
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInsights into the molecular interactions between urease subunit gamma from MRSA and drugs: an integrative approach by STD-NMR and molecular docking studies†
Anum Fatima a,
M. Iqbal Choudhary*abc,
Shezaib Siddiquia,
Humaira Zafar*a,
Kaifeng Hud and
Atia-tul Wahab*a
a,
M. Iqbal Choudhary*abc,
Shezaib Siddiquia,
Humaira Zafar*a,
Kaifeng Hud and
Atia-tul Wahab*a
aDr Panjwani Center for Molecular Medicine and Drug Research, International Center for Chemical and Biological Sciences, University of Karachi, Karachi-75270, Pakistan. E-mail: atia.tulwahab@iccs.edu; iqbal.choudhary@iccs.edu; hamramalik@gmail.com
bH. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi-75270, Pakistan
cDepartment of Biochemistry, Faculty of Science, King Abdulaziz University, Jeddah-22254, Saudi Arabia
dState Key Laboratory of Southwestern Chinese Medicine Resources, Chengdu University of Traditional Chinese Medicine, Chengdu, Sichuan-611137, China
First published on 1st October 2024
Abstract
Staphylococcus aureus, an important human pathogen, is developing resistance against a wide range of antibiotics. The antibiotic resistance in S. aureus has created the need to identify new drug targets, and to develop new drugs candidates. In the current study, urease subunit gamma from Methicillin Resistant Staphylococcus aureus (MRSA 252) was studied as a potential drug target, through protein–ligand interactions. Urease is the main virulence factor of MRSA, it catalyzes the conversion of urea into ammonia that is required for the survival of bacteria during acid stress. Its subunits and accessory proteins can serve as targets for drug discovery and development. Present study describes the cloning, expression, and purification of urease subunit gamma from MRSA 252. This was followed by screening of 100 US-FDA approved drugs against this protein using STD-NMR spectroscopy and among them, 15 drugs showed significant STD effects. In silico studies predicted that these drugs interacted mainly via non-covalent interactions, such as hydrogen bond, aromatic hydrogen bonding, π–π stacking, π–cation interactions, salt bridges, and halogen bonding. The thermal stability of UreA in the presence of these interacting drugs was evaluated using differential scanning fluorimetry (DSF), which revealed a significant effect on the Tm of UreA. Additionally, the inhibitory effects of these drugs on urease activity were assessed using a urease inhibition assay with Jack bean urease. The results showed that these drugs possess enzyme inhibitory activity, potentially impacting the survival of S. aureus. These hits need further biochemical and mechanistic studies to validate their therapeutic potential against the MRSA infections.
Introduction
Staphylococcus aureus, a clinically important pathogen, has acquired resistance to a wide range of antibiotics. To overcome this resistance, there is an urgent need to identify new drug targets, and thereby development of more effective drugs.1,2S. aureus has acquired antibiotic resistance through various mechanisms, such as drug target modification, efflux pumps activation, limiting the drug uptake, target overproduction, and drug inactivation.3 Therefore, identification of bacterial proteins, essential for the survival and pathogenesis of the bacteria, may serve as the drug targets for the discovery of new antibiotics.4
Urease is one of the main contributing factors in the pathogenesis of S. aureus strains.5–7 As in 90% strains of MRSA, urease enzyme is essential to provide alkaline environment against acid stress, and thereby facilitates S. aureus to maintain homeostasis.7,8 S. aureus encodes ureABCEFGD gene cluster for urease enzyme, where, ureA, ureB, and ureC genes encodes to γ, β, and α subunits, respectively, that form the apoenzyme.5 While ureEFGD genes encodes urease accessory proteins that form the urease pre-activation complex. This complex is essential for the urease enzyme activity,9 as it provides nickel to the active site of the enzyme to perform its activity.10 Urease catalyzes the hydrolysis of urea into ammonia and carbonic acid.11,12 Ammonia serves as nitrogenous source for bacterial growth during acid stress,13 and is further protonated into ammonium hydroxide ions, and increases the pH of the environment, which can cause damage to host tissues as well.14
In the current study, urease subunit γ from MRSA 252 was investigated as a potential drug target through protein–ligand interactions. Urease subunit γ is a part of the bacterial urease apoenzyme.15 The apoenzyme further requires accessory proteins assembly to be functional, and to perform its catalytic activity.9,15 We hypothesized that drugs interaction with urease subunit γ may inhibit the recruitment of the accessory proteins, thus limiting the urease enzyme activity.
Saturation transfer difference (STD)-NMR is a powerful analytical technique to identify the protein–ligand interactions.16 Quantitative analysis of STD spectra can also map the binding epitopes of the ligands for their interactions with the protein.17,18 In this study, 100 US-FDA approved drugs (ligands) were analyzed against the urease subunit gamma by using STD-NMR spectroscopy. The drugs showing interaction with protein on STD-NMR experiments were further studied by molecular docking studies. The molecular docking predicts the interactions of these drugs with the protein. The combination of STD-NMR and molecular docking provides comprehensive valuable insights for the identification of the potential drug candidates against the urease subunit gamma. Moreover, these protein–ligand interactions can help to inhibit the urease enzyme activity, and negatively regulate the MRSA growth during acid stress. Furthermore, there is a need to study the potential of these interacting drugs as new leads for the treatment of MRSA infections.
Methodology
Cloning of urease subunit γ into the pET-25b vector
Genomic DNA of MRSA 252 was isolated using MagPrep® Bacterial Genomic DNA Kit (Cat # 71256), and urease subunit γ gene FASTA sequence was retrieved from the National Center for Biotechnology Information (NCBI). Using Oligo perfect designing tools, and Integrated DNA technology, urease subunit γ gene was amplified by PCR (Tables S1 and S2†) using the forward primer (5ʹCCGCATATGTTGCATTTCACAACGAGAGC-3ʹ), and reverse primer (5ʹGCGAAGCTTTTAAACATAGGGTGATTACTGTGA-3ʹ) with the restriction sites, NdeI, and HindIII, for the purpose to clone the gene into pET25-b vector. Agarose gel electrophoresis was performed by using Fermentas, GenRuler DNA ladder, and Fermatas 6× sample loading dye. Using DNA Gel Extraction Kit, DNA was extracted and its concentration was measured by NanoDrop 2000 spectrophotometer (Thermo Fisher, USA). GenElute™ Plasmid Miniprep Kit was used to isolate the commercially available blank pET25b vector. Restriction sites were produced using NEB HindIII, and NEB NdeI enzymes in the pET25b vector and the amplified PCR products (gene), with incubation of 2 h at 37 °C, followed by 20 min heat deactivation at 80 °C (Table S3†). Qiagen Mini Elute Reaction Cleanup Kit was used to purify the gene product and the digested vector. T4 DNA ligase was used to ligate the urease subunit γ gene into the pET25b vector at 24 °C for 1.5 h using Invitrogen kit protocol (Cat. No. 15224-017) (Table S4†), and stored at −20 °C prior to transform in E. coli DH5α.Transformation of urease subunit γ: pET25b clone in E. coli DH5α, and E. coli BL21 (DE3) cells
After ligation of gene into the cloning vector, the plasmid was transformed into the E. coli DH5α, and E. coli BL21(DE3) cells according to the given CaCl2 heat shock transformation guidelines. Transformed culture was then plated on ampicillin (100 μg mL−1) (Biobasic Inc., Canada) supplemented L.B. agar plates, and incubated overnight at 37 °C. The positive colonies were picked and sequenced, allowed to grow in L.B. media and glycerol stocks were made. Further, colony PCR (Tables S5 and S6†) and DNA sequencing by Sanger's method (Macrogen Inc., Korea) was performed for the confirmation of positive clones.Expression of urease subunit γ
E. coli BL21(DE3) cells transformed positive clones were grown in L.B. media for the expression of urease subunit γ. After induction with 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) at O.D.600 ∼ 0.6, it was incubated for another 18–20 h at 18 °C and 180 rpm before harvesting by centrifugation at 8000 rpm for 15 min at 4 °C, and the harvested cells was stored at −80 °C until next use.Purification of urease subunit γ protein
Cell lysis was performed by ultra-sonication using buffer of pH 6.6 (20.0 mM Bis-Tris, 20.0 mM NaCl, and 1.0 mM PMSF). The buffer was selected based on protein's pI value i.e. 5.68, and the lysate was then centrifuged for 40 min at 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 × g, 4 °C. The supernatant was loaded on the equilibrated anion exchange column (Hi-Trap Q column, GE Healthcare, U.K.), and the protein was eluted with the linear gradient using 1.0 M NaCl.
600 × g, 4 °C. The supernatant was loaded on the equilibrated anion exchange column (Hi-Trap Q column, GE Healthcare, U.K.), and the protein was eluted with the linear gradient using 1.0 M NaCl.
The eluted fractions were analyzed by SDS-PAGE and further concentrated till 5 mL. Then the sample was loaded onto 75 pg Hi-load 26/600 Superdex column (GE Healthcare, U.K.) and further purified using pH 6.5 buffer (20 mM sodium phosphate, 20 mM NaCl). The eluted protein fractions were concentrated, and quantified by using NanoDrop (Thermo Fisher Scientific, USA).
Compound library and screening procedure
For mixture analysis, a total of 100 compounds (US-FDA approved drugs) from the in-house library of the PCMD Molecular Bank were screened by grouping them into 20 mixtures (Table S7†). Mixtures were prepared by combining 5 mM of each compound dissolved in deuterated buffer (20 mM sodium phosphate, 20 mM NaCl, pH 6.5). The mixtures were screened at 1 mM while 50 μM of urease subunit γ was used. By employing STD-NMR experiments, mixtures were screened for potential binders against urease subunit γ from MRSA 252. This strategy efficiently reduced the experimental time to screen the library of compounds. Further, the compounds interacting in mixture were analyzed individually by STD-NMR experiments.STD-NMR studies
The STD-NMR experiments were recorded on Bruker Avance NEO 600 MHz spectrometer (Switzerland), equipped with a cryogenically cooled probe and an automated SampleCase™. Topspin 4.5 NMR was used to process spectra. stdiffesgp.3 Pulse program from Bruker library was used to perform the STD-NMR experiments by employing excitation sculpting for water suppression.19 512 Scans for mixtures and 2048 scan for individual interacting compounds were recorded to analyze the STD effect. All the experiments were recorded at 298 K by giving the selective saturation (on-resonance) at 45.062 Hz for 5 s with an interpulse delay of 10 s and the off-resonance was set at −20,000 Hz. For STD-NMR screening, first the experiments were recorded in the absence of protein (control), and then in the presence of protein. By subtracting the on-resonance spectra from the off-resonance spectra, difference spectrum was obtained. Amplification factor for the ligand protons, which showed the interaction with protein, was calculated by using the formula:where, Io and Isat are signal intensities of protons in the off- and on-resonance STD-NMR spectrum, respectively.
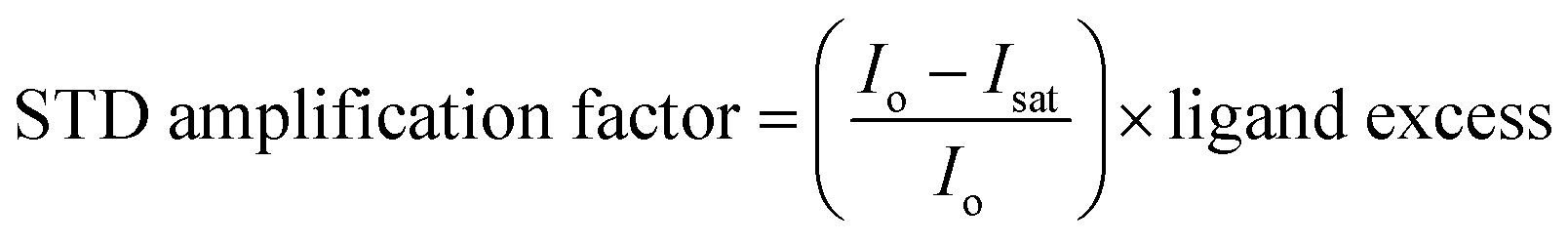
Homology modeling
Since, the single-crystal X-ray structure of urease subunit gamma was not found in the protein data bank (PDB), template based homology modeling was performed using the online available software Swiss-Model.20Molecular docking studies
Molecular docking was performed using Maestro Schrödinger (2023-2) Glide module 6.9 for predicting the protein ligand interactions at the atomic level. The homology model was built using the crystal structure of urease subunit gamma (PDB ID: 4FUR). The protein preparation wizard was used to prepare and minimize the protein. OPLS4e force field assigned the missing protons and partial charges and PROPKA predicts the protein pKa. Using the prime module, the missing loops and zero order bonds were assigned. The LigPrep tool was used to prepare the ligands by assigning relevant protonation states, and altering their torsions. Ligands ionization and tautomeric states were generated by using Epik prime tool. The results were analyzed by using the Glide_XP module for the best docked pose.Binding energy estimation by MM-GBSA analysis
MM-GBSA (Molecular mechanics-generalized Born surface area) is commonly used to estimate the ligand binding affinity. In order to predict the binding affinity of the ligands, prime MM-GBSA tool in Schrödinger (2023-2) was used. This tool re rank the Glide XP module docked conformations, and estimates the relative binding energy of the ligands. The more negative binding energy (kcal mol−1) reflects stronger binding affinity of ligand.MD simulation studies
MD simulation studies were performed by using the Desmond molecular dynamics within the Schrödinger package for a comprehensive investigation of stability of ligand–protein complexes. Initially, the selected complexes were assigned to an SPC (simple point charge) water box, extending 10 Å beyond the complex's atoms. To neutralize charges, counter ions (33 Na+ and 29 Cl−) were added, and the salt concentration was set to 0.15 M sodium, and chloride ions to approximate physiological conditions. The MD simulation in the NPT ensemble was conducted at a temperature of 300 K and 1.63 bar pressure over 100 ns. The OPLS-3e force field was employed, and plots and figures were generated using the Desmond simulation interaction diagram tool of Maestro.21Thermal shift assay (TSA)
Thermal shift assay was performed to analyze the effect of the drugs on the melting temperature of UreA. All the samples were prepared in 20 mM sodium phosphate buffer with 20 mM NaCl and pH 6.5. Each sample contained UreA (20 μM), ligand (drug) (2 mM), and Sypro®Orange dye 2 μL (1:1000; Invitrogen Life Technology, USA). Samples were prepared, and heated from 20 °C to 95 °C at rate of 0.3 °C min−1 using Bio-Rad Real-time PCR machine (Bio-Rad Laboratories, USA). During the temperature increment, HEX channel was used to measure the sypro-orange fluorescence intensity as a function of temperature. By comparing the melting temperature (Tm) at which half of the protein population is unfolded, differences in the thermal stability of protein were determined in the absence (control) and presence of drug molecule. Furthermore, the dissociation constant of nicotinamide was determined by observing the change in protein melting temperature at different concentrations. The concentrations used for the determination of Kd were 0.01 mM to 100 mM (0.01, 0.02, 0.05, 0.1, 0.2, 0.5, 0.7, 1, 2, 5, 7, 10, 20, 50, 70, and 100 mM) with 20 μM UreA.
Urease inhibition assay
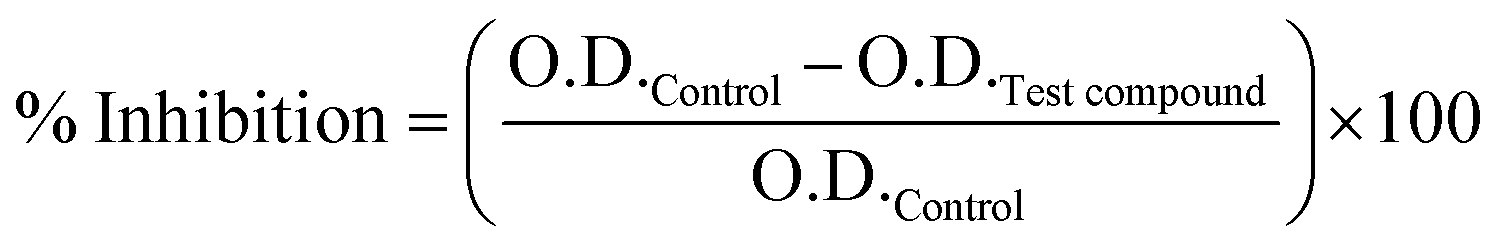
Urease inhibition assay was performed to analyze the enzyme inhibitory activity of the drug molecules that has shown interactions in STD-NMR studies. Jack bean (Canavalia ensiformis) urease was used in this study, because urease subunit gamma (MRSA 252) was not enough to evaluate the urease inhibitory activity. As it is one of the subunit of urease enzyme, the catalytic activity requires the whole enzyme. Jack bean urease is a fully functional enzyme with 49% sequence similarity with the urease subunit gamma (Table S7†).Jack bean urease (2.92 mg) was dissolved in 2.5 mL of sodium phosphate buffer (20 mM, pH 6.8). Urease enzyme (25 μL) was incubated with 5 μL of the drug (0.5 mM), and incubated for 15 min at 30 °C. Further, 55 μL of substrate urea (0.1 M urea in phosphate buffer) was added, and incubated for 15 min at 30 °C. After 15 min incubation, 45 μL phenol reagent (1% w/v phenol, 0.005% w/v sodium nitroprusside), and 70 μL of alkali reagent were added (0.5% w/v sodium hydroxide, and 0.1% sodium hypochlorite) in the reaction mixture. All the reactions were performed in triplicates, and the absorbance of the reaction mixtures, measured at 630 nm, was used to calculate the % inhibition of the drug molecules:
Results and discussion
Urease subunit γ gene of methicillin-resistant Staphylococcus aureus was cloned in pET25-b vector, and expressed by using recombinant DNA technology22 using the T7-based expression system of E. coli BL21(DE3) cells. The expressed protein was purified by anion exchange and gel filtration chromatography. A total of 100 drugs (grouped into 20 mixtures) were screened against urease subunit γ using STD-NMR experiments. Fifteen drugs from 11 mixtures have shown interactions with urease subunit γ, and these were further validated individually by STD-NMR experiments, showing that they have the ability to interact with urease subunit γ. Further, molecular docking and MMGBSA analysis was performed to analyze the interactions between urease subunit γ and drug molecule in their physiochemical state, and to predict the binding affinity of drugs, respectively.Cloning, expression and purification
The urease subunit γ gene was successfully cloned in pET25b vector (Fig. S1†), and transformed in the E. coli BL21(DE3) expression cells (Fig. S2†). The expression of the urease subunit gamma was found to be high, and on SDS-PAGE gel as it appears in the soluble lane at the estimated molecular weight of 11.2 kDa (Fig. S3†). Initially, the expressed protein was purified using anion exchange chromatography (Hi-Trap Q column) (Fig. S4†). Following, size exclusion chromatography on 75 pg 26/600 Superdex column, the protein was eluted at 162 mL, and appeared to be trimeric in nature (Fig. S5†). The yield of the purified urease subunit gamma was 24 mg mL−1 in 1 L of culture.STD-NMR studies
Conventional drug development is a long and costly process that takes 10–12 years, and two billion dollars investment for a drug to be approved by drug regulatory authorities. Drug repurposing is a smart approach that identify the new therapeutic uses of an old drug. This approach has many advantages over conventional drug discovery, including time, and cost efficiency. Hence, in the current study US FDA-approved drugs, available in the Drugs Bank of Dr Panjwani Center of Molecular Medicine and Drug Research (PCMD), were used to analyze their binding affinities with the purified urease gamma subunit of MRSA.Various biophysical techniques are used to study the protein–ligand interactions, such as Circular Dichroism (CD), Isothermal Titration Calorimetry (ITC), and Surface Plasmon Resonance (SPR) and many others. Saturation Transfer Difference-Nuclear Magnetic Resonance (STD-NMR) is a robust approach to identify the ligands with moderate to weak affinity. The technique is ligand based, and work on the principle of nuclear Overhauser effect (NOE). The protein is selectively saturated and the magnetization is transferred via spin diffusion to the whole protein, and also to the bound ligand. As soon as ligand is dissociated, the magnetization is detected in the solvent (buffer system).
The drugs were screened by using STD-NMR technique in the form of mixtures, and the ligands that showed binding with protein were further analyzed individually to map the ligand proximity to the protein i.e. group epitope mapping (GEM). Protons of the ligand which receive the highest degree of saturation indicate their close proximity to the protein. For GEM analysis, the proton receiving the highest STD integral value was set to be 100%, while the other protons' STD integrals were normalized against the most intense signal.23 From 20 mixtures, drugs from mixtures 1–2, 6–9, 11–13, 15, and 20 showed their interactions with protein, identified as the potential binders, and further evaluated individually. Finally, 15 drugs (Table 1) showed interactions with the receptor protein (urease subunit γ).
| Compound | Drug name | Structure |
|---|---|---|
| 1 | Nicotinamide | 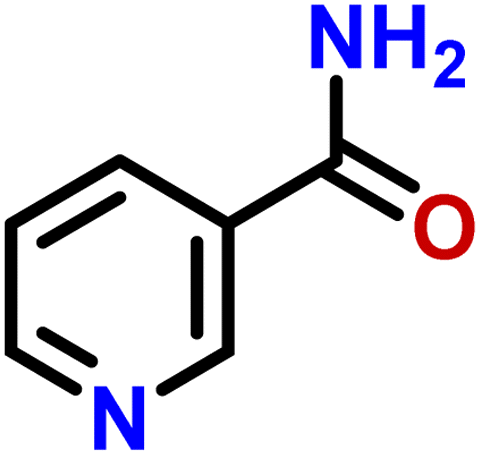 |
| 2 | Drotaverine HCl | 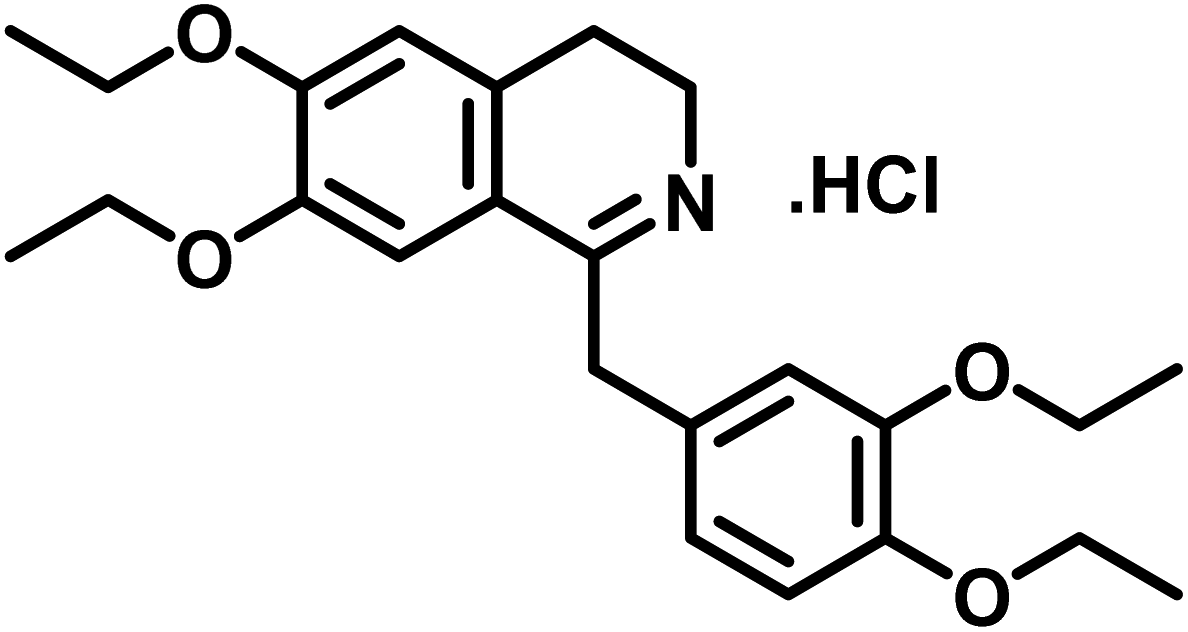 |
| 3 | Clidinium bromide | 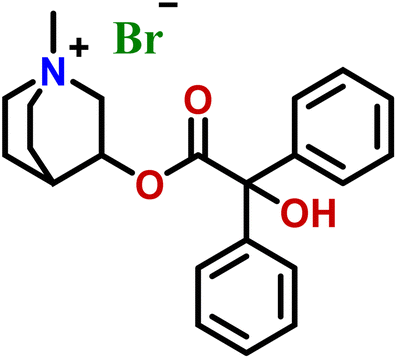 |
| 4 | Sulfanilamide | 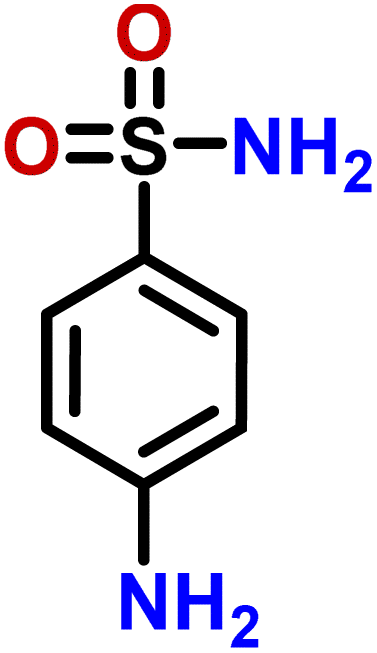 |
| 5 | Nicorandil | 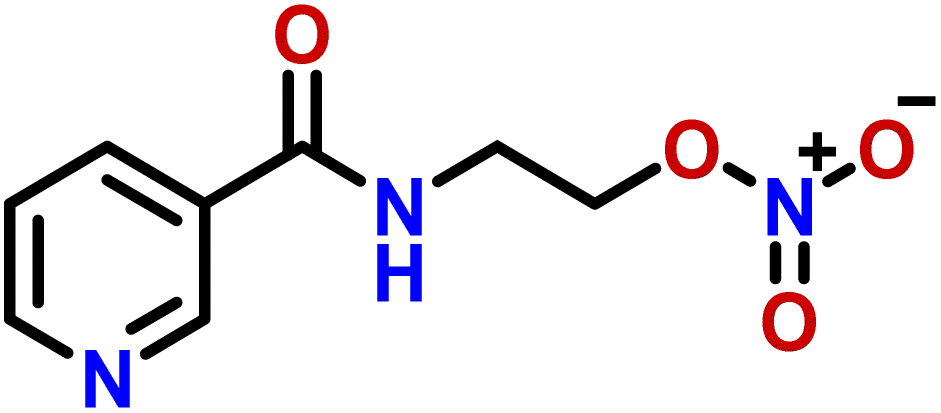 |
| 6 | Hydroxychloroquine sulphate | 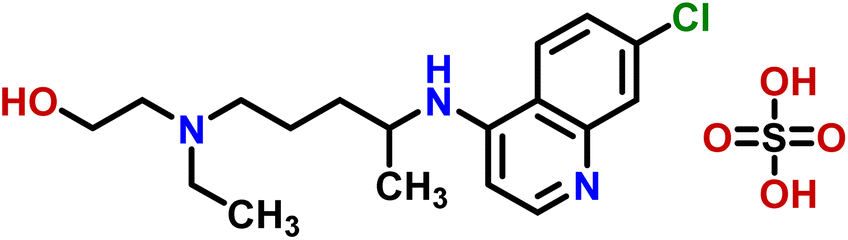 |
| 7 | Glucosamine HCl |  |
| 8 | Ephedrine |  |
| 9 | Penicillin G sodium |  |
| 10 | Tramadol hydrochloride |  |
| 11 | Nicotinic acid |  |
| 12 | Salbutamol sulfate |  |
| 13 | Bupropion hydrochloride |  |
| 14 | Tenofovir disoproxil |  |
| 15 | Rasagiline mesylate | 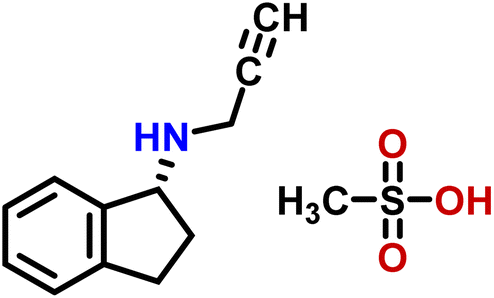 |
Compound 1 (nicotinamide), an active soluble form of vitamin B3, possess antioxidant and neuroprotective properties, and used for the treatment of vitamin B3 deficiency (pellagra), acne, non-melanoma skin cancer, skin aging, skin discoloration, and other pathological conditions.24 In this compound, H-2 of the aromatic ring received the maximum 100% saturation from the protein, while H-6 and H-5 received the 66 and 45% relative saturation, respectively (Fig. 1). The GEM analysis revealed that the H-2 lie in the closer proximity of protein.
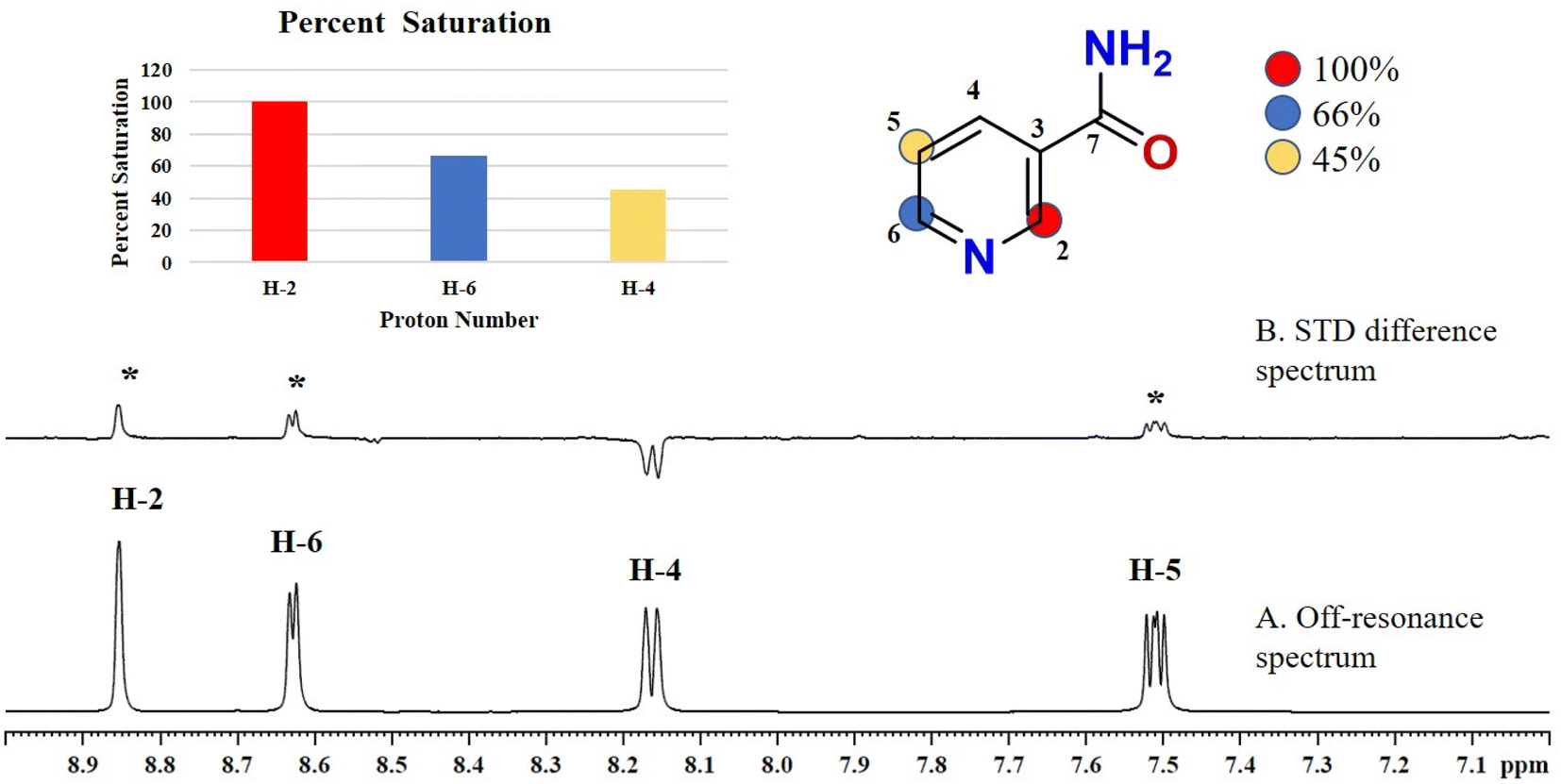 | ||
| Fig. 1 STD-NMR spectra of drug nicotinamide (1): (A) reference spectrum of the drug. (B) STD difference spectrum of nicotinamide in presence of urease subunit gamma, indicating the interacting protons of drug (protons interacting with protein are marked with ‘*’). (C) Graphical representation of percent saturation of drug proton receiving from protein. | ||
Compound 2 (drotaverine hydrochloride) is an anti-spasmodic drug used to relieve smooth muscles' spasmodic pain.25 Its GEM analysis indicated that CH2-18 received the highest saturation from the protein. While the rest of the protons were assigned relative saturation. CH2-20, CH2-22 and CH2-24 received 71.6% saturation from the protein. Further, the aromatic H-8, H-5, CH2-13/CH2-16 and CH2-17 received 79.8, 83.3, 56.3 and 40.2% saturation from the protein, respectively (Fig. 2). CH2-3 and CH2-4 received 21.3 and 15.4% relative saturation, respectively, from the protein. The methyl's, CH3-19, CH3-21/CH3-23 and CH3-25 received 44.3, 43, and 31% saturation, respectively. This GEM analysis revealed that CH2-18 is closer to the protein proximity attaining the highest saturation from the protein.
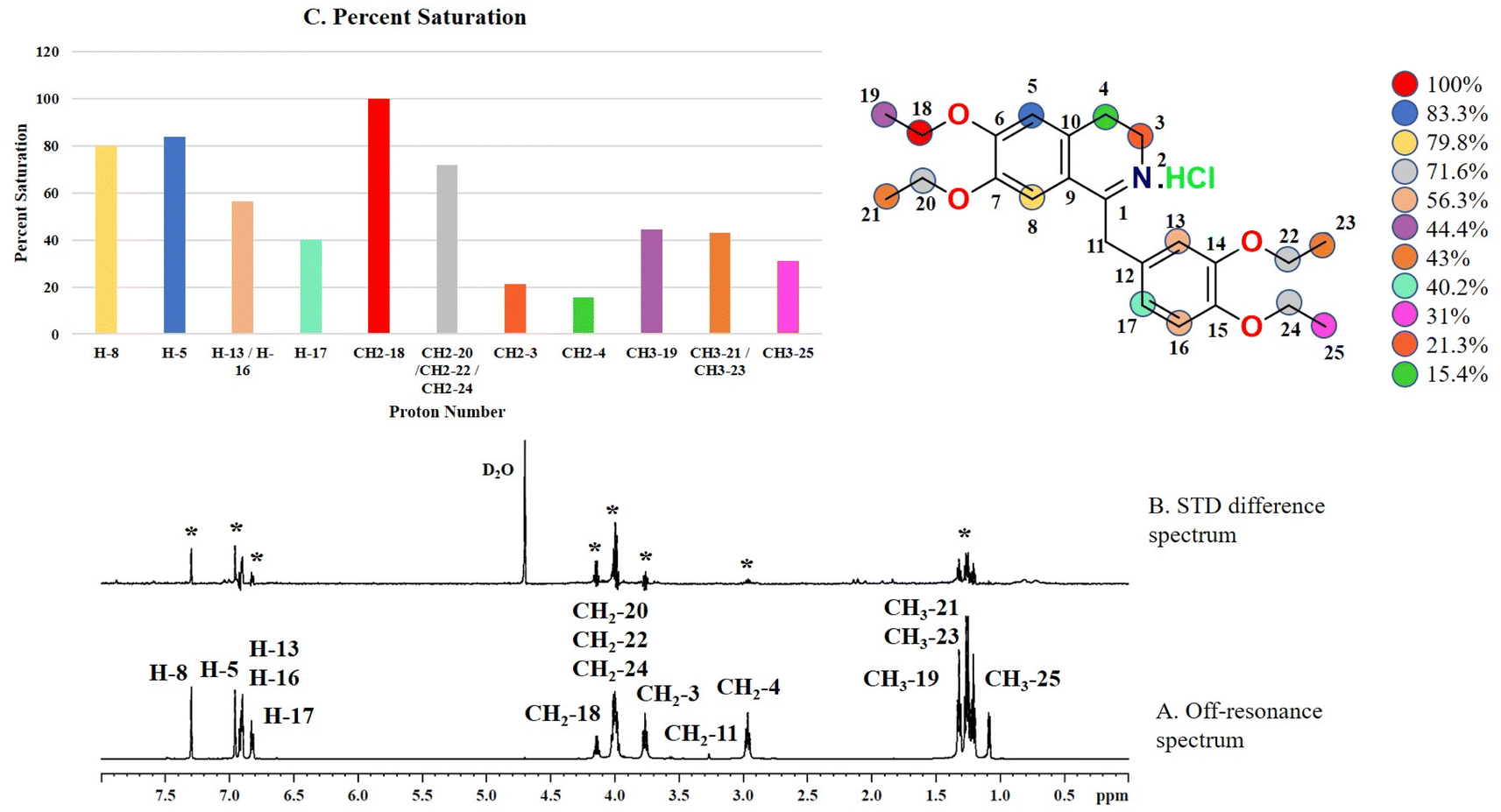 | ||
| Fig. 2 STD-NMR spectra of drug drotaverine HCl (2): (A) reference spectrum of the drug. (B) STD difference spectrum of drotaverine HCl in the presence of urease subunit gamma, indicating the interacting protons of drug (protons interacting with protein are marked with ‘*’). (C) Graphical representation of percent saturation of drug protons receiving from protein. | ||
Compound 3 (clidinium bromide), an anticholinergic drug, used as an antispasmodic agent to relieve abdominal cramps by inhibiting acetylcholine muscarinic action.26 GEM analysis indicated that the protons of the aromatic rings (H-4/H-5/H-6/H-7/H-8 and H-10/H-11/H-12/H-13/H-14) received the maximum saturation, and were in closer proximity to the protein (Fig. 3).
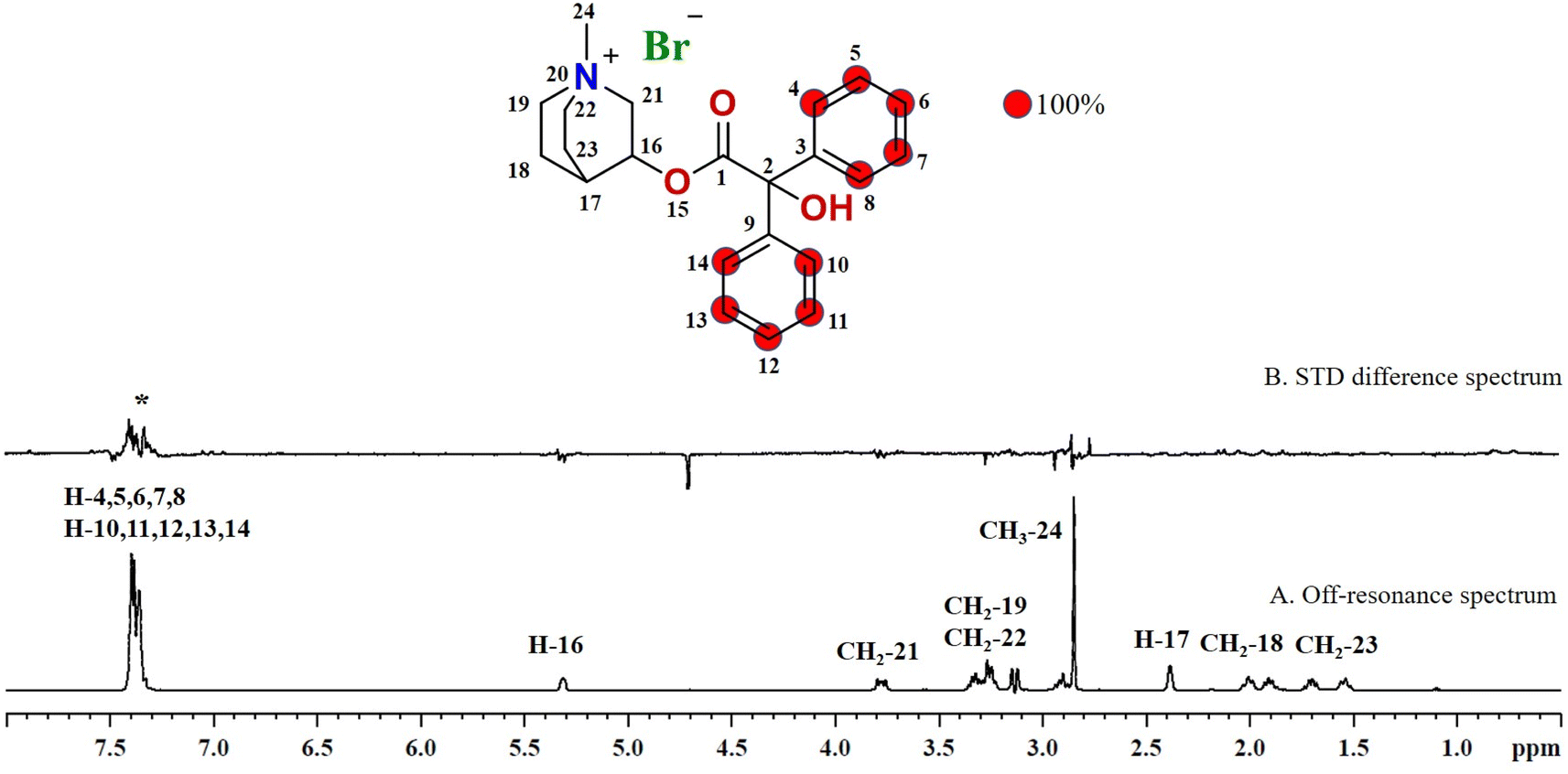 | ||
| Fig. 3 STD NMR spectra of drug clidinium bromide (3): (A) reference spectrum of the drug. (B) STD difference spectrum of clidinium bromide in presence of urease subunit gamma, indicating the interacting protons of drug (protons interacting with protein are marked with ‘*’). | ||
Compound 4 (sulfanilamide), a precursor of sulfonamide drugs, is a synthetic antimicrobial agent used for inhibiting folic acid synthesis in bacteria to prevent their growth and multiplication.27 As compound 4 is a small molecule with single heterocyclic ring, all the aromatic protons showed interactions with protein in STD difference spectrum. The GEM analysis indicated that H-3 and H-5 received 100% saturation indicating their closer proximity with the protein (Fig. 4). While H-2 and H-6 received 92% relative saturation from protein.
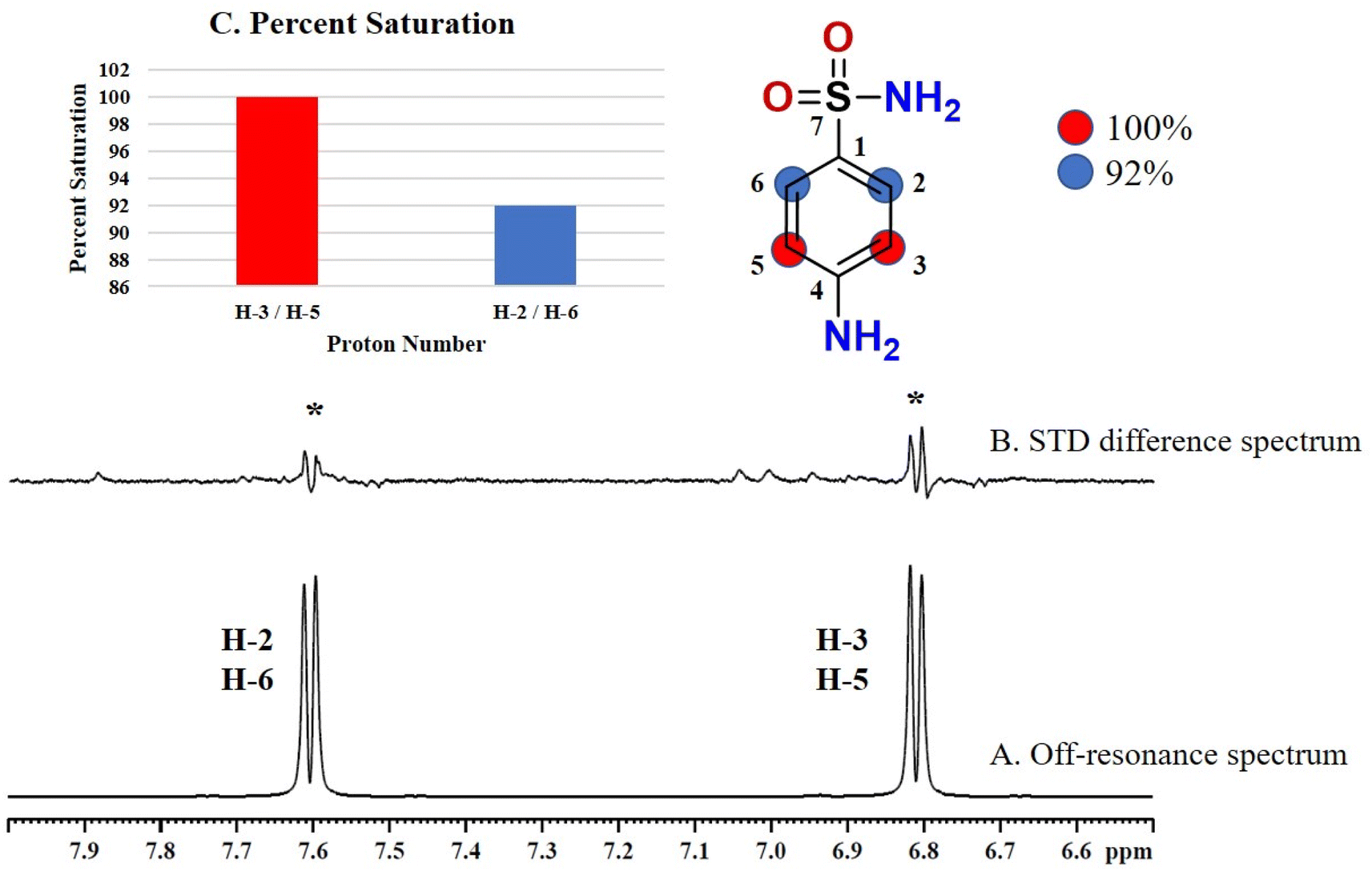 | ||
| Fig. 4 STD NMR spectra of drug sulfanilamide (4): (A) reference spectrum of the drug. (B) STD difference spectrum of sulfanilamide in presence of urease subunit gamma, indicating the interacting protons of drug (protons interacting with protein are marked with ‘*’). (C) Graphical representation of percent saturation of drug protons receiving from protein. | ||
Compound 5 (nicorandil) is a vasodilator drug used for the treatment of angina. It works by relaxing the vascular smooth muscles to control high blood pressures.28 In this compound, the GEM analysis indicated that the aromatic ring H-2 received 100% saturation from the protein (Fig. S7†). In comparison, H-6 and H-5 received 63.5 and 31.3% saturation, respectively. The results indicated that the H-2 is in closer proximity of the protein.
Compound 6 (hydroxychloroquine sulphate) is an antirheumatic drug used for the treatment of malaria, autoimmune, and viral diseases.29 It also possesses antibacterial activity, and act by alkalizing the bacterial intracellular organelles thereby decreasing the bacterial survival and multiplication rate.30 Through GEM analysis, it was found that the H-8 of the aromatic ring received 100% saturation from the protein (Fig. S8†). Whereas, H-6, H-2, and H-5 received 49, 42 and 34.3% relative saturation from the protein, respectively. Thus the analysis indicated the close proximity of H-8 with the protein.
Compound 7 (glucosamine HCl) is an anti-inflammatory drug used for the treatment of rheumatoid arthritis and osteoporosis.31 This antirheumatic drug has also been reported for antibacterial activity against the S. aureus.32 The GEM analysis revealed that the H-2 received 100% saturation from the protein (Fig. S9†). Whereas, the H-7 and H-7′ received 67 and 54% relative saturation from protein, respectively. The STD spectrum indicated that the H-2 is in the closest proximity of the protein.
Compound 8 (ephedrine) is an adrenergic agonist drug, used for the treatment of hypotension, asthma, and nasal decongestants.33 It also has a significant antimicrobial effect on S. aureus growth.34 The GEM analysis of this compound revealed that H-3, H-4, and H-5 received highest saturation, and were in closer proximity of the protein (Fig. S10†). The aliphatic H-9 received 37%, and the aromatic ring H-2 and H-6 received about 30% relative saturation from the protein.
Compound 9 (penicillin G sodium) is an antimicrobial agent used to inhibit the bacterial cell walls synthesis, leading to the destruction of susceptible bacteria during infections.35 According to the GEM analysis, H-6 was in closer proximity to the protein, receiving the highest saturation, while the H-5 received 59% relative saturation (Fig. S11†).
Compound 10 (tramadol hydrochloride) is a synthetic opioid analgesic used to relieve severe to moderate pain, it binds with the opioid receptors in the central nervous system.36 This opioid analgesic is also reported for its in vitro activity against the S. aureus.37 The GEM analysis of the compound 10 indicated that H-8 and H-12 of aromatic ring received the highest saturation from the protein, while H-10 has received 35.9% relative saturation from protein. This indicated that H-8 and H-12 were closer to the protein (Fig. S12†).
Compound 11 (nicotinic acid) is a form of vitamin B3, used for the treatment of pellagra. It has the ability to maintain the human cholesterol level.38 It can also facilitate the neutrophils to perform their functions more efficiently at the site of infection.39 The drug showed weak STD interaction, as only H-5 of the aromatic ring showed STD effects, while rest of the protons did not show any STD signals (Fig. S13†).
Compound 12 (salbutamol sulfate) is a β2-adrenergic receptor agonist used as bronchodilator for the treatment of asthma and COPD. It relaxes the smooth muscles of the lungs airways.40 In 2015, Vandevelde et al. reported for the first time that the salbutamol may contribute in the biofilm eradication of the S. pneumococcus.41 For compound 12, the GEM analysis indicated that aromatic H-3 received the highest saturation, indicating its closer proximity to the protein (Fig. S14†). While, H-5 received 74.9% relative saturation.
Compound 13 (bupropion hydrochloride) acts as an antidepressant by increasing the level of dopamine and serotonin in the brain, and also helps smokers to overcome their nicotine addiction.42 According to GEM analysis, H-2 of the aromatic ring is in closer vicinity of the protein by receiving the highest saturation, and H-4 and H-5 received 43.3 and 30.3% relative saturation, respectively (Fig. S15†).
Compound 14 (tenofovir disoproxil) is a nucleoside reverse transcriptase inhibitor, used for the treatment of HIV, and hepatitis B infections.43 The GEM analysis revealed that the methylene protons, such as CH2-14 and CH2-19 and H-16/H-21 were in closer proximity as they received the maximum saturation, while the H-8 received 11.8% and CH3-17, CH3-18, CH3-22 and CH3-23 received 9.8% relative saturation from protein (Fig. S16†).
Compound 15 (rasagiline mesylate), a monoamine oxidase B, selective irreversible inhibitor, is used for the treatment of Parkinson's disease by maintaining the dopamine levels in the CNS.44 In this compound, maximum saturation was received by the H-12 i.e. 100% and was used to normalize the other protons (Fig. S17†). In comparison, the aromatic ring H-1, and H-3 received 56.6 and 29.3%, respectively. Whereas, the H-2 and H-4 of aromatic ring received 27.2% relative saturation. The GEM analysis showed that the H-12 was in the close proximity of the protein.
According to GEM analysis of the interacting drugs, it is predicted that mostly the protons of the aromatic ring have high affinity and interact with the urease subunit gamma. While the protons at the aliphatic region showed a weak interaction with the protein.
Molecular docking studies
Molecular docking studies were performed for the prediction of protein–ligand interactions at atomic level. Since 3D structure of urease gamma subunit has not been elucidated till date, the homology model was built using crystal structure of urease subunit gamma 2 from Brucella melitensis biovar Abortus 2308 (PDB ID: 4FUR), that showed 52% similarity with the urease subunit gamma of MRSA252.Molecular docking studies provided further insights about the interactions of drug molecules with various amino acids of the urease subunit gamma. The docking scores were in the range of −6.024 to −3.673. All the drugs interacted via various non-covalent interactions with enzyme, such as hydrogen, aromatic hydrogen bonds, π–π stacking, π–cationic interactions, and salt bridge formation (Table 2).
| Drug | Name | Amino acid | Molecular interactions with protein | Docking score | Binding energy, kcal mol−1 |
|---|---|---|---|---|---|
| 1 | Nicotinamide | Thr85 | Hydrogen bond | −5.979 | −28.81 |
| 2 | Drotaverine HCl | Thr85 | Aromatic H-bond | −3.673 | −67.40 |
| 3 | Clidinium bromide | Lys10, Arg23 | π–cationic interactions | −5.086 | −62.07 |
| Asn31 | Hydrogen bond | ||||
| Glu7 | Aromatic H-bond | ||||
| 4 | Sulfanilamide | Glu7, Ala16 | Hydrogen bond | −6.311 | −26.89 |
| 5 | Nicorandil | Lys10, Arg23, Thr85 | Hydrogen bond | −4.320 | −33.02 |
| Lys10 | Salt bridge | ||||
| 6 | Hydroxy-chloroquine sulphate | Glu34, Thr85 | Hydrogen bond | −5.907 | −50.69 |
| Leu44, Thr85 | Aromatic H-bond | ||||
| Glu34 | Salt bridge | ||||
| 7 | Glucosamine hydrochloride | Glu7, Arg23, Asn31, Glu34 | Hydrogen bond | −4.713 | −26.69 |
| 8 | Ephedrine | Glu7 | Hydrogen bond | −6.204 | −40.53 |
| Phe3 | π–π stacking interactions | ||||
| Arg23 | π–cationic interactions | ||||
| Ala16 | Aromatic H-bond | ||||
| 9 | Penicillin G sodium | Lys10, Asn31, Thr85 | Hydrogen bond | −6.565 | −49.57 |
| Lys10 | Salt bridge | ||||
| Phe3 | π–π stacking interactions | ||||
| 10 | Tramadol HCl | Arg23 | π–cationic interactions | −5.930 | −62.14 |
| 11 | Nicotinic acid | Lys10 | Hydrogen bond | −5.368 | −22.05 |
| Lys10 | Salt bridge | ||||
| Leu44 | Aromatic H-bond | ||||
| 12 | Salbutamol sulfate | Glu34, Thr85 | Hydrogen bond | −6.236 | −58.77 |
| Glu34 | Salt bridge | ||||
| 13 | Bupropion HCl | Thr85 | Halogen bond | −5.113 | −53.79 |
| 14 | Tenofovir disoproxil | No interaction | −4.299 | −64.59 | |
| 15 | Rasagiline mesylate | Phe3 | π–π stacking interactions | −4.969 | −43.53 |
| Ala16 | Aromatic H-bond | ||||
| Arg23 | π–cationic interactions |
Compounds 1 (nicotinamide), and 2 (drotaverine hydrochloride) interacted with Thr85 via hydrogen and aromatic hydrogen bonds, respectively (Fig. S18, and S19†). Compound 3 (clidinium bromide) interacted with Asn31 and Glu7 via hydrogen, and aromatic hydrogen bonding (Fig. 5). The phenyl rings of the compound 3 interacted with the Lys10 and Arg23 via π–cation interactions.
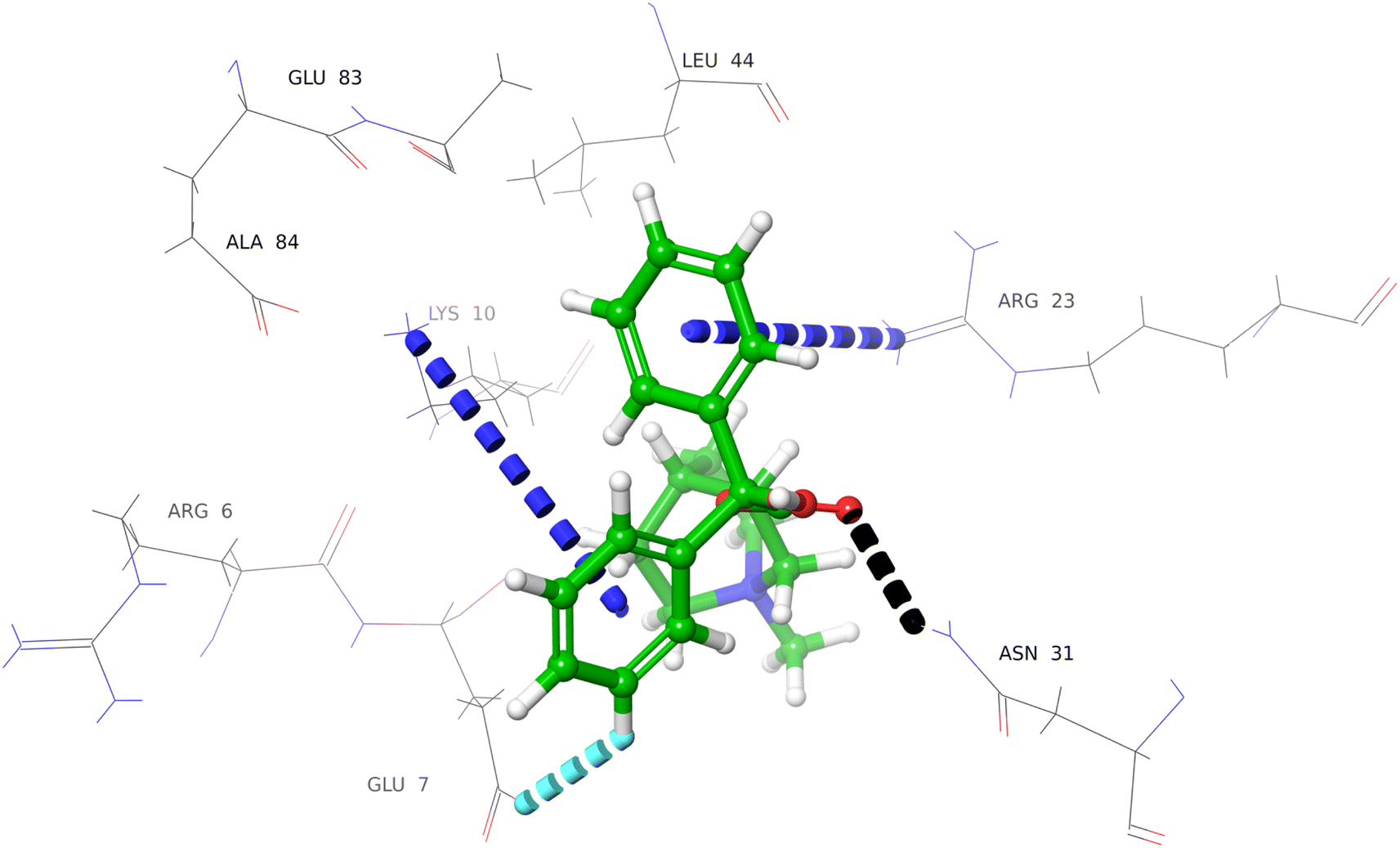 | ||
| Fig. 5 Docked pose of clidinium bromide (3) with urease subunit gamma. Protein–ligand interaction depicted in 3D representation (dotted lines), representing aromatic hydrogen bonding (light blue), π–cationic interaction (blue), and hydrogen bonding (black). | ||
Compound 4 (sulfanilamide) interacted via hydrogen bonds with the Glu7 and Ala16 residues of urease subunit gamma (Fig. S20†). Compound 5 (nicorandil) interacted with Lys10, Arg23, and Thr85 by hydrogen bonding, and salt bridge were formed with the Lys10 (Fig. S21†). Compound 6 (hydroxychloroquine sulphate) form hydrogen and aromatic hydrogen bonds with Glu34 and Thr85, Leu44 and Thr85, respectively (Fig. S22†). It also interacted with Glu34 by forming a salt bridge. Compound 7 (glucosamine HCl) showed interactions with Glu7, Arg23, Asn31, and Glu34 by hydrogen bonding (Fig. S23†). Compound 8 (ephedrine) interacted via hydrogen and aromatic hydrogen bond with Glu7, Ala16, respectively (Fig. 6). The phenyl ring interacted with Phe3, and Arg23 via π–π stacking, and π–cationic interactions, respectively.
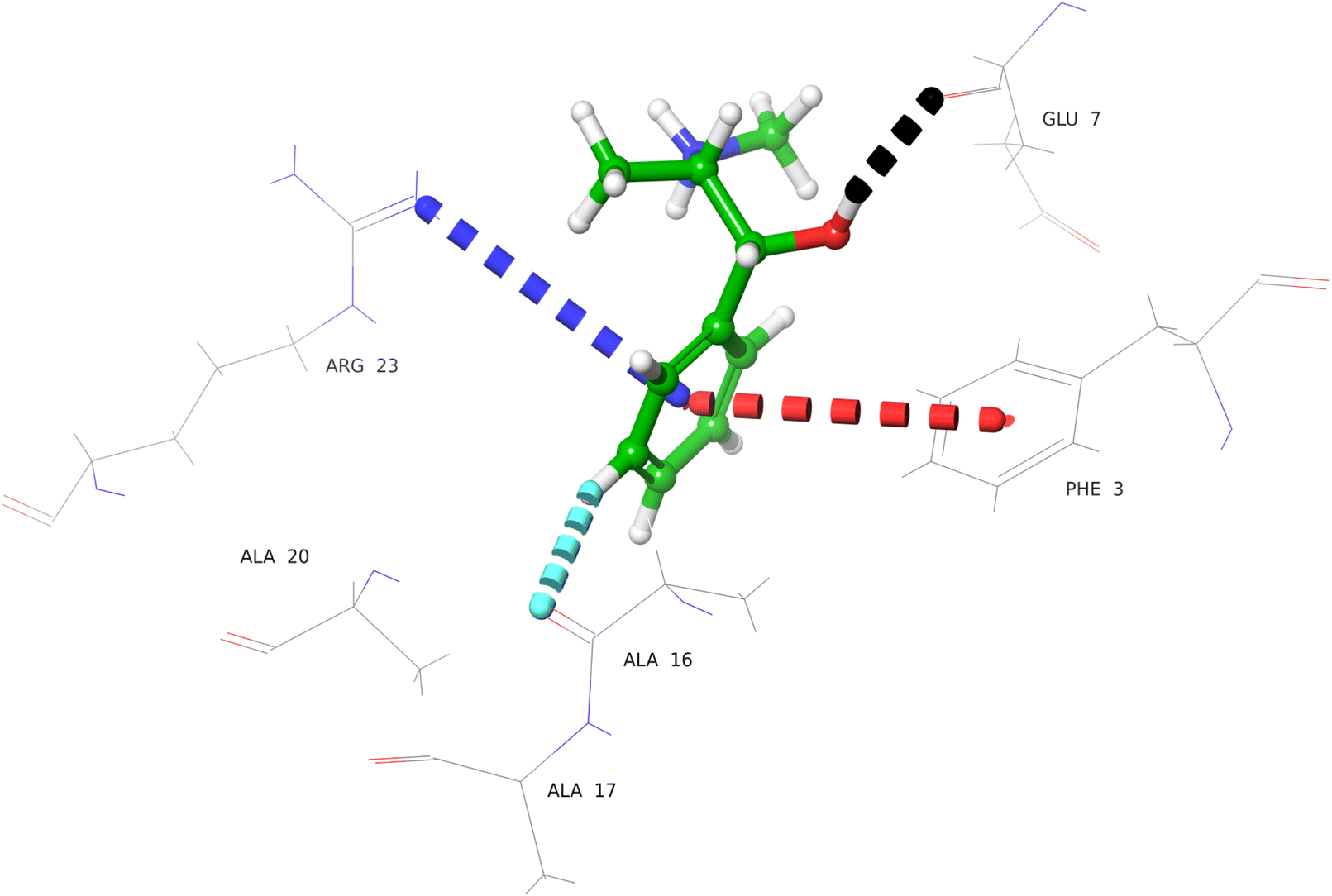 | ||
| Fig. 6 Docked pose of ephedrine (8) with urease subunit gamma. Protein–ligand interaction depicted in 3D representation (dotted lines), representing hydrogen bonding (black), π–cationic interaction (blue), aromatic hydrogen bonding (light blue), and π–π stacking (red). | ||
Compound 9 (penicillin G sodium) interacted with Lys10, Asn31 and Thr85 residues via hydrogen bond (Fig. S24†). The carboxylic oxygen of the drug formed a salt bridge with Lys10, while the phenyl ring interacted with Phe3 by π–π stacking interactions. Compound 10 (tramadol hydrochloride) interacted with the Arg23 via π–cation interaction (Fig. S25†). Compound 11 (nicotinic acid) interacted with Lys10 by hydrogen bonding, and by forming salt bridge and via aromatic interaction with the Leu44 (Fig. S26†).
Compound 12 (salbutamol sulphate) interacted with Glu34 and Thr85 residues of the protein by hydrogen bonds, and also by forming a salt bridge with Glu34 (Fig. S27†). Compound 13 (bupropion hydrochloride) interacted via forming a halogen bond with the Thr85 (Fig. S28†). Compound 14 (tenofovir disoproxil) was able to bind in the urease binding pocket via hydrophobic interactions (Fig. S29†). Compound 15 (rasagiline mesylate) interacted via π–π stacking, and by π–cationic interactions with Phe3, and Arg23, respectively (Fig. S30†). An aromatic hydrogen bond was also formed with Ala16.
Hence, the molecular docking studies further supported the interactions of these drugs with urease subunit gamma. All the drugs interacted mainly via non-covalent interactions, such as hydrogen bond, aromatic hydrogen bonding, π–π stacking, π–cation interactions, salt bridges and halogen bonding. These non-covalent interactions of drugs were formed with the binding site residues of the urease subunit gamma, mostly the Thr85, Lys10, Arg23 and Glu7. Further, MMGBSA analysis was performed to rank the ligands dock pose, and to predict their binding affinity. The binding energy of the ligands were estimated to be high in the range of −67.40 to −22.05 kcal mol−1. Among various drugs, compounds 2, 3, 6, 10, and 12–14 showed higher binding energies for their interactions with urease subunit gamma.
MD simulation studies
Drugs 2, 3, 6, 10, and 12–14 that showed higher binding affinities in docking studies were subjected to MD simulation studies. The stability of UreA–drug complex in a dynamic environment was analyzed through 100 ns molecular dynamics (MD) simulation studies. The RMSD (root mean square deviation) plots of the protein ligand complexes indicated that drugs 2, 3, 9, 10, and 12–14 formed stable complexes with UreA. The drug–UreA complexes of these drugs were stabilized with time evolution and no marked fluctuations were observed during the MD simulation time (Fig. 7, and S31†). Whereas, the UreA–drug complexes (6 and 8) did not stabilize with time evolution and marked fluctuations were observed, indicating that the ligand has diffused out from its binding site (Fig. S32†). Furthermore, the protein–ligand contact maps of the complexes represented various intermolecular interactions (Fig. 8, and S33†). The histogram of the complexes indicated that the protein–ligand interacted mainly by water bridges, H-bond, and ionic interactions. The protein residues Lys10, Glu34, Asn34, and Thr85 were mostly involved for their interactions with binding ligands.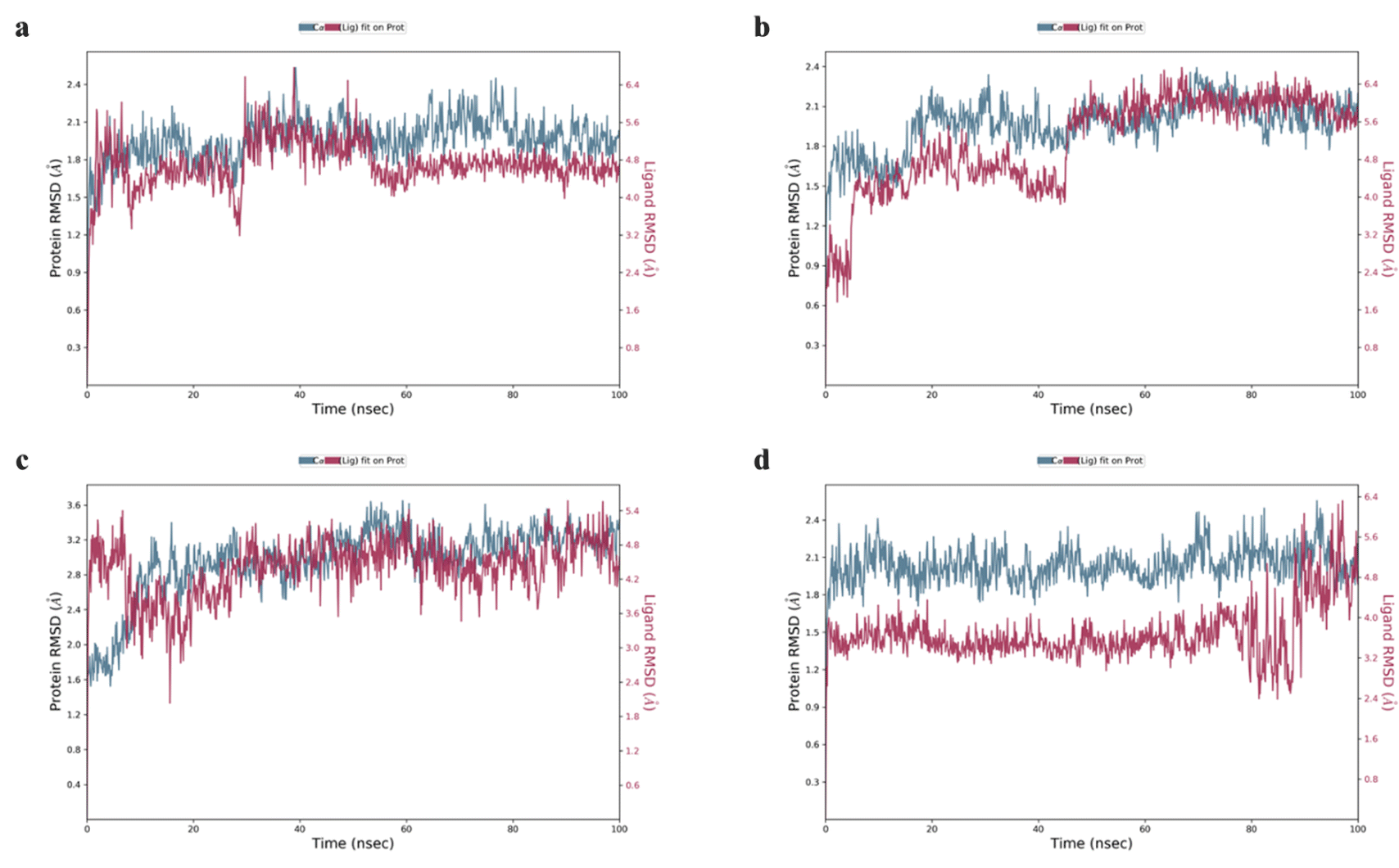 | ||
| Fig. 7 RMSD plots of (a) clidinium bromide (3), (b) tramadol HCl (10), (c) salbutamol sulphate (12), and (d) bupropion HCl (13) with UreA protein. | ||
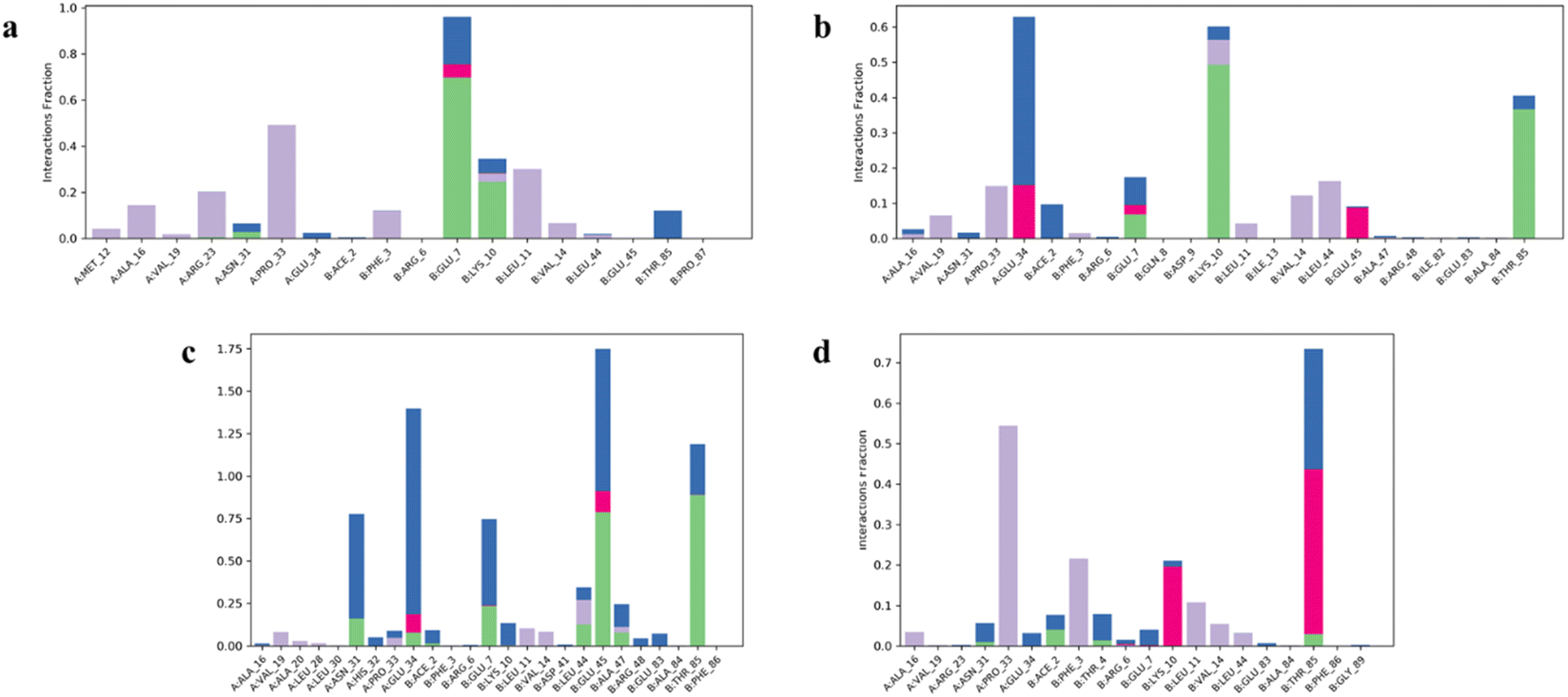 | ||
| Fig. 8 Histogram of (a) clidinium bromide (3), (b) tramadol HCl (10), (c) salbutamol sulphate (12), and (d) bupropion HCl (13) interactions with UreA protein binding site residues. | ||
Thermal shift assay (TSA)
The temperature at which half of a protein unfolds is known as melting temperature (Tm). Tm is used to analyze a protein's thermal profile. When a ligand binds to protein, the free energy of protein–ligand binding may change the melting temperature of the protein. This change in the melting temperature, in the presence of ligand, corresponds to protein–ligand interactions. All the drugs that exhibited interactions with UreA in STD-NMR spectra have shown effect on the melting temperature of UreA. Tm of UreA was determined to be 66.5 °C, without drug. Whereas, all the drugs molecule causes a decrease in UreA Tm between −0.6 to −3.4 °C (Fig. 9). These variations in Tm of UreA during the assay may indicate some conformational changes in its structure. Furthermore, the dissociation constant (Kd) for nicotinic acid was determined to be 70 mM when melting temperature was plotted against the nicotinic acid log concentration (Fig. S34†).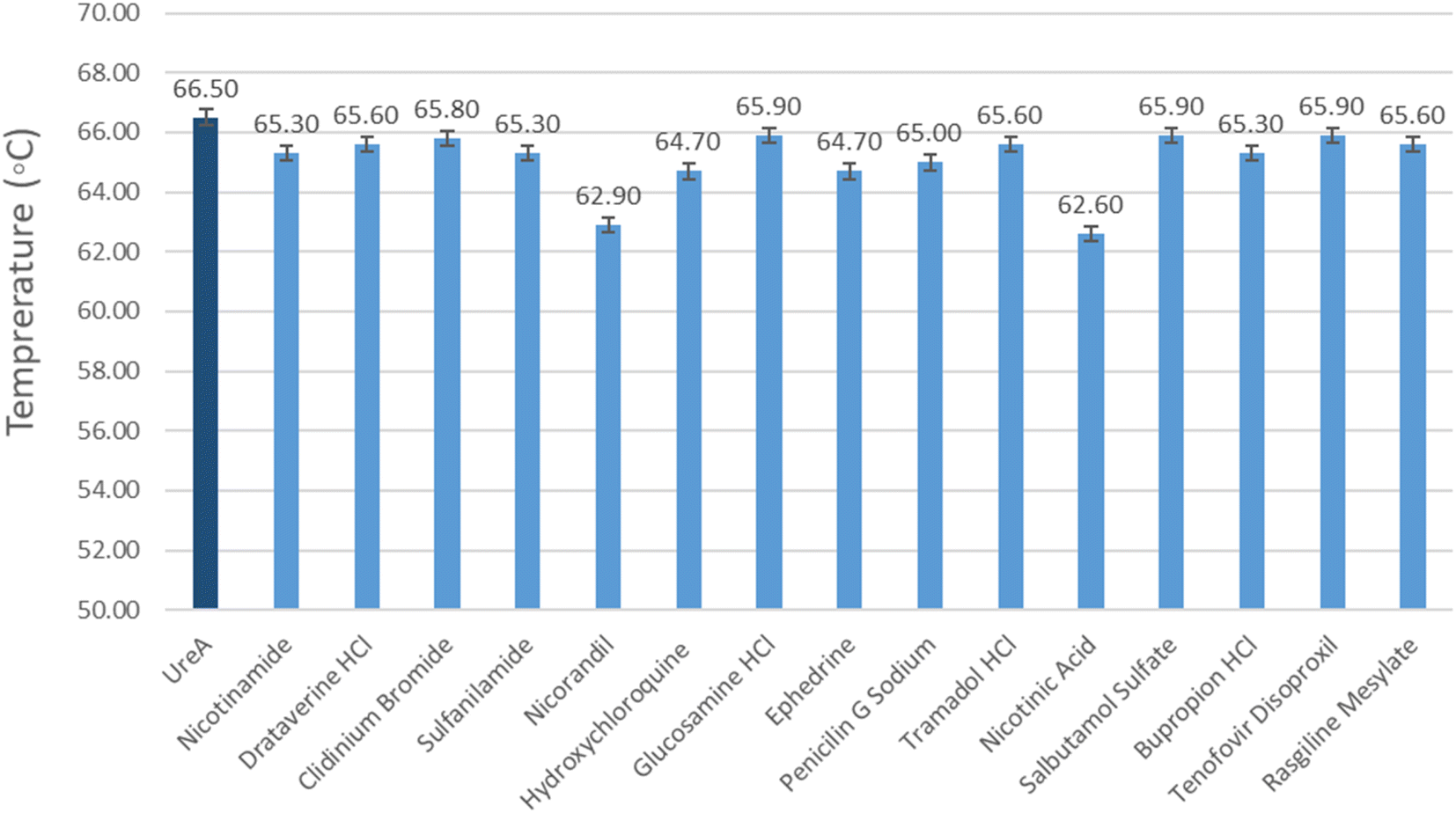 | ||
| Fig. 9 Graphical representation of variations in Tm values of UreA determined by DSF upon addition with the drug molecule. (Dark blue bars indicating the Tm value of UreA (20 μM) without drugs, light blue bars indicate the variations in Tm value when drug molecule is added). | ||
Urease inhibition assay
Since, the drugs showed interactions with urease subunit gamma, they were further analyzed for their urease inhibitory activity in vitro. The sequence similarity of urease subunit gamma (MRSA252) with Jack bean (Canavalia ensiformis) urease is 49%, hence the commercially available Jack bean urease was used to assess the urease inhibitory activity. Table 3 presents the mean % inhibition of the urease enzyme with the drug molecules. Comparing to positive control (Jack bean urease), drugs 6, 7, 8, 10, 12, 14, and 15 showed inhibition of the urease enzyme. Among them drugs 8, and 15 showed a significant inhibitory activity against Jack bean urease. Hence we propose on the basis of STD-NMR and in vitro inhibitory activity that compounds 8 and 15 can inhibit the urease activity, and most likely activity of urease gamma subunits.| Compound no. | Drug name | Mean % inhibition |
|---|---|---|
| 1 | Nicotinamide | −23.5 ± 6.65 |
| 2 | Drotaverine HCl | 17.6 ± 2.35 |
| 3 | Clidinium bromide | −4.8 ± 1.02 |
| 4 | Sulfanilamide | 4.5 ± 3.58 |
| 5 | Nicorandil | −12.2 ± 13.47 |
| 6 | Hydroxychloroquine sulphate | 63.7 ± 2.04 |
| 7 | Glucosamine hydrochloride | 54.2 ± 1.68 |
| 8 | Ephedrine | 72.0 ± 1.60 |
| 9 | Penicillin G sodium | 5.3 ± 3.32 |
| 10 | Tramadol HCl | 66.0 ± 0.39 |
| 11 | Nicotinic acid | −42.2 ± 6.23 |
| 12 | Salbutamol sulfate | 59.4 ± 0.00 |
| 13 | Bupropion HCl | 9.1 ± 2.32 |
| 14 | Tenofovir disoproxil | 66.0 ± 2.04 |
| 15 | Rasagiline mesylate | 71.1 ± 0.45 |
In this study, we have specifically targeted the urease subunit gamma with US-FDA approved drugs, through STD-NMR spectroscopic techniques. While 15 drugs showed interactions with the selected target (UreA), there is always a possibility of off-target effect. This off-target effect may appear as interactions with other proteins of cellular pathways in human host. This may lead to potential adverse effects. However, we did not analyze the potential off-target effect of drugs in this study. Regardless of these limitations, our study has provided some insights into potential of these drugs as likely inhibitors of newly identified UreA subunit. This subunit was thus initially studied as potential target for the drug discovery and development studies against MRSA infections. However, to validate these results further mechanistic and inhibitory studies are required.
Conclusion
The current study was an effort to identify new drug targets. As a result, urease gamma subunit of MRSA was selected for antimicrobial agents, and targeted to identify potential inhibitors. The outcomes of the study includes cloning, expression, and purification of urease gamma subunit in E. coli expression system. The purified enzyme was used to screen various drugs as potential binders of the enzyme by using STD-NMR technique. Fifteen drugs were identified as hits against the urease enzyme gamma subunit, and they also showed non-covalent interactions with the enzyme in molecular docking studies. These hits were further studied via in vitro inhibition assay, and by mechanistic techniques, against urease gamma, subunit of MRSA. Cumulatively results indicate that drugs 8 and 15 have the potential to inhibit urease subunit gamma and can serve as initial hits for further research as drug candidates against MRSA infections.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge The Searle Company, Ltd Pakistan, for the financial support for several research projects in drug discovery field.References
- T. A. Taylor, and C. G. Unakal, in: StatPearls, StatPearls Publishing, Treasure Island, FL, USA, 2020 Search PubMed.
- A. S. Lee, H. de Lencastre, J. Garau, J. Kluytmans, S. Malhotra-Kumar, A. Peschel and S. Harbarth, Nat. Rev. Dis. Prim., 2018, 4, 1–23, DOI:10.1038/nrdp.2018.33.
- H. W. Boucher and G. R. Corey, Clin. Infect. Dis., 2008, 46, S344–S349, DOI:10.1086/533590.
- T. A. Taylor, and C. G. Unakal, in StatPearls, StatPearls Publishing, Treasure Island, FL, USA, 2022 Search PubMed.
- S. Murchan, H. M. Aucken, G. L. O'Neill, M. Ganner and B. D. Cookson, J. Clin. Microbiol., 2004, 42, 5154–5160, DOI:10.1128/jcm.42.11.5154-5160.2004.
- C. Zhou, F. Bhinderwala, M. K. Lehman, V. C. Thomas, S. S. Chaudhari, K. J. Yamada, K. W. Foster, R. Powers, T. Kielian and P. D. Fey, PLoS Pathog., 2019, 15(1), e107538, DOI:10.1371/journal.ppat.1007538.
- S. Jing, X. Kong, L. Wang, H. Wang, J. Feng, L. Wei, Y. Meng, C. Liu, X. Chang, Y. Qu, J. Guan, H. Yang, C. Zhang, Y. Zhao and W. Song, Microbiol. Spectr., 2022, 10, 02340, DOI:10.1128/spectrum.02340-21.
- H. L. Mobley and R. P. Hausinger, Microbiol. Rev., 1989, 53, 85–108, DOI:10.1128/mr.53.1.85-108.1989.
- O. D. Merloni, B. Zambelli, F. Agostini, M. Bazzani, F. Musiani and S. Ciurli, Biochim. Biophys. Acta, Proteins Proteomics, 2014, 1844, 1662–1674, DOI:10.1016/j.bbapap.2014.06.016.
- Y. S. Nim and K. B. Wong, Inorganics, 2019, 7, 85, DOI:10.3390/inorganics7070085.
- H. L. Mobley, M. D. Island and R. P. Hausinger, Microbiol. Rev., 1995, 59, 451–480, DOI:10.1128/mr.59.3.451-480.1995.
- K. Oki, K. Washio, D. Matsui, S. Kato, Y. Hirata and M. Morikawa, Biosci. Biotechnol. Biochem., 2010, 74, 583–589, DOI:10.1271/bbb.90796.
- M. Vitolo, World J. Pharm. Pharm. Sci., 2022, 11, 96–135, DOI:10.20959/wjpps20223-21380.
- S. Murchan, H. M. Aucken, G. L. O'neill, M. Ganner and B. D. Cookson, J. Clin. Microbiol., 2004, 42, 5154–5160, DOI:10.1128/jcm.42.11.5154-5160.2004.
- P. Z. Konieczna, M. Kwinkowski, B. Kolesinska, J. Fraczyk, Z. Kaminski and W. Kaca, Curr. Protein Pept. Sci., 2012, 13, 789–806, DOI:10.2174/138920312804871094.
- M. Mayer and B. Meyer, Angew. Chem., Int. Ed., 1999, 38, 1784–1788, DOI:10.1002/(SICI)1521-3773(19990614)38:12<1784::AID-ANIE1784>3.0.CO;2-Q.
- O. Cala and I. Krimm, J. Med. Chem., 2015, 58, 8739–8742, DOI:10.1021/acs.jmedchem.5b01114.
- L. Hall, A. Sohail, E. J. Cabrita, C. MacDonald, T. Stockner, H. H. Sitte, J. Angulo and F. MacMillan, Sci. Rep., 2020, 10, 16483, DOI:10.1038/s41598-020-73443-z.
- U. Salar, A. Wahab and M. I. Choudhary, Biochimie, 2022, 201, 148–156, DOI:10.1016/j.biochi.2022.06.004.
- M. B. Waterhouse, S. Bienert, G. Studer, G. Tauriello, R. Gumienny, F. T. Heer, T. A. P. de Beer, C. Rempfer, L. Bordoli, R. Lepore and T. Schwede, Nucleic Acids Res., 2018, 46, 296–303, DOI:10.1093/nar/gky427.
- H. B. Banks, Y. Cao, A. Cho, W. Damm, R. Farid, A. Felts, T. Halgren, D. Mainz, J. Maple, R. Murphy, D. Philipp, M. Repasky, L. Zhang, B. Berne, R. Friesner, E. Gallicchio and R. Levy, J. Comput. Chem., 2005, 26, 1752–1780, DOI:10.1002/jcc.20292.
- Z. Vajo, J. Fawcett and W. C. Duckworth, Endocr. Rev., 2001, 22, 706–717, DOI:10.1210/edrv.22.5.0442.
- B. Meyer, J. Am. Chem. Soc., 2001, 123, 6108–6017, DOI:10.1021/ja0100120.
- H. M. Rolfe and J. Cosmet, Dermatol., 2014, 13, 324–328, DOI:10.1111/jocd.12119.
- R. R. Rai and S. Nijhawan, Saudi J. Gastroenterol., 2021, 27, 136–143, DOI:10.4103/sjg.SJG_266_20.
- S. M. El-Megharbel, M. S. Refat, G. Pu, X. Yu, G. Pu and F. Xi, Spectrosc. Spectr. Anal., 2021, 41, 3316–3320, DOI:10.3964/j.issn.1000-0593(2021)10-3316-05.
- C. R. Vanoni, J. P. Winiarski, G. R. Nagurniak, H. A. Magosso and C. L. Jost, Electroanalysis, 2019, 31, 867–875, DOI:10.1002/elan.201800832.
- H. Ashour, M. H. Elsayed, S. Elmorsy and I. A. Harb, Clin. Exper. Pharmacol. Physiol., 2020, 47, 1791–1797, DOI:10.1111/1440-1681.13395.
- M. Perrone, V. Laquintana, A. A. Lopedota, A. Cutrignelli, A. Lopalco, M. Franco, A. Pepe, S. Fontana and N. Denora, Data Brief, 2020, 33, 106575, DOI:10.1016/j.dib.2020.106575.
- D. M. Carrasco, J. I. Ayala and J. P. Casals, Actas Dermosifiliogr, 2022, 113, T166–T175, DOI:10.1016/j.ad.2022.01.012.
- H. W. Chiu, L. H. Li, C. Y. Hsieh, Y. K. Rao, F. H. Chen, A. Chen, S. M. Ka and K. F. Hua, Sci. Rep., 2019, 9, 1–3, DOI:10.1038/s41598-019-42130-z.
- S. Wu, J. Hu, L. Wei, Y. Du, X. Shi and L. Zhang, Food Chem., 2014, 148, 196–203, DOI:10.1016/j.foodchem.2013.10.044.
- C. G. Chen, S. Hou, J. Lv, Y. Zhang, H. Zhang and J. Xu, Appl. Catal. B Environ., 2020, 266, 118614, DOI:10.1016/j.apcatb.2020.118614.
- S. Tulgar, E. A. Alasehir and O. Selvi, Rev. Bras. Anestesiol., 2018, 68, 69–74, DOI:10.1016/j.bjane.2017.06.004.
- J. Zhang, Z. Xiong, J. Wei, Y. Song, Y. Ren, D. Xu and B. Lai, Chem. Eng. J., 2020, 383, 123144, DOI:10.1016/j.cej.2019.123144.
- M. Vazzana, T. Andreani, J. Fangueiro, C. Faggio, C. Silva, A. Santini, M. L. Garcia, A. M. Silva and E. B. Souto, Biomed. Pharmacother., 2015, 70, 234–238, DOI:10.1016/j.biopha.2015.01.022.
- Z. Tamanai-Shacoori, V. Shacoori, A. Jolivet-Gougeon, J. M. V. Van, M. Repère, P. Y. Donnio and M. Bonnaure-Mallet, Anesth. Analg., 2007, 105, 524–527, DOI:10.1213/01.ane.0000267525.51017.b8.
- E. T. B. Gille, K. Ahmed and S. Offermanns, Annu. Rev. Pharmacol. Toxicol., 2008, 48, 79–106, DOI:10.1146/annurev.pharmtox.48.113006.094746.
- K. Paruch, A. Biernasiuk, D. Khylyuk, R. Paduch, M. Wujec and Ł. Popiołek, Int. J. Mol. Sci., 2022, 23, 2823, DOI:10.3390/ijms23052823.
- M. S. Ferdynand and A. Nokhodchi, Drug Deliv. Transl. Res., 2020, 10, 1418–1427, DOI:10.1007/s13346-020-00707-6.
- M. Vandevelde, P. M. Tulkens, G. G. Muccioli and F. V. Bambeke, J. Antimicrob. Chemother., 2015, 70, 1713–1726, DOI:10.1093/jac/dkv032.
- M. Manjushree and H. D. Revanasiddappa, Spectrochim. Acta, Part A, 2019, 209, 264–273, DOI:10.1016/j.saa.2018.10.047.
- M. L. Cottrell, K. H. Yang, H. M. A. Prince, C. Sykes, N. White, S. Malone, E. S. Dellon, R. D. Madanick, N. J. Shaheen, M. G. Hudgens, J. Wulff, K. B. Patterson, J. A. E. Nelson and A. D. M. Kashuba, J. Infect. Dis., 2016, 214, 55–64, DOI:10.1093/infdis/jiw077.
- P. Patel, A. Pol, D. Kalaria, A. A. Date, Y. Kalia and V. Patravale, Eur. J. Pharm. Biopharm., 2021, 165, 66–74, DOI:10.1016/j.ejpb.2021.04.026.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra01732c |
| This journal is © The Royal Society of Chemistry 2024 |