
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDetection of monkeypox virus based on a convenient and sensitive single-step RPA-CRISPR/Cas12a strategy†
Tao Yu‡
ab,
Zhen Rong‡ b,
Zhixia Gu‡c,
Hongjuan Weib,
Yunxiang Wangb,
Rui Song*c,
Shengqi Wang*b and
Shumei Wang*d
b,
Zhixia Gu‡c,
Hongjuan Weib,
Yunxiang Wangb,
Rui Song*c,
Shengqi Wang*b and
Shumei Wang*d
aSchool of Pharmacy, Guangdong Pharmaceutical University, Guangzhou, Guangdong 510006, China
bBioinformatics Center of AMMS, Beijing 100850, China. E-mail: sqwang@bmi.ac.cn
cBeijing Ditan Hospital, Capital Medical University, Beijing 100102, China. E-mail: songruii@hotmail.com
dSchool of Chinese Materia Medica, Guangdong Pharmaceutical University, Guangzhou, Guangdong 510006, China. E-mail: gdpuwsm@126.com
First published on 7th May 2024
Abstract
The global outbreak of monkeypox virus (MPXV) has highlighted the need for rapid molecular diagnostics techniques. In this study, a single-step recombinase polymerase amplification (RPA)-CRISPR/Cas12a system was developed for rapid and sensitive detection of MPXV. The limit of detection of this assay was 1 copy per μL of extracted nucleic acids. A heating lysis method was integrated to further simplify the sample processing workflow and shorten the assay time to 40 min from sample to result. The reaction mixture can be lyophilized to improve its accessibility in resource-limited settings. The analysis results of the proposed single-step RPA-CRISPR/Cas12a assay for clinical MPXV positive and negative samples were 100% consistent with standard PCR assay. These results demonstrate the feasibility and efficiency of this method for rapid and accurate MPXV detection in real-world settings, showcasing its potential utility in urgent and practical settings.
1. Introduction
Monkeypox (Mpox) is a zoonotic viral disease caused by Mpox virus (MPXV), which has been recently reported in multiple countries. Despite the ongoing global COVID-19 pandemic, Mpox has now surfaced in non-endemic regions in 2022, presenting a new global threat.1 MPXV is an enveloped, double-stranded DNA virus belonging to the Orthopoxvirus genus of the Poxviridae family, with a genome size of approximately 197 kbps. Under electron microscopy, MPXV appears elliptical, oval, or brick-shaped and measures about 200 nm × 250 nm in size.2 MPXV consists of two distinct genetic clades: the more virulent Central African [Congo Basin (CB)] clade and the milder West African (WA) clade.1 Mpox is generally a self-limiting disease, with mortality rates of 10.6% for the CB lineage and 3.6% for the WA lineage, but certain high-risk groups, including newborns, children, pregnant women, and immunocompromised individuals, are more susceptible to severe symptoms and death following infection.3 The rapid detection of infected hosts is critical for surveillance and diagnostic efforts, which help reduce virus transmission by identifying infected cases as early as possible.Identifying Mpox on the basis of symptoms alone is challenging due to the similarity of clinical presentations caused by different orthopoxviruses.4,5 Early detection can help identify infected individuals and alert them for timely isolation and treatment, ultimately reducing the spread of the virus and lessening the effect of any outbreak.6 The need to develop diagnostic techniques with high sensitivity, accuracy, and rapid detection rates to minimize transmission is increasing. The gold standard for MPXV detection is real-time PCR owing to its high sensitivity and specificity, but it requires specialized and bulky equipment. This limitation has restricted its accessibility in resource-constrained areas where MPXV is endemic, thus reducing its utility in point-of-care applications.7 Therefore, a simple point-of-care test is crucial for the rapid and convenient diagnosis of MPXV infections.
The clustered regularly interspaced short palindromic repeat associated (CRISPR/Cas) system is a promising tool for nucleic acid testing. The advent of CRISPR/Cas has provided brand new rapid diagnostic methods with high specificity and programmability.8 Cas12a is usually used to detect DNA by trans-cleavage mechanism when CRISPR-derived RNA (crRNA) recognizes the complementary target. The ability of trans-cleavage ssDNA fluorescent probes is several times that of cis-cleavage targets.9–11 This detection method could be used to distinguish target sequences of concentrations in the picomolar range. New nucleic acid detection technologies for different purposes have been developed on the basis of this mechanism, combined with other gene amplification methods, such as specific high-sensitivity enzymatic reporter unlocking (SHERLOCK),12 DNA endonuclease-targeted CRISPR trans reporter (DETECTR),10 HOLMESv2 [improved version of HOLMES (One-Hour Low-cost Multipurpose highly Efficient System)],13 and other diagnostic platforms, to improve sensitivity. On the one hand, the advantages of loop-mediated isothermal amplification (LAMP) are high sensitivity, good specificity, and simultaneous recognition of multiple target sequences by using a set of four primers.14 On the other hand, LAMP primers are difficult to design, and the reaction temperature is high (65 °C). However, this temperature deactivates Cas12a, indicating that the temperature should be reduced after the LAMP reaction for a period of time before adding the Cas12a detection system. Recombinase polymerase amplification (RPA) combines isothermal recombinase-driven primer targeting template and strand displacement DNA synthesis, and it can react between 37 °C and 42 °C, even at room temperature, making it possible to react with CRISPR-Cas12a in one tube.15
Various single-step methods described in literatures have been used to separate RPA from CRISPR detection reagents,16,17 including adding CRISPR detection reagents to the tube cap,18,19 using physical devices to separate the reagents, and isolating RPA reagents from CRISPR detection reagents by using glycerol viscosity and sucrose density.20,21 Techniques for directly combining the two components by using photocleavable complementary ssDNA to block crRNA are available.22 RPA and Cas12a reagents can be pre-mixed with the double crRNAs without protospacer adjacent motif (PAM) site limitation and a crRNA with suboptimal PAM site.23–25 These CRISPR analysis systems can simplify the operation workflow and make it more accessible at resource-limited settings.
In this study, we developed a single-step RPA-CRISPR/Cas12a assay for the ultrasensitive detection of MPXV. The specific crRNA was designed and screened by comparing the relevant regions of the F3L gene related to monkeypox of four orthopoxviruses, without the restriction of conventional PAM sites. RPA was combined with CRISPR-Cas12a to construct a fast and ultrasensitive single-step MPXV detection and diagnosis system (Fig. 1). The whole reaction can be completed within 40 min by using lyophilized reagents, and the sensitivity of viral DNA detection was 5 copies in a single tube. Therefore, this platform has the potential for convenient and accurate diagnosis of Mpox at point of care needs.
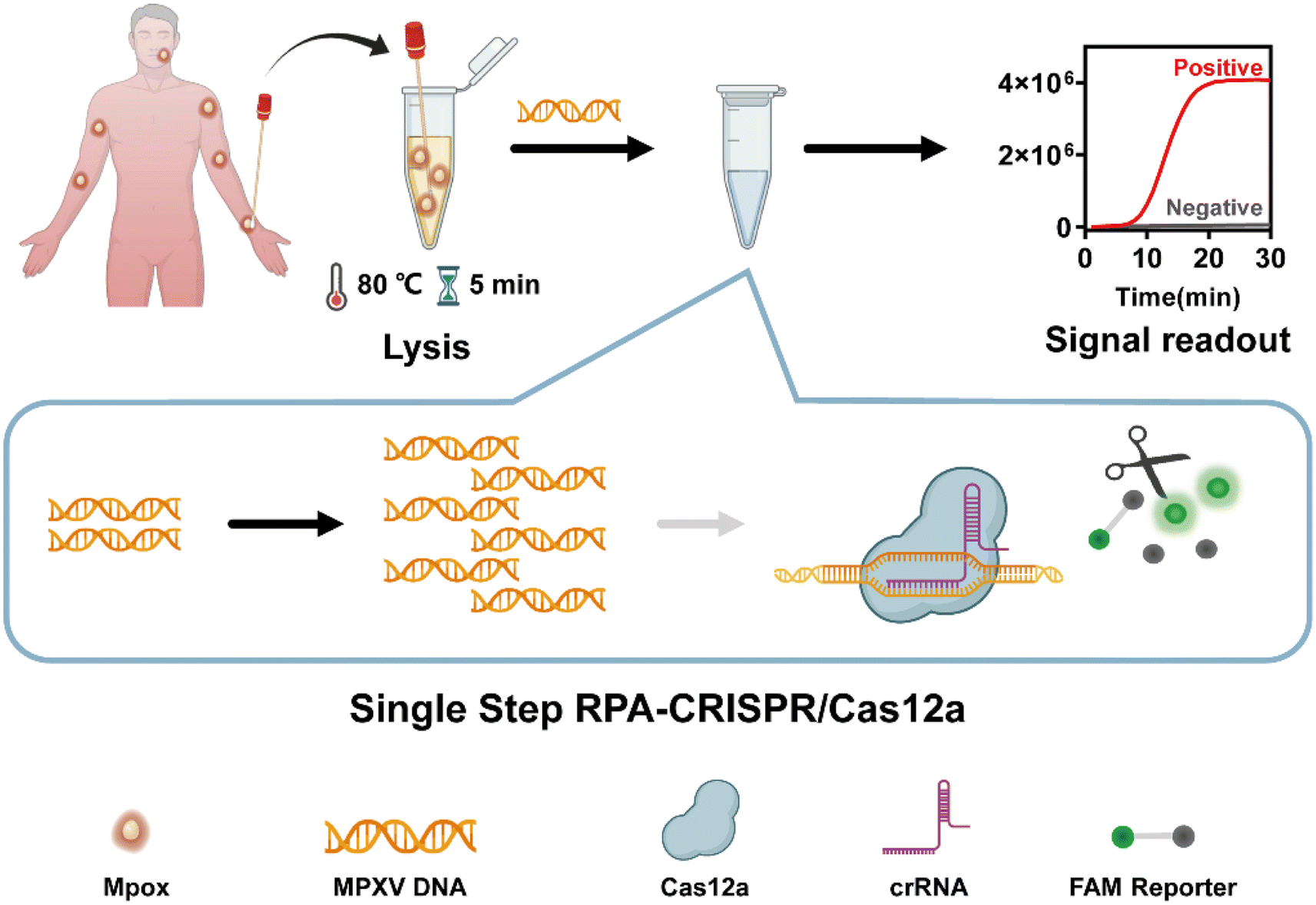 | ||
| Fig. 1 Principle and operation for MPXV detection using single-step RPA-CRISPR/Cas12a. | ||
2. Experimental section
2.1 Reagents and materials
All reagents used in this study are listed in Table S1.†2.2 DNA extraction
DNA was extracted from the MPXV F3L gene pseudovirus with a TIANamp Virus DNA/RNA Kit in accordance with the manufacturer's instructions. The extracted DNA was stored at −80 °C until use. The DNA of MPXV pseudovirus was diluted to different concentrations with DNA Diluent for daily use and stored at −20 °C.Pseudoviruses of cowpox virus (CPXV) F3L gene were extracted and stored using the same method. Two plasmids, one containing fragments of the vaccinia virus (VACV) E3L gene and the other containing fragments of the variola virus (VARV) E3L gene, were solubilized in a solution of 10 mM Tris and 1 mM EDTA (pH 8.0). They were then quantified by a Qubit Flex Fluorometer (Thermo, USA) with Qubit 1× dsDNA HS Assay Kits and converted to copies per μL. The DNA of these three species was diluted to the appropriate concentration and stored at −80 °C until specificity detection.
In this study, extraction-free nucleic acid releasers were used to release MPXV, saving time compared to the time-consuming clinical use of extraction kits. The pseudovirus (purchased at the National Institute of Metrology, China) was diluted with 10 mM Tris and 1 mM EDTA. An equal volume of saliva was added to the dilution to create a mock sample. The mock sample was then mixed with the screening lysis buffer at a ratio of 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and lysed by heating to release nucleic acids.
:1 and lysed by heating to release nucleic acids.
2.3 Design and preparation of crRNA by in vitro transcription
The crRNA comprised a handle region (UAAUUUCUACUAAGUGUAGAU) and a spacer region that could complement the target sequence. The handle region facilitated recognition and binding by the LbCas12a protein. Four orthopoxviruses F3L gene fragments were aligned using DNAMAN, removing crRNAs that have been used in literature and designing two crRNAs with a recognition length of 20 bp in each of the two specific small fragment regions, as shown in Fig. 2A. In vitro transcription (IVT) was employed to prepare the crRNA for this study, involving an annealing reaction, transcription, and subsequent RNA purification following a concise three-step procedure. The ssDNA templates for crRNA IVT are shown in Table S2.†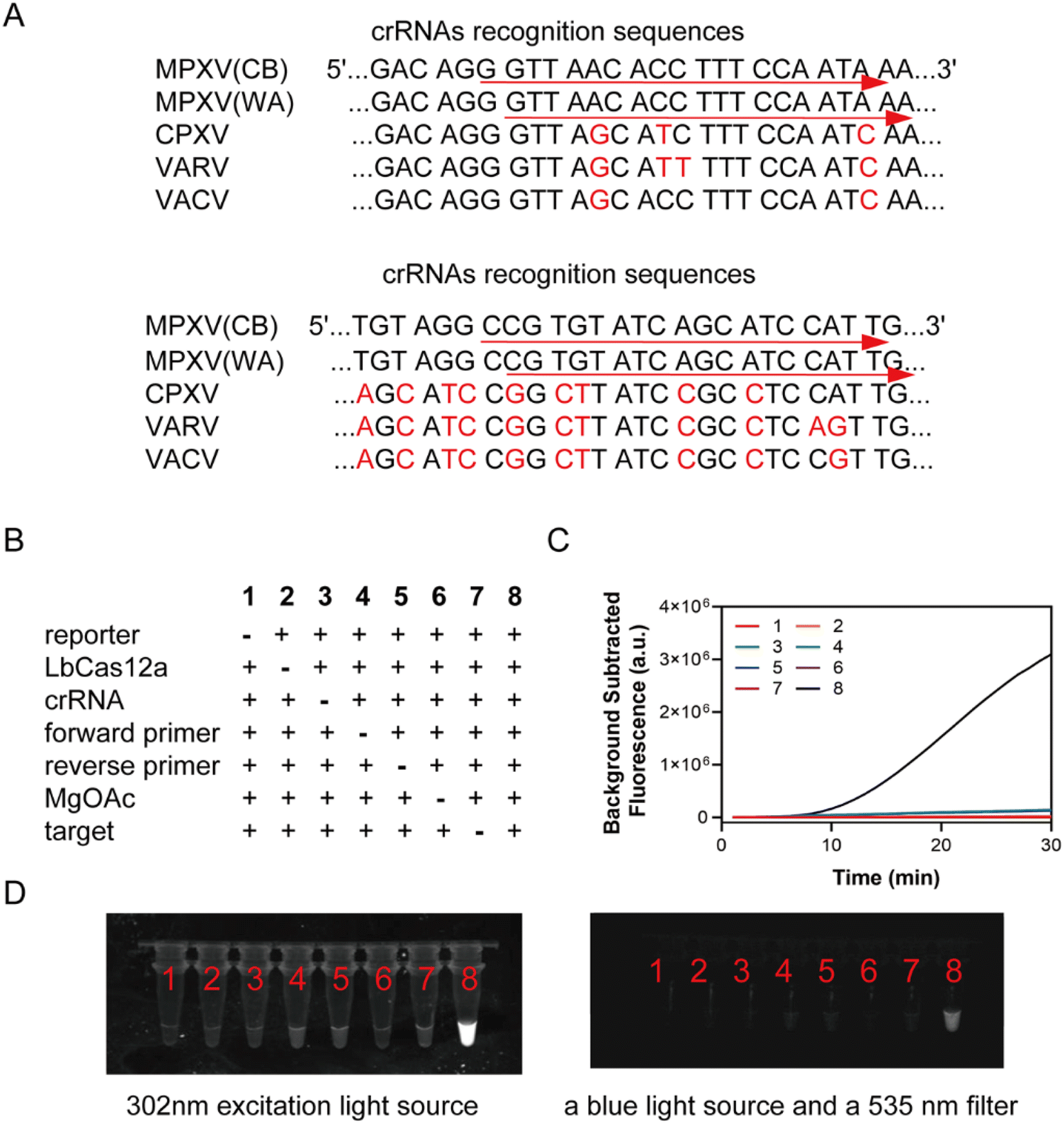 | ||
| Fig. 2 (A) Comparison of crRNA recognition sequences of F3L gene in MPXV, VARV, VACV, and CPXV. (B) Eight combinations of ingredients (excluding RPA rehydration buffer). (C) Real-time fluorescence curves for eight combinations. (D) Photographs taken after 30 min of reaction by using a 302 nm excitation light source (left), followed by fluorescence images taken using a blue light source and a 535 nm filter (right). | ||
First, 10 μL annealing reaction containing 10 μM synthesized ssDNA templates with an upstream T7 promoter, 10 μM T7-3G IVT primer (Table S1†), and 1× Standard Taq buffer was denatured for 5 min at 95 °C, followed by gradient annealing to 4 °C with a ramp rate at 0.1 °C s−1 in a PCR thermocycler (Thermo, USA). Next, the products of the annealing reaction were used as templates for synthesizing crRNA overnight with a HiScribe T7 Quick High Yield RNA synthesis kit at 37 °C. Then, the transcription product was treated with DNase I at 37 °C for 20 min and purified with a RNA Clean & Concentrator-5 Kit in accordance with the manufacturer's instructions. Finally, the purified crRNA was quantified by a Qubit Flex Fluorometer using Qubit RNA Broad Range Assay Kits. Finally, the crRNAs generated by IVT were diluted at an appropriate concentration and stored at −20 °C for further use.
2.4 Design and screen of RPA primers
RPA primers were constructed in accordance with the guidelines set out in the TwistAmp Assay Design Manual. Initially, a pair of primers capable of amplifying four regions of crRNA recognition was designed and then screened for the most effective crRNA. Four forward and four reverse primers were designed to contain the crRNA with the best efficiency. The primer details are shown in Table S2.† According to the screening results, the best primer pair was identified for the next condition optimization.2.5 RPA-CRISPR single-step assay
The RPA-CRISPR single-step reaction contains the RPA and CRISPR-Cas12 reagents. The RPA rehydration buffer was used instead of the reaction buffer for LbCas12a to improve the amplification results. Eight component combinations were made, as shown in Fig. 2B–D. Only combination 8, containing reporter molecule LbCas12a, crRNA, forward primer, reverse primer, magnesium acetate, and target, showed significantly enhanced fluorescence signals, which could be observed with a gel imager.Single-plex lyophilized RPA powder from TwistDx was dissolved by a solution containing 50 μL rehydration buffer (provided in TwistDx RPA kit); 1 μL of 100 μM FAM-CCCCCC-BHQ1 fluorescence reporter; 0.5 μL of 10 μM LbCas12a; 2 μL of 10 μM crRNA; 4 μL of 10 μM RPA forward primers; 4 μL of 10 μM RPA reverse primers; and 6 μL RNase/DNase-free water. Then, the resuspending mixture was divided into five 13.5 μL individual reactions. The reaction was finally activated with 5 μL DNA template and 1.5 μL of 280 mM magnesium acetate (MgOAc). The reaction was conducted at 37 °C for 30 min on an Applied Biosystems 7500 real-time PCR instrument with the FAM signal reading at 1 min intervals. Fluorescence was observed using a torch after the end of the reaction.
2.6 Optimization of RPA-CRISPR single-step reaction
Previous research indicated that the concentration of primers, LbCas12a enzyme, the ratio of LbCas12a to crRNA, MgOAc, and temperature have a remarkable effect on the RPA-CRISPR reaction system. Therefore, a concentration optimization process was conducted to identify the ideal reaction conditions for improving the established single-step RPA-CRISPR/Cas12a reaction system.In this study, optimization of only one condition parameter at a time was used to determine the ideal reaction conditions. Four primer concentrations (300, 400, 500, and 600 nM) were tested, and the most effective concentration was selected to optimize the next parameters based on experimental results. The concentration of LbCas12a, crRNA, magnesium acetate, and temperature were subsequently optimized. Noteworthy, the concentration and ratio of LbCas12a to crRNA have a significant effect on the optimal balance between RPA amplification and CRISPR detection. LbCas12a and crRNA combination could recognize the target, and a single change in the concentration of the enzyme could lead to a change in the ratio of the two. So, the ratio of LbCas12a to crRNA was first fixed, and the enzyme concentration was screened.
