
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZn(OTf)2-catalyzed intra- and intermolecular selenofunctionalization of alkenes under mild conditions†
Cong Qia,
Zhaogong Lua,
Yuyang Gub,
Xiaofeng Baoa,
Biao Xiong a and
Gong-Qing Liu*a
a and
Gong-Qing Liu*a
aSchool of Pharmacy, Nantong Key Laboratory of Small Molecular Drug Innovation, Nantong University, Nantong, 226019, People's Republic of China. E-mail: gqliu@ntu.edu.cn
bSchool of Medicine, Nantong University, Nantong 226019, People's Republic of China
First published on 22nd July 2024
Abstract
Zn(OTf)2-catalyzed intra- and intermolecular selenofunctionalization of alkenes was achieved with electrophilic N-phenylselenophthalimide. This method provides straightforward and efficient access to various seleno-substituted heterocycles and vicinal Se heteroatom-disubstituted molecules under mild conditions. This reaction is compatible with various substrates/functional groups, and preliminary studies on the reaction mechanistic were also conducted.
Introduction
Selenium (Se), a biological trace element, is essential for animal and human health. A deficiency or excess intake of Se can result in severe symptoms and is associated with various diseases.1 Furthermore, organoselenium compounds have emerged as valuable building blocks in organic synthesis and are employed as radicals, electrophiles and nucleophiles to construct a wide range of chemical bonds.2 Specifically, seleno-substituted heterocycles and vicinally functionalized selenides are highly notable due to their significance as chemical reagents, building blocks, and biologically active molecules (Scheme 1).3 Hence, numerous attempts have been made to develop a synthetic route for accessing these compounds. | ||
| Scheme 1 Vital seleno-substituted heterocycles and vicinally functionalized selenides. | ||
Among them, the intra- and intermolecular selenofunctionalization of olefins is considered one of the most straightforward methods because a selenium moiety can be simultaneously introduced with other synthetically valuable functionalities across a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, generating seleno-substituted heterocycles and vicinal Se, heteroatom-disubstituted molecules, in an atom-economical manner.4 Typically, this process occurs via the formation of a seleniranium intermediate using electrophilic selenium reagents,5 followed by the nucleophilic attack of suitable reagents. However, these organoselenium reagents are often undesirable and inconvenient to use. Their main drawbacks arise from their moisture-sensitive nature, the release of malodorous and highly toxic vapors, and the formation of various reactive by-products, during certain reactions (e.g., HCl from PhSeCl). Selenofunctionalization can also be achieved through utilizing readily available and bench-stable diselenides as selenium reagents. Various oxidants,6 photochemical,7 and electrochemical approaches8 have been used to produce selenium electrophilic species from the corresponding diselenides in these cases. Recently, we reported that hypervalent iodine,9 N–F reagents,10 and photochemical processes11 can activate diselenides to produce highly reactive selenium species, which can induce intra- and intermolecular selenofunctionalization to produce functionalized Se-containing molecules (Scheme 2a).
C bond, generating seleno-substituted heterocycles and vicinal Se, heteroatom-disubstituted molecules, in an atom-economical manner.4 Typically, this process occurs via the formation of a seleniranium intermediate using electrophilic selenium reagents,5 followed by the nucleophilic attack of suitable reagents. However, these organoselenium reagents are often undesirable and inconvenient to use. Their main drawbacks arise from their moisture-sensitive nature, the release of malodorous and highly toxic vapors, and the formation of various reactive by-products, during certain reactions (e.g., HCl from PhSeCl). Selenofunctionalization can also be achieved through utilizing readily available and bench-stable diselenides as selenium reagents. Various oxidants,6 photochemical,7 and electrochemical approaches8 have been used to produce selenium electrophilic species from the corresponding diselenides in these cases. Recently, we reported that hypervalent iodine,9 N–F reagents,10 and photochemical processes11 can activate diselenides to produce highly reactive selenium species, which can induce intra- and intermolecular selenofunctionalization to produce functionalized Se-containing molecules (Scheme 2a).
 | ||
| Scheme 2 Previous work and current work. | ||
N-Phenylselenophthalimide (N-PSP), which was originally reported by K. C. Nicolaou and coworkers,12 is a convenient source of electrophilic selenium for intra- and intermolecular selenofunctionalization because this compound is an odorless and colorless crystalline solid that can be readily obtained from potassium phthalimide and phenylselenenyl chloride. Additionally, the poor nucleophilicity of the phthalimide counteranion can eliminate chemoselectivity issues caused by competition between the desired nucleophiles and the phthalimide. Thus, various Lewis and Brønsted acids, such as TiCl4,13 FeCl3,14 BF3,15 Ca(NTf2)2,16 phosphoric acid,17 TMSOTf18 and p-TsOH,19 and base20 have been used to promote selenofunctionalization reactions with N-PSP. Despite these advances, a general and modular method for the preparation of various vicinally functionalized selenides is still lacking. Furthermore, to the best of our knowledge, intramolecular selenoamination of N-alkenyl sulfonamides with N-PSP leading to selenomethylpyrrolidine has not been accomplished. Therefore, an efficient strategy to achieve the selenofunctionalization of alkenes is highly desirable and intensively sought after.
Zinc is an essential trace element for humans. Deficiencies in zinc may cause many diseases in adults and can lead to growth retardation, delayed sexual maturation, infection susceptibility, and diarrhea in children.21 Zinc is also a notably attractive element in synthetic chemistry due to its abundance, low cost, lack of toxicity, and environmentally benign properties.22 Additionally, zinc displays Lewis acid interactions with a variety of functional groups. In previous studies, we revealed that zinc salts interact with CC double bonds, ketones, and imines.23 To demonstrate the powerful flexibility of zinc catalyst in the construction of a C–Se bond, herein, we wish to report a Zn(OTf)2-catalyzed intra- and intermolecular selenofunctionalization of alkenes with electrophilic N-PSP as a continuation of our interest in the selenofunctionalization of different alkenes (Scheme 2b).9–11,24
Results and discussion
Initially, we investigated the reactions between 1a and N-PSP 2a with various catalysts in CH2Cl2 at room temperature (Table 1). During the initial screening of diverse zinc catalysts, including ZnCl2, ZnBr2, ZnI2, and Zn(OTf)2, we found that Zn(OTf)2 was optimal for this reaction and afforded the corresponding phenylselenomethylpyrrolidine 3a in 85% yield (entries 1–4). We rationalized this outcome with sufficient Lewis acidity of Zn(OTf)2. Other metals, such as AgOTf, MnBr2, CuCl and InBr3, did not improve the yields (entries 5–8). Subsequent solvent screening experiments indicated that CH2Cl2 was the best solvent in terms of the reaction yield obtained (entries 9–13). Control experiments verified that the zinc catalyst was essential for the reaction, as the yield significantly decreased in its absence (entry 14).
|
|||
|---|---|---|---|
| Entry | Cat. | Solvent | Isolated yield (%) |
| a Reaction conditions: 1a (0.20 mmol, 1.00 equiv.), 2a (0.20 mmol, 1.00 equiv.), cat. (0.010 mmol, 0.05 equiv.), solvent (2 mL), r.t., 5 h. | |||
| 1 | ZnCl2 | CH2Cl2 | 45 |
| 2 | ZnBr2 | CH2Cl2 | 50 |
| 3 | ZnI2 | CH2Cl2 | 51 |
| 4 | Zn(OTf)2 | CH2Cl2 | 85 |
| 5 | AgOTf | CH2Cl2 | 67 |
| 6 | MnBr2 | CH2Cl2 | 33 |
| 7 | CuCl | CH2Cl2 | 45 |
| 8 | InBr3 | CH2Cl2 | 39 |
| 9 | Zn(OTf)2 | MeOH | 12 |
| 10 | Zn(OTf)2 | Acetone | 47 |
| 11 | Zn(OTf)2 | THF | 55 |
| 12 | Zn(OTf)2 | CH3CN | 17 |
| 13 | Zn(OTf)2 | Hexane | 20 |
| 14 | — | CH2Cl2 | 15 |
Having established the optimized reaction conditions, we evaluated the scope of the alkene aminoselenation reaction. As indicated in Scheme 3, both N-aryl and N-alkyl sulfonamides underwent smooth 5-exo cyclization to furnish the corresponding selenomethylpyrrolidines (3b–3f). Remarkably, we observed that the Thorpe–Ingold effect was not necessary for reactivity, as substrates bearing different gem-disubstituents or without substituents in the backbone exhibited similar reactivity (3g–3i vs. 3j). N-Tosyl o-allyl aniline (1k) was an effective substrate, generating indoline 3k. We were pleased to find that a more geometrically challenging 5-endo cyclization could be achieved with unsaturated amine 1l, which afforded bridged ring skeleton 3l with high diastereoselectivity. Di- and trisubstituted alkenes were efficiently converted into their corresponding pyrrolidines with good yields (3m–3n). Furthermore, 1-sulfonamido-5-hexene substrate was tolerated, although a lower yield was obtained (3o), possibly due to unfavorable entropy of the current cyclization.25 Differing from the PhSeX-mediated cyclization of unsaturated amines that produces a mixture of (phenylselanyl)pyrrolidines and halopyrrolidines,26 the current reaction was found to exclusively afford selenoaminated products.
 | ||
| Scheme 3 Scope of olefinic sulfonamides. Reaction conditions: 1 (0.20 mmol), 2a (0.20 mmol), Zn(OTf)2 (0.010 mmol) and CH2Cl2 (2 mL), air, r.t., 5 h. | ||
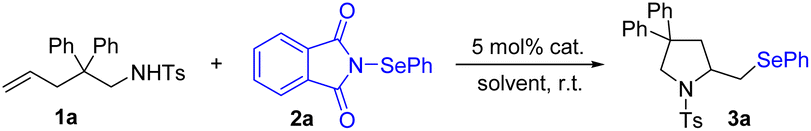
Although intermolecular alkene selenoamination is valuable for the synthesis of β-amino selenides, few methods possess a broad scope and wide applicability. This is largely due to the inherent challenges of chemoselectivity and regioselectivity issues as well as amine oxidation. Despite these challenges, significant progress has been made (often within restricted substrate classes),7a,8b,27 in the areas of three-component alkene selenoamination. During the preparation of this manuscript, Wang, Yi and Hong et al. reported a Ca(NTf2)2-catalyzed intermolecular selenoamination of alkenes with N-PSP.16 Encouraged by the above results, we hoped to expand the general utility of this catalyst system to include intermolecular examples and access biologically relevant amines. We chose challenging anilines as nucleophiles as they are susceptible to oxidation at nitrogen under previously reported oxidation selenofunctionalization conditions, thus limiting their application in this process. Fortunately, a broad range of anilines were found to be effective nucleophiles under the title conditions (Scheme 4). Remarkably, a diverse array of functional groups, such as halides, nitro, nitrile, thioether and pinacol borate, were well tolerated (4a–4i), providing an opportunity for further modifications to access biologically active selenium-containing compounds. This method is not limited to primary amines, as secondary anilines can also be aminated effectively (4j–4k). Additionally, the approved drugs benzocaine and sulfamethoxazole afforded products 4l and 4m, respectively.
 | ||
| Scheme 4 Scope of amines. Reaction conditions: styrene (0.20 mmol), 2a (0.20 mmol), amine (0.20 mmol), Zn(OTf)2 (0.010 mmol) and CH2Cl2 (2 mL), air, r.t., 5 h. | ||
Next, we examined a variety of challenging substrates, namely, electronically deactivated styrenes, under our optimized reaction conditions. As illustrated in Scheme 5, a variety of electron-deficient styrenes substituted with single CN, NO2, CO2Me and CF3 groups reacted efficiently (5a–5d). Impressively, styrenes bearing up to two halides or up to five fluorine atoms on the aromatic ring were well tolerated (5e–5g).
 | ||
| Scheme 5 Scope of styrenes. Reaction conditions: alkene (0.20 mmol), 2a (0.20 mmol), 4-chloroaniline (0.20 mmol), Zn(OTf)2 (0.010 mmol) and CH2Cl2 (2 mL), air, r.t., 5 h. | ||
Encouraged by these results, we further explored the versatility of our method by investigating different alcohols as nucleophiles because the resulting β-alkoxyl selenides are valuable for synthetic chemists. Through the adjacent alkoxyl group, these molecules can be attached to solid-phase carriers for the development of recyclable heterogeneous catalysts.28 Moreover, the neighboring oxygen and selenium groups can effectively coordinate with transition metals, leading to the creation of novel metal complexes with potentially distinctive catalytic activity.29 As shown in Scheme 6, a variety of benzyl alcohols proved to be effective nucleophiles under the optimized conditions (6a–6c). Furthermore, natural alcohols that are more structurally complicated, such as L-(−)-menthol, geraniol and cholesterol, also afforded the desired β-alkoxy selenides in good yields (6d–6f). This result highlights the powerful ability of this catalytic system to create novel and potentially bioactive organoselenium compounds from complex natural products.
 | ||
| Scheme 6 Scope of alcohols. Reaction conditions: styrene (0.20 mmol), 2a (0.50 mmol), alcohol (0.20 mmol), Zn(OTf)2 (0.010 mmol) and CH2Cl2 (2 mL), air, r.t., 5 h. | ||
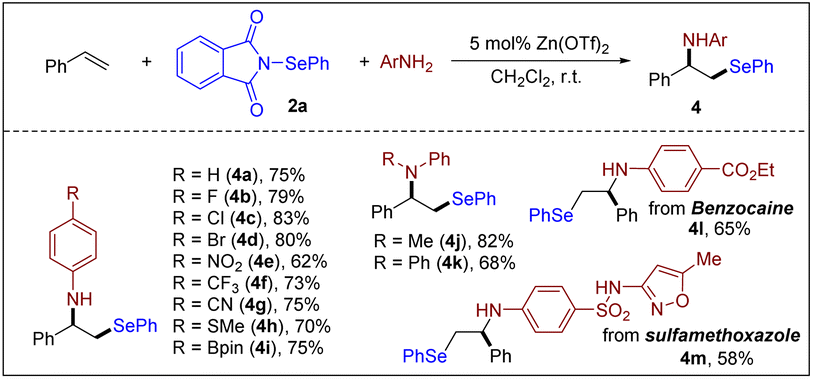
In the investigation of different nucleophiles, a trace amount of β-hydroxy selenide (ca. 5%) was detected as a byproduct. This product was formed by nucleophilic attack of adventitious water at the episelenonium ion intermediate. Given the importance of β-hydroxy selenides as valuable intermediates in the synthesis of allylic alcohols, olefins, vinyl, and heterocyclic compounds,30 we modified the solvent system to include a combination of CH2Cl2 and water. This adjustment was made to facilitate the straightforward synthesis of β-hydroxy selenides. As shown in Scheme 7, hydroxyselenenylation of styrene and aliphatic alkenes in a mixture of CH2Cl2 and H2O proceeded smoothly, resulting in β-hydroxy selenides in good yields with exclusive Markovnikov selectivity.
 | ||
Scheme 7 Synthesis of β-hydroxy selenides. Reaction conditions: alkene (0.20 mmol), 2a (0.20 mmol), Zn(OTf)2 (0.010 mmol) and CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O (50:1, 2 mL), air, r.t., 5 h. :H2O (50:1, 2 mL), air, r.t., 5 h. | ||
To further determine the mechanism of this reaction, 2.0 equivalents of 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) and 2,6-di-tert-butyl-4-methylphenol (BHT) were introduced as radical trapping agents, and the reaction proceeded smoothly under the title conditions (Scheme 8), which clearly suggested that the reaction did not involve a free radical pathway.
 | ||
| Scheme 8 Control experiments. | ||
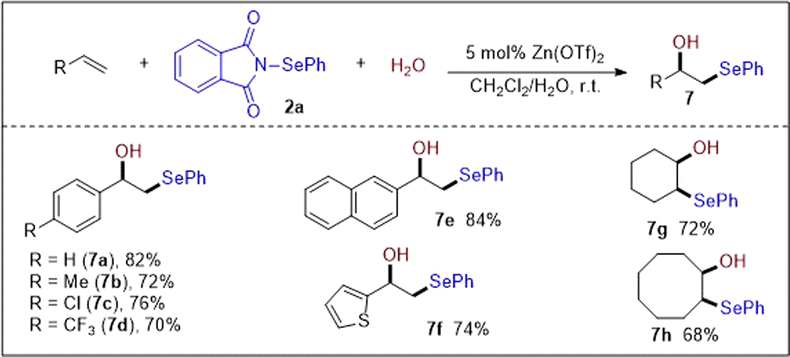
Based on the above investigations and reported literature,13,14,16 a plausible reaction mechanism was proposed, as shown in Scheme 9. Initially, Zn(OTf)2 activates N-PSP 2a by chelating to the amide carbonyl group to form intermediate A, which undergoes electrophilic attack on the CC bond and results in the formation of episelenonium ion B. Subsequently, the ring-opening of B by nucleophilic attack generates cation C. Finally, deprotonation of intermediate C by species D affords the desired selenation products with the release of Zn(OTf)2.
 | ||
| Scheme 9 Proposed plausible mechanistic pathway. | ||
Conclusions
In conclusion, we have reported a Zn(OTf)2-catalyzed selenofunctionalization of alkenes with N-phenylselenophthalimide for the assembly of vicinally functionalized selenoderivatives. Compared with previous approaches, the current method features ambient conditions and application of environmentally benign zinc catalysis. Moreover, this protocol achieves excellent substrate/functional group tolerance, and is suitable for the late-stage functionalization of complex molecules of biological importance. Future efforts will be directed toward the development of asymmetric variants of this reaction with chiral ligands.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This study was supported by the Science and Technology Plan Projects of Nantong (JC22022097, JC2023035) and the Large Instruments Open Foundation of Nantong University (KFJN2365).Notes and references
- (a) C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255–6285 Search PubMed; (b) W. Hou and H. Xu, J. Med. Chem., 2022, 65, 4436–4456 CrossRef CAS; (c) M. Zhou, S. Han, O. Aras and F. An, Curr. Med. Chem., 2021, 28, 6358–6374 CrossRef CAS PubMed.
- (a) B.-Y. Fan, X. Jiang, Y.-X. Li, W.-L. Wang, M. Yang, J.-L. Li, A.-D. Wang and G.-T. Chen, Med. Res. Rev., 2022, 42, 2025–2066 CrossRef CAS; (b) J. M. Sonego, S. I. de Diego, S. H. Szajnman, C. Gallo-Rodriguez and J. B. Rodriguez, Chem.–Eur. J., 2023, 29, e202300030 CrossRef CAS PubMed; (c) A. A. Heredia, L. M. Bouchet, W. D. Castro-Godoy and J. E. Argüello, Tetrahedron, 2023, 148, 133667 CrossRef CAS; (d) K.-N. Song, Y.-J. Lu, C.-J. Chu, Y.-N. Wu, H.-L. Huang, B.-Y. Fan and G.-T. Chen, J. Nat. Prod., 2021, 84, 2664–2674 CrossRef CAS.
- (a) X. Liu, Y. Liang, J. Ji, J. Luo and X. Zhao, J. Am. Chem. Soc., 2018, 140, 4782–4786 CrossRef CAS; (b) L. B. Marx and J. W. Burton, Chem.–Eur. J., 2018, 24, 6747–6754 CrossRef CAS PubMed; (c) H. Xu, X. Su, M.-b. Guo, R. An, Y.-h. Mou, Z. Hou and C. Guo, Eur. J. Med. Chem., 2020, 198, 112360 CrossRef CAS PubMed; (d) N. Arumugam, R. Raghunathan, V. Shanmugaiah and N. Mathivanan, Bioorg. Med. Chem. Lett., 2010, 20, 3698–3702 CrossRef CAS PubMed; (e) R. M. da Rosa, B. C. Piccoli, F. D. A. da Silva, L. Dornelles, J. B. T. Rocha, M. S. Sonego, K. R. Begnini, T. Collares, F. K. Seixas and O. E. D. Rodrigues, MedChemComm, 2017, 8, 408–414 RSC; (f) K. Song, M. Li, Y. Yang, Z. Zhang, Q. Zhu, J. Liu and A. Wang, J. Pharm. Pharmacol., 2022, 74, 337–350 CrossRef PubMed; (g) F. Ran, Y. Liu and G. Zhao, Med. Chem. Res., 2022, 31, 594–604 Search PubMed.
- (a) K. Sun, X. Wang, C. Li, H. Wang and L. Li, Org. Chem. Front., 2020, 7, 3100–3119 RSC; (b) J. Liu, J.-P. Wan and Y. Liu, Org. Chem. Front., 2024, 11, 597–630 RSC; (c) L. Lu, D. Huang, Z. Wang, X. Wang and X. Wu, Adv. Synth. Catal., 2023, 365, 2310–2331 Search PubMed.
- (a) A. Toshimitsu, T. Aoai, H. Owada, S. Uemura and M. Okano, J. Org. Chem., 1981, 46, 4727–4733 CrossRef CAS; (b) A. Toshimitsu, T. Aoai, H. Owada, S. Uemura and M. Okano, J. Chem. Soc., Chem. Commun., 1980, 412–413 RSC; (c) T. Y. Luh, W. H. So, K. S. Cheung and S. W. Tam, J. Org. Chem., 1985, 50, 3051–3053 CrossRef CAS.
- (a) Z. Zhao, Y. Zhang, Y. Shao, W. Xiong, R. Li and J. Chen, J. Org. Chem., 2020, 85, 15015–15025 CrossRef CAS; (b) P. S. Hellwig, A. M. Barcellos, R. Cargnelutti, T. Barcellos and G. Perin, J. Org. Chem., 2022, 87, 15050–15060 CrossRef CAS; (c) J.-D. Fang, X.-B. Yan, L. Zhou, Y.-Z. Wang and X.-Y. Liu, Adv. Synth. Catal., 2019, 361, 1985–1990 CrossRef CAS; (d) L. Xing, Y. Zhang, B. Li and Y. Du, Org. Lett., 2019, 21, 3620–3624 CrossRef CAS.
- (a) B. Huang, Y. Li, C. Yang and W. Xia, Green Chem., 2020, 22, 2804–2809 Search PubMed; (b) Q.-B. Zhang, P.-F. Yuan, L.-L. Kai, K. Liu, Y.-L. Ban, X.-Y. Wang, L.-Z. Wu and Q. Liu, Org. Lett., 2019, 21, 885–889 CrossRef CAS; (c) D. V. Patil, Y. T. Hong, H. Y. Kim and K. Oh, Org. Lett., 2022, 24, 8465–8469 CrossRef CAS PubMed; (d) H. Chen, R. Ding, H. Tang, Y. Pan, Y. Xu and Y. Chen, Chem.–Asian J., 2019, 14, 3264–3268 CrossRef CAS PubMed.
- (a) X.-J. Meng, P.-F. Zhong, Y.-M. Wang, H.-S. Wang, H.-T. Tang and Y.-M. Pan, Adv. Synth. Catal., 2020, 362, 506–511 CrossRef CAS; (b) L. Sun, Y. Yuan, M. Yao, H. Wang, D. Wang, M. Gao, Y.-H. Chen and A. Lei, Org. Lett., 2019, 21, 1297–1300 CrossRef CAS PubMed; (c) X. Cheng, B. Hasimujiang, Z. Xu, H. Cai, G. Chen, G. Mo and Z. Ruan, J. Org. Chem., 2021, 86, 16045–16058 CrossRef CAS; (d) P. Zhou, H. Jiao, K. Niu, H. Song, Y. Liu and Q. Wang, ACS Sustainable Chem. Eng., 2023, 11, 2607–2612 CrossRef CAS; (e) S. Zeng, Y. Zeng, H. Wang, P. Sun and Z. Ruan, J. Org. Chem., 2024, 89, 4074–4084 CrossRef CAS PubMed; (f) B. Li, Y. Zhou, Y. Xu, X. Li, Z. Li, L. Gu, W. Ma and R. Mei, J. Org. Chem., 2023, 88, 15414–15427 CrossRef CAS PubMed.
- (a) P.-F. Wang, W. Yi, Y. Ling, L. Ming, G.-Q. Liu and Y. Zhao, Chin. Chem. Lett., 2021, 32, 2587–2591 CrossRef CAS; (b) Z.-P. Liang, W. Yi, P.-F. Wang, G.-Q. Liu and Y. Ling, J. Org. Chem., 2021, 86, 5292–5304 CrossRef CAS.
- (a) Y.-Q. Jiang, Y.-H. Wang, C.-F. Zhou, Y.-Q. Zhang, Y. Ling, Y. Zhao and G.-Q. Liu, J. Org. Chem., 2022, 87, 14609–14622 CrossRef CAS PubMed; (b) C.-F. Zhou, Y.-Q. Zhang, Y. Ling, L. Ming, X. Xi, G.-Q. Liu and Y. Zhang, Org. Biomol. Chem., 2022, 20, 420–426 RSC; (c) Y.-H. Wang, Y.-Q. Zhang, C.-F. Zhou, Y.-Q. Jiang, Y. Xu, X. Zeng and G.-Q. Liu, Org. Biomol. Chem., 2022, 20, 5463–5469 RSC.
- (a) G.-Q. Liu, W. Yi, P.-F. Wang, J. Liu, M. Ma, D.-Y. Hao, L. Ming and Y. Ling, Green Chem., 2021, 23, 1840–1846 RSC; (b) G.-Q. Liu, C.-F. Zhou, Y.-Q. Zhang, W. Yi, P.-F. Wang, J. Liu and Y. Ling, Green Chem., 2021, 23, 9968–9973 RSC.
- (a) K. C. Nicolaou, D. A. Claremon, W. E. Barnette and S. P. Seitz, J. Am. Chem. Soc., 1979, 101, 3704–3706 CrossRef CAS; (b) K. C. Nicolaou, Tetrahedron, 1981, 37, 4097–4109 CrossRef CAS.
- E. Tang, W. Wang, Y. Zhao, M. Zhang and X. Dai, Org. Lett., 2016, 18, 176–179 CrossRef CAS.
- L. Lv and Z. Li, J. Org. Chem., 2018, 83, 10985–10994 CrossRef CAS.
- M.-H. Liao, M. Zhang, D.-H. Hu, R.-H. Zhang, Y. Zhao, S.-S. Liu, Y.-X. Li, W.-L. Xiao and E. Tang, Org. Biomol. Chem., 2020, 18, 4034–4045 RSC.
- M. Kuang, H. Li, Z. Zeng, H. Gao, Z. Zhou, X. Hong, W. Yi and S. Wang, Org. Lett., 2023, 25, 8095–8099 CrossRef CAS.
- J. Y. See, H. Yang, Y. Zhao, M. W. Wong, Z. Ke and Y.-Y. Yeung, ACS Catal., 2018, 8, 850–858 CrossRef CAS.
- E. Tang, Y. Zhao, W. Li, W. Wang, M. Zhang and X. Dai, Org. Lett., 2016, 18, 912–915 CrossRef CAS.
- (a) X. Zhao, Z. Yu, T. Xu, P. Wu and H. Yu, Org. Lett., 2007, 9, 5263–5266 CrossRef CAS; (b) P. Wu, L. Wang, K. Wu and Z. Yu, Org. Lett., 2015, 17, 868–871 CrossRef CAS PubMed.
- (a) H. Joshi and S. Sathyamoorthi, J. Org. Chem., 2022, 87, 5017–5028 CrossRef CAS PubMed; (b) S. E. Denmark and W. R. Collins, Org. Lett., 2007, 9, 3801–3804 CrossRef CAS PubMed.
- N. Roohani, R. Hurrell, R. Kelishadi and R. Schulin, J. Res. Med. Sci., 2013, 18, 144 Search PubMed.
- X.-F. Wu and H. Neumann, Adv. Synth. Catal., 2012, 354, 3141–3160 CrossRef CAS.
- (a) G.-Q. Liu and Y.-M. Li, Tetrahedron Lett., 2011, 52, 7168–7170 CrossRef CAS; (b) G.-Q. Liu, W. Li, Y.-M. Wang, Z.-Y. Ding and Y.-M. Li, Tetrahedron Lett., 2012, 53, 4393–4396 CrossRef CAS; (c) J. Hou, L. Xue, T. Yu, J. Li, S. Yu, C. Yao and Y.-M. Li, Chem. Commun., 2023, 59, 7096–7099 RSC.
- (a) P. Qu, Y.-Q. Jiang, Y.-H. Wang and G.-Q. Liu, Green Chem., 2023, 25, 7485–7507 RSC; (b) Y.-H. Wang, Y.-Q. Jiang, Y.-Q. Zhang, Y. Ling, L. Ming and G.-Q. Liu, Chem.–Eur. J., 2023, 29, e202300530 CrossRef CAS PubMed.
- G. Illuminati and L. Mandolini, Acc. Chem. Res., 1981, 14, 95–102 CrossRef CAS.
- F. Outurquin, X. Pannecoucke, B. Berthe and C. Paulmier, Eur. J. Org Chem., 2002, 2002, 1007–1014 CrossRef.
- (a) F.-H. Cui, Y. Hua, Y.-M. Lin, J. Fei, L.-H. Gao, X. Zhao and H. Xia, J. Am. Chem. Soc., 2022, 144, 2301–2310 CrossRef CAS; (b) K. Sun, X. Wang, Y. Lv, G. Li, H. Jiao, C. Dai, Y. Li, C. Zhang and L. Liu, Chem. Commun., 2016, 52, 8471–8474 RSC; (c) R. Wang, N. Zhang, Y. Zhang, B. Wang, Y. Xia, K. Sun, W. Jin, X. Li and C. Liu, Green Chem., 2023, 25, 3925–3930 RSC.
- (a) R. T. Taylor and L. A. Flood, J. Org. Chem., 1983, 48, 5160–5164 CrossRef CAS; (b) Y. Wang, L. Yu, B. Zhu and L. Yu, J. Mater. Chem. A, 2016, 4, 10828–10833 RSC.
- (a) B. Manimaran, A. Vanitha, M. Karthikeyan, B. Ramakrishna and S. M. Mobin, Organometallics, 2014, 33, 465–472 CrossRef CAS; (b) F. Saleem, G. K. Rao, A. Kumar, G. Mukherjee and A. K. Singh, Organometallics, 2013, 32, 3595–3603 CrossRef CAS.
- (a) K. B. Sharpless and R. F. Lauer, J. Am. Chem. Soc., 1973, 95, 2697–2699 CrossRef CAS; (b) W. Dumont, D. Van Ende and A. Krief, Tetrahedron Lett., 1979, 20, 485–488 CrossRef; (c) M. Tiecco, L. Testaferri, A. Temperini, L. Bagnoli, F. Marini and C. Santi, Chem.–Eur. J., 2004, 10, 1752–1764 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04266b |
| This journal is © The Royal Society of Chemistry 2024 |