
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSustainable methods for the carboxymethylation and methylation of ursolic acid with dimethyl carbonate under mild and acidic conditions†
Nuttapong Kadsanita,
Pattamabhorn Worsawata,
Chadamas Sakonsinsiri b,
Con R. McElroycd,
Duncan Macquarried,
Pakin Noppawane and
Andrew J. Hunt*a
b,
Con R. McElroycd,
Duncan Macquarried,
Pakin Noppawane and
Andrew J. Hunt*a
aMaterials Chemistry Research Center (MCRC), Department of Chemistry and Centre of Excellence for Innovation in Chemistry, Faculty of Science, Khon Kaen University, Khon Kaen, 40002, Thailand. E-mail: andrew@kku.ac.th
bDepartment of Biochemistry, Faculty of Medicine, Khon Kaen University, Khon Kaen, 40002, Thailand
cSchool of Chemistry, University of Lincoln, Brayford Pool Campus, Lincoln, LN6 7TS, UK
dGreen Chemistry Centre of Excellence, Department of Chemistry, University of York, Heslington, York, YO10 5DD, UK
eDepartment of Chemistry, Faculty of Science, Mahasarakham University, Maha Sarakham, 44150, Thailand
First published on 24th May 2024
Abstract
Ursolic acid is a triterpene plant extract that exhibits significant potential as an anti-cancer, anti-tumour, and anti-inflammatory agent. Its direct use in the pharmaceutical industry is hampered by poor uptake of ursolic acid in the human body coupled with rapid metabolism causing a decrease in bioactivity. Modification of ursolic acid can overcome such issues, however, use of toxic reagents, unsustainable synthetic routes and poor reaction metrics have limited its potential. Herein, we demonstrate the first reported carboxymethylation and/or methylation of ursolic acid with dimethyl carbonate (DMC) as a green solvent and sustainable reagent under acidic conditions. The reaction of DMC with ursolic acid, in the presence of PTSA, ZnCl2, or H2SO4–SiO2 yielded the carboxymethylation product 3β-[[methoxy]carbonyl]oxyurs-12-en-28-oic acid, the methylation product 3β-methoxyurs-12-en-28-oic acid and the dehydration product urs-2,12-dien-28-oic acid. PTSA demonstrated high conversion and selectivity towards the previously unreported carboxymethylation of ursolic acid, while the application of formic acid in the system led to formylation of ursolic acid (3β-formylurs-12-en-28-oic acid) in quantitative yields via esterification, with DMC acting solely as a solvent. Meanwhile, the methylation product of ursolic acid, 3β-methoxyurs-12-en-28-oic acid, was successfully synthesised with FeCl3, demonstrating exceptional conversion and selectivity, >99% and 99%, respectively. Confirmed with the use of qualitative and quantitative green metrics, this result represents a significant improvement in conversion, selectivity, safety, and sustainability over previously reported methods of ursolic acid modification. It was demonstrated that these methods could be applied to other triterpenoids, including corosolic acid. The study also explored the potential pharmaceutical applications of ursolic acid, corosolic acid, and their derivatives, particularly in anti-inflammatory, anti-cancer, and anti-tumour treatments, using molecular ADMET and docking methods. The methods developed in this work have led to the synthesis of novel molecules, thus creating opportunities for the future investigation of biological activity and the modification of a wide range of triterpenoids applying acidic DMC systems to deliver novel active pharmaceutical intermediates.
Introduction
Ursolic acid (1) is a natural product, which can be extracted from a wide variety of herbs, plants and fruits, in addition to agricultural waste streams such as apple peel.1 Ursolic acid and its derivatives have demonstrated several important health benefits and potentially significant pharmacological effects for humans including known anticancer,2 and antitumor properties.3 A significant volume of research has indicated preventative outcomes and therapeutic effects in relation to cancer, obesity/diabetes, cardiovascular disease, brain disease, liver disease, and muscle wasting (sarcopenia) through prescribing ursolic acid or its derivatives. However, the limited solubility of ursolic acid leads to both poor bioavailability, and its rapid metabolism, thus reducing its suitability for the pharmaceutical industry.Recent studies have focused on modifying the structure of ursolic acid to improve its bioavailability and potential as an active pharmaceutical ingredient (Scheme 1). Tu et al. synthesized several ursolic acid derivatizes by reaction of the C-3 hydroxyl or C-28 carboxylic acid groups.4 It was found that anticancer bioactivity is more effective against the growth of NTUB1 cell when esters are formed on either the hydroxy or carboxylic acid of ursolic acid.4 In fact, the formation of isopropyl ester at C-28 or the succinyl at C-3-OH were amongst the most effective. However, this study utilized benzene as a solvent in the esterification method, which is classified as toxic, carcinogenic and mutagenic.5
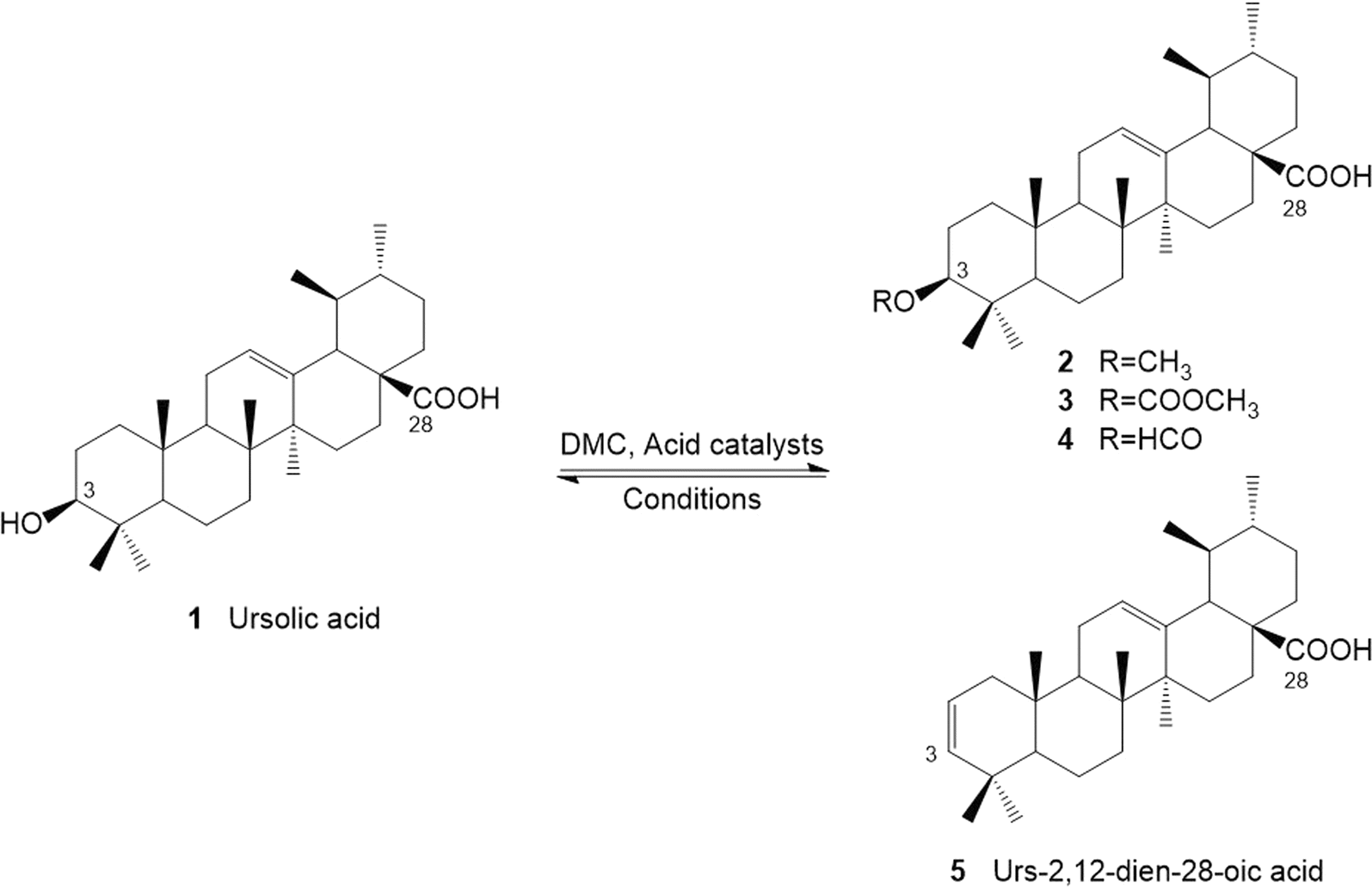 | ||
| Scheme 1 Reaction of ursolic acid and DMC under acidic conditions. | ||
The C-3 modification of ursolic acid has been widely achieved using an anhydride reacted at C-3-OH with DMAP.6 Moreover, acetylation has been used as protecting group at C-3 or use of halogen substitution post esterification at C-28 has enabled the introduction of imidazole functionality leading to enhanced bio-activity of ursolic acid derivatives.7 Application of DMAP is widely used in the synthesis of ursolic acid derivatives. In 2016, Sahni and co-worker reported isolating ursolic acid from an acetone extract of a hybrid eucalyptus.8 A total of 6 compounds were synthesised with esters and amide functionality at C-3 and C-28 positions to attenuate the neuro-protective potential of ursolic acid. Ursolic acid was treated with acetic anhydride, butyryl chloride, or propyl chloride at the C-3 position in the presence of DMAP and THF. 3-O-Acetylursolic acid, 3-O-propyrate ursolic acid and 3-O-butyrate ursolic acid were obtained in 85%, 61%, and 49% yield, respectively. From the results, it was found that the yield decreased when the carbon number of the substituent groups increased, most likely due to the steric hindrance of the alkyl group.8 DMAP is classified as toxic to the environment and human organs.9,10 In addition, solvents such as THF, dichloromethane, and benzene have been used in the synthesis of ursolic acid derivatives, once again several of these solvents used have been classified as toxic to the environment or hazardous.11 Other modification by Nascimento et al. demonstrated the synthesis of ursolic acid derivatives at C-3 via esterification with formic acid and 70% perchloric acid. The resulting ursolic acid derivative (3β-formloxyurs-12-en-28-oic acid) was shown to have anti-bacterial properties, demonstrating high activity against Escherichia coli. However, as shown by the low yield of the desired product (28%), their methodology did not improve the efficiency of the synthesis of ursolic acid derivatives.12
Methylation reactions at the C-3 hydroxyl have been investigated. Methylation using iodomethane in THF in the presence of NaH occurred in around 59% yield.13 The methylated derivative displayed inhibition of nitric oxide production activity on lipopolysaccharide-induced RAW247 cells. Additionally, it demonstrated lower toxicity than ursolic acid. However, iodomethane is categorised as a suspected carcinogen and exposure to it is extremely dangerous.14–16 Traditional reagents used for methylation reactions and some carboxymethyl include iodomethane, dimethyl sulfate, tetramethylammonium chloride, and diazomethane are toxic, hazardous, or unsustainable.14,17–19 The use of toxic reagents to modify the ursolic acid and produce derivatives negates any positive effect of producing bioactive molecules for use as pharmaceuticals. Efficient synthesis needs to be improved to enhance the number of useful ursolic acid derivatives. As such, substitution of such reagents and solvents, with more sustainable reaction chemistry should be a priority for investigation.
Carboxymethylation and methylation with dimethyl carbonate (DMC), is one such strategy for addressing the replacement of unfavourable methodologies. DMC has been utilized as a sustainable reagent and green solvent for methylation and carboxymethylation reactions, due to its low toxicity,20 and the ability to be produced from bio-based feedstocks, methanol and carbon dioxide.21 Typically carboxymethylation and methylation reactions of DMC with alcohols are carried out in the presence of a base.22 However, as ursolic acid has a carboxylic acid group on the C-28 carbon of the triterpenoid structure, this can react with typical bases used in the DMC process, thus leading to acid base neutralisation/deactivation and/or unwanted by-products. Recent research has demonstrated the potential for Brønsted and Lewis acid-catalysed carboxymethylation and methylation reactions of alcohols with DMC.23 Catalysts such as p-toluenesulfonic acid (PTSA), H2SO4, AlCl3 and FeCl3 promoted quantitative conversions and good selectivity of primary aliphatic alcohols toward for carboxymethylation products. For secondary alcohols it was found that the carboxymethylation products were obtained when using PTSA and AlCl3, and dehydration products obtained by FeCl3 and H2SO4. FeCl3 was also demonstrated to be effective for the methylation of cyclohexanol.23 To date the methylation reaction of the C-3 hydroxyl group on ursolic acid with DMC has yet to be investigated. Carboxymethylation of alcohols with DMC has also been achieved with solid acids (sulfonated mesoporous polymer (MP-SO3H)), with high conversion and selectivity of various alcohol such as ethanol, iso-propanol and 1-hexanol.24 Such developments offer an opportunity to create selective routes for the modification of ursolic acid at the C-3 hydroxyl with DMC.
Herein, for the first time ursolic acid was modified by the carboxymethylation and/or methylation reactions with the acid-catalysed dimethyl carbonate (DMC) chemistry. The effect of Brønsted, Lewis and heterogenous acids on the carboxymethylation and/or methylation reactions was also studied. Furthermore, the carboxymethylation reactions of ursolic acid were optimized under mild and sustainable conditions. It is important to note that an additional two terpene compound substrates were studied under optimal conditions to highlight the potential for the use of such modifications to be used on a wider class of compounds. These modification methods have the potential to create sustainable compounds with active pharmacological properties and various applications, opening new avenues in the field.
Experimental
Materials and reagents
Ursolic acid (95% purified), DMC (dimethyl carbonate), and PTSA (para-toluene sulfonic acid) were purchased from Sigma-Aldrich. HCOOH (formic acid) and AlCl3 (aluminium chloride) were purchased from Loba Chemie. H2SO4 (sulfuric acid) was purchased from RCI Labscan. FeCl3 (ferric(III)chloride) was purchased from Ajax Finechem. ZnCl2 (zinc(II)chloride) was purchased from Kemaus. Cyclohexanol was purchased from CARLO ERBA Reagents. HCl (hydrochloric acid) was purchased from QReC New Zealand. Zeolite was purchased from Alfa Aesar. SiO2–OSO3H (silica sulfate) and SiO2–HClO4 (silica perchlorate) was self-synthesized follow previously reported.25,26 Silica gel 60 (230–400 mesh size) was purchased from Merck.Acid screening of cyclohexanol modification by DMC
1 equivalent (11.70 μL) of cyclohexanol and 5 equivalents of acid were added to 1.5 mL of dimethyl carbonate in a 25 mL Teflon lined stainless steel autoclave. The reaction was heated to 150 °C for 6 hours. The solution was diluted, and products were characterized by using 1H-NMR and GC-MS.Ursolic acid modification by DMC
50 mg (1 eq.) of ursolic acid and 5 equivalents of the acid were added to 1.5 mL of dimethyl carbonate (∼160 eq.) in a one-necked 10 mL round-bottom glass flask containing a magnetic stirrer bar and molecular sieve. The reaction was run at 90 °C for 24 hours. On the high temperature (150 °C) heating method, the reaction was run in 25 mL Teflon auto-cleave stainless steel at 150 °C for 6 hours. The resulting solution was extracted with 50% water/ethyl acetate to remove the acid. Na2SO4 was added to the organic layer to remove water, then Na2SO4 was removed by filtration and the solvent was removed by evaporation in vacuo to obtain the mix of products. The ursolic acid ester products were isolated by column chromatography with the gradient 1% to 60% ratio of ethyl acetate/hexane as a mobile phase ratio. The products were characterized by using 1H-NMR, 13C-NMR and MS.In silico ADMET properties evaluation
The compounds underwent screening for drug-likeness according to Lipinski's rule using SwissADME.27 Subsequently, compounds that met the criteria for drug-likeness were assessed for their Absorption, Distribution, Metabolism, Excretion, and Toxicity (ADMET) properties. Pharmacokinetic properties were evaluated using the pKCSM online tools (https://biosig.unimelb.edu.au/pkcsm/prediction).28In silico molecular docking
Before conducting docking calculations on the selected compound, preliminary validation was carried out to ensure that ligands bound correctly within the protein binding site pocket. The 3D cocrystal structure of the EGFR kinase domain complexed with AFN941 (PDB: 6ITW)29 was aligned with a ligand-free protein structure (PDB: 2GS2) to generate the apo form of EGFR kinase with AFN941.30 Subsequently, AFN941 was redocked into the active site of EGFR kinase domain, with coordinates X = 49.759486 Å, Y = 1.645234 Å, and Z = −21.709539 Å. Similarly, chimaeric Bcl2-xL (PDB: 2W3L) (X = 39.805667 Å, Y = 26.935452 Å, and Z = −12.414476 Å), kappaB kinase beta (PDB: 3RZF) (X = 90.978742 Å, Y = −23.192645 Å, and Z = 54.212806 Å), and murine COX-2 S530T mutant (PDB: 5FDQ) (X = 33.671488 Å, Y = 28.029791 Å, and Z = 71.919628 Å) were redocked into their respective active sites, with specific coordinates provided. Docking and screening of compounds against these targets were performed using Autodock Vina,31 with a grid spacing of 1 Å and a box size of 20 × 20 × 20 Å, while default values were applied for other parameters.The three-dimensional (3D) structures of the apo form of the EGFR kinase domain (PDB: 2GS2),29 chimaeric Bcl2-xL (PDB: 2W3L),32 kappaB kinase beta (I4122) (PDB: 3RZF),32 and murine COX-2 S530T mutant (PDB: 5FDQ)32 were obtained from the Protein Data Bank (https://www.rcsb.org). Hydrogen atoms and Kollmann united atom partial atomic charges were added using ADT Tools.33 Ligand 3D structures were constructed based on molecular docking studies, and their protonation states were assigned using MarvinSketch software at pH 7.4. Both protein and ligand structures were converted into “PDBQT” format using ADT Tools. Docking calculations for the compounds were carried out using the same protocols as the validation process. The docked conformation with the lowest binding energy for each ligand was then selected, and their protein–ligand interactions were analysed using BIOVIA Discovery Studio 2020.34
Results and discussion
Acid screening for DMC reactions with cyclohexanol
In this work, carboxymethylation and/or methylation reactions occurred on C-3-OH of ursolic acid. Therefore, cyclohexanol was selected as a model compound for reagent screening of various acid-reagents for the carboxymethylation and/or methylation reactions, due to the ease of analysis by GC. The most effective acid catalysts demonstrating high conversions for each category of acid (Lewis acids, Brønsted acid and heterogeneous solid acids), were selected for investigation of the carboxymethylation and/or methylation reactions with ursolic acid (Table 1).| Reagent | Conversion (%) | GC-MS selectivity (%) | ||
|---|---|---|---|---|
| Methylation product | Carboxymethylation product | Other | ||
| a Conditions: cyclohexanol (1 eq.), acid (5 eq.), DMC (160 eq.), 150 °C for 6 h. | ||||
| Brønsted acid system | ||||
| Formic acid | 69 | 0.0 | 10.1 | 89.9 |
| PTSA | 95.3 | 11.9 | 85.2 | 2.9 |
| H2SO4 | 99.7 | 2.8 | 1.0 | 96.2 |
| HCl | 53.1 | 0.0 | 78.3 | 21.7 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
||||
| Lewis acid system | ||||
| ZnCl2 | 87.9 | 0.0 | 87.2 | 12.8 |
| FeCl3 | 100 | 58.3 | 1.1 | 40.6 |
| AlCl3·6H2O | 55.3 | 0.0 | 36.1 | 63.9 |
|
||||
| Heterogenous acid system | ||||
| H2SO4–SiO2 | 93.0 | 5.3 | 85.9 | 8.8 |
| HClO4–SiO2 | 45.1 | 0.00 | 89.3 | 10.7 |
| Zeolite β (25:1 SiO2:Al2O3) |
38.3 | 0.00 | 51.6 | 48.4 |
In Brønsted acid, PTSA and H2SO4 show high conversions of 95% and >99% respectively, while lower conversions were observed using formic acid and HCl, 69% and 53%, respectively. PTSA and HCl presented high selectivity towards the carboxymethylation product, however, use of formic acid resulted in high selectivity to the formylation product, while H2SO4 promoted dehydration to the respective alkene (full results are presented in the ESI, Tables S2 and S3†). This result is consistent with previous results where H2SO4 promotes the dehydration of secondary alcohols in DMC at 90 °C, while the use of PTSA with secondary alcohols and DMC promotes carboxymethylation.23 Formic acid has been demonstrated to act as both reagent and reactant in the high selectivity formylation of cyclohexanol with DMC as the solvent. Such results are comparable to other methods for formylation of alcohol with formic acid under solvent-free conditions catalysed by free I2 or I2 from Fe(NO3)3·9H2O/NaI.35 Where, formylation of cyclohexanol demonstrated a 90% isolated yield within 1 hour at room temperature, which is somewhat higher than 69% conversion of cyclohexanol with formic acid in the presence of DMC reported in this current work.
High conversion was obtained using H2SO4–SiO2, while lower conversions were observed with HClO4–SiO2 and zeolite β. However, both acid supported on silicas demonstrated high selectivity towards the carboxymethylation product. The possible mechanism was consistent with a Brønsted acid process. Similar results were reported by Kempanna et al., which show high conversion and selectivity of alcohol to carboxymethylation product using a sulfonated mesoporous polymer.24
While the Lewis acids, ZnCl2 and FeCl3 promoted high conversion, only moderate conversions were observed when using AlCl3. Interestingly, high selectivity towards methylation was observed with FeCl3. These results are consistent with previously reported methods for the methylation of cyclohexanol using stoichiometric FeCl3 with DMC at 90 °C for 24 hours.23
Based on the screening results, PTSA, ZnCl2 and H2SO4–SiO2 demonstrated the greatest promise for promoting the carboxymethylation reaction. Moreover, FeCl3 also demonstrated promise for promoting the methylation reaction, while formylation occurred by using formic acid. Thus, these acids were selected for further investigation with ursolic acid and DMC for carboxymethylation, methylation and/or formylation reactions.
Carboxymethylation of ursolic acid by Brønsted acids
PTSA was selected as a Brønsted acid for the carboxymethylation and/or methylation reaction of ursolic acid with DMC (Table 2). PTSA loading was investigated using stoichiometric 1, 5 and 10 equivalents under different heating regimes (90 °C and 150 °C). 5 equivalents of PTSA showed highest conversion of around 80%, significantly lower than the conversion of cyclohexanol due to the sterically hindered ursolic acid secondary alcohol. Lower conversions of ursolic acid were observed with stoichiometric and 10 equivalents of PTSA. This indicated 5 equivalents of PTSA was the optimum amount of acid to use with ursolic acid due to high complexation of acid and DMC. This is consistent with the mechanism previously reported by Jin et al.23 and it is shown in ESI (Fig. S8†). A possible esterification reaction on C-28 carboxylic acid group (Scheme 1) and dehydration of C-3 hydroxyl group enhances % conversion but decreases selectivity towards the desired C-3 carboxymethylation product.| Reagent | Temperature (°C) | Conversion (%) | Isolated yield of 3 (%) | Selectivity (%) | |||
|---|---|---|---|---|---|---|---|
| 2 | 3 | 5 | Other | ||||
| PTSA (1 eq.) | 90 | 38.6 | 11.0 | 0 | 29.7 | 29.2 | 41.1 |
| 150 | 51.2 | 36.1 | 0 | 69.7 | 23.4 | 6.9 | |
| PTSA (5 eq.) | 90 | 74 | 45.5 | 0 | 59.8 | 33.3 | 6.9 |
| 150 | 79.8 | 32.8 | 0 | 40.9 | 55.1 | 4.0 | |
| PTSA (10 eq.) | 90 | 25.4 | 2.1 | 0 | 8.2 | 56.3 | 35.5 |
| 150 | 51.6 | 15.3 | 0 | 29.3 | 58.0 | 12.7 | |
The alkene product resulting from dehydration of the alcohol at the C-3 ursolic acid was obtained as a major product in 5 eq. and 10 eq. at 150 °C. The similar results have been observed in the cyclohexanol work reported by Kanakikodi et al.24 The proposed mechanism of ursolic acid dehydration is presented in ESI (Fig. S9†).
The high temperature (150 °C) heating conditions resulted in higher conversions than refluxing at 90 °C, due to the harsher conditions of the reaction. However, higher selectivity is generally observed at 90 °C, due to reduced side reactions under the more stable conditions. In fact, it has been reported that the quantitative decarboxymethylation of the carboxymethylation products can take place under higher temperature conditions.36 On other hand, high amount of PTSA led to a by-product in the form of a tosylate generated at the C-3 of ursolic acid, leading to reduced selectivity at under reflux heating conditions. This was most evident with 10 equivalents of PTSA at 90 °C, indicating that the side reaction can take place at low temperatures and ambient pressure. Similar results were found in the high temperature (150 °C) system but with lower conversions and selectivity to the carboxymethylation product. Therefore, 5.0 equivalents PTSA with reflux heating conditions (90 °C) were selected as appropriate conditions to use in the carboxymethylation of ursolic acid with DMC. Importantly, this is the first time the synthesis of 3β-[[methoxy]carbonyl]oxyurs-12-en-28-oic acid has been reported in the literature. Ursolic acid and other triterpenes can now be carboxymethylated using DMC chemistry, such structures warrant further investigation, this will be fully investigated in a follow on paper, including assessing bioactivity esterification reactions of ursolic acid at C-3 reported in the literature widely use DMAP, this has been classified as a toxic substance.37 Therefore, this work demonstrated a promising and potentially greener method for C-3 ursolic acid modification with DMC. Additionally, other dialkyl carbonates could be used in place of DMC to produce a library of novel compounds.
Other Brønsted acids investigated include the application of formic acid (Table 3), this was used to study the carboxymethylation and/or methylation of ursolic acid with DMC. However, 3β-formylurs-12-en-28-oic acid was the major product produced via esterification. It was found that DMC only participated as a solvent in the reaction and was not protonated by the formic acid. In fact, the nucleophile of ursolic acid reacted to carbonyl group of formic acid to yield the 3β-formylurs-12-en-28-oic acid. To confirm this hypothesis the reaction between only ursolic acid and excess formic acid was studied, the results demonstrated 100% conversion of ursolic acid and 100% selectivity to 3β-formylurs-12-en-28-oic acid without DMC. Similar outcomes were seen in the ursolic acid study published by Tkachev et al.38 3β-formylurs-12-en-28-oic acid was previously synthesised from using ursolic acid with formic acid by stirring at 60 °C for 6 hours. The product was taken up in benzene and run through a silica gel column to give formyl ursolic acid at 90% yield. However, benzene is classified as a toxic reagent and as such the work presented in this current study demonstrates great promise as a sustainable greener method for modification of ursolic acid, in quantitative yield and requiring no complex work up.
| Reagent/reactant | Temperature (°C) | Conversion (%) | Isolated yield of 4 (%) | Selectivity (%) | |
|---|---|---|---|---|---|
| 4 | Other | ||||
| Formic acid (5 eq.) | 90 | 82.2 | 69.1 | 85.5 | 14.5 |
| 150 | 58.4 | 78.4 | 99.3 | 0.7 | |
| Formic acid (5 eq.) (without DMC) | 90 | 100 | 100 | 100 | 0 |
Previous studies have demonstrated a 28% yield 3β-formylurs-12-en-28-oic acid by using HCO2H, perchloric acid at 60 °C for 4 hours.12 In the current study, a solvent free reaction carried out at 90 °C for 24 hours using ursolic acid (500 mg, 1.1 mmol) and HCO2H (5 eq.) gave 100% conversion and selectivity without the need for a catalyst or reagent. However, when DMC was used as the solvent under both reflux and high temperature (150 °C) heating conditions, the conversions were observed to be lower. High temperatures (150 °C) resulted in selectivity of 99% towards the 3β-formylurs-12-en-28-oic acid, but moderate conversion was observed because of decomposition of formic acid. Here water produced during esterification caused formic acid to hydrolyse, producing carbon dioxide and additional water. In contrast, this did not occur under reflux conditions, which corresponded to both high conversions and good selectivity.
Carboxymethylation and/or methylation of ursolic acid by Lewis acids
With ZnCl2, the results show a good conversion of ursolic acid and selectivity towards the carboxymethylation product. When using 5.0 or 10.0 equivalents of ZnCl2 under 90 °C heating conditions, it was found that no reaction took place. For the nucleophilic attack of the hydroxy group of ursolic acid on DMC, which was crucial in terms of selectivity toward the carboxymethylation product or methylation products, it is essential to form an intermediate between ZnCl2 and DMC shown in Scheme 2.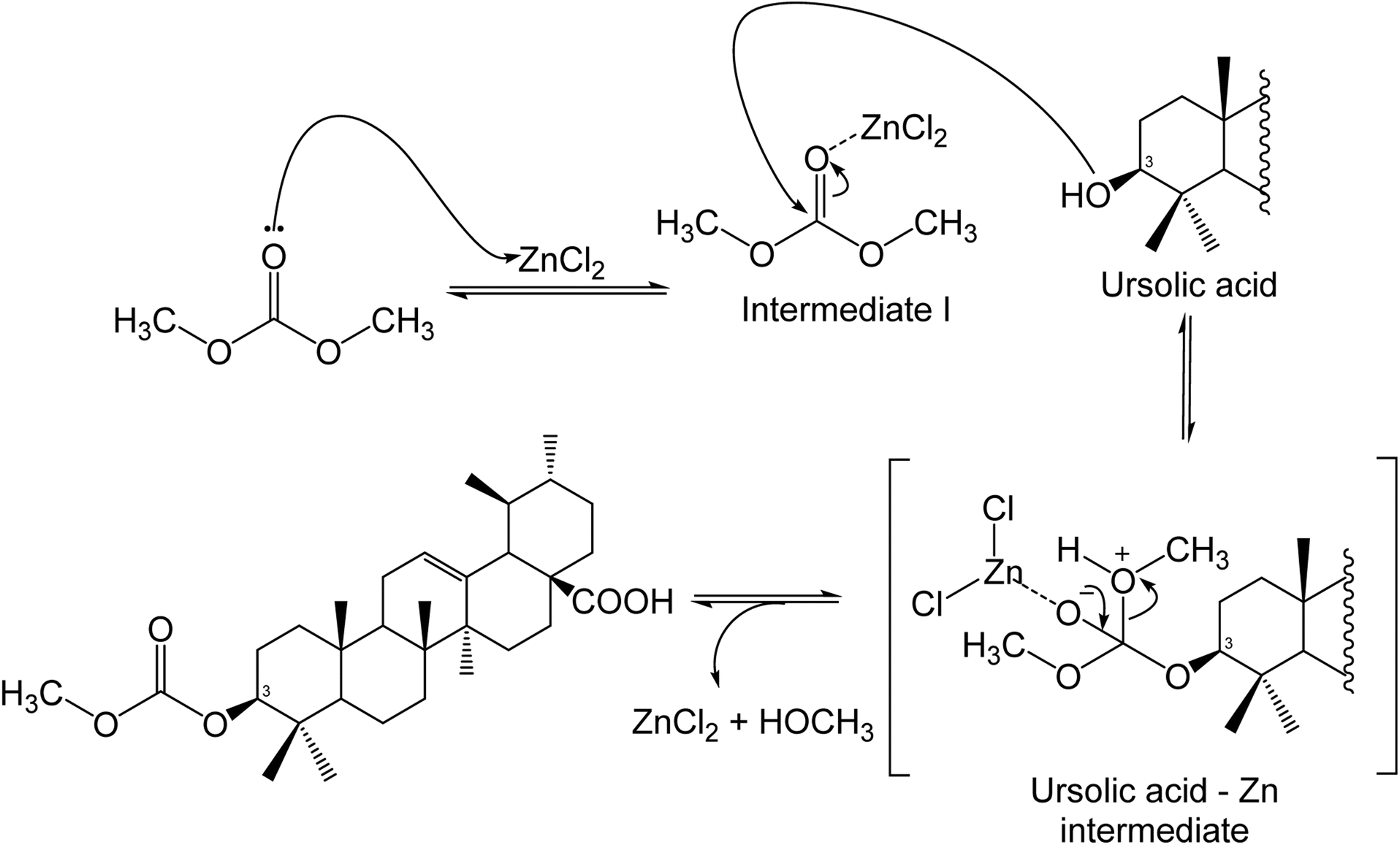 | ||
| Scheme 2 Possible mechanism of carboxymethylation of ursolic acid with DMC by ZnCl2. | ||
50% conversion of ursolic acid was achieved under high temperature conditions (150 °C), with a 21.9% selectivity for carboxymethylation (Table 4). When the amount of ZnCl2 was increased to 10 equivalents, conversion increased to 80.8% with 32.6% selectivity (Table 4). As shown in Scheme 2, a high loading of ZnCl2 resulted in an intermediate that coupled with DMC and ursolic acid.
| Reagent | Temperature (°C) | Conversion (%) | Isolated yield of 3 (%) | Selectivity (%) | ||
|---|---|---|---|---|---|---|
| 3 | 5 | Other | ||||
| ZnCl2 (5 eq.) | 90 | 0 | 0 | 0 | 0 | 0 |
| 150 | 50.0 | 11.9 | 23.6 | 70.1 | 6.3 | |
| ZnCl2 (10 eq.) | 90 | 0 | 0 | 0 | 0 | 0 |
| 150 | 80.8 | 25.2 | 35.1 | 45.3 | 19.6 | |
With FeCl3, results show >99% conversion of ursolic acid but with low selectivity to the carboxymethylation product (Table 5). In fact, this simple method demonstrated that the FeCl3 was highly selectivity toward the methylation product, 3β-methoxyurs-12-en-28-oic acid (99% selective at 150 °C for 6 hours). This was a highly important and promising result for the modification of ursolic acid. Kwon et al.,13 demonstrated a 59% yield of 3β-methoxyurs-12-en-28-oic acid by using CH3I, NaH and THF. As discussed CH3I as a methylating agent comes with issues and it has recently been classified as a toxic reagent,39 while solvents such as THF are also problematic due to their potential for peroxide formation.40 Thus, this work had been shown to be an excellent green method for the methylation of ursolic acid with the highest yield.
| Reagent | Conversion (%) | Isolated yield of 2 (%) | Selectivity (%) | |
|---|---|---|---|---|
| 2 | 3 | |||
| FeCl3 (0.5 eq.) | 52.6 | 46.6 | 87.7 | 12.3 |
| FeCl3 (1 eq.) | >99 | >99 | >99 | <1 |
| FeCl3 (5 eq.) | >99 | >99 | >99 | <1 |
| FeCl3 (10 eq.) | >99 | >99 | >99 | <1 |
The mechanism of methylation reaction is presented in Scheme 3. The interaction between FeCl3 and DMC leads to coordinated of the carbonyl group and –OCH3 group. Due to the higher reduction potentials of FeCl3 (the reduction potentials of Fe3+ and Zn2+ are −0.037 and −0.761, respectively),41 methylation is the desired reaction pathway with this Lewis acid. Here the 5-ligand coordinated iron intermediate42 makes the methyl group more positive, which leads to a highly efficient methylation reaction. This is consistent with previous reports that used Lewis acids with DMC to form stabilised intermediates that promote methylation pathways.23,43
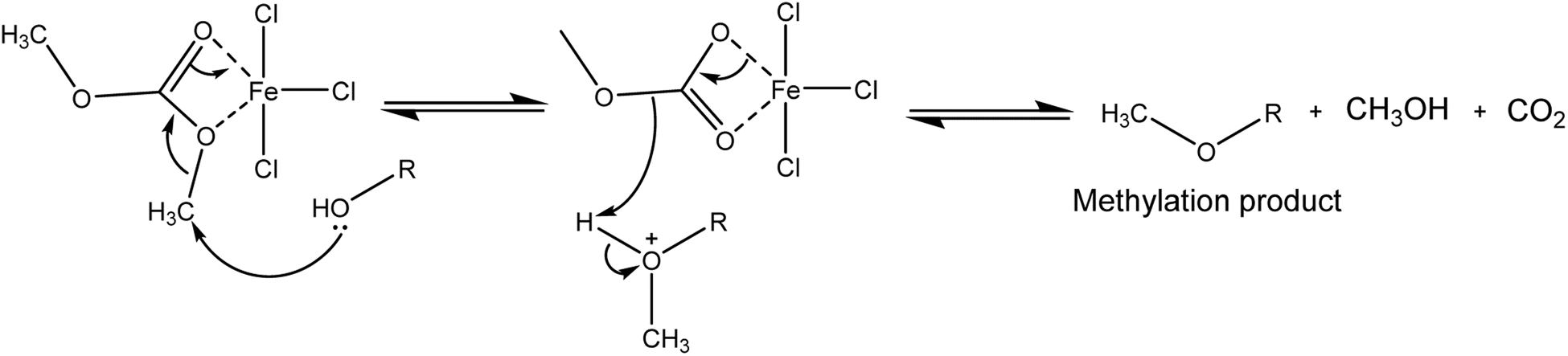 | ||
| Scheme 3 Possible mechanism of methylation of ursolic acid with DMC by FeCl3. | ||
Carboxymethylation of ursolic acid by heterogeneous solid acids
The reaction of ursolic acid in the presence of H2SO4–SiO2 showed high conversion but poor selectivity toward carboxymethylation product (Table 6). This is due to H2SO4 bound to the silica surface leading to dehydration reaction, consistent with the reaction between cyclohexanol and free H2SO4. Dehydration to the alkene was the major product potentially due to the carboxylic acid of ursolic acid interacting with the polar surface of H2SO4–SiO2 thus bringing the C-3 alcohol into greater and longer contact with the sulphuric acid group leading to dehydration.44 5 eq. of H2SO4–SiO2 at 150 °C demonstrated higher yields and selectivity toward carboxymethylation reaction compared to other conditions, potentially due to the increased temperature and pressure leading to greater interaction of DMC with the heterogenous acid over ursolic acid. However, increasing the amount of acid at 150 °C, led to lower yields and selectivity toward carboxymethylation reaction, but high selectivity towards the dehydration product, consistent with previous work on the dehydration of alcohol with H2SO4.23 Moreover, lower selectivity occurred under high temperature conditions (150 °C), limited methylation product was observed at high temperature. Similar results have been reported in methylation studies performed by Selva and Tundo.45 This is consistent with the possible mechanism reported by Kempanna's work (Fig. S11†).24| Reagent | Temperature (°C) | Conversion (%) | Isolated yield of 3 (%) | Selectivity (%) | ||
|---|---|---|---|---|---|---|
| 3 | 5 | Other | ||||
| H2SO4–SiO2 (5 eq.) | 90 | 87.5 | 17.4 | 19.9 | 74.5 | 5.6 |
| 150 | 78.3 | 23.2 | 29.7 | 40.5 | 29.8 | |
| H2SO4–SiO2 (10 eq.) | 90 | 78.4 | 18.6 | 23.6 | 54.1 | 22.3 |
| 150 | 69.8 | 9.1 | 13.1 | 77.5 | 9.4 | |
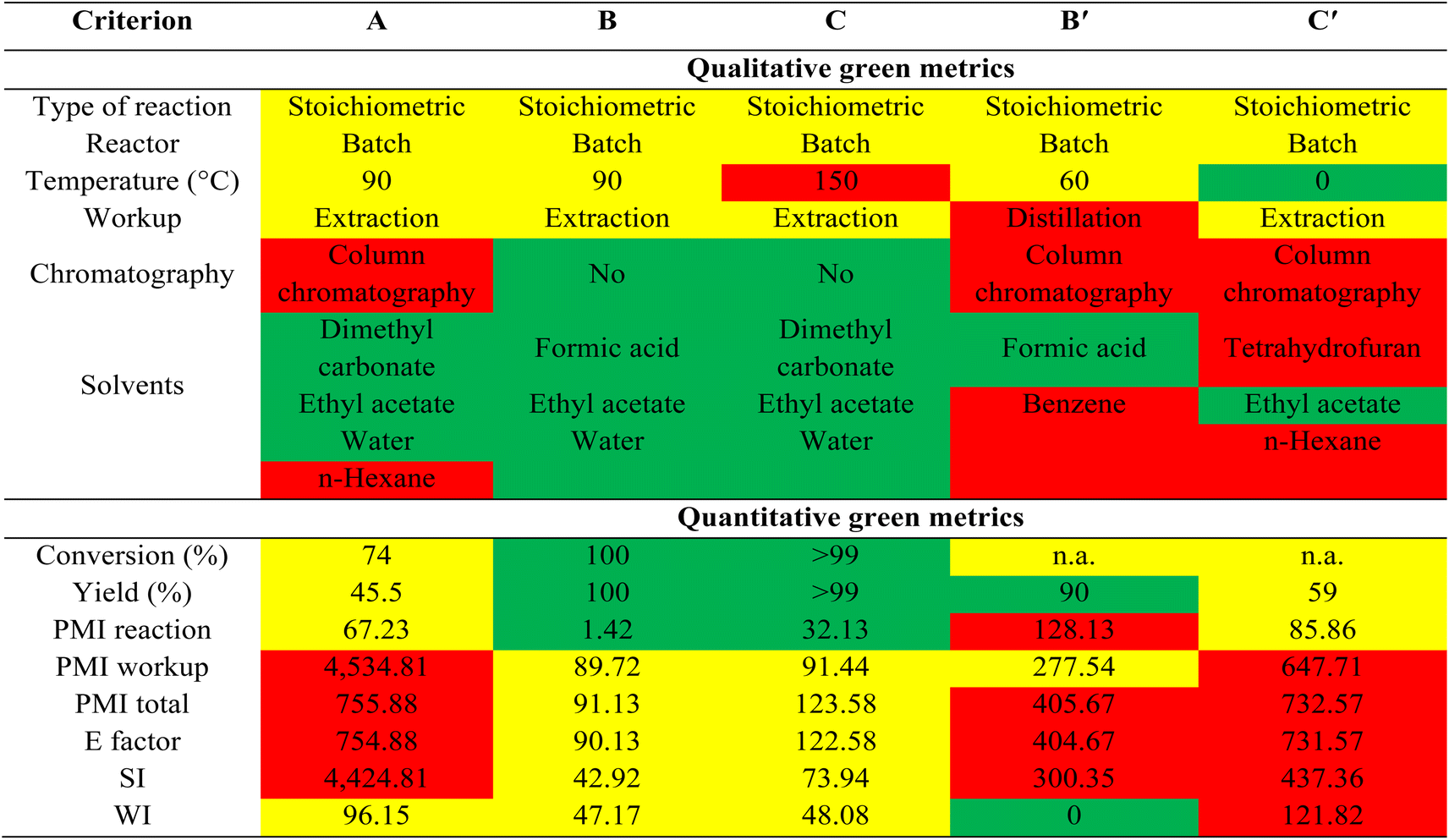
A qualitative and quantitative comparison of green metrics46–48 between our current reactions (B: HCO2H, formylation, and C: FeCl3, methylation) and literature methods (B′: HCO2H, formylation,30 and C′: NaH, MeI, methylation13) (Table 7 and ESI†) clearly highlights the advantages of our methods over those previously reported. The high temperature used in process B (methylation) is a minor limitation, as are the excess equivalents of acid used in all the processes. Attempts were made to reduce the amounts of acid required in carboxymethylation and methylation reactions using DMC, however this led to a drop in conversions and selectivity. Importantly, previous methods for methylation and formylation of ursolic acid required extensive workup and chromatographic processes, utilizing substantial amounts of sometimes hazardous solvents (benzene, tetrahydrofuran, or n-hexane), leading to high process mass intensity (PMI), E-factor, solvent intensity (SI), and water intensity (WI). In contrast, the more sustainable procedures outlined in this work (using FeCl3 and formic acid) achieve high conversion and yield, entail minimal solvent workup, eliminate the need for chromatography, and employ green solvents/reagents (DMC or formic acid). This results in significantly improved PMI, E-factor, SI, and WI compared to earlier studies. The carboxymethylation process of ursolic acid using PTSA in this study was not included in the comparison of green metric calculations due to its novelty and lack of comparable literature reports. Its green metrics are detailed in Table 7. The primary drawback of this method is the low yield of the product, necessitating chromatography for purification. The use of large volumes of solvents such as n-hexane in chromatography results in high values of PMI, E factor, and SI, highlighting a limitation of the method, in addition to hexane's toxicity. Therefore, future efforts should focus on enhancing product yield and eliminating the need for chromatographic separation. Nevertheless, the overall methods developed in this work would be preferable the modification of terpenes/terpenoids by organic chemists and the green credentials of such procedures may make their exploitation plausible in industrial applications.
To explore the broader applicability of the processes, the substrate scope was broadened to include two terpenes with hydroxyl groups, including corosolic acid and menthol, representing triterpene and terpene categories, respectively. These compounds can undergo modification through carboxymethylation and methylation reactions using DMC under optimal conditions as determined in the ursolic acid study. Corosolic acid and menthol were subjected to reactions with DMC under three different optimal acid conditions (PTSA, FeCl3, and formic acid) to establish a connection between the results obtained from ursolic acid and other terpene classes. The reactions of menthol with PTSA and formic acid showed high conversion rates, high yields, and selectivity towards carboxymethylation and formylation products, respectively (Table S4, ESI†). However, when FeCl3 was used, moderate yields and selectivity towards methylation products were observed, as shown in Table S4 (ESI†). These outcomes align with the results of the cyclohexanol and ursolic acid reactions, particularly under PTSA and formic acid conditions. Regarding corosolic acid, the results under PTSA and FeCl3 acid conditions demonstrated good conversion but low selectivity towards carboxymethylation and methylation, due to the production of multiple desired products, as the carboxymethylation and methylation reactions can occur at both the C-2 or C-3 positions, as detailed in the ESI.† As such, tuning the selectivity of this method in terpenes with multiple hydroxyl groups would be an exciting challenge. This study does mark the first carboxymethylation of corosolic acid, showcasing a green and sustainable modification approach for this compound. For methylation of corosolic acid, previous studies have reported methoxylation at C-2 through the hydrolysis of guajanoic acid under basic conditions. Formylation of corosolic acid was achieved using formic acid, similar to the ursolic acid study, resulting in high conversion and selectivity without the need for DMC. The study involving corosolic acid and menthol demonstrates the efficient and sustainable modification of other terpenes with hydroxyl groups through carboxymethylation, methylation, and formylation reactions.
In drug discovery, in silico approaches play a crucial role by offering cost-effective means to identify potential drug candidates and minimize the need for animal testing.49,50 To assess the drug-likeness of compounds like ursolic acid, corosolic acid, and their derivatives, SwissADME,27 a free online tool that applies Lipinski's rule of five was utilised (Table S9†).51,52
Lipinski's rule and ADMET prediction
As demonstrated in Table S9 (ESI†), while some ursolic acid and corosolic acid derivatives exceeded the conventional 500 Da limit for optimal oral bioavailability,52 they remained within the modern threshold of <700 Da.53 Overall, most properties aligned with the extended Lipinski's rule of five, with only minor deviations such as slightly elevated logP values (>7.5).54 Notably, the main concern at this stage revolves around the relatively high lipophilicity of these compounds, a characteristic inherited from their parent systems. When more than one rules outlined by Lipinski are violated, it indicates potential issues with absorption, permeability, and subsequently, bioavailability.55,56 Although the majority of compounds in our study adhere to Lipinski's rules, indicating a high likelihood of penetrating cell membranes and achieving favourable theoretical oral bioavailability, there are exceptions. Carboxymethylation derivatives of ursolic acid (compound 3) and corosolic acid (compounds 9–11, Fig. S12, ESI†) violated Lipinski's rules due to their high molecular weight and logP. However, it's important to note their low water solubility, permeability, and subsequently, bioavailability, which may limit their clinical application as oral drugs. Additionally, ursolic acid is classified as a class IV drug in the Biopharmaceutics Classification System (BCS), indicating low oral bioavailability and poor permeability.57
It is crucial to emphasize the significance of evaluating ADMET properties in the drug discovery process. This assessment helps identify compounds with unfavourable ADME characteristics early on, reducing the likelihood of compounds failing in clinical trials.58–60 Predicted ADMET properties of ursolic acid, corosolic acid, and their derivatives are presented in Tables S10–S12.†
The oral absorption of drugs relies on their ability to traverse the gastrointestinal tract (GIT) walls.61 Hence, we assessed the absorption profiles of all compounds based on Caco-2 permeability and human intestinal absorption. In our predictive model, a compound is deemed to have high Caco-2 permeability when Papp > 0.90, and good intestinal absorbance when the value exceeds 30%.28 Our results indicated that ursolic acid (1), corosolic acid (6, Fig. S12†), their dehydration products (compounds 5, 7 and 8), and dimethylated corosolic acid (compound 14, Fig. S12†) exhibited high Caco-2 permeability, with all compounds showing substantial predicted intestinal absorption (>80%) (Table S10†). Caco-2 cells, derived from human epithelial colorectal adenocarcinoma, serve as an effective model for mimicking gastrointestinal epithelium, making them a validated system for oral absorption studies.62–64 Human intestinal absorption, reflecting a compound's bioavailability and absorption, is assessed from cumulative excretion in bile, urine, and feces.65 Thus, our data suggest that these compounds may effectively traverse the GIT membrane, supporting Lipinski's RO5 analysis. Additionally, skin permeability was predicted, with all compounds meeting the criterion of skin permeability (logKp < −2.5),28 suggesting topical administration as a viable alternative for absorption. However, based on the predicted values (logKp about −2.7), it can be concluded that they exhibited only slightly low to moderate skin permeability. While these predictions align with those found in existing literature,66–69 it's important to emphasize that these triterpenes still encounter absorption and permeation challenges. Drug absorption can be affected by efflux proteins in cell membranes, such as Pgp, an ATP-binding cassette transporter that pumps drugs out of intestinal cells.70–73 Our predictions revealed that all compounds, except for corosolic acid, are non-substrates for Pgp I and non-inhibitors of Pgp I. While they do not interact with Pgp I to block drug efflux, some derivatives such as the carboxymethylation derivatives of ursolic acid (compound 3) and corosolic acid (compounds 8–11, Fig. S12, ESI†), the methylation derivative of corosolic acid (compound 12, Fig. S12†), and the formylation derivatives of corosolic acid (compounds 15 and 16, Fig. S12†) may inhibit and block Pgp II, potentially inhibiting multidrug resistance (Table S10†). While triterpenes like ursolic acid and its derivatives encounter challenges with oral absorption, Ren et al. explored pulmonary administration as a promising alternative. Their investigation revealed that the bioavailability of ursolic acid significantly increased following pulmonary administration compared to intragastric administration.74
The distribution of all compounds was evaluated based on their blood–brain barrier (BBB) permeability and central nervous system (CNS) penetration (Table S11†). The BBB serves as a crucial physiological barrier composed of endothelial cells that regulate the passage of compounds from the blood to the CNS, providing protective properties.61 According to the pkCSM model, compounds with a logBB > 0.3 can readily cross the BBB, while those with logBB < −1 face difficulty in reaching the brain.28 Most compounds displayed intermediate BBB permeability values (−1 < logBB < 0.3), but some showed high permeability (e.g., formylation derivative of corosolic acid, compound 15), indicating potential distribution into the brain, with compound 15 being particularly adept at crossing the BBB. The liposolubility of a compound may aid in traversing this barrier and potentially exerting effects at the CNS level. Additionally, CNS permeability, measured as the permeability-surface area product (logPS), can be determined through in situ brain perfusions with direct compound injection into the carotid artery. A logPS > −2 indicates CNS penetration, while a logPS < −3 suggests inability to penetrate the CNS. As indicated in Table S11,† all compounds are capable of CNS penetration except for methylation and dehydration derivatives of ursolic acid (compounds 2 and 5) showed moderate to high results.
The evaluation of liver first-pass metabolism characteristics depends on the interaction with various microsomal enzymes, notably cytochrome P450 (CYP450).61 These enzymes, primarily located in the liver, are responsible for much of the initial drug metabolism, with CYP3A4 playing a pivotal role in approximately 50% of xenobiotic metabolism in humans.75,76 ADMET predictions revealed that all compounds are non-substrates for the CYP2D6 isoform and non-inhibitors of CYP2C9, CYP2C19, CYP2D6, and CYP3A4 isoforms. However, they are substrates for the CYP3A4 isoform (Table S11†). These findings suggest that the compounds may not undergo metabolism by the selected CYP isoforms, remaining chemically inert as they are unable to activate the enzymes as substrates except for CYP3A4 isoform. Additionally, they may not interfere with the function of CYP isoforms or disrupt the metabolism of other drugs, as they were not identified as inhibitors. Reports indicate that ursolic acid is primarily metabolized by CYP3A in rat liver microsomes. In human liver microsomes and recombinant human CYP450 enzymes, its metabolism is mainly mediated by CYP3A4 and CYP2C9.74 Therefore, it is important to note that biotransformation by metabolic enzymes in the liver remains one of the primary reasons for the low oral bioavailability of drugs. However, based on the in vivo findings of Ren et al.,74 it was observed that the metabolism of ursolic acid in lung microsomes was significantly lower compared to that in liver microsomes. This reduced metabolism suggests that pulmonary administration could be a promising approach for the application of ursolic acid.
Excretion, an essential pharmacokinetic parameter, elucidates the process of eliminating intact drug molecules or their metabolites from the body, thus influencing the duration of drug presence and volume of distribution.59,75,77 This process was assessed through total clearance and categorizing compounds as renal organic cation transporter 2 (OCT2) substrates (Table S12†). Total clearance lacks a defined range; higher values indicate faster excretion.28 Moreover, all compounds are not OCT2 substrates. OCT2, a renal uptake transporter, governs the renal clearance of drugs.78 The compound's ability to bind to this protein is indicative of its clearance rate, crucial for determining the dosing regimen to achieve plasma steady state.79 According to the findings, all compounds do not interact or bind with this transporter, suggesting they may not be excreted via this pathway.
Drug toxicity is a significant concern in drug discovery and development.80 To assess potential toxicity, compounds were evaluated using pkCSM (Table S12†). Genetic toxicity, indicating mutagenicity, was evaluated through the AMES mutagenic test. A positive AMES test result suggests mutagenicity.28,81 All compounds showed negative results, suggesting they are likely non-mutagenic and non-carcinogenic. Cardiotoxicity was assessed by examining whether the compounds acted as hERG I and II inhibitors. hERG channels are crucial in cardiac repolarization,82–84 and their inhibition can cause QT interval prolongation and severe arrhythmias, leading to drug failure.28,85 None of the compounds inhibited hERG I or II, indicating their cardioprotective nature. Hepatotoxicity is another critical concern, often leading to medication withdrawal post-market.86 In silico analysis indicated that most compounds did not disrupt normal liver function, except for some, including ursolic acid (Table S12†). Additionally, none of the compounds exhibited skin sensitization, a potential adverse effect for dermally applied drugs.28 The toxic prediction profile indicates that these compounds are likely safe for oral administration, but these findings need validation through in vivo studies. While computational analysis offers a quick and cost-effective means of assessing ADMET properties, further experimental testing is necessary to verify these predictions.
Biological activity review and molecular docking of ursolic acid, corosolic acid and their derivatives
This brief review provides an overview of in vitro and in vivo studies investigating the bioactivity of ursolic acid, corosolic acid, and their derivatives, focusing on their anti-inflammatory, anti-tumour, and anti-cancer effects. Chronic inflammation, linked to various diseases, is controlled by molecular mediators like proinflammatory cytokines and enzymes. Ursolic acid and oleanolic acid, derived from Plantago major, demonstrated significant inhibition of COX-2, with ursolic acid showing superior activity.87 Synthesized derivatives of ursolic acid consisting of oxadiazole, triazolone, and piperazine moieties displayed enhanced anti-inflammatory properties.88 In addition, one derivative, 3β-methoxyurs-12-en-28-oic acid, exhibiting three times more potent inhibition of NO production than ursolic acid alone.13 Corosolic acid was found to inhibit NLRP3 inflammasome activation, suggesting its potential as an anti-inflammatory agent.89–92 In cancer treatment, both ursolic acid,93–99 and corosolic acid,93,100–108 have shown promise in inhibiting cancer cell proliferation and inducing apoptosis. Ursolic acid and corosolic acid have demonstrated anticancer effects across various cancer types through multiple mechanisms, highlighting their potential as therapeutic agents.Ursolic and corosolic acid have demonstrated remarkable anti-inflammatory, anti-tumour, and anti-cancer activities alongside low cytotoxicity. To understand these properties better, molecular docking studies were conducted with specific proteins associated with these activities. Ursolic acid, corosolic acid, and their derivatives were docked with COX-2 enzyme (PDB: 5FDQ) to rationalize the observed anti-inflammatory activity, and with EGFR kinase domain (PDB: 2GS2), chimaeric Bcl2-xL (breast cancer MCF-7, PDB: 2W3L), and kappaB kinase beta (PDB: 3RZF) to rationalize the observed anti-cancer and anti-tumour activity. These selected targets were based on various literature.88,109–111 Using Autodock Vina, all compounds were docked into the crystal structure of these proteins, validating the protocol by redocking the reference ligand. The results, detailed in Tables S13–S16,† revealed comparable binding energies between the derivatives and their parent compounds.
Notably, the formylated ursolic acid derivative (3β-formylurs-12-en-28-oic acid, compound 4) exhibited the lowest binding affinity (−7.3 kcal mol−1), slightly better than ursolic acid (−7.3 kcal mol−1), with the 5FDQ receptor pocket. Conversely, compound 6 (corosolic acid) demonstrated better binding affinity (−7 kcal mol−1) than its derivatives (−6.4 to −6.9 kcal mol−1). Within the 2W3L receptor pocket, corosolic acid displayed the lowest binding affinity at −7.3 kcal mol−1, slightly better than its derivatives (−6.5 to −7.2 kcal mol−1). All ursolic acid derivatives, except for the carboxymethylation product (3β-[[methoxy]carbonyl]oxyurs-12-en-28-oic acid, compound 3), showed better results than their parent compound. Compound 10 interacted with the 2GS2 and 3RZF receptor pockets with binding affinities of −8 and −7.6 kcal mol−1, respectively, the lowest among the tested compounds. The dehydration product of ursolic acid (compound 5) showed the same binding affinity as ursolic acid at −7.6 kcal mol−1, while others exhibited slightly lower affinities (−7 to −7.4 kcal mol−1) with the 2GS2 receptor pockets. Other corosolic acid derivatives displayed slightly lower binding affinities than corosolic acid (−7.9 kcal mol−1), ranging from −6.5 to −7.8 kcal mol−1 with the 2GS2 receptor pocket. The dehydration product of ursolic acid (compound 5) showed slightly better binding affinity than ursolic acid at −7.1 kcal mol−1 compared to −6.9 kcal mol−1, while others exhibited slightly lower affinities (−6 to −6.8 kcal mol−1) with the 3RZF receptor pocket. For corosolic acid, the dehydration product (compound 7) showed the same binding affinity as ursolic acid at −7.1 kcal mol−1, while others exhibited slightly lower affinities (−6.3 to −6.9 kcal mol−1) with the 2GS2 receptor pocket. These docking results, depicted in Fig. 1, highlighted the significance of hydrophobic interactions and hydrogen bonds in the COX-2 enzyme (PDB: 5FDQ) relating to anti-inflammatory activity, EGFR kinase domain (PDB: 2GS2), chimaeric Bcl2-xL (breast cancer MCF-7, PDB: 2W3L), and kappaB kinase beta (PDB: 3RZF) linked to anti-cancer and anti-tumour activities of compounds with the lowest binding affinity against each target.
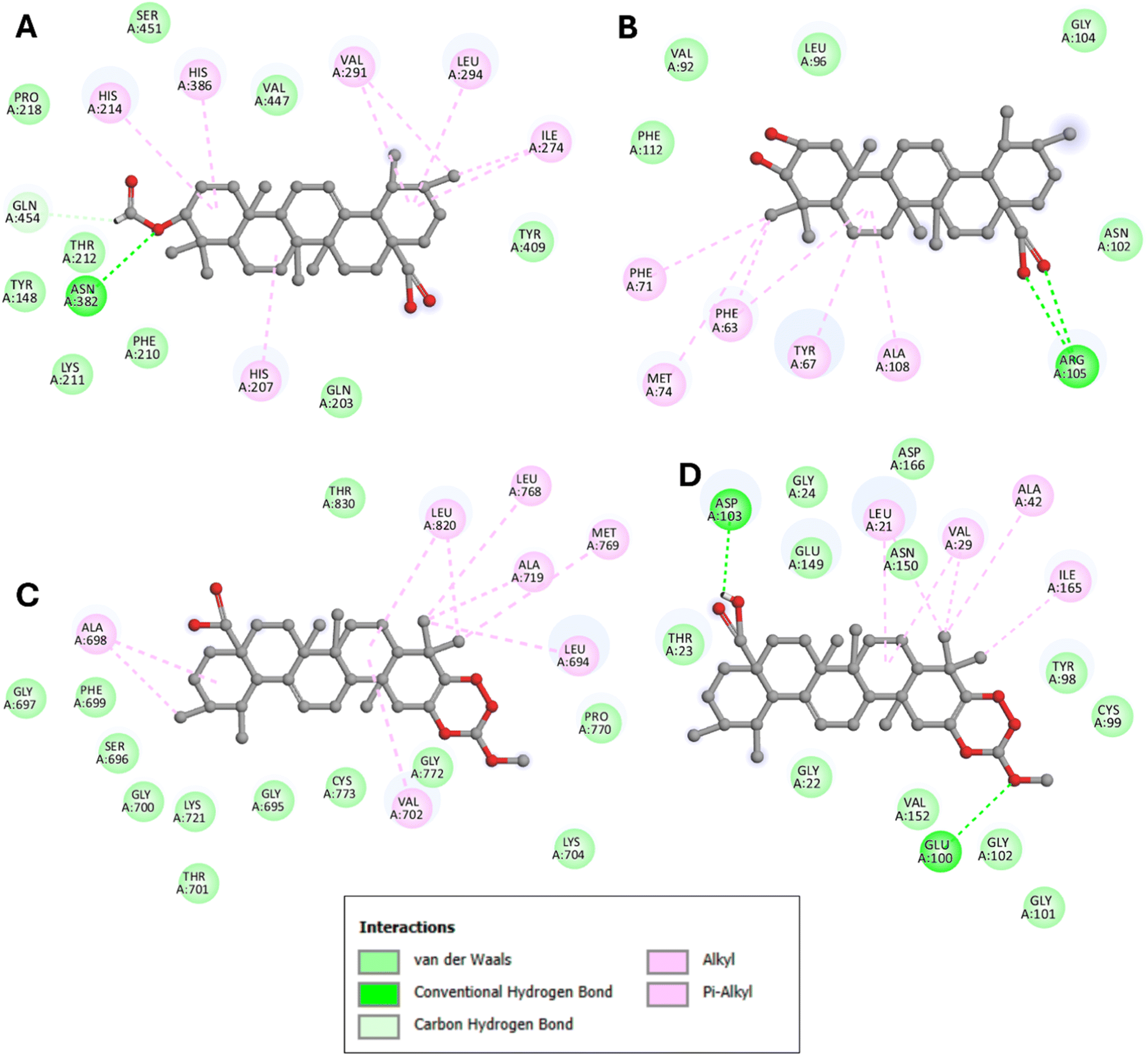 | ||
| Fig. 1 2D view of the binding conformations and hydrogen bond interactions of: (A) compound 4 (3β-formylurs-12-en-28-oic acid) at the active site of 5FDQ, (B) compound 6 (corosolic acid) at the active site of 2W3L, and compound 10 (carboxymethylation) product of corosolic acid, (ESI (Fig. S12†)) at the (C) 2GS2 and (D) 3RZF active sites. | ||
ADMET has highlighted the limitations of the triterpenoid derivatives, specifically the potential limited oral uptake and rapid metabolism of these compounds in the human body. Molecular docking studies with specific proteins associated with anti-inflammatory, anti-tumour, and anti-cancer activities, alongside low cytotoxicity have demonstrated great promise with enhanced activity of several derivatives. This study demonstrates the significant potential of using carbonate chemistry (DMC) to modify triterpenoids. The sustainable methods developed in this work could be used for the carboxymethylation or methylation modification of a wide range of natural products containing a hydroxyl groups and could extend beyond triterpenoids. These methods could also be utilised with various dialkyl carbonate reagents (i.e. diethyl carbonate or diphenyl carbonate to name but a few) to develop a library of carbonates from triterpenoids, with the focus to overcome the limitations of uptake, rapid metabolism and provide further enhancement of biological activity.
Conclusions
This research reports the first sustainable carboxymethylation and/or methylation of ursolic acid via an acidic mediated process using DMC as reactant and solvent. The acids p-toluene sulfonic acid (PTSA), formic acid, ZnCl2, FeCl3 and H2SO4–SiO2 all exhibited promise as reagents in the carboxymethylation and/or methylation of cyclohexanol with DMC. These acids were successfully applied to the modification of ursolic acid. In the carboxymethylation reaction of ursolic acid, 5.0 equivalents loading of PTSA demonstrated the greatest performance at 90 °C, with 74% conversions and 59.8% selectivity toward the carboxymethylation product, 3β-[[methoxy]carbonyl]oxyurs-12-en-28-oic acid. Importantly, this work demonstrated the first synthesis of this ursolic acid carboxymethylation product. This compound will be assessed along with other alkyl carbonate products and other triterpenoids in bioassay screening in a follow-on paper. It was noted that lower conversions were observed when increasing the amount of acid. ZnCl2, FeCl3 and H2SO4–SiO2 yielded both carboxymethylation and methylation products. 3β-Formylurs-12-en-28-oic acid was produced via the esterification of ursolic acid and formic acid in quantitative yield promising a new highly efficient formylation method. The use of FeCl3 led to >99% conversion of ursolic acid and 99% selective to the methylation product, 3β-methoxyurs-12-en-28-oic acid. The coordination of FeCl3 with carbonyl group and –OCH3 group of DMC made the 5-ligand coordinated iron intermediate, which led to a highly efficient methylation reaction. Different interactions of DMC and Lewis acids leads to different reaction, in which FeCl3 gives highly efficient methylation while ZnCl2 favours carboxymethylation. These highly promising result offer a selective and potentially sustainable route to this modification of ursolic acid. Significantly, menthol and corosolic acid were used to demonstrate the applicability of the methods using the three optimised acidic conditions, and the results obtained demonstrated a good correlation with those observed for ursolic acid. Moreover, this work shows the green and sustainable methods developed in this work could lead to new biologically active compounds with properties such as anti-inflammatory, anti-tumour, and anti-cancer activities. This work represents a sustainable route for the modification of triterpenoids such as ursolic acid and corosolic acid.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project is funded by National Research Council of Thailand (NRCT) (Grant number: N42A650240). Mr Nuttapong Kadsanit would like to thank the support of Institute for the Promotion of Teaching Science and Technology for his Development and Promotion of Science and Technology Talents Project scholarship. The financial support of the Material Chemistry Research Center, Khon Kaen University and the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Ministry of Higher Education, Science, Research and Innovation are also gratefully acknowledged.References
- H. Yamaguchi, T. Noshita, Y. Kidachi, H. Umetsu, M. Hayashi, K. Komiyama, S. Funayama and K. Ryoyama, J. Health Sci., 2008, 54, 654–660 Search PubMed.
- S. lei Yan, C. yin Huang, S. tzy Wu and M. Chin Yin, Toxicol. In Vitro, 2010, 24, 842–848 CrossRef PubMed.
- M. C. Liu, S. J. Yang, L. H. Jin, D. Y. Hu, W. Xue and S. Yang, Med. Chem. Res., 2016, 25, 2267–2279 CrossRef CAS.
- H. Y. Tu, A. M. Huang, B. L. Wei, K. H. Gan, T. C. Hour, S. C. Yang, Y. S. Pu and C. N. Lin, Bioorg. Med. Chem., 2009, 17, 7265–7274 CrossRef CAS PubMed.
- A. Yardley-Jones, D. Anderson and D. V. Parke, Br. J. Ind. Med., 1991, 48, 437–444 CAS.
- X. Yang, Y. Li, W. Jiang, M. Ou, Y. Chen, Y. Xu, Q. Wu, Q. Zheng, F. Wu, L. Wang, W. Zou, Y. J. Zhang and J. Shao, Chem. Biol. Drug Des., 2015, 86, 1397–1404 CrossRef CAS PubMed.
- A. S. Leal, R. Wang, J. A. R. Salvador and Y. Jing, Bioorg. Med. Chem., 2012, 20, 5774–5786 CrossRef CAS PubMed.
- R. Sahni, V. Parcha, Y. Dobhal and A. Maithani, Pharm. Biol. Eval., 2016, 3, 126–134 Search PubMed.
- R. K. Sharma and R. Bandichhor, Green Processing and Synthesis, 2018, vol. 7, pp. 387–388 Search PubMed.
- C. Gmbh and C. Kg, J. Wound Care, 2000, 9, 420 CrossRef.
- F. P. Byrne, S. Jin, G. Paggiola, T. H. M. Petchey, J. H. Clark, T. J. Farmer, A. J. Hunt, C. Robert McElroy and J. Sherwood, Sustainable Chem. Processes, 2016, 4, 1–24 CrossRef.
- P. G. G. Do Nascimento, T. L. G. Lemos, A. M. C. Bizerra, A. M. C. Arriaga, D. A. Ferreira, G. M. P. Santiago, R. Braz-Filho and J. G. M. Costa, Molecules, 2014, 19, 1317–1327 CrossRef PubMed.
- T. H. Kwon, B. Lee, S. H. Chung, D. H. Kim and Y. S. Lee, Bull. Korean Chem. Soc., 2009, 30, 119–123 CrossRef CAS.
- I. Iniesta, M. Radon and C. Pinder, Pract. Neurol., 2013, 13, 393–395 CrossRef PubMed.
- S. J. Barbee, J. J. Stone and R. J. Hilaski, Am. Ind. Hyg. Assoc. J., 1995, 56, 74–76 CrossRef CAS PubMed.
- M. D. Schwartz, A. O. Obamwonyi, J. D. Thomas, J. F. Moorhead and B. W. Morgan, Am. J. Ind. Med., 2005, 47, 550–556 CrossRef CAS PubMed.
- J. C. R. Rippey and M. I. Stallwood, Emerg. Med. J., 2005, 22, 878–879 CrossRef CAS PubMed.
- J. Fowles, R. Boatman, J. Bootman, C. Lewis, D. Morgott, E. Rushton, J. Van Rooij and M. Banton, Crit. Rev. Toxicol., 2013, 43, 811–828 CrossRef CAS PubMed.
- J. Ellis, New Sci., 2001, 172, 59 Search PubMed.
- S. H. Pyo, J. H. Park, T. S. Chang and R. Hatti-Kaul, Curr. Opin. Green Sustainable Chem., 2017, 5, 61–66 CrossRef.
- P. Tundo, L. N. He, E. Lokteva and C. Mota, Chem. Beyond Chlorine, 2016, 1–608 Search PubMed.
- S. Ramesh, F. Devred and D. P. Debecker, Appl. Catal., A, 2019, 581, 31–36 CrossRef CAS.
- S. Jin, A. J. Hunt, J. H. Clark and C. R. McElroy, Green Chem., 2016, 18, 5839–5844 RSC.
- K. S. Kanakikodi, S. R. Churipard, A. B. Halgeri and S. P. Maradur, Sci. Rep., 2020, 10, 1–12 CrossRef PubMed.
- D. Kumar, M. Sonawane, B. Pujala, V. K. Jain, S. Bhagat and A. K. Chakraborti, Green Chem., 2013, 15, 2872 RSC.
- S. Rasheed, D. N. Rao, A. S. Reddy, R. Shankar and P. Das, RSC Adv., 2015, 5, 10567–10574 RSC.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
- D. E. V. Pires, T. L. Blundell and D. B. Ascher, J. Med. Chem., 2015, 58, 4066–4072 CrossRef CAS PubMed.
- L. Shi, Z. Gao, T. Zhang, H. Zhang and Y. Dong, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2019, 75, 153–158 CrossRef CAS PubMed.
- X. Zhang, J. Gureasko, K. Shen, P. A. Cole and J. Kuriyan, Cell, 2006, 125, 1137–1149 CrossRef CAS PubMed.
- J. Eberhardt, D. Santos-Martins, A. F. Tillack and S. Forli, J. Chem. Inf. Model., 2021, 61, 3891–3898 CrossRef CAS PubMed.
- J. Porter, A. Payne, B. de Candole, D. Ford, B. Hutchinson, G. Trevitt, J. Turner, C. Edwards, C. Watkins, I. Whitcombe, J. Davis and C. Stubberfield, Bioorg. Med. Chem. Lett., 2009, 19, 230–233 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- Biovia, Dassault Syst. 3D Exp. Co.
- R. Amin, K. Ardeshir, A. N. Heidar Ali and T. R. Zahra, Chin. J. Catal., 2011, 32, 60–64 CrossRef CAS.
- M. Selva, E. Militello and M. Fabris, Green Chem., 2008, 10, 73–79 RSC.
- 4-Dimethylaminopyridine – SDS No. 32942968, Molekula Ltd, UK, 2015, https://molekula.com/catalog/download/1122-58-3/32942968-4-dimethylaminopyridine-dmap/safety-data-sheet, accessed 3rd April 2024.
- A. V. Tkachev, A. Yu. Denisov, Y. V. Gatilov, I. Yu. Bagryanskaya, S. A. Shevtsov and T. V. Rybalova, Tetrahedron, 1994, 50, 11459–11488 CrossRef CAS.
- M. Guo and S. Gao, J. Environ. Qual., 2009, 38, 513–519 CrossRef CAS PubMed.
- D. E. Clark, Chem. Health Saf., 2001, 8, 12–21 CrossRef CAS.
- W. M. Haynes, D. R. Lide and T. J. Bruno, CRC Handbook of Chemistry and Physics, 2014 Search PubMed.
- V. Vershinin, H. Forkosh, M. Ben-Lulu, A. Libman and D. Pappo, J. Org. Chem., 2021, 86, 79–90 CrossRef CAS PubMed.
- P. Worsawat, P. Noppawan, C. Croise, N. Supanchaiyamat, C. R. McElroy and A. J. Hunt, Org. Biomol. Chem., 2023, 21, 1070–1081 RSC.
- S. A. Salami, M. Manyeruke, X. Siwe-Noundou and R. W. M. Krause, Int. J. Mol. Sci., 2022, 23, 9529 CrossRef CAS PubMed.
- M. Selva and P. Tundo, J. Org. Chem., 2006, 71, 1464–1470 CrossRef CAS PubMed.
- S. Michałek, L. Gurba-Bryśkiewicz, W. Maruszak, M. Zagozda, A. M. Maj, Z. Ochal, K. Dubiel and M. Wieczorek, RSC Adv., 2022, 12, 33605–33611 RSC.
- R. A. Sheldon, ACS Sustain. Chem. Eng., 2018, 6, 32–48 CrossRef CAS.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2015, 18, 288–296 RSC.
- S. Brogi, T. C. Ramalho, K. Kuca, J. L. Medina-Franco and M. Valko, Front. Chem., 2020, 8, 1–5 CrossRef PubMed.
- A. S. Rifaioglu, H. Atas, M. J. Martin, R. Cetin-Atalay, V. Atalay and T. Doǧan, Briefings Bioinf., 2019, 20, 1878–1912 CrossRef CAS PubMed.
- C. A. Lipinski, F. Lombardo, B. W. Dominy and P. J. Feeney, Adv. Drug Delivery Rev., 2001, 46, 3–26 CrossRef CAS PubMed.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- P. Matsson, B. C. Doak, B. Over and J. Kihlberg, Adv. Drug Delivery Rev., 2016, 101, 42–61 CrossRef CAS PubMed.
- B. C. Doak, B. Over, F. Giordanetto and J. Kihlberg, Chem. Biol., 2014, 21, 1115–1142 CrossRef CAS PubMed.
- A. Ariffin, N. A. Rahman, W. A. Yehye, A. A. Alhadi and F. A. Kadir, Eur. J. Med. Chem., 2014, 87, 564–577 CrossRef CAS PubMed.
- T. Shearer, K. Smith, D. Diaz, C. Asher and J. Ramirez, Comb. Chem. High Throughput Screening, 2005, 8, 89–98 CrossRef CAS PubMed.
- O. Michalak, M. Cybulski, W. Szymanowski, A. Gornowicz, M. Kubiszewski, K. Ostrowska, P. Krzeczyński, K. Bielawski, B. Trzaskowski and A. Bielawska, Int. J. Mol. Sci., 2023, 24, 8875 CrossRef CAS PubMed.
- C. Merlot, Drug Discovery Today, 2010, 15, 16–22 CrossRef CAS PubMed.
- S. Alqahtani, Expert Opin. Drug Metab. Toxicol., 2017, 13, 1147–1158 CrossRef CAS PubMed.
- R. F. Jayeeta Ghosh, M. S. Lawless, M. Waldman and V. Gombar, In Silico Methods for Predicting Drug Toxicity, Springer New York, New York, NY, 2016, vol. 1425 Search PubMed.
- C. M. Nisha, A. Kumar, A. Vimal, B. M. Bai, D. Pal and A. Kumar, J. Mol. Graphics Modell., 2016, 65, 100–107 CrossRef CAS PubMed.
- S. B. Ferreira, T. B. Dantas, D. de Figuerêdo Silva, P. B. Ferreira, T. R. de Melo and E. de Oliveira Lima, Curr. Top. Med. Chem., 2019, 18, 2186–2196 CrossRef PubMed.
- K. Horie, F. Tang and R. T. Borchardt, Pharm. Res., 2003, 20, 161–168 CrossRef CAS PubMed.
- J. Hodgson, Nat. Biotechnol., 2001, 19, 722–726 CrossRef CAS PubMed.
- G. Widiyarti, A. Sundowo, M. Megawati and T. Ernawati, Indones. J. Pharm. Sci. Technol., 2019, 1, 1 CrossRef.
- S. Vardhan and S. K. Sahoo, Comput. Biol. Med., 2020, 124, 103936 CrossRef CAS PubMed.
- R. K. Singla, L. Scotti and A. K. Dubey, Curr. Neuropharmacol., 2017, 15, 1100–1106 CAS.
- K. Kalani, H. S. Cheema, H. Tripathi, F. Khan, M. P. Daroker and S. K. Srivastava, RSC Adv., 2015, 5, 32133–32143 RSC.
- J. P. James, P. D. Ail, L. Crasta, R. S. Kamath, M. H. Shura and T. J. Sindhu, J. Health Allied Sci., 2024, 14, 190–201 CrossRef.
- P. Stenberg, C. A. S. Bergström, K. Luthman and P. Artursson, Clin. Pharmacokinet., 2002, 41, 877–899 CrossRef CAS PubMed.
- J. Hunter and B. H. Hirst, Adv. Drug Delivery Rev., 1997, 25, 129–157 CrossRef CAS.
- H. Suzuki and Y. Sugiyama, Eur. J. Pharm. Sci., 2000, 12, 3–12 CrossRef CAS PubMed.
- J. Taipalensuu, H. Törnblom, G. Lindberg, C. Einarsson, F. Sjöqvist, H. Melhus, P. Garberg, B. Sjöström, B. O. Lundgren and P. Artursson, J. Pharmacol. Exp. Ther., 2001, 299, 164–170 CAS.
- C. Ren, D. Kong, C. Ning, H. Xing, Y. Cheng, Y. Zhang, Y. Lu, N. Li, X. Chen and D. Zhao, J. Pharm. Sci., 2021, 110, 905–913 CrossRef CAS PubMed.
- K. M. Honorio, T. L. Moda and A. D. Andricopulo, Med. Chem., 2013, 9, 163–176 CrossRef CAS PubMed.
- A. P. Li, Drug Discovery Today, 2001, 6, 357–366 CrossRef CAS PubMed.
- J. Lin, D. Sahakian, S. de Morais, J. Xu, R. Polzer and S. Winter, Curr. Top. Med. Chem., 2005, 3, 1125–1154 CrossRef PubMed.
- J. Ekowati, N. W. Diyah, K. A. Nofianti, I. S. Hamid and S. Siswandono, J. Math. Fundam. Sci., 2018, 50, 203–219 CrossRef CAS.
- N. Shehzadi, K. Hussain, M. Islam, N. I. Bukhari, M. T. Khan, M. Salman, S. Z. Siddiqui, A. U. Rehman and M. A. Abbasi, Lat. Am. J. Pharm., 2016, 35, 1991–1997 CAS.
- K. H. Bleicher, H. J. Böhm, K. Müller and A. I. Alanine, Nat. Rev. Drug Discovery, 2003, 2, 369–378 CrossRef CAS PubMed.
- C. Xu, F. Cheng, L. Chen, Z. Du, W. Li, G. Liu, P. W. Lee and Y. Tang, J. Chem. Inf. Model., 2012, 52, 2840–2847 CrossRef CAS PubMed.
- M. C. Sanguinetti, C. Jiang, M. E. Curran and M. T. Keating, Cell, 1995, 81, 299–307 CrossRef CAS PubMed.
- S. Wang, Y. Li, L. Xu, D. Li and T. Hou, Curr. Top. Med. Chem., 2013, 13, 1317–1326 CrossRef CAS PubMed.
- Z. Zhou, Q. Gong, B. Ye, Z. Fan, J. C. Makielski, G. A. Robertson and C. T. January, Biophys. J., 1998, 74, 230–241 CrossRef CAS PubMed.
- F. Li, H. Wang, Y. Wang, S. Feng, B. Hu, X. Zhang, J. Wang, W. Li and M. Cheng, J. Biomol. Struct. Dyn., 2020, 38, 5401–5418 CrossRef CAS PubMed.
- D. M. Bissell, G. J. Gores, D. L. Laskin and J. H. Hoofnagle, Hepatology, 2001, 33, 1009–1013 CrossRef CAS PubMed.
- T. Ringbom, L. Segura, Y. Noreen, P. Perera and L. Bohlin, J. Nat. Prod., 1998, 61, 1212–1215 CrossRef CAS PubMed.
- Z. Y. Wei, K. Q. Chi, K. S. Wang, J. Wu, L. P. Liu and H. R. Piao, Bioorg. Med. Chem. Lett., 2018, 28, 1797–1803 CrossRef CAS PubMed.
- Y. Yamaguchi, K. Yamada, N. Yoshikawa, K. Nakamura, J. Haginaka and M. Kunitomo, Life Sci., 2006, 79, 2474–2479 CrossRef CAS PubMed.
- Y. Li, Z. H. Zhou, M. H. Chen, J. Yang, J. Leng, G. S. Cao, G. Z. Xin, L. F. Liu, J. P. Kou, B. L. Liu, P. Li and X. D. Wen, Antioxid. Redox Signaling, 2016, 24, 893–908 CrossRef CAS PubMed.
- M. P. Bhatt, Y. C. Lim, Y. M. Kim and K. S. Ha, Diabetes, 2013, 62, 3851–3862 CrossRef CAS PubMed.
- S. J. Kim, J. Y. Cha, H. S. Kang, J. H. Lee, J. Y. Lee, J. H. Park, J. H. Bae, D. K. Song and S. S. Im, BMB Rep., 2016, 49, 276–281 CrossRef CAS PubMed.
- V. Benetou, A. Lagiou and P. Lagiou, F1000Research, 2015, 4, 1–10 Search PubMed.
- N. Sultana, Int. J. Biochem. Res. Rev., 2017, 16, 1–35 CrossRef CAS.
- G. D. Kim, Prev. Nutr. Food Sci., 2021, 26, 434–444 CrossRef CAS PubMed.
- Z. Wang, P. Zhang, H. Jiang, B. Sun, H. Luo and A. Jia, Molecules, 2022, 27, 1–13 CrossRef.
- W. L. Liao, Y. F. Liu, T. H. Ying, J. C. Shieh, Y. T. Hung, H. J. Lee, C. Y. Shen and C. W. Cheng, Int. J. Mol. Sci., 2022, 24, 366 CrossRef PubMed.
- S. Rashid, B. A. Dar, R. Majeed, A. Hamid and B. A. Bhat, Eur. J. Med. Chem., 2013, 66, 238–245 CrossRef CAS PubMed.
- Y. Ren and A. D. Kinghorn, Planta Med., 2019, 85, 802–814 CrossRef CAS PubMed.
- S. M. Woo, S. U. Seo, K. J. Min, S. S. Im, J. O. Nam, J. S. Chang, S. Kim, J. W. Park and T. K. Kwon, Int. J. Mol. Sci., 2018, 19, 1–13 Search PubMed.
- L. Zhang, J. N. Wang, J. M. Tang, X. Kong, J. Y. Yang, F. Zheng, L. Y. Guo, Y. Z. Huang, L. Zhang, L. Tian, S. F. Cao, C. H. Tuo, H. L. Guo and S. Y. Chen, Mol. Biol. Rep., 2012, 39, 5085–5093 CrossRef CAS PubMed.
- C. Y. Ku, Y. R. Wang, H. Y. Lin, S. C. Lu and J. Y. Lin, PLoS One, 2015, 10, 1–17 Search PubMed.
- K. J. Nho, J. M. Chun and H. K. Kim, Food Chem. Toxicol., 2013, 56, 8–17 CrossRef CAS PubMed.
- C. Li, Z. Yang, C. Zhai, W. Qiu, D. Li, Z. Yi, L. Wang, J. Tang, M. Qian, J. Luo and M. Liu, Mol. Cancer, 2010, 9, 1–13 Search PubMed.
- J. Park, J. Lee, M. Lee, E. Cha, S. Kim and J. Sul, Mol. Med. Rep., 2018, 18, 2880–2888 CAS.
- M. Jia, Y. Xiong, M. Li and Q. Mao, Oncol. Res. Featur. Preclin. Clin. Cancer Ther., 2020, 28, 371–383 Search PubMed.
- J. Yang, R. Wu, W. Li, L. Gao, Y. Yang, P. Li and A. N. Kong, Mol. Carcinog., 2018, 57, 512–521 CrossRef CAS PubMed.
- B. Ma, H. Zhang, Y. Wang, A. Zhao, Z. Zhu, X. Bao, Y. Sun, L. Li and Q. Zhang, J. Exp. Clin. Cancer Res., 2018, 37, 1–16 CrossRef PubMed.
- M. I. Umah and I. G. M. Sanjaya, Int. J. Curr. Sci. Res. Rev., 2023, 06, 3772–3782 Search PubMed.
- D. Y. Kang, N. Sp, J.-M. Lee and K.-J. Jang, Biomedicines, 2021, 9, 297 CrossRef CAS PubMed.
- Y.-Q. Meng, C.-D. Xu, T.-T. Yu, W. Li, Q.-W. Li and X.-X. Li, J. Asian Nat. Prod. Res., 2020, 22, 359–369 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02122c |
| This journal is © The Royal Society of Chemistry 2024 |