
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProbing the mechanism of guest–framework bonding interactions through a first-principles study on the structural and electronic properties of type-II clathrate AxSi136 (A = Na, K, Rb; 0 ≤ x ≤ 24) under pressure
Dong Xue *a,
Yanbin Dengb and
Charles W. Mylesc
*a,
Yanbin Dengb and
Charles W. Mylesc
aDepartment of Physics, Langfang Normal University, Langfang 065000, China
bDepartment of Physics, Shangrao Normal University, Shangrao 334001, China
cDepartment of Physics & Astronomy, Texas Tech University, Lubbock, Texas 79409-1051, USA
First published on 25th June 2024
Abstract
The role of noncovalent bonding, including multiatomic interactions (van der Waals-like forces) and ionic characteristics, in the intermetallic clathrate AxSi136 (A = Na, K, Rb; 0 < x ≤ 24) is qualitatively discussed. Using the local density approximation (LDA) to density functional theory (DFT), we investigated the effect of different guest filling and pressure parameters on the structural and electronic properties of these materials. In the context of the rigid-band model, we first noted that the competition between van der Waals-like multiatomic interactions and ionicity due to the extent of charge transfer responsible for guest–framework complexes accounts for the nonmonotonic structural response upon guest filling in AxSi136 (0 ≤ x ≤ 8), which is in good agreement with previous experimental findings as well as theoretical predictions. In comparison with computational work initiated under zero temperature and pressure conditions, the DFT calculations at high pressure (P = 3 GPa) show no apparent variation with respect to the electronic structure. Regarding the A16Si136 compound, the encapsulated sodium atoms residing in the 20-atom cage cavity act as centers of somewhat localized electrons compared with the alkaline metal sites inside Si28 cage voids. Moreover, the substitution of heavier guest atoms (e.g., Rb) for all the Na atoms in Na8Si136 yields less significant charge transfer between the guest and framework constituents. The net effect of quickly increasing multiatomic interactions and slowly decreasing ionic bonding between the encapsulated atom and Si28 cage may prevent the entire lattice configuration from contracting in a more rapid way when guest species are tuned from Na to Rb in AxSi136 (A = Na, Rb; 0 < x ≤ 8) with increased composition x. In other words, the coulombic attraction due to ionic bonding slightly outweighs the repulsive interaction between the Rb atom and Si28 cage. In addition, the determined formation energy per conventional unit cell in K8Si136, Rb8Si136 and Na12Si136 attains a minimum value, demonstrating the stabilizing effect of guests incorporated into “oversized” cage cavities.
I. Introduction
Metastable materials with clathrate phases have recently emerged and become formidable in the field of thermoelectric (TE) applications.1–5 Because it is capable of efficiently converting waste heat into recycled power, TE clathrate serves as a competitive candidate for harvesting energy.6–10 In the group-14 clathrate, inorganic open-framework atoms (C, Si, Ge or Sn), indexed as the minimal “building blocks”, are in a fourfold coordinated sp3 bonding configuration. Group-14 clathrate structures are generally identified as type I or II according to their different stoichiometric compositions and polyhedral cage characteristics. Among these two expanded-volume phases, relatively few results have been obtained for the type II structure, which crystallizes in the cubic structure labeled Fd![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m (space group no. 227) under ambient conditions.11–13 Type II binary clathrate compounds are normally described by the general formula ALxM136 (AL = alkaline metal or alkaline-earth metal; M = Si, Ge, Sn; 0 < x ≤ 24). Here, the “guest” impurity denoted by AL can be filled into two different sized cages: 20-atom (M20, dodecahedron) and 28-atom (M28, hexakaidecahedron) cages. These two categories of cages are arranged periodically in a 2
m (space group no. 227) under ambient conditions.11–13 Type II binary clathrate compounds are normally described by the general formula ALxM136 (AL = alkaline metal or alkaline-earth metal; M = Si, Ge, Sn; 0 < x ≤ 24). Here, the “guest” impurity denoted by AL can be filled into two different sized cages: 20-atom (M20, dodecahedron) and 28-atom (M28, hexakaidecahedron) cages. These two categories of cages are arranged periodically in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio in the 136-atom unit cell. Moreover, 34 tetrahedrally bonded atoms are situated within the primitive unit cell of the “guest-free” clathrate, forming a face-centered cubic (FCC) structure. The larger cubic unit cell (M136) can also be visualized as the supercell (SC) form of such a 34-atom unit cell. On the other hand, the conventional unit cell containing 136 atoms is “computationally equivalent” to its primitive counterpart. In other words, both of these unit cells are exactly and completely equivalent descriptions of the infinite lattice. In addition, the computation time length, which approximately scales with N2lnN (N: total number of atoms in the unit cell) within the framework of the periodic DFT method, is significantly prolonged when considering Si136 rather than Si34. Therefore, we chose to perform density functional theory (DFT) calculations on the basis of the AxSi34 (A = Na, K, Rb; 0 ≤ x ≤ 6) format instead of the chemical stoichiometry AxSi136 (A = Na, K, Rb; 0 ≤ x ≤ 24) throughout this entire paper.
:1 ratio in the 136-atom unit cell. Moreover, 34 tetrahedrally bonded atoms are situated within the primitive unit cell of the “guest-free” clathrate, forming a face-centered cubic (FCC) structure. The larger cubic unit cell (M136) can also be visualized as the supercell (SC) form of such a 34-atom unit cell. On the other hand, the conventional unit cell containing 136 atoms is “computationally equivalent” to its primitive counterpart. In other words, both of these unit cells are exactly and completely equivalent descriptions of the infinite lattice. In addition, the computation time length, which approximately scales with N2lnN (N: total number of atoms in the unit cell) within the framework of the periodic DFT method, is significantly prolonged when considering Si136 rather than Si34. Therefore, we chose to perform density functional theory (DFT) calculations on the basis of the AxSi34 (A = Na, K, Rb; 0 ≤ x ≤ 6) format instead of the chemical stoichiometry AxSi136 (A = Na, K, Rb; 0 ≤ x ≤ 24) throughout this entire paper.
AxSi136, a novel representative of guest–host complexes, has attracted increasing attention from researchers.14–19 However, the detailed mechanism that governs the guest–framework bonding interactions in AxSi136 remains unresolved. The stabilizing effect of alkaline metal (Na, K, Rb) filling Si28 cages (0 < x ≤ 8) as a preferential choice leads to a nonmonotonic structural response, which is qualitatively described by lattice contraction instead of expansion in our earlier study.20 Accordingly, experimental results have shown a nonmonotonic structural response to guest filling in NaxSi136 (x = 0, 4, 8). Specifically, lattice contraction occurs as sodium is initially and solely introduced into Si28 (28-atom cage) voids while leaving all 20-atom cage cavities intact. These novel findings motivated us to examine dissimilar guest–framework bonding interactions for Na@Si28 (sodium “impurity” inside the silicon hexakaidecahedron) and Na@Si20 (sodium “impurity” inside the silicon dodecahedron) in the current work. Furthermore, such guest–host complexes are held together in unique structural relationships through noncovalent bonding between guest atoms and framework constituents. Commonly mentioned types of noncovalent interactions include ionic bonding, hydrogen bonding and van der Waals forces (multiatomic interactions).21 Probing the impact of guest–framework bonding interactions on the structural and electronic properties of materials, which is of great interest, has become an important field because it can be fairly useful in material design. Moreover, the discovery of superconductivity in type I metal-doped NaxBaySi46 has led to a similar curiosity about type II “cage-structured” clathrate compounds with identical minimal “building blocks” originating from Si.22 For instance, H. Kawaji et al. observed a critical temperature (TC) of approximately 4 K for (Na,Ba)xSi46.23 In addition, the presence of metallic guests inside (Ba1−xSrx)8Si46 (0 ≤ x ≤ 1) can modify the electronic structure of bare Si46, resulting in the formation of a high density of states (DOS) peak around the Fermi energy (EF) level. It is then expected that the localized sharp peak of the DOS at EF contributes to the increasing TC of the superconducting phase. In addition, research concerning the structural and electronic behaviors of AxSi136 under pressure has rarely been conducted to the best of our knowledge. According to recent studies, G. K Ramachandran et al. examined the phase transformation of Si136 in the pressure range of 0 to 12 GPa.24 It is therefore of much interest to examine the impact of pressure on charge transfer from the guest to the silicon framework upon guest doping in this paper.
