
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and characterization of tetra substituted 2,3-dihydrothiazole derivatives as DNA and BSA targeting agents: advantages of the visible-light-induced multicomponent approach†
Ranjana Aggarwal *ab,
Naman Jaina and
Gyan Prakash Dubeya
*ab,
Naman Jaina and
Gyan Prakash Dubeya
aDepartment of Chemistry, Kurukshetra University, Kurukshetra 136119, Haryana, India
bCSIR-National Institute of Science Communication and Policy Research, New Delhi 110012, India. E-mail: ranjana67in@yahoo.com; ranjanaaggarwal67@gmail.com; Tel: +91-9896740740
First published on 22nd July 2024
Abstract
This report describes the visible-light-induced one-pot multicomponent regioselective synthesis of a series of 5-aroyl-3-((arylidene)amino)-2-((arylidene)hydrazono)-4-methyl-2,3-dihydrothiazoles as DNA and BSA targeting agents. The multicomponent condensation of thiocarbohydrazide and aldehydes with α-bromo-1,3-diketones, generated in situ by the bromination of unsymmetrical 1,3-diketones with NBS using white LED light as an environmental friendly source in the presence of EtOAc solvent furnished the titled 2,3-dihydrothiazole derivatives in excellent yields. The exact regioisomeric structure was identified unambiguously by employing multinuclear 2D-NMR spectroscopy [1H–13C] HMBC; [1H–13C] HMQC and [1H–15N] HMBC. Furthermore, the binding characteristics of the synthesized 2,3-dihydrothiazole derivatives were assessed with double-stranded calf-thymus DNA duplex (ct-DNA) and bovine serum albumin (BSA). Initial screening of all the synthesized 2,3-dihydrothiazole derivatives using various in silico techniques including molecular reactivity analysis, Lipinski rule and molecular docking, concluded 5-(4′-chlorobenzoyl)-3-((4′′-methoxybenzylidene)amino)-2-(4′′′-methoxybenzylidene)hydrazono)-4-methyl-2,3-dihydrothiazole derivative 6a as the most suitable compound for studying binding interaction with DNA and BSA. Additionally, to illustrate the ex vivo binding mode of 6a with DNA and BSA, several spectroscopic techniques viz. UV-visible, circular dichroism (CD), steady-state fluorescence and competitive displacement assays were carried out.
1. Introduction
Among the current research on mainstream azoles, the heteroaromatic thiazole nucleus gained extensive appreciation in drug development as a result of its fascinating implication in pharmaceuticals. The presence of N and S atoms enables the thiazole ring to interact with various biological receptors and help it to impart its therapeutic effect.1 As a consequence, the thiazole ring exhibits tremendous biological and chemotherapeutic significance such as anticancer,2 anti-inflammatory,3 anthelmintic,4 analgesic,5 antimicrobial,6 anti-HIV,7 antioxidant,8 antimalarial,9 antibiotic10 etc. Thiazole core also signifies its importance in various naturally occurring bioactive compounds such as Thiamin,11 Thiamine pyrophosphate,12 Epothilone,13 Mycothiazole14 etc. Apart from this, the thiazole core marks its prominence in other fields of science as well for instance as polymers, photosensitizers, semiconductors, fluorescent dyes, and dye-sensitized solar cells.Deoxyribonucleic acid (DNA) is one of the most important biomacromolecules as it contains the genetic information that controls the growth of almost all living organisms.15,16 Any modification in the basic functioning of DNA molecules may cause several types of diseases in the human body namely cystic fibrosis, sickle cell disease, genetic disorders, Huntington's disease etc. Recent research has revealed that the thiazole ring can preferentially interact with DNA, altering its functioning and thus being used to treat nucleic acid-dependent diseases. The small organic molecules can interact with DNA through non-covalent bonding mainly following one of the three possible mechanisms: intercalation, groove binding and electronic interactions.17 Thus, researchers have shifted their efforts to the design and synthesis of novel thiazole derivatives that can target the DNA molecule. Fig. 1 comprises the rationale drug designing of molecule under consideration and thiazole core-bearing compounds having the capability to interact with DNA.18–23
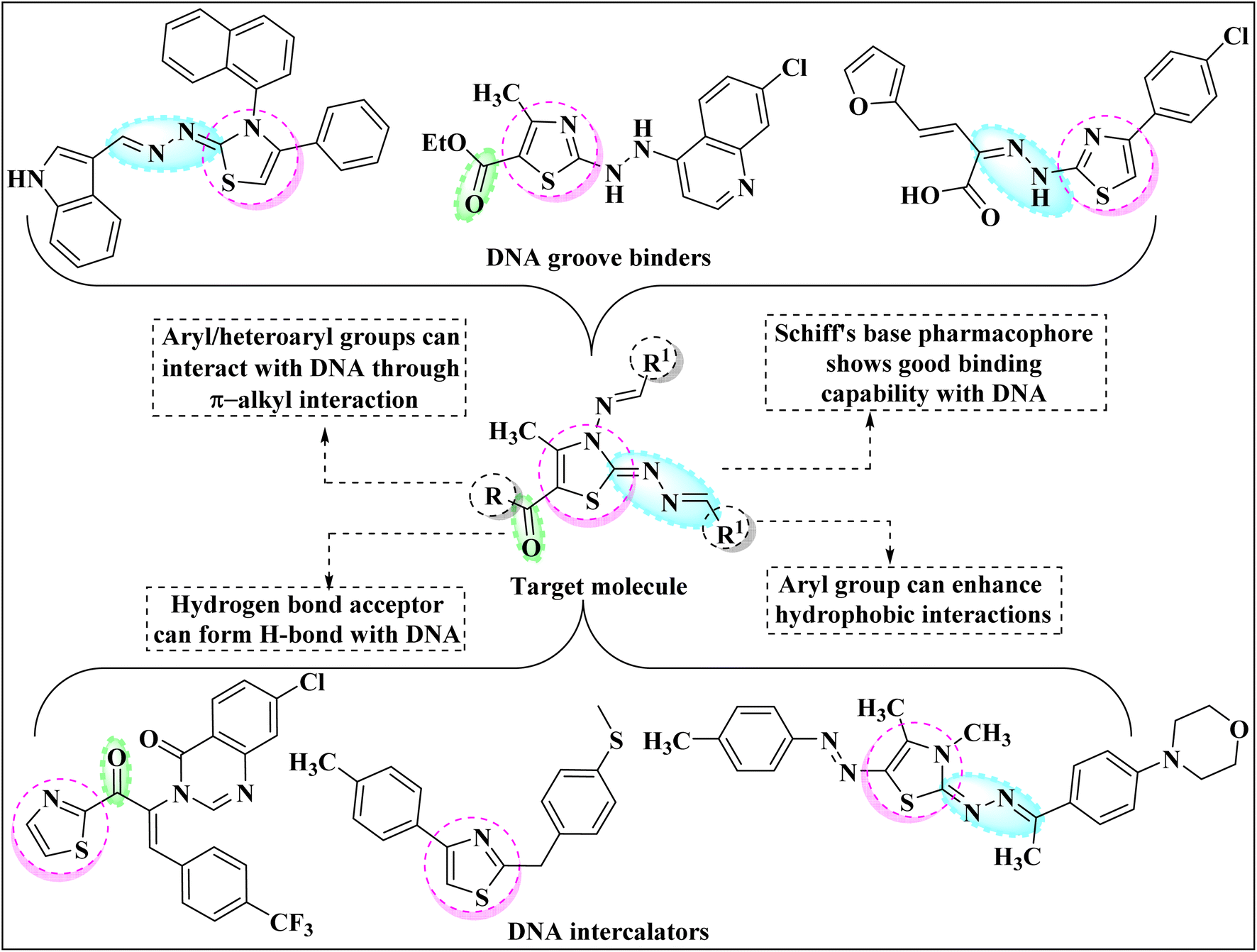 | ||
| Fig. 1 Potential of thiazole core as DNA binder and rational drug design. | ||
Serum albumins, also known as transporter proteins, are a type of biological receptor that has been extensively studied due to their diverse applications in the binding, transport, and distribution of exogenous and endogenous substances in vivo.24 Serum albumins play a vital role in drug developments as they are the first proteins which comes in contact with the substance after entering the organism. The interaction studies of small molecules with albumin proteins help in the better understanding of pharmacology and pharmacodynamics properties of novel compounds. Bovine Serum Albumins (BSA) exhibit 75% structural resemblance to Human Serum Albumins, thus widely used as a probe for studying the interactions of small molecules with albumin proteins. The BSA protein mainly consists of three binding sites namely domains I, II and III and each domain is further divided into two subdomains A and B. Recent literature survey reveals that fatty acids prefer to bind to sites IB, IIIA and IIIB whereas small heterocyclic compounds mostly bind to sub-domains IIA and IIIA.25
Our research group has been enthusiastically indulged in the synthesis of aza-heterocycles using green protocols such as solvent free grinding,26,27 use of green solvents28,29 and multi-component approach.30,31 Also, our research group has investigated the use of visible light as a sustainable energy source for the synthesis of aza-heterocycles, primarily azoles and exploring the possible biological applications of synthesized compounds as DNA binding agents.32–36 In light of widespread applications associated with thiazole core and continuing our previous research efforts, it was envisioned to undertake the one-pot multicomponent regioselective synthesis of 5-aroyl-3-((arylidene)amino)-2-((arylidene)hydrazono)-4-methyl-2,3-dihydrothiazole derivatives. The reaction was designed to follow a multicomponent pathway in which thiocarbohydrazide and aldehydes were condensed with α-bromo-1,3-diketones (generated in situ by the bromination of unsymmetrical 1,3-diketones with NBS) under visible light in the presence of EtOAc solvent. The final structure of the regioisomer was identified unambiguously using multinuclear 2D NMR techniques viz. [1H–13C] HMBC; [1H–13C] HMQC and [1H–15N] HMBC.
Furthermore, the biological potential of the synthesized 2,3-dihydrothiazole derivatives was investigated as DNA and BSA targets using various in silico studies including molecular reactivity analysis, Lipinski rule, molecular docking and several spectroscopic techniques viz. UV-visible, circular dichroism (CD), steady-state fluorescence and competitive displacement assay.
2. Results and discussion
2.1 Chemistry
Recently Vedula et al., reported the synthesis of coumarin-linked thiazole derivatives via one-pot multicomponent reaction of thiocarbohydrazide, aldehydes (2 eq.) and substituted 3-(2-bromoacetate) coumarins in the presence of ethanol as a solvent under microwave irradiation using a catalytic amount of acetic acid.37Taking a lead from here and in continuation of our research work related to the study of the regioselectivity pattern of the reaction of unsymmetrical 1,3-diketones with bi/tri nucleophilic systems herein, we intend to conduct the synthesis of 5-aroyl-3-((arylidene)amino)-2-((arylidene)hydrazono)-4-methyl-2,3-dihydrothiazole. For this purpose, 4-methoxybenzaldehyde 2a and 1-(4-chlorophenyl)butane-1,3-dione 4a were chosen as the model substrates to achieve the optimum reaction conditions for the synthesis of the title compounds. The reaction optimization was achieved testing two pathways, Path A and Path B. Scheme 1 depicts the model reaction for the regioselective synthesis of tetra substituted 2,3-dihydrothiazole following two pathways, Path A and Path B.
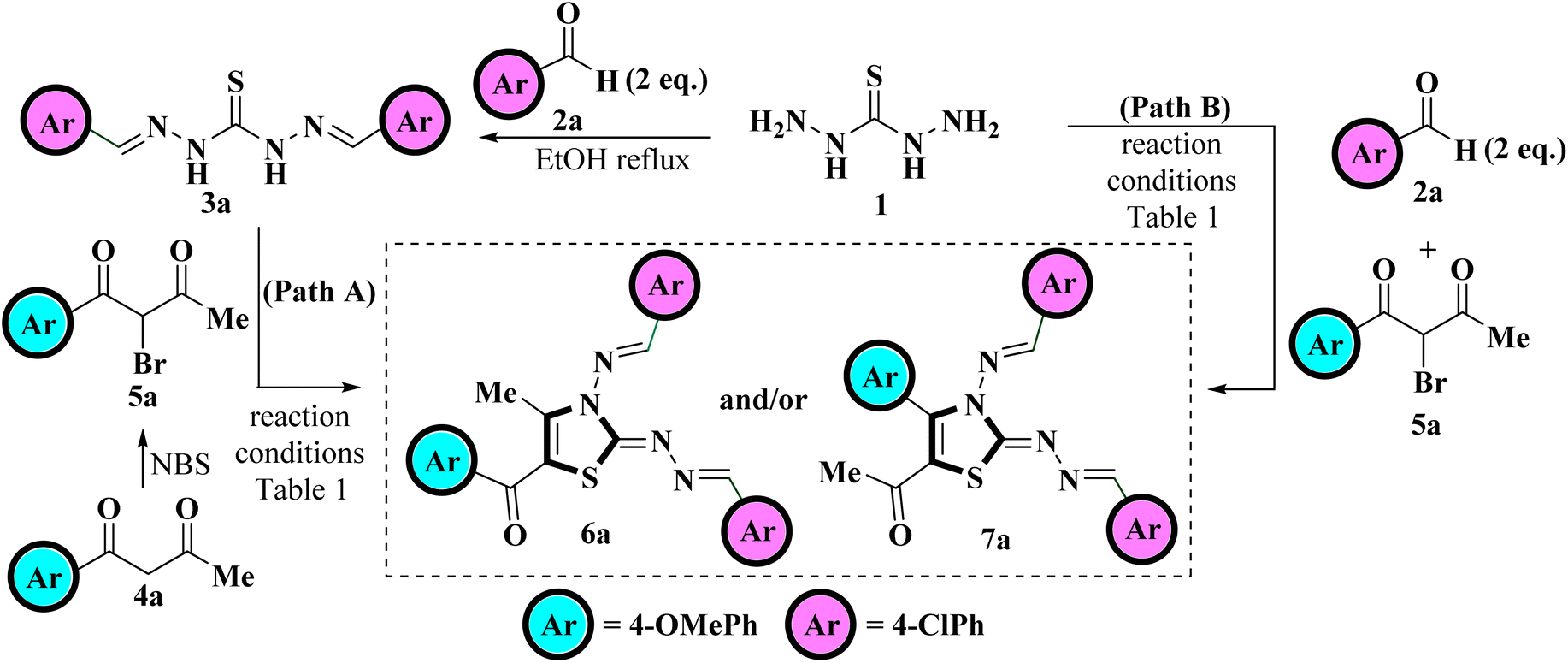 | ||
| Scheme 1 Model reaction for the synthesis of tetra substituted 2,3-dihydrothiazole. | ||
Path A involved the step-wise protocol in which thiocarbohydrazide 1 was first allowed to condensed with 4-methoxy benzaldehyde 2a under ethanol reflux to generate a hydrazine analogous intermediate 3a. The reaction was found to be completed within short time spam of 30 minutes as indicated by TLC. The intermediate 3a was then subjected to react with 2-bromo-1-(4-chlorophenyl)butane-1,3-dione 5a, generated in situ through bromination of 4-chlorophenylbutane-1,3-dione 4a with mild brominating reagent NBS in polar aprotic (DCM, THF, MeCN, EtOAc) as well as polar protic (EtOH, MeOH, H2O) solvents under various conditions, including conventional heating and visible light irradiation. A careful monitoring of the reaction progress evidenced that the reaction does not proceed at all in DCM even at elevated temperatures (Table 1, entries 1 and 2) but traces were obtained when the reaction was carried out in THF and MeCN (Table 1, entries 3–5). The reaction was then monitored in EtOAc at elevated temperature and a careful monitoring of the reaction progress through TLC highlights the complete consumption of the reactants within 6 h but furnished the product in low yield (Table 1, entry 6). The reaction was then monitored in polar protic solvents at elevated temperatures but yield of the product was found to be deprived further (Table 1, entries 7–9). To obtain a higher product yield, the reaction was also investigated under visible light irradiation. Astonishingly, the reaction was observed to be completed in a shorter time as indicated by the TLC, where EtOAc was found to provide the desired product with the highest yield (Table 1, entries 10–13). Thus, visible-light-mediated reactions have an advantage over conventional heating because of the short reaction time along with the high product yield.
| Entry | Solvent | Reaction conditionsa | Timeb/c | Yieldd/e/f (%) |
|---|---|---|---|---|
| a Reaction conditions: reaction of 1 (1 mmol), 2a (2 mmol) with 4a (1 mmol) proceeded under a different set of reaction conditions. RT = room temperature.b Time taken for step-wise protocol (Path A).c Time taken for one-pot multicomponent protocol (Path B).d NR = no reaction.e Isolated yield of step-wise reaction (Path A).f Isolated yield of one-pot multicomponent reaction (Path B). | ||||
| 1 | DCM | RT | 36 hb/c | NRd |
| 2 | DCM | Reflux | 6 hb/c | NRd |
| 3 | THF | RT | 36 hb/c | NRd |
| 4 | THF | Reflux | 6 h]b/c | Tracese/f |
| 5 | MeCN | Reflux | 6 hb/c | Tracese/f |
| 6 | EtOAc | Reflux | 6 hb/3 hc | 42e/64f |
| 7 | MeOH | Reflux | 6 hb/4 hc | 35e/48f |
| 8 | EtOH | Reflux | 6 hb/4 hc | 38e/54f |
| 9 | H2O | Reflux | 6 hb/5 hc | 30e/42f |
| 10 | MeOH | White LED (9W) | 50 min.b/40 min.c | 53e/78f |
| 11 | EtOH | White LED (9W) | 45 min.b/40 min.c | 60e/83f |
| 12 | H2O | White LED (9W) | 60 min.b/50 min.c | 40e/68f |
| 13 | EtOAc | White LED (9W) | 40 min.b/30 min.c | 72e/88f |
After examining the reaction conditions for the step-wise protocol (Path A), we investigate the course of the reaction using a one-pot multicomponent approach (Path B), in which thiocarbohydrazide 1, 4-methoxybenzaldehyde 2a, and 2-bromo-1-(4-chlorophenyl)butane-1,3-dione 5a were allowed to react together in a one-pot. No significant change was observed when the reaction was carried out in DCM, THF and MeCN (Table 1, entries 1–5), whereas considerable enhancement in the product yield was noticed when the reaction was carried out in EtOAc and polar protic solvents (Table 1, entries 6–13) with EtOAc being the most promising solvent when subjected to white LED light yielding 88% product (Table 1, entries 13). Also, reaction was found to be completed in lesser time in one-pot multicomponent strategy than step-wise protocol (Table 1). Thus one-pot multicomponent approach was found to be superior to the step-wise procedure where the reaction proceeds smoothly in polar aprotic EtOAc solvent when irradiated with white LED light and furnished the target product in higher yields.
After getting optimized reaction conditions in hand the next task was to extend the substrate scope of this one-pot multicomponent approach under visible light. To meet the objective, thiocarbohydrazide 1, was allowed to condense with various aromatic aldehydes 2a–c and α-bromo-1,3-diketones 5a–f (generated in situ by the bromination of unsymmetrical 1,3-diketones 4a–f with NBS) under irradiation of white LED light (9W) using EtOAc as a solvent (Scheme 2). This multicomponent approach furnished the target compounds at room temperature in excellent yields in just 30–50 minutes which was later on identified as 5-aroyl-3-((arylidene)amino)-2-((arylidene)hydrazono)-4-methyl-2,3-dihydrothiazole 6a–n Table 2. The reaction exhibits great functional group tolerance with variously substituted electron-donating as well as electron-withdrawing aldehydes 2a–c and unsymmetrical 1,3-diketones 4a–f. It has been noticed that the presence of an electron-donating group at the para position of the phenyl ring either on aldehyde or diketone furnished the desired product in high yields in comparison to the electron-withdrawing groups. However, the presence of an electron-donating group at the meta position of the phenyl ring or heteroaryl thienyl ring in diketone diminished the product yield.
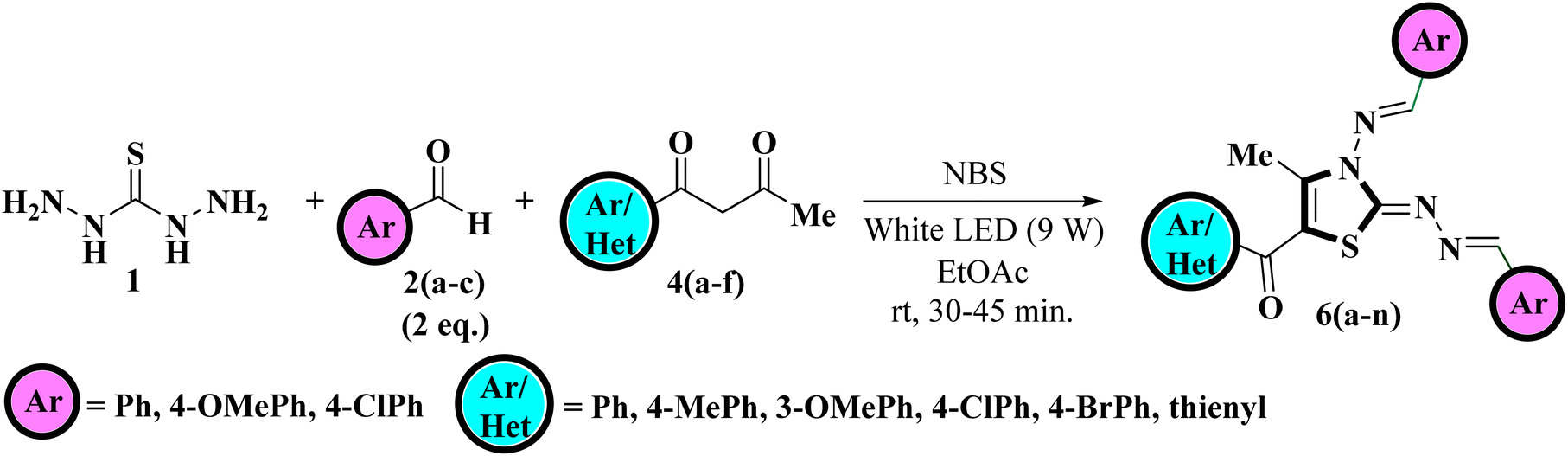 | ||
| Scheme 2 Visible-light-mediated one-pot multicomponent approach for the regioselective synthesis of title compounds. | ||
| Sr, no. | Structure | Compound |
|
Yield (%) |
|---|---|---|---|---|
| 1 |  |
6a | 4-ClPh | 88 |
| 2 | 6b | 4-BrPh | 90 | |
| 3 | 6c | Ph | 92 | |
| 4 | 6d | 4-MePh | 93 | |
| 5 | 6e | Thienyl | 85 | |
| 6 |  |
6f | 4-BrPh | 87 |
| 7 | 6g | Ph | 89 | |
| 8 | 6h | 4-MePh | 92 | |
| 9 | 6i | 3-OMePh | 84 | |
| 10 | 6j | Thienyl | 85 | |
| 11 |  |
6k | 4-BrPh | 88 |
| 12 | 6l | 4-MePh | 87 | |
| 13 | 6m | 3-OMePh | 83 | |
| 14 | 6n | Thienyl | 82 |

The IR spectrum of resulted compound 6a displayed two characteristic absorption bands at 1612 cm−1 and 1592 cm−1 corresponding to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N and carbonyl group respectively. 1H NMR spectrum of the compound 6a displayed three singlets in the aliphatic region at δ 2.52 ppm, δ 3.88 ppm and δ 3.85 ppm integrated for three protons each corresponding to one set of methyl protons and two sets of methoxy protons along with two more singlets in the aromatic region at δ 10.13 ppm and δ 8.36 ppm integrated for one proton each consistent to two methine protons. Also, a regular pattern of twelve protons in the aromatic region was observed incorporated from one equivalent of 1-(4-chlorophenyl)butane-1,3-dione 4a and two equivalents of 4-methoxybenzaldehyde 2a indicating the success of the reaction. Additionally, three sharp singlets in the aliphatic region at δ 55.5 ppm, δ 55.4 ppm and δ 15.6 ppm and one in the aromatic region at δ 187.3 ppm in the 13C NMR spectrum corresponding to one methyl, two methoxy and one carbonyl carbon respectively along with a set of seventeen desired carbon signals in the aromatic region confirms the successful condensation of 1 and 2a with 4a.
N and carbonyl group respectively. 1H NMR spectrum of the compound 6a displayed three singlets in the aliphatic region at δ 2.52 ppm, δ 3.88 ppm and δ 3.85 ppm integrated for three protons each corresponding to one set of methyl protons and two sets of methoxy protons along with two more singlets in the aromatic region at δ 10.13 ppm and δ 8.36 ppm integrated for one proton each consistent to two methine protons. Also, a regular pattern of twelve protons in the aromatic region was observed incorporated from one equivalent of 1-(4-chlorophenyl)butane-1,3-dione 4a and two equivalents of 4-methoxybenzaldehyde 2a indicating the success of the reaction. Additionally, three sharp singlets in the aliphatic region at δ 55.5 ppm, δ 55.4 ppm and δ 15.6 ppm and one in the aromatic region at δ 187.3 ppm in the 13C NMR spectrum corresponding to one methyl, two methoxy and one carbonyl carbon respectively along with a set of seventeen desired carbon signals in the aromatic region confirms the successful condensation of 1 and 2a with 4a.
The IR data, together with 1H and 13C NMR, provide conclusive evidence in favour of the completion of the reaction; however, these techniques alone were insufficient to identify the correct regioisomeric structure of the resulting product. Based on our previous experience with regioselective synthesis of aza-heterocycle, heteronuclear 2D NMR techniques such as [1H–13C] HMBC; [1H–13C] HMQC and [1H–15N] HMBC were utilized to identify the exact regioisomeric structure. The [1H–13C] HMBC spectrum of the resulted compound 5-(4′-chlorobenzoyl)-3-((4′′-methoxybenzylidene)amino)-2-(4′′′-methoxybenzylidene) hydrazono)- 4-methyl-2,3-dihydrothiazole 6a exhibits correlation peaks of the methyl protons (δ 2.52 ppm) with C-4 (δ 148.6 ppm) and C-5 (δ 111.3 ppm) indicative of the presence of methyl group at C-4 position of thiazole core. Also, the carbonyl carbon appeared at δ 187.3 ppm displays cross peaks with the 2′/6′- proton (δ 7.74–7.70 ppm) of the phenyl ring signifying the presence of carbonyl carbon next to the phenyl ring. Likewise, [1H–15N] HMBC spectrum demonstrates the cross peaks of the N atom (δ 306.0 ppm) with methyl protons (δ 2.52 ppm) at C-4 position. Furthermore, the diazo N atoms at N-9 (δ 346.5 ppm) and N-10 (δ 278.1 ppm) shows correlation peaks with methylene protons (δ 8.36 ppm) at C-7 position.28 Lastly, the N atom (δ 278.2 ppm) displays cross peaks with methine protons (δ 10.13 ppm) at C-6 position. These outcomes are in great agreement with our previous results related to the regioselective synthesis of thiazole derivative where carbonyl carbon was found next to the aryl ring and methyl group at C-4 position of the thiazole ring.26,36 Thus, the structure of regioisomer 6a was confirmed as 5-(4′-chlorobenzoyl)-3-((4′′-methoxybenzylidene)amino)-2-(4′′′-methoxybenzylidene)hydrazon-o)-4-methyl-2,3-dihydrothiazole. A similar correlation pattern of [1H–13C] HMBC; [1H–13C] HMQC and [1H–15N] HMBC were noticed for 5-(4′-bromobenzoyl)-3-((4′′-methoxybenzylidene)amino)-2-(4′′′-methoxybenzylidene)hydrazono)-4-methyl-2,3-dihydrothiazole 6b as shown in Fig. 2.
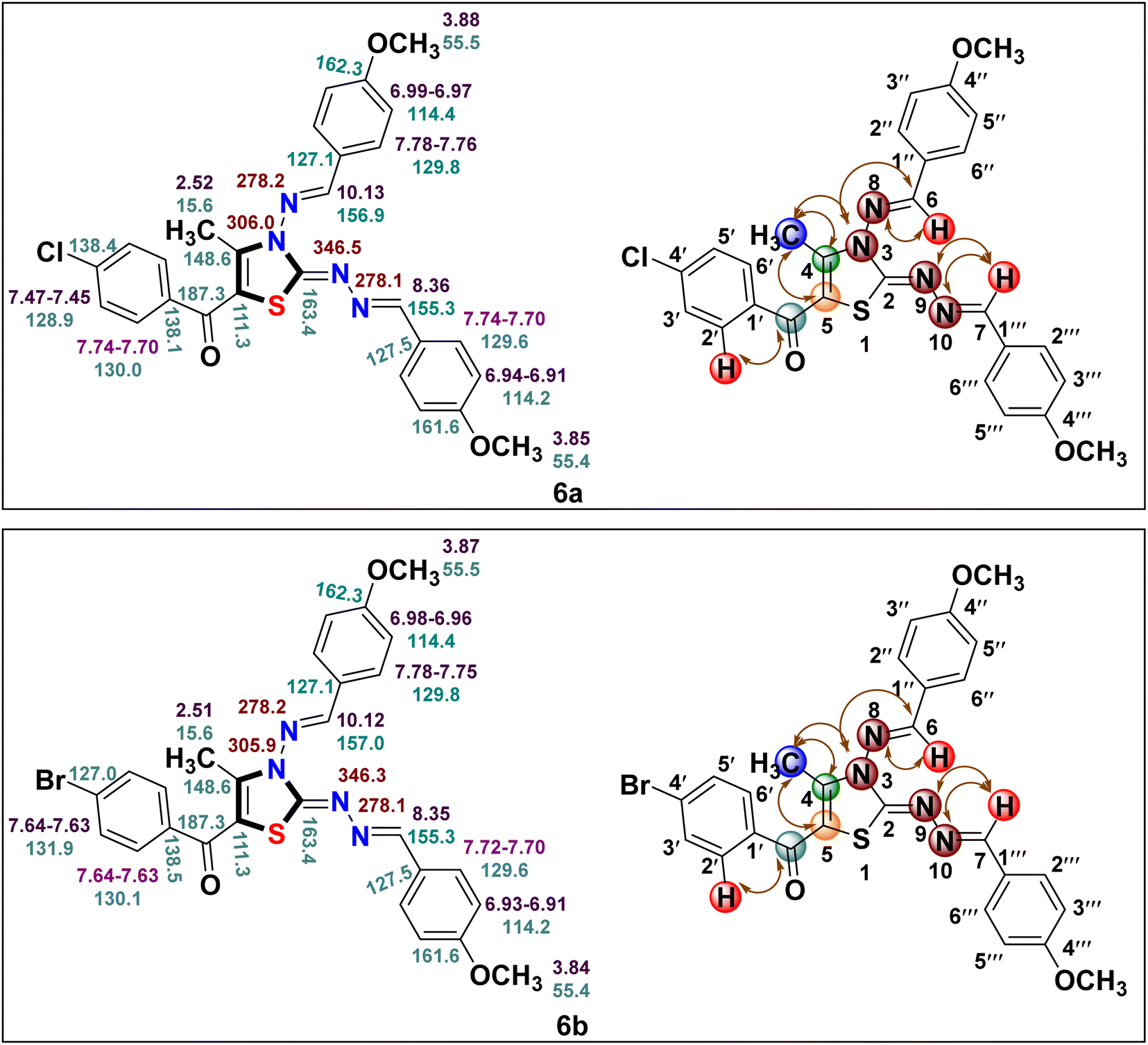 | ||
| Fig. 2 1H (in purple), 13C (in blue) and 15N (in brown) chemical shifts for compounds 6a and 6b along with correlation representation. | ||
The plausible mechanistic pathway for the regioselective synthesis of title 5-aroyl-3-((arylidene)amino)-2-((arylidene)hydrazono)-4-methyl-2,3-dihydrothiazole derivatives is illustrated in Scheme 3. Primarily, in situ formation of hydrazine analogous intermediate 3 was taken place by the reaction of variously substituted aldehydes 2 and thiocarbohydrazide 1. The homolytic bond fission of C–Br bond of α-bromo-1,3-diketones 5 and S–H bond of hydrazine analogous 3 results in the formation of two free radical intermediates 8 and 9 respectively due to the exposure of visible light.38 These two free radicals were then combined via electron sharing to form S-alkylated intermediate 10. Following that, the nucleophilic attack of a lone pair of electrons over the N atom take place on the carbonyl carbon. As two electrophilic carbonyl centres of different electrophilicities are present thus two paths are possible Path a and Path b. However, attack take place at the less sterically hindered carbonyl carbon next to the methyl group yields the cyclized intermediate 11, this ruled out the possibility of formation of other regioisomer 12. Finally, cyclised regioisomer 11 underwent removal of water to furnished the final 2,3-dihydrothiazole derivatives 6.
 | ||
| Scheme 3 Mechanistic pathway for the one-pot multicomponent approach towards regioselective synthesis of 2,3-dihydrothiazole derivatives 6. | ||
2.2 Biological assay
| Comd. | EHOMO | ELUMO | ΔE | IP | EA | η | S | χ | μ | ω |
|---|---|---|---|---|---|---|---|---|---|---|
| 6a | −3.569 | −2.283 | 1.286 | 3.569 | 2.283 | 0.643 | 0.777 | 2.926 | −2.926 | 6.657 |
| 6b | −3.692 | −2.285 | 1.407 | 3.692 | 2.285 | 0.703 | 0.710 | 2.988 | −2.988 | 6.347 |
| 6c | −4.190 | −2.290 | 1.900 | 4.190 | 2.290 | 0.950 | 0.526 | 3.240 | −3.240 | 5.525 |
| 6d | −3.994 | −2.289 | 1.705 | 3.994 | 2.289 | 0.852 | 0.586 | 3.141 | −3.141 | 5.788 |
| 6e | −2.626 | −2.269 | 0.357 | 2.626 | 2.269 | 0.178 | 2.801 | 2.447 | −2.447 | 16.780 |
| 6f | −3.783 | −2.284 | 1.499 | 3.783 | 2.284 | 0.749 | 0.667 | 3.033 | −3.033 | 6.139 |
| 6g | −4.322 | −2.952 | 1.370 | 4.322 | 2.952 | 0.685 | 0.729 | 3.637 | −3.637 | 9.655 |
| 6h | −4.063 | −2.287 | 1.776 | 4.063 | 2.287 | 0.888 | 0.563 | 3.175 | −3.175 | 5.676 |
| 6i | −4.178 | −2.298 | 1.880 | 4.178 | 2.298 | 0.940 | 0.531 | 3.238 | −3.238 | 5.577 |
| 6j | −2.992 | −2.282 | 0.710 | 2.992 | 2.282 | 0.355 | 1.408 | 2.637 | −2.637 | 9.794 |
| 6k | −3.676 | −2.282 | 1.394 | 3.676 | 2.282 | 0.697 | 0.717 | 2.979 | −2.979 | 6.366 |
| 6l | −3.978 | −2.285 | 1.693 | 3.978 | 2.285 | 0.846 | 0.590 | 3.131 | −3.131 | 5.792 |
| 6m | −4.100 | −2.286 | 1.814 | 4.100 | 2.286 | 0.907 | 0.551 | 3.193 | −3.193 | 5.620 |
| 6n | −2.535 | −2.256 | 0.281 | 2.537 | 2.256 | 0.140 | 3.558 | 2.396 | −2.396 | 20.438 |
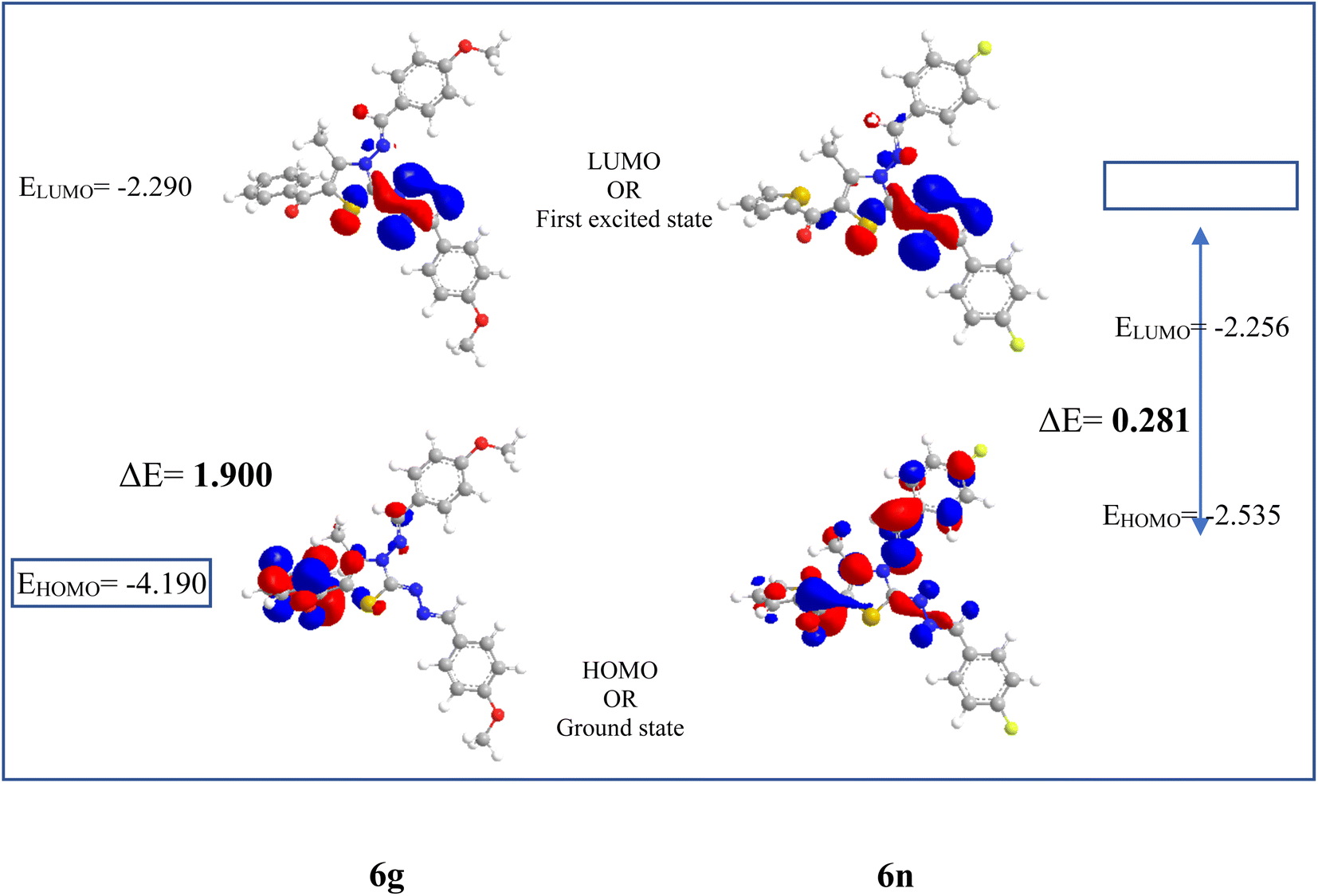 | ||
| Fig. 3 Frontier orbitals and energy gap between them for the compounds 6c and 6n. | ||
Furthermore, Pearson and Parr describe some descriptors viz., chemical hardness (η = 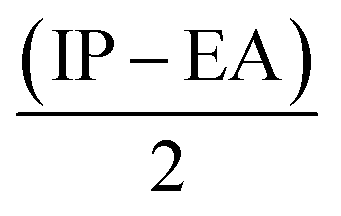 ), chemical softness (S =
), chemical softness (S = 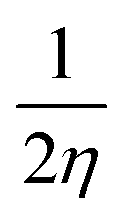 ), electronegativity (χ =
), electronegativity (χ =  ), chemical potential (μ =
), chemical potential (μ = 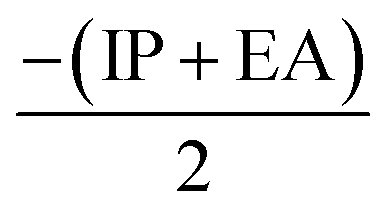 ) and electrophilicity index (ω =
) and electrophilicity index (ω =  ) known as Global reactivity descriptors40 and are highlighted in the Table 3.
) known as Global reactivity descriptors40 and are highlighted in the Table 3.
From Table 3 it was concluded that compound 6c has highest value of hardness (η = 0.950 eV) thus be the most stable and inert among all the tested compounds whereas, 6n was found to exhibits the highest value of softness (S = 3.558 eV) indicates the lesser stability and more reactivity of the ligand. Furthermore, the electronegativity was found to be highest for the compound 6g (χ = 3.637 eV), indicates the high reducing potential of the ligand 6g. The electrophilicity index, ability of a ligand to accept an electron, was found to largest for the compound 6n (ω = 20.438 eV).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (logarithm of partition coefficient between water and n-octanol) helps to determine the compound hydrophilicity and ligands with CLogP < 5 were found to exhibit good oral-bioavailability but unfortunately all the synthesized derivatives have slightly higher value of CLogP. Also, all the synthesized derivatives have a LogS value over −3.0, indicates the poor solubility of the compounds in water. Furthermore, LogKp values, a parameter used to describe the skin permeability are within the standard range of −1.0 to −8.0. Thus, majority of the synthesized compounds have the values of Lipinski rule of 5 within the desired range with few derivatives showing one or two violations. However, compounds with two or more violations (6k, 6l and 6m) of the Lipinski rule are found to have poor bioavailability.42
P (logarithm of partition coefficient between water and n-octanol) helps to determine the compound hydrophilicity and ligands with CLogP < 5 were found to exhibit good oral-bioavailability but unfortunately all the synthesized derivatives have slightly higher value of CLogP. Also, all the synthesized derivatives have a LogS value over −3.0, indicates the poor solubility of the compounds in water. Furthermore, LogKp values, a parameter used to describe the skin permeability are within the standard range of −1.0 to −8.0. Thus, majority of the synthesized compounds have the values of Lipinski rule of 5 within the desired range with few derivatives showing one or two violations. However, compounds with two or more violations (6k, 6l and 6m) of the Lipinski rule are found to have poor bioavailability.42
| C. No. | TPSA | MW | cLogP | nHBA | nHBD | RB | Violations | LogS |
LogKp |
|---|---|---|---|---|---|---|---|---|---|
| 6a | 105.8 | 519.02 | 5.8 | 6 | 0 | 8 | 1 | −7.1 | −4.9 |
| 6b | 105.8 | 563.47 | 5.8 | 6 | 0 | 8 | 1 | −7.4 | −5.1 |
| 6c | 105.8 | 484.58 | 5.2 | 6 | 0 | 8 | 0 | −6.5 | −5.1 |
| 6d | 105.8 | 498.61 | 5.5 | 6 | 0 | 8 | 0 | −6.8 | −5.0 |
| 6e | 134.0 | 490.61 | 5.2 | 6 | 0 | 8 | 0 | −6.5 | −5.1 |
| 6f | 87.3 | 503.42 | 5.2 | 4 | 0 | 6 | 0 | −6.5 | −5.1 |
| 6g | 87.3 | 424.53 | 5.2 | 4 | 0 | 6 | 0 | −6.5 | −5.1 |
| 6h | 87.3 | 438.55 | 5.2 | 4 | 0 | 6 | 0 | −6.5 | −5.1 |
| 6i | 96.5 | 454.55 | 5.2 | 5 | 0 | 7 | 0 | −6.5 | −5.1 |
| 6j | 115.6 | 430.55 | 5.2 | 4 | 0 | 6 | 0 | −6.5 | −5.1 |
| 6k | 87.3 | 539.40 | 6.4 | 6 | 0 | 6 | 2 | −7.6 | −4.8 |
| 6l | 87.3 | 474.53 | 6.4 | 6 | 0 | 6 | 2 | −6.7 | −4.8 |
| 6m | 96.5 | 490.53 | 5.8 | 7 | 0 | 7 | 2 | −6.7 | −4.8 |
| 6n | 115.6 | 466.54 | 5.8 | 6 | 0 | 6 | 1 | −6.7 | −4.8 |
For the docking analysis of the synthesized compounds with ct-DNA, structure of the DNA was required which was obtained from the protein data bank (PDB ID:1bna) having a sequence [d (CGCGAATTCGCG)2]. The binding affinities of all the synthesized derivatives with ct-DNA along with their three-dimensional poses and interactive binding sites are displayed in Table 5. All the derivatives were found to bind with DNA in the minor groove through non-covalent interactions including H-bond, pi-anion, pi–pi stacking and pi-donor. From the outcomes it has been witnessed that bromo derivatives (6b, 6f and 6k) were fail to interact with ct-DNA due to large size of bromine atom whereas, thienyl derivatives (6e, 6j and 6n) were found to have low binding affinities for ct-DNA. Furthermore, compound 6a was found to exhibit maximum binding affinity for the ct-DNA molecule.
| Cmpd. | 3D plot of binding interactions | Binding affinity (kcal mol−1) | H-bond | Residual nitrogenous base interactions | |
|---|---|---|---|---|---|
| Hydrophobic/pi–cation/pi–anion/pi–alkyl interactions | van der Walls interactions | ||||
| 6a | 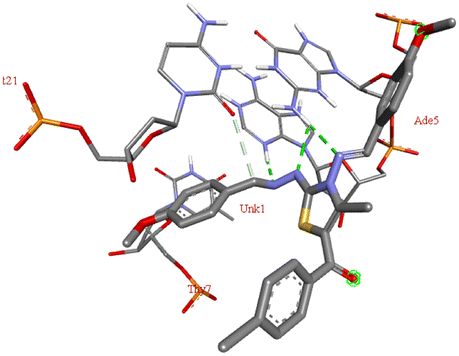 |
−8.4 | DG A:4, DA A:5 | DT A:7, DC B:21 | — |
| 6b | No interactions found | −7.0 | — | — | — |
| 6c | 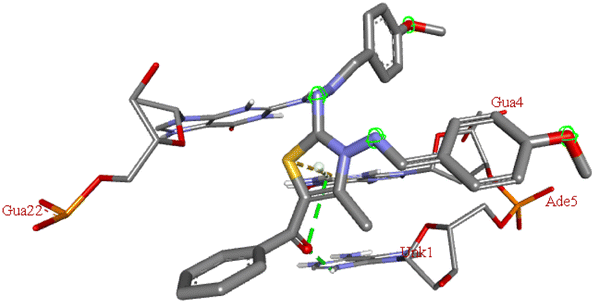 |
−7.3 | DG A:4, DA A:5, DG B:22 | — | — |
| 6d |  |
−6.7 | DC A:3, DG B:22 | DG A:4 | — |
| 6e | 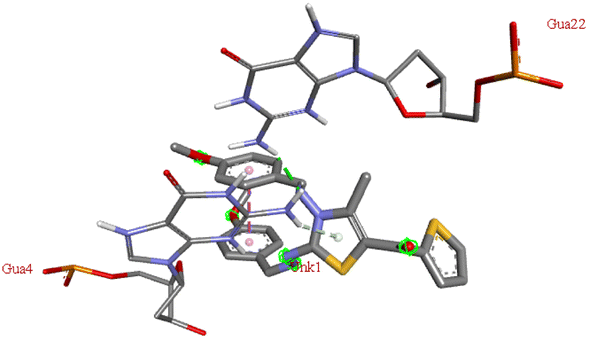 |
−6.1 | DG B:22 | DG A:4 | — |
| 6f | No interactions found | −6.8 | — | — | — |
| 6g | 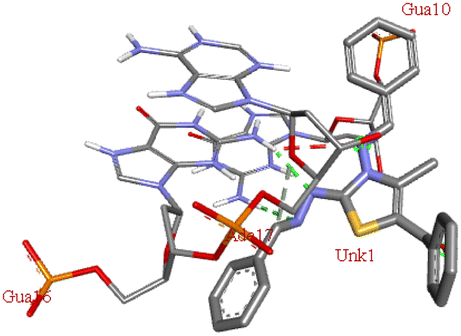 |
−7.8 | DG A: 10, DG B:16 | DA B:17 | — |
| 6h | 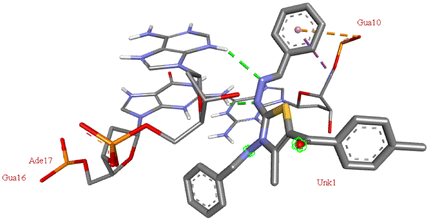 |
−8.1 | DG B: 16, DA B:17 | DG A:10 | — |
| 6i | 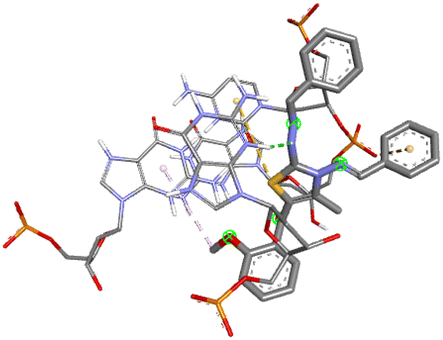 |
−7.6 | DG B: 16 | DC A:11, DG A:12 | — |
| 6j |  |
−6.0 | DG A: 4 | DC B:21 | — |
| 6k | No interactions found | −5.8 | — | ||
| 6l |  |
−7.8 | DG B:22 | DG A:4, DC B:23 | — |
| 6m | 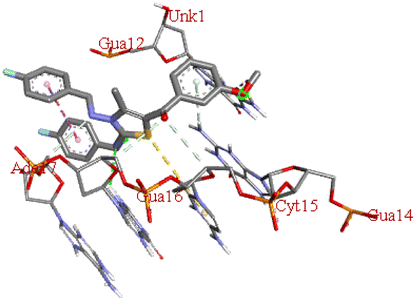 |
−7.8 | DG B: 16 | DG A:12, DG B:14, DC B:15, DA B:17 | — |
| 6n | 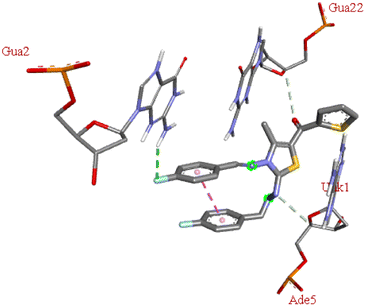 |
−6.6 | DG A:2 | DA A:5, DG B:22 | — |
The compound 5-(4′-chlorobenzoyl)-3-((4′′-methoxybenzylidene)amino)-2-(4′′′-methoxybenzylidene) hydrazono)-4-methyl-2,3-dihydrothiazole 6a was found to exhibit maximum binding affinity −8.4 kcal mol−1 for the ct-DNA molecule. Furthermore, the N atoms of the hydrazinyl residue and imino group of the compound 6a were found to participate in H-bonding with guanidine and adenine residues present at 4th and 5th position of the DNA sequence respectively. Fig. 4 displays the lowest energy 2d and 3d poses of the interaction between ligand 6a and DNA.
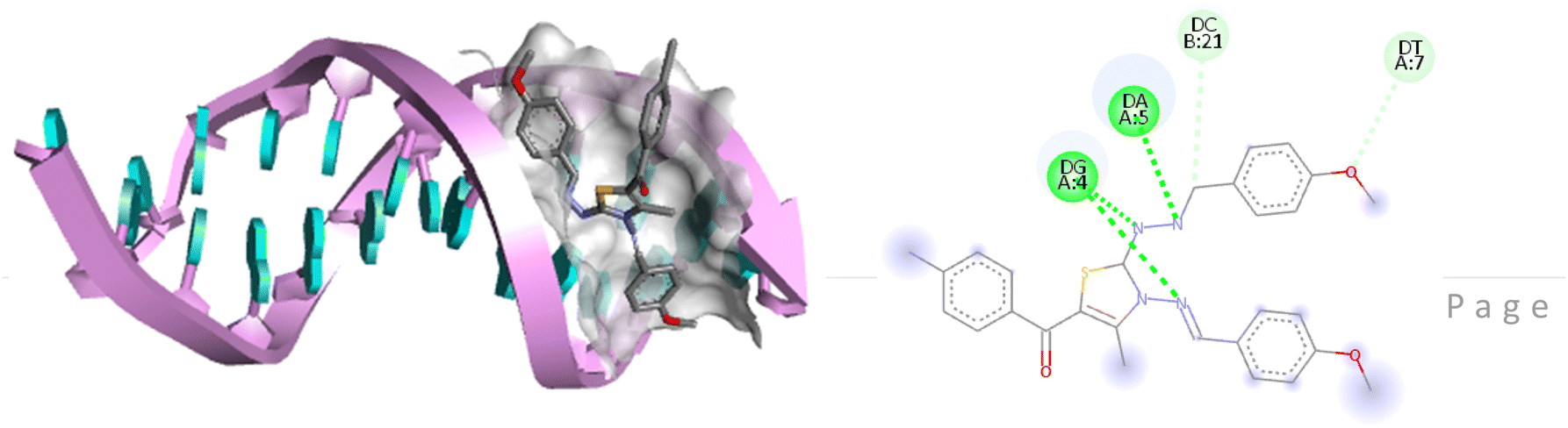 | ||
| Fig. 4 2D and 3D poses of the interaction of ligand 6a with DNA protein. | ||
Although, compound 6a shows one violation of Lipinski rule due to its molecular weight exceeding 500 Da, but high binding affinity with DNA and lesser stability/high reactivity (ΔE = 1.286 eV) of 6a after thienyl derivatives make it a good candidate for further binding studies with DNA using various spectroscopic techniques such as UV-absorption, fluorescence, and CD spectroscopy.
Thus, UV-visible analysis was carried out to identify the interactions between test ligand 6a and ct-DNA. In the process, UV-visible spectra of a fixed concentration of DNA in Tris–HCl (67 μM, pH 7.2) were examined in the absence as well as the presence of increasing concentration of ligand 6a (0–20 μM) in the spectral range of 200–500 nm at room temperature. An absorption maximum at 260 nm was observed for the native DNA which shows an increment with an increasing concentration of ligand 6a. From the Fig. 5, it is evident that a hyperchromic shift in the absorption maxima occurs with increasing concentration of ligand 6a suggesting the groove binding mode ligand.
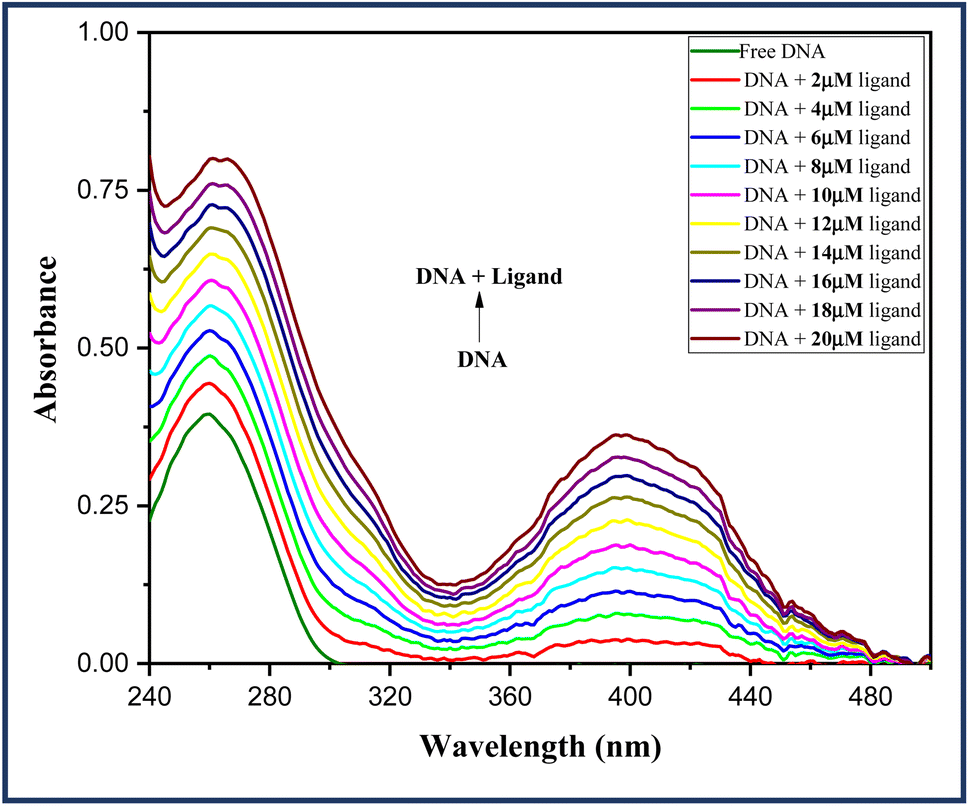 | ||
| Fig. 5 Absorption spectra of DNA (67 μM) in the absence and presence of an increasing concentration of ligand 6a (0–20 μM). | ||
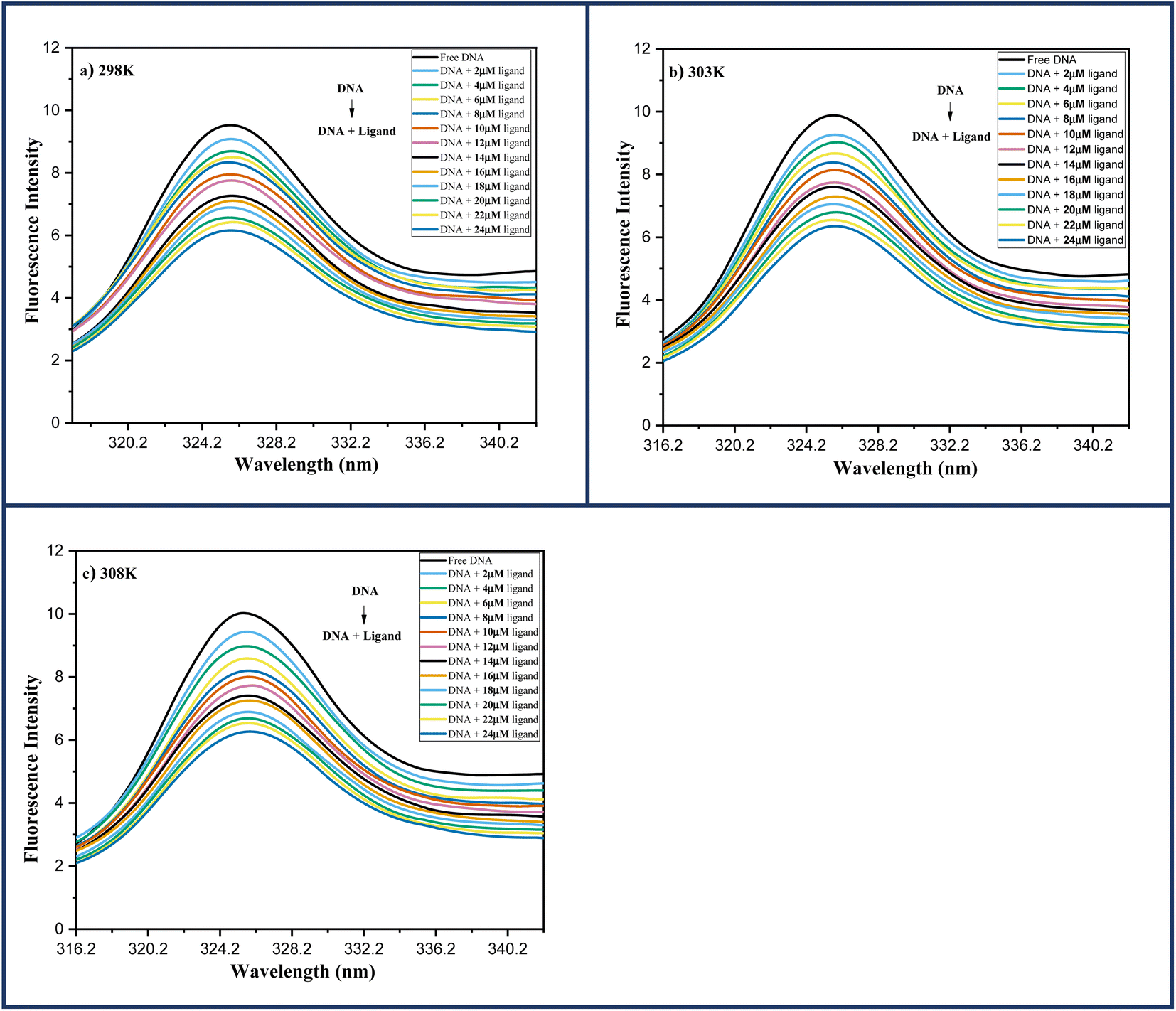 | ||
| Fig. 6 Fluorescence spectra of DNA-6a (67 μM) complex system with increasing ligand concentration 6a (0–24 μM) at (a) 298 K; (b) 303 K; (c) 308 K. | ||
Several molecular mechanisms are responsible for the quenching of the emission intensity of DNA with increasing ligand concentration but mainly depend upon two mechanisms namely static quenching and dynamic quenching. Static quenching is referred as a ground state phenomenon in which a complex is formed between fluorophore and quencher whereas, dynamic quenching is an excited state phenomenon that arises due to the collision or diffusion between fluorophore and quencher.44 The extent of quenching is determined in terms of a constant known as the Stern–Volmer constant using eqn (1).
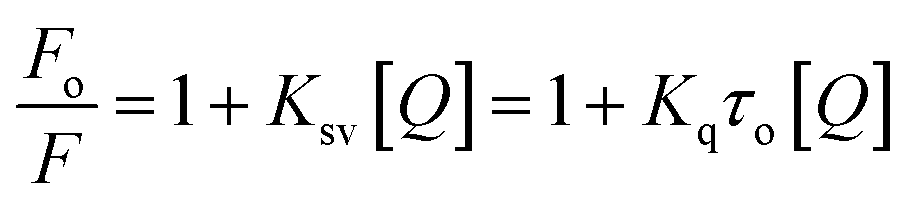 | (1) |
In order to determine the mechanism of quenching followed during the interaction between DNA and ligand 6a, Stern–Volmer plots were utilized at three different temperatures: 298, 303 and 308 K by using the fluorescence intensity at 325 nm (Fig. 7a). A straight-line graph was plotted between the ratio of fluorescence intensity at 325 nm in the absence (Fo) and presence (F) of ligand 6a against the increasing molar concentration [Q] of ligand 6a by keeping the intercept constant at 1. The slope of the plot provides the values of the Stern–Volmer constant Ksv and the quenching constant Kq Table 6. It has been observed that in the case of dynamic mechanism, the quenching constants show an increment with the rise in temperature as high-temperature favours faster diffusion and results in more collisions between fluorophore and quencher whereas, in the case of static mechanism the quenching constants decrease with a rise in temperature as the complex of fluorophore and quencher dissociates at higher temperature. From Table 6, it is evident that the quenching constants Ksv and Kq goes on decreasing with an increase in temperature suggesting the static mechanism to be followed during the interaction between DNA and ligand 6a.
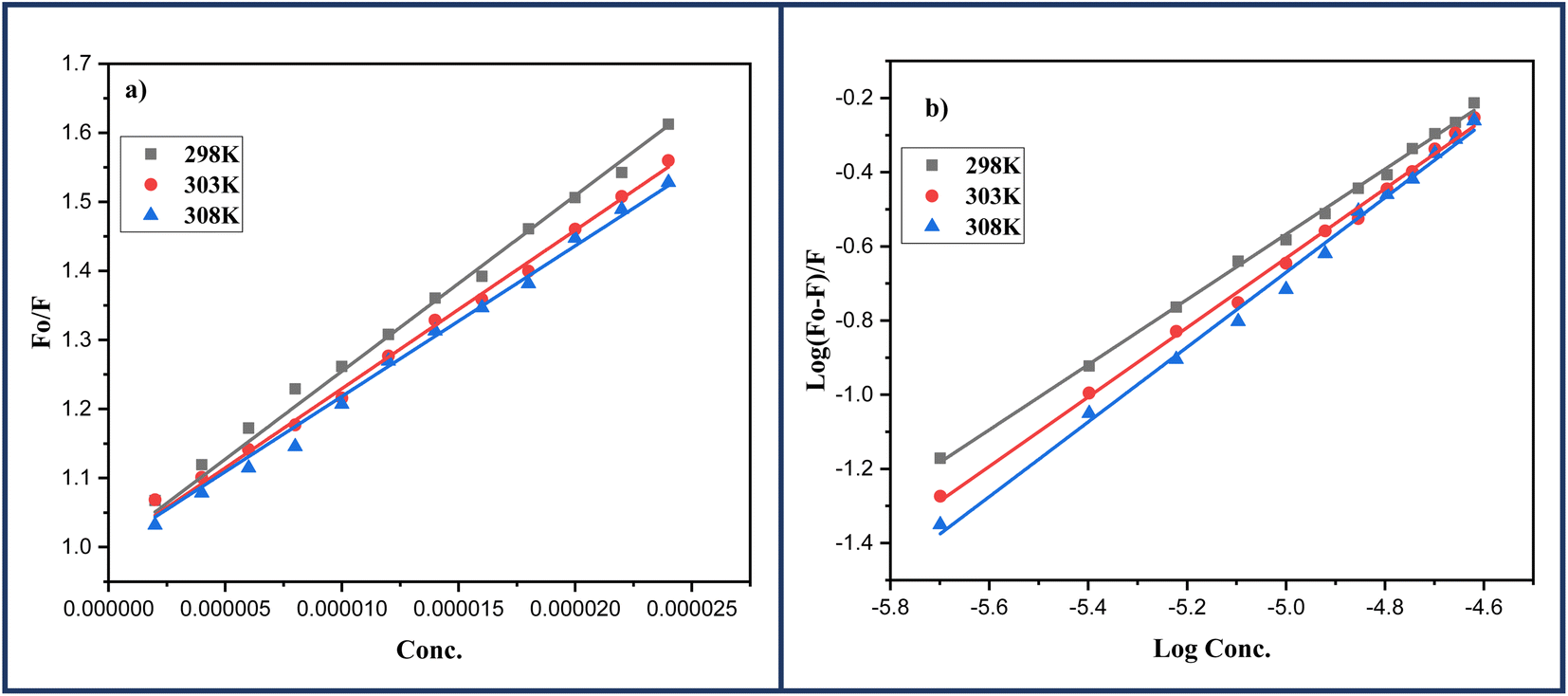 | ||
| Fig. 7 (a) Stern–Volmer; (b) Scatchard's plots for the DNA-6a complex systems at varying three different temperatures: 298, 303 and 308 K respectively. | ||
| T (K) | Ksv × 104 (M−1) | Kq × 1012 (M−1 s−1) | LogKb |
Kb × 104 (M−1) | n | ΔG° (kJ mol−1) | ΔH° (kJ mol−1) | ΔS° (J mol−1 K−1) |
|---|---|---|---|---|---|---|---|---|
| 298 | (2.54 ± 0.02) | (2.54 ± 0.02) | 4.37 ± 0.11 | 2.34 | 1.00 ± 0.02 | −24.9 | −94.9 | −235.05 |
| 303 | (2.29 ± 0.02) | (2.29 ± 0.02) | 4.05 ± 0.08 | 1.12 | 0.93 ± 0.01 | −23.5 | ||
| 308 | (2.18 ± 0.02) | (2.18 ± 0.02) | 3.82 ± 0.05 | 0.66 | 0.87 ± 0.01 | −22.5 |
2.2.5.1 Identification of binding constant and number of binding sites. How effectively a quencher can interact with fluorophore can be identified in terms of binding constant (Kb) using double-logarithmic Scatchard's eqn (2).45 Furthermore, the same equation can also be used to calculate the number of binding sites n.
 | (2) |
A linear regression was obtained when the logarithmic ratio of the difference in fluorescence intensity of fluorophore in the absence and presence of quencher 6a to the fluorescence intensity of fluorophore in the presence of quencher 6a was plotted against the molar concentration of the quencher 6a (Fig. 7b). The slope and intercept of this linear regression help to determine the number of binding sites n and binding constants Kb respectively at three different temperatures: 298, 303 and 308 K and are presented in the Table 6. The value of binding constant Kb at all three temperatures is of the order 104 which is in great agreement with the literature value indicating the grove binding mode of ligand 6a with DNA.46 Furthermore, the number of binding sites nearly 1 at all three temperatures indicates 1:1 binding stoichiometry of the ligand 6a with DNA which also indicates only one mode of binding i.e., grove binding.
Also, limit of detection (LOD) was calculated for each titration and the results are highlighted in Table S1.†
2.2.5.2 Identification of binding forces. When a drug interacts with a biomacromolecule, certain types of forces exhibit between them namely hydrophobic force, hydrogen bond, van der Waals interaction or electrostatic interaction.47 The sign of thermodynamic parameters, specifically change in enthalpy (ΔH°) and change in entropy (ΔS°) provide access to identify these forces. The following three scenarios have been found to aid in predicting the binding forces displayed by ligands and biomacromolecules during the interaction process.
(i) ΔH° > 0 & ΔS° > 0 shows hydrophobic interactions
(ii) ΔH° < 0 & ΔS° > 0 represents electrostatic interactions
(iii) ΔH° < 0 & ΔS° < 0 demonstrates van der Waals interaction or hydrogen bonding.
For this purpose, van't Hoff eqn (3) was utilized in order to determine the values of ΔH° & ΔS°.
|
ΔG° = −2.303RTLogKb = ΔH° − TΔS°
| (3) |
The slope and the intercept of linear regression obtained by the plot of the logarithm of binding constant Kb and reciprocal of temperature (1/T) provide the value of ΔH° & ΔS° respectively (ESI Fig. S1†). The negative values of both the thermodynamic parameters (ΔH° = −94.9 kJ mol−1 & ΔS° = −235.05 J mol−1 K−1), indicating that hydrogen bonding or van der Waals interaction has occurred between ligand 6a and DNA during the interaction process Table 6.
Apart from this, the spontaneity of the binding process at all three studied temperatures: 298, 303 and 308 K was also determined in the terms of the change in Gibbs free energy (ΔG°). The negative values of the ΔG° at all three temperatures indicate the spontaneity of the binding process Table 6. ESI Fig. S1† depicts the graphical representation of thermodynamic parameters ΔH°, ΔS° and ΔG°.
In this experiment, the emission spectra of DNA-Ho (70:1) and DNA-EtBr (10:1) complexes in Tris–HCl (pH 7.2) were recorded in the absence and presence of increasing concentrations of ligand 6a (0–35 μM). Both the spectra were recorded at room temperature in the spectral range of 360–600 nm for the DNA-Ho complex and 500–700 nm for DNA-EtBr by keeping the excitation wavelength fixed at 343 nm and 475 nm respectively. Fig. 8 clearly shows that the emission spectra of the DNA-EtBr complex do not alter as the concentration of ligand 6a increases, ruling out the possibility of ligand 6a acting as an intercalator. Furthermore, the DNA-HO complex emission spectra show a gradual decrease with increasing ligand concentration 6a, confirming the grove binding mode of ligand 6a with DNA.
 | ||
| Fig. 8 DNA-Hoechst and DNA-EtBr complexes competitive displacement plots in the absence and in the presence of an increasing concentration 6a (0–35 μM) in DMSO at pH = 7.2. | ||
For this purpose, the CD spectrum of pure DNA in Tris–HCl (67 μM, pH 7.2) was recorded in the absence as well as in the presence of an increasing concentration of ligand 6a (0.2–1.0 μM) in the spectral range of 220–300 nm. There has been no evident change in either the negative or positive peak (ESI Fig. S2†). Thus, CD spectroscopy provides conclusive evidence in favour of grove binding mode of ligand 6a with DNA and ruled out the possibility of intercalation binding.
| Cmpd | 3D plot | Binding affinity (kcal mol−1) | H-bond | Residual amino acid interactions | |
|---|---|---|---|---|---|
| Hydrophobic/pi–cation/pi–anion/pi–alkyl interactions | van der Walls interactions | ||||
| 6a | 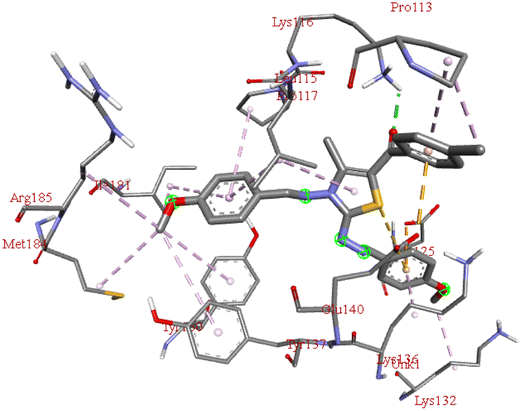 |
−9.6 | LYS B:116 | LYS B:132, LYS B:136, TYR B:137, PRO B:117, MET B:184, TYR B:160, ILE B:181, ARG B:185, LEU B:115, PRO B:113, GLU B:140, GLU B:125 | — |
| 6b | No interactions found | −7.2 | — | — | — |
| 6c | 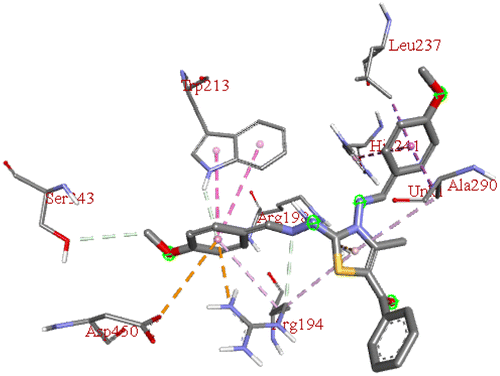 |
−9.1 | ARG A:198 | ASP A:450, ARG A:194, TRP A:213, SER A:343, HIS A:241, ALA A:290, LEU A:237 | — |
| 6d | 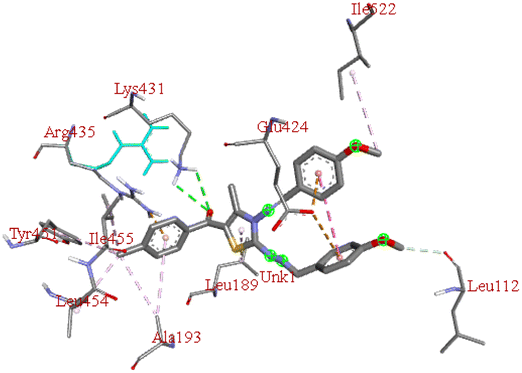 |
−7.9 | LYS B:431 | GLU B:424, ILE B:522, LEU B:112, ILE B:455, TYR B:451, LEU B:189, LEU B:454, ALA B:193, ARG B:435 | — |
| 6e | 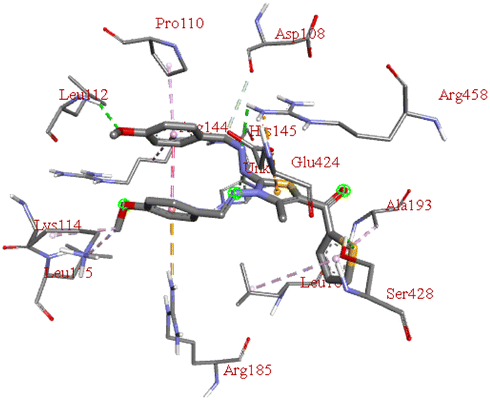 |
−7.5 | LEU B:112, SER B:428, ARG B:458 | GLU B:424, ARG B:185, LEU B:115, LYS B:114, ARG B:144, ASP B:108, PRO B:110, ALA B:193, HIS B:145, LEU B:189 | — |
| 6f | No interactions found | −7.0 | — | — | — |
| 6g | 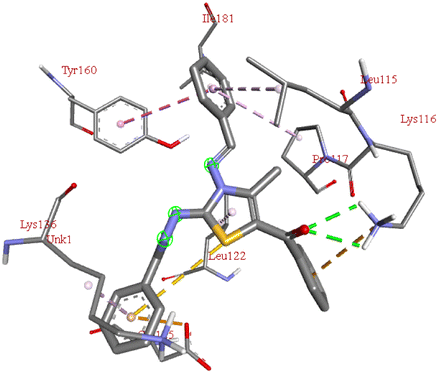 |
−9.2 | LYS B:116 | TYR B:160, ILE B:181, LEU B:115, PRO B:117, LEU B:122, LYS B:136, GLU B:125 | — |
| 6h | 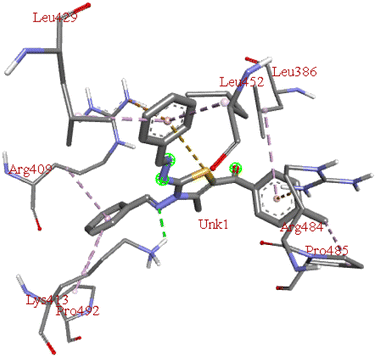 |
−8.7 | LYS A:413 | PRO A: 485, ARG A:484, LEU A:386, LEU A:452, LEU A:429, ARG A:409, PRO A:492 | — |
| 6i | 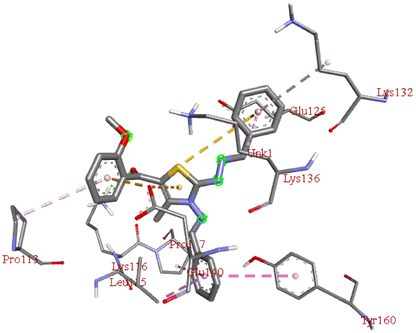 |
−8.8 | LYS B:116 | PRO B:113, LYS B:132, LYS B:136, GLU B:140, TYR B:160, LEU B:115, PRO B:117, GLU B:125 | — |
| 6j | 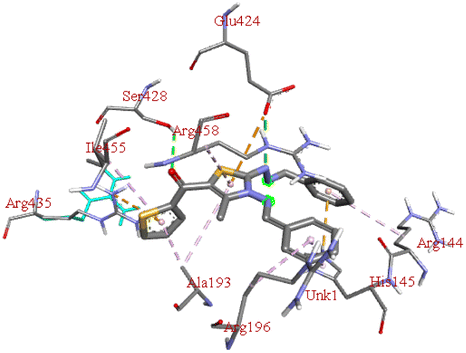 |
−7.8 | GLU B:424, SER B:428 | ARG B:144, ARG B:196, HIS B:145, ARG B:458, ALA B:193, ILE B:455, ARG B:435 | — |
| 6k | No interactions found | −7.4 | — | — | — |
| 6l |  |
−9.2 | HIS B:145 | GLU B:424, LEU B:189, LYS B:431, ARG B:435, ALA B:193, LEU B:454, ARG B:458, ARG B:196 | — |
| 6m | 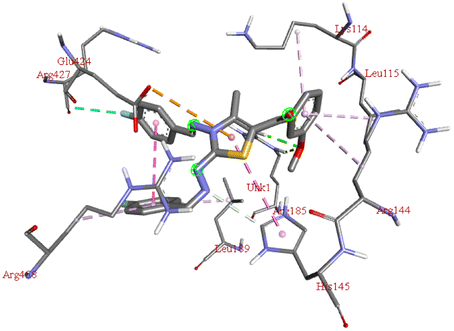 |
−8.3 | ARG B:185 | LYS B:114, LEU B:115, ARG B:144, HIS B:145, GLU B:424, ARG B:427, ARG B:458, LEU B: 189 | — |
| 6n |  |
−7.6 | ARG B:196, TYR B:147 | LYS B:204, VAL B:461, ASP B:108, ALA B:200, LYS B: 465, SER B:104, LEU B:103, GLU B:100 | — |
From the values of binding affinity, we can say that the synthesized compounds bind more effectively with protein than DNA. However, the outcomes of the protein binding are almost similar to that of DNA binding where bromo derivatives (6b, 6f and 6k) again fail to interact with BSA protein. Also, thienyl derivatives (6e, 6j and 6n) were found to have low binding affinities for protein similar to ct-DNA. Furthermore, almost all the derivatives bind with the protein through non-covalent interaction with B chain of the protein with few exceptions where derivatives 6c and 6h were found to interact with protein through chain A.
Among all the tested derivatives 5-(4′-chlorobenzoyl)-3-((4′′-methoxybenzylidene)amino)-2-(4′′′-methoxybenzylidene) hydrazono)-4-methyl-2,3-dihydrothiazole 6a was observed to exhibits the highest binding affinity −9.6 kcal mol−1. Ligand 6a binds with BSA protein through H-bonding with LYS B:116 and also possesses other non-covalent interactions such as hydrophobic, pi-cation, pi-anion, pi–alkyl interactions with other amino acid residues viz., LYS B:132, LYS B:136, TYR B:137, PRO B:117, MET B:184, TYR B:160, ILE B:181, ARG B:185, LEU B:115, PRO B:113, GLU B:140, GLU B:125. Fig. 9 displays the lowest energy 2d and 3d poses of the interaction between ligand 6a and BSA protein.
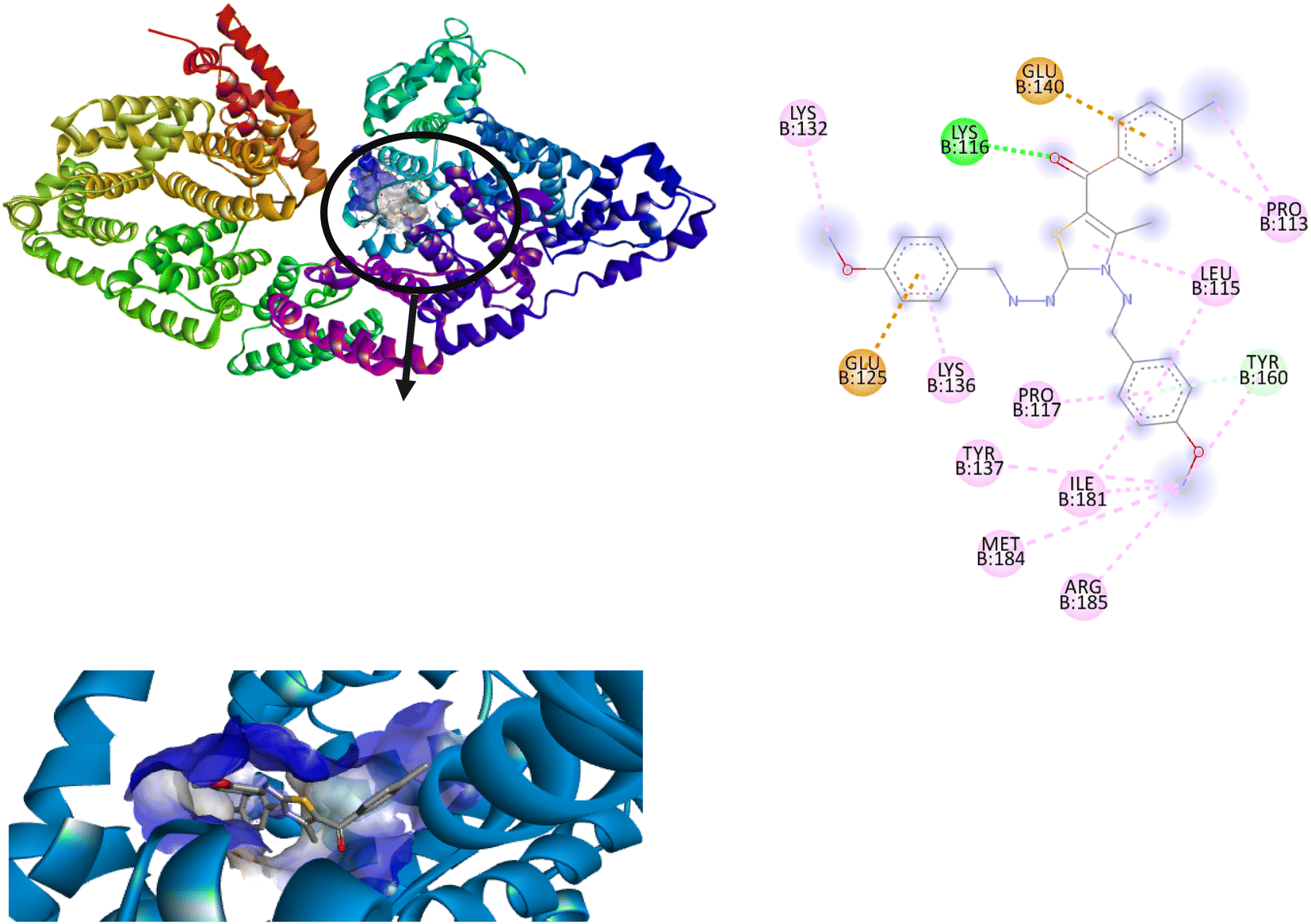 | ||
| Fig. 9 2D and 3D poses of the interaction of ligand 6a with BSA protein. | ||
Through molecular docking the binding interactions of the ligands not only with BSA but also with other biologically relevant proteins like trypsin and glutathione were investigated. The most suitable thiazole derivative 6a was docked with trypsin (PDB ID: 2zq1) and glutathione (PDB ID: 3dk9). The outcomes reveals that the binding affinity of the thiazole derivatives for trypsin and glutathione are −6.3 and −8.9 kcal mol−1 respectively. The 2D poses of the interaction of ligand 6a with trypsin and glutathione are presented in Fig. S3† given below.
The affinity of the synthesized thiazole derivatives for BSA is highest, indicating a strong interaction and supporting its role as a BSA targeting agent but the values for trypsin and glutathione also suggest potential interactions with these proteins as well. Thus, in conclusion it has been proposed that the synthesized thiazole derivatives are not specific for BSA protein only but can also interact with other proteins as well specifically trypsin and glutathione.
Again, compound 6a was selected for further in vitro analysis with BSA protein using various spectroscopic techniques due to high binding affinity and lesser stability/high reactivity.
 | ||
| Fig. 10 (a) Absorption spectra of BSA protein (13 μM) in the absence and in the presence of an increasing ligand 6a (0–16 μM) and (b) Job's plot for BSA-6a complex system. | ||
Furthermore, the method of continuous variation also acknowledged as Job's plot was employed to determine the binding stoichiometry of ligand and protein.50 In this experiment, ten different solutions of BSA in Phosphate Buffer saline (13 μM, pH 7.4) and ligand 6a were prepared by keeping the final concentration constant at 20 μM and absorption spectra of these solutions were monitored at 280 nm. A maximum at 0.5 was obtained from a graph between the mole fractions of ligand 6a and absorbance at 280 nm indicating the 1:1 binding stoichiometry of ligand 6a with BSA (Fig. 10b).
:9:0.5, indicating that tryptophan residue is more important among these aromatic amino acids. As a result, the presence of any foreign ligand in the vicinity of these amino acids alters the intensity of emission of native BSA protein detected by the instrument.In this experiment, a fixed concentration of BSA in Phosphate Buffer saline (13 μM, pH 7.4) was titrated with increasing ligand concentration 6a (0–20 μM) and the data was collected in the spectral range of 300–500 nm while keeping the excitation wavelength (λex) fixed at 290 nm. The emission maxima at 342 nm gradually decreased with increasing ligand concentration and was finally quenched at a higher concentration of ligand 6a, indicating preferential binding of ligand 6a with BSA protein. Further, the same procedure was carried out at three different temperatures: 298, 303, and 308 K with similar results Fig. 11.
 | ||
| Fig. 11 Fluorescence spectra of BSA (13 μM) protein in the increasing ligand concentration 6a (0–20 μM) at (a) 298 K; (b) 303 K; (c) 308 K. | ||
Furthermore, a straight-line graph was obtained at three different temperatures: 298, 303, and 308 K using Stern–Volmer eqn (1) where the ratio of fluorescence intensity at 342 nm in the absence (Fo) and presence (F) of ligand 6a was plotted against the increasing molar concentration [Q] of ligand 6a by keeping the intercept constant at 1 Fig. 12a. The quenching constants Ksv and Kq were calculated from this linear regression slope and are displayed in the Table 8. As the quenching constants Ksv and Kq show a decrement with the rise in temperature suggesting the static quenching mechanism is to be followed during the interaction process between ligand 6a and BSA protein.
 | ||
| Fig. 12 (a) Stern–Volmer; (b) Scatchard's plots for the BSA-6a complex systems at varying three different temperatures: 298, 303 and 308 K respectively. | ||
| T (K) | Ksv × 104 (M−1) | Kq × 1012 (M−1 s−1) | LogKb |
Kb × 104 (M−1) | n | ΔG° (kJ mol−1) | ΔH° (kJ mol−1) | ΔS° (J mol−1 K−1) |
|---|---|---|---|---|---|---|---|---|
| 298 | (3.95 ± 0.09) | (3.95 ± 0.09) | 5.27 ± 0.22 | 18.62 | 0.98 ± 0.02 | −30.0 | −156.1 | −423.5 |
| 303 | (3.64 ± 0.13) | (3.64 ± 0.13) | 4.64 ± 0.19 | 4.36 | 0.96 ± 0.03 | −26.9 | ||
| 308 | (3.31 ± 0.06) | (3.31 ± 0.06) | 4.36 ± 0.18 | 2.29 | 1.16 ± 0.04 | −25.7 |
2.2.10.1 Identification of binding constant and number of binding sites. To determine the binding constant Kb and the number of binding sites n double-logarithmic Scatchard's eqn (2) was employed. A straight-line graph was obtained when the logarithmic ratio of the difference in fluorescence intensity of fluorophore in the absence and presence of quencher 6a to the fluorescence intensity of fluorophore in the presence of quencher 6a was plotted against the molar concentration of the quencher 6a (Fig. 12b). The values of binding constant Kb and the number of binding sites n obtained from this linear regression at three temperatures: 298, 303, and 308 K are highlighted in the Table 8. The value of binding constant Kb comes out to be of the order 104 at all three temperatures indicative of moderate binding of ligand 6a with BSA protein. Also, the number of binding sites was nearly found to be equal to 1 which is in great agreement with the outcomes of Job's plot.
Also, limit of detection (LOD) was calculated for each titration and the results are highlighted in Table S1.†
2.2.10.2 Identification of binding forces. Van't Hoff eqn (3) was utilized in order to identify the binding forces exhibited during the interaction process between ligand 6a and BSA protein. It is evident (ESI Fig. S4†) that the straight-line regression attained from the plot of the logarithm of binding constant Kb and reciprocal of temperature (1/T). The thermodynamic parameters ΔH° & ΔS° calculated from the slope and the intercept of the plot respectively are displayed in the Table 8. The negative values of both the parameters (ΔH° = −156.1 kJ mol−1 & ΔS° = −423.5 J mol−1 K−1), represents either van der Waals interaction or hydrogen bonding has occurred between ligand 6a and BSA protein during the binding process.
Also, the change in Gibbs free energy (ΔG°) was calculated at all three temperatures: 298, 303, and 308 K in order to identify the spontaneity of the binding process. The negative values of the ΔG° at all three temperatures indicate the spontaneity of the binding process between ligand 6a and the BSA protein Table 8. Fig. S4† depicts the graphical representation of thermodynamic parameters ΔH°, ΔS° and ΔG°.
For the process, the fluorescence quenching experiments were carried out in which emission spectra of a pre-treated solution of BSA and site markers in 1:1 with increasing concentrations of ligand 6a (0–20 μM) were recorded and displayed in (ESI Fig. S5†). The data so obtained was used to determine the binding constant Kb Table 9. As the value of the binding constant affected much in the case of phenylbutazone as compared to Ibuprofen hence ligand 6a is supposed to bind in the same fashion as phenylbutazone does (Table 9). Thus, on the outcomes of site markers, it is concluded that ligand 6a binds in Sudlow's Site I of BSA.
| System | Kb (M−1) |
|---|---|
| BSA + 6a | 18.62 × 104 |
| BSA + IB + 6a | 16.81 × 104 |
| BSA + PB + 6a | 6.48 × 104 |
 | ||
| Fig. 13 CD spectra of BSA in the absence as well as in the presence of increasing concentration of ligand 6a. | ||
The CD results can be expressed in terms of mean residue ellipticity (MRE) (eqn (4))
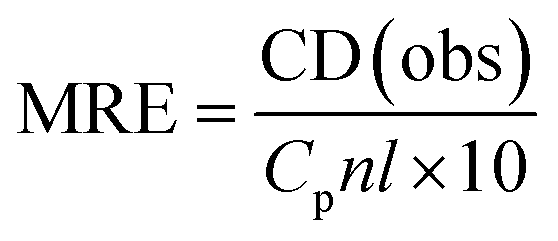 | (4) |
MRE can be used to calculate the α-helix content of BSA using following equation:
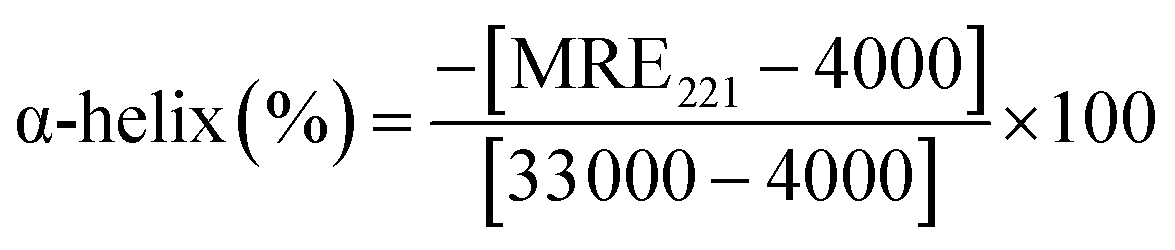 | (5) |
000 is the MRE value of a pure α-helix at 221 nm.
Table 10 displayed the related decrease in ellipticity, indicative of helicity disruption or denaturation of protein structure by increasing concentration of ligand 6a. The unfolding of the α-helix of protein occurs at the rate directly proportional to the concentration of 6a. Thus, it can be concluded that at higher ligand concentration the BSA structure opens up i.e., denaturation of protein happens.
| 6a (μM) | 0.2 | 0.4 | 0.6 | 0.8 | 1.0 |
|---|---|---|---|---|---|
| % Helicity | 49.63 | 46.79 | 39.65 | 32.51 | 26.83 |
3. Conclusion
In nutshell, regioselective synthesis of 5-aroyl-3-((arylidene)amino)-2-((arylidene)hydrazono)-4-methyl-2,3-dihydrothiazole derivatives 6a–n has been successfully achieved in excellent yields by the one-pot reaction of thiocarbohydrazide 1 and aldehydes 2 with α-bromo-1,3-diketones 5 (generated in situ by the bromination of unsymmetrical 1,3-diketones 4(a–f) with NBS) using white LED light (9 W) as a green energy source. Out of the two possible regioisomers the exact one was identified using multinuclear 2D-NMR [1H–13C] HMBC; [1H–13C] HMQC and [1H–15N] HMBC spectroscopic techniques. On the basis of in silico studies including molecular reactivity analysis, Lipinski rule and molecular docking 2,3-dihydrothiazole derivative 6a was selected for binding studies with DNA and BSA biomolecules using UV-absorption spectroscopy, fluorescence and CD spectroscopy. The steady-state fluorescence supports the static mechanism that occurs during interactions of compound 6a with DNA and BSA. The competitive displacement assay and circular dichroism analysis have also demonstrated compound 6a to bind with DNA through grove binding mode. Furthermore, site-marker assay indicates ligand 6a to bind with BSA at Sudlow's site I (in subdomain IIA). The theoretical outcomes of in silico studies agree very well with experimental ex vivo results. We hope that the outcomes of this study will help and motivate scientists to further explore the biological potential of 2,3-dihydrothiazole derivatives.4. Experimental
4.1 General method of synthesis
1,3-Diketones and thiocarbohydrazide were prepared according to the literature procedure. Aldehydes and NBS were purchased commercially.4.1.1.1 6a. 5-(4′-chlorobenzoyl)-3-((4′′-methoxybenzylidene)amino)-2-((4′′′-methoxybenzylidene) hydrazono)- 4-methyl-2,3-dihydrothiazole. Yellow solid; M. pt. 176 °C; yield: 88%; IR (KBr) νmax (cm−1): 1592 (C
O), 1612 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.13 (s, 1H, 6-CH), 8.36 (s, 1H, 7-CH), 7.78–7.76 (d, 2H, 3J = 8.8 Hz, Ph 2′′, 6′′-H), 7.74–7.70 (m, 4H, Ph 2′, 5′, 2′′′, 6′′′-H), 7.47–7.45 (d, 2H, 3J = 8.4 Hz, Ph 3′, 6′-H), 6.99–6.97 (d, 2H, 3J = 8.8 Hz, Ph 3′′, 5′′-H), 6.94–6.91 (d, 2H, 3J = 8.8 Hz, Ph 3′′′, 5′′′-H), 3.88 (s, 3H, 4′′-OCH3), 3.85 (s, 3H, 4′′′-OCH3), 2.52 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 187.3, 163.4, 162.3, 161.6, 156.9, 155.3, 148.6, 138.4, 138.1, 130.0, 129.8, 129.6, 128.9, 127.5, 127.1, 114.4, 114.2, 111.3, 55.5, 55.4, 15.6; HRMS (EI) m/z: 519.1260 [M+1]+, 521.1225 [M+3]+ (3:1); Anal. Calcd. for C27H23ClN4O3S: C, 62.48; H, 4.47; N, 10.80% Found: C, 62.45; H, 4.45; N, 10.79%.
4.1.1.2 6b. 5-(4′-bromobenzoyl)-3-((4′′-methoxybenzylidene)amino)-2-((4′′′-methoxybenzylidene) hydrazono)-4-methyl-2,3-dihydrothiazole. Pale Yellow solid; M. pt. 179 °C; yield: 90%; IR (KBr) νmax (cm−1): 1594 (C
O), 1613 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.12 (s, 1H, 6-CH), 8.35 (s, 1H, 7-CH), 7.78–7.75 (d, 2H, 3J = 8.8 Hz, Ph 2′′, 6′′-H), 7.72–7.70 (d, 2H, 3J = 8.8 Hz, Ph 2′′′, 6′′′-H), 7.64–7.63 (d, 4H, 3J = 4 Hz, Ph 2′, 3′, 5′, 6′-H), 6.98–6.96 (d, 2H, 3J = 8.8 Hz, Ph 3′′, 5′′-H), 6.93–6.91 (d, 2H, 3J = 8.8 Hz, Ph 3′′′, 5′′′-H), 3.87 (s, 3H, 4′′-OCH3), 3.84 (s, 3H, 4′′′-OCH3), 2.51 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 187.3, 163.4, 162.3, 161.6, 157.0, 155.3, 148.6, 138.5, 131.9, 130.1, 129.8, 129.6, 127.5, 127.1, 126.9, 114.4, 114.2, 111.3, 55.5, 55.4, 15.6; HRMS (EI) m/z: 563.0759 [M+1]+, 565.0734 [M+3]+ (1:1); Anal. Calcd. for C27H23BrN4O3S: C, 57.55; H, 4.11; N, 9.94% Found: C, 57.52; H, 4.09; N, 9.93%.
4.1.1.3 6c. 5-benzoyl-3-((4′′-methoxybenzylidene)amino)-2-((4′′′-methoxybenzylidene)hydrazono)-4-methyl-2,3-dihydrothiazole. Orange solid; M. pt. 165 °C; yield: 92%; IR (KBr) νmax (cm−1): 1596 (C
O), 1610 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.15 (s, 1H, 6-CH), 8.36 (s, 1H, 7-CH), 7.77–7.75 (m, 4H, Ph 2′, 6′, 2′′, 6′′-H), 7.72–7.70 (m, 2H, Ph 2′′′, 6′′′-H), 7.58–7.55 (m, 1H, Ph 4′-H), 7.50–7.46 (m, 2H, Ph 3′, 5′-H), 6.97–6.96 (d, 2H, 3J = 4 Hz, Ph 3′′, 5′′-H), 6.92–6.91 (d, 2H, 3J = 4 Hz, Ph 3′′′, 5′′′-H), 3.87 (s, 3H, 4′′-OCH3), 3.84 (s, 3H, 4′′′-OCH3), 2.47 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 188.7, 163.8, 162.2, 161.5, 156.7, 155.1, 148.1, 139.8, 132.1, 129.7, 129.5, 128.6, 128.5, 127.6, 127.2, 114.4, 114.2, 112.0, 55.5, 55.5, 15.7; HRMS (EI) m/z: 485.1697 [M+1]+; Anal. Calcd. for C27H24N4O3S: C, 66.92; H, 4.91; N, 11.56% Found: C, 66.89; H, 4.90; N, 11.54%.
4.1.1.4 6d. 3-((4′′-methoxybenzylidene)amino)-2-((4′′′-methoxybenzylidene)hydrazono)-4-methyl-5-(4′-methylbenzoyl)-2,3-dihydrothiazole. Pale Yellow solid; M. pt. 188 °C; yield: 93%; IR (KBr) νmax (cm−1): 1591 (C
O), 1608 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.14 (s, 1H, 6-CH), 8.37 (s, 1H, 7-CH), 7.78–7.76 (d, 2H, 3J = 8.8 Hz, Ph 2′′, 6′′-H), 7.73–7.68 (m, 4H, Ph 2′, 5′, 2′′′, 6′′′-H), 7.29–7.27 (d, 2H, 3J = 8.4 Hz, Ph 3′, 6′-H), 6.98–6.96 (d, 2H, 3J = 8.8 Hz, Ph 3′′, 5′′-H), 6.93–6.91 (d, 2H, 3J = 8.8 Hz, Ph 3′′′, 5′′′-H), 3.88 (s, 3H, 4′′-OCH3), 3.85 (s, 3H, 4′′′-OCH3), 2.49 (s, 3H, 4-CH3), 2.44 (s, 3H, 4′-CH3); 13C NMR (100 MHz, CDCl3) δ: 188.4, 163.8, 162.2, 161.5, 154.9, 147.5, 142.9, 137.0, 129.7, 129.5, 129.2, 128.8, 127.7, 127.2, 114.4, 114.2, 55.5, 55.4, 21.7, 15.6; HRMS (EI) m/z: 499.1772 [M+1]+; Anal. Calcd. for C28H26N4O3S: C, 67.45; H, 5.26; N, 11.24% Found: C, 67.43; H, 5.23; N, 11.23%.
4.1.1.5 6e. 3-((4′′-methoxybenzylidene)amino)-2-((4′′′-methoxybenzylidene)hydrazono)-4-methyl-5-thienoyl-2,3-dihydrothiazole. Black solid; M. pt. 175 °C; yield: 85%; IR (KBr) νmax (cm−1): 1586 (C
O), 1609 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.12 (s, 1H, 6-CH), 8.37 (s, 1H, 7-CH), 7.94–7.93 (dd, 1H, 3J = 3.6 Hz, 4J = 0.8 Hz, thienyl 3′-H), 7.79–7.77 (d, 2H, 3J = 8.8 Hz, Ph 2′′, 6′′-H), 7.74–7.72 (d, 2H, Ph 2′′′, 6′′′-H), 7.67–7.66 (dd, 1H, 3J = 4.8 Hz, 5J = 0.8 Hz, thienyl 5′-H), 7.18–7.16 (dd, 1H, 3J = 4.8 Hz, 3J = 3.6 Hz, thienyl 4′-H), 6.99–6.96 (d, 2H, 3J = 8.8 Hz, Ph 3′′, 5′′-H), 6.94–6.92 (d, 2H, 3J = 8.8 Hz, Ph 3′′′, 5′′′-H), 3.88 (s, 3H, 4′′-OCH3), 3.85 (s, 3H, 4′′′-OCH3), 2.69 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 179.0, 163.1, 162.2, 161.5, 156.8, 155.0, 148.7, 145.2, 133.4, 132.5, 129.8, 129.5, 128.0, 127.6, 127.2, 114.4, 114.2, 109.5, 55.5, 55.4, 15.3; HRMS (EI) m/z: 491.1237 [M+1]+; Anal. Calcd. for C25H22N4O3S2: C, 61.21; H, 4.52; N, 11.42% Found: C, 61.19; H, 4.49; N, 11.40%.
4.1.1.6 6f. 3-((benzylidene)amino)-2-(benzylidene)hydrazono)-5-(4′-bromobenzoyl)-4-methyl--2,3-dihydrothiazole. Pale Yellow solid; M. pt. 176 °C; yield: 87%; IR (KBr) νmax (cm−1): 1597 (C
O), 1610 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.27 (s, 1H, 6-CH), 8.43 (s, 1H, 7-CH), 7.83–7.78 (m, 4H, Ph 2′′, 6′′, 2′′′, 6′′′-H), 7.65 (s, 4H, Ph 2′, 3′, 5′, 6′-H), 7.47–7.41 (m, 6H, Ph 3′′, 4′′, 5′′, 3′′′, 4′′′, 5′′′-H), 2.54 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 187.4, 164.3, 157.0, 155.9, 148.4, 138.4, 134.7, 134.5, 132.0, 131.4, 130.6, 130.2, 129.0, 128.8, 128.1, 128.1, 127.2, 111.8, 15.7; HRMS (EI) m/z: 503.0543 [M+1]+, 505.0543 [M+3]+ (1:1); Anal. Calcd. for C25H19BrN4OS: C, 59.65; H, 3.80; N, 11.13% Found: C, 59.63; H, 3.79; N, 11.10%.
4.1.1.7 6g. 5-benzoyl-3-((benzylidene)amino)-2-((benzylidene)hydrazono)-4-methyl-2,3-dihydrothiazole. Yellow solid; M. pt. 150 °C; yield: 89%; IR (KBr) νmax (cm−1): 1597 (C
O), 1613 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.29 (s, 1H, 6-CH), 8.43 (s, 1H, 7-CH), 7.84–7.82 (m, 2H, Ph 2′, 6′-H), 7.79–7.76 (m, 4H, Ph 2′′, 6′′, 2′′′, 6′′′-H), 7.61–7.56 (m, 1H, Ph 4′-H), 7.52–7.49 (m, 2H, Ph 3′, 5′-H), 7.48–7.46 (m, 3H, Ph 3′′, 4′′, 5′′-H), 7.42–7.39 (m, 3H, Ph 3′′′, 4′′′, 5′′′-H), 2.50 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 188.8, 164.7, 156.7, 155.7, 147.8, 139.6, 134.7, 134.6, 132.3, 131.2, 130.5, 128.9, 128.8, 128.6, 128.6, 128.1, 128.0, 112.5, 15.7; HRMS (EI) m/z: 425.1441 [M+1]+; Anal. Calcd. for C25H20N4OS: C, 70.73; H, 4.75; N, 13.20% Found: C, 70.71; H, 4.72; N, 13.19%.
4.1.1.8 6h. 3-((benzylidene)amino)-2-((benzylidene)hydrazono)-4-methyl-5-(4′-methylbenzoyl)-2,3-dihydrothiazole. Pale Yellow solid; M. pt. 187 °C; yield: 92%; IR (KBr) νmax (cm−1): 1593 (C
O), 1605 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.29 (s, 1H, 6-CH), 8.43 (s, 1H, 7-CH), 7.84–7.77 (m, 4H, Ph 2′′, 6′′, 2′′′, 6′′′-H), 7.72–7.70 (d, 2H, 3J = 8 Hz, Ph 2′, 6′-H), 7.47–7.46 (m, 3H, Ph 3′′, 4′′, 5′′-H), 7.40–7.39 (m, 3H, Ph 3′′′, 4′′′, 5′′′-H), 7.30–7.28 (d, 2H, 3J = 8 Hz, Ph 3′, 5′-H), 2.51 (s, 3H, 4-CH3), 2.45 (s, 3H, 4′-CH3); 13C NMR (100 MHz, CDCl3) δ: 188.5, 156.6, 155.6, 143.2, 134.8, 134.7, 131.2, 130.4, 129.3, 128.9, 128.9, 128.8, 128.7, 128.1, 128.0, 21.8, 15.6; HRMS (EI) m/z: 439.1587 [M+1]+; Anal. Calcd. for C26H22N4OS: C, 71.21; H, 5.06; N, 12.78% Found: C, 71.20; H, 5.04; N, 12.75%.
4.1.1.9 6i. 3-((benzylidene)amino)-2-((benzylidene)hydrazono)-5-(3′-methoxybenzoyl)-4-methyl--2,3-dihydrothiazole. Pale Yellow solid; M. pt. 137 °C; yield: 84% IR (KBr) νmax (cm−1): 1599 (C
O), 1610 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.29 (s, 1H, 6-CH), 8.44 (s, 1H, 7-CH), 7.87–7.75 (m, 4H, Ph 2′′, 6′′, 2′′′, 6′′′-H), 7.48–7.46 (m, 3H, Ph 5′, 4′′, 4′′′-H), 7.42–7.37 (m, 5H, Ph 6′, 3′′, 5′′, 3′′′, 5′′′-H), 7.28 (m, 1H, Ph 2′-H), 7.13–7.11 (m, 1H, Ph 4′-H), 3.88 (s, 3H, 3′-OCH3), 2.51 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 188.5, 176.2, 159.8, 155.7, 143.0, 140.9, 134.6, 131.2, 130.4, 129.7, 128.9, 128.8, 128.1, 128.0, 126.0, 121.1, 118.8, 112.9, 55.6, 15.7; HRMS (EI) m/z: 455.1564 [M+1]+; Anal. Calcd. for C26H22N4O2S: C, 68.70; H, 4.82; N, 12.33% Found: C,68.68; H, 4.81; N, 12.32%.
4.1.1.10 6j. 3-((benzylidene)amino)-2-((benzylidene)hydrazono)-4-methyl-5-thienoyl-2,3dihydrothiazole. Brown solid; M. pt. 171 °C; yield: 85%; IR (KBr) νmax (cm−1): 1587 (C
O), 1605 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.28 (s, 1H, 6-CH), 8.45 (s, 1H, 7-CH), 7.95 (s, 1H, thienyl 3′-H), 7.85–7.80 (m, 4H, Ph 2′′, 6′′, 2′′′, 6′′′-H), 7.69–7.68 (d, 1H, 3J = 4 Hz, thienyl 5′-H), 7.48 (s, 3H, Ph 3′′, 4′′, 5′′-H), 7.41 (s, 3H, Ph 3′′′, 4′′′, 5′′′), 7.18 (m, 1H, thienyl 4′-H), 2.71 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 179.1, 164.0, 156.7, 155.7, 148.4, 145.0, 134.7, 134.6, 133.7, 132.7, 131.3, 130.5, 128.9, 128.8, 128.1, 128.0, 15.3; HRMS (EI) m/z: 431.0994 [M+1]+; Anal. Calcd. for C23H18N4OS2: C, 64.16; H, 4.21; N, 13.01% Found: C, 64.14; H, 4.19; N, 13.00%.
4.1.1.11 6k. 5-(4′-bromobenzoyl)-3-((4′′-fluorobenzylidene)amino)-2-((4′′′- benzylidene)hydrazono)-4-methyl-2,3-dihydrothiazole. Orange solid; M. pt. 178 °C; yield: 88%; IR (KBr) νmax (cm−1): 1586 (C
O, 1609 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.24 (s, 1H, 6-CH), 8.38 (s, 1H, 7-CH), 7.84–7.80 (m, 2H, Ph 2′′′, 6′′′-H), 7.78–7.74 (m, 2H, Ph 2′′, 6′′-H), 7.65–7.64 (d, 4H, 3J = 4 Hz, Ph 2′, 3′, 5′, 6′-H), 7.18–7.14 (m, 2H, Ph 3′′′, 5′′′-H), 7.12–7.08 (m, 2H, Ph 3′′, 5′′-H), 2.53 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 187.4, 165.9, 165.5, 164.3, 163.4, 155.7, 154.6, 148.1, 138.2, 131.9, 130.9, 130.9, 130.7, 130.7, 130.2, 130.1, 130.0, 129.9, 129.8, 127.2, 116.3, 116.1, 115.9, 111.8, 15.6; 19F NMR (300 MHz, CDCl3) δ: −107.88, −109.16; HRMS (EI) m/z: 539.0349 [M+1]+, 541.0322 [M+3]+ (1:1); Anal. Calcd. for C25H17BrF2N4OS: C, 55.67; H, 3.18; N, 10.39% Found: C, 55.65; H, 3.15; N, 10.38%.
4.1.1.12 6l. 3-((4′′-fluorobenzylidene)amino)-2-((4′′′-fluorobenzylidene)hydrazono)-4-methyl-5-(4′-methylbenzoyl)-2,3-dihydrothiazole. Yellow solid; M. pt. 151 °C; yield: 87%; IR (KBr) νmax (cm−1): 1598 (C
O), 1615 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.25 (s, 1H, 6-CH), 8.38 (s, 1H, 7-CH), 7.83–7.79 (m, 2H, Ph 2′′′, 6′′′-H), 7.77–7.74 (m, 2H, Ph 2′′, 6′′-H), 7.61–7.59 (d, 2H, 3J = 8 Hz, Ph 2′, 6′-H), 7.30–7.28 (d, 2H, 3J = 8 Hz, Ph 3′, 5′-H), 7.17–7.13 (m, 2H, Ph 3′′′, 5′′′-H), 7.11–7.06 (m, 2H, Ph 3′′, 5′′-H), 2.49 (s, 3H, 4-CH3), 2.45 (s, 3H, 4′-CH3); 13C NMR (100 MHz, CDCl3) δ: 188.4, 165.8, 165.4, 164.7, 163.3, 162.9, 155.2, 154.2, 147.1, 143.2, 136.7, 131.0, 131.0, 130.9, 130.8, 130.0, 129.9, 129.8, 129.8, 129.3, 128.9, 116.2, 116.0, 115.8, 112.5, 21.8, 15.6; 19F NMR (300 MHz, CDCl3) δ: −108.20, −109.43; HRMS (EI) m/z: 475.1407 [M+1]+; Anal. Calcd. for C26H20F2N4OS: C, 65.81; H, 4.25; N, 11.81% Found: C, 65.79; H, 4.22; N, 11.80%.
4.1.1.13 6m. 3-((4′′-fluorobenzylidene)amino)-2-((4′′′-fluorobenzylidene)hydrazono)-5-(3′-methoxy benzoyl)-4-methyl--2,3-dihydrothiazole. Pale Yellow solid; M. pt. 158 °C; yield: 83%; IR (KBr) νmax (cm−1): 1590 (C
O), 1603 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.24 (s, 1H, 6-CH), 8.39 (s, 1H, 7-CH), 7.83–7.80 (m, 2H, Ph 2′′′, 6′′′-H), 7.78–7.74 (m, 2H, Ph 2′′, 6′′-H), 7.42–7.38 (t, 1H, 3J = 8 Hz, Ph 5′-H), 7.36–7.34 (m, 1H, Ph 6′-H), 7.27 (m, 1H, Ph 2′-H), 7.17–7.07 (m, 5H, Ph 4′, 3′′, 5′′, 3′′′, 5′′′-H), 3.87 (s, 3H, 3′-OCH3), 2.68 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 188.4, 165.9, 165.4, 164.7, 163.4, 162.95, 159.8, 155.6, 154.3, 147.6, 140.8, 131.0, 130.9, 130.8, 130.7, 130.0, 129.9, 129.9, 129.8, 129.7, 121.0, 118.8, 116.3, 116.0, 115.8, 113.0, 112.6, 55.6, 15.6; 19F NMR (300 MHz, CDCl3) δ: −108.03, −109.34; HRMS (EI) m/z: 491.1378 [M+1]+; Anal. Calcd. for C26H20F2N4O2S: C, 63.66; H, 4.11; N, 11.42% Found: C, 63.64; H, 4.09; N, 11.41%.
4.1.1.14 6n. 3-((4′′-fluorobenzylidene)amino)-2-((4′′′-fluorobenzylidene)hydrazono)-4-methyl-5-thienoyl −2,3-dihydrothiazole. Brown solid; M. pt. 175 °C; yield: 82%; IR (KBr) νmax (cm−1): 1583 (C
O), 1606 (CN); 1H NMR (400 MHz, CDCl3) δ: 10.23 (s, 1H, 6-CH), 8.38 (s, 1H, 7-CH), 7.93–7.92 (d, 1H, 3J = 4 Hz, thienyl 3′-H), 7.84–7.80 (m, 2H, Ph 2′′′, 6′′′-H), 7.79–7.75 (m, 2H, Ph 2′′, 6′′-H), 7.69–7.68 (d, 1H, 3J = 4 Hz, thienyl 5′-H), 7.18–7.08 (m, 5H, thienyl 4′-H, Ph 3′′, 5′′, 3′′′, 5′′′-H), 2.68 (s, 3H, 4-CH3); 13C NMR (100 MHz, CDCl3) δ: 179.0, 165.9, 165.4, 164.0, 163.4, 162.9, 155.3, 154.3, 148.2, 144.9, 133.7, 132.7, 131.0, 130.95 130.8, 130.8, 130.0, 129.9, 129.9, 129.8, 128.0, 116.3, 116.1, 116.0, 115.8, 110.0, 15.3; 19F NMR (300 MHz, CDCl3) δ: −108.07, −109.30; HRMS (EI) m/z: 467.0804 [M+1]+; Anal. Calcd. for C23H16F2N4OS2: C, 59.22; H, 3.46; N, 12.01% Found: C, 59.20; H, 3.45; N, 11.99%.
Data availability
Data for this article, including experimental data, (1H, 13C, [1H–13C] HMBC; [1H–13C] HMQC and [1H–15N] HMBC) for final compounds, Van't Hoff plot, CD spectrum for DNA, fluorescence spectra of BSA-IB and BSA-PB complexes and a general experimental section for both synthesis and binding studies have been included as part of the ESI.†Conflicts of interest
The authors declare no competing interest.Acknowledgements
We are highly thankful to Haryana State Council for Science, Innovation and Technology (HSCSIT), Panchkula for financial assistance to Naman Jain as project fellow.References
- K. Chen, X. Yao, T. Tang, L. M. Chen, C. Xiao, J. Y. Wang, H. F. Chen, Z. X. Jiang, Y. Liu and X. Zheng, Thiazole-based and thiazolidine-based protein tyrosine phosphatase 1B inhibitors as potential anti-diabetes agents, Med. Chem. Res., 2021, 30, 519–534 CrossRef CAS.
- T. I. de Santana, M. de O. Barbosa, P. A. T. de M. Gomes, A. C. N. da Cruz, T. G. da Silva and A. C. L. Leite, Synthesis, anticancer activity and mechanism of action of new thiazole derivatives, Eur. J. Med. Chem., 2018, 144, 874–886 CrossRef PubMed.
- J. Jacob P and S. L. Manju, Identification and development of thiazole leads as COX-2/5-LOX inhibitors through in-vitro and in-vivo biological evaluation for anti-inflammatory activity, Bioorg. Chem., 2020, 100, 103882 CrossRef CAS PubMed.
- K. K. Bansal, J. K. Bhardwaj, P. Saraf, V. K. Thakur and P. C. Sharma, Synthesis of thiazole clubbed pyrazole derivatives as apoptosis inducers and anti-infective agents, Mater. Today Chem., 2020, 17, 100335 CrossRef CAS.
- G. Kumar and N. P. Singh, Synthesis, anti-inflammatory and analgesic evaluation of thiazole/oxazole substituted benzothiazole derivatives, Bioorg. Chem., 2021, 107, 104608 CrossRef CAS.
- I. Althagafi, N. El-Metwaly and T. A. Farghaly, New series of thiazole derivatives: synthesis, structural elucidation, antimicrobial activity, molecular modeling and MOE docking, Molecules, 2019, 24, 1–23 CrossRef.
- A. Rauf, M. K. Kashif, B. A. Saeed, N. A. Al-Masoudi and S. Hameed, Synthesis, anti-HIV activity, molecular modeling study and QSAR of new designed 2-(2-arylidenehydrazinyl)-4-arylthiazoles, J. Mol. Struct., 2019, 1198, 126866 CrossRef CAS.
- M. Mic, A. Pîrnău, C. G. Floare, G. Marc, A. H. Franchini, O. Oniga, L. Vlase and M. Bogdan, Synthesis and molecular interaction study of a diphenolic hidrazinyl-thiazole compound with strong antioxidant and antiradical activity with HSA, J. Mol. Struct., 2021, 1244, 7–10 CrossRef.
- S. Sahu, S. K. Ghosh, P. Gahtori, U. Pratap Singh, D. R. Bhattacharyya and H. R. Bhat, In silico ADMET study, docking, synthesis and antimalarial evaluation of thiazole-1,3,5-triazine derivatives as Pf-DHFR inhibitor, Pharmacol. Rep., 2019, 71, 762–767 CrossRef CAS PubMed.
- Y. A. Ammar, S. M. A. A. El-Hafez, S. A. Hessein, A. M. Ali, A. A. Askar and A. Ragab, One-pot strategy for thiazole tethered 7-ethoxy quinoline hybrids: synthesis and potential antimicrobial agents as dihydrofolate reductase (DHFR) inhibitors with molecular docking study, J. Mol. Struct., 2021, 1242, 130748 CrossRef CAS.
- C. P. Suffridge, L. M. Bolaños, K. Bergauer, A. Z. Worden, J. Morré, M. J. Behrenfeld and S. J. Giovannoni, Exploring Vitamin B1 Cycling and Its Connections to the Microbial Community in the North Atlantic Ocean, Front. Mar. Sci., 2020, 7, 1–16 CrossRef.
- E. Bugiardini, S. Pope, R. G. Feichtinger, O. V. Poole, A. M. Pittman, C. E. Woodward, S. Heales, R. Quinlivan, H. Houlden, J. A. Mayr, M. G. Hanna and R. D. S. Pitceathly, Utility of whole blood thiamine pyrophosphate evaluation in TPK1-related diseases, J. Clin. Med., 2019, 8, 991 CrossRef CAS.
- H. Cheng, H. Huang and G. Huang, Synthesis and antitumor activity of epothilone B, Eur. J. Med. Chem., 2018, 157, 925–934 CrossRef CAS PubMed.
- R. J. Morrison, F. W. Van Der Mei, F. Romiti and A. H. Hoveyda, A Catalytic Approach for Enantioselective Synthesis of Homoallylic Alcohols Bearing a Z-Alkenyl Chloride or Trifluoromethyl Group. A Concise and Protecting Group-Free Synthesis of Mycothiazole, J. Am. Chem. Soc., 2020, 142, 436–447 CrossRef CAS PubMed.
- Y. J. Luo, B. L. Wang, S. B. Kou, Z. Y. Lin, K. L. Zhou, Y. Y. Lou and J. H. Shi, Assessment on the binding characteristics of dasatinib, a tyrosine kinase inhibitor to calf thymus DNA: insights from multi-spectroscopic methodologies and molecular docking as well as DFT calculation, J. Biomol. Struct. Dyn., 2020, 38, 4210–4220 CrossRef CAS.
- A. K. Das, S. I. Druzhinin, H. Ihmels, M. Müller and H. Schönherr, Colorimetric and Fluorimetric DNA Detection with a Hydroxystyryl–Quinolizinium Photoacid and Its Application for Cell Imaging, Chem. –Eur. J., 2019, 25, 12703–12707 CrossRef CAS.
- P. Sangeetha, C. Sankar and K. Tharini, New thiazoldinone substituted 2,6-diarypiperidin-4-one: synthesis, crystal structure, spectral characterization, binding mode with calf thymus DNA, J. Mol. Struct., 2019, 1198, 126899 CrossRef.
- J. Wang, M. F. Ansari and C. H. Zhou, Identification of Unique Quinazolone Thiazoles as Novel Structural Scaffolds for Potential Gram-Negative Bacterial Conquerors, J. Med. Chem., 2021, 64, 7630–7645 CrossRef CAS PubMed.
- M. H. El-Wakil, A. F. El-Yazbi, H. M. A. Ashour, M. A. Khalil, K. A. Ismail and I. M. Labouta, Discovery of a novel DNA binding agent via design and synthesis of new thiazole hybrids and fused 1,2,4-triazines as potential antitumor agents: computational, spectrometric and in silico studies, Bioorg. Chem., 2019, 90, 103089 CrossRef CAS PubMed.
- T. A. Farghaly, A. M. Abo Alnaja, H. A. El-Ghamry and M. R. Shaaban, Synthesis and DNA binding of novel bioactive thiazole derivatives pendent to N-phenylmorpholine moiety, Bioorg. Chem., 2020, 102, 104103 CrossRef CAS.
- J. E. F. Alves, J. F. de Oliveira, T. R. C. de Lima Souza, R. O. de Moura, L. B. de Carvalho Júnior, M. do C. Alves de Lima and S. M. V. de Almeida, Novel indole-thiazole and indole-thiazolidinone derivatives as DNA groove binders, Int. J. Biol. Macromol., 2021, 170, 622–635 CrossRef CAS.
- P. F. da S. Santos-Junior, I. J. dos S. Nascimento, E. C. D. da Silva, K. L. C. Monteiro, J. D. de Freitas, S. de Lima Lins, T. M. S. Maciel, B. C. Cavalcanti, J. de B. José de, F. C. de Abreu, I. M. Figueiredo, J. C. C. Santos, C. do Ó. Pessoa, E. F. da Silva-Júnior, J. X. de Araújo-Júnior and T. M. de Aquino, Synthesis of hybrids thiazole-quinoline, thiazole-indole and their analogs:in vitroanti-proliferative effects on cancer cell lines, DNA binding properties and molecular modeling, New J. Chem., 2021, 45, 13847–13859 RSC.
- S. Eryılmaz, E. Türk Çelikoğlu, Ö. İdil, E. İnkaya, Z. Kozak, E. Mısır and M. Gül, Derivatives of pyridine and thiazole hybrid: synthesis, DFT, biological evaluation via antimicrobial and DNA cleavage activity, Bioorg. Chem., 2020, 95, 103476 CrossRef.
- X. Su, L. Wang, Y. Xu, L. Dong and H. Lu, Study on the binding mechanism of thiamethoxam with three model proteins:spectroscopic studies and theoretical simulations, Ecotoxicol. Environ. Saf., 2021, 207, 111280 CrossRef CAS.
- B. C. Yallur, U. Katrahalli, P. M. Krishna and M. D. Hadagali, BSA binding and antibacterial studies of newly synthesized 5,6-Dihydroimidazo[2,1-b]thiazole-2-carbaldehyde, Spectrochim. Acta, Part A, 2019, 222, 117192 CrossRef CAS PubMed.
- R. Aggarwal, M. Hooda, N. Jain, D. Sanz, R. M. Claramunt, B. Twamley and I. Rozas, An efficient, one-pot, regioselective synthesis of 2-aryl/hetaryl-4-methyl-5-acylthiazoles under solvent-free conditions, J. Sulfur Chem., 2022, 43, 12–21 CrossRef CAS.
- R. Aggarwal, M. Hooda, P. Kumar and G. Sumran, Solvent-free, Silica Catalyzed Intriguing Conversion of 3- Hydrazonobutan-2-one Oxime into Biacetyl Bis-hydrazone Schiff Bases, Lett. Org. Chem., 2021, 18, 532–537 CrossRef CAS.
- R. Aggarwal, M. Hooda, P. Kumar, S. Kumar, S. Singh and R. Chandra, An expeditious on-water regioselective synthesis of novel arylidene-hydrazinyl-thiazoles as DNA targeting agents, Bioorg. Chem., 2023, 136, 106524 CrossRef CAS.
- R. Aggarwal, M. Hooda, P. Kumar and M. C. Torralba, Visible-light-mediated regioselective synthesis of novel thiazolo[3,2-: B] [1,2,4]triazoles: advantageous synthetic application of aqueous conditions, Org. Biomol. Chem., 2022, 20, 584–595 RSC.
- R. Aggarwal, S. Kumar and G. Singh, Multi-component reaction to access a library of polyfunctionally substituted 4 , 7- dihydropyrazolo [ 3 , 4- b ] pyridines, Synth. Commun., 2019, 1–13 Search PubMed.
- R. Aggarwal, One-pot synthesis and in vitro bioactivity of novel 4-aminopyrazolo [ 3 , 4- b ] pyridine derivatives as potential antimicrobial compounds, Med. Chem. Res., 2024, 33, 117–126 CrossRef CAS.
- R. Aggarwal, N. Jain, S. Sharma, P. Kumar, G. P. Dubey, H. Chugh and R. Chandra, Visible-light driven regioselective synthesis, characterization and binding studies of 2-aroyl-3-methyl-6,7-dihydro-5H-thiazolo[3,2-a]pyrimidines with DNA and BSA using biophysical and computational techniques, Sci. Rep., 2021, 11, 1–18 CrossRef.
- R. Aggarwal, M. Hooda, P. Kumar, N. Jain, G. P. Dubey, H. Chugh and R. Chandra, Visible-Light-Prompted Synthesis and Binding Studies of 5,6-Dihydroimidazo[2,1- b]thiazoles with BSA and DNA Using Biophysical and Computational Methods, J. Org. Chem., 2022, 87, 3952–3966 CrossRef CAS.
- R. Aggarwal, N. Jain, G. P. Dubey, S. Singh and R. Chandra, Visible Light-Prompted Regioselective Synthesis of Novel 5-Aroyl/hetaroyl-2′,4-dimethyl-2,4′-bithiazoles as DNA- and BSA-Targeting Agents, Biomacromolecules, 2023, 24, 4798–4818 CrossRef CAS PubMed.
- R. Aggarwal and M. Hooda, Visible-light promoted serendipitous synthesis of 3,5-diaryl-1,2,4-thiadiazoles via oxidative dimerization of thiobenzamides, J. Sulfur Chem., 2022, 43, 242–251 CrossRef CAS.
- R. Aggarwal, S. Sharma, M. Hooda, D. Sanz, R. M. Claramunt, B. Twamley and I. Rozas, Visible-light mediated regioselective approach towards synthesis of 7-aroyl-6-methyl-[1,2,4]triazolo[3,4-b][1,3,4]thiadiazines, Tetrahedron, 2019, 75, 130728 CrossRef CAS.
- S. Mamidala, S. R. Peddi, R. K. Aravilli, P. C. Jilloju, V. Manga and R. R. Vedula, Microwave irradiated one pot, three component synthesis of a new series of hybrid coumarin based thiazoles: antibacterial evaluation and molecular docking studies, J. Mol. Struct., 2021, 1225, 129114 CrossRef CAS.
- R. Raveesha, A. M. Anusuya, A. V. Raghu, K. Yogesh Kumar, M. G. Dileep Kumar, S. B. Benaka Prasad and M. K. Prashanth, Visible-light-mediated regioselective synthesis of novel thiazolo[3,2-: B] [1,2,4]triazoles: advantageous synthetic application of aqueous conditions, Comput. Toxicol., 2022, 21, 100202 CrossRef CAS.
- V. K. Choudhary, A. K. Bhatt, D. Dash and N. Sharma, DFT calculations on molecular structures, HOMO–LUMO study, reactivity descriptors and spectral analyses of newly synthesized diorganotin(IV) 2-chloridophenylacetohydroxamate complexes, J. Comput. Chem., 2019, 40, 2354–2363 CrossRef CAS.
- J. Haribabu, V. Garisetti, R. E. Malekshah, S. Srividya, D. Gayathri, N. Bhuvanesh, R. V. Mangalaraja, C. Echeverria and R. Karvembu, Design and synthesis of heterocyclic azole based bioactive compounds: molecular structures, quantum simulation, and mechanistic studies through docking as multi-target inhibitors of SARS-CoV-2 and cytotoxicity, J. Mol. Struct., 2022, 1250, 131782 CrossRef CAS.
- R. Raveesha, A. M. Anusuya, A. V. Raghu, K. Yogesh Kumar, M. G. Dileep Kumar, S. B. Benaka Prasad and M. K. Prashanth, Discovery of novel JAK2 and EGFR inhibitors from a series of thiazole-based chalcone derivatives, Comput. Toxicol., 2022, 21, 100202 CrossRef CAS.
- A. B. Umar, A. Uzairu, S. Uba and G. A. Shallangwa, In silico Studies of some potential anti-cancer agents on M19-MEL cell line, Moroccan J. Chem., 2021, 9, 260–273 CAS.
- H. Mahaki, H. Tanzadehpanah, O. K. Abou-Zied, N. H. Moghadam, A. Bahmani, S. Salehzadeh, D. Dastan and M. Saidijam, Cytotoxicity and antioxidant activity of Kamolonol acetate from Ferula pseudalliacea, and studying its interactions with calf thymus DNA (ct-DNA) and human serum albumin (HSA) by spectroscopic and molecular docking techniques, Process Biochem., 2019, 79, 203–213 CrossRef CAS.
- Z. Nan, C. Hao, X. Ye, Y. Feng and R. Sun, Interaction of graphene oxide with bovine serum albumin: a fluorescence quenching study, Spectrochim. Acta, Part A, 2019, 210, 348–354 CrossRef CAS PubMed.
- M. Szymańska, M. Insińska-Rak, G. Dutkiewicz, G. N. Roviello, M. A. Fik-Jaskółka and V. Patroniak, Thiophene-benzothiazole dyad ligand and its Ag(I) complex – Synthesis, characterization, interactions with DNA and BSA, J. Mol. Liq., 2020, 319, 1–10 CrossRef.
- S. Singhal, P. Khanna and L. Khanna, Synthesis, DFT studies, molecular docking, antimicrobial screening and UV fluorescence studies on ct-DNA for novel Schiff bases of 2-(1-aminobenzyl) benzimidazole, Heliyon, 2019, 5, e02596 CrossRef.
- K. P. Thakor, M. V. Lunagariya, B. S. Bhatt and M. N. Patel, Fluorescence and absorption studies of DNA–Pd(II) complex interaction: synthesis, spectroanalytical investigations and biological activities, Luminescence, 2019, 34, 113–124 CrossRef CAS PubMed.
- S. Kumar and M. S. Nair, Deciphering the interaction of flavones with calf thymus DNA and octamer DNA sequence (CCAATTGG)2, RSC Adv., 2021, 11, 29354–29371 RSC.
- S. Salehzadeh, F. Hajibabaei, N. H. Moghadam, S. Sharifinia, S. Khazalpour and R. Golbedaghi, Binding Studies of Isoxsuprine Hydrochloride to Calf Thymus DNA Using Multispectroscopic and Molecular Docking Techniques, J. Fluoresc., 2018, 28, 195–206 CrossRef CAS.
- P. R. Patlolla, N. Desai, S. Gupta and B. Datta, Interaction of a dimeric carbocyanine dye aggregate with bovine serum albumin in non-aggregated and aggregated forms, Spectrochim. Acta, Part A, 2019, 209, 256–263 CrossRef CAS PubMed.
- K. Karami, N. Jamshidian and M. Zakariazadeh, Synthesis, characterization and molecular docking of new C,N-palladacycles containing pyridinium-derived ligands: DNA and BSA interaction studies and evaluation as anti-tumor agents, Appl. Organomet. Chem., 2019, 33, 3747–3751 Search PubMed.
- J. Gu, S. Zheng, H. Zhao and T. Sun, Investigation on the interaction between triclosan and bovine serum albumin by spectroscopic methods, J. Environ. Sci. Health, Part B, 2020, 55, 52–59 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI consists of additional experimental data, (1H, 13C, [1H–13C] HMBC; [1H–13C] HMQC and [1H–15N] HMBC) for final compounds, Van't Hoff plot, CD spectrum for DNA, fluorescence spectra of BSA-IB and BSA-PB complex and general experimental section for both synthesis and binding studies. See DOI: https://doi.org/10.1039/d4ra02331e |
| This journal is © The Royal Society of Chemistry 2024 |