
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of dimeric 1,2-benzothiazine 1,1-dioxide scaffolds: molecular structures, Hirshfeld surface analysis, DFT and enzyme inhibition studies†
Muqudis Fatimaa,
Waseeq Ahmad Siddiqui *a,
Muhammad Iqbal Choudharyb,
Adnan Ashraf*c,
Shanawer Niazd,
Muhammad Asam Razae,
Seikh Mafiz Alamf,
Muhammad Ashfaqg,
Muhammad Nawaz Tahirg and
Kholood Ahmed Dahloush
*a,
Muhammad Iqbal Choudharyb,
Adnan Ashraf*c,
Shanawer Niazd,
Muhammad Asam Razae,
Seikh Mafiz Alamf,
Muhammad Ashfaqg,
Muhammad Nawaz Tahirg and
Kholood Ahmed Dahloush
aInstitute of Chemistry, University of Sargodha, Sargodha 40100, Pakistan. E-mail: waseeq786@gmail.com
bInternational Center for Chemical and Biological Sciences, H. E. J Research Institute of Chemistry, University of Karachi, Karachi, Pakistan
cDepartment of Chemistry, The University of Lahore, Lahore, Pakistan. E-mail: adnanashraf7772@gmail.com
dDepartment of Physics, Thal University Bhakkar, Bhakkar, 30000, Pakistan
eDepartment of Chemistry, University of Gujrat, Gujrat, Pakistan
fDepartment of Chemistry, Aliah University, New Town, Kolkata 700 156, India
gDepartment of Physics, University of Sargodha, Sargodha-40100, Pakistan
hDepartment of Chemistry, College of Science, King Saud University, P. O. Box 2455, Riyadh 11451, Saudi Arabia
First published on 28th May 2024
Abstract
1,2-Benzothiazines are bioactive compounds with diverse pharmacological properties. We report here the synthesis of a series of dimers containing 1,2-benzothiazine scaffolds as potential pharmacophores. The characterization of compounds was done using analytical techniques such as FT-IR, 1H NMR, and elemental analyses. The molecular structures of the compounds (5–8) were confirmed by X-ray crystallography. The molecular interactions in compounds (5–8) were determined by Hirshfeld Surface Analysis (HSA). Density functional theory (DFT) investigations were carried out to calculate vibrational properties, NMR behaviour, dipole moments, molecular electrostatic potential (MEP), frontier molecular orbital (FMO), natural bonding orbital (NBO) analysis and global reactivity descriptors. The global reactivity descriptors indicated the charge transfer reactions and stabilized as follows: 8 > 7 > 6 > 5. In FMO analysis a substantial HOMO–LUMO gap, ranging from 4.43 to 5.12 eV, with high LUMO values was observed for all compounds, while the highest value for linear polarizability was found in compound 8. The in vitro and in silico studies confirm that compound 8 is more active toward AChE and BChE enzymes.
Introduction
Acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) are members of the cholinesterases (ChEs) family, which specifically hydrolyses carboxylic ester into choline. AChE hydrolyzes neurotransmitter acetylcholine, and BChE uses butyrylcholine as the substrate.1,2 In the central nervous system, AChE is found in cholinergic synapses and neuromuscular junctions. The acetylcholine receptors are activated at postsynaptic membranes in the presence of AChE, which hydrolyzes the acetylcholine into choline and acetate. The AChE is necessarily required for the normal functioning of the central and peripheral nervous system because it controls the termination of synaptic transmission and inhibits the continuous nerve firings at the end of nerves.3,4 It is also present in the form of Yt antigen known as Cartwright in red blood cell membranes which determines the type of blood group. The single gene product i.e., AChE is expressed as various splicing forms in different tissues. There are two active subsites in AChE i.e., esteratic and anionic sites.5 The quaternary amine of acetylcholine preferably binds to the anionic subsite and actylcholine hydrolyzed to acetate and choline at esteratic subsite, leading to the formation of acyl-enzyme and free choline.6 The regeneration of the free enzyme and byproduct acetic acid is liberated by the nucleophilic attack of water molecules on acyl enzyme.On the other hand, BChE controls the acylamidase activity and hydrolyses the acyl amides, and BChE is usually distributed in tissues. The neurons in the human brain are distinct and linked with AChE and are also present in glial cells.7,8 The AChE and BChE accelerate the Aβ peptide assembly associated with Alzheimer's disease, enhancing neurotoxicity.9
AChE inhibitors including quinazoline, Esrine, rivastigmine, tacrine, galantamine and donepezil are used in drug discovery as reference compounds. Different classes of bioactive compounds have been investigated as AChE inhibitors, which may target active sites of the enzymes. The molecular modelling studies bust the design of enzyme inhibitors which offer the potential to overcome different diseases. Therefore, in search for new enzyme inhibitors, various bioactive compounds including quinolines, coumarins, chalcones, pyrroles, pyrrolidines, triazoles, etc. were investigated, resulting in better enzyme inhibition activity and different binding modes.10 Among bioactive compounds, 1,2-benzothiazine scaffolds11 contain interesting structural features i.e., non-aromatic thiazine ring fused with aromatic fragment.12 These compounds belong to an important class of pharmaceuticals with anti-inflammatory activity. The benzothiazine based compounds have been investigated for their potential biological applications13 including, anti-HIV,14 antibacterial,15 antifungal, antioxidant,16 antitumor17 and alkaline phosphate inhibitors.18 Interestingly, despite their broad spectrum of biological applications, a limited number of 1,2-benzothiazine derivatives were evaluated for their enzyme inhibition potential.
In continuation of our work on developing sulfonamide and 1,2-benzothiazine functionalized heterocyclic ring systems, here we report the dimeric 1,2-benzothiazine compounds. The 1,2-benzothiazine precursors were reacted in the presence of silver oxide under a specific set of conditions to isolate dimeric 1,2-benzothiazine scaffolds. The available analytical techniques confirmed the successful synthesis and molecular structures. The crystal structures are being used in DFT to calculate the different parameters. The data extracted from the DFT studies was found to be in line with the experimentally obtained data. The HSA indicated the molecular interactions available in the 1,2-benzothiazine heterocyclic ring system. These compounds were further evaluated to find out the potential inhibition of enzymes. Docking studies further supported the biological potential to find out the possible binding interactions of the tested compounds with the active pocket of the targeted enzyme.
Results and discussion
The 1,2-benzothiazine derivatives were synthesised as per our previous procedure which were further dimerized in the presence of silver oxide to isolate compounds 5–8. The syntheses of the 1,2-benzothiazine based dimers was initially confirmed by NMR spectroscopy as described in Fig. S2–S5 and Table S1.† The 1H NMR spectra of the dimmers 5–8 clearly indicated the absence of OH peak which was initially present at 12.04–12.08 ppm in the precursor compounds.19,20 The aromatic protons were present at 8.11–7.50 ppm while the other thiazine ring proton signals resonated at 7.88–7.62 ppm after dimer formation in compounds 5–8. Furthermore, a slight downfield shifting from 3.95–2.94 to 3.96–2.77 ppm in methyl ester protons H-12 was observed. In addition, the methyl protons of H-13 in compound 5 shift from 3.95 to 3.05 ppm and a new signal appeared at 3.77 ppm which confirms the formation of dimmer. In compound 6 shifting of peaks of methylene protons from 3.51 to 3.58–3.54 whereas in the dimers, same proton signal was found at 3.52–3.50 ppm. In compound 7, the benzyl ring protons shifted from 7.11–6.89 to 7.27–7.15 and the second benzyl ring protons appeared at 7.05–6.95 ppm. In compound 8, the benzyl ring protons 6.95–6.61 to 7.24–7.15 ppm and new signals for the other benzyl ring protons were found at 7.05–6.95 ppm. The theoretical studies also suggested the formation of the targeted compounds as similar pattern of the peaks has been observed from Gaussian result (Fig. S6, S7 and Table S1†).The synthesis of the compounds 5–8 was confirmed by FT-IR (Fig. S9, S10, and Table S2†). In compounds 5–8 aromatic and aliphatic CH stretching vibrations were observed at 3031–3005 cm−1 and 2956–2928 cm−1, respectively. The deformation of the CH with a medium strength peak was observed at 1442–1440 cm−1 in experimental data. In reactant compounds carbonyl group signal was observed at 1667–1645 cm−1 while in all dimer molecules three different carbonyl stretching signals were observed between 1778–1710, 1745–1656 and 1663–1646 cm−1.21 The disappearance of the hydroxyl group signal in compounds 5–8 which was initially present at 3000–2600 cm−1 in reactants and the appearance of new carbonyl signals clearly indicate the formation of the compounds 5–8. The formation of these compounds was also supported by the presence of ether C–O group at 1265–1238 cm−1. The sulphonamide asymmetric and symmetric vibrations were found at 1365–1327 cm−1 and 1160–1122 cm−1.22,23 Furthermore, FTIR of the synthesized compounds were also computed from output files of the DFT studies. It was found a strong relation between experimental and theoretical studies (Fig. S8–S10, and Table S2†).
Molecular structures 5–8
The prominent difference between the crystal structures of compounds 5–8 lies at attachment with N-atoms of the thiazine ring system (Fig. 1). The noticeable similarity of compound 5 with compound 7 is that both are monoclinic crystal systems whereas the compound 6 and 8 are crystallized in the lowest symmetry crystal system. The geometrical parameters of compounds 5–8 are aline with the reported literature structures.24,25 In compound 5, the phenyl ring A is inclined at 14.7 (2)° with respect to thiazine ring B (C1/C6–C8/N1/S2), whereas the phenyl ring C (C12–C17) is inclined at 20.8 (2)° with respect to thiazine ring D (C12/C17–C19/N2/S1) (Table S3†).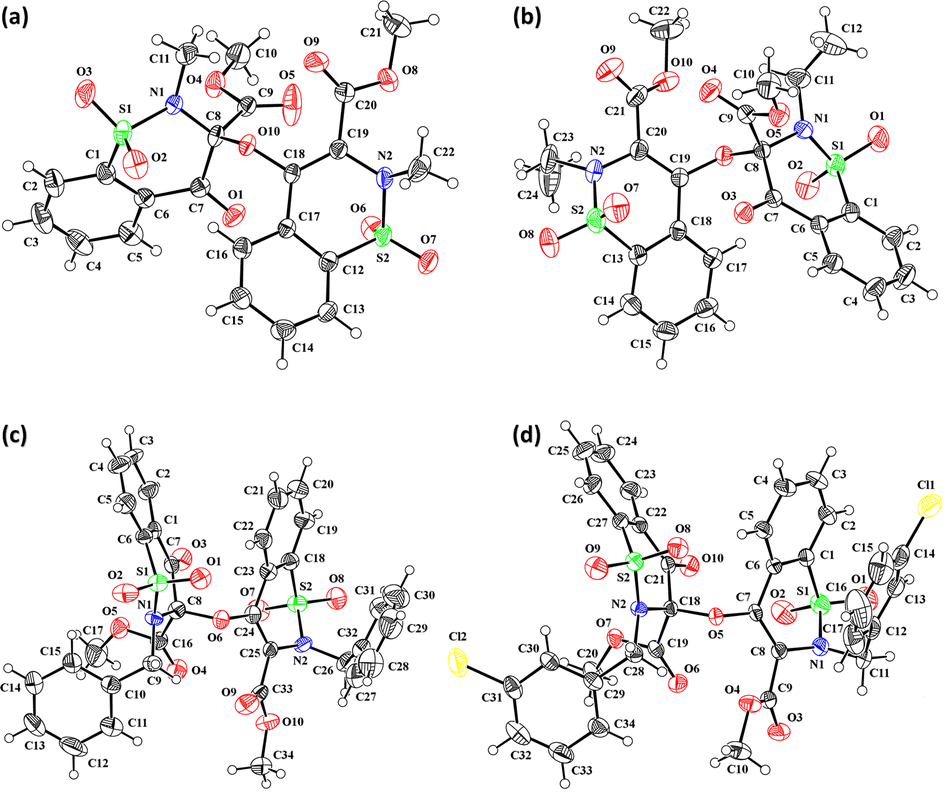 | ||
| Fig. 1 ORTEP diagram of compound (a) 5, (b) 6, (c) 7, (d) 8 at a probability level of 40%. | ||
In compound 6, the dihedral angles between rings are 35.6 (9)° and 22.9 (1)°, and in compound 7 are 27.8 (2)° and 15.8 (2)°. The corresponding angles in compound 8 are 21.4 (1)° and 11.8 (1)°. In compound 5, the nitrogen and sulfur atoms of thiazine ring B are away from the mean plane defined by (C1/C6–C8) at 0.2382 (8) and 0.5951 (8) Å, respectively. In contrast, the nitrogen and sulfur atoms of thiazine ring D, are deviated from the mean plane defined via (C12/C17–C19) by 0.1660 (9) and 0.6303 (9) Å, respectively. The deviations of nitrogen and sulfur atoms from the corresponding planar part of thiazine rings in compound 6 are 0.4628 (1), 0.2644 (3), 0.1173 (4) and 0.6635 (4) Å while in compound 7 are 0.3878 (1), 0.3014 (1), 0.3025 (1) and 0.4872 (1) Å. The corresponding deviations in compound 8 are 0.2562 (5), 0.5645 (5), 0.3529 (5) and 0.4138 (5) Å. The geometry around S-atoms is a distorted tetrahedron in compounds 5–8. In compound 5, the first methyl ester group E (C9/C10/O4/O5) and second methyl ester group F (C20/C21/O8/O9) are inclined at 86.9 (2)° and 21.1 (2)° relative to the attached thiazine rings. The dihedral angles among corresponding parts of compound 6 are 67.8 (1)° and 22.8 (2)°, and in compound 7, they are 74.8 (3)° and 13.7 (2)°. In compound 8, the dihedral angles are 19.1 (2)° and 76.1 (1)°. The molecular geometry was stabilized by intramolecular C–H⋯O bonding in the case of compounds 5–8. The molecules of compound 5 form a layer structure in the bc plane by forming C–H⋯O bonding and one such layer of molecules is shown in Fig. S11a.† The non-carbonyl O-atom of the group E and the O atom of the sulfonyl group (S2/O6/O7) are involved in H-bonding. The molecules of compound 6 are observed to be connected in the form of chains via C–H⋯O bonding, where carbonyl O-atom (O3) acts as H-bond acceptor, and no other O-atom participated in H-bonding (Fig. S11b†). The connection of molecules in the form of chains is also noticed in compound 7 via C–H⋯O bonding (Fig. S11c†). The C–H⋯O and C–H⋯Cl bonding is involved in stabilizing compound 8 (Fig. S11d†). The π⋯π interactions in phenyl rings played a key role in the stabilization of supramolecular assembly of the compounds 5–8 (Fig. S12†). The interactions in compound 8 are stronger than those in compounds 5–7 as the distance describing the interactions is smaller in compound 8 than in compounds 5–7.
Hirshfeld surface analysis
In recent days, the plan of the research groups associated with the supramolecular chemistry of the crystals is to unravel the major properties of the crystals. That decider is how the molecules are reacting to each other. HSA can explain the molecular environment in the crystals structures and we have done the same analysis for compounds 5–8 by using Crystal Explorer 21.5.26 The design of the Hirshfeld surface based on normalized distances (dnorm) can distinctively discriminate short contacts from more extended contacts through three distinct colours (red, white and blue). Short and long contacts are shown by red and blue spots on the surface whereas white spots showed equal contacts (distance = sum of the van der Waals radii).27–29 The red spots around particular CH and O-atoms for the surface of compounds 5–8 (Fig. 2a–d) described the short contact. Most of the surface in the case of compounds 5–8 is surrounded by H-atoms and O-atoms indicating C–H⋯O interactions (Table S4†). In compound 8, H⋯Cl contacts are also observed but these contacts are not short enough to be considered as H-bonding. The influence of the Cl-atom in the crystal packing of compound 8 is mainly observed by H⋯Cl contacts; however, some weak C⋯Cl contacts are also noticed.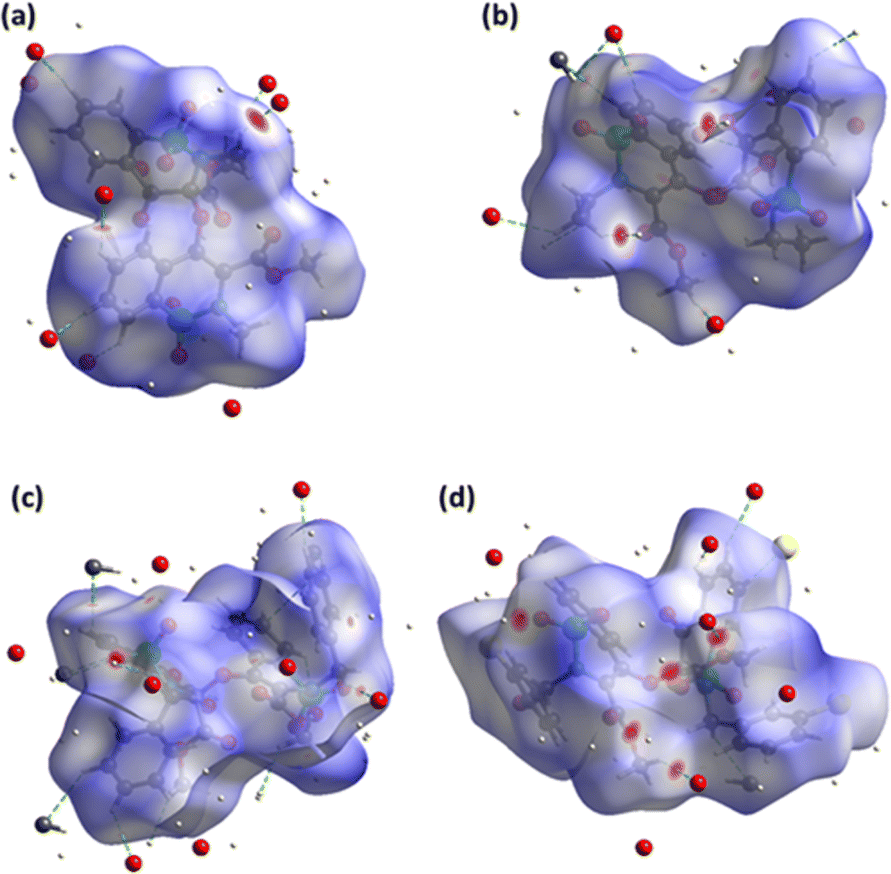 | ||
| Fig. 2 Hirshfeld surface plotted over dnorm for compound (a) 5, (b) 6, (c) 7, (d) 8. | ||
The supramolecular assembly was stabilized by π⋯π interactions in compounds 5–8, irrespective of H-bonding interactions which are recognized by the design of the surface based on the shape index by noticing the consecutive red and blue triangles around the rings (Fig. S13†).
The conventional way of exploring supramolecular assembly can be renewed by 2D plots as the fragmentation of the crystal packing interactions through these plots.30–32 Imperative 2D plots alongside their contribution in the supramolecular assembly of compounds 5–8 are shown in Fig. 3. The highest contributor in the supramolecular assembly of compound 5 is H⋯O while in other compounds, H⋯H contacts are exceptional. In the case of compound 7, H⋯C contacts have larger contribution compared to other compounds. H⋯C contacts have exactly same contribution in compounds 5 and 6. In compound 6, H⋯O contacts are more dominating as compared to other compounds. In compound 8, H⋯Cl contacts have a 12.8% contribution but the plot showed that the other compounds have no such contact. Enrichment ratio results are listed in Tables S5–S8† for compounds 5–8, respectively which are the information provider related to the pair having highest tendency to form crystal packing interactions.33 Carbon–carbon pair has highest tendency to form crystal packing interactions in compound 5 whereas for compounds 6 and 7, the oxygen–hydrogen pair has the highest tendency to form crystal packing interactions. The presence of chlorine in compound 8 makes it different from other compounds as the carbon–chlorine pair is most favourable in compound 8.
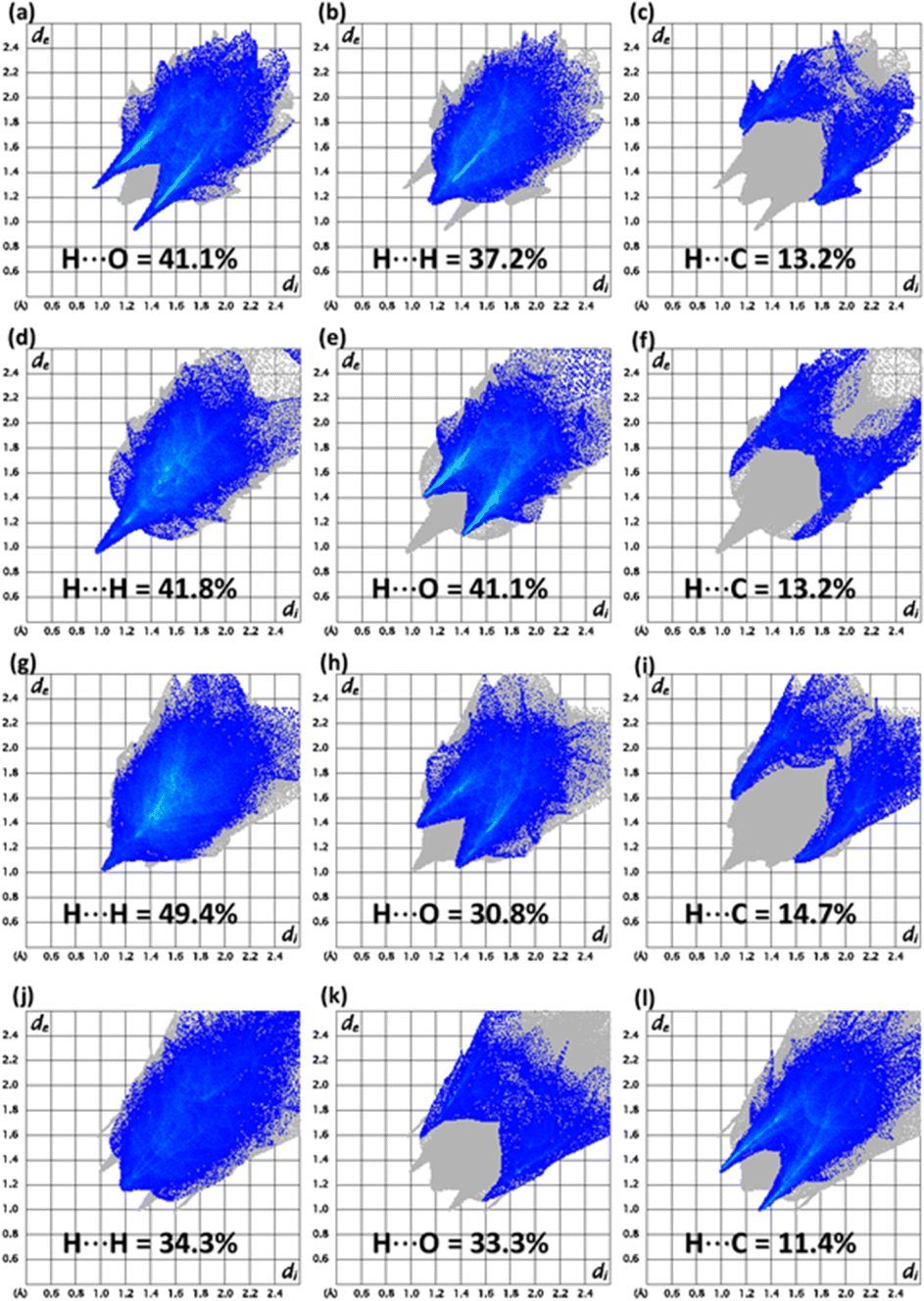 | ||
| Fig. 3 Important 2D plots for (a–c) 5, (d–f) 6, (g–i) 7, (j–l) 8. | ||
The pressure bearing ability of the crystal can be explored by the surface designed for voids through the concept based on procrystal electron density.34–36 The grey coloured regions are the voids and such surfaces for compounds 5–8 are shown in Fig. 4. Smaller voids in crystal mean more pressure bearing ability and better mechanical properties. The void volume in compounds 5–8 is 142.26, 185.85, 405.40 and 281.97 Å3, respectively. The voids devoured 12.4%, 14.6%, 13.1% and 16.6% in compounds 5–8, respectively. The study showed that the compounds have ability to bear significant pressure as the space devoured by voids is much smaller than the space devoured by molecules.
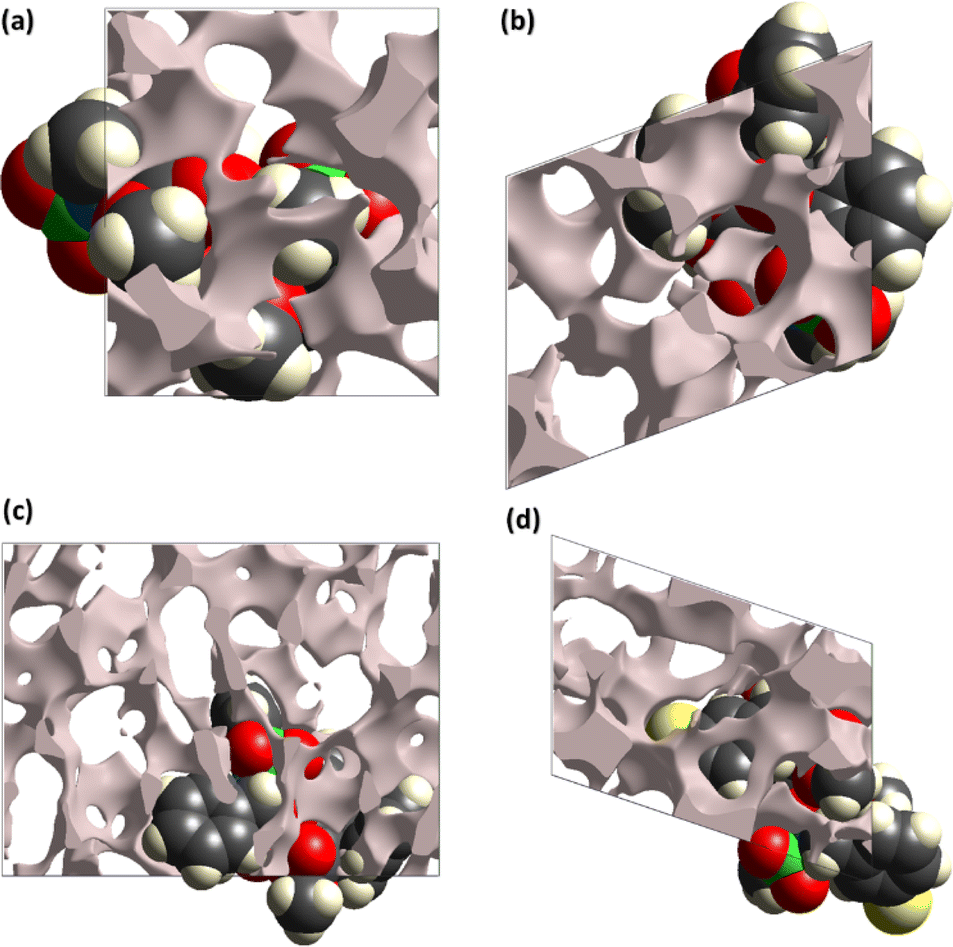 | ||
| Fig. 4 Surfaces containing voids in (a) 5, (b) 6, (c) 7, (d) 8. | ||
The interactions among molecules can be elaborated in terms of the interaction energy between molecular pairs.37–40 The calculations are performed at HF/3-21G electron density level and a cluster of 3.8 Å around the reference molecule (molecule of asymmetric unit) is engaged in calculations. The interaction energy comes from the four contributions, 1 – electrostatic coulomb, 2 – dispersion, 3 – polarization, 4 – repulsion. The dispersion and polarization energies are always attractive whereas coulomb energy can be attractive or repulsive. The interaction energy results are tabulated in Tables S9–S12† for the compounds 5–8, respectively.
The common result for compounds 5–8 is that the attractive interaction energy is minimum for the pair with maximum distance apart. Total interaction energy is attractive for the pairs of compounds 5–8 except for one pair of compound 8 with intermolecular distance of 12.15 Å in which the repulsion is dominant over the attractive contributions.
The major contribution of the total energy comes from the dispersion energy for compounds 5–8. Total interaction is largest for the pair with distances 7.40, 8.82, 8.11 and 8.85 Å in compounds 5–8, respectively. One out of 8 pairs of compound 5 has zero repulsion energy whereas for other compounds, greater than one pairs have zero repulsion energy. For visual inspection of the competition among the types of energies for stabilization of pairs, energy frameworks are formed for compounds 5–8 (Fig. 5). The center to center connection of molecules is made by cylinders in which width is directly relates to the strength of the interaction. The width of cylinders for dispersion energy is larger than that of cylinders for coulomb energy, which shows that the dispersion energy is the major contributor to the stabilization of the supramolecular assembly of compounds 5–8 compared to coulomb energy.
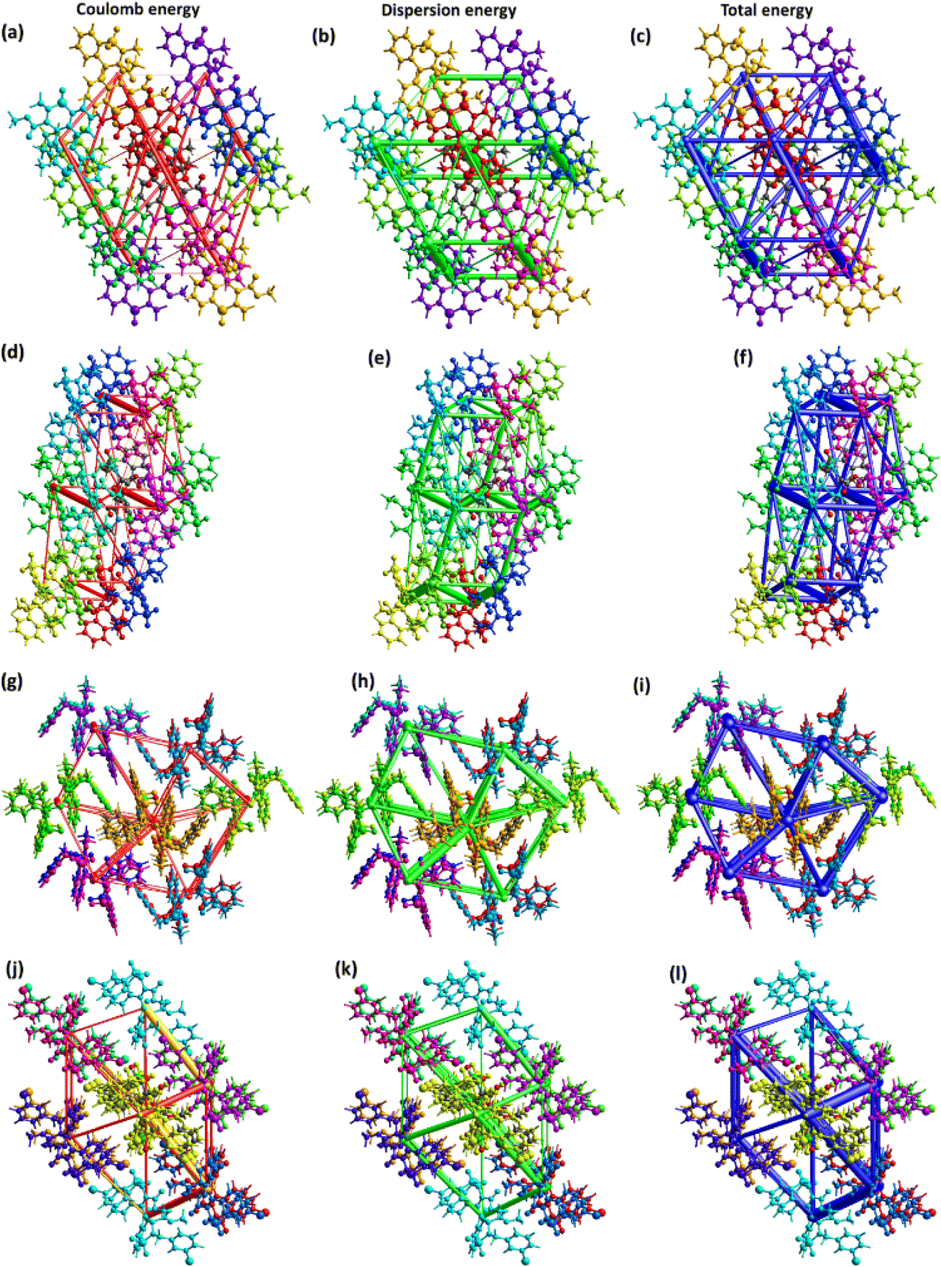 | ||
| Fig. 5 Energy frameworks for compounds 5–8. | ||
Natural bonding orbitals (NBO) analysis
The NBO analysis was used to determine the electron density distribution and hyperconjugation interactions (Fig. S14, and Table S13†). In 5, the π → π* interactions π(C18–C20) → π*(C15–C16) and π(C18–C20) → π*(C15–C16) represented the maximum stabilization energies at 23.23 and 23.56 kcal mol−1. The π(O11–C48) → π*(C46–C47) and π(O3–C25) → π*(C22–C24) interactions resulted in the minimum stabilization energies at 3.21 and 4.96 kcal mol−1. The resonance generated interactions LP2(O6) → π*(O7–C27) and LP2(O10) → π*(C11–C48) with maximum stabilization energies were found at 51.16 and 47.69 kcal mol−1. The lowest stabilization energy was compelled by LP2(O6) → π*(C28–C29) and LP1(O11) → π*(C47–C48) at 3.37 and 3.32 kcal mol−1.In 6, the π → π* interactions π(C44–C46) → π*(C39–C48) and π(C22–C24) → π*(C15–C16) have shown maximum stabilization energies at 24 and 23.92 kcal mol−1 and the interactions π(O11–C51) → π*(C49–C50) and π(O5–C25) → π*(C22–C24) with minimum stabilization at 3.64 and 5.04 kcal mol−1. The resonance generated interaction LP2(O7) → π*(O6–C27), LP2(C12) → π*(O11–C51) with maximum stabilization energy at 52.29 and 47.17 kcal mol−1, respectively. These interactions LP2(O7) → π*(C28–C29), LP2(O7) → π*(C28–C31) with minimum stabilization energy at 3.36 and 3.37 kcal mol−1.
In 7, the π → π* interactions π(C22–C24) → π*(C15–C16) and π(C51–C53) → π*(C46–C15) have shown maximum stabilization energies at 23.89 and 24.34 kcal mol−1 and the interactions π(O5–C25) → π*(C22–C24) and π(O11–C72) → π*(C56–C57) with minimum stabilization at 4.88 and 3.63 kcal mol−1. The resonance generated interaction LP2(O7) → π*(O6–C41), LP2(O8) → π*(O7–C29) with maximum stabilization energy at 52.50 and 44.82 kcal mol−1 respectively. These interactions LP2(O12) → σ*(C73–C74), LP2(O7) → σ*(C42–H43) with minimum stabilization energy at 3.35 and 3.05 kcal mol−1.
In 8, the π → π* interactions π(C22–C24) → π*(C17–C26) and π(C54–C55) → π*(C61–C63) have shown maximum stabilization energies at 24.36 and 23.98 kcal mol−1 and the interactions π(O7–C29) → π*(C27–C28) and π(O14–C53) → π*(C54–C55) with minimum stabilization at 4.81 and 5.04 kcal mol−1. The resonance generated interaction LP2(O8) → π*(O7–C29), LP2(O11) → π*(O10–C48) with maximum stabilization energy at 44.82 and 54.04 kcal mol−1 respectively. These interactions LP3(O5) → σ*(S3–O6), LP2(O11) → σ*(C49–H50) with minimum stabilization energy at 3.02 and 3.14 kcal mol−1.
Frontier molecular orbital (FMO) analysis
The FMO analysis is significantly used to understand different properties of the chemical compounds including, optoelectronic, kinetic stability, adsorption, chemical hardness and chemical softness of the chromophores. These properties are entirely dependent on the HOMO–LUMO energy difference. The higher energy gap indicates the harder nature of the compounds, higher stability and lower polarizability. The computed results for the compounds 5–8 are given in Table S14, Fig. S15 and S16.† The calculated HOMO energies are −7.50, −7.48, −6.48 and −6.99 eV for the compounds 5–8 while the LUMO energies for these compounds are −2.38, −2.38, −2.34 and −2.56 eV with band gap 5.12, 5.10, 4.50 and 4.43 eV, respectively. The difference in energies (HOMO–LUMO) for the entitled compounds decreases in a sequential way. The decreasing order 5 > 6 > 7 > 8 of the energy difference indicated that the stability and hardness decreased from compound 5 to 8. The chromophores with stabilized LUMO significantly impacted the biological activities of the compounds. The observed activity is mainly because of the presence of a higher electronegative atom in benzothiazine ring system. The electronic cloud for HOMO/LUMO in all compounds is present on the dimeric oxygen atom and one of the benzothiazine molecules. The electronic charge resulted in higher activity because of the strong electron withdrawing effect of the dimeric oxygen and thiazine ring, which stabilized the LUMO orbitals.Molecular electrostatic potential (MEP)
MEP is a magical tool for the illustration of electrostatic potential in the molecules on the basis of colour mapping, as shown in Fig. 6. It is considered an excellent tool to describe the size, shape, electrostatic potential of the compounds and the relationship between molecular structure and physicochemical properties. In MEP diagram the red colour region indicates the electron deficit region and blue colour indicates the presence of an excess of electrons while green colour region represents the neutral part. The MEP diagram clearly depicted that all the oxygen atoms present in 1,2-benzothiazine functionalized dimers are found in the red colour region which indicates the presence of negative potential. The benzothiazine ring benzene and the groups attached to nitrogen atoms are electron-deficient in nature.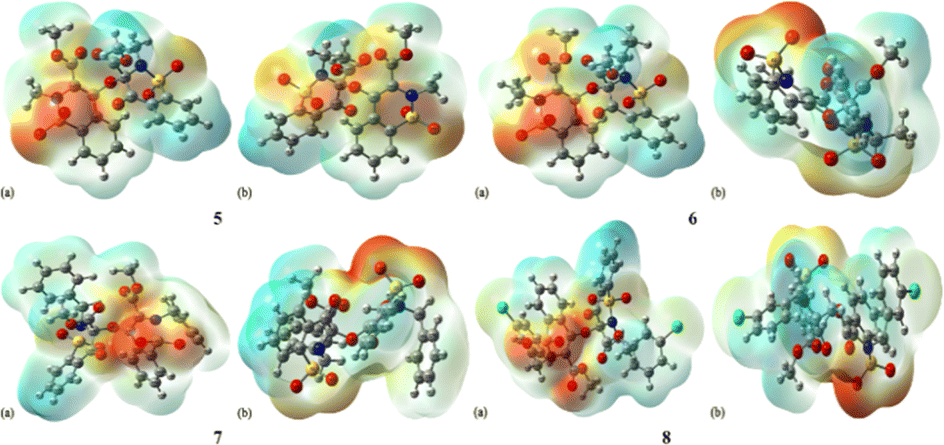 | ||
| Fig. 6 MEP for the compounds 5–8, (a) front, (b) back. | ||
Global reactivity descriptors
The global properties are presented in Table S15† which indicate the stability and reactivity of the synthesized compounds 5–8. The higher ionization potential of the compound 8 indicated higher stability and the compounds 5 and 6 were found least stable with the same value. The compound 8 has a higher affinity for electrons and compound 7 has the least affinity for the electrons. The greater negative value −5.05 eV of the chemical potential also supports the higher stability of the compound 8 and the order of stability is 8 > 7 > 6 > 5. The compound is harder among all synthesized dimeric compounds because of a higher global hardness value i.e., 1.53 eV and compounds 5 and 6 are equally soft with global softness of 0.36 eV and also most polarized. The electrophilicity values indicated that compound 7 is a good nucleophile with a lower value of 7.73 eV and compound 8 is a good electrophile with higher value of 8.34 eV.Dipole moment and linear polarizability
The B3LYP/LANL2DZ was utilized to determine the dipole moment and linear polarizability of the compounds 5–8 (Table S16†). The observed data indicated that the dipole moment value of 2.649 D for the compound 6 is higher as compared to compound 8 (2.640 D) and the lowest dipole moment value of 2.412 D is observed in 5. In compounds 6 (2.431 D) and 7 (1.082 D) the dipole moment value is higher along the y-axis and in the case of 5 (−0.884 D) and 8 (−0.078 D), the higher value is observed along x-axis and z-axis. The linear polarizability was found higher along αxy i.e., 427.666 and 489.878 a. u. in compounds 6 and 7, respectively. In compound 5, the higher polarizability 376.595 a. u. was found along αxx and in 8 the higher value 479.778 a. u. was observed along αxz.Acetylcholine and butyrylcholine esterase inhibition
The inhibition of enzymes by organic molecules is a fundamental concept within the fields of biochemistry and pharmacology, carrying significant implications for drug development and the understanding of biochemical processes.41 The inhibitory effects of organic molecules on enzymes may appear through a range of mechanisms, encompassing competitive, non-competitive, and uncompetitive inhibition. The acetylcholine and butyrylcholine inhibition potential is presented in Table 1 and it is clear from the results that compound 8 has depicted the highest inhibition potential against both enzymes. The enzyme inhibition activity was 88.67 ± 1.9% and 63.44 ± 2.2% against AChE and BChE, respectively. The compounds 5, 6 and 7 exhibited 63.91 ± 2.4%, 71.43 ± 1.8% and 64.92 ± 1.6% potential, respectively toward AChE. The order of inhibitory potential against BChE was 7 > 6 > 5.| Comp. | Inhibition (%) | Molecular docking | ||
|---|---|---|---|---|
| AChE | BChE | AChE | BChE | |
| 5 | 63.91 ± 2.4 | 52.81 ± 1.1 | −9.3895 | −9.3933 |
| 6 | 71.43 ± 1.8 | 52.95 ± 1.3 | −12.7492 | −9.8450 |
| 7 | 64.92 ± 1.6 | 54.40 ± 1.9 | −7.8997 | −11.5605 |
| 8 | 88.67 ± 1.9 | 63.44 ± 2.2 | −14.6809 | −13.2676 |
In the present case we have studied the effect of substitution at position-2 of the 1,2-benzothiaze scaffolds. Our idea is that a change in substituent at position-2 should also be associated with a corresponding response in the enzyme inhibition activities of the molecule. Herein, the increased inhibition with the 3-chloro-benzyl substituent at position-2 (compound 8) has been found in line to our idea and suggests to bring about further changes in the newly proposed work in order to increase their medicinal applications. The concept of enzyme inhibition through docking, which holds significant importance in computational biology and drug discovery, assumes a central position in comprehending and manipulating complex biological processes. The present methodology utilizes molecular docking software as a means for predicting the binding interactions that occur between small molecules, commonly referred to as ligands, and target enzymes. The potential inhibitors are identified via a series of simulations by evaluating the geometry and energies of the ligand.42 The docking techniques provide the opportunity to gain valuable insights regarding complex process of enzyme inhibition in the diverse compounds and such approaches provide substantial guidance in the design and development of innovative pharmacophores.43 The synthesized molecules were docked with the respective PDB files of the AChE and BChE using free online software, and their binding scores were presented in Table 1. It was observed that the compound 8 exhibited highest docking score among the tested compounds. The docking interactions of the compound 8 against AChE and BChE are presented in Fig. 7 and 8, respectively. The hydrogen bond interactions were observed with Tyr70 and Tyr121 due to carbonyl and sulfonamide oxygen while pi–pi interactions were exhibited by 8 with Trp84, Trp279, Trp334 and Phe331 (Fig. 7). The compound 8 also showed a good score in docking studies and depicted remarkable interactions with the different amino acid residues located on the active site of the BChE. The oxygen atoms of the carbonyl and sulfonamide moieties depicted hydrogen bond interactions with Thr120 and Ser198, respectively. Similarly, pi–pi interactions were observed with His438, Tyr332 and Ala328 while pi–alkyl with Gly121, Phe329 and Thr120 as shown in Fig. 8.
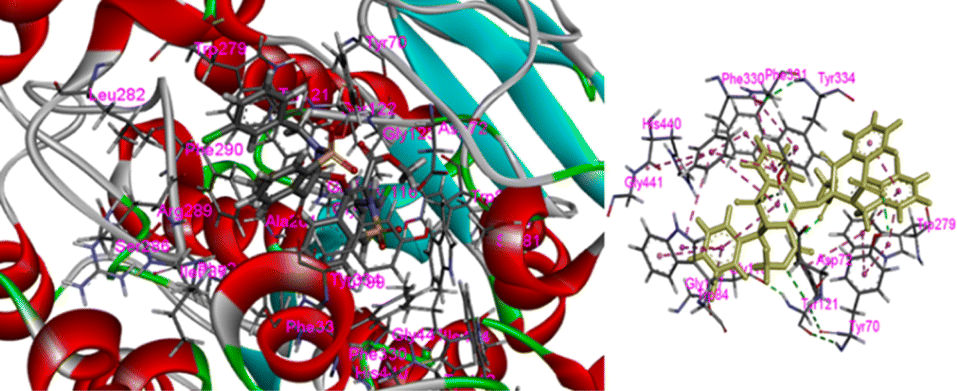 | ||
| Fig. 7 Best docking pose of the 8 on AChE. | ||
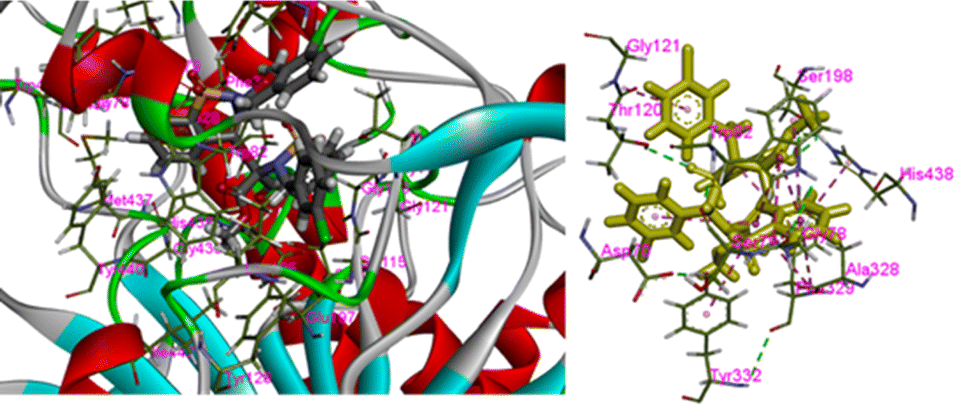 | ||
| Fig. 8 Best docking pose of the 8 on BChE. | ||
Experimental
Reagents and instruments used
The chemicals silver oxide, dimethyl sulfate, ethyl iodide, benzyl chloride and 3-chloro benzyl chloride were purchased from Sharlu, Spain. Chloro acetic acid from Daejung Korea. Sodium saccharin was purchased from the local market (Cuckoo brand, China). The solvents used in this research were distilled as per literature procedures. The precursors 1–4 were synthesized by adopting reported procedures.18–20,44 Bruker DRX (400 MHz), NMR spectrometer was used to record 1H NMR spectra at 25 °C. The ATR spectrophotometer (Prestige-21), Shimadzu, Japan was used to isolate FT-IR spectra. The Vario (EL-III) CHN analyser, Germany was used to determine elemental composition.Synthetic of compounds 5–8
Synthesis of dimer compounds was followed from the literature procedure.45 The equimolar quantities of the precursor 1–4 and silver(I) oxide were mixed in dry acetone (30 mL) and stirred for about 45 h at 65 °C. The silver mirror appeared around the flask; the yellow residue was obtained by evaporating. The obtained solids were washed with acetone/dichloromethane mixture to isolate pure compounds (Fig. S1†) and recrystallized to get white crystals (Scheme 1).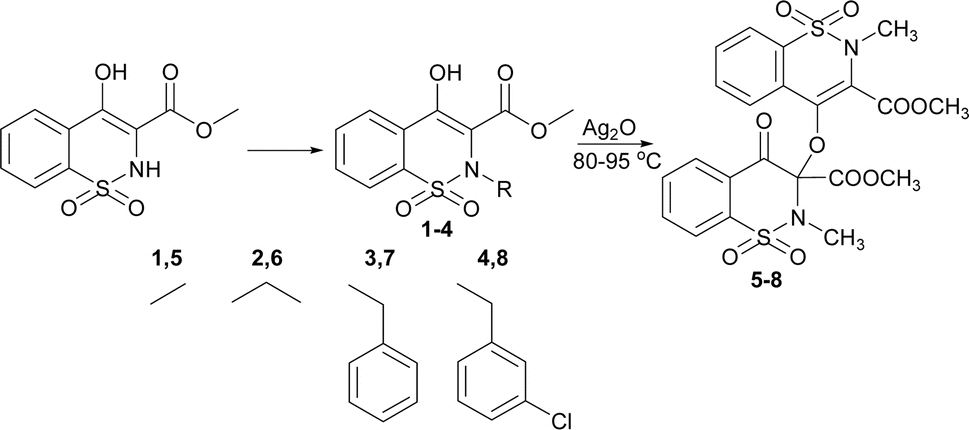 | ||
| Scheme 1 Synthetic route of the targeted compounds. | ||
The compound 5 was synthesized by adopting general procedure using 1 (5.0 g, 18.58 mmol) and silver(I) oxide (4.29 g, 18.58 mmol). Yield: 74% (4.01 g, white crystals), m. p.: 104–106 °C. Elemental analysis: found: C, 49.50; H, 3.81; N, 4.99; calcd for C22H20N2O10S2: C, 49.25; H, 3.76; N, 5.22. FT-IR (KBr disc, cm−1): 2956, 2359 (CH str.), 1778, 1745 (C
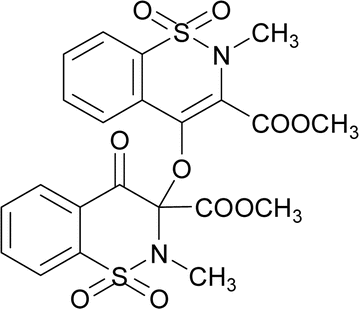
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O str.), 1328, 1265 (C–O str.), 1192, 1160 (SO2), 1148 (C–N str.). 1H NMR (400.13 MHz, CDCl3, 25 °C): δ = 8.11–8.04 (m, 1H, H-5), 7.97–7.92 (m, 1H, H-6), 7.88–7.62 (m, 4H, H-5′, H-6′, H-7′, H-8′), 7.59–7.50 (m, 2H, H-7, H-8), 3.88 (s, 3H, H-12), 3.77 (s, 3H, H-12′), 3.05 (s, 3H, H-13), 2.99 (s, 3H, H-13′).
O str.), 1328, 1265 (C–O str.), 1192, 1160 (SO2), 1148 (C–N str.). 1H NMR (400.13 MHz, CDCl3, 25 °C): δ = 8.11–8.04 (m, 1H, H-5), 7.97–7.92 (m, 1H, H-6), 7.88–7.62 (m, 4H, H-5′, H-6′, H-7′, H-8′), 7.59–7.50 (m, 2H, H-7, H-8), 3.88 (s, 3H, H-12), 3.77 (s, 3H, H-12′), 3.05 (s, 3H, H-13), 2.99 (s, 3H, H-13′).The compound 6 was synthesized by adopting general procedure using 2 (5.0 g, 17.60 mmol) and silver oxide (4.06 g, 17.60 mmol). Yield: 60% (3.4 g, white crystals), m. p.: 137–139 °C. Elemental analysis: found: C, 51.09; H, 4.37; N, 4.79; calcd for C24H24N2O10S2: C, 51.06; H, 4.28; N, 4.96. FT-IR (KBr disc, cm−1): 2948, 2346 (CH str.), 1744, 1705 (C
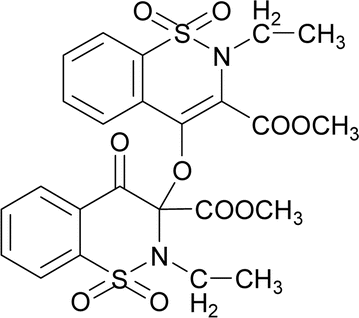 O str.), 1318, 1245 (C–O str.), 1162, 1140 (SO2), 1128 (C–N str.). 1H NMR (400.13 MHz, CDCl3, 25 °C): δ = 8.06 (dd, 3J(H5,H6) = 8 Hz, 4J(H5,H7) = 1 Hz, 1H, H-5), 8.03 (td, 3J(H7,H8)/(H7,H6) = 8 Hz, 4J(H7,H5) = 1 Hz, 1H, H-6), 7.94–7.82 (m, 2H, H-7, H-8), 7.83–7.65 (m, 4H, H-5′, H-6′, H-7′, H-8′), 3.94 (s, 3H, H-12), 3.80 (s, 3H, H-12′), 3.58 (d, 2J(H13α,H13β) = 14 Hz, 1H, H-13β), 3.54 (d, 2J(H13α,H13β) = 14 Hz, H-13α), 3.52 (d, 2J(H13′α,H13′β) = 14 Hz, 1H, H-13′β), 3.50 (d, 2J(H13′α,H13′β) = 14 Hz, H-13′α), 1.32–1.25 (m, 3H, H-14), 0.82–0.76 (m, 3H, H-14′).
O str.), 1318, 1245 (C–O str.), 1162, 1140 (SO2), 1128 (C–N str.). 1H NMR (400.13 MHz, CDCl3, 25 °C): δ = 8.06 (dd, 3J(H5,H6) = 8 Hz, 4J(H5,H7) = 1 Hz, 1H, H-5), 8.03 (td, 3J(H7,H8)/(H7,H6) = 8 Hz, 4J(H7,H5) = 1 Hz, 1H, H-6), 7.94–7.82 (m, 2H, H-7, H-8), 7.83–7.65 (m, 4H, H-5′, H-6′, H-7′, H-8′), 3.94 (s, 3H, H-12), 3.80 (s, 3H, H-12′), 3.58 (d, 2J(H13α,H13β) = 14 Hz, 1H, H-13β), 3.54 (d, 2J(H13α,H13β) = 14 Hz, H-13α), 3.52 (d, 2J(H13′α,H13′β) = 14 Hz, 1H, H-13′β), 3.50 (d, 2J(H13′α,H13′β) = 14 Hz, H-13′α), 1.32–1.25 (m, 3H, H-14), 0.82–0.76 (m, 3H, H-14′).The compound 7 was synthesized by adopting a general procedure using 3 (5.0 g, 14.50 mmol) and silver oxide (3.34 g, 14.50 mmol). Yield: 51% (3.99 g, white crystals), m. p.: 145–147 °C. Elemental analysis: found: C, 58.99; H, 4.07; N, 4.01.; calcd for C34H28N2O10S2: C, 59.29; H, 4.10; N, 4.07. FT-IR (KBr disc, cm−1): 2952 (CH str.), 1777, 1735 (C
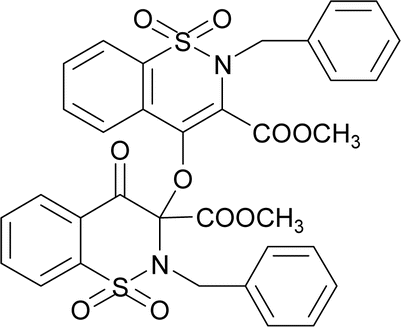 O str.), 1346, 1238 (C–O str.), 1168, 1109 (SO2), 1023 (C–N str.). 1H NMR (400.13 MHz, CDCl3, 25 °C): δ = 8.06 (dd, 3J(H5,H6) = 8 Hz, 4J(H5,H7) = 1 Hz, 1H, H-5), 7.85 (td, 3J(H7,H8)/(H7,H6) = 8 Hz, 4J(H7,H5) = 1 Hz, 1H, H-6), 7.78–7.72 (m, 2H, H-7, H-8), 7.70–7.63 (m, 2H, H-7′, H-8′), 7.35 (td, 3J(H7,H8)/(H7,H6) = 8 Hz, 4J(H7,H5) = 1 Hz, 1H, H-6′), 7.27 (dd, 3J(H5,H6) = 8 Hz, 4J(H5,H7) = 1 Hz, 1H, H-5), 7.27–7.15 (m, 5H, H-15, H-16,H-17,H-18, H-19), 7.05–6.95 (m, 3H, H-15′, H-16′, H-17′), 6.89–6.84 (m, 2H, H-18′, H-19′), 5.27 (d, 2J(H13α,H13β) = 14 Hz, 1H, H-13β), 4.94 (d, 2J(H13α,H13β) = 14 Hz, H-13α), 4.67 (d, 2J(H13′α,H13′β) = 14 Hz, 1H, H-13′β), 4.59 (d, 2J(H13′α,H13′β) = 14 Hz, H-13′α), 3.93 (s, 3H, H-12), 2.77 (s, 3H, H-12′).
O str.), 1346, 1238 (C–O str.), 1168, 1109 (SO2), 1023 (C–N str.). 1H NMR (400.13 MHz, CDCl3, 25 °C): δ = 8.06 (dd, 3J(H5,H6) = 8 Hz, 4J(H5,H7) = 1 Hz, 1H, H-5), 7.85 (td, 3J(H7,H8)/(H7,H6) = 8 Hz, 4J(H7,H5) = 1 Hz, 1H, H-6), 7.78–7.72 (m, 2H, H-7, H-8), 7.70–7.63 (m, 2H, H-7′, H-8′), 7.35 (td, 3J(H7,H8)/(H7,H6) = 8 Hz, 4J(H7,H5) = 1 Hz, 1H, H-6′), 7.27 (dd, 3J(H5,H6) = 8 Hz, 4J(H5,H7) = 1 Hz, 1H, H-5), 7.27–7.15 (m, 5H, H-15, H-16,H-17,H-18, H-19), 7.05–6.95 (m, 3H, H-15′, H-16′, H-17′), 6.89–6.84 (m, 2H, H-18′, H-19′), 5.27 (d, 2J(H13α,H13β) = 14 Hz, 1H, H-13β), 4.94 (d, 2J(H13α,H13β) = 14 Hz, H-13α), 4.67 (d, 2J(H13′α,H13′β) = 14 Hz, 1H, H-13′β), 4.59 (d, 2J(H13′α,H13′β) = 14 Hz, H-13′α), 3.93 (s, 3H, H-12), 2.77 (s, 3H, H-12′).The compound 8 was synthesized by adopting general procedure using 4 (5.0 g, 13.10 mmol) and silver oxide (3.02 g, 13.10 mmol). Yield: 53% (4.1 g, white crystals), m. p.:136–138 °C. Elemental analysis: found: C, 53.10; H, 3.41; N, 3.60 calcd. for C34H26Cl2N2O10S2: C, 53.90; H, 3.46; N, 3.70. FT-IR (KBr disc, cm−1): 2928 (CH str.), 1771, 1726 (C
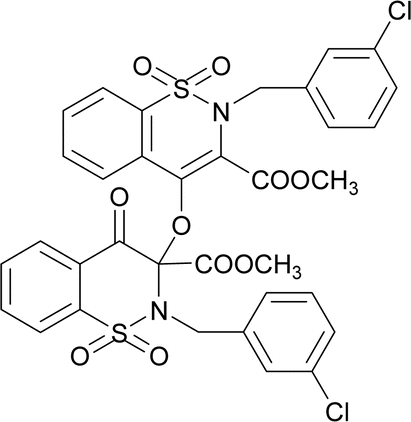 O str.), 1292, 1245 (C–O str.), 1164, 1111 (SO2), 1123 (C–N str.). 1H NMR (400.13 MHz, CDCl3, 25 °C): δ = 8.09 (dd, 3J(H5,H6) = 8 Hz, 4J(H5,H7) = 1 Hz, 1H, H-5), 7.88 (td, 3J(H7,H8)/(H7,H6) = 8 Hz, 4J(H7,H5) = 1 Hz, 1H, H-6), 7.80–7.75 (m, 2H, H-7, H-8), 7.74–7.65 (m, 2H, H-7′, H-8′), 7.42 (td, 3J(H7,H8)/(H7,H6) = 8 Hz, 4J(H7,H5) = 1 Hz, 1H, H-6′), 7.29 (dd, 3J(H5,H6) = 8 Hz, 4J(H5,H7) = 1 Hz, 1H, H-5′), 7.24–7.15 (m, 4H, H-15, H-17, H-18, H-19), 7.05–6.95 (m, 2H, H-15′, H-17′), 6.93–6.89 (m, 1H, H-18′), 6.79–6.74 (m, 1H, H-19′), 5.24 (d, 2J(H13α,H13β) = 14 Hz, 1H, H-13β), 4.88 (d, 2J(H13α,H13β) = 14 Hz, H-13α), 4.74 (d, 2J(H13′α,H13′β) = 14 Hz, 1H, H-13′β), 4.68 (d, 2J(H13′α,H13′β) = 14 Hz, H-13′α), 3.96 (s, 3H, H-12), 2.91 (s, 3H, H-12′).
O str.), 1292, 1245 (C–O str.), 1164, 1111 (SO2), 1123 (C–N str.). 1H NMR (400.13 MHz, CDCl3, 25 °C): δ = 8.09 (dd, 3J(H5,H6) = 8 Hz, 4J(H5,H7) = 1 Hz, 1H, H-5), 7.88 (td, 3J(H7,H8)/(H7,H6) = 8 Hz, 4J(H7,H5) = 1 Hz, 1H, H-6), 7.80–7.75 (m, 2H, H-7, H-8), 7.74–7.65 (m, 2H, H-7′, H-8′), 7.42 (td, 3J(H7,H8)/(H7,H6) = 8 Hz, 4J(H7,H5) = 1 Hz, 1H, H-6′), 7.29 (dd, 3J(H5,H6) = 8 Hz, 4J(H5,H7) = 1 Hz, 1H, H-5′), 7.24–7.15 (m, 4H, H-15, H-17, H-18, H-19), 7.05–6.95 (m, 2H, H-15′, H-17′), 6.93–6.89 (m, 1H, H-18′), 6.79–6.74 (m, 1H, H-19′), 5.24 (d, 2J(H13α,H13β) = 14 Hz, 1H, H-13β), 4.88 (d, 2J(H13α,H13β) = 14 Hz, H-13α), 4.74 (d, 2J(H13′α,H13′β) = 14 Hz, 1H, H-13′β), 4.68 (d, 2J(H13′α,H13′β) = 14 Hz, H-13′α), 3.96 (s, 3H, H-12), 2.91 (s, 3H, H-12′).Density functional theory assessments
DFT-based geometry optimization for compounds 5–8 was conducted utilizing the B3LYP46 hybrid functional, which combines Becke's 3-parameter exchange functional with Lee, Yang, and Parr's correlation functional. The calculations were performed employing the LANL2DZ basis set,47 which includes dispersion correction of Grimme's DFT-D3 approach along with Becke-Johnson damping.48 Additionally, stringent convergence criteria for both self-consistent field (SCF) energies and the electron density were applied throughout all computational steps. In the context of DFT calculations, the initial geometries for compounds 5–8 were derived from their respective crystal structures. Subsequent geometry optimizations of these complexes were conducted in the gas phase, employing the B3LYP/LANL2DZ level of theory. The ground state structure was verified through frequency analysis, which resulted in positive vibrational frequencies and hence was found to be stable. In Fig. S14† visual representations of the optimized structures for compounds 5–8 are provided.The compounds 5–8 went through various analyses, including Nuclear Magnetic Resonances, Infrared spectra, Frontier Molecular Orbitals, Natural Bond Orbitals, Molecular Electrostatic Potentials, Global Reactivity Descriptors, Dipole Moments and Linear Polarizabilities. All theoretical calculations were performed with the Turbomole 7.0.1 program package and GAMESS software.49,50 Therefore, the Avogadro51 computer program facilitated the input and output files and results. Theoretical FT-IR data for compounds 5–8 were obtained in the gas phase using the B3LYP/LANL2DZ level of theory however theoretical chemical shift values for 1H NMR were computed using the same level of theory with the PCM method in DMSO. Finally, Multiwfn v3.3.9 was utilized to process raw data,52 and the results were corroborated using GaussSum v2.2.53
Acetylcholine and butyrylcholine esterase inhibition
In vitro assessment of the compounds 5–8 was performed using spectrophotometric method.54 The 100 μL enzyme (acetylcholine and butyrylcholine esterase) was mixed with the compound (100 μL) and waited for 10 minutes. The respective substrate of 50 μL and DTNB of 50 μL were added, followed by the addition of 1000 μL of phosphate buffer. The mixture was allowed to incubate at 37 °C for thirty minutes. The absorbance was measured at 405 nm with a UV/VIS spectrophotometer against positive and negative blank. The percentage inhibition was checked according to reputed formulae.55,56Docking studies
Docking studies for the compounds 5–8 were performed with online free software according to the reported method.57–60 The AChE and BChE with PDB codes 1EVE and 1P0I, respectively were utilized for docking purposes. The 2D ligand enzyme interactions were established via the ligand interaction module. The Discovery Studio Visualizer was utilized to view the results of docking studies including graphical surface and interactions.Conclusions
1,2-Benzothiazines are the core nuclei of the well-known NSAIDs which are capable of showing a variety of bioactivities. The derivatives of the 1,2-benzothiazine are utilized to synthesize benzothiazine-based dimers in the presence of silver dioxide. The synthesized 1,2-benzothiazine dimers were characterized with state-of-the-art techniques and molecular structures of all the dimers were confirmed by X-ray diffraction analysis. The molecular structures of 5–8 showed that the supramolecular assembly is stabilized through C–H⋯O bonding. The Hirshfeld analysis confirmed the presence of intermolecular interactions and showed that H⋯O contacts are the key contributor to the stabilization of the supramolecular assembly of compound 5 whereas in the rest of compounds, H⋯H contact is the largest contributor. Void analysis showed that the compounds are expected to bear a significant amount of pressure. The interaction energy calculations showed that the major contribution to the stabilization of the supramolecular assembly comes from dispersion energy in the case of compounds 5–8. Quantum chemical investigations were conducted for the compounds 5–8, and a remarkable concordance was observed between the computed results and experimental data. This alignment encompassed various aspects, including structural parameters, NMR behaviour, and IR analysis. Furthermore, the outcomes of studies involving Natural Bond Orbitals (NBO), and global reactivity descriptors consistently pointed toward the enhanced stability of compounds 5–8. Moreover, the substantial band gap, as revealed during Frontier Molecular Orbital (FMO) analysis, wherein a significant energy gap between the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO) was observed, strongly implied that these complexes have the potential to serve as effective agents in biological applications.Author contributions
Writing – original draft; M. F, A. A. Methodology, W. A. S., formal analysis, M. I. C., S. N., M. A., M. N. T., S. M. A., supervision, W. A. S., M. I. C., conceptualization; A. A., writing – review & editing; M. A. R., S. M. A., visualization; software, M. A. R., M. N. T. validation; A. A.Conflicts of interest
All the authors declare no conflict of interest.Acknowledgements
The authors are grateful to the Researchers Supporting Project Number (RSP2024R388) for funding this research, King Saud University, Riyadh, Saudi Arabia. The authors thank the Higher Education Commission of Pakistan for IRSIP grant to M. F.Notes and references
- M. Pohanka, Bratisl. Lek. Listy, 2013, 114, 726–734 CAS.
- M. Pohanka, Biomedical Papers of the Medical Faculty of Palacky University in Olomouc, 2011, vol. 155 Search PubMed.
- L. S. Schneider, Dialogues in clinical neuroscience, 2000, vol. 2, pp. 111–128 Search PubMed.
- D. M. Quinn, Chem. Rev., 1987, 87, 955–979 CrossRef CAS.
- M. Brufani and L. Filocamo, Alzheimer Disease: from Molecular Biology to Therapy, 1997, pp. 171–177 Search PubMed.
- D. Nachmansohn and I. B. Wilson, Adv. Enzymol. Relat. Areas Mol. Biol., 1951, 12, 259–339 CAS.
- M. B. Colovic, D. Z. Krstic, T. D. Lazarevic-Pasti, A. M. Bondzic and V. M. Vasic, Curr. Neuropharmacol., 2013, 11, 315–335 CrossRef CAS PubMed.
- P. Taylor and S. Lappi, Biochemistry, 1975, 14, 1989–1997 CrossRef CAS PubMed.
- F. J. Carvajal and N. C. Inestrosa, Front. Mol. Neurosci., 2011, 4, 19 CAS.
- S. C. Peitzika and E. Pontiki, Molecules, 2023, 28, 1084 CrossRef CAS PubMed.
- U. S. Ali, W. A. Siddiqui, A. Ashraf, M. A. Raza, K. M. Batoo, M. Imran, S. E. Shirsath, M. Ashfaq, M. N. Tahir and S. Niaz, J. Mol. Struct., 2024, 137824 CrossRef CAS.
- F. Aman, A. M. Asiri, W. A. Siddiqui, M. N. Arshad, A. Ashraf, N. S. Zakharov and V. A. Blatov, CrystEngComm, 2014, 16, 1963–1970 RSC.
- M. Naeem, A. Ashraf, M. Imran, W. A. Siddiqui, G. Muhammad, A. Saleem, U. Younas, F. Ali, M. A. Raza and L. Mitu, Comments Inorg. Chem., 2024, 1–25 CrossRef CAS.
- A. Imani, S. Soleymani, R. Vahabpour, Z. Hajimahdi and A. Zarghi, Iran. J. Pharm. Res., 2021, 20, 1 CrossRef CAS PubMed.
- R. Reetu, R. Gujjarappa and C. C. Malakar, Asian J. Org. Chem., 2022, 11, e202200163 CrossRef CAS.
- S. S. Panga, R. Tamatam, P. Adivireddy, P. Venkatapuram, S. K. Narra and K. Paturu, Res. Chem. Intermed., 2019, 45, 3053–3075 CrossRef CAS.
- W. D. Guerra, D. Lucena-Agell, R. Hortigüela, R. A. Rossi, J. Fernando Diaz, J. M. Padron and S. M. Barolo, ChemMedChem, 2021, 16, 3003–3016 CrossRef CAS PubMed.
- A. Ashraf, S. A. Ejaz, S. U. Rahman, W. A. Siddiqui, M. N. Arshad, J. Lecka, J. Sévigny, M. E. M. Zayed, A. M. Asiri and J. Iqbal, Eur. J. Med. Chem., 2018, 159, 282–291 CrossRef CAS PubMed.
- A. Ashraf, F. Aman, S. Movassaghi, A. Zafar, M. Kubanik, W. A. Siddiqui, J. Reynisson, T. Söhnel, S. M. Jamieson and M. Hanif, Organometallics, 2019, 38, 361–374 CrossRef CAS.
- F. Aman, M. Hanif, W. A. Siddiqui, A. Ashraf, L. K. Filak, J. h. Reynisson, T. Söhnel, S. M. Jamieson and C. G. Hartinger, Organometallics, 2014, 33, 5546–5553 CrossRef CAS.
- S. Zainab, W. A. Siddiqui, M. A. Raza, A. Ashraf, M. Pervaiz, F. Ali, U. Younas, A. Saleem, M. Ashfaq and M. N. Tahir, J. Mol. Struct., 2023, 1284, 135316 CrossRef CAS.
- W. A. S. Shehnaz, A. Ashraf, M. Ashfaq, M. N. Tahir and S. Niaz, Appl. Organomet. Chem., 2023, 37, e7077 CrossRef CAS.
- W. A. Siddiqui, M. A. Raza, A. Ashraf, M. Ashfaq, M. N. Tahir and S. Niaz, J. Mol. Struct., 2023, 136603 Search PubMed.
- M. N. Arshad, O. Şahin, M. Zia-ur-Rehman, M. Shafiq, I. U. Khan, A. M. Asiri, S. B. Khan and K. A. Alamry, J. Chem. Crystallogr., 2013, 43, 671–676 CrossRef CAS.
- D. Krishna Swaroop, N. Ravi Kumar, P. Nagender, G. Jitender Dev, N. Jagadeesh Babu and B. Narsaiah, Eur. J. Org. Chem., 2018, 2018, 413–417 CrossRef CAS.
- P. R. Spackman, M. J. Turner, J. J. McKinnon, S. K. Wolff, D. J. Grimwood, D. Jayatilaka and M. A. Spackman, J. Appl. Crystallogr., 2021, 54, 1006–1011 CrossRef CAS PubMed.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19–32 RSC.
- A. R. Raza, S. L. Rubab, M. Ashfaq, Y. Altaf, M. N. Tahir, M. F. u. Rehman, T. Aziz, M. Alharbi and A. F. Alasmari, Molecules, 2023, 28, 5024–5044 CrossRef CAS PubMed.
- S. M. A. Mashhadi, M. H. Bhatti, E. Jabeen, U. Yunus, M. Ashfaq, M. Akhtar, M. N. Tahir, S. M. Alshehri, S. Ahmed and S. C. Ojha, ACS Omega, 2023, 8, 30186–30198 CrossRef CAS PubMed.
- J. J. McKinnon, D. Jayatilaka and M. A. Spackman, MedChemComm, 2007, 3814–3816, 10.1039/B704980C.
- E. Alaman, A. A. Ağar, M. N. Tahir, M. Ashfaq, E. B. Poyraz and N. Dege, J. Struct. Chem., 2023, 64, 1314–1328 CrossRef CAS.
- A. N. Malik, M. N. Tahir, A. Ali, M. Ashfaq, M. Ibrahim, A. E. Kuznetsov, M. A. Assiri and M. Y. Sameeh, ACS Omega, 2023, 8, 25034–25047 CrossRef CAS PubMed.
- C. Jelsch, K. Ejsmont and L. Huder, IUCrJ, 2014, 1, 119–128 CrossRef CAS PubMed.
- M. J. Turner, J. J. McKinnon, D. Jayatilaka and M. A. Spackman, CrystEngComm, 2011, 13, 1804–1813 RSC.
- Q. M. Aliyeva, M. N. Tahir, M. Ashfaq, K. S. Munawar, S. Y. Rahmanova, U. M. Hasanova, A. A. Rustamova, H. F. Mammadova and E. M. Movsumov, J. Struct. Chem., 2023, 64, 995–1006 CrossRef CAS.
- O. Simsek, M. Ashfaq, M. N. Tahir, S. Ozturk and E. Agar, J. Struct. Chem., 2023, 64, 942–953 CrossRef CAS.
- C. F. Mackenzie, P. R. Spackman, D. Jayatilaka and M. A. Spackman, IUCrJ, 2017, 4, 575–587 CrossRef CAS PubMed.
- M. J. Turner, S. Grabowsky, D. Jayatilaka and M. A. Spackman, J. Phys. Chem. Lett., 2014, 5, 4249–4255 CrossRef CAS PubMed.
- M. Kurbanova, M. Ashfaq, M. N. Tahir, A. Maharramov, N. Dege, N. Ramazanzade and E. B. Cinar, J. Struct. Chem., 2023, 64, 437–449 CrossRef CAS.
- M. Kurbanova, M. Ashfaq, M. N. Tahir, A. Maharramov, N. Dege and A. Koroglu, J. Struct. Chem., 2023, 64, 302–313 CrossRef CAS.
- V. Poroikov, Biochem. (Mosc.) Suppl. B: Biomed. Chem., 2020, 14, 216–227 CrossRef.
- T. Xu, H. Zhao, M. Wang, A. Chow and M. Fang, Anal. Chem., 2021, 93, 3072–3081 CrossRef CAS PubMed.
- W. Hussain, N. Rasool and Y. D. Khan, Curr. Drug Discovery Technol., 2021, 18, 463–472 CrossRef CAS.
- N. Ahmad, M. Zia-ur-Rehman, H. L. Siddiqui, M. F. Ullah and M. Parvez, Eur. J. Med. Chem., 2011, 46, 2368–2377 CrossRef CAS PubMed.
- M. S. Park and S. K. Kwon, Bull. Korean Chem. Soc., 2000, 21, 1249–1250 CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- R. Ahlrichs, M. Bär, M. Häser, H. Horn and C. Kölmel, Chem. Phys. Lett., 1989, 162, 165–169 CrossRef CAS.
- G. M. Barca, C. Bertoni, L. Carrington, D. Datta, N. De Silva, J. E. Deustua, D. G. Fedorov, J. R. Gour, A. O. Gunina and E. Guidez, J. Chem. Phys., 2020, 152, 154102 CrossRef CAS PubMed.
- D. Marcus, H. Donald, E. Curtis and D. Lonie, J. Cheminf., 2012, 4, 17 Search PubMed.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- N. M. O'boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- M. Danish, M. A. Raza, H. Khalid, U. Iftikhar and M. N. Arshad, Appl. Organomet. Chem., 2021, 35, e6033 CrossRef CAS.
- M. A. Raza, K. Fatima, Z. Saqib, J. K. Maurin and A. Budzianowski, J. Mol. Struct., 2019, 1195, 712–722 CrossRef CAS.
- M. A. Raza, S. H. Sumrra, K. Javed, Z. Saqib, J. K. Maurin and A. Budzianowski, J. Mol. Struct., 2020, 1219, 128609 CrossRef.
- M. A. Raza and K. Fatima, J. Phys. Org. Chem., 2020, 33, e4076 CrossRef CAS.
- M. Danish, A. Bibi, M. A. Raza, M. N. Arshad and A. M. Asiri, J. Mol. Struct., 2022, 1259, 132711 CrossRef CAS.
- M. Danish, A. Bibi, M. A. Raza, N. Noreen, M. N. Arshad and M. A. Aisri, Acta Chim. Slov., 2020, 67, 785–798 CrossRef CAS.
- N. Dege, M. A. Raza, O. E. Doğan, T. Ağar and M. W. Mumtaz, J. Iran. Chem. Soc., 2021, 18, 2345–2368 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2295137–2295140. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02009j |
| This journal is © The Royal Society of Chemistry 2024 |