The interest in structural stability issues, electronic features, and guest–framework bonding interaction mechanisms of type II clathrate compounds is rooted not only in the field of experimenters but also within the scope of theoretical explorers. As early as 1965, scientists managed to synthesize the first-generation silicon clathrates Na8Si46 and NaxSi136 (x < 11), which paved the way for subsequent investigations within this field.25 Combined with unprecedented progress in high-performance computing facilities, an increasing amount of theoretical work focusing on clathrate compounds has become widely recognized in recent decades. An early example of this is the calculational study on the electronic structure of NaxSi136 (x = 0, 4, 8, 16, 24) by Smelyansky et al. in 1997.26 In this paper, the authors verified the prominent metallic character of NaxSi136 when the composition x of the Na metal exceeded 8. In addition, the lattice geometry changes very little as the Na composition x is tuned directly from 3 to 24 according to previous work. In addition to AxM136 and AxM46, ternary and quaternary type II clathrate compounds as well as their type VIII counterparts have drawn significant attention from many research groups.27 For instance, first-principle work relating to the determination of the electronic and vibrational features of Rb7.3Na16Ga20Si116, Cs8Na16Ga21Si115, Cs8Ga8Si128, and Rb8Ga8Si128 has been presented by means of density functional theory (DFT).28,29 Furthermore, structural and thermoelectric analyses with respect to Rb8Na16Si136 and Cs8Na16Si136 were also experimentally performed.30 However, the detailed mechanism accounting for why framework contraction occurs upon impurity guest doping (x changes from 0 to 4 and to 8) in AxSi136 (A = Na, K, Rb; 0 < x ≤ 24) still awaits intensive exploration. In other words, it is necessary and intriguing to investigate how chemical bonding, which is closely related to guest–host interactions in AxSi136, affects the structural and electronic characteristics of the material of interest. Additionally, motivated by some of the above experimental studies on alkali metal-containing clathrate, we performed a theoretical investigation confirming the structural stability of the clathrate A8Si136, in which all the hexakaidecahedron cages preferentially encapsulate the guest (A), while the other dodecahedron cages appear to be empty even under high pressure conditions.18,30–33
Recently, the search for state-of-the-art thermoelectric clathrates has prevailed. Despite the novelty associated with the cage structure, the quantitative parameter that marks a promising clathrate candidate for power generation is a high dimensionless figure of merit ZT (>1). Thermoelectric material normally possesses the dimensionless figure of merit defined as: ZT = TS2σ/κ where, Z is the figure of merit, T is the temperature, S is the Seebeck coefficient of the material, σ is the electrical conductivity and κ is the total thermal conductivity of the material. Many synthesized clathrate compounds (e.g., Eu8Ga16Ge30 and Sr8Ga16Ge30) that meet the “phonon glass electron crystal” (PGEC) criteria are known to exhibit semiconducting or intermetallic behavior while possessing excellent ZT values.34 The encapsulated “guest” atoms inside the “cage-structured” voids can vibrate resonantly with framework atoms, thus contributing to scattering heat-carrying acoustic phonons and enhancing ZT. Apart from such resonant vibrations from the phonon spectrum standpoint, the guest–framework bonding interaction is still not precisely understood due to the lack of systematically detailed exploration of the relevant electronic band structure, total and projected electronic density of states, etc. In this paper, we perform ab initio calculations to inspect these unique properties of stabilized AxSi136 compounds from an electronic point of view. The effects of pressure and guest filling on relevant electronic structures are also discussed.
II. Methodology
Our calculational work implemented by the Vienna Ab initio Simulation Package (VASP) is within the framework of density functional theory.35–37 As initially proposed by Hohenberg, Kohn, and Sham, the DFT method has experienced many refinements to reduce the discrepancy between the predicted ground state properties and their experimental counterpart results. Consequently, the local density approximation (LDA) or generalized gradient approximation (GGA) was appropriately incorporated into the exchange–correlation functional, while no d or f electrons need to be considered at the current stage. These approximations help increase the accuracy as the computational time increases. In this paper, only the LDA to density functional theory is employed to approximate the exchange–correlation effect, and the projected augmented wave (PAW) method is used to determine the electronic properties, including the band structure, total and projected density of states, bulk modulus, elastic constant, and other ground state properties in the chosen range of external pressures. In this paper, we also presented calculations of the total free energy and formation enthalpy concerning the phase stability of AxSi136. Normally, obtaining more accurate results requires us to choose PAW potentials rather than ultrasoft pseudopotentials. This is because of two obvious factors: first, the radial cutoffs of the core remain smaller than the radii used for the ultrasoft pseudopotentials; second, the PAW potentials reconstruct the exact valence wave functions with all nodes in the core region. In other words, using PAW potentials can guarantee the achievement of accuracy while reducing the computational effort. According to the outermost electron configuration of Si (3s23p2), these electrons are selected as the valence electrons. It is widely recognized that alkaline metal atoms in A-doped silicon clathrates preferentially occupy the 8b Wyckoff position.The first calculation step involved obtaining a geometrically optimized configuration, under which the electronic structure was then calculated. The ionic positions are optimized by minimizing the total binding energy, provided that a fixed unit cell volume has already been built. This self-consistent process is carried out repeatedly for a range of cell volumes. Group theory is simultaneously applied by the simulation package to reduce the computational cost of acquiring energy and force. Finally, the LDA energy versus volume is fit into the 3rd Birch–Murnaghan equation of state (EOS), resulting in the structural and geometrical parameters at the equilibrium state. Moreover, the optimized lattice constant can be determined according to the global minimum energy generated from the Birch–Murnaghan EOS.
Due to the large number of atoms in the unit cell of the AxSi136 compounds, the Monkhorst–Pack scheme was used before setting up the k-points in the Brillouin zone in the 4 × 4 × 4 mode. Moreover, the Methfessel–Paxton scheme was also used. The energy cutoff parameters of the PAW potentials are carefully selected to ensure the accuracy of the calculations for each clathrate compound of interest. In other words, 250 eV was determined to be reliably effective for the entire calculational work. The energy convergence is set to 5 × 10−6 eV per atom, and the interatomic interaction force convergence standard is set to 0.1 eV nm−1. Moreover, the inner crystal stress convergence standard is set to 0.02 GPa, while the maximum atomic displacement convergence standard is given by 5 × 10−5 nm. A conjugate-gradient (CG) algorithm is constructed to relax the ions into their instantaneous ground state before the electronic feature details are revealed.
III. Results and discussion
A. Structural properties
The AxSi136 binary clathrate system possesses a conventional unit cell, which faces the center cubic of Fdm symmetry. In total, 136 silicon atoms are tetrahedrally coordinated and are pertinent to two types of polyhedral cages: eight hexakaidecahedra [51264] and sixteen dodecahedra [512]. The interaction between neighboring silicon atoms in the bare Si136 configuration is covalent. Although it is possible to have both [51264] and [512] occupied after guest atoms are chemically “inserted”, the order of which type of polyhedron should be filled upon the doping process is not random. In other words, many early studies, including those involving power X-ray diffraction data, verified that hexakaidecahedron cages are preferentially occupied by sodium until they are full.18 Moreover, as the composition x (x > 8) exceeds 8, the dodecahedron cages can begin to encapsulate the remaining x − 8 guest atoms. Some early work reported that the order of guest filling concerning two different polyhedral cages is unique. This is believed to correlate with the degree of occupation of Si136 antibonding states.38 A detailed analysis of this topic is presented below. Furthermore, the preferential cage occupancy is accompanied by an unusual structural response, leading to lattice framework contraction rather than expansion occurring once x changes from 0 to 4 and to 8. To our knowledge, this work is the first to indicate that the dominant attractive interaction due to ionic bonding between positively charged guest and cage network atoms that carry negative charges outweighs the multiatomic bonding (van der Waals like) between the entire guest atom and cage cavity Si28 in the case of NaxSi136 (x = 4, 8). This ionic character here is distinguished from the conventionally defined ionic bond between the monovalent cation and the monovalent anion in the NaCl structure. Instead, the charge transferred from sodium to the framework atoms is assumed to be uniformly distributed among the silicon network atoms, leading to the resultant ionic bonding between the Na atom and its surrounding cage cavity. In other words, the net interaction due to multiatomic bonding and ionic bonds is believed to be attractive, causing unusual lattice contraction. The replacement of heavy guest (e.g., Rb) by Na in AxSi136 (0 < x ≤ 8) with a fixed composition can lead to a smaller size difference between the guest and Si28 framework, indicating the existence of stronger multiatomic bond repulsions while slightly weakening the extent of charge transfer between Rb and host atoms in the same cage cavity. Eventually, competition between repulsive multiatomic bonding and attractive ionic bonding causes the whole lattice of RbxSi136 (x = 4, 8) to contract in a less rapid way (see Fig. 2(a) and (c)), indicating that net guest–host interactions are still attractive. For each A16Si136 (A = Na, K, Rb), the ionic bonding strength remains higher in Si28 cages than in their Si20 counterparts. This is because of the relatively low amount of charge transfer between the guest (A) and the constituent atoms of the Si20 cages compared with the charge transfer behavior inside the Si28 cage cavities. In short, the competition between multiatomic interactions and ionic bonding influences the net effect of various noncovalent bonding interactions. In addition to intensive discussions on the above issues, this paper initially examines the impact of pressure on the structural and electronic properties of the AxSi136 system.
For the sake of capturing the optimized lattice constant, the energy versus volume curve is acquired by means of the 3rd Birch–Murnaghan equation of state at first stage. Fig. 1 shows the energy versus volume curve for the different A8Si136 (A = Na, K, Rb) clathrates in the presence of P = 0 GPa. It is seen that the energy per atom is slightly less for Na8Si136 when compared to the other two compounds.
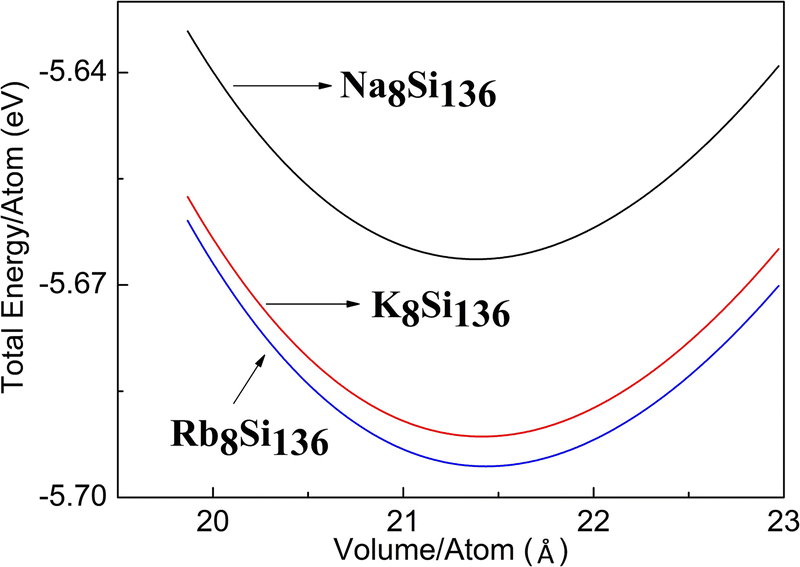 | ||
| Fig. 1 Equation of state (E vs. V) of AxSi136 (A = Na, K, Rb). | ||
Using intuitive language, we determined the lattice constant for the cage-structured system AxSi136 under pressures of P = 0 GPa and P = 3 GPa, as shown in Fig. 2. The “guest-free” A0Si136 structure undergoes configurational compression at pressures greater than 0 GPa. Trends for the variation in the lattice constant have also been theoretically observed for each fixed alkaline composition in the subdiagrams labeled (a), (b), and (c). For the case of NaxSi136, lattice contraction as Na is introduced solely into the Si28 cages for NaxSi136 (x = 4, 8) repeatedly occurs at nonzero pressure, which is analogous to our previous calculations obtained at P = 0 GPa.20 As seen from the numerical results in Fig. 2(a), our computed minimum lattice constant for NaxSi136 is 14.548 Å at P = 0 GPa and 14.474 Å at P = 3 GPa. Early experimental report has pointed out the occupancies of Na@Si20 and Na@Si28, motivating us to perform our current calculational work.18 This is to say, the oversized cages are filled first when x < 8 and the smaller cages are then filled when x > 8. Specifically, sodium occupancy of Si20 cages remains to be zero as total Na content is between 0 and about 6.8 according to such report.18 Meanwhile, a strong preferred occupation by Na of the oversized Si28 cages is observed as the normalized Na occupancy rises to about 0.96 with increased total Na content from 0 to 8. Meanwhile, almost all Si20 cages remain to be empty. Consequently, we conduct our computational work on determining the lattice parameter variation as a function of guest content, while assuming all the Si28 cages should be preferentially occupied before the Na atoms fill into Si20 cages.
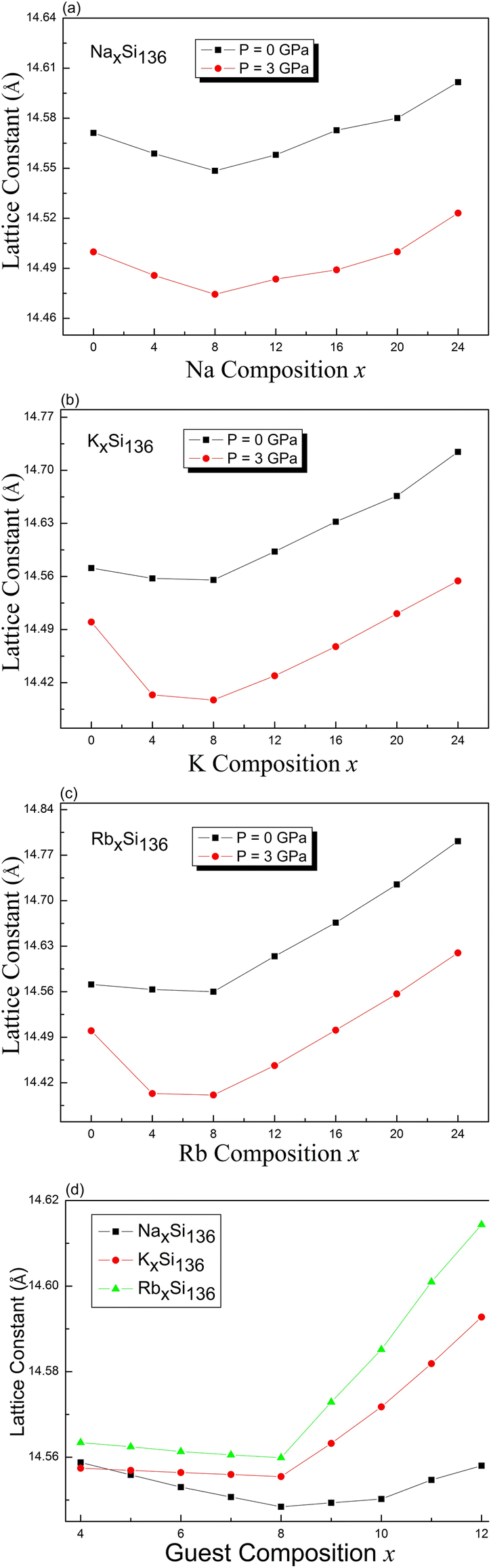 | ||
| Fig. 2 Predicted x dependence of the lattice constant for the type-II clathrate-based compounds (a) NaxSi136 (b) KxSi136 (c) RbxSi136 under different pressures with 0 ≤ x ≤ 24 and (d) AxSi136 (A = Na, K, Rb; 4 ≤ x ≤ 12) at P = 0 GPa. | ||
Once the sodium atoms are entirely substituted by potassium or rubidium, the lattice contraction shows no prominent feature compared with the lattice variation at P = 3 GPa for a composition of x = 4, 8, as shown in Fig. 2(b) and (c). In other words, the increased atomic weight and expanded volume of guest impurities might significantly weaken the attractive interactions between the guest and Si framework as x is tuned from 0 to 4 and to 8. Fig. 2(d) illustrates the trends for the variation in the lattice constant calculated at zero pressure, in the presence of increased composition value from 4 to 12 in a detailed manner. The minimum lattice parameter for each material is still found when x equals to 8. In addition to these, our calculated lattice parameter of Rb12Si136 at zero pressure remains at 14.6144 Å, which is slightly lower than the experimental value (14.7142 Å) for Rb11.1(1)Si136.39 The equilibrium volume of Si136 at zero pressure gives a cubic lattice constant of 14.5712 Å, about 0.38% smaller than the experimental value of 14.6269 Å.40
To quantitatively verify the preferential occupation of hexakaidecahedron cages by guests upon gradual filling (Δx = 4), the formation energy per unit cell (Ef) for AxSi136 (4 ≤ x ≤ 12) at zero pressure is listed in Table 1. This quantity has the following format:
| Ef(AxSi136) = EAxSi136 − 4EA − EAx−4Si136 |
| Compound | Pressure (GPa) | A = Na | A = K | A = Rb |
|---|---|---|---|---|
| A4Si136 | 0 GPa | −4.3632 | −7.3544 | −8.2292 |
| A8Si136 | 0 GPa | −5.3408 | −8.0464 | −8.4220 |
| A12Si136 | 0 GPa | −5.5072 | −5.4392 | −4.5584 |
B. Electronic band structure
DFT calculations of the band structure at nonzero pressure were performed to examine the impact of high pressure on the electronic properties of Na8Si136. Fig. 2 shows the pressure impact of the reduced atomic volume for the clathrate series NaxSi136, while an unusual structural response still exists according to the associated XRD experiment.18 For Fig. 3, the symmetric points of the Brillouin zone (BZ) are given by Γ, X, U|K, L and W. The Fermi level is chosen to remain at 0 eV as an energy reference. The overall structure of Na8Si136 at P = 0 GPa is extremely similar to that of Na8Si136 at P = 3 GPa on an energy scale between −2.5 eV and 1.5 eV. The pressure of 3 GPa nearly causes the top of the valence band (VB) to change invariably compared to that of the P = 0 GPa case. However, a small exception exists in the L–W and W–X directions, where the trivial energy discrepancy at the top of the VB is more prominent than that in the other directions. Moreover, the bottom part of the conduction band (CB) between approximately −0.25 eV and 0 eV is almost unaffected by the pressure variation. In addition, the calculated band character is always metallic because the Fermi level lies in the CB for the P = 0 GPa and P = 3 GPa cases.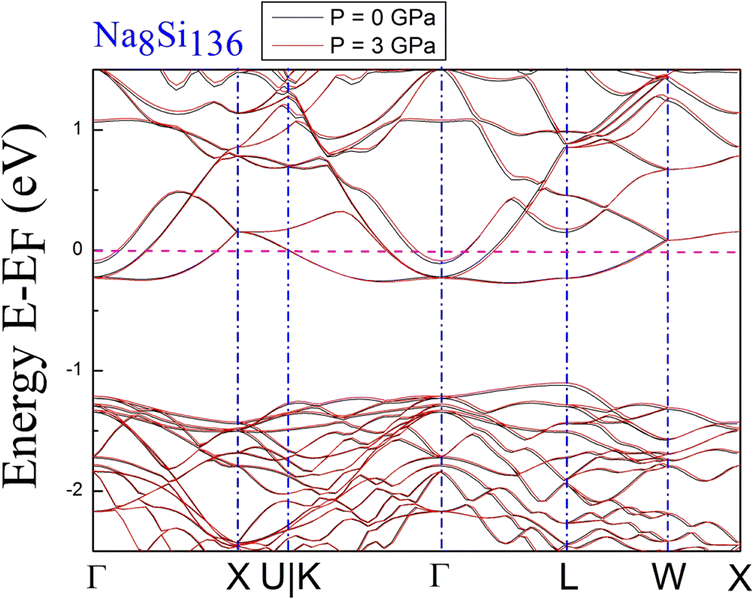 | ||
| Fig. 3 Predicted electronic band structures of Na8Si136 under P = 0 GPa and P = 3 GPa conditions. | ||
Each subdiagram labeled (a)–(d) in Fig. 4 shows the band structure of stoichiometric A8Si136 (A = K, Rb) under zero- and high-pressure conditions. Analogous to previous BS characteristics for Na8Si136, the Fermi level is also crossed by several dispersive bands arising from the CB along the Γ–X, Γ–L and Γ–U|K lines in Fig. 4.
 | ||
| Fig. 4 Predicted electronic band structures of (a) and (b) K8Si136 and (c) and (d) Rb8Si136 between P = 0 GPa and P = 3 GPa. | ||
The results clearly demonstrate the metallic character of the compound, whose features are not affected by changes in the external pressure. In addition, our BS calculations indicate that the antibonding states of the pure silicon framework Si136 are above the bandgap, as shown in Fig. 5. In contrast, the presence of encapsulated Na atoms in Na8Si136 (see Fig. 3) plays no prominent role in varying the VB structure, leading to qualitative agreement with the so-called “rigid band model”.41 In other words, the band behavior of guest-containing binary clathrate remains nearly the same as that of its “parent” Si136 framework. It is intriguing to investigate how “doped” guests influence the occupation of antibonding states due to the presence of the outermost electrons of alkaline metals under various pressure conditions. Specifically, we present in more detail the calculations to determine to what extent the guest atoms donate to the Si framework with respect to A8Si136 (A = K, Rb) under the designated pressure conditions. Furthermore, the A8Si136 clathrates were found to be metallic with a “pseudogap” that connects the top of the VB to the bottom of the CB, as shown in Fig. 3 and 4. The calculated “pseudogap” is approximately 1.06 eV for K8Si136 at P = 0 GPa and approximately 1.01 eV at P = 3 GPa. For Rb8Si136, the calculated “pseudogap” remains at approximately 1.08 eV at zero pressure and approximately 1.05 eV at 3 GPa. Fig. 5 also shows that the direct band gap of Si136 is approximately 1.21 eV. Moreover, the location of the valence band maximum along with the location of the conduction band minimum is found to be at the same site, which is denoted by the high symmetry point L.
 | ||
| Fig. 5 Predicted electronic band structure and electronic density of states of Si136 at P = 0 GPa. | ||
C. Electronic density of states
To examine the effect of guest filling on dissimilar guest–host interactions, we show in Fig. 6–9 the total and projected electronic density of states of AxSi136 (A = Na, K, Rb; 0 ≤ x ≤ 16) obtained at T = 0 K. It is widely noted that the crystal structure and chemical stoichiometry of such silicon-based binary clathrates can be described by the Zintl concept. Metallic behavior might be macroscopically manifested by excess electrons originating from guest atoms when coping with the chemical notation of AxSi136. One widely recognized fact that needs to be mentioned here is that the multiatomic interactions between the alkaline guest and the “oversized” Si28 cage are basically repulsive. This is because the radius of the incorporated guest element is close to the cage radius of the Si28 cavity. In other words, the separation between the guest atom and the cage framework atom is always smaller than the atomic radius of the encapsulated atom. When the distance between neighboring atoms is smaller than approximately 0.4 nanometers, the forces are repulsive in nature. The radius of the Si28 cage in Si136 is approximately 2.72 Å, while the atomic radius of the alkaline element (Na, K, Rb) ranges from 1.91 Å to 2.38 Å and to 2.55 Å.42,43 Furthermore, the ionic radius of cation A+ (A = Na, K, Rb) is in the range of 0.97 Å and 1.48 Å.43 It is known that multiatomic interactions depend on how tight-fitting a guest atom is inside the cage cavity. For instance, there appears to be less repulsive interaction for Na@Si28 than for Rb@Si28. Similarly, the Si20 cage cavity is significantly repelled by encapsulated alkaline metal atoms such as Na, K, and Rb compared to the same guest incorporated into the “oversized” cage counterpart when considering AxSi136 (x = 12, 16, 20, 24). However, the net guest–host interaction becomes repulsive at the current stage in these materials. In other words, the resultant competition between repulsive multiatomic bonding and attractive ionic bonding causes the Si20 cage to expand, leading to the expansion of the whole lattice of AxSi136 (x = 12, 16, 20, 24). It is also well known that the van der Waals-like repulsive force increases rapidly as the separation between guest atoms and cage framework atoms is gradually reduced.In general, multiatomic interactions between guest atoms and silicon framework constituents are always repulsive. On the other hand, ionic bonds constructed due to charge transfer between guest and network atoms tend to deflate the relevant cage geometry. Consequently, the competition between ionic and multiatomic Na–Si (or K–Si, Rb–Si) interactions within the identical cage cavity (Si20, Si28) of the framework plays an essential role in affecting the whole lattice volume change. Fig. 6 shows the electronic density of states (EDOS) and p-orbital projected density of states (p-PDOS) for Si136, Na8Si136, and Rb8Si136 in the lower portion of the conduction band. The EDOS profiles can be qualitatively described in the context of the rigid-band model. The EDOS profiles for the guest-containing clathrates are nearly the same as those of the empty framework when considering the lower portion of the CB. A small exception can be identified by the sharp peak found around the tail of the CB. This rigid-band behavior means that the guest–framework interaction is predominantly ionic in nature. The multiatomic repulsion between guest atoms and cage cavities is outweighed by such ionicity. Furthermore, there is charge transfer from the guests into the host conduction states. Specifically, the nonzero EDOS at the Fermi level in guest-containing materials is mainly attributed to the p-orbital of Si, indicating charge transfer from the alkaline metal to the framework. In the case of Rb8Si136, the extent of charge transfer seems to be less significant than that in Na8Si136. This is also verified in Fig. 7 and 8, where the s-orbital PDOS for Rb is strongly suppressed in the vicinity of the Fermi level in contrast to the s-orbital PDOS for Na in a similar region. Additionally, the p-orbital PDOS of the silicon framework at the Fermi level is more strongly increased in the Rb8Si136 case than in the Na8Si136 case. In general, Fig. 7–9 show the total electronic density of states along with the projected density of states for Na8Si136, Rb8Si136, and Na16Si136 at P = 0 GPa. From Fig. 7, the VB can be divided into two typical parts. First, the density of states corresponding to the energy window between −12 and −7 eV is predominantly attributed to the 3s orbital states of silicon atoms. Second, the contribution stemming from the 3p states of Si seems to be nonnegligible in the same energy interval. The increase in the EDOS from approximately −5 to −1.2 eV is mainly attributed to the 3p states of Si rather than the 3s states. Moreover, the lower part of the CB is composed of 3s and 3p states of Na as well as 3p states of Si.
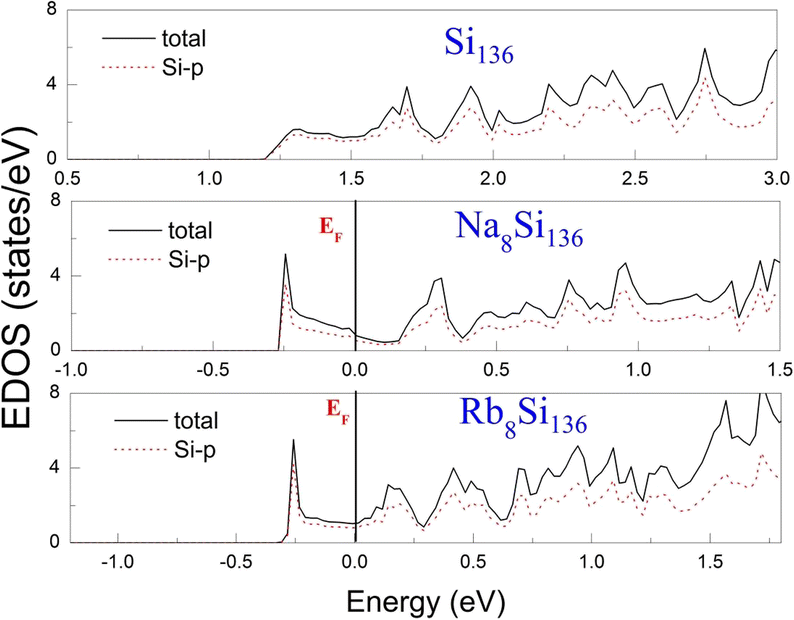 | ||
| Fig. 6 Predicted total EDOS and p-orbital projected EDOS of Si136, Na8Si136 and Rb8Si136 at P = 0 GPa. | ||
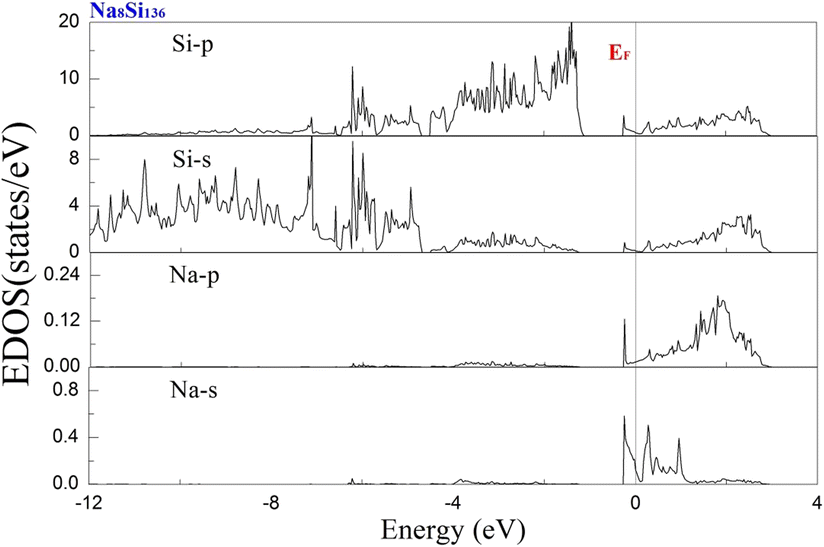 | ||
| Fig. 7 Predicted projected EDOS of Na8Si136 at P = 0 GPa. | ||
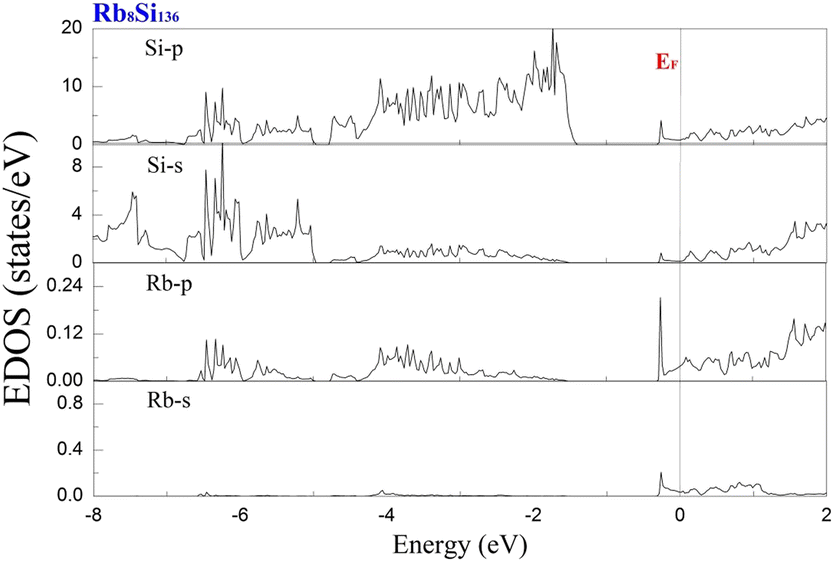 | ||
| Fig. 8 Predicted projected EDOS of Rb8Si136 at P = 0 GPa. | ||
Unlike in the case of Na8Si136, Fig. 8 shows that the charge transfer from guests to silicon framework atoms is weakened due to the considerably reduced s-orbital PDOS for Rb at the Fermi level compared to the s-orbital PDOS for Na in Fig. 7. Although the ionic attraction between Rb cations and negatively charged framework atoms is believed to cause the lattice volume to collapse, the rapidly decreased size difference may cause the “oversized” Si28 cage voids to expand in a more significant manner due to the encapsulation of rubidium rather than sodium. In comparison with NaxSi136 (0 ≤ x ≤ 8), the resultant guest–framework interaction, which is attractive in nature, is believed to hinder the extent of lattice volume collapse, leading to a slight decrease in the lattice constant for RbxSi136 (0 ≤ x ≤ 8) with increasing composition x as a whole (see Fig. 2(c)). Generally, unit cell contraction accompanied by an increase in composition (0 < x ≤ 8) can be attributed to an overall attractive interaction that arises from competition between ionic interactions and multiatomic interactions in AxSi136 (A = Na, K, Rb).
To investigate the guest–host interaction behavior in Si20 cages, the calculated electronic DOS of Na16Si136 is shown in Fig. 9. The EDOS profile concerning the lower part of the CB can thus be clearly divided into contributions from Na guests residing at the 8b Wyckoff sites and contributions from equal amounts of Na atoms situated at the 16c Wyckoff sites. The notation “sum” in Fig. 9 yields the summation of the above mentioned contributions from Na guests residing inside different types of cage polyhedra. In the vicinity of the Fermi level, the EDOS contributions due to the incorporation of the s-orbital of Na atoms into Si20 cages remain much lower than those of Na inside “oversized” cages. In other words, the local effect of Na was more pronounced in the Si20 cages than in the Si28 cages. In other words, significant charge transfer occurs from the guest to framework atoms of Si28 cage cavities. The free radius of the Si20 cage is approximately equal to 1.99 Å, which is considerably smaller than that of the Si28 cage.41 In other words, a significantly decreased size difference between the Na atom and cage cavity may result in extremely strong repulsion. Compared with the resultant attractive interaction between Na and the Si28 cage cavity, the net guest–host interaction involving the Si20 cage becomes repulsive. Similarly, our subsequent computed work also verified that significant charge transfer from K to Si28 framework atoms occurs in comparison with the extent of charge transfer in the Si20 cage of K16Si136. The extremely reduced size difference between the Rb atom and Si20 cavity is believed to be the main factor that causes prominent lattice expansion to occur, which can be quantitatively observed in Fig. 2(b) and (c). It is also speculated that the ability of a guest atom to fit inside its cage may help to affect the extent of charge transfer from the guest to framework atoms. The smaller the size difference between the guest and cage cavity is, the weaker the intensity of charge transfer from the guest to framework atoms.
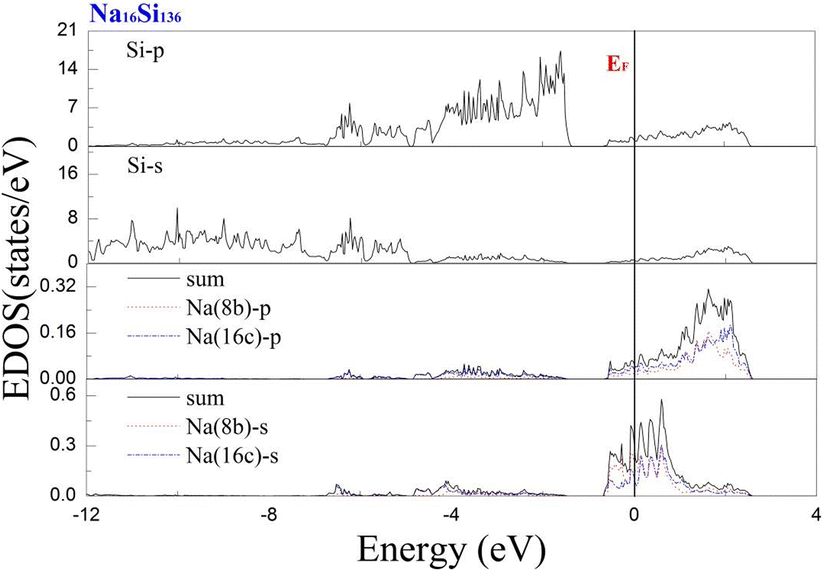 | ||
| Fig. 9 Predicted projected EDOS of Na16Si136 at P = 0 GPa. | ||
In order to reveal the real-space information when taking guest–framework interaction into consideration, we extract the charge density difference at P = 0 GPa in Fig. 10 from our DFT calculation regarding Na12Si136 and Rb12Si136, respectively. The inset figure of each subdiagram indirectly demonstrates the charge distribution between the single guest atom and its surrounding cage framework. Also, two distinct colors have been utilized to explain such issue in a qualitative manner. In the presence of Si28 cage, the zone manifested by blue color remains to be the region where the central guest atom lose the electrons. Meanwhile, the zone manifested by yellow color remains to be the region where the cage framework atoms acquire the transferred electrons. This is to say, the ionic bonding nature can be detected when considering Na(or Rb)@Si28 case. In contrast to this, the somewhat covalent bonding formation due to resultant competition between multiatomic repulsive interaction and Coulomb attractive force is observed, when dealing with Na(or Rb)@Si20 case. In order words, the charges arising from the central metallic atom along with the cage framework atoms accumulate in the vicinity between the guest and the edge of Si20 cage cavity. It is also seen that the ionic bonding strength is slightly weakened for Rb residing in the Si28 cage, in contrast to Na encapsulated in the Si28 cage. This correlates with the fact that smaller size difference between the guest and cage cavity leads to weaker intensity of charge transfer from the guest to framework atoms.
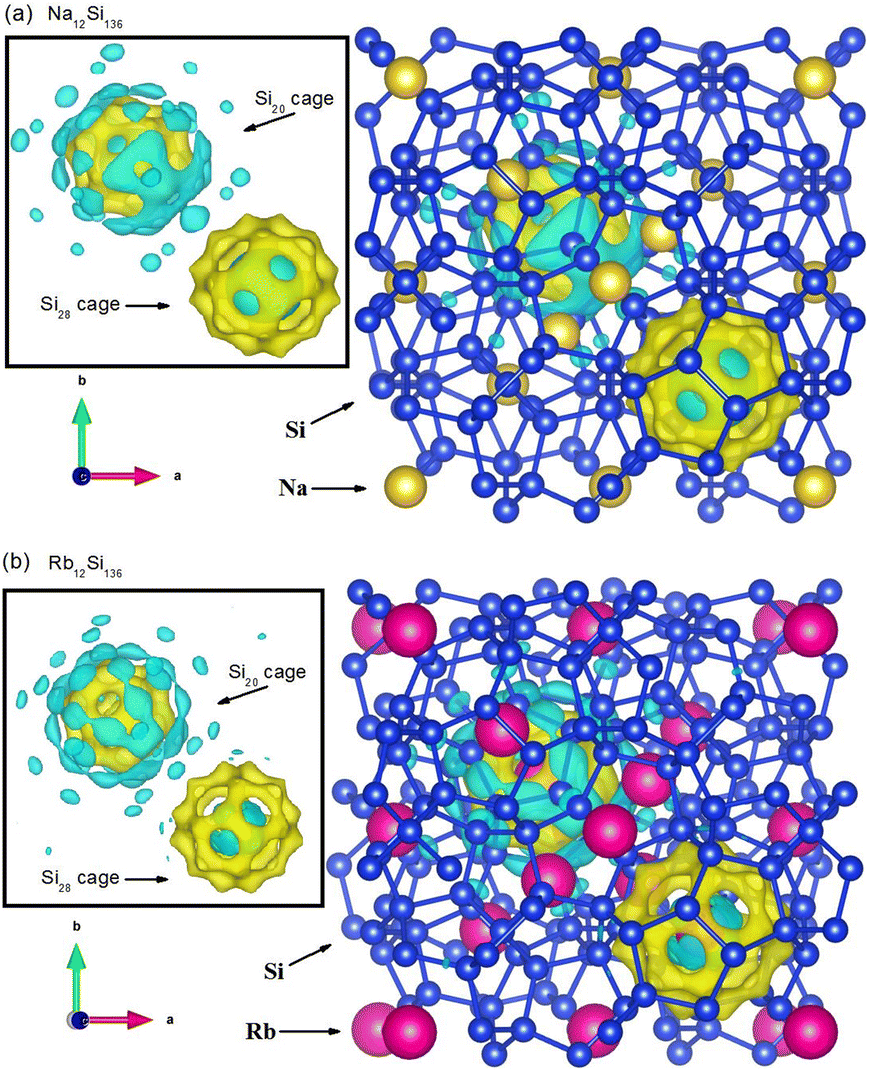 | ||
| Fig. 10 Contour of charge density difference for (a) Na12Si136 and (b) Rb12Si136. | ||
To determine the effect of elevated pressure on guest–framework interactions in different cage cavities, we implemented calculations to reveal the electronic structures of A16Si136 (A = Na, Rb) at P = 3 GPa and T = 0 K. The DFT-determined projected electronic density of states under nonzero pressure are clearly illustrated in Fig. 11. Additionally, the projected EDOS for Rb16Si136 at zero pressure is given for comparison. There is no apparent difference in the EDOS profiles of Na16Si136 and Rb16Si136 at different pressures (see Fig. 9 and 11) when taking the p-orbital contributions of Si atoms to the top of the valence band (−4 to −1.2 eV) structure into account. The influence of elevated pressure on the s-orbital projected density of states for Na atoms is negligibly small. The extent of charge transfer from Na to the Si28 cage framework atoms is greater than that from Na to the Si20 cage constituents at P = 3 GPa. The s-orbital projected EDOS profile at the Fermi level for Rb16Si136 in Fig. 11 indicates essential characteristics. Specifically, the contributions to the projected EDOS due to guests localized in different cage polyhedra remain almost the same under the two pressure conditions. This is, however, contrary to the s-orbital projected EDOS at EF for Na16Si136 under both pressure conditions. Finally, Table 2 lists the quantitative calculations of the total and projected EDOS at EF with respect to the AxSi136 (x = 4, 8, 12) clathrates.
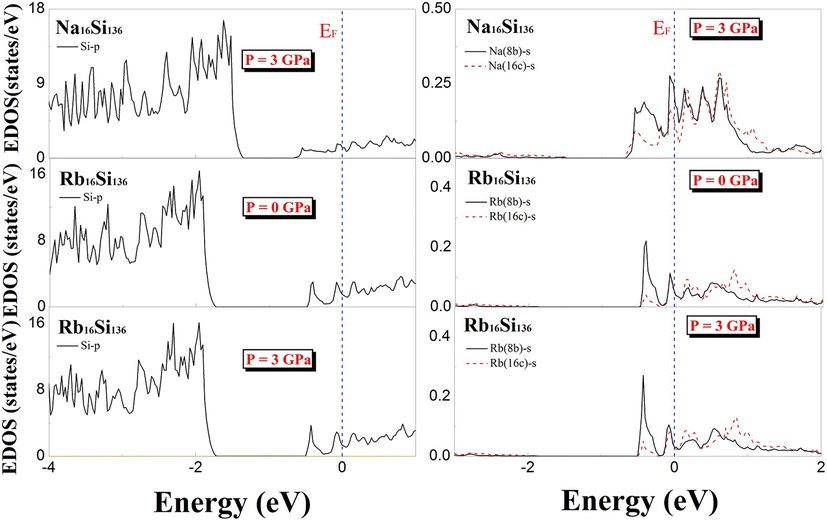 | ||
| Fig. 11 Predicted projected EDOS of Na16Si136, Rb16Si136 at P = 3 GPa and projected EDOS of Rb16Si136 at P = 0 GPa for comparison. | ||
| EDOS | P = 0 GPa | P = 3 GPa | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NaxSi136 | KxSi136 | RbxSi136 | NaxSi136 | KxSi136 | RbxSi136 | |||||||
| A-s (states per eV) | x = 4 | 0.58 | x = 4 | 0.168 | x = 4 | 0.046 | x = 4 | 0.535 | x = 4 | 0.156 | x = 4 | 0.043 |
| x = 8 | 0.123 | x = 8 | 0.138 | x = 8 | 0.036 | x = 8 | 0.112 | x = 8 | 0.129 | x = 8 | 0.033 | |
| x = 12 | 0.259 | x = 12 | 0.115 | x = 12 | 0.041 | x = 12 | 0.267 | x = 12 | 0.074 | x = 12 | 0.044 | |
| Si-p (states per eV) | x = 4 | 2.093 | x = 4 | 1.234 | x = 4 | 1.193 | x = 4 | 2.094 | x = 4 | 1.213 | x = 4 | 1.216 |
| x = 8 | 0.536 | x = 8 | 0.836 | x = 8 | 0.807 | x = 8 | 0.526 | x = 8 | 0.834 | x = 8 | 0.809 | |
| x = 12 | 1.136 | x = 12 | 0.945 | x = 12 | 1.538 | x = 12 | 1.197 | x = 12 | 0.846 | x = 12 | 1.679 | |
| N(EF) | x = 4 | 3.176 | x = 4 | 1.712 | x = 4 | 1.516 | x = 4 | 3.116 | x = 4 | 1.666 | x = 4 | 1.459 |
| x = 8 | 0.801 | x = 8 | 1.215 | x = 8 | 1.029 | x = 8 | 0.768 | x = 8 | 1.199 | x = 8 | 1.024 | |
| x = 12 | 1.827 | x = 12 | 1.776 | x = 12 | 2.535 | x = 12 | 1.855 | x = 12 | 1.457 | x = 12 | 2.544 | |
Table 2 shows the total density of states at the Fermi level N(EF) for each AxSi136 (x = 4, 8, 12) under zero pressure. These values are similar to those of the same compositional material, AxSi136, at P = 3 GPa. Additionally, the contributions of the guest atom s states and Si p states to the EDOS at the Fermi level are also evaluated in Table 2. By considering the ratio of the contribution of Si p states to the contribution of guest s states of each binary compound at the two given pressures, we conclude that the strength of the charge transfer from the guest to the Si20 framework decreases to a certain extent in the presence of A12Si136 compared to that in the cases of A4Si136 and A8Si136. Similarly, the ratio of the Si p state contribution to N(EF), which is the total EDOS, experiences a large increase as the compositional value x varies from 4 to 8. Moreover, such characteristics have a weak dependence on external pressure changes. This also indicates that significant charge transfer occurs in the presence of guest atoms, which are incorporated into large cages (Si28).
IV. Conclusions
We used LDA to study the mechanism of guest–framework bonding interactions by investigating the structural and electronic properties of AxSi136 clathrates. We noted that ionic attraction dominates the guest–framework bonding interactions in “oversized” Si28 cage polyhedra. Moreover, significant charge transfer from the guest to framework atoms is believed to increase the extent of ionic bonding, causing the cage framework to contract in a certain manner. For the same alkaline metal, such as Na or K, the extent of charge transfer is normally greater in the 28-atom cage than in the 20-atom cage. These characteristics are also irrelevant to elevated pressure (from P = 0 GPa to P = 3 GPa). Considering the Na guests that are entirely substituted by heavier Rb atoms in Na8Si136 as an example, we have found that less significant charge transfer from Rb to Si framework constituents is available. Moreover, the size difference between the effective guest and cage volume begins to decrease, resulting in a much stronger repulsion between Rb and the Si28 cage. Compared with the lattice constant of “guest-free” Si136, ionic and repulsive multiatomic interactions found in the Rb4Si136 and Rb8Si136 cases can lead to a slight decrease in the lattice parameter. In other words, the size difference between the effective guest and cage volume, along with the ionicity due to charge transfer, has a combined impact on the guest–framework bonding interaction. Specifically, the repulsive interaction originating from the size difference between the alkaline metal radius and the Si20 cage radius outweighs the ionic bonding due to charge transfer. Consequently, the whole lattice of AxSi136 (x = 12, 16, 20, 24) clathrates expands with continuously increasing composition value x. From the theoretical point of view, the replacement of Na by heavy guest atoms (K or Rb) in AxSi136 (x = 12, 16, 20, 24) can increase the strength of repulsive multiatomic interactions, while slightly weakening the strength of ionic attraction when taking the fixed composition into account. Moreover, the repulsion due to the decreased size difference between the encapsulated guest and Si20 cage increases more rapidly than the decreased ionic attraction due to weakened charge transfer, resulting in prominent lattice expansion. In addition, the role of pressure and the effect of guest atom character on the structural stability and electronic structures of these clathrates are intensively discussed. It should be emphasized that the band structure, EDOS, and PDOS corresponding to the top portion of the valence band have a very weak dependence on the guest filling and pressure (0 ≤ P ≤ 3 GPa) of the same series of AxSi136 clathrates.Data availability
The datasets used and analysed during the current study are available from the corresponding author on reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The equipment of the “Milky Way II” at Supercomputer Center in Lvliang is acknowledged for providing access to high performance computing technologies. This work was supported in part by the PhD & Postdoc Research Starup Foundation of Langfang Normal University (XBQ202122).References
- M. Christensen, S. Johnsen, F. Juranyi and B. B. Iversen, J. Appl. Phys., 2009, 105, 073508 CrossRef.
- A. R. Khabibullin, T. D. Huan, G. Nolas and L. M. Woods, Acta Mater., 2017, 131, 475–481 CrossRef CAS.
- G. S. Nolas, J. L. Cohn, G. A. Slack and S. B. Schujman, Appl. Phys. Lett., 1998, 73, 178 CrossRef CAS.
- Y. Imai and M. Imai, J. Alloys Compd., 2011, 509, 3924–3930 CrossRef CAS.
- A. M. Guloy, R. Ramlau, Z. Tang, W. Schnelle, M. Baitinger and Y. Grin, Nature, 2006, 43, 320–323 CrossRef PubMed.
- G. S. Nolas, G. A. Slack and S. B. Schujman, Semicond. Semimetals, 2000, 69, 255–300 Search PubMed.
- G. S. Nolas, B. C. Chakoumakos, B. Mahieu, G. J. Long and T. J. R. Weakley, Chem. Mater., 2000, 12, 1947–1953 CrossRef CAS.
- L. Qiu, I. P. Swainson, G. S. Nolas and M. A. White, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 035208 CrossRef.
- K. Suekuni, T. Takasu, T. Hasegawa, N. Ogita, M. Udagawa, M. A. Avila and T. Takabatake, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 205207 CrossRef.
- T. Tadano, Y. Gohda and S. Tsuneyuki, Phys. Rev. Lett., 2015, 114, 095501 CrossRef PubMed.
- M. C. Schäfer and S. Bobev, J. Am. Chem. Soc., 2013, 135, 1696–1699 CrossRef PubMed.
- K. Wei, X. Zeng, T. M. Tritt, A. R. Khabibullin, L. M. Woods and G. S. Nolas, Materials, 2016, 9, 732 CrossRef PubMed.
- M. Beekman and G. S. Nolas, J. Mater. Chem., 2008, 8, 842–851 RSC.
- J. Hubner, Y. Prots, W. Schnelle, M. Bobnar, M. Konig, M. Baitinger, P. Simon, W. Carrillo-Cabrera, A. Ormeci, E. Svanidze, Y. Grin and U. Schwarz, Chem.–Eur. J., 2019, 25, 1–10 CrossRef.
- J. Chen, A. S. Arvij, X. Zheng, S. Y. Rodriguez and J. H. Ross, J. Alloys Compd., 2014, 593, 261 CrossRef CAS.
- M. Jing and S. Dong, J. Nat. Gas Chem., 2005, 14, 238–242 CAS.
- X. Connetable and X. Blase, Appl. Surf. Sci., 2004, 226, 289–297 CrossRef.
- M. Beekman, E. N. Nenghabi, K. Biswas and C. W. Myles, Inorg. Chem., 2010, 49, 5338–5340 CrossRef CAS PubMed.
- Y. Li, J. C. T. Garcia, N. Chen and L. Liu, J. Appl. Phys., 2013, 113, 203908 CrossRef.
- D. Xue, C. W. Myles and C. Higgins, Materials, 2016, 9, 691 CrossRef PubMed.
- J. Lipkowski and H. Schneider, J. Coord. Chem., 2021, 74, 1–16 CrossRef.
- S. Yamanaka, Springer Ser. Mater., 2014, vol. 199, pp. 193–226 Search PubMed.
- H. Kawaji, H. Horie, S. Yamanaka and M. Ishikawa, Phys. Rev. Lett., 1995, 74, 1427 CrossRef CAS PubMed.
- G. K. Ramachandran, P. F. McMillan, S. K. Deb, M. Somayazulu, J. Gryko, J. Dong and O. F. Sankey, J. Phys.: Condens.Matter, 2000, 12, 4013 CrossRef CAS.
- J. S. Kaspe, P. Hagenmuller, M. Pouchard and C. Cros, Science, 1965, 1713–1714 CrossRef PubMed.
- V. I. Smelyansky and J. S. Tse, Chem. Phys. Lett., 1997, 264, 459–465 CrossRef CAS.
- Y. Kono, N. Ohya, T. Taguchi, K. Suekuni, T. Takabatake, S. Yamamoto and K. Akai, J. Appl. Phys., 2010, 107, 123720 CrossRef.
- S. Bobev, J. Meyers, V. Fritsch and Y. Yamasaki, 25th International Conference on Thermoelectrics, 2006 Search PubMed.
- K. Biswas and C. W. Myles, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 245205 CrossRef.
- S. Bobev and S. Sevov, J. Solid State Chem., 2000, 153, 92–105 CrossRef CAS.
- S. Stefanoski, C. D. Malliakas, M. G. Kanatzidis and G. S. Nolas, Inorg. Chem., 2012, 51, 8686–8692 CrossRef CAS PubMed.
- S. Stefanoski and G. S. Nolas, Cryst. Growth Des., 2011, 11, 4533–4537 CrossRef CAS.
- H. Morito, H. Yamane, R. Y. Umetsu and K. Fujiwara, Crystals, 2021, 11, 808 CrossRef CAS.
- S. Paschen, V. Pacheco, A. Bentien and A. Sanchez, Phys. B, 2003, 328, 39–43 CrossRef CAS.
- G. Kresse and J. Furthmuller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef CAS PubMed.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef PubMed.
- K. Laasonen, R. Car, C. Lee and D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 43, 6796 CrossRef CAS PubMed.
- K. Biswas, C. W. Myles, M. Sanati and G. S. Nolas, J. Appl. Phys., 2008, 104, 033535 CrossRef.
- I. Veremchuk, M. Beekman, I. Antonyshyn and W. Schnelle, Materials, 2016, 9, 593 CrossRef PubMed.
- K. Biswas and C. W. Myles, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 115113 CrossRef.
- G. K. Ramachandran, J. Dong, O. F. Sankey and P. F. McMillan, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 63, 033102 CrossRef.
- E. Reny, P. Gravereau, C. Cros and M. Pouchard, J. Mater. Chem., 1998, 8, 2839–2844 RSC.
- C. W. Myles, J. Dong and O. F. Sankey, Phys. Status Solidi B, 2003, 239, 26–34 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |