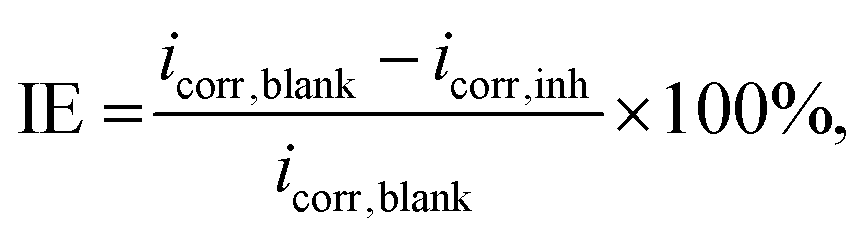DOI:
10.1039/D4RA02159B
(Paper)
RSC Adv., 2024,
14, 11157-11168
A data-driven QSPR model for screening organic corrosion inhibitors for carbon steel using machine learning techniques†
Received
21st March 2024
, Accepted 2nd April 2024
First published on 8th April 2024
Abstract
Machine learning (ML) techniques have shown great potential for screening corrosion inhibitors. In this study, a data-driven quantitative structure–property relationship (QSPR) model using the gradient boosting decision tree (GB) algorithm combined with the permutation feature importance (PFI) technique was developed to predict the corrosion inhibition efficiency (IE) of organic compounds on carbon steel. The results showed that the PFI method effectively selected the molecular descriptors most relevant to the IE. Using these important molecular descriptors, an IE predictive model was trained on a dataset encompassing various categories of organic corrosion inhibitors for carbon steel, achieving RMSE, MAE, and R2 of 6.40%, 4.80%, and 0.72, respectively. The integration of GB with PFI within the ML workflow demonstrated significantly enhanced IE predictive capability compared to previously reported ML models. Subsequent assessments involved the application of the trained model to drug-based corrosion inhibitors. The model demonstrates robust predictive capability when validated on available and our own experimental results. Furthermore, the model has been employed to predict IE for more than 1500 drug compounds, suggesting five novel drug compounds with the highest predicted IE on carbon steel. The developed ML workflow and associated model will be useful in accelerating the development of next-generation corrosion inhibitors for carbon steel.
1. Introduction
Carbon steel is the most widely used metallic material in industry owing to its unique mechanical properties, availability, and low cost.1 However, the significant weakness of carbon steel is its poor corrosion resistance when exposed to aggressive environments, such as acidic solutions, which are used for various processes such as cleaning, pickling, descaling, and well acidizing.1,2 To prevent the corrosion of carbon steel, different methods have been used, including the use of corrosion inhibitors. Organic corrosion inhibitors showed good efficiency and have great potential.3,4 However, their toxicity and environmental pollution are issues of great concern. The search for less toxic, environmentally friendly, and renewable corrosion inhibitors has become a research focus in this field.5
The search for new organic corrosion inhibitors involves a vast chemical space that includes millions of potential compounds. Experimental scientists often rely on insights from previous research to select potential compounds and conduct experiments. Such empirical trial-and-error studies are time-consuming and costly. To this end, computer-aided approaches have also been used, such as density functional theory (DFT) calculations, molecular dynamics (MD) simulation, and machine learning.6–9
Currently, ML techniques have shown great potential to generate IE predictive models for screening new inhibitors.10 Several ML models have been developed to predict the IE of organic compounds on carbon steel. Zhao et al.11 used the support vector machine (SVM) algorithm to build a QSPR model for predicting the IE of amino acids using a dataset of 19 compounds. The study used the inhibitor molecular quantum chemically-derived descriptors and adsorption energies on the Fe surface to predict the IE. With similar methods, Li et al.12 proposed a QSPR model for predicting the IE of benzimidazole derivatives on Q235 carbon steel using a dataset of 20 compounds. Ser et al.13 employed an artificial neural network (ANN) algorithm to construct an IE predictive model for pyridine and quinoline compounds on steel using a dataset of 40 compounds from the input data, including the inhibitor molecular descriptors and the adsorption energies on the Fe surface. Recently, Quadri et al. used an ANN algorithm to build an IE predictive model for pyridazines,14 ionic liquids,15 and pyrimidines16 on steel. In these studies, the input data includes five main descriptors selected from many quantum chemically-derived and cheminformatics-derived descriptors. As can be seen, the above studies focused on small datasets, and each dataset contains one or two categories of compounds. Moreover, the calculations for quantum chemically-derived descriptors such as frontier orbital energies and molecule–surface interaction energies are time-consuming and unsuitable for screening a large number of compounds.
Recently, Dai et al.17 proposed an IE predictive model for cross-category organic compounds on carbon steel based on a three-level direct message passing neural network (3L-DMPNN) model using a dataset containing 270 organic inhibitors with molecular structure information that integrates atomic-level, chemical bond-level, and molecular-level features. This 3L-DMPNN model showed good prediction performance, with a RMSE of 7.8%. The results also showed that cheminformatics-derived descriptors can be good input features for IE predictive models on large datasets because these descriptors can be calculated easily and quickly. However, a large number of descriptors are used without assessing the influence of each type of descriptor, making the model lack interpretability. Notably, some studies showed that the feature selection also affects the model's predictive performance. For example, Winkler et al.18,19 used a Bayesian regularized neural network with sparse Bayesian feature selection methods to determine the descriptors that are correlated with the IE from the large descriptor pool. Their results showed that the models with few selected descriptors performed best. Recent works by Li et al.20 and Schiessler et al.21 also showed that using a large number of descriptors gives less performance compared to choosing a suitable group of descriptors for machine learning models such as SVM, kernel ridge regression (KR)20 or deep learning models using NN.21 It has been shown that feature selection can increase the performance of the IE predictive model while at the same time improving its interpretability.
Moreover, within the realm of corrosion inhibition predictive models, it is noteworthy that while NN and SVM are commonly employed algorithms, other potent machine learning methodologies, such as GB, are underutilized. A recent investigation conducted by Akrom et al.,22 which assessed the predictive capabilities of multiple algorithms for IE in diazine derivatives using a dataset of 100 compounds, highlighted that the GB algorithm exhibited superior performance. This underscores the significance of algorithm selection in potentially enhancing the predictive performance of IE models.
In this study, an IE predictive model was developed by integrating the GB algorithm with the PFI method, utilizing a dataset comprising 317 organic inhibitors for carbon steel in a 1 M HCl solution. Initially, the importance of molecular descriptors was estimated by the PFI method. Subsequently, various models utilizing input datasets, incorporating distinct descriptor groups distinguished by the highest PFI index, were evaluated to identify the optimal descriptor group. Additionally, the performances of models utilizing molecular descriptors were compared with those employing molecular fingerprints or a combination of features, aiming to identify the most effective IE predictive model. The performance of the proposed ML workflow, integrating the GB algorithm and PFI method, was validated across different published datasets to assess its comparative performance against other ML models. Furthermore, the correlation between selected molecular descriptors and IE was scrutinized. Finally, the constructed IE predictive model was applied to prospectively screen potential inhibitors for carbon steel in 1 M HCl solution from drug compounds.
2. Materials and methods
The workflow employed for the construction of the IE predictive model in this study is illustrated in Fig. 1. This comprehensive workflow includes data collection, feature generation, feature selection, model selection, and prediction. The detailed descriptions of these processes are provided in the subsequent sections.
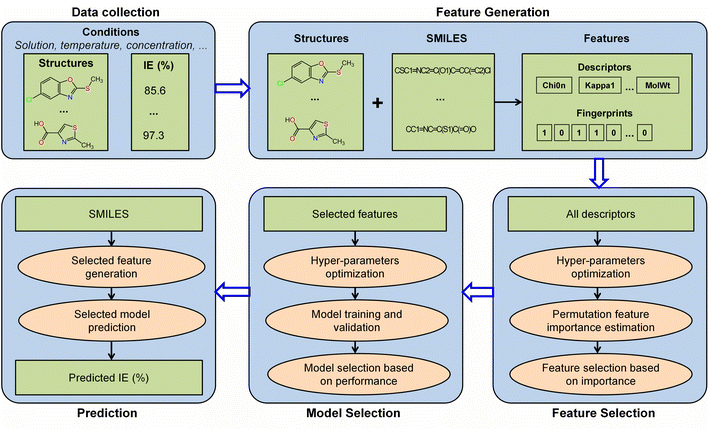 |
| | Fig. 1 Machine learning workflow for the construction of the IE predictive model. | |
2.1. Data
2.1.1. Experimental data. The primary aim of this study is to introduce an advanced ML workflow, coupled with an effective ML model, for the prediction of IE across diverse categories of organic substances. Hence, we combined two datasets constructed in recent publications14,17 to form a larger dataset encompassing diverse organic compound categories. This dataset (denoted as Fe–HCl-317) includes electrochemical impedance spectroscopy (EIS) measured IEs of 317 organic inhibitors for carbon steel in a 1 M HCl solution at 25–30 °C with a concentration of inhibitors of 1 mmol L−1. The distributions of the number of compounds according to IE and molecular weight are shown in Fig. S1 in ESI.† It can be seen that the IEs of the collected inhibitors are predominantly distributed between 60% and 100% (Fig. S1a†), while the molecular weights exhibit a predominant distribution within the range of 100 to 500 g mol−1 (Fig. S1b†). The details of the Fe–HCl-317 dataset are given in Data availability statements.
2.1.2. Molecular descriptors. In this study, 208 two-dimensional (2D) descriptors were employed, calculated using RDKit software23 and referred to as 2Ddes features, to characterize the molecular structure. The 2Ddes features include 4 partial charge descriptors, 4 molecular property descriptors, 22 topological and connectivity descriptors, 21 Lipinski descriptors, 59 MOE-type descriptors, 8 BCUT2D descriptors, 4 Estate descriptors, 85 constitutional descriptors, and 1 Quantitative Estimation of Drug-Likeness (QED) descriptor. The comprehensive list of 2Ddes feature set is given in Table S1 in ESI.†
2.1.3. Molecular fingerprints. Morgan fingerprints with a radius of 2 and a size of 2048 bit (equivalent to ECFP4 extended-connectivity fingerprints)24 were also used to represent the molecule. This feature group is denoted as ECFP4.
2.2. Methods
2.2.1. ML algorithm. In this study, the GB algorithm was used to build the machine learning model. GB is a powerful supervised ensemble learning algorithm. GB uses the gradient information from the existing weak learner to train the new weak learner, and then uses a sum function to aggregate the model prediction.25 The GB algorithm used in this study is integrated into GradientBoostingRegressor, a regression model in the scikit-learn Python package,26 using squared error as the loss function. In each iteration, GB calculates the gradient of the loss function with respect to the predictions made by the current ensemble. It then fits a new decision tree to the negative gradient, which is essentially the residual error left by the current model. In addition, we also compare the performance of GB with some other typical machine learning algorithms such as linear regression (LR), KR, and random forest (RF), which are integrated into LinearRegression, KernelRidge, and RandomForestRegressor models in scikit-learn, respectively.
2.2.2. Hyper-parameters turning. The grid search cross-validation (CV) method was used to select the optimal value for the model's hyper-parameters. This method uses a K-fold CV process to evaluate the model's performance on all possibilities in a discrete hyper-parameter space to search optimal values.27 Details of the K-fold CV method will be presented below. In this study, there are four hyper-parameters surveyed, including n_estimators, min_samples_split, min_samples_leaf, and max_depth. These are essential hyper-parameters of the model using the GB algorithm.28 The considered values and optimal values of hyper-parameters for the four typical models, which will be presented in the results section, are given in Table 1. The optimal values of hyper-parameters for other considered models are listed in Table S2 in ESI.†
Table 1 The considered and optimal values of hyper-parameters for models
| Hyper-parameter |
Considered values |
Optimal valuea |
| Model A |
Model B |
Model C |
Model D |
| Model A: GB/2Ddes, Model B: GB/ECFP4, Model C: GB/2Ddes40, Model D: GB/2Ddes40-ECFP4. These models are described below. |
| n_estimators |
50, 100, 200, 400, 600 |
600 |
200 |
400 |
200 |
| min_samples_split |
2, 3, 4, 5, 6 |
2 |
2 |
2 |
2 |
| min_samples_leaf |
1, 2, 3, 4, 5 |
5 |
5 |
2 |
5 |
| max_depth |
1, 2, 3, 4, 5 |
4 |
4 |
3 |
3 |
2.2.3. Feature importance. To optimize the predictive performance of the model, the permutation feature importance (PFI) method29 was employed for the selection of descriptors most correlated with inhibition efficiency. First, the hyper-parameters of the model using the 2Ddes features will be selected by the method presented above. Then, this model is used to estimate the importance of descriptors using the PFI method. In this method, the importance of a descriptor, denoted as the PFI index, is quantified by the reduction of the model's score when randomly shuffling the data for that descriptor while keeping the data for other descriptors unchanged. The permutation process was iterated 10 times for each descriptor in this study to establish the distribution range and mean values of the PFI index. Upon assessing the importance of molecular descriptors, datasets comprising the top N descriptors with the highest mean PFI indexes were employed to refine the model's performance. The model utilizing the selected top N descriptors is denoted as GB/2DdesN.
2.2.4. Evaluation metrics. The performance of the model is evaluated by using the 10-fold CV method based on three metrics: root-mean-square error (RMSE), mean absolute error (MAE), and coefficient of determination (R2). First, the initial data set will be shuffled randomly and then divided equally into ten folds in the order, using one of the ten folds as the evaluation set and the remaining 9 folds as the training set.The metrics RMSE, MAE, and R2 are determined by the following equations:
| |
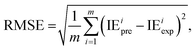 | (1) |
| |
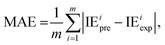 | (2) |
| |
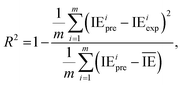 | (3) |
where IE
iexp and IE
ipre are the experimental and predicted IE of the
ith sample, respectively.
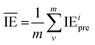
is the average value of the predicted IE.
2.2.5. Quantum chemical calculations. To validate the corrosion inhibition capability based on the electronic structure of the new compounds predicted by the machine learning model, the distributions and energies of highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were calculated on ORCA program30,31 using the B3LYP correlation-exchange functional32 and the Def2-TZVP basis set.33 The distributions of HOMO and LUMO were visualized using the VESTA software.34