2.7 Preparation of ready-to-use tube with lyophilized reagents
Single-plex lyophilized RPA powder from TwistDx was dissolved by a solution containing fluorescence reporter; LbCas12a; crRNA; forward primers; reverse primers; 12.5 μL mixture of lyophilized protective agent containing 10% trehalose, 15% mannitol, and 2% glycine; and replenish with RNase/DNase-free water to 50 μL. A 10 μL aliquot of this mixture was added to the bottom of a 200 μL PCR tube. PCR tubes containing reagents were placed in an aluminum 96-well ice box and frozen at −80 °C for 2 h, followed by lyophilization in a pre-cooled vacuum lyophilizer in accordance with the prescribed program. The freeze procedure was as follows: the lyophilized reagents were subjected to a temperature treatment starting at −40 °C for 6 h, followed by heating to −30 °C at a rate of 1 °C min−1, vacuuming to a pressure of 0.1 mbar, and holding for 6 h. Following this step, the temperature was increased to 10 °C at the same rate and held for 2 h, and finally heated to 25 °C with a holding time of 30 min to complete the process. The lyophilized reagents were then removed and stored at −20 °C for future use.2.8 Stability testing of lyophilized reagents
Storage environments were simulated under light protection at three different temperatures of 37 °C, 4 °C, and −20 °C to assess the stability of the lyophilized reagents. The lyophilized reagents were then taken at intervals of 0, 1, 2, and 4 weeks and tested for extracted nucleic acid at a concentration of 20 copies per μL. Samples were divided into five tubes of sufficient volume and stored at −80 °C. One tube of nucleic acid was used per experiment to minimize degradation of the nucleic acid.2.9 Extraction-free optimization
The extraction-free nucleic acid technique is a time-saving method, and for this reason, the composition of the extraction-free lysate, the lysis temperature, and the lysis time were optimized. NaOH and Triton X-100 were selected as the main components of the extraction-free lysate, and eight groups of lysate combinations were established. The volume ratio of the simulated sample to the lysate was 2:1. The composition of the eight combinations is presented in Table S3.† The kit extraction and without-extraction direct detection were employed as controls. Subsequently, the optimal lysate combination was selected for lysis temperature optimization experiments. Five temperatures were used: 37 °C, 50 °C, 65 °C, 80 °C, and 95 °C. Finally, the lysis time was optimized and set to four times: 3 min, 5 min, 8 min, and 10 min.
2.10 Evaluation of single-step RPA-CRISPR fluorescent assay
First, serial dilutions of extracted MPXV F3L nucleic acids (1 to 1000 copies per μL) were used to test the sensitivity of single-step RPA-CRISPR/Cas12a. After adding 5 μL of extracted DNA template and 2 μL of 280 mM magnesium acetate to the cap of the tube, the mixture was centrifuged and thoroughly mixed with 13 μL of reaction mix at the bottom of the tube. Subsequently, the tubes containing the mixing reagents were transferred to the device and incubated at 41 °C for 30 min. Next, the test was repeated using lyophilized premixed reagents. Finally, mock samples with pseudoviruses were tested using extraction-free lysates and lyophilized premixed reagents. To assess the specificity of this method, gene fragments with significant similarities to other three orthopoxviruses, VACV, VARV, and CPXV, were used.2.11 Assay performance on clinical samples
Clinical samples for this study were collected from the Beijing Ditan Hospital, and negative samples were collected from healthy male volunteers, with informed written consent from all participants. In brief, rash fluid swab was collected and stored via disposable virus sampling tube. The clinical samples were tested following the guidelines approved by the Ethics Committee of Beijing Ditan Hospital (approval ID: DTEC-YJ2023-001-02). The samples randomly numbered by one person, pre-treated by a second person, and then tested by a third person. The results were compared with those obtained using the conventional real-time PCR.For real-time PCR assay, 0.4 μL of 10 μM forward primer, 0.4 μL of 10 μM reverse primer, 0.2 μL of 10 μM TaqMan probe, 0.8 μL AK Taq Master PCR Mix (25×), 4 μL of 5× AK Taq Buffer (with Mg2+), 9.2 μL RNase/DNase-free water, and 5 μL of the target were mixed to a final volume of 20 μL. Next, amplification was performed in duplicate on a Applied Biosystems 7500 real-time PCR system. The thermal cycling program was as follows: 95 °C for 2 min 30 s, followed by 45 cycles of 94 °C for 30 s and 55 °C for 30 s.
2.12 Data analysis
All bar charts, graphs, and scatter plots in this study were created by GraphPad Prism 8. All gel images were captured by Tanon 4600 gel imaging system. The scheme was created by https://http://www.biorender.com and PowerPoint. All the fluorescence signal were collected by ABI 7500.3. Results
3.1 Screen of crRNA and RPA primers
A pair of primers were designed to amplify four crRNA recognition regions to screen for the most efficient crRNA. The experimental results are shown in Fig. 3A, where only the use of crRNA1 and crRNA3 produced fluorescence signals. The fluorescence curve of crRNA1 was on top of the other curves, indicating that crRNA1 was the most efficient. Therefore, crRNA1 was chosen for the next experiment. A pair of primers was selected as F4-R1 on the basis of the criteria of the earliest appearance of the fluorescence curve and the strongest fluorescence intensity, as shown in Fig. 3B and S1.†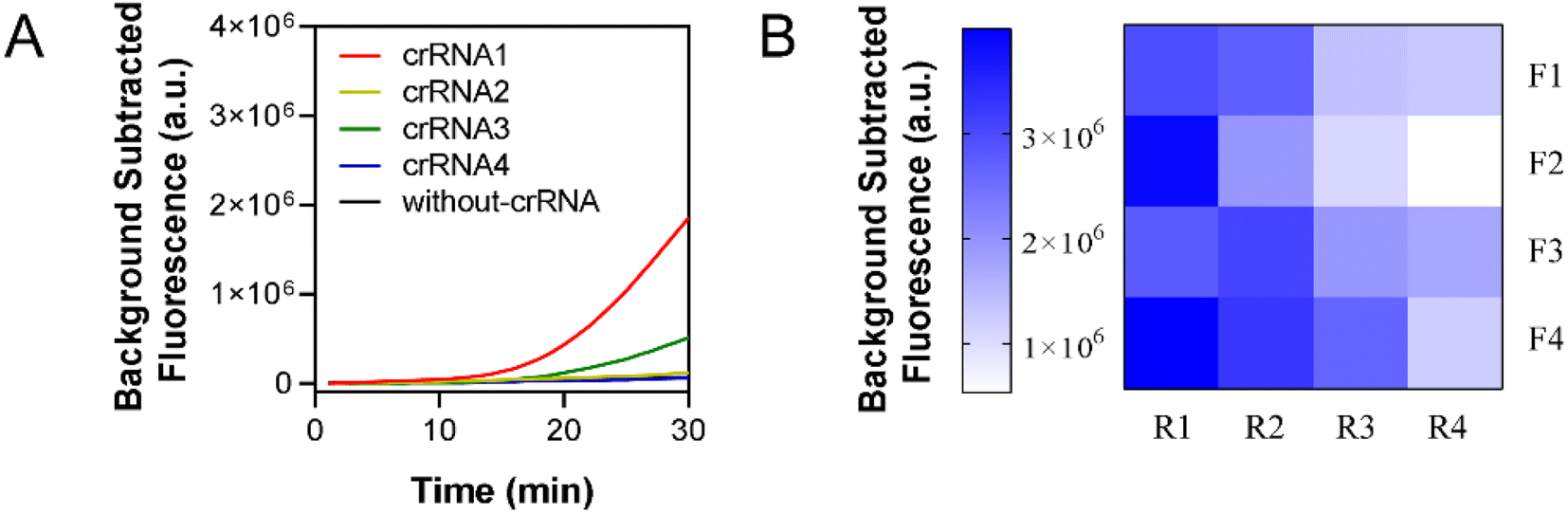 | ||
| Fig. 3 Screen of crRNA, RPA primers (A) real-time fluorescence data collected using a one-step RPA-CRISPR system. Four crRNAs and a positive control without crRNA were used in sequence. Fluorescence was measured every min at 37 °C, and the number of target was 1000 copies. (B) Graph of fluorescence values for 16 pairwise combinations of four forward primers and four reverse primers with background removed at 30 min. | ||
3.2 Optimization of RPA-CRISPR single-step reaction
To improve the sensitivity of the assay, the single-step RPA-CRISPR/Cas system was optimized. Fig. 4A illustrates that the real-time fluorescence curve showed an initial appearance at 400 nM, resulting in the highest fluorescence value when the background was removed at 30 min. The results are shown in Fig. 4B, with 50 nM being the optimal LbCas12a concentration. Next, the concentration of fixed enzyme was 50 nM, and the concentration of crRNA was varied, Fig. 4C shows that the crRNA concentration of 100 nM was more effective than 50 nM. A higher concentration of crRNA did not produce faster reaction efficiency, and a 1:2 ratio of LbCas12a to crRNA was used to consider cost savings. According to recent literature reports, increasing the concentration of magnesium acetate within a certain range can improve the reaction efficiency of the one-step method. Several concentrations were tested, and the optimal concentration of magnesium acetate was selected as 28 mM in Fig. 4D. The conclusions were similar to those in literature.26,27 Finally, the reaction temperature was optimized to a range of 25–42 °C for the RPA reaction and 25–48 °C for the LbCas12a reaction. Normally, 37 °C is used for CRISPR detection. The temperature was increased by a gradient of 2 °C, as shown in Fig. 4E and F. Higher temperatures accelerated the reaction rate, the efficiency of the reaction decreased sharply after exceeding the optimal temperature for RPA, and the reaction temperature was determined to be 41 °C. Fig. S2† shows the fluorescence curves in real time for the optimized conditions.
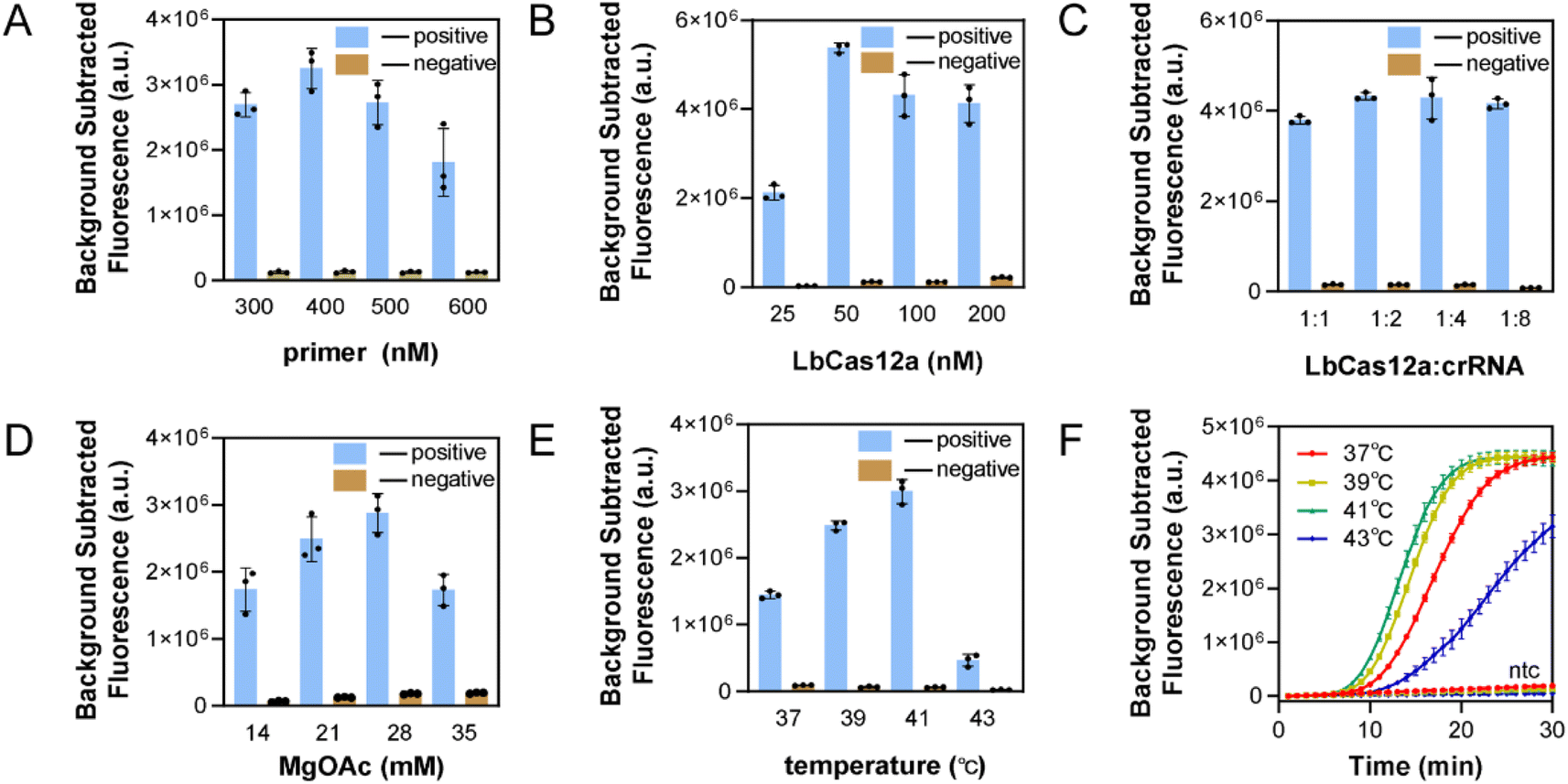 | ||
| Fig. 4 Optimization results for single-step RPA-CRISPR reaction conditions. (A) Effect of primer concentration. (B) Effect of CRISPR/Cas12a RNP concentration. (C) Effect of LbCas12a to crRNA ratios. (D) Effect of MgOAc concentration. (E) Effect of reaction temperature. This graphs used fluorescence values at 15 min instead of 30 min with the background signal removed. (F) Fluorescence profiles recorded in real time at different reaction temperatures. | ||
In summary, the optimized optimal primer concentration was 400 nM, the LbCas12a concentration was 50 nM, the ratio of LbCas12a to crRNA was 1:2, the concentration of magnesium acetate was 28 mM, and the reaction temperature was 41 °C.
3.3 Stability testing of lyophilized reagents
The background fluorescence values were removed after 30 min, as shown in Fig. S3.† No significant decrease was observed in the fluorescence value of the lyophilized reagent stored at 4 °C and protected from light for 4 weeks, indicating the stability of the lyophilized system when stored at 4 °C for a short period of time. The lyophilized reagent stored at 37 °C for 1 week did not show any significant loss of value. Thus, it was able to withstand transport for a short period of time and a short distance without the need for cold chain.3.4 Extraction-free optimization
The results are shown in Fig. S4.† The graph shows that buffer 5 outperformed the other combinations, and the lysis effect was similar at 80 °C and 90 °C. So, 80 °C was chosen as the lysis temperature. The lysis effect was found to be similar at 3, 5, 8, and 10 min. However, the lysis time was extended to 5 min to account for the complexity of the clinical samples.3.5 Evaluation of single-step RPA-CRISPR fluorescent assay
Finally, the sensitivity and specificity of the single-step CRISPR/Cas12a-assisted RPA method were tested under the above optimal conditions. Fig. 5A and S5A† show the detection of DNA target with concentrations as low as 1 copy per μL. In addition, as shown in Fig. 5B and S5B,† the lyophilized reagents resulted in the detection of extracted nucleic acids at concentrations as low as 1 copy per μL. As only a small number of clinical samples infected with MPXV could be obtained, the sensitivity of the method was assessed by adding the F3L pseudovirus to saliva to simulate the sample and test it using an extraction-free approach. Lysis buffer 5 was added to the mock sample and heated in a metal or water bath at 80 °C or higher for 5 min to release nucleic acids by following the above-described no-extraction technique. Then, 5 μL of the lysed sample was taken and added to the lyophilized reagent. The reagent was supplemented with rehydration buffer, water, and magnesium acetate; mixed; and allowed to react for 30 min at 41 °C. The results are shown in Fig. 5C and S5C,† where one of the three replicates was tested negative for a 1 copy per μL sample. Fig. S6A† shows that when the simulated samples of 1 and 5 copies per μL were replicated 20 times, 75% of the 1 copy per μL samples were judged positive and 100% of the 5 copies per μL samples were detected. Therefore, the proposed technique can accurately detect F3L pseudoviruses in saliva at 5 copies per μL. In Fig. S6B,† a four-parameter curve is fitted to the fluorescence values and the simulated sample concentration. This allows for an approximate determination of the input sample concentration. Furthermore, F3L gene fragments of MPXV and three other orthopoxviruses, including vaccinia virus, variola virus, and cowpox virus (Table S4†), were detected to validate the assay specificity of this assay. As shown in Fig. 5D and S5D,† only MPXV was detected, indicating the superior specificity of the assay.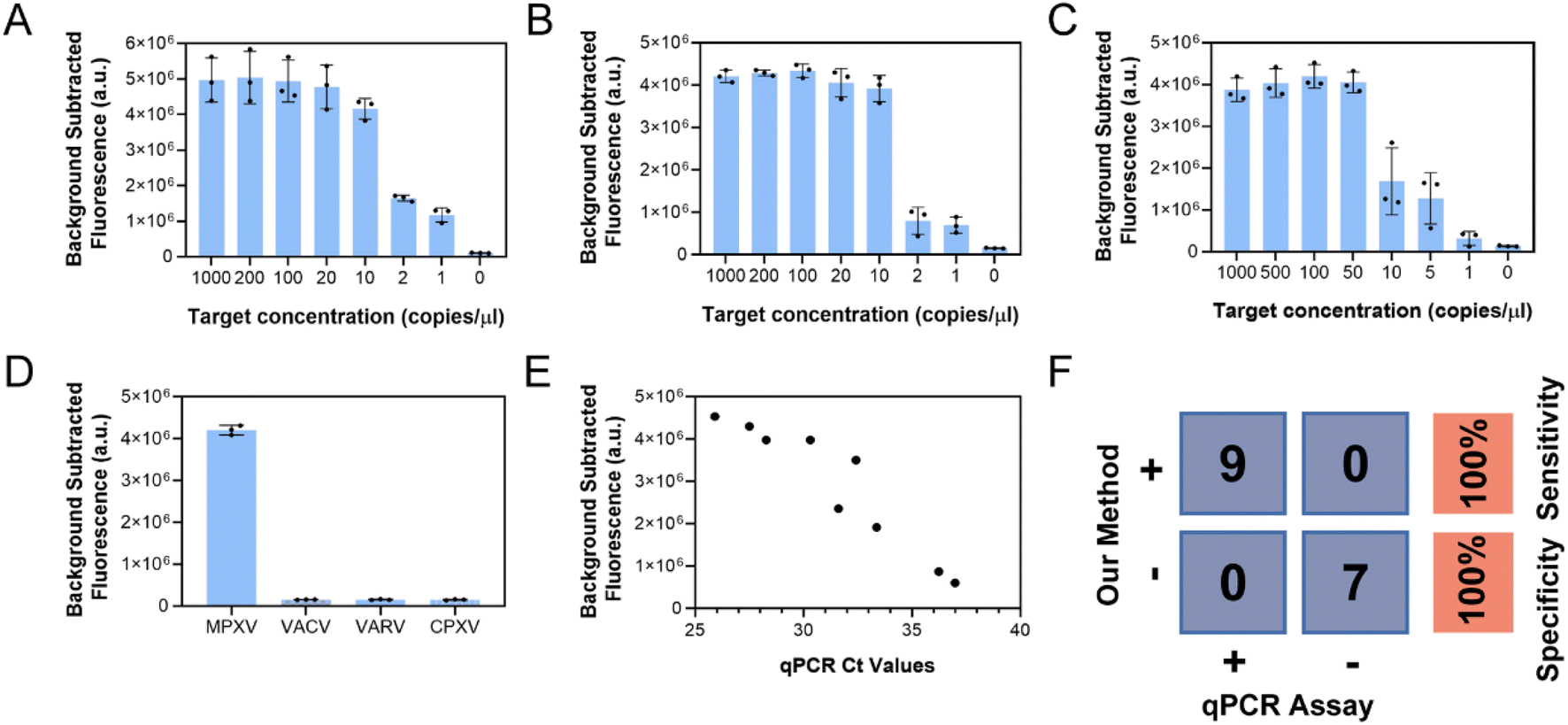 | ||
| Fig. 5 Evaluation of single-step RPA-CRISPR fluorescent assay and performance on clinical samples. (A) Fluorescence curves generated for the detection of different concentrations of MPXV F3L DNA by using freshly prepared reagents. (B) Fluorescence curves generated for the detection of different concentrations of MPXV F3L DNA by using lyophilized reagents. (C) Fluorescence curves generated to detect different concentrations of MPXV F3L pseudovirus, ranging from 1000 copies per μL to 1 copy per μL. The reagents used were lyophilized, and detection was performed using saliva samples. (D) Lyophilized reagents used to detect four orthopoxviruses: MPXV F3L gene (103 copies per μL), VACV E3L gene (104 copies per μL), VARV E3L gene (104 copies per μL), and CPXV F3L gene (104 copies per μL). (E) Scatter plot generated to plot the Ct value of the qPCR results against the corresponding fluorescence value of the removed background signal detected by one-step RPA-CRISPR/Cas12a assay. (F) Statistical analysis performed on the results of 40 samples by using the proposed method and qPCR. | ||
3.6 Assay performance on clinical samples
The clinical feasibility of this single-step RPA-CRISPR assay was validated using 16 clinical samples. As shown in Fig. S7,† real-time PCR results indicated that 9 clinical samples were MPXV positive and 7 clinical samples were MPXV negative. The scatter diagram (Fig. 5E) shown that the fluorescence signal intensity for CRISPR assay was correlated with the Ct value the qPCR results. The assay specificity and sensitivity of the proposed single-step RPA-CRISPR/Cas12a assay were shown in Fig. 5F.4. Discussion
Single-step RPA-CRISPR assays are gaining popularity in the CRISPR/Cas-based diagnostic field owing to their simplicity. However, designing and optimizing these methods remain challenging and inefficient, primarily due to a lack of understanding of the underlying mechanisms. In this study, a single-step RPA-CRISPR detection strategy was used for DNA sequence identification via crRNA without the need for PAM sequences (5′-TTTN-3′). A significant improvement of this study is the use of commercial enzymes that do not require optimization of the LbCas12a protein sequence and the design of no-PAM crRNAs could also be considered when performing a single-step RPA-CRISPR assay to detect dsDNA viruses.28According to recent reports, most MPXV assays using isothermal amplification and CRISPR detection require a two-step reaction due to the competition between isothermal amplification and the conventional crRNA design principles for CRISPR detection in a single tube. In the two-step process, the product is first amplified in a separate tube and then transferred to the CRISPR/Cas system for fluorescence detection. This approach, which involves opening the amplification reaction tube, increases the possibility of cross-contamination and predisposes clinical samples to false-positive results. Alternatively, the spatial isolation of the procedure can be used, with the CRISPR detection reagent on the cap of the tube and the RPA amplification reagent on the bottom of the tube, first amplifying for 10–20 min, followed by inverting and mixing the amplification products and detection reagent, and collecting the fluorescence detection for 10–30 min. Although essentially a two-step method, it is classified as a single-tube method to avoid cross-contamination. The reagent configuration is cumbersome and not suitable for clinical batch testing, and detection reagents occasionally drop to the bottom of the tube prematurely.
The proposed method involves incubating all components, including the RPA mixture and the crRNA/LbCas12a mixture, in a test tube, followed by detecting fluorescence during amplification, which can detect 1 copy per μL of extracted nucleic acid or 5 copies per μL of pseudovirus in saliva. Extraction-free lysate and lyophilized reagents are used to simplify the detection steps and reagent configuration. The lyophilized reagents can be stably stored at 4 °C for at least 4 weeks, making them suitable for on-site testing in resource-limited environments. The assay performances of isothermal amplification and CRISPR assays were summarized in Table S5,† 19,25,29–38 which indicated the proposed single-step RPA-CRISPR/Cas12a assay exhibited a relative superior performance.
5. Conclusions
In summary, a convenient and sensitive single-step RPA-CRISPR/Cas12a assay was developed for point-of-care testing of MPXV. This assay can be integrated with rapid sample lysis processing method to simplify analysis workflow. The assay can detected MPXV pseudoviruses in saliva as low as 5 copies per μL within 40 min. The clinical feasibility was validated by testing MPXV clinical samples. Besides, the reagents can be lyophilized to improve its accessibility to end-user at resource-limited settings without cold chain for reagent distribution. This simple and sensitive single-step RPA-CRISPR assay holds considerable potential for point-of-care testing applications at resource-limited settings.Data availability
Data will be made available on request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.References
- R. M. Jiang, Y. J. Zheng, L. Zhou, L. Z. Feng, L. Ma, B. P. Xu, H. M. Xu, W. Liu, Z. D. Xie, J. K. Deng, L. J. Xiong, W. J. Luo, Z. S. Liu, S. N. Shu, J. S. Wang, Y. Jiang, Y. X. Shang, M. Liu, L. W. Gao, Z. Wei, G. H. Liu, L. Gang, W. Xiang, Y. X. Cui, G. Lu, M. Lu, X. X. Lu, R. M. Jin, Y. Bai, L. P. Ye, D. C. Zhao, A. D. Shen, X. Ma, Q. H. Lu, F. X. Xue, J. B. Shao, T. Y. Wang, Z. Y. Zhao, X. W. Li, Y. H. Yang and K. L. Shen, World J. Pediatr., 2023, 19, 231–242 CrossRef CAS PubMed.
- C. T. Cho and H. A. Wenner, Bacteriol. Rev., 1973, 37, 1–18 CrossRef CAS PubMed.
- H. Ejaz, K. Junaid, S. Younas, A. E. Abdalla, S. N. A. Bukhari, K. O. A. Abosalif, N. Ahmad, Z. Ahmed, M. A. Hamza and N. Anwar, J. Infect. Public Heal., 2022, 15, 1156–1165 CrossRef PubMed.
- Z. Jezek, M. Szczeniowski, K. M. Paluku and M. Mutombo, J. Infect. Dis., 1987, 156, 293–298 CrossRef CAS PubMed.
- A. Lewis, A. Josiowicz, S. M. Hirmas Riade, M. Tous, G. Palacios and D. M. Cisterna, Emerging Infect. Dis., 2022, 28, 2123–2125 CrossRef PubMed.
- B. D. Kevadiya, J. Machhi, J. Herskovitz, M. D. Oleynikov, W. R. Blomberg, N. Bajwa, D. Soni, S. Das, M. Hasan, M. Patel, A. M. Senan, S. Gorantla, J. McMillan, B. Edagwa, R. Eisenberg, C. B. Gurumurthy, S. P. M. Reid, C. Punyadeera, L. Chang and H. E. Gendelman, Nat. Mater., 2021, 20, 593–605 CrossRef CAS PubMed.
- P. Srivastava and D. Prasad, 3 Biotech, 2023, 13, 200 CrossRef PubMed.
- K. Pardee, A. A. Green, M. K. Takahashi, D. Braff, G. Lambert, J. W. Lee, T. Ferrante, D. Ma, N. Donghia, M. Fan, N. M. Daringer, I. Bosch, D. M. Dudley, D. H. O'Connor, L. Gehrke and J. J. Collins, Cell, 2016, 165, 1255–1266 CrossRef CAS PubMed.
- S. Y. Li, Q. X. Cheng, J. K. Liu, X. Q. Nie, G. P. Zhao and J. Wang, Cell Res., 2018, 28, 491–493 CrossRef CAS PubMed.
- J. S. Chen, E. Ma, L. B. Harrington, M. Da Costa, X. Tian, J. M. Palefsky and J. A. Doudna, Science, 2018, 360, 436–439 CrossRef CAS PubMed.
- D. C. Swarts and M. Jinek, Mol. Cell, 2019, 73, 589–600 CrossRef CAS PubMed.
- M. J. Kellner, J. G. Koob, J. S. Gootenberg, O. O. Abudayyeh and F. Zhang, Nat. Protoc., 2019, 14, 2986–3012 CrossRef CAS PubMed.
- L. Li, S. Li, N. Wu, J. Wu, G. Wang, G. Zhao and J. Wang, ACS Synth. Biol., 2019, 8, 2228–2237 CrossRef CAS PubMed.
- T. Notomi, H. Okayama, H. Masubuchi, T. Yonekawa, K. Watanabe, N. Amino and T. Hase, Nucleic Acids Res., 2000, 28, e63 CrossRef CAS PubMed.
- O. Piepenburg, C. H. Williams, D. L. Stemple and N. A. Armes, PLoS Biol., 2006, 4, e204 CrossRef PubMed.
- F. Hu, Y. Liu, S. Zhao, Z. Zhang, X. Li, N. Peng and Z. Jiang, Biosens. Bioelectron., 2022, 202, 113994 CrossRef CAS PubMed.
- B. Wang, R. Wang, D. Wang, J. Wu, J. Li, J. Wang, H. Liu and Y. Wang, Anal. Chem., 2019, 91, 12156–12161 CrossRef CAS PubMed.
- Y. Xiong, G. Cao, X. Chen, J. Yang, M. Shi, Y. Wang, F. Nie, D. Huo and C. Hou, Appl. Microbiol. Biotechnol., 2022, 106, 4607–4616 CrossRef CAS PubMed.
- Q. Chen, I. Gul, C. Liu, Z. Lei, X. Li, M. A. Raheem, Q. He, Z. Haihui, E. Leeansyah, C. Y. Zhang, V. Pandey, K. Du and P. Qin, J. Med. Virol., 2023, 95, e28385 CrossRef CAS PubMed.
- K. Yin, X. Ding, Z. Li, H. Zhao, K. Cooper and C. Liu, Anal. Chem., 2020, 92, 8561–8568 CrossRef CAS PubMed.
- M. Lin, H. Yue, T. Tian, E. Xiong, D. Zhu, Y. Jiang and X. Zhou, Anal. Chem., 2022, 94, 8277–8284 CrossRef CAS PubMed.
- Y. Chen, X. Xu, J. Wang, Y. Zhang, W. Zeng, Y. Liu and X. Zhang, Anal. Chem., 2022, 94, 9724–9731 CrossRef CAS PubMed.
- X. Ding, K. Yin, Z. Li, R. V. Lalla, E. Ballesteros, M. M. Sfeir and C. Liu, Nat. Commun., 2020, 11, 4711 CrossRef CAS PubMed.
- L. Xia, J. Yin, J. Zhuang, W. Yin, Z. Zou and Y. Mu, Anal. Chem., 2023, 95, 4744–4752 CrossRef CAS PubMed.
- F. Li, S. Liu, B. Luo, M. Huang, Y. Teng and T. Wang, Microbiol. Spectrum, 2023, 11, e0423322 CrossRef PubMed.
- W. Feng, H. Peng, J. Xu, Y. Liu, K. Pabbaraju, G. Tipples, M. A. Joyce, H. A. Saffran, D. L. Tyrrell, S. Babiuk, H. Zhang and X. C. Le, Anal. Chem., 2021, 93, 12808–12816 CrossRef CAS PubMed.
- J. Wei, W. Wang, Q. Yu, M. Zhang, F. Xue, B. Fan, T. Zhang, Y. Gao, J. Li, X. Meng and B. Pang, Sens. Actuators, B, 2023, 390, 133950 CrossRef CAS PubMed.
- K. Lin, J. Guo, X. Guo, Q. Li, X. Li, Z. Sun, Z. Zhao, J. Weng, J. Wu, R. Zhang and B. Li, Anal. Chim. Acta, 2023, 1248, 340938 CrossRef CAS PubMed.
- S. D. Davi, J. Kissenkotter, M. Faye, S. Bohlken-Fascher, C. Stahl-Hennig, O. Faye, O. Faye, A. A. Sall, M. Weidmann, O. G. Ademowo, F. T. Hufert, C. P. Czerny and A. Abd El Wahed, Microbiol. Infect. Dis., 2019, 95, 41–45 CrossRef CAS PubMed.
- I. Iizuka, M. Saijo, T. Shiota, Y. Ami, Y. Suzaki, N. Nagata, H. Hasegawa, K. Sakai, S. Fukushi, T. Mizutani, M. Ogata, M. Nakauchi, I. Kurane, M. Mizuguchi and S. Morikawa, J. Med. Virol., 2009, 81, 1102–1108 CrossRef CAS PubMed.
- J. Feng, G. Xue, X. Cui, B. Du, Y. Feng, J. Cui, H. Zhao, L. Gan, Z. Fan, T. Fu, Z. Xu, S. Du, Y. Zhou, R. Zhang, H. Fu, Z. Tian, Q. Zhang, C. Yan and J. Yuan, Microbiol. Spectrum, 2022, 10, e0271422 CrossRef PubMed.
- C. Yu, L. Zuo, J. Miao, L. Mao, B. Selekon, E. Gonofio, E. Nakoune, N. Berthet and G. Wong, Viruses, 2022, 15, 84 CrossRef PubMed.
- L. Mao, J. Ying, B. Selekon, E. Gonofio, X. Wang, E. Nakoune, G. Wong and N. Berthet, Viruses, 2022, 14, 2112 CrossRef CAS PubMed.
- M. Singh, C. S. Misra, G. Bindal, S. S. Rangu and D. Rath, J. Med. Virol., 2023, 95, e28974 CrossRef CAS PubMed.
- F. Zhao, Y. Hu, Z. Fan, B. Huang, L. Wei, Y. Xie, Y. Huang, S. Mei, L. Wang, L. Wang, B. Ai, J. Fang, C. Liang, F. Xu, W. Tan and F. Guo, Cells Rep. Methods, 2023, 3, 100620 CrossRef CAS PubMed.
- F. Zhao, P. Wang, H. Wang, S. Liu, M. Sohail, X. Zhang, B. Li and H. Huang, Anal. Methods, 2023, 15, 2105–2113 RSC.
- R. Yang, S. Xie, B. Zhou, M. Guo, J. Fan, F. Su, Z. Ji, Y. Chen and B. Li, ACS Appl. Mater. Interfaces, 2024, 16, 9890–9899 CrossRef CAS PubMed.
- S. J. Low, M. T. O'Neill, W. J. Kerry, M. Krysiak, G. Papadakis, L. W. Whitehead, I. Savic, J. Prestedge, L. Williams, J. P. Cooney, T. Tran, C. K. Lim, L. Caly, J. M. Towns, C. S. Bradshaw, C. Fairley, E. P. F. Chow, M. Y. Chen, M. Pellegrini, S. Pasricha and D. A. Williamson, Lancet Microbe, 2023, 4, e800–e810 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02049a |
| ‡ T. Yu, Z. Rong and Z.-X. Gu contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |