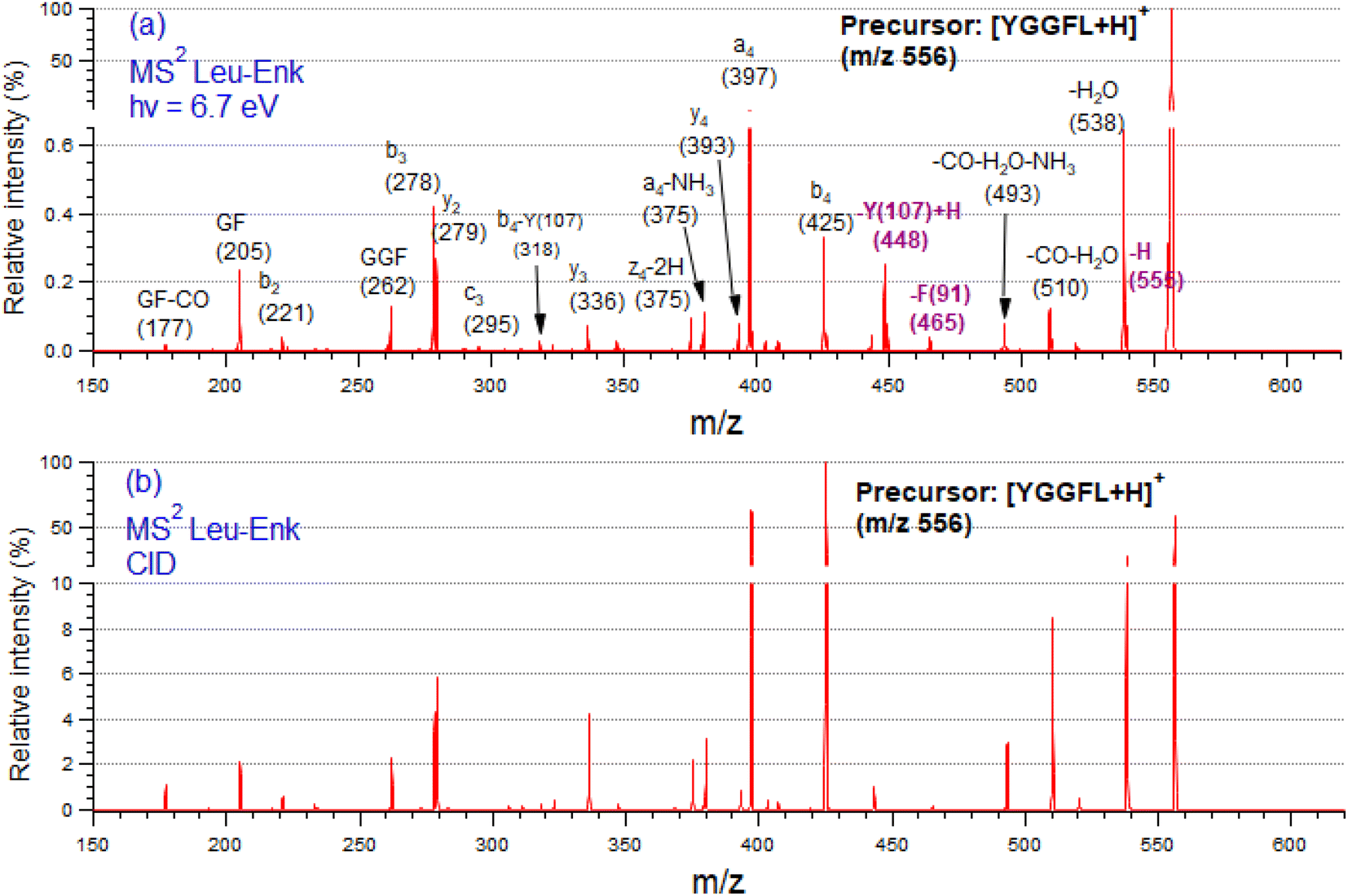 | ||
| Fig. 2 Tandem mass spectra (MS2) of protonated leucine-enkephalin peptide recorded (a) at the photon energy of 6.7 eV and (b) collision induced dissociation spectrum. The assignment of the fragments is given in the figure. | ||
The fragmentation pattern of [YGGFL-H]+ after UV light irradiation (Fig. 2a) is dominated by the backbone fragments a4, b4, b3 and y2, as well as internal ions and neutral losses, mainly water. As already pointed out,34 the fragmentation pattern of protonated LeuEnk below ionization energy appears very similar to that produced by CID (Fig. 2b), suggesting that upon electronic excitation, IC to a vibrationally hot GS is the dominant deactivation mechanism. Photo-specific fragments like Y and F side chain losses (m/z 448, and 465, respectively) usually observed in photofragmentation of peptides containing these amino acids are also present in the FUV-PD spectrum. H loss (m/z 555), another specific photofragment, is also observed. The presence of these fragments suggests a competition between fragmentation directly from the ES and fragmentation from a vibrationally hot GS after IC.
3.3. Photoabsorption spectra
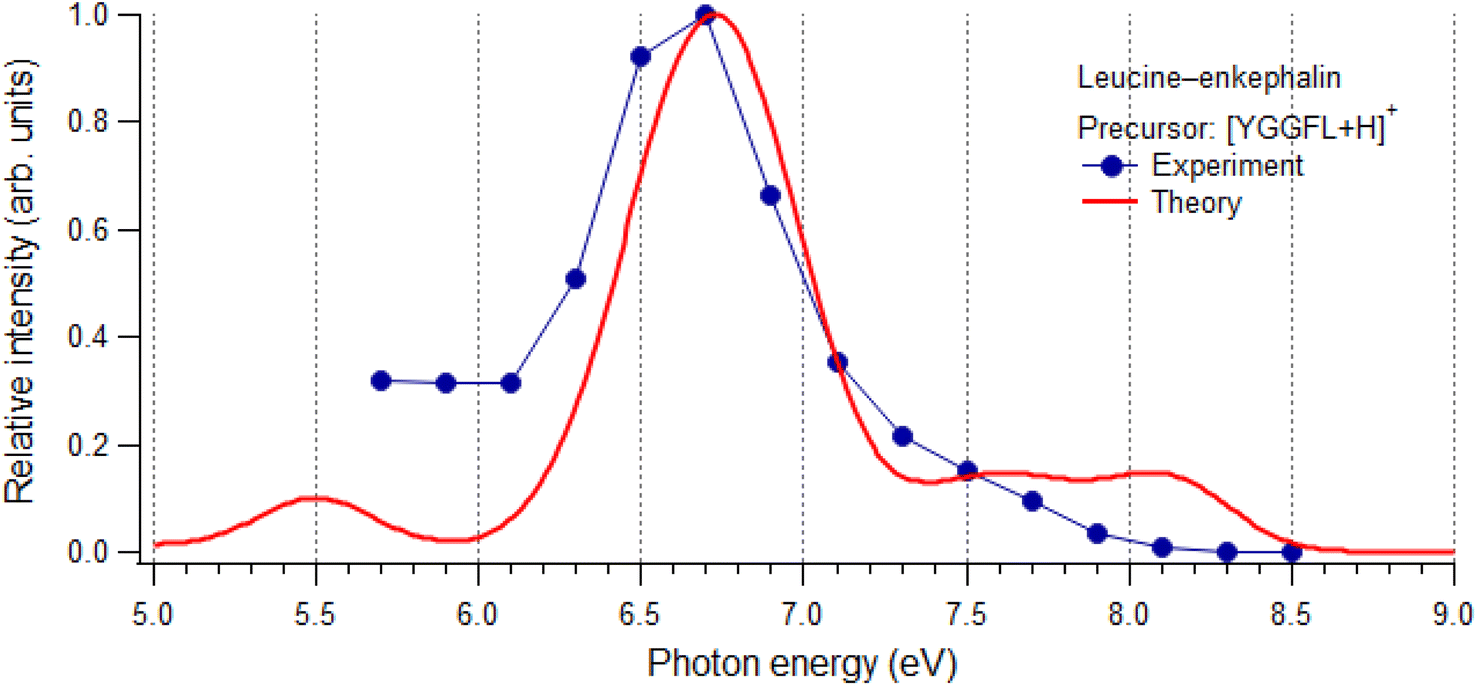 | ||
| Fig. 3 The experimental total ion yield, obtained as the sum over all resolved fragments in the m/z 150–620 range, compared to the normalized calculated absorption spectra (TD-CAM-B3LYP/cc-pVDZ). | ||
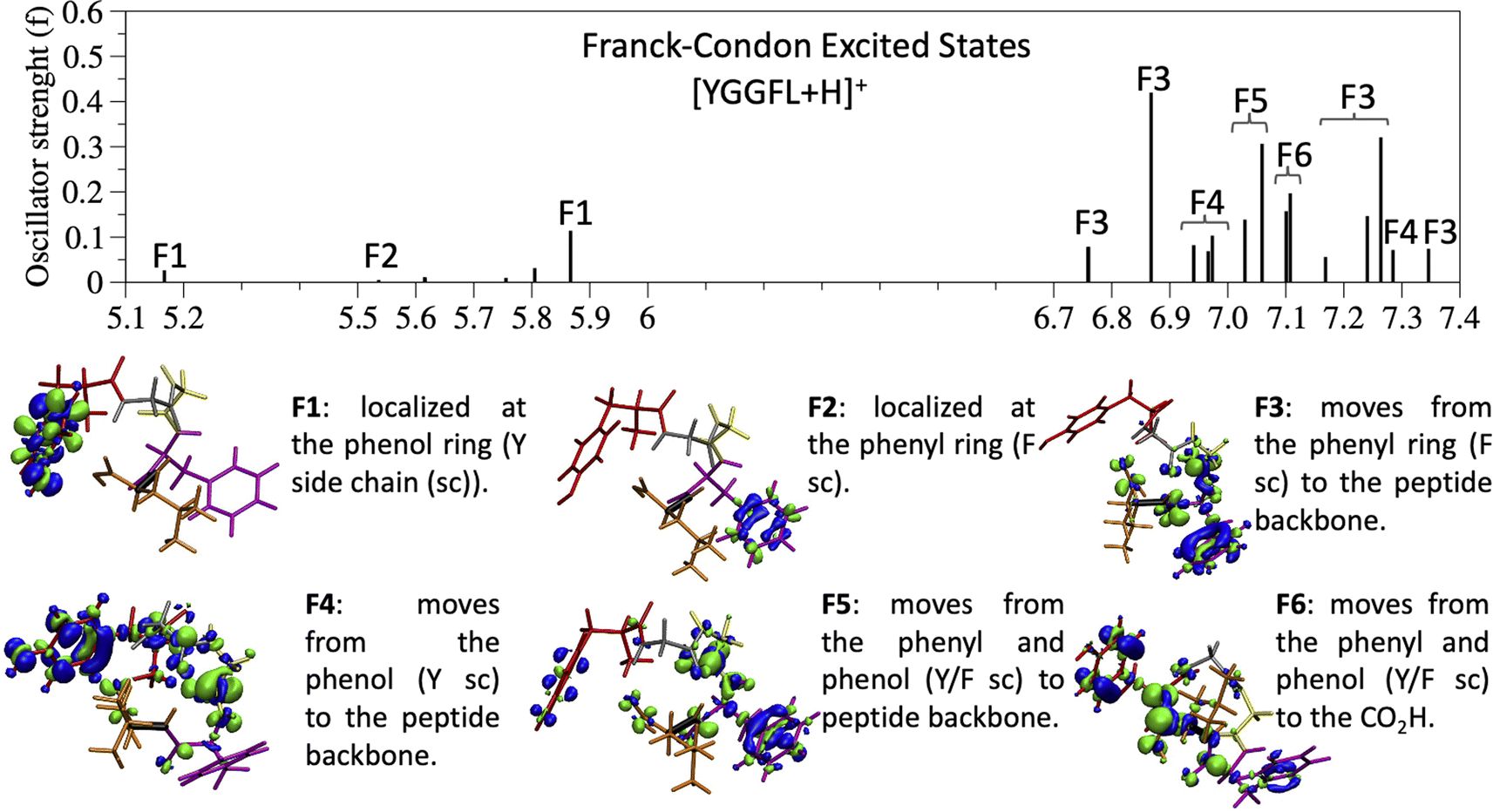 | ||
| Fig. 4 Brightest excited states (oscillator strength f > 0.05) up to 8 eV and description of the families used to classify the excited states at the Franck–Condon region with an example of the electron density difference for each family showing how the electron density changes when going from the ground to the excited state. The family of each excited state is indicated on the graph. | ||
3.4. Photodissociation mechanisms
Since the experimental spectrum (recall Fig. 2a) shows specific photo-fragments (m/z 555, H loss; m/z 448 Y side chain loss + H and m/z 465 F side chain loss), we mapped the possible relaxation mechanisms from the FC region by optimizing the selected brightest ES (Table S2‡ contains additional information). The optimizations lead to seven different minima on the S1 surface plus one minimum in the S2 ES. Similarly to the FC region, the types of S1 minima can also be classified according to where the electron density is localized. The geometry and electronic density difference of each of these minima is shown in Fig. 5. In all of them, the electronic density is localized on the X peptide bond (min-PBX), on the aromatic ring of the Y or F side chains, or in the COOH (min-HT: one H of the N-terminal NH3+ is transferred to the C-terminal). Min1-HT is the most stable minimum by far (more than 1 eV). The relative energies of the other minima are within a 0.5 eV range.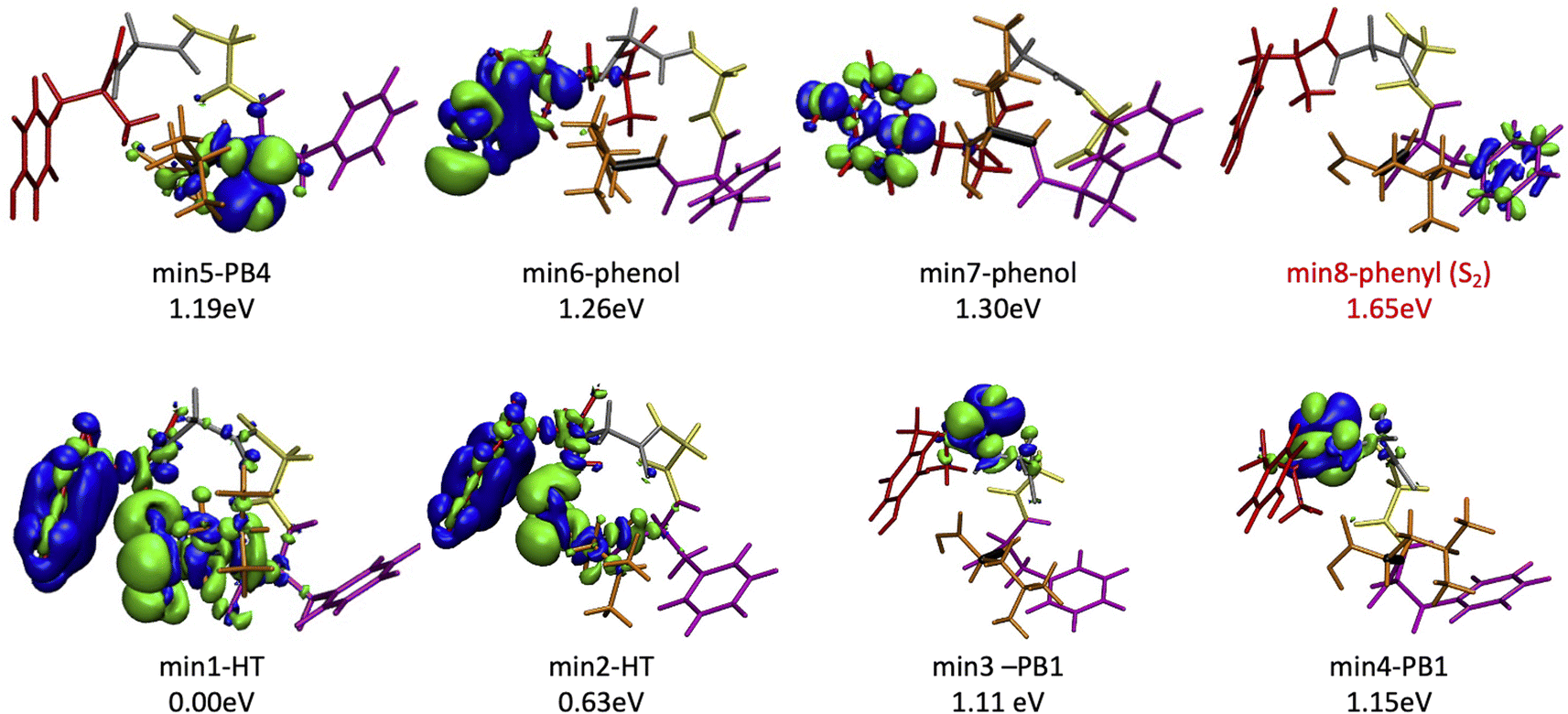 | ||
| Fig. 5 Optimized geometries, electron density differences and relative energies for the S1 and S2 minima. | ||
Given the high density of excited states accessible in the FC region and minima found in the S1 surface, and in the absence of a dynamic study, we have attempted to estimate which minimum is more likely to be populated. With this objective, we define the quantity % fave. To compute % fave we assigned the value 100% to the oscillator strength of the brightest state at the FC region (S16 with f = 0.42) and computed the corresponding percentage, relative to this maximum, for each of the states. Then we calculated the average of the % f of all the states leading to a given minimum. Thus, the highest the % fave, the brightest are the states leading to that minimum. The computed values are shown in Table 1 together with other quantities like number of states leading to each minimum and the oscillator strength of the brightest of those minima. From the values of % fave (Table 1) we expect min1-HT to be the most populated, since the most intense FC bright states lead to this minimum and the larger number of states (six). Furthermore, it is the most stable S1 minimum by far (>1 eV). After min1-HT, min5-PB4 and min6-phenol will be the second most populated being 1.19 and 1.26 eV higher in energy than min1-HT. We will discuss possible deactivation mechanism from these minima.
| Minimum | Rel. E [eV] | #States | % fave | fmax | Vert. E [eV] |
|---|---|---|---|---|---|
| Min1-HT | 0.00 | 6 | 43 | 0.42 | 0.99 |
| Min2-HT | 0.63 | 1 | 20 | 0.08 | 2.19 |
| Min3-PB1 | 1.11 | 4 | 9 | 0.11 | 3.75 |
| Min4-PB1 | 1.15 | 1 | 17 | 0.07 | 3.75 |
| Min5-PB4 | 1.19 | 5 | 36 | 0.32 | 3.67 |
| Min6-phenol | 1.26 | 1 | 35 | 0.15 | 1.00 |
| Min7-phenol | 1.30 | 2 | 7 | 0.06 | 4.82 |
| Min8-phenyl | 1.65 | 2 | 2 | 0.07 | 5.19 |
Previous studies have shown that some fragmentation channels could proceed through triplet states.67,68 To preliminary assess the possibility of intersystem crossing (ISC) from these minima we have estimated the spin orbit coupling (SOC) terms using the single particle Breit–Pauli operator with an effective charge approximation as implemented in the PySOC code.69 The values for the SOC and vertical energies computed at each S1 minima are reported in Tables S2–S10 of the ESI.‡ For the minima with large values of SOC (min3, min4, min5, and min6) we then optimized the T1 state reaching in all cases to a T1 minimum (Table S11‡). In all the cases we observe small conformational rearrangement and no structural changes suggesting a fragmentation process originating from these structures, with the only exception of min6 (vide infra). In particular, there is no sign of Cα–Cβ bond weaking at any of the studied T1 minimum. In all of them Cα–Cβ bond distance is 1.54 Å, exactly the same value as in the GS minimum, min0.
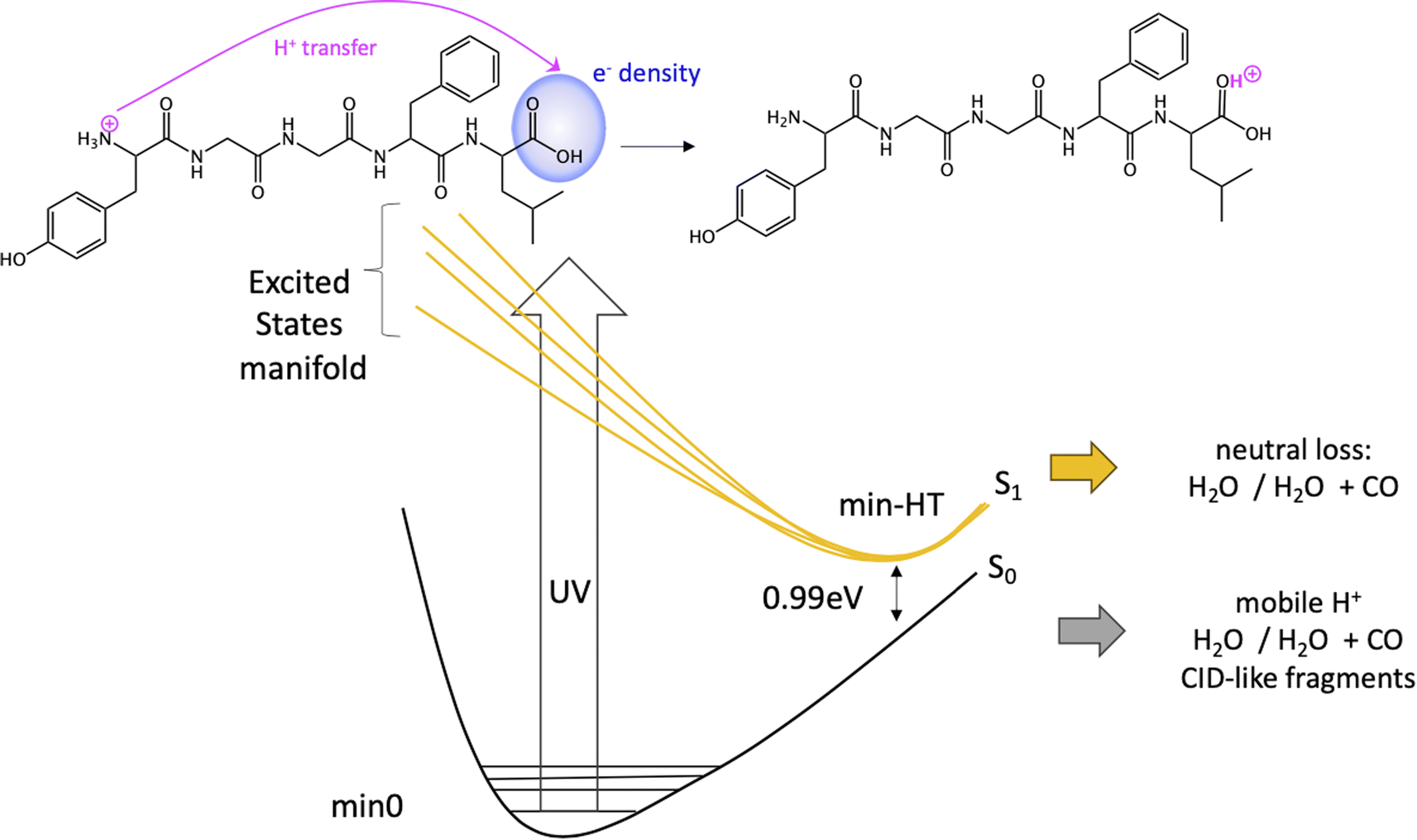 | ||
| Scheme 1 Fragmentation mechanisms from min-HT. | ||
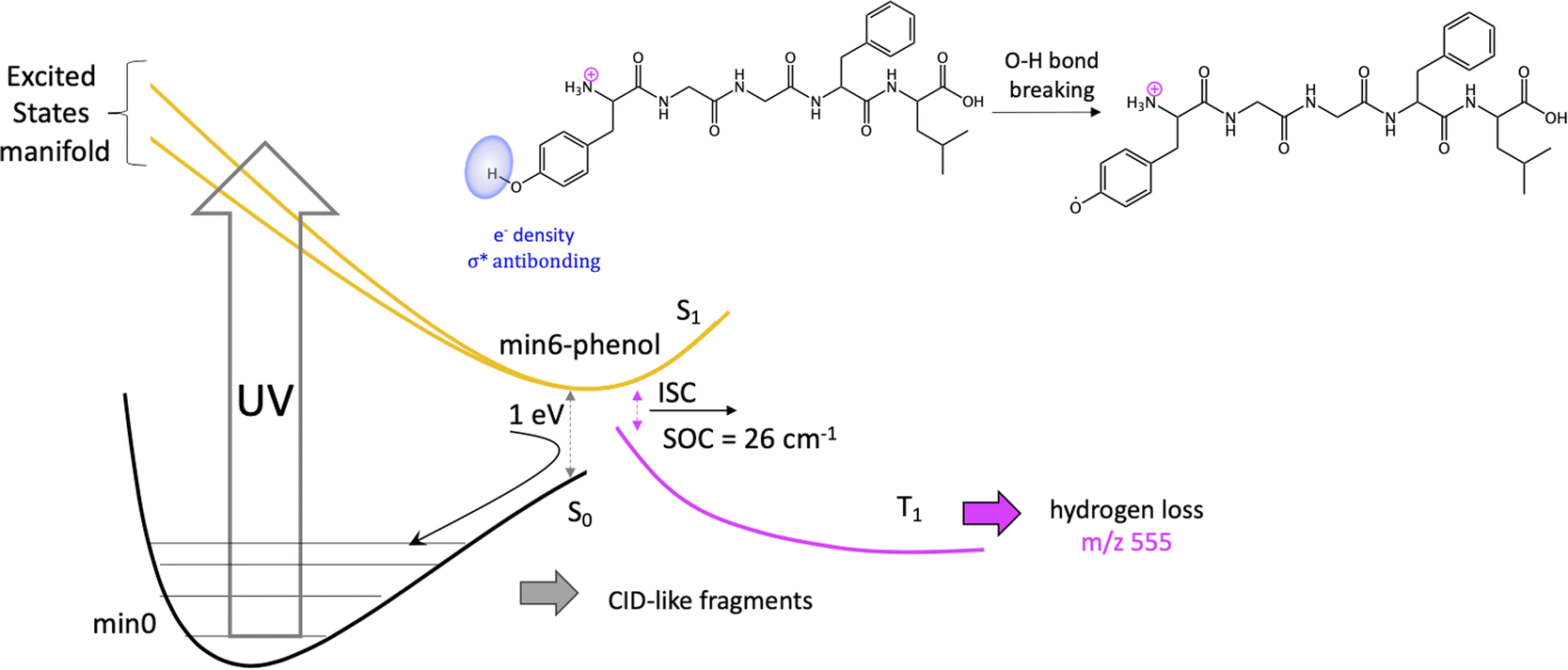 | ||
| Scheme 2 Fragmentation mechanism from min6-phenol. | ||
It should be noticed that the mechanisms proposed to explain H loss from charged amino acids as TyrH+ or short, charged peptides, is different to the one we proposed here for protonated LeuEnk, since in the other cases the lost H comes from the charged NH3+ group.43,72 This can be explained by the high sensitivity of the nonradiative deactivation processes to the backbone environment of the aromatic ring. Considering protonated LeuEnk geometry, where the peptide chain is long enough to allow the peptide to fold in such a way that the NH3+ charge is solvated by COOH group and the carbonyl groups of two peptide bonds (recall Fig. 1), the phenol environment resembles more of the neutral tyrosine than of protonated Tyr or small protonated peptides containing Tyr, where the NH3+ group is interacting with the electronic cloud of the ring. Thus, it is logical that the H loss mechanism resembles more that proposed for neutral aromatic amino acids.73
The other minimum, min7-phenol, is slightly higher in energy (1.30 eV). The excess of electronic density (green surface) of the min7-phenol resembles the ππ* state proposed to explain fragmentation for protonated Tyr and Phe43 and the neurotransmitters noradrenaline71 and adrenaline.46 The proposed mechanism implies an ESPT from the ammonium to the aromatic ring (of Tyr in this case), followed by Cα–Cβ bond breaking, with the corresponding fragment (Y side chain) bearing an extra hydrogen as compared to the mass of the aromatic chromophore cation, i.e., a mass difference with MH+ of 108 instead of 107. This mechanism will lead to the observation of the photofragment m/z 448 (Scheme 3).
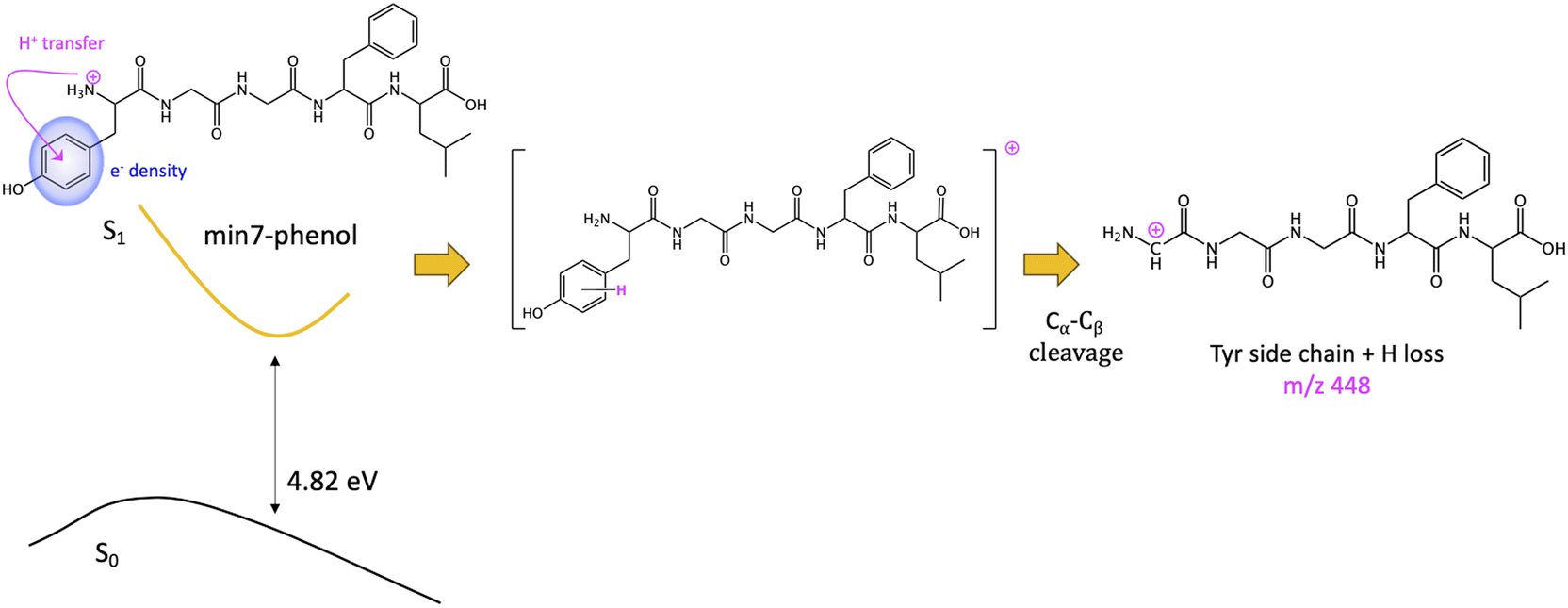 | ||
| Scheme 3 Fragmentation mechanism from min7-phenol. | ||
A similar mechanism will be in place to explain F side chain loss (m/z 465). However, it is worth to note that the minima with electron density localized at the F side chain (min8-phenyl) is the highest in energy and belongs to the S2 surface. In addition, it has the lowest value of % fave (only 2 excited states, with very low oscillator strength, lead to this minimum). This is consistent with the low intensity observed for this channel.
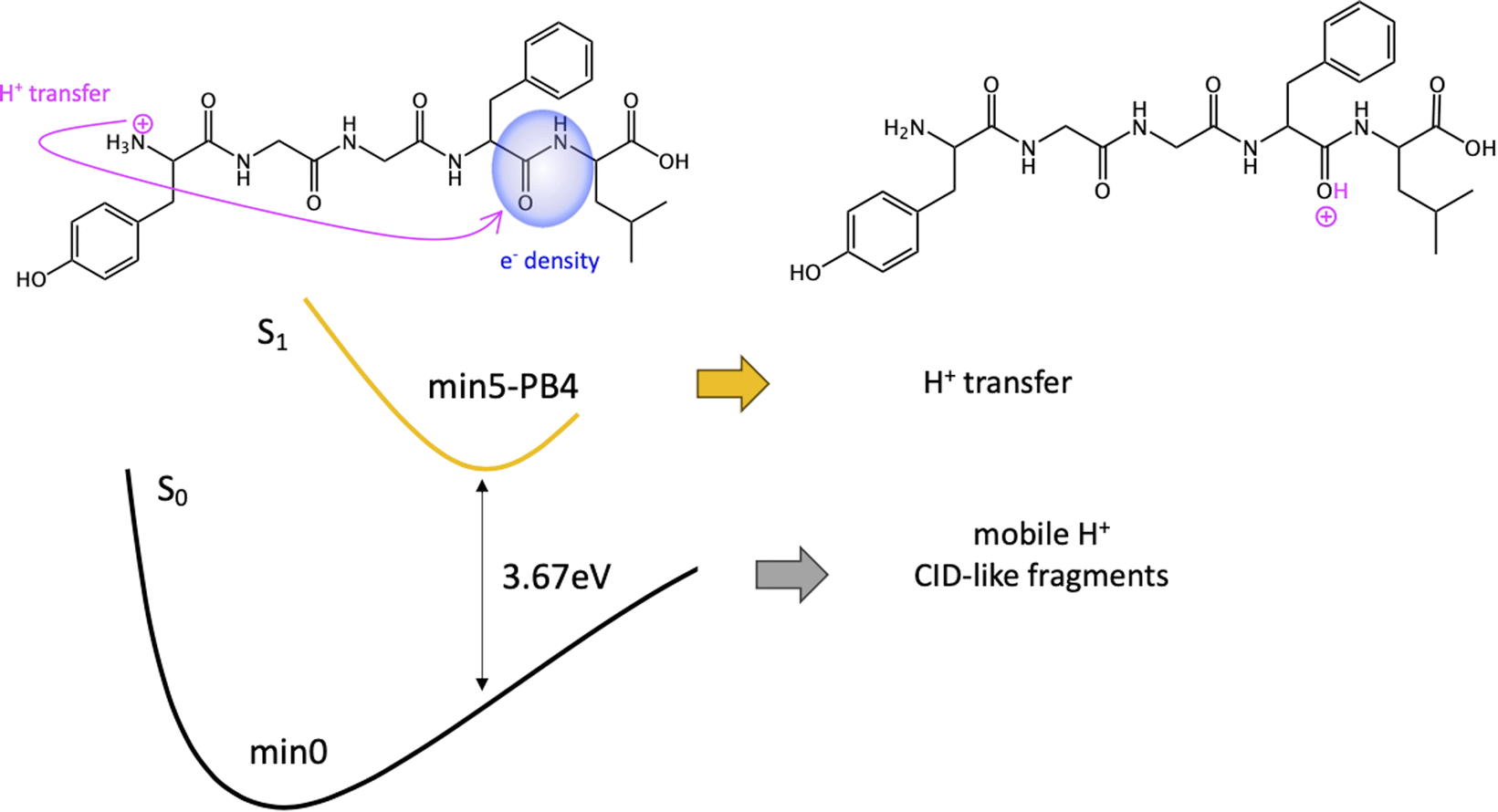 | ||
| Scheme 4 Fragmentation mechanisms from min5-PB4. | ||
4. Conclusions
In conclusion, we have performed a combined experimental and theoretical study of FUV-PD of a protonated pentapeptide, leucine-enkephalin, isolated in the gas-phase. TD-DFT calculations were performed to investigate in detail excitation of [YGGFL + H]+ and the evolution of the excited states.Calculations suggest that the main fragmentation mechanism starts by an electron transfer from the aromatic ring of Y/F side chain to the carboxylic acid, rapidly followed by a barrierless excited state proton transfer (ESPT) from the NH3+ terminal to the COOH forming min-HT. From this minimum, CO and CO + H2O loss could be observed. Also, IC to the GS could take place, where the activated H+ can move to the different basic positions on the peptide backbone triggering fragmentation as proposed by the mobile-proton model usually employed to explain CID fragmentation.
We also proposed a mechanism for the specific photofragments. In the singlet PES, the channel leading to the photofragment m/z 448 (loss of Tyr side chain + H), would be triggered by an ESPT to Tyr aromatic ring driven by a light-induced electron transfer to the aromatic ring and followed by Cα–Cβ bond cleavage. Another relevant channel would be the light-induced electron transfer from the aromatic ring (F/Y residues) to the carbonyl of the 4th peptide bond. This charge transfer might trigger a proton transfer from the ammonium group to the negatively charged CO followed by fragmentation at this amide bond (b4/a4 ions). Nevertheless, this will lead to the same fragments as in CID experiments.
On the other hand, since some of the singlet minima present large SOC, and therefore triplet population cannot be discarded, we complete the study considering possible photofragmentation mechanisms in the triplet manifold. However, the T1 minima optimized from this region of the PES show small structural changes but for min6, leading to H loss. For this channel, calculations suggest that the H is lost from phenol's hydroxyl group of Tyr sice chain, as observed for the neutral peptide and in contrast with mechanism proposed for protonated Tyr. H loss in T1 PES is a barrierless process.
In summary, the fragmentation mechanisms proposed are mostly triggered by ESPT followed by (i) fragmentation in the excited state (Y + H side chain loss, H loss) or (ii) IC to a hot vibrationally GS where fragmentation takes place. The resemblance of FUV-PD and CID mass spectra support the assumption that the fragmentation mainly takes place from a vibrationally hot peptide in the GS as it happens in CID. Despite the initiation of the process through photon absorption rather than collisions, it is ultimately the proton transfer that triggers fragmentation. Previous studies have already pointed out the validity of the mobile proton model to explain UVPD results.74,75 If, following UV photon absorption, there is a charge transfer to the CO groups of peptide backbone with subsequent proton migration (ESPT), the final observed fragments will be the same as in a CID-like mechanism. However, when aromatic amino acids are present, proton might migrate to the side chain aromatic ring of these amino acids via ESPT, leading to the observation of specific photofragments (Cα–Cβ bond breaking) that do not appear in CID experiments.
The present work deepens the existing understanding of the complex cascading processes that govern photodissociation of a quite large biopolymer in the far ultraviolet range.
Author contributions
A. M., A. G. and L. N. conceived and design the experiment. A. R. M., M. R., A. G., F. C. and L. N. conducted the measurements. A. R. M. and M. R. treated the experimental data. A. M. S. and L. M. F. designed the theoretical study and performed and analyzed TD-DFT calculations. A. M. S., L. M. F. and A. R. M. co-wrote the first draft of the paper, which was then commented, corrected and improved by all coauthors.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
We thank the technical staff of SOLEIL for running smoothly the facility under project No. 20131031. AMS acknowledges Madrid Government (Comunidad de Madrid-Spain) under the Multiannual Agreement with UAM in the line Support to Young Researchers, in the context of the V PRICIT (SI3-PJI-2021-00463) and CCC-UAM for generous allocation of computing time. MR acknowledges support from the Ministry of education, science and technological development of Republic of Serbia under the grant OI 171020. AG acknowledges support from the Agence Nationale de la Recherche Scientifique, France, contract grant number: ANR-08-BLAN-0065. The collaborative work was performed within the framework MD-GAS CA18212 COST action. We are grateful to Ines Corral for discussions and help with calculations.References
- D. I. Pattison and M. J. Davies, Actions of Ultraviolet Light on Cellular Structures, Springer, New York, 2006 Search PubMed.
- R. P. Rastogi, Richa, A. Kumar, M. B. Tyagi and R. P. Sinha, Molecular mechanisms of ultraviolet radiation-induced DNA damage and repair, J. Nucleic Acids, 2010, 2010, 1–32 CrossRef PubMed.
- DNA Photodamage: from Light Absorption to Cellular Responses and Skin Cancer, ed. R. Improta and T. Douki, The Royal Society of Chemistr, 2021 Search PubMed.
- J. S. Taylor, Unraveling the Molecular Pathway from Sunlight to Skin Cancer, Acc. Chem. Res., 1994, 27, 76–82 CrossRef CAS.
- L. Martínez Fernández, F. Santoro and R. Improta, Nucleic Acids as a Playground for the Computational Study of the Photophysics and Photochemistry of Multichromophore Assemblies, Acc. Chem. Res., 2022, 55, 2077–2087 CrossRef PubMed.
- V. A. Bahamondes Lorca and S. Wu, Ultraviolet Light, Unfolded Protein Response and Autophagy, Photochem. Photobiol., 2023, 99, 498–508 CrossRef CAS PubMed.
- D. I. Pattison, A. S. Rahmanto and M. J. Davies, Photochem. Photobiol. Sci., 2012, 11, 38–53 CrossRef CAS PubMed.
- G. Giuliani, F. Melaccio, S. Gozem, A. Cappelli and M. Olivucci, QM/MM Investigation of the Spectroscopic Properties of the Fluorophore of Bacterial Luciferase, J. Chem. Theory Comput., 2021, 17, 605–613 CrossRef CAS PubMed.
- L. V. Schäfer, G. Groenhof, A. R. Klingen, G. M. Ullmann, M. Boggio-Pasqua, M. A. Robb and H. Grubmüller, Photoswitching of the fluorescent protein asFP595: Mechanism, proton pathways, and absorption spectra, Angew. Chem., Int. Ed., 2007, 46, 530–536 CrossRef PubMed.
- D. Polli, P. Altoè, O. Weingart, K. M. Spillane, C. Manzoni, D. Brida, G. Tomasello, G. Orlandi, P. Kukura, R. A. Mathies, M. Garavelli and G. Cerullo, Conical intersection dynamics of the primary photoisomerization event in vision, Nature, 2010, 467, 440–443 CrossRef CAS.
- S. Gozem, P. J. M. Johnson, A. Halpin, H. L. Luk, T. Morizumi, V. I. Prokhorenko, O. P. Ernst, M. Olivucci and R. J. D. Miller, Excited-State Vibronic Dynamics of Bacteriorhodopsin from Two-Dimensional Electronic Photon Echo Spectroscopy and Multiconfigurational Quantum Chemistry, J. Phys. Chem. Lett., 2020, 11, 3889–3896 CrossRef CAS PubMed.
- P. Nogly, T. Weinert, D. James, S. Carbajo, D. Ozerov, A. Furrer, D. Gashi, V. Borin, P. Skopintsev, K. Jaeger, K. Nass, P. Båth, R. Bosman, J. Koglin, M. Seaberg, T. Lane, D. Kekilli, S. Brünle, T. Tanaka, W. Wu, C. Milne, T. White, A. Barty, U. Weierstall, V. Panneels, E. Nango, S. Iwata, M. Hunter, I. Schapiro, G. Schertler, R. Neutze and J. Standfuss, Retinal isomerization in bacteriorhodopsin captured by a femtosecond x-ray laser, Science, 2018, 361, eaat0094 CrossRef PubMed.
- C. López-Camarillo, E. A. Ocampo, M. L. Casamichana, C. Pérez-Plasencia, E. Álvarez-Sánchez and L. A. Marchat, Protein kinases and transcription factors activation in response to UV-radiation of skin: Implications for carcinogenesis, Int. J. Mol. Sci., 2012, 13, 142–172 CrossRef PubMed.
- A. Gęgotek, S. Atalay and E. Skrzydlewska, UV induced changes in proteome of rats plasma are reversed by dermally applied cannabidiol, Sci. Rep., 2021, 11, 20666 CrossRef PubMed.
- L. Vanhaelewyn, D. Van Der Straeten, B. De Coninck and F. Vandenbussche, Front. Plant Sci., 2020, 11, 597642 CrossRef.
- S. Kataria, A. Jajoo and K. N. Guruprasad, Impact of increasing Ultraviolet-B (UV-B) radiation on photosynthetic processes, J. Photochem. Photobiol., B, 2014, 137, 55–66 CrossRef CAS PubMed.
- A. T. Slominski, M. A. Zmijewski, P. M. Plonka, J. P. Szaflarski and R. Paus, How UV Light Touches the Brain and Endocrine System Through Skin, and Why, Endocrinology, 2018, 159, 1992–2007 CrossRef CAS PubMed.
- P. H. Hart, S. Gorman and J. J. Finlay-Jones, Modulation of the immune system by UV radiation: More than just the effects of vitamin D?, Nat. Rev. Immunol., 2011, 11, 584–596 CrossRef CAS PubMed.
- J. J. Bernard, R. L. Gallo and J. Krutmann, Photoimmunology: how ultraviolet radiation affects the immune system, Nat. Rev. Immunol., 2019, 19, 688–701 CrossRef CAS PubMed.
- S. Takeuchi, T. Matsuda, R. Ono, M. Tsujimoto and C. Nishigori, Mitotic genes are transcriptionally upregulated in the fibroblast irradiated with very low doses of UV-C, Sci. Rep., 2016, 6, 29233 CrossRef CAS PubMed.
- O. Licht, D. Barreiro-Lage, P. Rousseau, A. Giuliani, A. R. Milosavljević, A. Isaak, Y. Mastai, A. Albeck, R. Singh, V. T. T. Nguyen, L. Nahon, L. Martínez-Fernández, S. Díaz-Tendero and Y. Toker, Peptide Bond Formation in the Protonated Serine Dimer Following Vacuum UV Photon-Induced Excitation, Angew. Chem., 2023, 135, e2022187 CrossRef.
- N. J. Green, J. Xu and J. D. Sutherland, Illuminating Life's Origins: UV Photochemistry in Abiotic Synthesis of Biomolecules, J. Am. Chem. Soc., 2021, 143, 7219–7236 CrossRef CAS PubMed.
- M. Claverie, C. McReynolds, A. Petitpas, M. Thomas and S. C. M. Fernandes, Marine-derived polymeric materials and biomimetics: An overview, Polymers, 2020, 12, 1002 CrossRef CAS.
- A. Sionkowska, Photochemical stability of collagen/poly(ethylene oxide) blends, J. Photochem. Photobiol., A, 2006, 177, 61–67 CrossRef CAS.
- B. A. Kerwin and R. L. Remmele, Protect from light: Photodegradation and protein biologics, J. Pharm. Sci., 2007, 96, 1468–1479 CrossRef CAS PubMed.
- T. P. Cleland, C. J. Dehart, R. T. Fellers, A. J. Vannispen, J. B. Greer, R. D. LeDuc, W. R. Parker, P. M. Thomas, N. L. Kelleher and J. S. Brodbelt, High-Throughput Analysis of Intact Human Proteins Using UVPD and HCD on an Orbitrap Mass Spectrometer, J. Proteome Res., 2017, 16, 2072–2079 CrossRef CAS PubMed.
- X. Dang and N. L. Young, Ultraviolet photodissociation enhances top-down mass spectrometry as demonstrated on green fluorescent protein variants, Proteomics, 2014, 14, 1128–1129 CrossRef CAS PubMed.
- S. Soorkia, C. Jouvet and G. Grégoire, UV Photoinduced Dynamics of Conformer-Resolved Aromatic Peptides, Chem. Rev., 2020, 120, 3296–3327 CrossRef CAS PubMed.
- A. Y. Pereverzev, I. Szabó, V. N. Kopysov, E. Rosta and O. V. Boyarkin, Gas-phase structures reflect the pain-relief potency of enkephalin peptides, Phys. Chem. Chem. Phys., 2019, 21, 22700–22703 RSC.
- J. S. Brodbelt, L. J. Morrison and I. Santos, Ultraviolet Photodissociation Mass Spectrometry for Analysis of Biological Molecules, Chem. Rev., 2020, 120, 3328–3380 CrossRef CAS PubMed.
- R. Parthasarathi, Y. He, J. P. Reilly and K. Raghavachari, New insights into the vacuum UV photodissociation of peptides, J. Am. Chem. Soc., 2010, 132, 1606–1610 CrossRef CAS PubMed.
- F. Canon, A. R. Milosavljević, L. Nahon and A. Giuliani, Action spectroscopy of a protonated peptide in the ultraviolet range, Phys. Chem. Chem. Phys., 2015, 17, 25725–25733 RSC.
- S. Bari, O. Gonzalez-Magaña, G. Reitsma, J. Werner, S. Schippers, R. Hoekstra and T. Schlathölter, Photodissociation of protonated leucine-enkephalin in the VUV range of 8-40 eV, J. Chem. Phys., 2011, 134, 024314 CrossRef CAS PubMed.
- M. L. J. Ranković, F. Canon, L. Nahon, A. Giuliani and A. R. Milosavljević, VUV action spectroscopy of protonated leucine-enkephalin peptide in the 6-14 eV range, J. Chem. Phys., 2015, 143, 244311 CrossRef.
- A. Giuliani, A. R. Milosavljević, F. Canon and L. Nahon, Contribution of synchrotron radiation to photoactivation studies of biomolecular ions in the gas phase, Mass Spectrom. Rev., 2014, 33, 424–441 CrossRef CAS PubMed.
- A. R. Milosavljević, V. Z. Cerovski, M. L. Ranković, F. Canon, L. Nahon and A. Giuliani, VUV photofragmentation of protonated leucine-enkephalin peptide dimer below ionization energy, Eur. Phys. J. D, 2014, 68, 68 CrossRef.
- C. J. Shaffer, K. Slováková and F. Tureček, Near-UV photodissociation of phosphopeptide cation-radicals, Int. J. Mass Spectrom., 2015, 390, 71–80 CrossRef CAS.
- C. Dehon, S. Soorkia, M. Pedrazzani, C. Jouvet, M. Barat, J. A. Fayeton and B. Lucas, Photofragmentation at 263 nm of small peptides containing tyrosine: The role of the charge transfer on CO, Phys. Chem. Chem. Phys., 2013, 15, 8779–8788 RSC.
- N. S. True, Effects of rapid intramolecular vibrational redistribution on molecular spectra at microwave frequencies, Chem. Phys. Lett., 1983, 101, 326–330 CrossRef CAS.
- R. R. Julian, The Mechanism Behind Top-Down UVPD Experiments: Making Sense of Apparent Contradictions, J. Am. Soc. Mass Spectrom., 2017, 28, 1823–1826 CrossRef CAS PubMed.
- D. Milesevic, D. Popat, P. Robertson and C. Vallance, Photodissociation dynamics of N,N-dimethylformamide at 225 nm and 245 nm, Phys. Chem. Chem. Phys., 2022, 24, 28343–28352 RSC.
- I. Antol, M. Eckert-Maksić, M. Barbatti and H. Lischka, Simulation of the photodeactivation of formamide in the nO–π and π–π* states: An ab initio on-the-fly surface-hopping dynamics study, J. Chem. Phys., 2007, 127, 234303 CrossRef PubMed.
- G. Féraud, M. Broquier, C. Dedonder, C. Jouvet, G. Grégoire and S. Soorkia, Excited State Dynamics of Protonated Phenylalanine and Tyrosine: Photo-Induced Reactions Following Electronic Excitation, J. Phys. Chem. A, 2015, 119, 5914–5924 CrossRef PubMed.
- T. N. V. Karsili, A. M. Wenge, S. J. Harris, D. Murdock, J. N. Harvey, R. N. Dixon and M. N. R. Ashfold, O-H bond fission in 4-substituted phenols: S1 state predissociation viewed in a Hammett-like framework, Chem. Sci., 2013, 4, 2434–2446 RSC.
- K. Ranka, N. Zhao, L. Yu, J. F. Stanton and N. C. Polfer, Radical Rearrangement Chemistry in Ultraviolet Photodissociation of Iodotyrosine Systems: Insights from Metastable Dissociation, Infrared Ion Spectroscopy, and Reaction Pathway Calculations, J. Am. Soc. Mass Spectrom., 2018, 29, 1791–1801 CrossRef CAS PubMed.
- J. Dezalay, M. Broquier, S. Soorkia, K. Hirata, S. I. Ishiuchi, M. Fujii and G. Grégoire, Excited-state proton transfer in protonated adrenaline revealed by cryogenic UV photodissociation spectroscopy, Phys. Chem. Chem. Phys., 2020, 22, 11498–11507 RSC.
- M. Pérot, B. Lucas, M. Barat, J. A. Fayeton and C. Jouvet, Mechanisms of UV photodissociation of small protonated peptides, J. Phys. Chem. A, 2010, 114, 3147–3156 CrossRef PubMed.
- J. Sztáray, A. Memboeuf, L. Drahos and K. Vékey, Leucine enkephalin - A mass spectrometry standard, Mass Spectrom. Rev., 2011, 30, 298–320 CrossRef PubMed.
- S. Dörner, L. Schwob, K. Schubert, M. Girod, L. MacAleese, C. L. Pieterse, T. Schlathölter, S. Techert and S. Bari, The influence of the methionine residue on the dissociation mechanisms of photoionized methionine-enkephalin probed by VUV action spectroscopy, Eur. Phys. J. D, 2021, 75, 142 CrossRef.
- T. Tabarin, R. Antoine, M. Broyer and P. Dugourd, Specific photodissociation of peptides with multi-stage mass spectrometry, Rapid Commun. Mass Spectrom., 2005, 19, 2883–2892 CrossRef CAS PubMed.
- A. Herburger, C. Van Der Linde and M. K. Beyer, Photodissociation spectroscopy of protonated leucine enkephalin, Phys. Chem. Chem. Phys., 2017, 19, 10786–10795 RSC.
- A. R. Milosavljević, C. Nicolas, J. Lemaire, C. Dehon, R. Thissen, J. M. Bizau, M. Réfrégiers, L. Nahon and A. Giuliani, Photoionization of a protein isolated in vacuo, Phys. Chem. Chem. Phys., 2011, 13, 15432–15436 RSC.
- A. R. Milosavljević, C. Nicolas, J. F. Gil, F. Canon, M. Réfrégiers, L. Nahon and A. Giuliani, VUV synchrotron radiation: A new activation technique for tandem mass spectrometry, J. Synchrotron Radiat., 2012, 19, 174–178 CrossRef PubMed.
- L. Nahon, N. De Oliveira, G. A. Garcia, J. F. Gil, B. Pilette, O. Marcouillé, B. Lagarde and F. Polack, DESIRS: A state-of-the-art VUV beamline featuring high resolution and variable polarization for spectroscopy and dichroism at SOLEIL, J. Synchrotron Radiat., 2012, 19, 508–520 CrossRef CAS PubMed.
- A. R. Milosavljević, C. Nicolas, J. F. Gil, F. Canon, M. Réfrégiers, L. Nahon and A. Giuliani, Nucl. Instrum. Methods Phys. Res., Sect. B, 2012, 279, 34–36 CrossRef.
- T. Yanai, D. P. Tew and N. C. Handy, A new hybrid exchange-correlation functional using the Coulomb-attenuating method (CAM-B3LYP), Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- T. Körzdörfer and J. L. Brédas, Organic electronic materials: Recent advances in the dft description of the ground and excited states using tuned range-separated hybrid functionals, Acc. Chem. Res., 2014, 47, 3284–3291 CrossRef PubMed.
- C. Adamo and D. Jacquemin, The calculations of excited-state properties with time-dependent density functional theory, Chem. Soc. Rev., 2013, 42, 845–856 RSC.
- F. J. A. Ferrer, J. Cerezo, E. Stendardo, R. Improta and F. Santoro, Insights for an accurate comparison of computational data to experimental absorption and emission spectra: Beyond the vertical transition approximation, J. Chem. Theory Comput., 2013, 6, 2072–2082 CrossRef PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, M. J. Frisch and D. J. Fox, Gaussian, 16, Revision A.03, Gaussian Inc., Wallingford CT, 2016 Search PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing noncovalent interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J. P. Piquemal, D. N. Beratan and W. Yang, NCIPLOT: A program for plotting noncovalent interaction regions, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- K. Biemann, Sequencing of peptides by tandem mass spectrometry and high-energy collision-induced dissociation, Methods Enzymol., 1990, 193, 455–479 CAS.
- N. L. Burke, J. G. Redwine, J. C. Dean, S. A. McLuckey and T. S. Zwier, UV and IR spectroscopy of cold protonated leucine enkephalin, Int. J. Mass Spectrom., 2015, 378, 196–205 CrossRef CAS.
- C. M. Tseng, M. F. Lin, Y. L. Yang, Y. C. Ho, C. K. Ni and J. L. Chang, Photostability of amino acids: Photodissociation dynamics of phenylalanine chromophores, Phys. Chem. Chem. Phys., 2010, 12, 4989–4995 RSC.
- S. Soorkia, C. Dehon, S. K. S, M. Pérot-Taillandier, B. Lucas, C. Jouvet, M. Barat and J. A. Fayeton, Ion-induced dipole interactions and fragmentation times: Cα–Cβ chromophore bond dissociation channel, J. Phys. Chem. Lett., 2015, 6, 2070–2074 CrossRef CAS PubMed.
- C.-M. Tseng, M.-F. Lin, Y. L. Yang, Y. C. Ho, C.-K. Ni and J.-L. Chang, Photostability of amino acids: photodissociation dynamics of phenylalanine chromophores, Phys. Chem. Chem. Phys., 2010, 12, 4989 RSC.
- A. V. Zabuga, M. Z. Kamrath, O. V. Boyarkin and T. R. Rizzo, Fragmentation mechanism of UV-excited peptides in the gas phase, J. Chem. Phys., 2014, 141, 154309 CrossRef PubMed.
- X. Gao, S. Bai, D. Fazzi, T. Niehaus, M. Barbatti and W. Thiel, Evaluation of Spin-Orbit Couplings with Linear-Response Time-Dependent Density Functional Methods, J. Chem. Theory Comput., 2017, 13, 515–524 CrossRef CAS PubMed.
- A. R. Dongré, J. L. Jones, Á. Somogyi and V. H. Wysocki, Influence of peptide composition, gas-phase basicity, and chemical modification on fragmentation efficiency: Evidence for the mobile proton model, J. Am. Chem. Soc., 1996, 118, 8365–8374 CrossRef.
- H. Wako, S. I. Ishiuchi, D. Kato, G. Féraud, C. Dedonder-Lardeux, C. Jouvet and M. Fujii, A conformational study of protonated noradrenaline by UV-UV and IR dip double resonance laser spectroscopy combined with an electrospray and a cold ion trap method, Phys. Chem. Chem. Phys., 2017, 19, 10777–10785 RSC.
- G. Grégoire, C. Jouvet, C. Dedonder and A. L. Sobolewski, Ab initio Study of the Excited-State Deactivation Pathways of Protonated Tryptophan and Tyrosine, J. Am. Chem. Soc., 2007, 129, 6223–6231 CrossRef PubMed.
- A. L. Sobolewski, W. Domcke, C. Dedonder-Lardeux and C. Jouvet, Excited-state hydrogen detachment and hydrogen transfer driven by repulsive 1π–σ* states: A new paradigm for nonradiative decay in aromatic biomolecules, Phys. Chem. Chem. Phys., 2002, 4, 1093–1100 RSC.
- E. M. Solovyeva, A. Y. Pereverzev, M. V. Gorshkov and O. V. Boyarkin, Ultraviolet Photodissociation of Peptides: New Insight on the Mobile Proton Model, J. Exp. Theor. Phys., 2020, 130, 626–632 CrossRef CAS.
- L. A. Macias, S. N. Sipe, I. C. Santos, A. Bashyal, M. R. Mehaffey and J. S. Brodbelt, Influence of Primary Structure on Fragmentation of Native-Like Proteins by Ultraviolet Photodissociation, J. Am. Soc. Mass Spectrom., 2021, 32, 2860–2873 CrossRef CAS PubMed.
Footnotes |
| † Correspondence and requests for materials should be addressed to A. M. S. and A. R. M. |
| ‡ Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01690d |
| § Present address: Institute of Physical Chemistry J. 3Heyrovsky, CAS, Dolejškova 3, 18223 Prague 8, Czech Republic. |
| This journal is © The Royal Society of Chemistry 2024 |