
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceN-heterocyclic carbene catalyzed [2 + 3] annulation reaction for the synthesis of trifluoroethyl 3,2′-spirooxindole γ-lactam†
Yiru Pu‡
a,
Maolin Wang‡c,
Wanrong Tian‡a,
Xian Gea,
Dikai Zhud,
Chuanqi Wanga,
Yingjie Zenga,
Feiyan Tao*d,
Yun Deng*a and
Jun Lu *ab
*ab
aState Key Laboratory of Southwestern Chinese Medicine Resources, School of Pharmacy, Chengdu University of Traditional Chinese Medicine, Chengdu, 611137, China. E-mail: dengyun2000@hotmail.com; ljaaa111@163.com
bInstitute for Advancing Translational Medicine in Bone & Joint Diseases, School of Chinese Medicine, Hong Kong Baptist University, Hong Kong SAR 999077, China
cClinical Research Center, The First Affiliated Hospital of Shantou University Medical College, Shantou, Guangdong Province 515000, China
dResearch and Development Centre, China Tobacco Sichuan Industrial Co. Ltd, Chengdu, 610066, China. E-mail: taofeiyan@163.com
First published on 10th June 2024
Abstract
Asymmetric catalytic processes promoted by N-heterocyclic carbenes (NHCs) hold great potential for the sustainable preparation of chiral molecules. However, catalyzing the reactions by manipulating the reactive intermediates is challenging. We report herein that the known NHC-catalyzed [3 + 2] annulation reaction between ketimine and enal can also be turned into a [2 + 3] annulation reaction for the highly enantioselective direct synthesis of trifluoroethyl 3,2′-spirooxindole γ-lactams (4) through timely catalysis of the intermediates. DFT calculations revealed that this transformation included the key step of the nucleophilic attack of the Breslow intermediate M2 derived from NHC and enal (2) to the unattacked ketimine (1). Our study demonstrates that it is possible to tune the desired selectivities through the dynamic catalysts of the reactive intermediates.
N-heterocyclic carbenes (NHCs)1 have been extensively adopted as an asymmetric synthetic catalyst for the synthesis of chiral heterocycles. The use of NHCs results in various powerful intermediates such as Breslow,2 enolate,3 homoenolate,4 and unsaturated acylazolium,5 which could undergo further [3 + 2],6 [4 + 2]7 annulation and more reactions. The challenge of controlling the modes of NHC-catalyzed reactions lies in the manipulation of the intermediates for the annulation reactions.
Spiro[hydroxyindole-3,2′-pyrrolidine], a γ-lactam containing skeleton bearing a quaternary chiral center, has been recognized as an essential backbone in many natural products and biologically active molecules.8 The spirooxindole scaffold exhibits considerable anti-cancer activity and has dual inhibitory activity targeting the p53-MDM2/Bcl2.9 The synthesis of the structure has therefore received considerable attention. In 2012, Chi et al. reported the synthesis of γ-lactam by NHCs catalysis (Scheme 1a),10 but did not mention the reaction mechanism. To enhance the bioactivity of γ-lactam, the fluoroalkyl group is usually introduced into the motif. In 2015, Wang et al. synthesized 5′-CF3 spiro[hydroxyindole-3,2′-pyrrolidine] from ketimine and cinnamaldehyde catalyzed by a chiral proline and benzoic acid (Scheme 1b).11 Later, Han et al. reported the synthesis of trifluoroethyl 3,2′-spirooxindole γ-lactam from 3-((2,2,2-trifluoroethyl)amino)indolin-2-one and cinnamaldehyde catalyzed by chiral prolines in which the 3-aminooxindoles were pre-synthesized from N-2,2,2-trifluoroethyl isatin ketamine (Scheme 1c).12
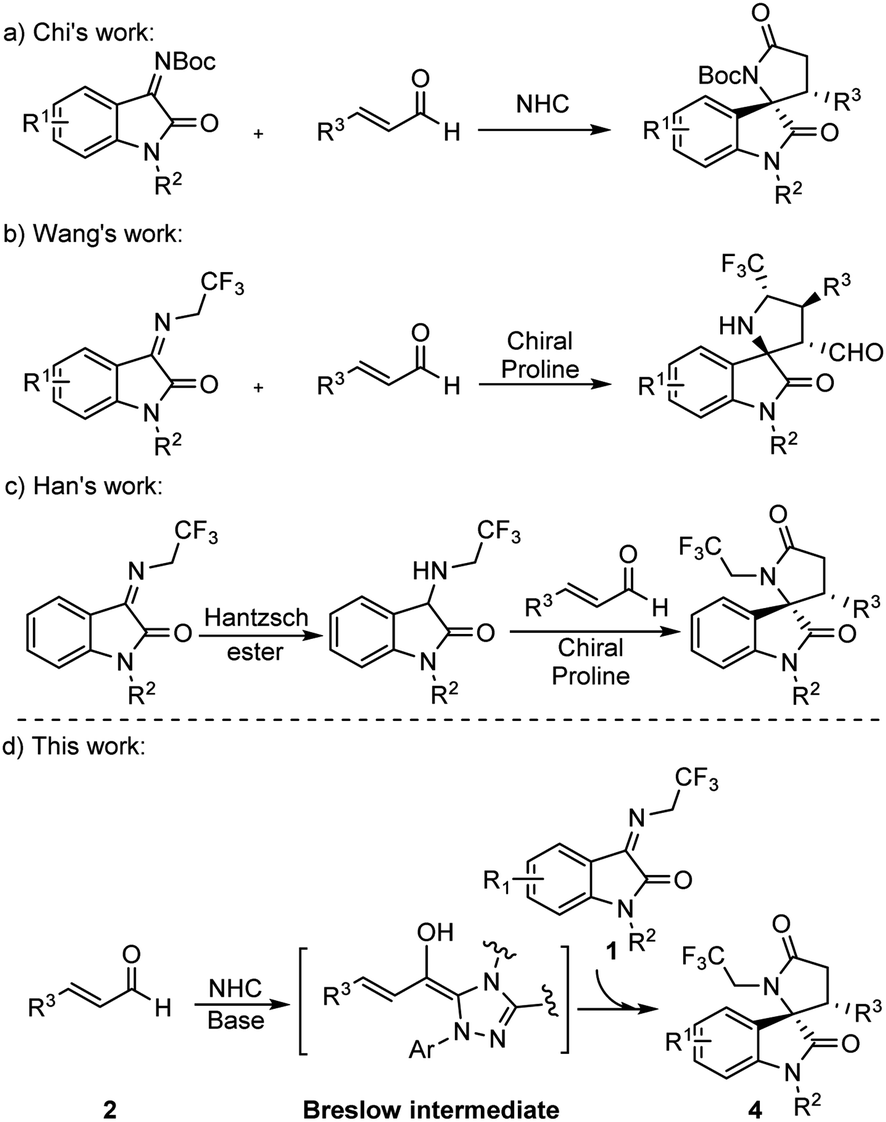 | ||
| Scheme 1 Synthesis of γ-lactam. | ||
Continuing our efforts to discover molecules with medication significance, we set our design strategy to synthesize trifluoroethyl 3,2′-spirooxindole γ-lactam.
Since Chi et al. used NHC catalyzing ketimine and enal synthesized spirocyclic γ-lactam, we initiated our study for the synthesis of trifluoroethyl 3,2′-spirooxindole γ-lactam 4 by following Chi et al.’s procedure, in which NHC was added into the mixture of ketimine 1 and cinnamaldehyde 2. Unfortunately, 5 was generated dominantly instead of the target product 4 (Table 1, entry 1). We rationalize that this is due to the deprotonation of the methylene position in the CF3CH2-functionalized ketimine in the presence of the base, leading to its functioning as a nucleophile to attack the electrophile cinnamaldehyde through an [3 + 2] annulation reaction that yielded 5. To obtain 4, a [2 + 3] annulation reaction would be expected and the nucleophilicity of the enal and the ketimine must be reversed.
|
||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | NHC | Base | Solvent | Temp. (°C) | Yield (%)b | drc | ee of 4a (%)d | |
| 4a | 5a | |||||||
| a Entry 1 was performed with 1a (0.10 mmol), 2a (0.15 mmol), 3a (20 mol%) and DIPEA (2.0 equiv.) in solvent (1.0 mL) at 20 °C for 24 h under N2. Entry 2 was varied to co-incubation activation of 3a, DIPEA, and 2a for half an hour, followed by adding 1a.b Isolated yields of 4a and 5a.c dr values were determined by 1H NMR analysis of the crude product.d ee values were determined by HPLC using a chiral column. | ||||||||
| 1 | 3a | DIPEA | DCM | 20 | 8 | 64 | — | — |
| 2 | 3a | DIPEA | DCM | 20 | 52 | 22 | 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 :1 |
22 |
| 3 | 3b | DIPEA | DCM | 20 | 60 | 10 | 8:1 |
66 |
| 4 | 3c | DIPEA | DCM | 20 | 51 | 17 | 5:1 |
47 |
| 5 | 3d | DIPEA | DCM | 20 | 62 | 11 | 9:1 |
80 |
| 6 | 3e | DIPEA | DCM | 20 | 67 | 10 | 7:1 |
74 |
| 7 | 3f | DIPEA | DCM | 20 | 76 | 16 | 10:1 |
90 |
| 8 | 3g | DIPEA | DCM | 20 | 66 | 12 | 9:1 |
86 |
| 9 | 3f | K2CO3 | DCM | 20 | 28 | 46 | 7:1 |
62 |
| 10 | 3f | Et3N | DCM | 20 | 82 | 6 | 15:1 |
95 |
| 11 | 3f | Et3N | THF | 20 | 77 | — | 15:1 |
86 |
| 12 | 3f | Et3N | Toluene | 20 | 48 | 7 | 15:1 |
46 |
| 13 | 3f | Et3N | DCM | 0 | 80 | — | >20:1 |
98 |
| 14 | — | Et3N | DCM | 20 | — | 85 | — | — |
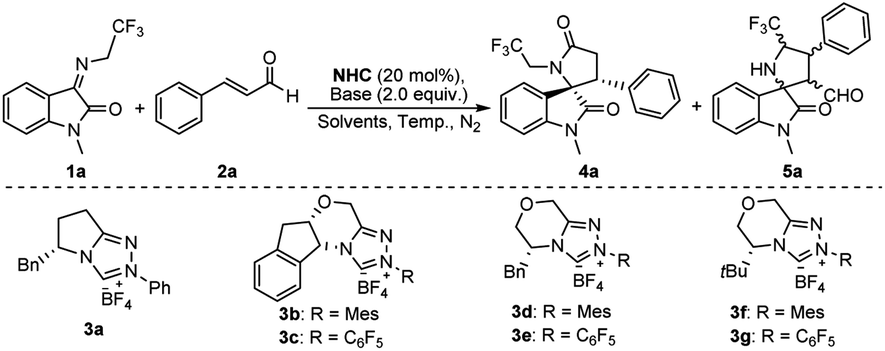
Recently, many types of NHC-catalyzed annulation reactions between enals and different coupling partners, such as alkenes, imines, and ketones, have been reported.13 Interestingly, the addition of the NHC catalyst to the enals can allow the inversion of the normal reactivity through the formation of Breslow intermediates that serve as the prenucleophiles.14 With this in mind, we speculate that the combination of cinnamaldehyde (enal) and NHC, followed by the addition of ketimine, could generate the Breslow intermediate (nucleophile) and leave the ketimine (electrophile) untacked by the base, thus opening the possibility of [2 + 3] annulation reaction entry for the asymmetric synthesis of trifluoroethyl 3,2′-spirooxindole γ-lactam (4).
Herein, studies on the synthesis of trifluoroethyl 3,2′-spirooxindole γ-lactam from enals and ketimines catalyzed by NHC are disclosed. Moreover, the mechanism of the [2 + 3] annulation reaction was discussed based on DFT calculation. To our knowledge, this study is the first example in which NHC-catalyzed reaction modes were employed by timely manipulation of the intermediates for the synthesis of trifluoroethyl 3,2′-spirooxindole γ-lactam.
Based on our hypothesis, we initiated the study by investigating the reaction between ketimine 1a and enal 2a catalyzed by the NHC precatalyst 3a in the presence of DIPEA. By simply changing the addition sequence by mixing 2 with 3f in the presence of DIPEA, followed by the addition of 1, the reaction selectivities were reversed (Table 1, entry 1 vs. entry 2). The optimization of the reaction conditions was started with the screening of the catalysts (Table 1, entries 2–8). Catalysts bearing N-mesityl (3b, 3d and, 3f) afforded better enantioselectivities for 4 than those with phenyl (3a) and pentafluorophenyl (3c, 3e and 3g), possibly due to the enhanced nucleophilicity of (mesityl)NHCs by the electron-donating mesityl in combination with their steric characters making them excellent catalysts for the [2 + 3] annulation reactions between ketimine and enal. 3f was found to be the favorable one that resulted in the desired product 4 of 76% yield and 90% enantioselectivity (Table 1, entry 7). Further optimization studies revealed that the yield and selectivities were sensitive to the bases (Table 1, entries 8–10). Et3N turned out to be the preferred base (Table 1, entry 10). Reactions conducted in THF and toluene gave reduced yields and ee. (Table 1, entries 11–12) To improve the reaction selectivities, temperature effects were tested. Gratifyingly, we found that at 0 °C, 5a was completely suppressed, resulting in the highest yield of 4 in 80% and with excellent dr and ee (Table 1, entry 13). However, the reaction at 20 °C without the addition of any catalyst resulted in 5 only, and no 4 was detected (Table 1, entry 14).