2.2.6. Experimental measurements. To validate the prediction performance of the proposed model, we conducted electrochemical experiments to evaluate the IE of four drug compounds (vidarabine, cytarabine, chlorothiazide, and idoxuridine) on Q235 steel in a 1 M HCl solution with an inhibitor concentration of 1 mmol L−1. The experiments were performed by the PGSTAT30 workstation using a three-electrode cell with a platinum electrode as the counter electrode and Ag/AgCl (saturated KCl) as the reference electrode at room temperature. The working electrode was the research electrode, made of Q235 steel. The steel electrode was immersed in the solution at open circuit potential (OCP) for 1800 s before performing a polarization curve test within the potential range of EOCP ± 250 mV and a scan rate of 1 mV s−1. The corrosion current density (icorr) can be obtained by linear extrapolation of the anodic and cathodic Tafel lines. The IE is calculated as follows:| |
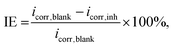 | (4) |
where icorr,blank and icorr,inh represent corrosion current densities in the absence and presence of inhibitors, respectively.
3. Results and discussion
3.1. Influence of molecular descriptors selection on model's performance
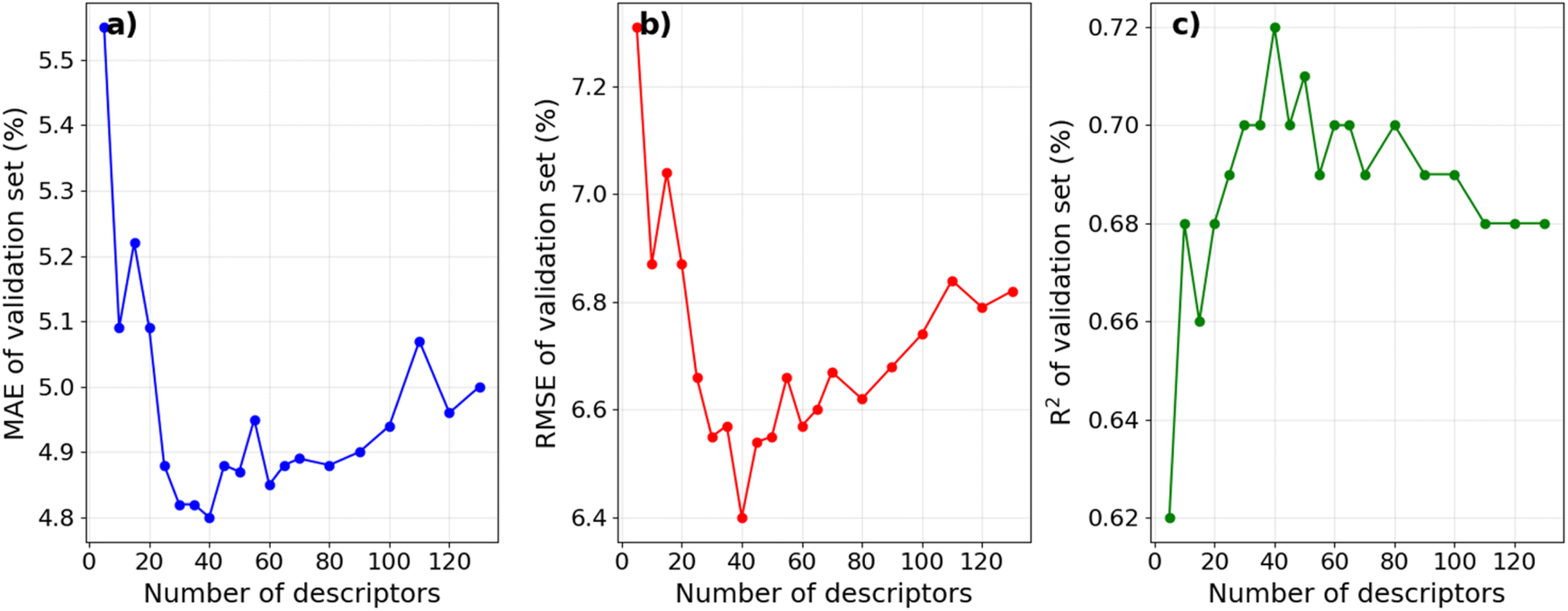
The influence of selecting molecular descriptors on the predictive performance of the model is illustrated in Fig. 2. It can be seen that the model's performance metrics exhibit significant variations with the number of selected descriptors. When employing less than the top 20 descriptors with the highest PFI indexes, the average MAE and RMSE values for the validation set in a 10-fold CV remain comparatively high, and the R2 value remains relatively low. This implies that an insufficient number of descriptors may hinder the model from capturing essential relationships influencing IE, indicating a state of under-fitting. Conversely, when the number of top descriptors exceeds 80, a notable decline in the model's predictive performance is observed, suggesting the potential inclusion of descriptors unrelated to IE in the dataset, leading to over-fitting.35,36 The model achieves its optimal predictive performance with approximately 40 descriptors. These findings align with the result of Li et al., who employed the recursive feature elimination method to select descriptors for an IE predictive model on an Mg alloy. Their results similarly indicated that a moderate number of descriptors is optimal for optimizing IE predictive performance.20
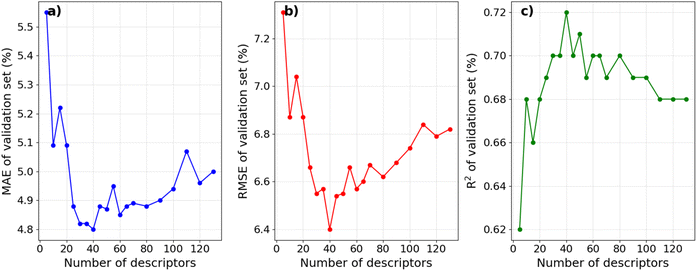 |
| | Fig. 2 Average MAE, RMSE, and R2 of the validation sets in the 10-fold CV depend on the number of selected top descriptors. | |
3.2. Influence of feature selection on model's performance
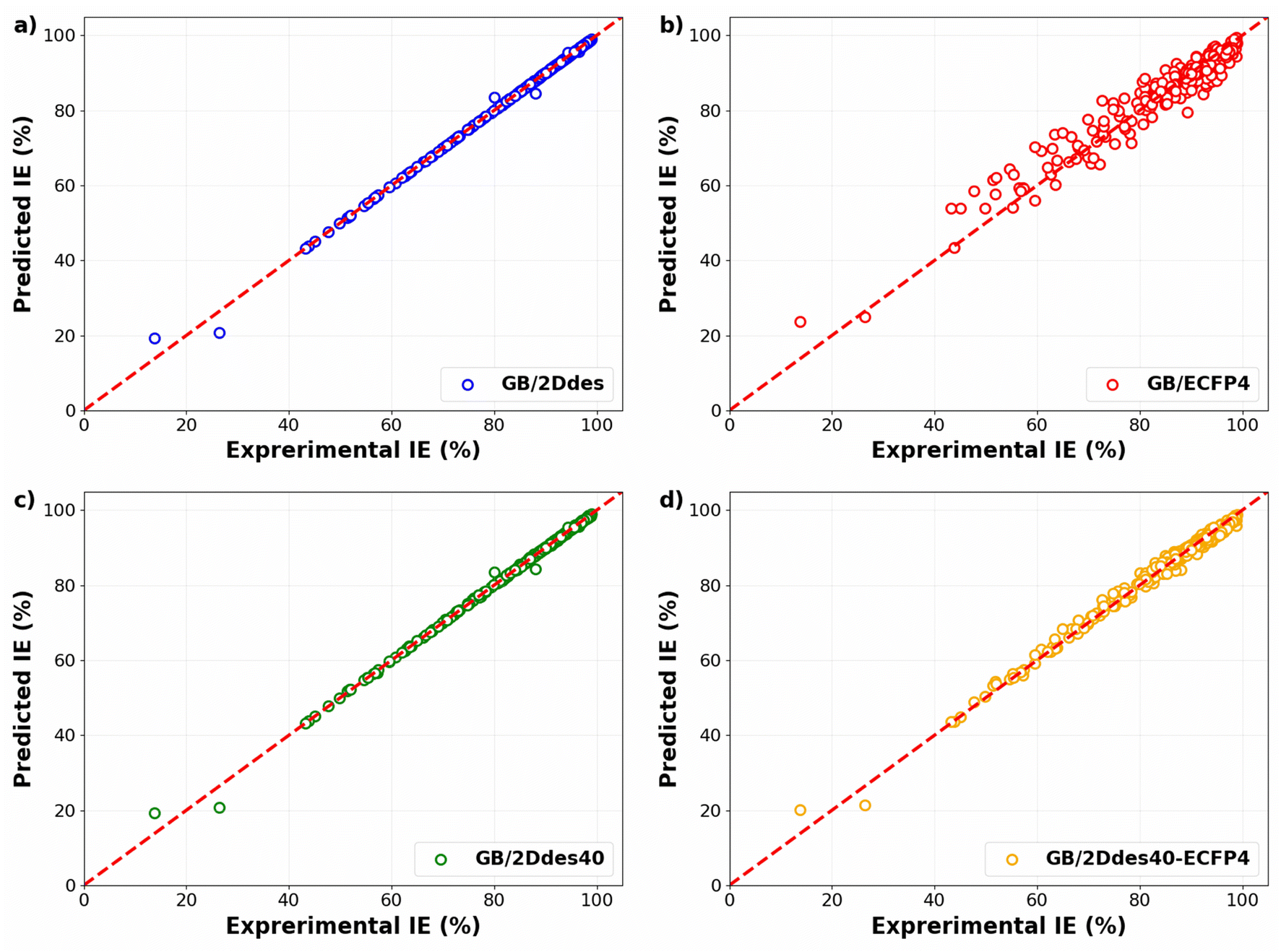
Fig. 3 compares the experimental IEs with the predicted IEs in the training sets of the 10-fold CV for four distinct models. These models are constructed on four different feature sets: 2D descriptors (GB/2Ddes model), ECFP4 fingerprints (GB/ECFP4 model), top 40 2D descriptors (GB/2Ddes40 model), and top 40 2D descriptors combined with ECFP4 fingerprints (GB/2Ddes40-ECFP4 model). It can be seen that all models exhibit commendable regression performance, with only the GB/ECFP4 model demonstrating slightly inferior performance compared to the other three models. However, it is crucial to emphasize that this result solely illustrates the regression performance on the training set and does not reflect the predictive performance of the models.
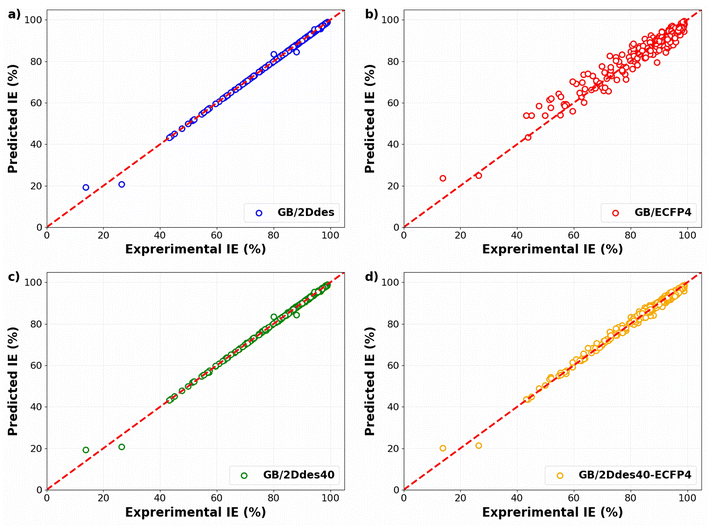 |
| | Fig. 3 Experimental versus predicted IEs in the training sets of the 10-fold CV as obtained by the models using different feature sets. | |
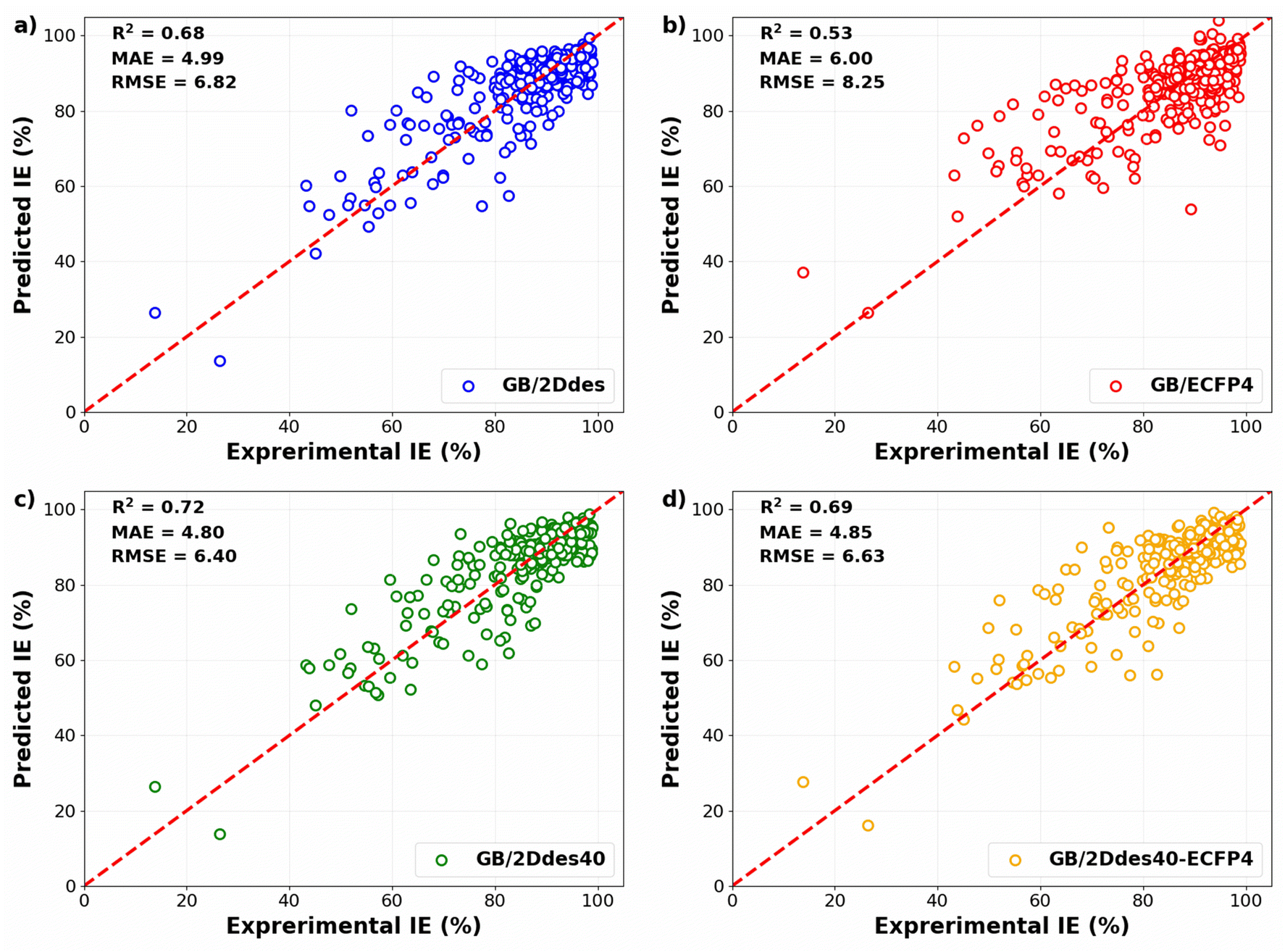
To evaluate the predictive capabilities of the models, the predicted IEs in the validation sets of the 10-fold cross-validation were compared with the corresponding experimental IEs, as illustrated in Fig. 4. All four models exhibit diminished errors at higher IE values, with more substantial errors observed at lower IE values. This observation may be attributed to the concentration of training data at high IE levels. A comparative analysis between Fig. 4a and b reveals that the utilization of molecular descriptors (the 2Ddes feature set) yields significantly superior performance compared to molecular fingerprints (the ECFP4 feature set). Notably, the model's predictive performance employing the top 40 descriptors (the 2Ddes40 feature set) outperformed others, achieving R2, MAE, and RMSE values of 0.72, 4.80%, and 6.40%, respectively (Fig. 4c). Furthermore, the model employing a feature set that combines the top 40 descriptors with molecular fingerprints (the 2Ddes40-ECFP4 feature set) was also examined. However, the performance of this model (Fig. 4d) was found to be inferior to that of the model utilizing only the top 40 descriptors.
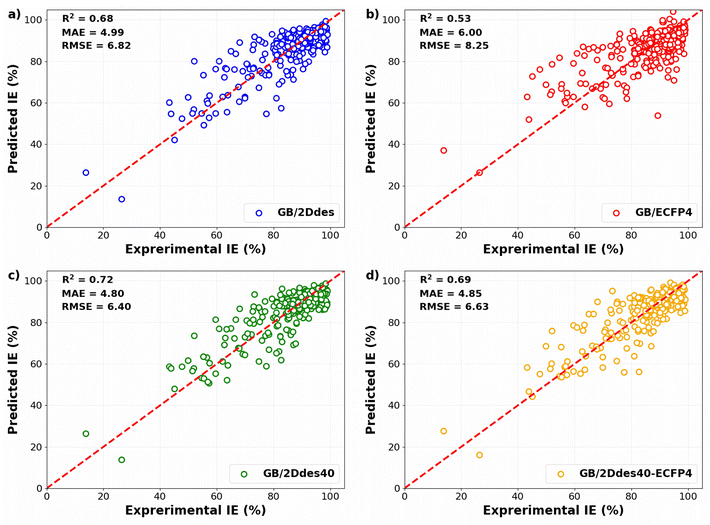 |
| | Fig. 4 Experimental versus predicted IEs in the validation set of the 10-fold CV obtained by the models using different feature sets. | |
Table 2 presents the performance of our model in comparison to the published ML models13–15,17,37 for predicting IE on carbon steel. We find that the GB/2Ddes40 model has better prediction performance than many published IE prediction models in the literature. It can be seen that feature selection is able to improve predictive accuracy on a small dataset, which is a challenge in predicting corrosion inhibition efficiency.38
Table 2 The comparison between the model performance of this work and the literature
| Dataset |
Number of compounds |
Validation method |
MAE (%) |
RMSE (%) |
R2 |
Ref. |
| Pyridines-quinolines |
41 |
5-Fold CV |
— |
16.74 |
— |
Ser et al.13 |
| Quinoxaline |
40 |
5-Fold CV |
— |
15.97 |
— |
Quadri et al.37 |
| Pyridazines |
20 |
5-Fold CV |
— |
14.69 |
— |
Quadri et al.14 |
| Ionic liquids |
30 |
5-Fold CV |
— |
10.01 |
— |
Quadri et al.15 |
| Organic compounds |
270 |
10-Fold CV |
5.30 |
7.82 |
0.41 |
Dai et al.17 |
| Organic compounds |
317 |
10-Fold CV |
4.80 |
6.40 |
0.72 |
This work |
3.3. Influence of ML algorithm
To compare the performance of different ML algorithms, we also conducted the same ML workflow for three typical algorithms, i.e., LR, KR, and RF. Table 3 shows that the GB algorithm has the best performance for the IE prediction compared to the remaining ones. This result agrees with the conclusion in a recent publication that the GB algorithm has the best performance when predicting the IE of diazine compounds.22
Table 3 The performance of the models using different ML algorithms
| ML algorithm |
Optimal feature set |
MAE (%) |
RMSE (%) |
R2 |
| LR |
2Ddes30 |
8.36 |
10.99 |
0.17 |
| KR |
2Ddes130 |
7.43 |
9.49 |
0.37 |
| RF |
2Ddes120 |
5.26 |
7.16 |
0.65 |
| GB |
2Ddes40 |
4.80 |
6.40 |
0.72 |
3.4. Feature importance and correlation
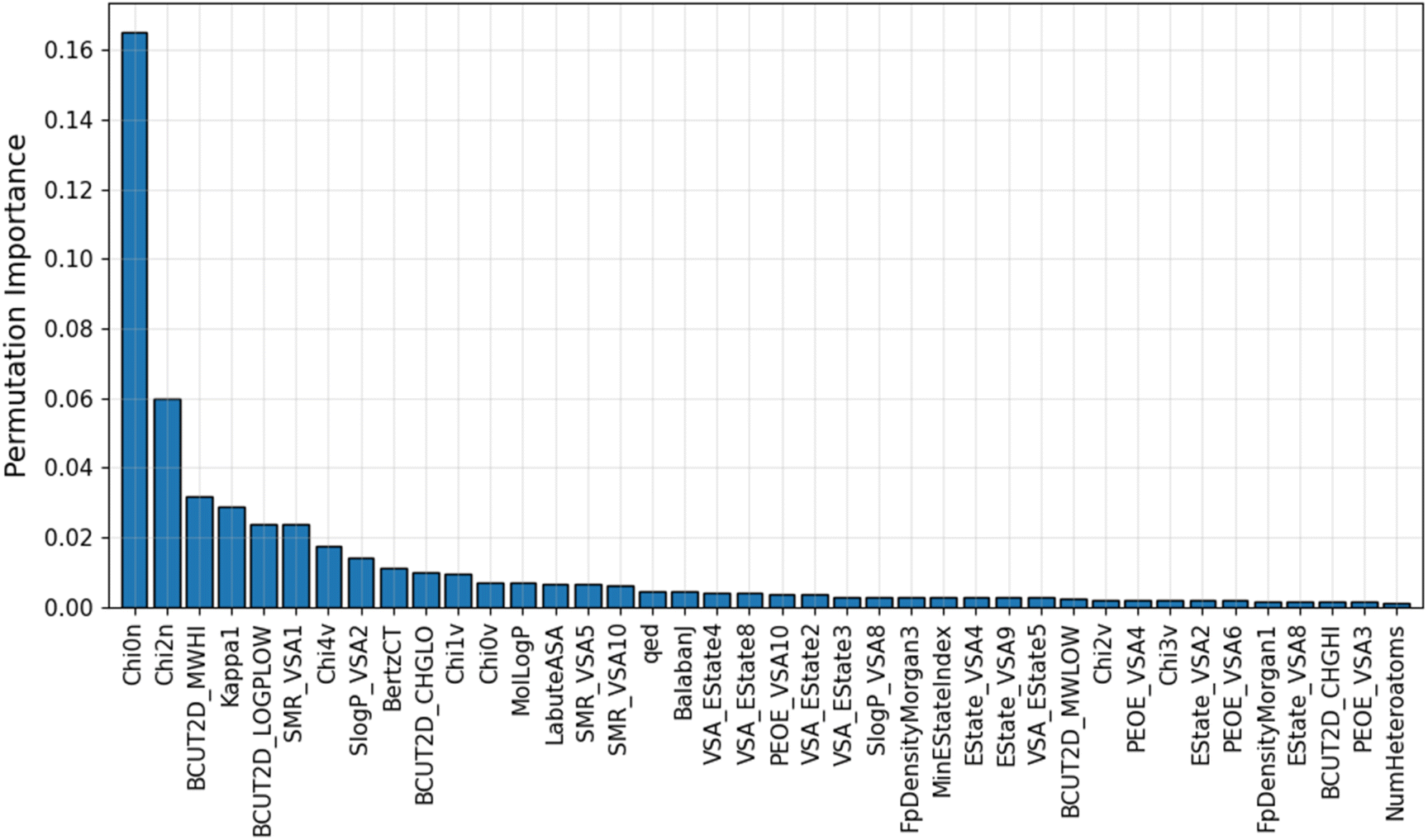
An advantage of the ML model developed in this study is that it uses a small optimal number of input features, which improves its predictive performance. This gives the IE predictive model better interpretability when characterizing these highly correlated input features. The PFI index of the top 40 most important descriptors used in the GB/2Ddes model is shown in Fig. 5. These descriptors include the topological type descriptors such as Chi, Kappa, BertCZ, and BalabanJ, the BCUT2D type descriptors, and the MOE type descriptors. Topological descriptors characterize the topological structure of the molecule but take into account the electronic character of the atoms in the molecule.39–41 The geometric structure and electronic properties are two important factors affecting the interaction between the molecule and the metal surface. Therefore, the topological descriptors are highly correlated with corrosion inhibition efficiency. The MOE descriptors represent the contribution of the sum vdW surface area (VSA) of the atoms to molecular properties such as molecular refraction (MR), octanol–water partition coefficient (logP), partial charges (PEOE), and electrotopological state (EState).42 MOE descriptors reflect many characteristics necessary to evaluate corrosion inhibition efficiency, such as hydrophilicity, hydrophobicity, polarity, electrostatic interaction, and steric effect.42,43 BCUT2D are also descriptors that show high efficiency when building QSAR/QSPR models thanks to their high diversity.39 Some research results showed that the BCUT2D descriptors can describe the interaction between molecules,44,45 and this may also be an important factor affecting the corrosion inhibition efficiency.
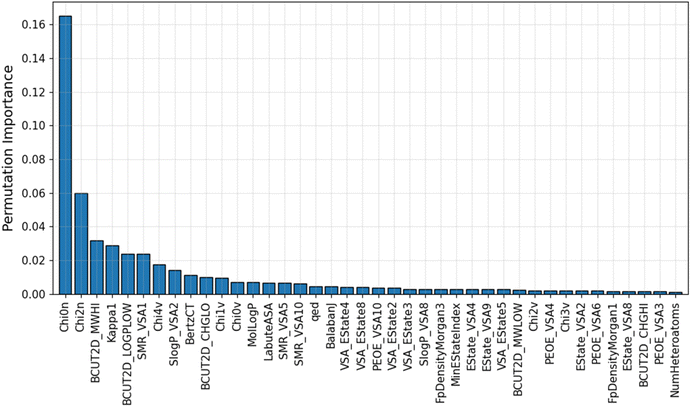 |
| | Fig. 5 Average PFI indexes of the top 40 most important descriptors after 10 permutations. | |
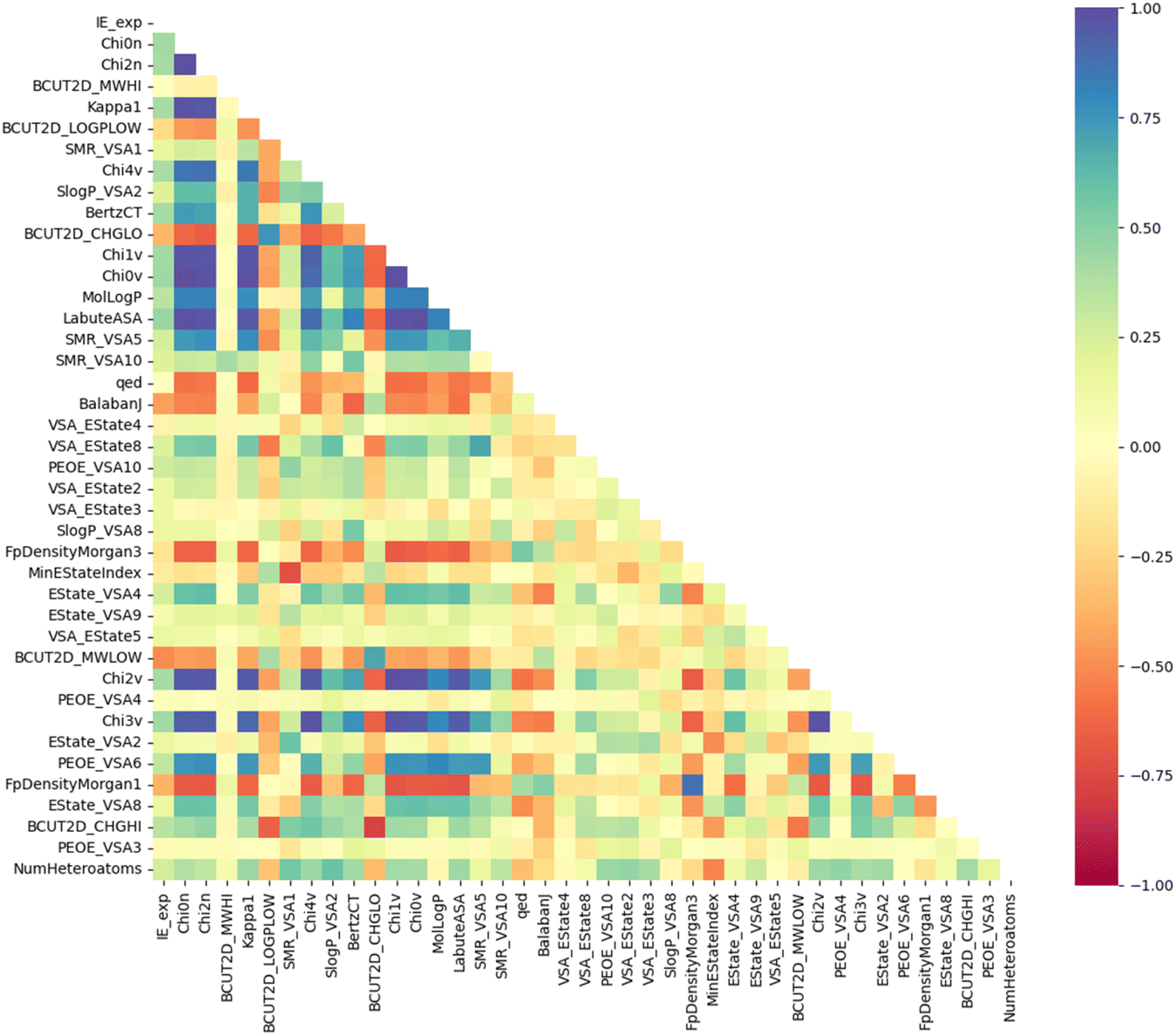
Moreover, Fig. 6 illustrates the Pearson correlation between the top 40 descriptors and experimental IE. It can be seen that most descriptors do not have a strong linear correlation with experimental IE, including descriptors with high PFI indexes such as Chi0n and Chi2n. The correlation between descriptors is also mainly concentrated at the weak correlation level. It can be seen that the IE has a non-linear correlation with the descriptors, and the synergy between a group of descriptors can create an effective prediction model.20
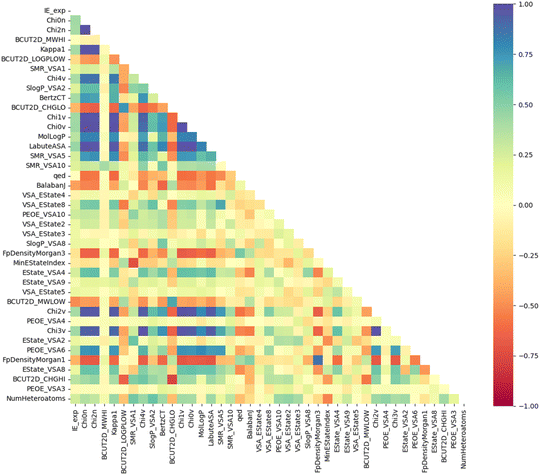 |
| | Fig. 6 Pearson correlation coefficients between the top 40 descriptors and IEs. | |
3.5. Testing on published datasets
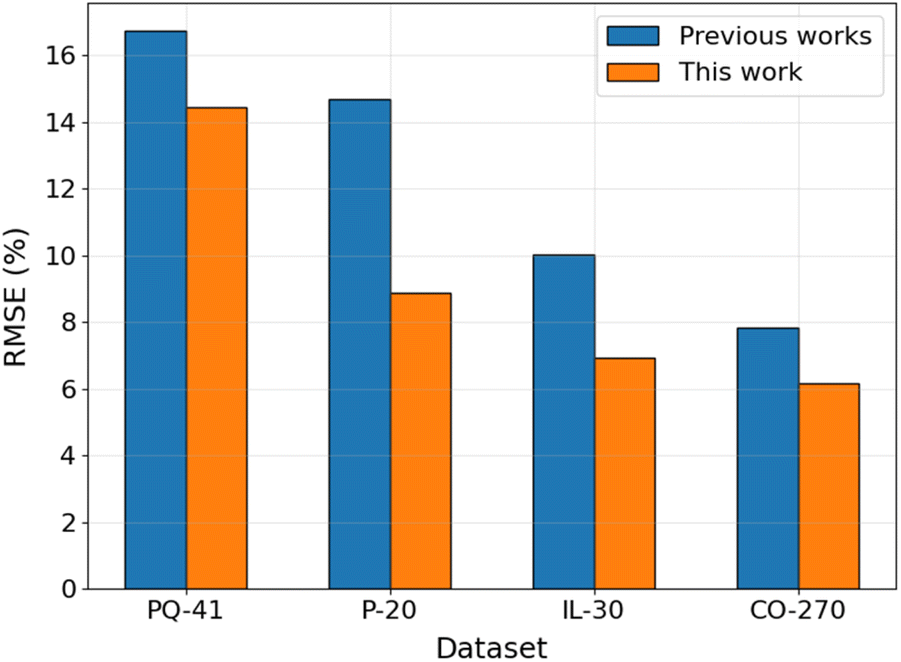
To assess the performance of the proposed ML workflow integrating the PFI method and the GB algorithm, its performance was examined on existing datasets to compare with the published ML models. The dataset characteristic, validation methodology, and ML algorithm details are listed in Table S3 in ESI.† The comparison of the model's RMSE is shown in Fig. 7. The results reveal that, when utilizing the same dataset and validation methodology, the model employing the GB algorithm and the top N descriptors with the highest PFI index (GB/2DdesN model) exhibits significantly superior predictive performance compared to published models, manifesting reductions in RMSE ranging from 14% to 39%. This improvement is evident not only on datasets comprising a limited number of compounds within the same category, such as the 41 pyridines and quinolines (PQ-41), the 20 pyridazines (P-20), and the 30 ionic liquids (IL-30) datasets, but also on larger datasets encompassing diverse types of organic compounds, exemplified by the 270 cross-category organic compounds (CO-270) dataset. Specifically, the utilization of the GB algorithm in conjunction with cheminformatics-derived descriptors selected by the PFI method outperforms the performance achieved by employing the NN algorithm with quantum chemically-derived descriptors and adsorption energies on the PQ-41 dataset.13 This superior performance extends to comparisons with the NN algorithm combined with five selected quantum chemically-derived and cheminformatics-derived descriptors on the P-20 and IL-30 datasets,14,15 as well as the integration of deep learning models, such as DMPNN, with cheminformatics-derived descriptors on the CO-270 dataset.17
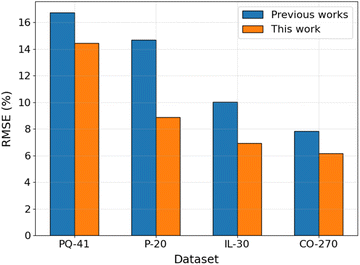 |
| | Fig. 7 Comparison of the performance of GB/2DdesN model with published models13–15,17 on different datasets. | |
Furthermore, Table S3† also presents the performance of models employing the GB algorithm with all molecular descriptors, without undergoing a feature selection step (GB/2Ddes models). The results reveal that the GB/2Ddes models exhibit commendable performance across three out of four datasets, with the only exception being a slightly lower performance than the GA-NN model on the PQ-41 dataset. This observation suggests that the GB algorithm stands as a robust choice for predicting IE when compared to other ML algorithms, consistent with the findings from a recent study by Akrom et al.22 Moreover, it is also evident that the application of the PFI feature selection method significantly enhances the performance of the GB algorithm across all four published datasets, similar to the findings observed on our Fe–HCl-317 dataset.
3.6. Model validation on drug compounds
A web tool named SMILES2IE-steel was developed using Streamlit (https://streamlit.io), a Python-based platform for developing web applications for machine learning and data science, using the GB/2Ddes40 model, which was trained on the Fe–HCl-317 dataset. This tool allows us to quickly predict the IE of organic inhibitors on carbon steel in a 1 M HCl solution by entering a list of molecular SMILES. The interface of SMILES2IE-steel is shown in Fig. S2 in ESI.†
The utilization of drug compounds as corrosion inhibitors has been a subject of extensive investigation over several years. These compounds exhibit potential as environmentally friendly and low-toxicity green inhibitors.46,47 Notably, pharmaceutical substances generally remain largely unaltered, even post the expiration date of most drugs, rendering them viable for application as corrosion inhibitors.48 The SMILES2IE-steel tool was employed to predict the IEs of ten previously published drug compounds. Fig. 8 (T1–T10 compounds) illustrates the close alignment between the IEs predicted by our model and the corresponding experimental values.49–58 Despite slight disparities in experimental conditions compared to the predicted conditions (detailed information listed in Table S4†), the results underscore the high reliability of the GB/2Ddes40 model in predicting IE for drug compounds. This robust predictive performance can be attributed to the structural similarities between many drug compounds and the compounds used to train the model, both being heterocyclic organic compounds containing elements such as N, O, and S.
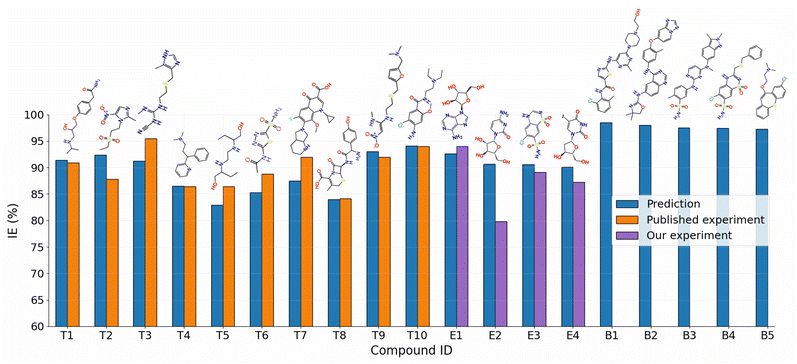 |
| | Fig. 8 Comparison of the predicted and experimental IEs for ten published drug compounds (T1–T10)49–58 and four unpublished drug compounds (E1–E4), along with the five new drug compounds (B1–B5) with the highest predicted IEs. | |
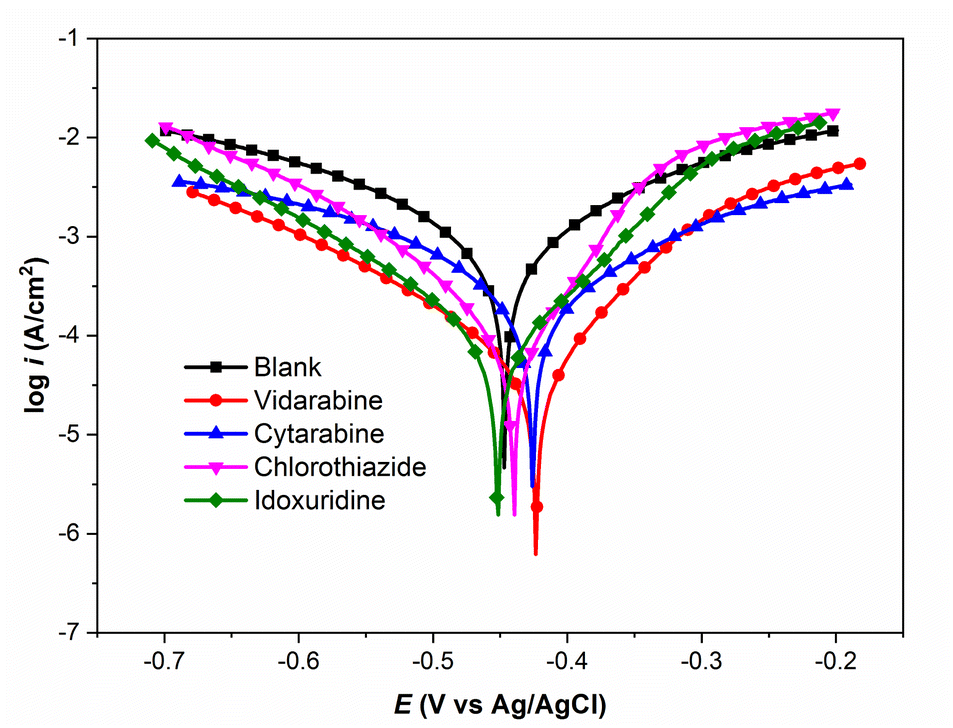
Our experimental results for corrosion inhibition of four unpublished drug compounds, vidarabine (E1), cytarabine (E2), chlorothiazide (E3), and idoxuridine (E4), on Q235 steel, are provided in Table 4 and Fig. 9. The comparison of experimental and predicted IEs for E1–E4 compounds is shown in Fig. 8. It can be seen that the prediction IEs of SMILES2IE-steel for vidarabine, chlorothiazide, and idoxuridine are close to our experimental values, with errors of 1.4%, 1.5%, and 2.9% respectively. Vidarabine presents the highest corrosion inhibition efficiency of 94.0%, showing that this is a good inhibitor for carbon steel in 1 M HCl solution. However, the experimental IE of cytarabine is much lower than its predicted IE, with an error of 10.9%. The large discrepancy between the experimental IE and the predicted IE for cytarabine is likely because the data set for our ML model does not include the structural features of cytarabine or because the standalone molecular descriptors do not fully reflect the interaction (or adsorption properties) between the molecules and the metal surface. Emphasize that adsorption properties are related to the effectiveness of compounds in inhibiting steel corrosion. However, calculating the adsorption of more than 300 organic compounds with relatively complex structures on steel surfaces is challenging. This calculation is beyond the scope of the present study. Note that standalone molecular properties also reflect the ability of molecules to interact with the metal surface to some extent through several topological descriptors such as Chi, Kappa, BertCZ, and BalabanJ, the BCUT2D type descriptors, which are used in the present work. Fast calculation is one of the advantages of the descriptors over adsorption properties. Additionally, outliers of prediction are not always bad, as adding these data will enrich the domain of the training dataset,20 and exploring deeper insights into how the outliers differ from the other existing data will help find better descriptors to enhance prediction accuracy. These are also the research contents that we are pursuing.
Table 4 Electrochemistry parameters, experimental IEs, and predicted IEs for Q235 steel in 1 M HCl solution in the absence and presence of 1 mmol L−1 drug compound
| ID |
Name |
Ecorr (mV vs. Ag/AgCl) |
icorr (µA cm−2) |
βc (mV dec−1) |
βa (mV dec−1) |
Exp. IE (%) |
Pre. IE (%) |
| |
Blank |
−445 |
431 |
−128 |
133 |
— |
— |
| E1 |
Vidarabine |
−421 |
26 |
−64 |
97 |
94.0 |
92.6 |
| E2 |
Cytarabine |
−424 |
87 |
−92 |
96 |
79.8 |
90.7 |
| E3 |
Chlorothiazide |
−432 |
47 |
−49 |
80 |
89.1 |
90.6 |
| E4 |
Idoxuridine |
−443 |
55 |
−77 |
110 |
87.2 |
90.1 |
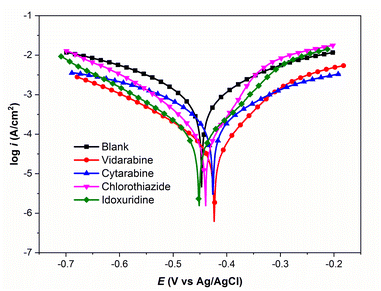 |
| | Fig. 9 Polarization curves for Q235 steel in 1 M HCl solution in the absence and presence of inhibitors. | |
3.7. Predicting new corrosion inhibitors
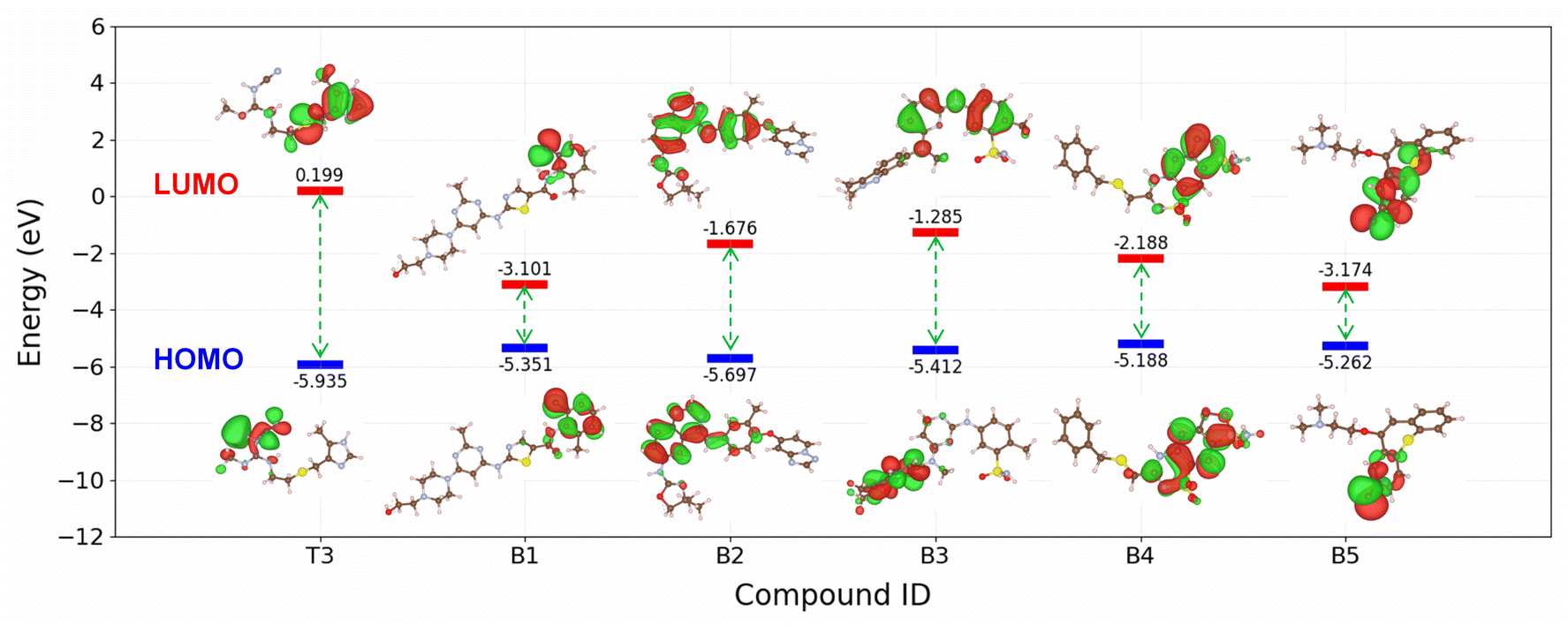
The SMILES2IE-steel tool was employed to predict the IE of a dataset comprising 1509 FDA-approved drug compounds collected from the Database of Digital Properties of Approved Drugs (DDPD).59 This web-based tool enables rapid IE prediction for the entire set of 1509 compounds within seconds. The distribution of the predicted IEs is shown in Fig. S3,† revealing a substantial proportion of drug compounds exhibiting high (≥90%) and very high (≥95%) corrosion inhibition efficiency. The five compounds with the highest predictive IEs are shown in Fig. 8 (B1–B5 compounds) and Table S5 in ESI.† Notably, these compounds are predicted to be more effective corrosion inhibitors compared to published compounds such as T1–T10. Structural analysis indicates that these compounds feature numerous heterocyclic atoms and aromatic rings. This structural attribute is expected to enhance the interaction between the molecules and the metal surface, thereby strengthening their corrosion inhibition capabilities.
To further analyse the electronic structure characteristics of B1–B5 molecules, we conducted calculations on key electronic properties such as HOMO and LUMO energies. As depicted in Fig. 10, these five new compounds exhibit significantly higher HOMO energies and notably lower LUMO energies compared to the T3 compound, which showed the highest IE among the ten compounds scrutinized in this study. Typically, higher HOMO values signify an increased capacity of the molecule to donate electrons to the metal surface, while lower LUMO values indicate a heightened ability to receive electrons from the metal surface.60,61 The electronic structure results indicate that compounds B1–B5 possess superior capabilities in both electron donation and acceptance from the metal surface compared to the T3 compound. This observation aligns with the predictions of our machine learning model, because a more effective interaction between the molecule and the surface corresponds to enhanced corrosion inhibition ability.62,63 It is noted that the model's prediction results still need to be confirmed with experimental measurements. However, these findings underscore the utility of the proposed machine learning model for the screening of novel corrosion inhibitor compounds for steel within the realm of drug compounds, presenting a promising avenue for the identification of potential green corrosion inhibitors.
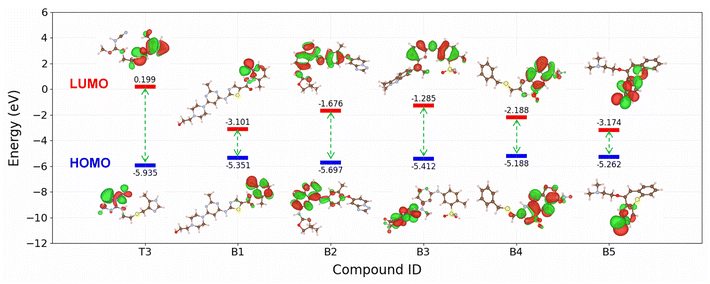 |
| | Fig. 10 Comparison of the HOMO and LUMO energies of B1–B10 with T3. | |
4. Conclusions
A novel QSPR model has been constructed for the prediction of corrosion inhibition efficiencies of organic compounds on carbon steel. The model was trained on a dataset of different types of organic inhibitors employing a novel ML workflow integrating the GB algorithm and PFI method. The PFI method effectively identifies a crucial descriptor group, primarily of topological, BCUT2D-type, and MOE-type descriptors. The model utilizing this descriptor group demonstrated optimal performance, achieving MAE, RMSE, and R2 values of 6.40, 4.80, and 0.72, respectively. Furthermore, the proposed ML workflow exhibited superior performance compared to other ML models across four published datasets, with RMSE reductions ranging from 14% to 39%. Model reliability was verified by comparing predictions with experimental data on drug compounds, a group of potential green corrosion inhibitors. The model was subsequently employed to screen novel drug compounds with high corrosion inhibition efficiencies, and the outcomes were elucidated through DFT calculations. The obtained results indicated that the proposed ML workflow and model have the potential for screening and developing next-generation corrosion inhibitors.
Data availability
The details of the Fe–HCl-317 dataset can be found at https://github.com/pth-lab/Fe-HCl-317-dataset.
Author contributions
Conceptualization (THP); investigation (THP); visualization (THP); writing – original manuscript (THP); methodology (THP and DNS), analysis (THP and DNS); supervision (DNS and PKL); writing – review & editing (DNS and PKL).
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We acknowledge Ho Chi Minh City University of Technology (HCMUT), VNU-HCM for supporting this study.
References
- H. Wei, B. Heidarshenas, L. Zhou, G. Hussain, Q. Li and K. (Ken) Ostrikov, Mater. Today Sustain., 2020, 10, 100044 CrossRef.
- M. Finšgar and J. Jackson, Corros. Sci., 2014, 86, 17–41 CrossRef.
- P. D. Desai, C. B. Pawar, M. S. Avhad and A. P. More, Vietnam J. Chem., 2023, 61, 15–42 CrossRef CAS.
- R. Aslam, G. Serdaroglu, S. Zehra, D. Kumar Verma, J. Aslam, L. Guo, C. Verma, E. E. Ebenso and M. A. Quraishi, J. Mol. Liq., 2022, 348, 118373 CrossRef CAS.
- C. Verma, E. E. Ebenso, M. A. Quraishi and C. M. Hussain, Mater. Adv., 2021, 2, 3806–3850 RSC.
- I. B. Obot, D. D. Macdonald and Z. M. Gasem, Corros. Sci., 2015, 99, 1–30 CrossRef CAS.
- A. Kokalj, Corros. Sci., 2021, 193, 109650 CrossRef CAS.
- D. Winkler, Metals (Basel), 2017, 7, 553 CrossRef.
- T. H. Pham, O. K. Le, V. Chihaia, P. K. Le and D. N. Son, J. Electrochem. Soc., 2023, 170, 111504 CrossRef.
- A. Kokalj, M. Lozinšek, B. Kapun, P. Taheri, S. Neupane, P. Losada-Pérez, C. Xie, S. Stavber, D. Crespo, F. U. Renner, A. Mol and I. Milošev, Corros. Sci., 2021, 179, 108856 CrossRef CAS.
- H. Zhao, X. Zhang, L. Ji, H. Hu and Q. Li, Corros. Sci., 2014, 83, 261–271 CrossRef CAS.
- L. Li, X. Zhang, S. Gong, H. Zhao, Y. Bai, Q. Li and L. Ji, Corros. Sci., 2015, 99, 76–88 CrossRef CAS.
- C. T. Ser, P. Žuvela and M. W. Wong, Appl. Surf. Sci., 2020, 512, 145612 CrossRef.
- T. W. Quadri, L. O. Olasunkanmi, E. D. Akpan, O. E. Fayemi, H.-S. Lee, H. Lgaz, C. Verma, L. Guo, S. Kaya and E. E. Ebenso, Mater. Today Commun., 2022, 30, 103163 CrossRef CAS.
- T. W. Quadri, L. O. Olasunkanmi, O. E. Fayemi, E. D. Akpan, H.-S. Lee, H. Lgaz, C. Verma, L. Guo, S. Kaya and E. E. Ebenso, Comput. Mater. Sci., 2022, 214, 111753 CrossRef CAS.
- T. W. Quadri, L. O. Olasunkanmi, O. E. Fayemi, H. Lgaz, O. Dagdag, E.-S. M. Sherif, E. D. Akpan, H.-S. Lee and E. E. Ebenso, J. Mol. Model., 2022, 28, 254 CrossRef CAS PubMed.
- J. Dai, D. Fu, G. Song, L. Ma, X. Guo, A. Mol, I. Cole and D. Zhang, Corros. Sci., 2022, 209, 110780 CrossRef CAS.
- D. A. Winkler, M. Breedon, A. E. Hughes, F. R. Burden, A. S. Barnard, T. G. Harvey and I. Cole, Green Chem., 2014, 16, 3349–3357 RSC.
- D. A. Winkler, M. Breedon, P. White, A. E. Hughes, E. D. Sapper and I. Cole, Corros. Sci., 2016, 106, 229–235 CrossRef CAS.
- X. Li, B. Vaghefinazari, T. Würger, S. V. Lamaka, M. L. Zheludkevich and C. Feiler, npj Mater. Degrad., 2023, 7, 64 CrossRef CAS.
- E. J. Schiessler, T. Würger, S. V. Lamaka, R. H. Meißner, C. J. Cyron, M. L. Zheludkevich, C. Feiler and R. C. Aydin, npj Comput. Mater., 2021, 7, 193 CrossRef.
- M. Akrom, S. Rustad, A. G. Saputro, A. Ramelan, F. Fathurrahman and H. K. Dipojono, Mater. Today Commun., 2023, 35, 106402 CrossRef CAS.
- G. Landrum, RDKit: Open-Source Cheminformatics, https://rdkit.org, accessed 18 April 2023 Search PubMed.
- D. Rogers and M. Hahn, J. Chem. Inf. Model., 2010, 50, 742–754 CrossRef CAS PubMed.
- J. H. Friedman, Ann. Stat., 2001, 29, 1189–1232 Search PubMed.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg and J. Vanderplas, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- Z. M. Alhakeem, Y. M. Jebur, S. N. Henedy, H. Imran, L. F. A. Bernardo and H. M. Hussein, Materials (Basel), 2022, 15, 7432 CrossRef CAS PubMed.
- L. Yang and A. Shami, Neurocomputing, 2020, 415, 295–316 CrossRef.
- A. Altmann, L. Toloşi, O. Sander and T. Lengauer, Bioinformatics, 2010, 26, 1340–1347 CrossRef CAS PubMed.
- F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2012, 2, 73–78 CrossRef CAS.
- F. Neese, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2022, 12, e1606 CrossRef.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297 RSC.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- Y. Liu, J. Wu, M. Avdeev and S. Shi, Adv. Theory Simul., 2020, 3, 1900215 CrossRef CAS.
- J. Wang, P. Xu, X. Ji, M. Li and W. Lu, Materials (Basel), 2023, 16, 3134 CrossRef CAS PubMed.
- T. W. Quadri, L. O. Olasunkanmi, O. E. Fayemi, H. Lgaz, O. Dagdag, E.-S. M. Sherif, A. A. Alrashdi, E. D. Akpan, H.-S. Lee and E. E. Ebenso, Arabian J. Chem., 2022, 15, 103870 CrossRef CAS.
- T. Sutojo, S. Rustad, M. Akrom, A. Syukur, G. F. Shidik and H. K. Dipojono, npj Mater. Degrad., 2023, 7, 18 CrossRef.
- L. H. Hall and L. B. Kier, Rev. Comput. Chem., 2007, 367–422 Search PubMed.
- A. T. Balaban, Chem. Phys. Lett., 1982, 89, 399–404 CrossRef CAS.
- S. H. Bertz, J. Am. Chem. Soc., 1981, 103, 3599–3601 CrossRef CAS.
- P. Labute, J. Mol. Graphics Modell., 2000, 18, 464–477 CrossRef CAS PubMed.
- A. E. Comesana, T. T. Huntington, C. D. Scown, K. E. Niemeyer and V. H. Rapp, Fuel, 2022, 321, 123836 CrossRef CAS.
- M. P. González, C. Terán, M. Teijeira, P. Besada and M. J. González-Moa, Bioorg. Med. Chem. Lett., 2005, 15, 3491–3495 CrossRef PubMed.
- D. T. Stanton, J. Chem. Inf. Comput. Sci., 1999, 39, 11–20 CrossRef CAS.
- G. Gece, Corros. Sci., 2011, 53, 3873–3898 CrossRef CAS.
- M. A. Quraishi and D. S. Chauhan, in Sustainable Corrosion Inhibitors II: Synthesis, Design, and Practical Applications, 2021, pp. 1–17 Search PubMed.
- N. Vaszilcsin, D.-A. Duca, A. Flueraş and M.-L. Dan, Stud. Univ. Babeş-Bolyai, Chem., 2019, 64, 17–32 CAS.
- G. Karthik and M. Sundaravadivelu, Egypt. J. Pet., 2016, 25, 183–191 CrossRef.
- I. Reza, A. Saleemi and S. Naveed, Pol. J. Chem. Technol., 2011, 13, 67–71 CrossRef.
- A. Singh, A. Gupta, A. K. Rawat, K. R. Ansari, M. A. Quraishi and E. E. Ebenso, Int. J. Electrochem. Sci., 2014, 9, 7614–7628 CrossRef.
- I. Ahamad, R. Prasad and M. A. Quraishi, Corros. Sci., 2010, 52, 3033–3041 CrossRef CAS.
- S. Dahiya, N. Saini, N. Dahiya, H. Lgaz, R. Salghi, S. Jodeh and S. Lata, Port. Electrochim. Acta, 2018, 36, 213–230 CrossRef CAS.
- L. P. Chaudhari and S. N. Patel, J. Bio- Tribo-Corrosion, 2019, 5, 20 CrossRef.
- A. S. Fouda, K. Shalabi and A. E-Hossiany, J. Bio- Tribo-Corrosion, 2016, 2, 18 CrossRef.
- S. K. Shukla and M. A. Quraishi, Mater. Chem. Phys., 2010, 120, 142–147 CrossRef CAS.
- R. S. A. Hameed, Port. Electrochim. Acta, 2011, 29, 273–285 CrossRef.
- Z. Golshani, S. M. A. Hosseini, M. Shahidizandi and M. J. Bahrami, Mater. Corros., 2019, 70, 1862–1871 CrossRef CAS.
- Q. Li, S. Ma, X. Zhang, Z. Zhai, L. Zhou, H. Tao, Y. Wang and J. Pan, Database, 2022, 2022, baab083 CrossRef PubMed.
- S. Hadisaputra, A. A. Purwoko, A. Hakim, N. Prasetyo and S. Hamdiani, ACS Omega, 2022, 7, 33054–33066 CrossRef CAS PubMed.
- M. Leng, Y. Xue, L. Luo and X. Chen, Comput. Theor. Chem., 2023, 1229, 114327 CrossRef CAS.
- D. Kumar, N. Jain, V. Jain and B. Rai, Appl. Surf. Sci., 2020, 514, 145905 CrossRef CAS.
- D. Kumar, V. Jain and B. Rai, Corros. Sci., 2020, 171, 108724 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc,
Phung K. Leab and
Do Ngoc Son
*abc,
Phung K. Leab and
Do Ngoc Son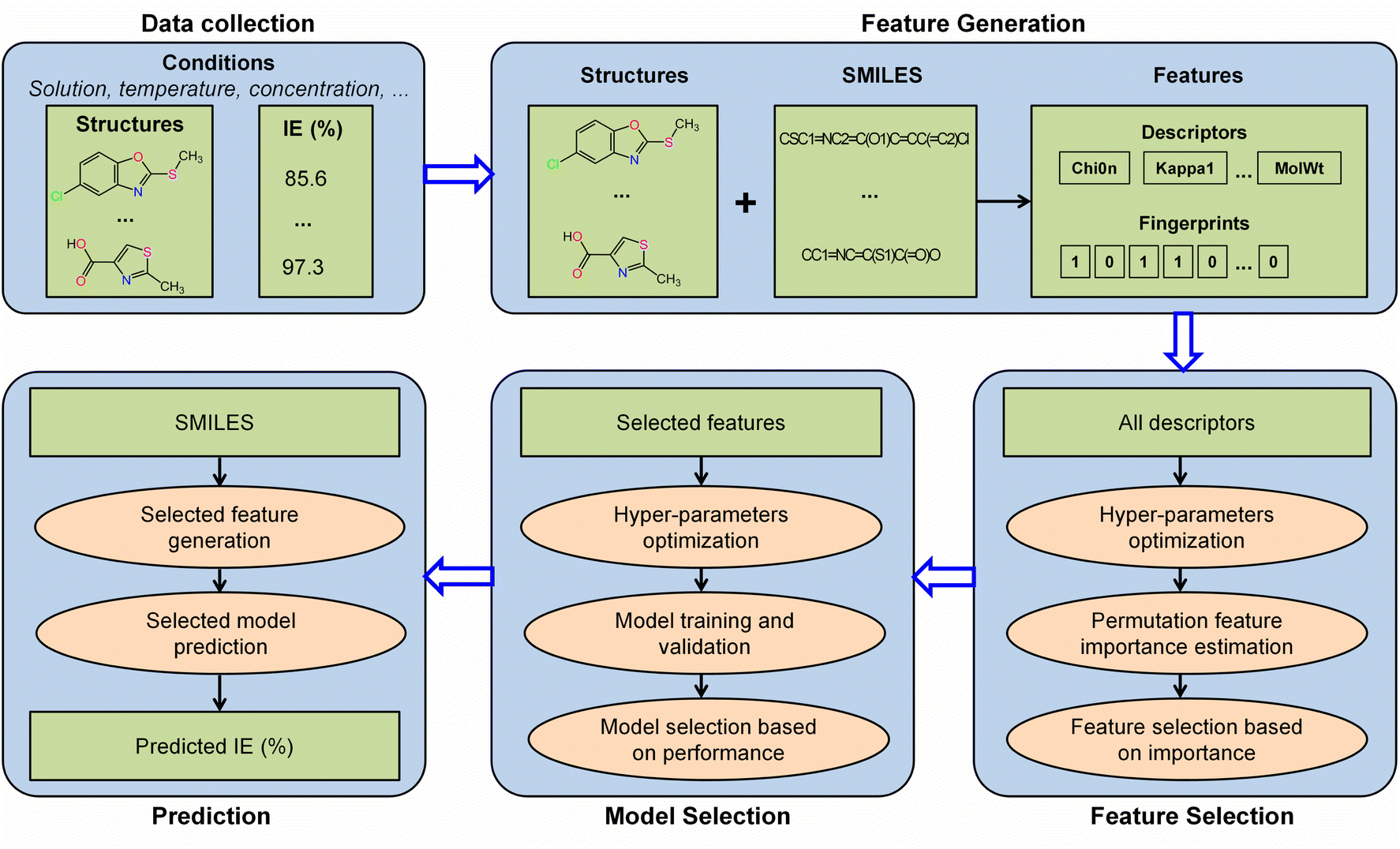
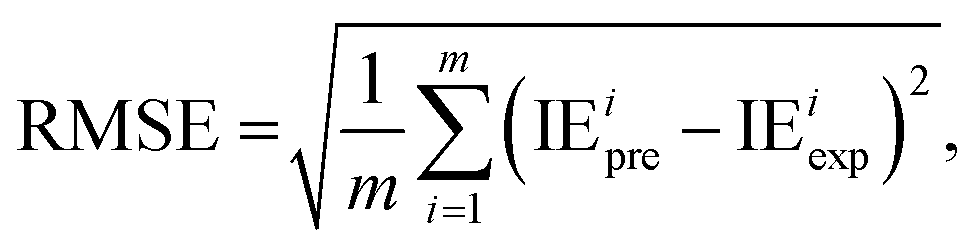
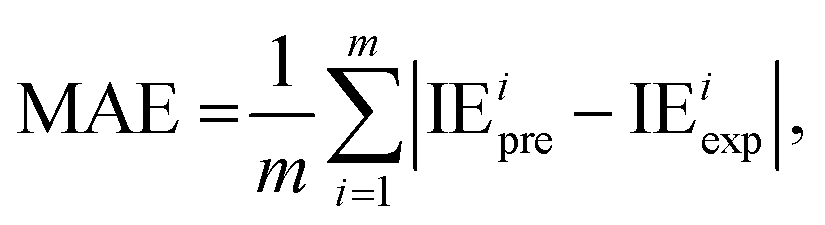
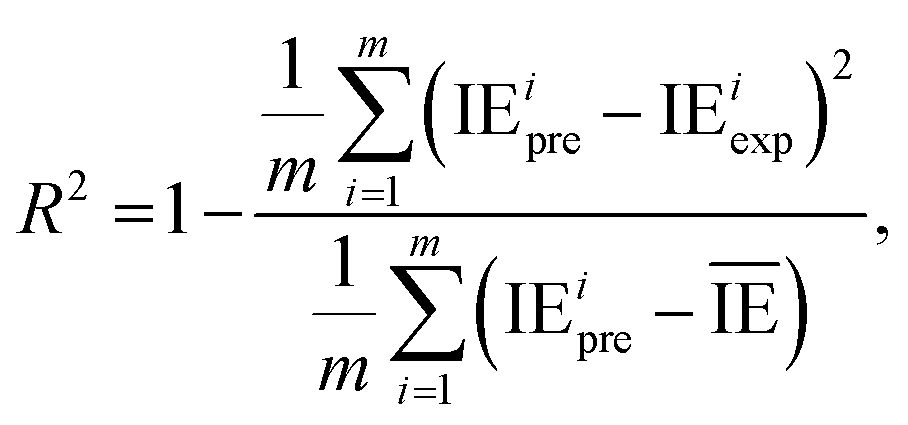
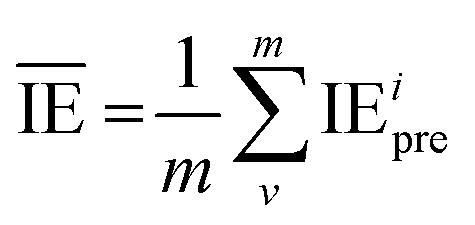 is the average value of the predicted IE.
is the average value of the predicted IE.