Under the optimum reaction conditions, we further investigated the universality of both substrate species (Table 2). Initially, the substituents at distinct positions on N-2,2,2-trifluoroethylisatinone imine (1) were examined, and the corresponding products (4a-4j) were achieved in moderate yields and excellent ee values, regardless of substituting on the aromatic ring R1 or the N atom R2 of the substrate. Of these, with the finest results for the derived product 4b, the chiral skeleton was obtained in 81% yield and up to 99% ee value. When N–CH3 was replaced with benzyl or allyl, the products (4f-4j) still maintained good yields and favorable enantioselectivities. Accordingly, a substrate investigation of the substituted α,β-unsaturated alkenals (2) revealed that the cross-reaction of the alkenals bearing electron-absorbing substituents with 1 featuring diverse substituents on the aromatic ring gives the appropriate indoles skeletons (4k-4n and 4t-4w) in 68–82% yields and 90–99% ee values. Despite the overall trend presenting moderate yields, partial compounds, such as 4l and 4w, exhibited ee values of up to 99%. Subsequently, experimental results proved that product ee values were sustained beyond 85% (4o-4s and 4x-4ab), even though a benzyl group with a large potential resistance was adopted at the N–R2 position. To explore the impact of other substituents at the R3-position of the alkenal, we opted for the reaction of 1d and the substrate of electron-donating (2-OCH3) substituents on the benzene ring to afford 4ac in moderate yield and high enantioselectivity. Moreover, the heterocyclic furanyl and alkyl groups proceeded smoothly when replacing the phenyl group in cinnamaldehyde and resulted in the required target (4ad, 4ae) in good yields and with excellent enantioselectivity. The absolute configuration of 4aa was determined as (3S, 3'R) using single crystal derivatization, based on which the absolute configurations of the other products could be extrapolated.
| a Reactions were performed with 1 (0.10 mmol), 2 (0.15 mmol), 3f (20 mol%) and Et3N (2.0 equiv.) in DCM (1.0 mL) at 0 °C for 24 h under N2. |
|---|
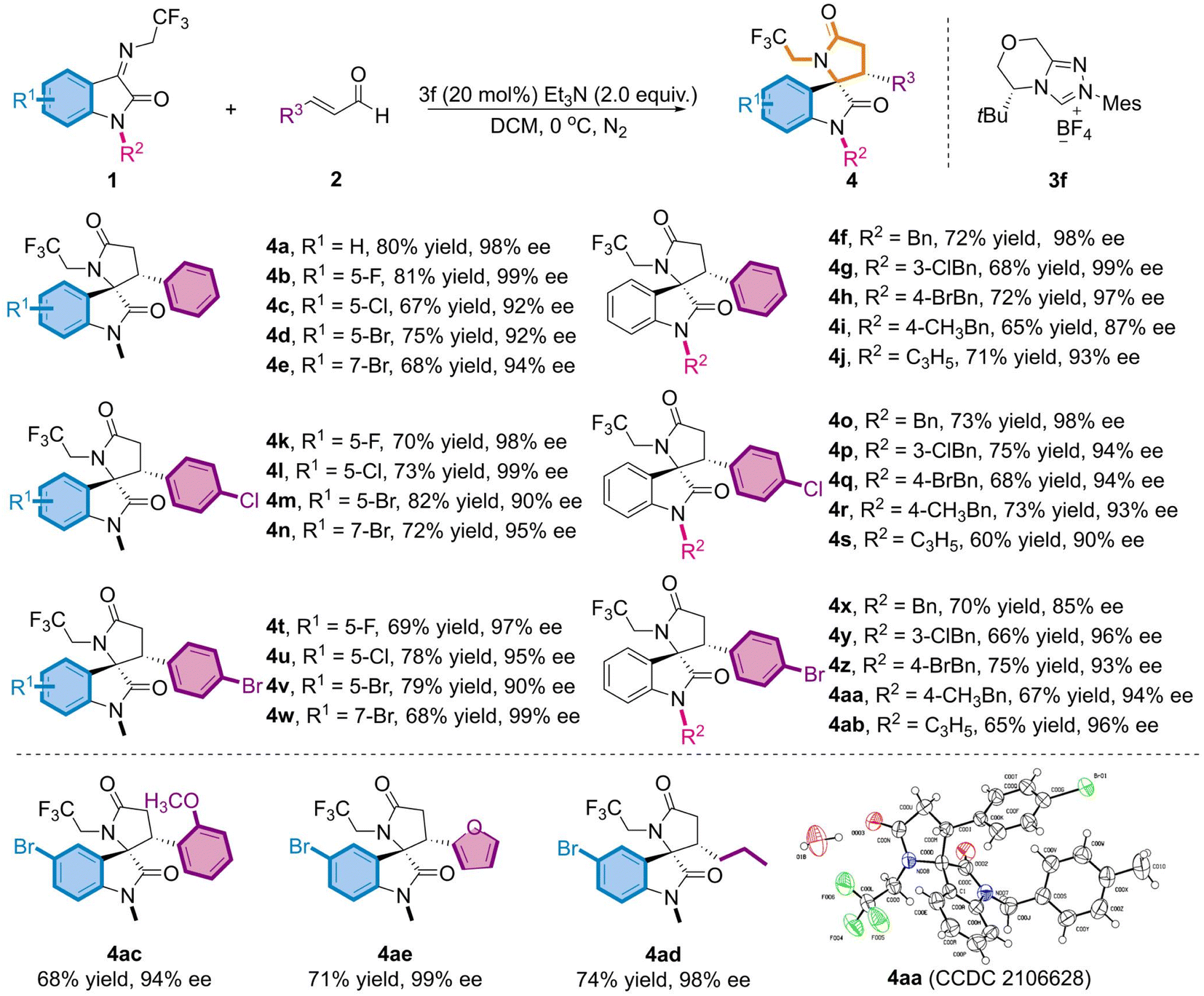 |
Based on the experimental results, although the formation of 4 through the NHC-catalyzed [2 + 3] annulation reactions from 1 and 2 can be concluded, the origin of the reaction selectivities modulated by the timely catalysts of the addition sequence of the substrates is unknown. Thus, we performed a theoretical investigation of the mechanism of the NHC-catalyzed [2 + 3] annulation reaction (Fig. 1). DFT calculations were performed for the model reaction (1a and 2a) with 3f as a catalyst, dichloromethane as the solvent, and a temperature of 25 °C. First, carbene-activated 3f was generated from the triazolocarbine tetrafluoroboronium salt (3f) in the presence of triethylamine. It subsequently underwent nucleophilic attack to the β-C of the cinnamaldehyde (2) and generated the zwitterionic intermediate M1 through the transition state TS1 with a Gibbs free energy barrier of 27.5 kcal mol−1. We deduce that the successive formation of the homoenolate intermediate M2 was from TS2, an Et3N-assisted 1,2-proton transfer process rather than TS2' (the direct proton transfer pathway) since the former path is favored by a 35.0 kcal mol−1 reduced energy barrier (32.8 kcal mol−1 vs. 67.8 kcal mol−1). The nucleophilic attack on M2 to 1a affords M3 via transition state TS3, in which a C–C bond is involved.
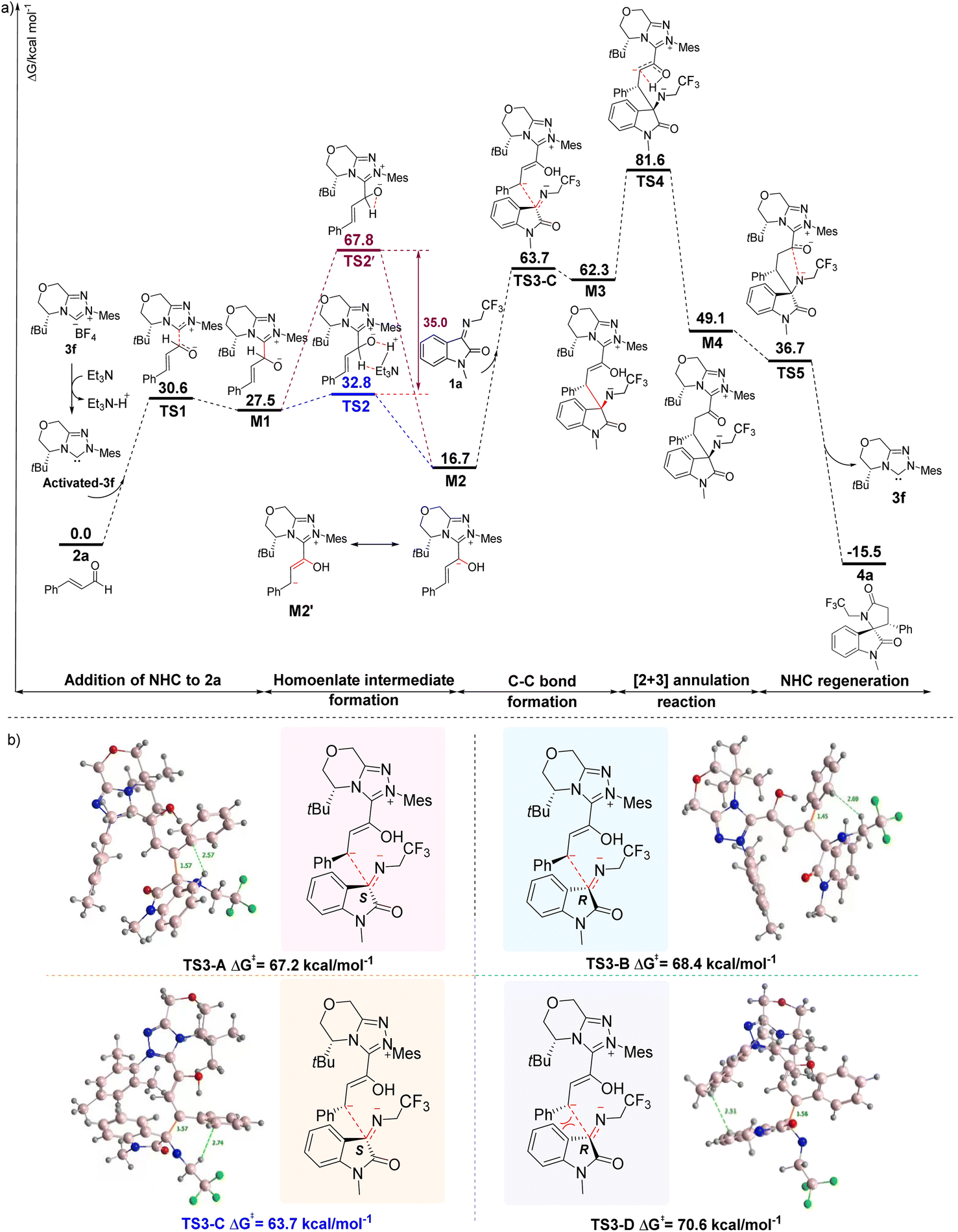 | ||
| Fig. 1 Postulated mechanism for the NHC-catalyzed [2 + 3] annulation reaction based on DFT calculations. (a). Energy profiles of detailed mechanistic processes. (b). Transition-state structures of the enantioselectivity-determining step. | ||
As M2 could approach 1a from either the re or si face, creating possible transition states of TS3-A, TS3-B, TS3-C, or TS3-D with a chiral center (C3) generated through the C–C bond formation (Fig. 1b). Computational results reveal that TS3-A and TS3-C with the S-conformation of C3 possess lower free energies than TS3-B and TS3-D of R-conformation on C3, indicating that the attack of M2 to 1a is more likely from the si face. Comparing TS3-A and TS3-C, the latter gives rise to less spatial resistance and lower Gibbs energy (67.2 vs. 63.7 kcal mol−1). Thus, TS3-C is taken as the transition state from M2 to M3. The subsequent step of transformation from M3 to M4, which involves a 1,3-proton transfer process would be the rate-determining step because the highest Gibbs energy transition state TS4 is involved, with an elevated energy of 32.5 kcal mol−1 relative to M3. The annulation intermediate TS5 is generated from M4 by the nitrogen anion attacking the carbonyl carbon. Finally, 4a is yielded through TS5 with the release of carbene 3f.
To verify the practicability of the catalytic reactions, we conducted reaction assays on all gram-level substrates. Analogously, the chiral 4l was obtained in the established conditions with 73% yield and 94% ee from the reaction performed with 1.0 g 1k, 0.90 g 2c and 10 mol% 3f (Scheme 2a). Moreover, the two carbonyl groups on chiral 4l could be conveniently converted to methylene by lithium aluminum hydride to give chiral 6l with yields and ee values of 81% and 94%, respectively, ensuring an unaffected chiral characteristic during the conversion (Scheme 2b).
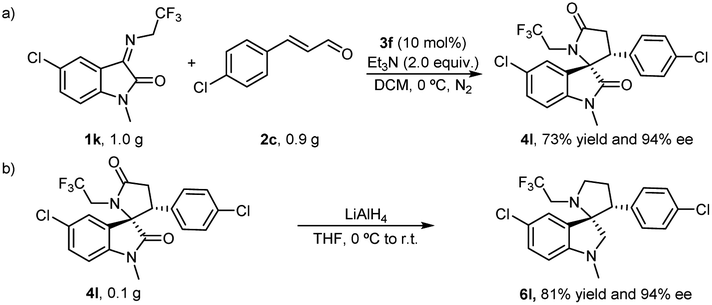 | ||
| Scheme 2 Synthetic applications. (a). Gram-scale experiment. (b). Synthetic transformations of (3S, 3′R)-4l. | ||
In summary, we have successfully performed a direct synthesis of trifluoroethyl 3,2′-spirooxindole γ-lactams (4) via the NHC-modulated asymmetric [2 + 3] annulation reaction of ketimines with in situ generated Breslow intermediate. Moreover, the origin of the selective [2 + 3] annulation reaction over the known [3 + 2] annulation reaction was revealed by DFT calculations. Diverse trifluoroethyl 3,2′-spirooxindole γ-lactams were prepared in good yields with excellent diastereo- and enantioselectivities. Further extension of this strategy to the synthesis of complex chiral molecules is in progress in our laboratory.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grant No.82273812), the “Thousand Talents Program” of Sichuan Province, the Health Commission of Sichuan Province (No.21PJ106), the National Interdisciplinary Innovation Team of Traditional Chinese Medicine (No.ZYYCXTD-D-202209), the Sichuan Outstanding Youth Fund Project of Chengdu University of Traditional Chinese Medicine (No. BJRC2020002), Shenzhen Commission of Science and Innovation programs (JCYJ20200109105613463).References
- E. Gras and S. Chassaing, Carbenes and Nitrenes, Wiley Online Library, The United States of America, 2019, pp. 219–249 Search PubMed.
- B. Ronald, On the Mechanism of Thiamine Action. IV.1 Evidence from Studies on Model Systems, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef.
- S. Mondal, A. Ghosh and A. T. N. Biju, Heterocyclic Carbene (NHC)-Catalyzed Transformations Involving Azolium Enolates, Chem. Rec., 2022, 22, e202200054 CrossRef CAS PubMed.
- (a) S. S. Sohn, E. L. Rosen and J. W. Bode, N-heterocyclic carbene-catalyzed generation of homoenolates: gamma-butyrolactones by direct annulations of enals and aldehydes, J. Am. Chem. Soc., 2004, 126, 14370–14371 CrossRef CAS PubMed; (b) V. Nair, S. Vellalath, M. Poonoth, R. Mohan and E. N. Suresh, heterocyclic carbene catalyzed reaction of enals and 1,2-dicarbonyl compounds: stereoselective synthesis of spiro gamma-butyrolactones, Org. Lett., 2006, 8, 507–509 CrossRef CAS PubMed; (c) M. Rommel, T. Fukuzumi and J. W. Bode, Cyclic ketimines as superior electrophiles for NHC-catalyzed homoenolate additions with broad scope and low catalyst loadings, J. Am. Chem. Soc., 2008, 130, 17266–17267 CrossRef CAS PubMed; (d) L. H. Sun, L. T. Shen and S. Ye, Highly diastereo- and enantioselective NHC-catalyzed [3+2] annulation of enals and isatins, Chem. Commun., 2011, 47, 10136–10138 RSC; (e) J. Dugal-Tessier, E. A. O'Bryan, T. B. Schroeder, D. T. Cohen and K. A. Scheidt, An N-heterocyclic carbene/Lewis acid strategy for the stereoselective synthesis of spirooxindole lactones, Angew Chem. Int. Ed. Engl., 2012, 51, 4963–4967 CrossRef CAS PubMed; (f) R. S. Menon, A. T. Biju and V. Nair, Recent advances in employing homoenolates generated by N-heterocyclic carbene (NHC) catalysis in carbon-carbon bond-forming reactions, Chem. Soc. Rev., 2015, 44, 5040–5052 RSC; (g) S. Mukherjee, S. Joseph, A. Bhunia, R. G. Gonnade, S. R. Yetra and A. T. Biju, Enantioselective synthesis of spiro γ-butyrolactones by N-heterocyclic carbene (NHC)-catalyzed formal [3 + 2] annulation of enals with 3-hydroxy oxindoles, Org. Biomol. Chem., 2017, 15, 2013–2019 RSC.
- S. Mondal, S. R. Yetra, S. Mukherjee and A. T. Biju, NHC-Catalyzed Generation of α,β-Unsaturated Acylazoliums for the Enantioselective Synthesis of Heterocycles and Carbocycles, Acc. Chem. Res., 2019, 52, 425–436 CrossRef CAS PubMed.
- (a) C. Guo, M. Schedler, C. G. Daniliuc and F. N. Glorius, heterocyclic carbene catalyzed formal [3+2] annulation reaction of enals: an efficient enantioselective access to spiro-heterocycles, Angew Chem. Int. Ed. Engl., 2014, 53, 10232–10236 CrossRef CAS PubMed; (b) X. Y. Chen, K. Q. Chen, D. Q. Sun and S. N. Ye, Heterocyclic carbene-catalyzed oxidative [3 + 2] annulation of dioxindoles and enals: cross coupling of homoenolate and enolate, Chem. Sci., 2017, 8, 1936–1941 RSC; (c) T. Azaz, H. Mourya, V. Singh, B. Ram and B. N. Tiwari, Heterocyclic Carbene-Catalyzed Enantioselective [3 + 2] Annulation of Enals with Vinyl Ketones, J. Org. Chem., 2023, 88, 1219–1226 CrossRef CAS PubMed.
- (a) J. Xu, S. Yuan and M. N. Miao, Heterocyclic Carbene Catalyzed [4 + 2] Annulation Reactions with in Situ Generated Heterocyclic ortho-Quinodimethanes, Org. Lett., 2016, 18, 3822–3825 CrossRef CAS PubMed; (b) X. Y. Chen, Q. Liu, P. Chauhan, S. Li, A. Peuronen, K. Rissanen and E. Jafari, N-Heterocyclic Carbene Catalyzed [4+2] Annulation of Enals via a Double Vinylogous Michael Addition: Asymmetric Synthesis of 3,5-Diaryl Cyclohexenones, Angew. Chem., Int. Ed. Engl., 2017, 56, 6241–6245 CrossRef CAS PubMed.
- (a) H. Z. Gui, Y. Wei and M. Shi, Recent Advances in the Construction of Trifluoromethyl-Containing Spirooxindoles through Cycloaddition Reactions, Chem.–Asian J., 2020, 15, 1225–1233 CrossRef CAS PubMed; (b) S. Nasri, M. Bayat and F. Mirzaei, Recent Strategies in the Synthesis of Spiroindole and Spirooxindole Scaffolds, Top. Curr. Chem., 2021, 379, 25 CrossRef CAS PubMed; (c) M. M. Li, H. Huang, Y. R. Pu, W. R. Tian, Y. Deng and J. Lu, A close look into the biological and synthetic aspects of fused pyrazole derivatives, Eur. J. Med. Chem., 2022, 243, 114739 CrossRef CAS PubMed; (d) S. S. Panda, A. S. Girgis, M. N. Aziz and M. S. Bekheit, Spirooxindole: A Versatile Biologically Active Heterocyclic Scaffold, Molecules, 2023, 28, 618 CrossRef CAS PubMed; (e) X. Zhang, Y. Chen, X. Li, H. Xu, J. R. Yang, C. C. Wang, C. Z. Zhang, Y. Deng, A. P. Lu, C. Zheng and J. Lu, Carrier-free self-assembled nanomedicine based on celastrol and galactose for targeting therapy of hepatocellular carcinoma via inducing ferroptosis, Eur. J. Med. Chem., 2024, 267, 116183 CrossRef CAS PubMed; (f) C. Q. Liu, Y. N. Cao, Y. Zuo, C. Z. Zhang, S. M. Ren, X. Zhang, C. Q. Wang, Y. J. Zeng, J. Ling, Y. L. Liu, Z. X. Chen, X. J. Cao, Z. Z. Wu, C. T. Zhang and J. Lu, Hybridization-based discovery of novel quinazoline-2-indolinone derivatives as potent and selective PI3Kα inhibitors, J. Adv. Res., 2024, 11(24), S2090 Search PubMed.
- (a) M. S. Islam, A. M. Al-Majid, E. N. Sholkamy, A. Barakat, M. Viale, P. Menichini, A. Speciale, F. Loiacono, M. Azam, V. P. Verma, S. Yousuf and M. Teleb, Optimized spirooxindole-pyrazole hybrids targeting the p53-MDM2 interplay induce apoptosis and synergize with doxorubicin in A549 cells, Sci. Rep., 2023, 13, 7441 CrossRef CAS PubMed; (b) B. R. Chahat and B. Kumar, p53 as a potential target for treatment of cancer: A perspective on recent advancements in small molecules with structural insights and SAR studies, Eur. J. Med. Chem., 2023, 247, 115020 CrossRef PubMed.
- H. Lv, B. Tiwari, J. Mo, C. Xing and Y. R. Chi, Highly enantioselective addition of enals to isatin-derived ketimines catalyzed by N-heterocyclic carbenes: synthesis of spirocyclic γ-lactams, Org. Lett., 2012, 14, 5412–5415 CrossRef CAS PubMed.
- M. X. Ma, Y. Y. Zhu, Q. T. Sun, X. Y. Li, J. H. Su, L. Zhao, Y. Y. Zhao, S. Qiu, W. J. Yan, K. R. Wang and R. Wang, The asymmetric synthesis of CF3-containing spiro[pyrrolidin-3,2’-oxindole] through the organocatalytic 1,3-dipolar cycloaddition reaction, Chem. Commun., 2015, 51, 8789–8792 RSC.
- S. J. Liu, Q. Mao, G. Zhan, R. Qin, B. H. Chen, J. Xue, M. L. Luo, Q. Zhao and B. Han, Stereoselective synthesis of trifluoroethyl 3,2’-spirooxindole γ-lactam through the organocatalytic cascade reaction of 3-((2,2,2-trifluoroethyl)amino)indolin-2-one, Org. Biomol. Chem., 2021, 19, 467–475 RSC.
- (a) S. R. Yetra, S. Mondal, S. Mukherjee, R. G. Gonnade and A. T. Biju, Enantioselective Synthesis of Spirocyclohexadienones by NHC-Catalyzed Formal [3+3] Annulation Reaction of Enals, Angew Chem. Int. Ed. Engl., 2016, 55, 268–272 CrossRef CAS PubMed; (b) H. Li, X. Li, Y. Yu, J. Li, Y. Liu, H. Li and W. Wang, Synthesis of Indolizines via Reaction of 2-Substitued Azaarenes with Enals by an Amine-NHC Relay Catalysis, Org. Lett., 2017, 19, 2010–2013 CrossRef CAS PubMed; (c) S. Shee, S. Barik, A. Ghosh and A. T. Biju, Synthesis of Functionalized Dihydrocoumarins by NHC-Catalyzed [3 + 3] Annulation of Enals with 2-Substituted Naphthoquinones, Org. Lett., 2021, 23, 8039–8044 CrossRef CAS PubMed; (d) T. Fan, J. Song and L. Z. Gong, Asymmetric Redox Allylic Alkylation to Access 3,3'-Disubstituted Oxindoles Enabled by Ni/NHC Cooperative Catalysis, Angew Chem. Int. Ed. Engl., 2022, 61, e202201678 CrossRef CAS PubMed.
- R. Song, Z. Jin and Y. R. Chi, NHC-catalyzed covalent activation of heteroatoms for enantioselective reactions, Chem. Sci., 2021, 12, 5037–5043 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 2106628. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02252a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |