
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMitigating diabetes associated with reactive oxygen species (ROS) and protein aggregation through pharmacological interventions
Giulia Bennici a,
Hanan Almahasheerb,
Mawadda Alghrablya,
Daniela Valensinc,
Arian Kolac,
Chrysoula Kokotidoude,
Joanna Lachowiczf and
Mariusz Jaremko*a
a,
Hanan Almahasheerb,
Mawadda Alghrablya,
Daniela Valensinc,
Arian Kolac,
Chrysoula Kokotidoude,
Joanna Lachowiczf and
Mariusz Jaremko*a
aDivision of Biological and Environmental Sciences and Engineering (BESE), King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia. E-mail: mariusz.jaremko@kaust.edu.sa
bDepartment of Biology, College of Science, Imam Abdulrahman Bin Faisal University (IAU), Dammam 31441-1982, Saudi Arabia
cDepartment of Biotechnology, Chemistry and Pharmacy, University of Siena, Via Aldo Moro 2, 53100 Siena, Italy
dDepartment of Materials Science and Technology, University of Crete, 70013 Heraklion, Crete, Greece
eInstitute of Electronic Structure and Laser (IESL) FORTH, 70013 Heraklion, Crete, Greece
fDepartment of Population Health, Division of Environmental Health and Occupational Medicine, Wroclaw Medical University, Mikulicza-Radeckiego 7, Wroclaw, PL 50-368, Poland
First published on 29th May 2024
Abstract
Diabetes mellitus, a complex metabolic disorder, presents a growing global health challenge. In 2021, there were 529 million diabetics worldwide. At the super-regional level, Oceania, the Middle East, and North Africa had the highest age-standardized rates. The majority of cases of diabetes in 2021 (>90.0%) were type 2 diabetes, which is largely indicative of the prevalence of diabetes in general, particularly in older adults (K. L. Ong, et al., Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021, Lancet, 2023, 402(10397), 203–234). Nowadays, slowing the progression of diabetic complications is the only effective way to manage diabetes with the available therapeutic options. However, novel biomarkers and treatments are urgently needed to control cytokine secretion, advanced glycation end products (AGEs) production, vascular inflammatory effects, and cellular death. Emerging research has highlighted the intricate interplay between reactive oxygen species (ROS) and protein aggregation in the pathogenesis of diabetes. In this scenario, the main aim of this paper is to provide a comprehensive review of the current understanding of the molecular mechanisms underlying ROS-induced cellular damage and protein aggregation, specifically focusing on their contribution to diabetes development. The role of ROS as key mediators of oxidative stress in diabetes is discussed, emphasizing their impact on cellular components and signaling. Additionally, the involvement of protein aggregation in impairing cellular function and insulin signaling is explored. The synergistic effects of ROS and protein aggregation in promoting β-cell dysfunction and insulin resistance are examined, shedding light on potential targets for therapeutic intervention.
1 Introduction
We provide a thorough analysis of the literature spanning from 2000 to 2023, concentrating on significant advancements and trends in the subject. We seek to offer a comprehensive study of the development of theories and approaches in this field by looking at research that spans more than 20 years about diabetes associations with ROS and protein aggregation.Diabetes, commonly referred to as diabetes mellitus (DM), is a fatal metabolic illness that affects millions of individuals globally. According to global data, diabetes will be the cause of 592 million deaths by 2035.1,2
Type 1 diabetes mellitus (T1DM), also known as insulin-dependent, constitutes 5–10% of all cases; type 2 diabetes (T2DM), non-insulin-dependent diabetes mellitus, is the most frequent form of diabetes, with a percentage of 90–95% of all cases.3,4 Gestational diabetes mellitus (GDM) occurs between 0.6% and 15% of pregnancy cases, and it is related to socioeconomic conditions and ethnicity.5
T2DM results in progressive and complex metabolic dysfunctions due to the anomalous secretion of insulin by pancreatic cells, the anomalous action of this hormone in the cells (insulin resistance), or possibly by both events.3,6
These dysfunctions include disorders of the heart, such as hypertension and strokes, foot ulcers, retinopathy-related visual impairment, renal failure, complications from nephropathy, and diseases of the nerve system that cause neuropathy, which is linked to osteoarthropathy.6
Nephropathy, retinopathy, neuropathy, and lower limb amputations are examples of microvascular complications from diabetes mellitus, while cardiovascular problems represent macrovascular complications.7 These mainly arise from the damage induced by Amadori and AGEs, as well as the released free radicals (Fig. 1).
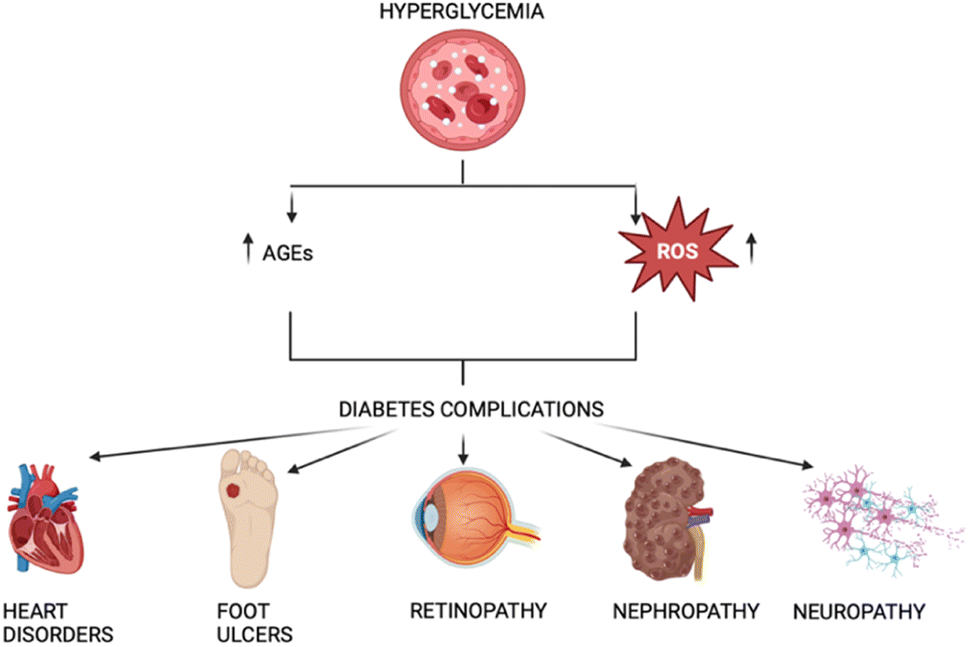 | ||
| Fig. 1 Correlation between AGEs, ROS, and diabetes. | ||
Recent advances in the field of diabetic complications were made in terms of medications and pathogenesis. Asadi S. et al. studied the relationship between these diseases and genetic variations of glutathione peroxidase (GPx) and methylenetetrahydrofolate reductase (MTHFR). They discovered that Gpx-1 TT and MTHFR genotypes were both linked to higher BMI values, diabetic patients with the MTHFR TT genotype had a considerably higher chance of developing neuropathy, and that patients with diabetes who have the GPx-1 CT genotype are at higher risk of developing retinopathy.8 In another study Ahmed et al. they investigated the Ischemia-modified albumin (IMA) which is formed when ischemia of the hypoxic tissue induces modification of circulating albumin and is a sensitive marker of tissue hypoxia and oxidative stress. In this case, they wanted to assess if there are any relationships between this marker and diabetes and its related complications, especially with neuropathy and retinopathy. The results showed that IMA levels were higher in T2DM patients, which presented neuropathy and retinopathy, resulting in a promising marker for the detection of diabetes-related complications.9
Concerning the diabetic nephropathy complication, recent advances have led to a novel potential drug called Finerenone, which may be able to reduce the damage to the kidney tissue and, hence, the collapse of the organ.10
AGEs are formed by a non-enzymatic reaction called the Maillard process, which occurs between ketones or aldehydes, the amino groups of proteins, nucleic acids and lipids. The process starts under hyperglycemic or oxidative stress conditions. Amadori modification rapidly progresses, and a significant amount of ROS are created during hyperglycemia. Moreover, excessive production of ROS has been linked to the onset of DM and its associated complications. Since oxidative stress is increased by hyperglycation, glycation and oxidation are inextricably linked (Fig. 2).11
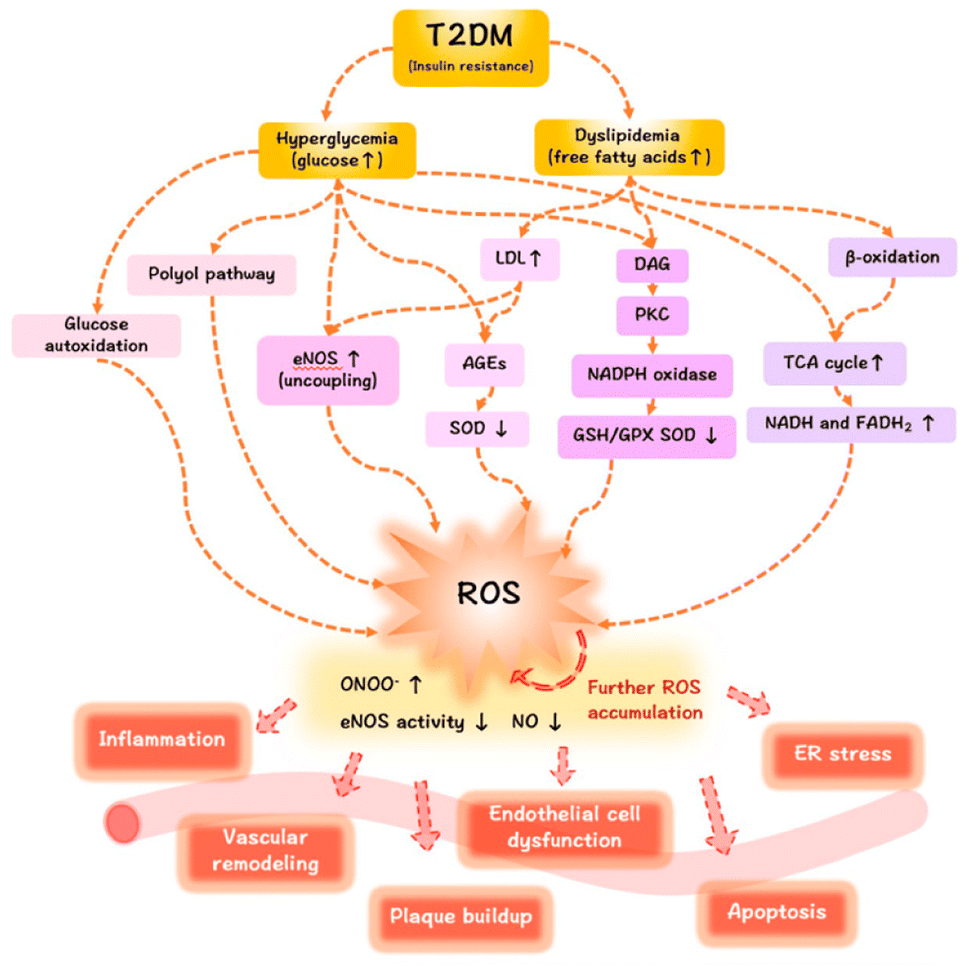 | ||
| Fig. 2 The mechanisms of ROS/oxidative stress generation in diabetes mellitus and the effects of ROS release on the vasculature.12 | ||
Hyperglycemia is known to activate the formation of diacylglycerol (DAG), protein kinase C (PKC), and nicotinamide adenine dinucleotide phosphate (NADPH)-oxidase, all of which contribute to generating ROS and oxidative stress in diabetes. Due to the subsequent interaction effects with the corresponding receptor (RAGE), AGEs may exacerbate the inflammatory reactions associated with diabetes, which are mediated by pathological cellular death (apoptosis and autophagy)13 (Fig. 3).
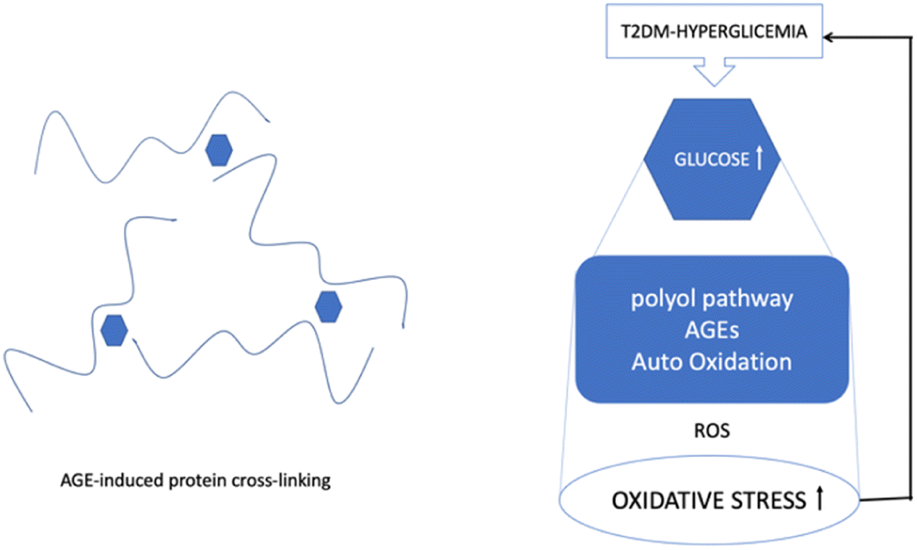 | ||
| Fig. 3 ROS, hyperglycemia, and protein cross-linking. | ||
T2DM is often associated with cognitive dysfunctions and late-onset Alzheimer's disease (LOAD) several epidemiological studies indicate that people suffering from T2DM have a higher risk of developing Alzheimer's disease (AD).14–16
Similar to AD, T2DM leads to decreased volumes of gray matter, white matter, and hippocampus and the gray matter loss mainly occurs in medial temporal, anterior, cingulate, and medial frontal lobes.17–20
The association between T2DM and AD is quite complex and not fully elucidated yet. However, both diseases share the following features:
●Impairment of glucose and energy metabolism.21
●Deposits of amyloidogenic peptides, Aβ (amyloid beta), and islet amyloid polypeptide (IAPP) in the brain and in the pancreatic islets of Langerhans of patients affected by AD and T2DM, respectively.22,23
●Accumulation of advanced glycated products and oxidative stress were observed in both T2DM and AD.24,25
●Inflammation, mitochondrial and synaptic dysfunction, and impairment of autophagy are common to both AD and T2DM26,27
●Dyshomeostasis of metal ions.28
2 Clinical experimentation therapies
In the context of diabetes, the role of Reactive Oxygen Species (ROS), Advanced Glycation End Products (AGEs), and protein aggregation are of significant interest due to their contribution to the pathophysiology of the disease.29 Hyperglycemia, a hallmark of diabetes, leads to increased oxidative stress and the formation of AGEs, which in turn can cause damage to various cellular structures and functions. Moreover, the interaction between ROS and AGEs can exacerbate the aggregation of proteins, contributing to the formation of insoluble fibrils that impair cellular function and viability. Protein aggregation is particularly detrimental in diabetes as it can lead to the dysfunction of crucial proteins involved in insulin signaling and glucose metabolism. Addressing these molecular mechanisms has become a focal point in diabetes research. Consequently, a number of drugs targeting the reduction of ROS, inhibition of AGE formation, and prevention of protein aggregation are under development or in clinical trials. These therapeutic agents aim to alleviate oxidative stress, minimize glycation, and stabilize protein structures, thereby offering potential to mitigate the progression of diabetic complications and improve patient outcomes. In the table below (Table 1), some of them are listed with their relative mechanism of action. In this section, some of the drugs included in Table 1 are described from the most to the least studied.| Drug | Mechanism of action |
|---|---|
| Aminoguanidine | Post-Amadori inhibitor |
| Pyridoxamine | Post-Amadori inhibitor |
| Benfotiamine | Post-Amadori inhibitor |
| ACEIs | Pre-Amadori and post-Amadori inhibitor |
| ARBs | Agonist of peroxisome proliferator-activated receptor gamma (PPARγ) |
| Statins | Stimulate RAGE shedding |
| Alagebrium (ALT-711) | AGE-breaker |
| TZDs | Agonist of PPARγ |
| ALT-946 | Post-Amadori inhibitor |
| OPB-9195 | Post-Amadori inhibitor |
| Tenilsetam | Post-Amadori inhibitor |
| LR-90 | Post-Amadori inhibitor |
| TM2002 | Transition metal ion chelator |
| sRAGE | Inhibits AGE-RAGE binding |
| PEDF | Reduces AGE-RAGE signal transduction, activation of PPARγ |
| IAPP targeting therapy | Targets IAPP hormone |
| NI203 | Antibody that targets IAPP aggregates |
| T3D-959 | PPAR delta-activating compound |
| NE3107 | Regulates nuclear factor kappa B/extracellular signal-regulated kinase (NF-κB/ERK) pathway |
2.1 Aminoguanidine
Aminoguanidine (AG) (Fig. 4) is a therapeutic nucleophilic hydrazine compound offering protection against diabetes-induced impairments in vascular dynamics31 and in addition, its use has the potential to expand in neurodegenerative diseases such as AD and Parkinson's.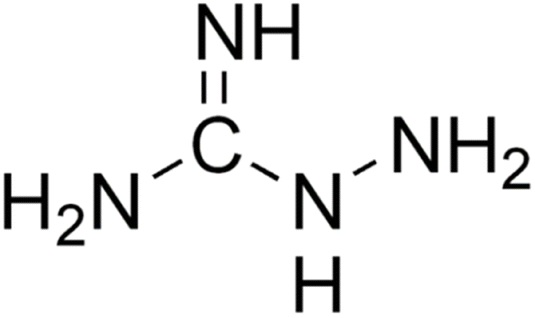 | ||
| Fig. 4 Aminoguanidine structure. | ||
Aminoguanidine is a low molecular weight compound, soluble in polar solvents, and a highly reactive reagent that can react with a wide variety of biological molecules such as glucose and pyruvate.32
It was commercially available under the name of Pimagedine and was used as a therapeutic drug for diabetic nephropathy33 but the development and clinical trials were discontinued due to serious adverse side such as myocardial infarction, congestive heart failure, anemia and gastrointestinal disturbance. Since AG is a competitive inhibitor, large quantities were required for each dose to be effective leading to subsequent cytotoxicity and withdrawal from the market.32,33
Although past clinical trials faced challenges due to adverse effects, further exploration of Aminoguanidine's therapeutic potential through preclinical studies and alternative formulations may provide valuable insights for future clinical development.
When proteins or lipids are exposed to sugars, they become glycated, and they form advanced glycation end products (AGEs); aminoguanidine prevents the formation of the AGEs. The glycated products of these reactions are common biomarkers of aging and the progression of degenerative diseases such as diabetes, AD, and atherosclerosis.
Aminoguanidine can trap reactive carbonyls formed during the Maillard reaction, especially Amadori intermediates, therefore inhibiting their further conversion into AGEs (Fig. 5).
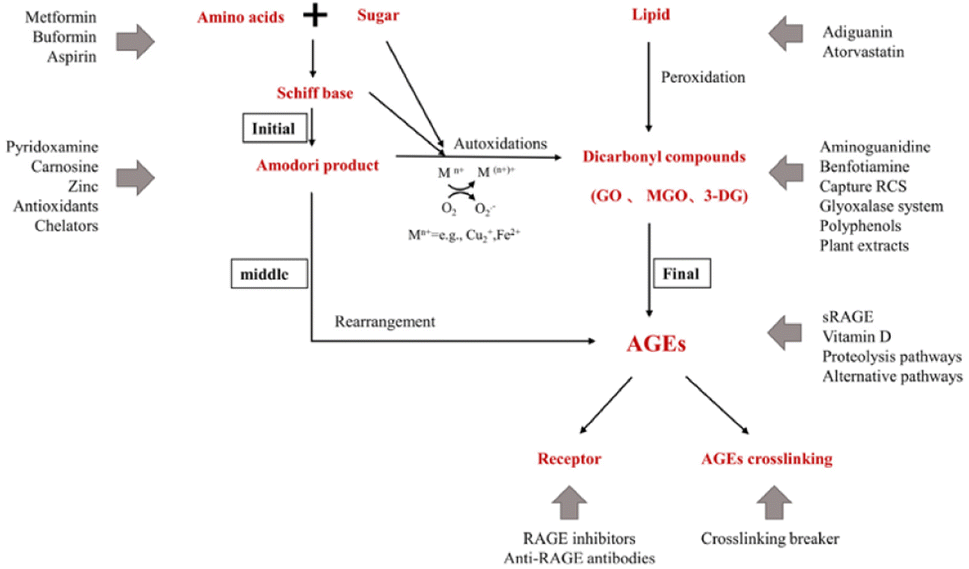 | ||
| Fig. 5 Action sites that inhibit the formation of AGEs in vivo. Mn+ refers to transition metals. Endogenous AGEs are formed through the Maillard reaction in three main stages: early, middle, and final.34 | ||
Proteins are very common targets of glycation and that reaction usually takes place at their N-terminal end, guanidinium group of arginine or amino-group of lysine. As a result, AGEs may induce cross-linking between proteins and the affected protein may be subject to loss of activity and/or misfolding and aggregation.35 Since AG is structurally similar to arginine, it can mimic the main target of AGEs and act as an alternate route of conjugation and glycation, protecting the functional proteins. Additional to that, AG can compete with Lys/Arg amino acid of a protein for glycose binding and protect the carbonyl formation on the protein.
AG also acts by reacting rapidly with α,β-dicarbonyl compounds such as glyoxal, methylglyoxal, and 3-deoxyglucosone. Moreover, aminoguanidine also inhibits the accumulation of advanced oxidation protein products (AOPP), produced as a result of excessive oxidative stress in diabetes.36
Regarding its therapeutic applications in other diseases, aminoguanidine has a protective role in the diabetes-induced impairment in vascular dynamics such as structural modification of collagen in the arterial wall and heart, caused by AGEs.37 Atheroprotective effects of aminoguanidine were also reported in animal models, where AG reduced the AGEs accumulation and atherosclerotic plaques in the abdominal and thoracic area of diabetic mice.
AGE accumulation seems to have also an effect on Parkinson's disease patients, who display disturbed sugar metabolism and alpha-synuclein protein glycation. Diabetes is a risk element for Parkinson's disease and other degenerative disorders and recent studies suggest that the risk factor can reach 23–38%.38,39 Alpha-synuclein (αSyn) is a long-lived protein and thus is subject to glycation. AGEs can accumulate to αSyn and increase its aggregation tensity and toxicity. Moreover, studies reported the co-localization in the substantia nigra of AGEs with αSyn in Lewy bodies and the presence of αSyn in a glycated form in the brain of Parkinson's disease patients.40,41 Interestingly, aminoguanidine acting as a methylglyoxal scavenger was shown to have a protective role against aggregation of αSyn.41
Since AG reduces oxidative stress and AGE levels and reduces ROS formation, In a study, Magdaleno et al.36 examined genes linked to antioxidants and oxidants that can control reactive oxygen species (ROS), as well as proteins sensitive to oxidative stress that control the expression of collagen type I. Their findings imply that AG either mitigates oxidative stress or lowers the amount of AGE-associated oxidative modifications, such as RAGE binding, to decrease collagen type I expression and myofibroblast activation. For example, different signaling pathways that modulate oxidative stress and contribute to cellular dysfunction associated with diabetes can be triggered by RAGE activation, depending on the cellular context and interacting ligand.
Here, they suggest that AG reduces oxidatively-associated cardiac fibrosis by attenuating the fibrogenic response in T2DM rats. Therefore, there is substantial evidence from this study to support the link between diabetes and cardiac fibrosis.36
Complexes formed by aminoguanidine and metals can slow down glycation and subsequent AGEs formation, by scavenging metal cations such as Fe(III) and Cu(II). These metal cations are ubiquitous in most alive organisms. It is highly considered that the mechanism of AGEs inhibition results mainly from the metal chelating rather than the carbonyl entrapment route since these metals can catalyse the auto-oxidation of the Amadori compounds. Additionally, the complex formation of copper ions with aminoguanidine, antagonizes and prevents the formation of complexes of Amadori products with Cu(II). Otherwise, the Amadori products would be degraded and AGEs would be formed.42
AG is capable of forming stable coordination compounds with redox active metal ions (Cu(II), Fe(III) and Fe(II)) under physiological conditions, with some of these complexes displaying antioxidant activity.43,44
2.2 Pyridoxamine
Pyridoxamine (PM) is a structural analog and one of the three natural inconvertible forms of vitamin B6, along with pyridoxal and pyridoxine (Fig. 6).45 It belongs also to the AGE inhibitor family, by suppressing post-Amadori products, AGEs formation and inhibiting advanced lipoxidation endproducts formation.46 Its inhibiting action towards oxidation of Amadori products that leads to AGE formation was first discovered in 199947 and since then it has found application in clinical and non-clinical models as a therapeutic molecule on diabetic nephropathy,48 vascular diseases and retinopathies.49 Pyridoxamine is the active ingredient of the FDA-approved drug pyridorin which finds application in preventing the progression of diabetic nephropathy.50,51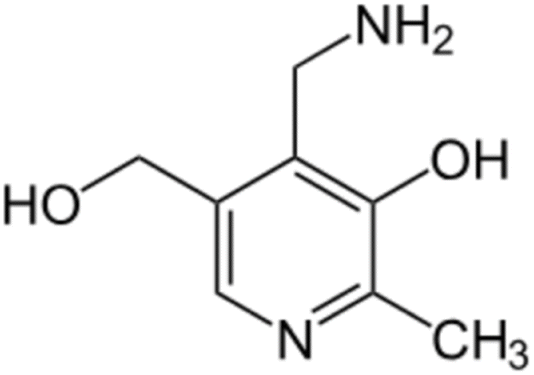 | ||
| Fig. 6 Pyridoxamine structure. | ||
Some of its biological activities, that have a therapeutic and inhibitory effect on the progression of diabetes disease are its ROS scavenging ability; its binding and trapping of the dicarbonyl intermediates produced during the glycation reaction, such as methylglyoxal (MGO), and its entrapment of toxic carbonyl compounds that derive after lipid or protein degradation.
Pyridoxamine as AG has metal chelating activity, and this could be one of the potential mechanisms that contribute to its action. It can form stable complexes with copper (Cu2+), zinc (Zn2+), and iron ions (Fe3+), which catalyze the oxidative reactions taking place in the protein glycation procedure. Additionally, PM can inhibit the production or can directly react with the hydroxyl radical from the Fenton reaction, in addition to binding with several carbonyl compounds.52
The binding interaction of Amadori with metal ions leads to the compounds autooxidation, degradation and the subsequent production of glycation end products. PM can compete against an Amadori compound on the binding Cu(II) or Fe(III) ions, preventing this autoxidation.52
PM has antioxidant activity as a ROS scavenger and reacts with reactive carbonyl compounds which are generated byproducts of protein glycation, thus inhibiting further protein damage.52
Pyridoxamine has been proven to have therapeutic action on both diabetic humans and animals49,53,54 as a potent inhibitor of post-Amadori products in vivo and in vitro.55
Administration of pyridoxamine at the early stages of diet-induced kidney nephropathy in mice showed significant improvement in insulin sensitivity and reduction of serum creatinine. AGEs produced by glycation can alter useful extracellular proteins such as collagen and elastin and interfere with different signalling pathways in the kidney. Binding of AGE's by PM, greatly decreases their levels and effect.48
The action of pyridoxamine is not limited to protection against diabetes but has shown also encouraging results against diabetes-associated damage. One such example is cognitive damage and recognition memory impairment linked with the long term effects of diabetes associated cognitive decline, (DACD).56
Another complication of diabetic mellitus is the delayed bone repair process, which is the result of the reactive methylglyoxal production and subsequent prevention of osteoblast differentiation. Pyridoxamine treatment improved the bone regeneration ability in hyperglycemic diabetic mice by restoring the MGO levels.57
2.3 IAPP targeting therapy
To tackle the challenges presented by IAPP aggregation, researchers are developing IAPP targeting therapies with several objectives. Firstly, these therapies aim to prevent IAPP misfolding by designing small molecules, peptides, or antibodies that inhibit the initial misfolding process, thus preventing the formation of toxic oligomers and fibrils. Secondly, efforts are being made to promote fibril dissociation by investigating compounds capable of breaking down existing IAPP amyloid fibrils into non-toxic monomers, thereby restoring normal β-cell function. Additionally, therapies are being explored to enhance β-cell protection by shielding them from the toxic effects of IAPP aggregates, utilizing antioxidants or anti-inflammatory agents to preserve insulin secretion capacity (Fig. 7).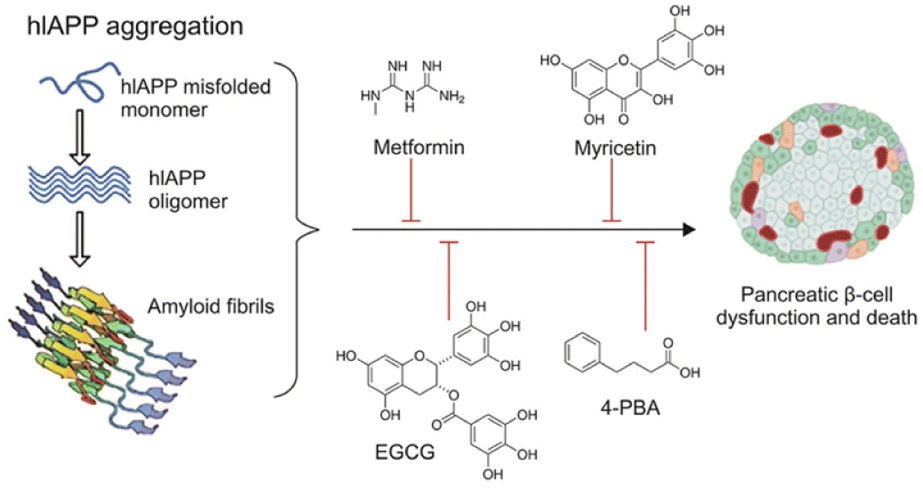 | ||
| Fig. 7 Schematic representation of hIAPP aggregation and common antidiabetic molecules and repurposed drugs with the potential to inhibit and/or reverse hIAPP aggregation in T2DM.58 | ||
By addressing the underlying mechanisms of IAPP aggregation and its detrimental effects, these therapies show promise in enhancing β-cell survival and function in individuals with diabetes. This potential improvement could lead to a slowdown of disease progression and better glucose control. In particular, Wren Therapeutics is developing an IAPP-targeting small molecule for the treatment of Type 2 diabetes and metabolic disease. This therapy is still at the discovery stage.59,60
2.4 NI203
It is a recombinant human monoclonal antibody that targets pathologic IAPP aggregates and neutralizes their toxicity. In preclinical models of type-2 diabetes NI203 supported normal beta cell functions resulting in re-stored insulin secretion and reduction of the type-2 diabetes symptoms. It is under development by Neurimmune, using its reverse translational medicine platform for the treatment of type 2 diabetes.612.5 T3D-959
This drug candidate activates two nuclear receptors that are central regulators of normal glucose and lipid metabolism in the brain. It is the first PPAR delta-activating compound, PPAR delta and PPAR gamma targets are central regulators of glucose and lipid metabolism. T3D-959 is orally delivered as a once-a-day medicine. It is currently in phase II clinical trial (NCT04251182).622.6 NE3107
This molecule has shown anti-inflammatory qualities in a number of preclinical disease models, including rheumatoid arthritis, diabetes mellitus, Parkinson's disease, and chronic obstructive pulmonary disease. TNFα, IFNγ, IL-1α, and transforming growth factor-beta are among the inflammatory cytokines that are less likely to be released when NE3107 inhibits the nuclear factor kappa B/extracellular signal-regulated kinase (NF-κB/ERK) pathway (Fig. 8). Limiting the NF-κB/ERK pathway reduces the production of pTau and Aβ, oxidative stress, and glial activation. Reducing hyperglycemia and hyperinsulinemia, NE3107 inhibits the activation of mitogen-activated protein kinases (MAPK), which is implicated in the mediation of insulin resistance.63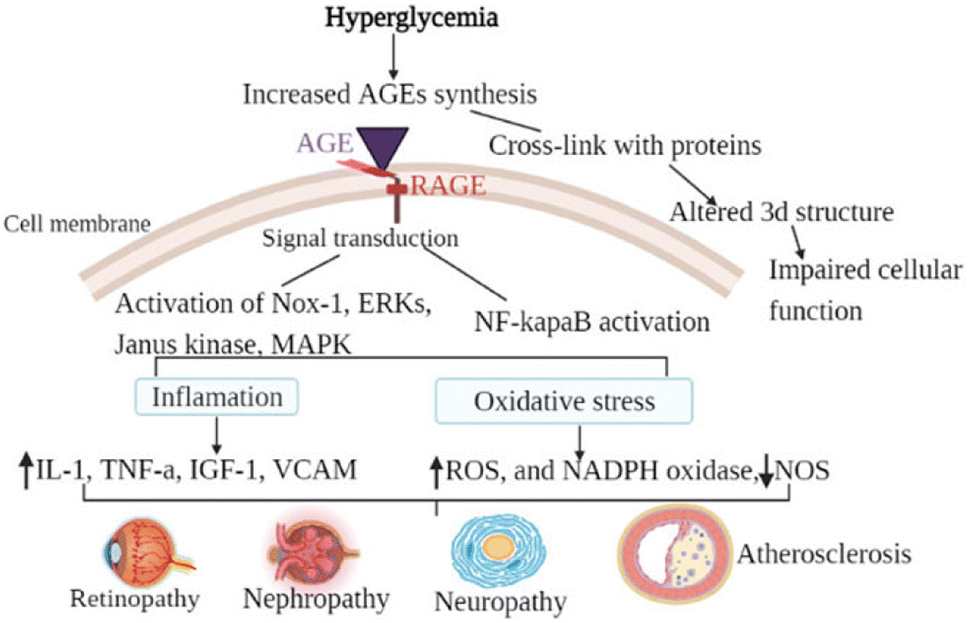 | ||
| Fig. 8 The general mechanism of advanced glycation end-products in diabetic vascular complications is due to the activation of multiple signal transduction pathways as a result of RAGE/AGE interaction or through cross-link formation with cellular proteins. Activation of RAGE leads to the activation of Nox-1, ERKs, Janus kinase, MAPK, and activation of NF-κB. When those pathways are activated, oxidative stress (decreased NOS, increased ROS, and increased NADPH oxidase) and inflammatory factors are activated. Cross-link (adduct) formation on the other hand altered the three-dimensional structure of protein consequently impairing cellular function.64 | ||
3 In use therapies
3.1 Exenatide
Exenatide, a mimetic of glucagon-like peptide-1 (GLP-1), is a synthetic form of exendin-4, a peptide of 39 amino acids (Fig. 9). GLP-1 is produced in the intestine after food intake and starts the production of insulin from the pancreas.65 People who present T2DM have a low level of GLP-1, thus an addition of this peptide might help to correct the disease.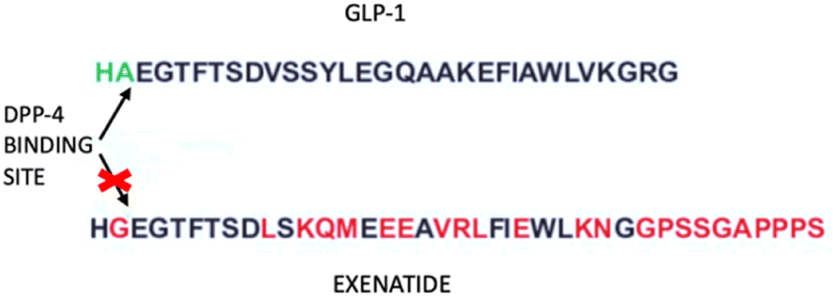 | ||
| Fig. 9 Exenatide and GLP-1 sequences. The arrows indicate the DPP-4 binding site, which lacks in the exenatide structure. | ||
Human GLP-1 has the problem of a very short half-life due to the enzymatic action of dipeptidyl dipeptidase IV, hence there is the need for a peptide that has a longer half-life and is resistant to the action of the enzyme. Scientists developed the Exenatide, an analog of GLP-1 that lacks the enzyme binding site.66 Exenatide operates through multiple mechanisms to lower glucose levels, including boosting insulin secretion, stimulating beta cell generation, increasing satiety, and setback the emptiness of the stomach. Its use is indicated for type 2 diabetes patients.67 Exenatide has a beneficial effect on oxidative stress and inflammation.68 This was demonstrated in a study by Jin-dan Wu et al. where they wanted to ascertain how exenatide affected oxidative stress and inflammatory markers in patients with T2DM whose disease was not adequately controlled with other drugs. The result was that exenatide treatment decreased body weight, body mass index (BMI), lowered hemoglobin A1c (HbA1c) and the seven-point glucose profile, decreased daily mean glucose, limited glycemic excursion, and decreased oxidative stress and inflammatory markers.69
Gastrointestinal side effects are the main consequences of this drug, with nausea as the most common symptom. Some patients may experience insulin-induced hypoglycemia when administered with other drugs, such as metformin.70
3.2 Metformin
Metformin (Fig. 10) is the first-line medication for T2DM, it is an oral medicine that belongs to the guanidine group, and it is mainly used as monotherapy. It increases the cell response to insulin and reduces glucose absorption and gluconeogenesis in the liver.71 The activation of AMP-activated protein kinase (AMPK) plays an important role in the anti-diabetic action of metformin. AMPK contributes to the reduction in hepatic gluconeogenesis and it increases glucose uptake in skeletal muscles.72,73 Metformin increases the amount and/or affinity of muscle and adipocyte insulin receptors, increases insulin receptor tyrosine kinase activity, enhances the transport of glucose and glycogen synthesis, and decreases both hepatic gluconeogenesis and glycogenolysis. Furthermore, metformin has been reported to decrease lipid oxidation and plasma-free fatty acid levels, resulting in the inhibition of the overactive Randle cycle. In patients with T2DM, metformin monotherapy decreases the fasting blood glucose levels by ∼60–70 mg dl−1 and HbA(1c) by 1.5–2.0%.74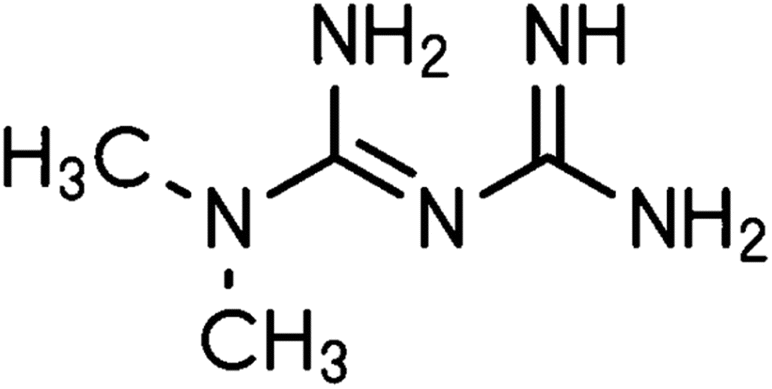 | ||
| Fig. 10 Metformin structure. | ||
It has been demonstrated that metformin lowers intracellular ROS. However, there is still much to learn about the exact mechanism underlying metformin's antioxidant effects. Metformin's cardiovascular-protective effects are mediated in part by activating the AMP-activated protein kinase (AMPK) pathway.75 In this study, they have demonstrated that by upregulating the expression of the antioxidant tioredoxin (Trx), metformin dramatically lowers the levels of ROS inside cells. Trx expression is induced by metformin via the AMPK-FOXO3 pathway. Therefore, one important mechanism for the antioxidant effects of metformin is the activation of the AMPK-FOXO-Trx axis.75
In another study by Ishibashi et al., they investigated the AGEs and their receptor (RAGE). These two play a role in tubulointerstitial damage in diabetic nephropathy. The current study indicates that metformin may prevent AGE-induced tubular cell apoptosis as well as inflammatory and fibrotic responses. This is likely achieved by lowering ROS production through suppression of RAGE expression via activation of AMP-activated protein kinase. By inhibiting the AGEs RAGE-ROS axis, metformin may prevent tubular cell damage in diabetic nephropathy.76
Even if this drug is the most used one, it was demonstrated it has some side effects, which are mainly gastrointestinal symptoms such as nausea and diarrhea. Additionally, some patients may experience a reduction of Vitamin B12, and hypoglycemia may occur when it is combined with insulin therapy.77
The worst side effect related to Metformin is lactic acidosis and the main causes of this metformin-associated adverse event are typically high plasma concentrations of the medication in patients with risk factors like renal insufficiency or secondary to pathological events that disrupt the lactate oxidative pathway, like hepatic disease, septicemia, shock, or any contraindication to metformin. The liver is thought to be the primary organ involved in both the development of lactic acidosis and the antidiabetic effect of metformin. The latter is brought on by suppressing the mitochondrial electron transport chain and preventing pyruvate from being converted to glucose, which raises lactate levels instead of oxidation.
Pancreatitis has been linked to metformin use, either as a result of drug overdose or renal insufficiency.78 In the recent study by Congiu et al.79 the intrinsic role of metformin in aggregation of amylin and its copper complex was shown.
3.3 Pramlintide
In 2005, an injectable medication, Pramlintide, received approval by the US FDA for diabetes treatment.80 It is a synthetic amylin variant proven to effectively reduce post-prandial glucose (PPG) levels, delay the emptying of stomach contents, and facilitate weight loss.81 Pramlintide is a synthetic equivalent of human amylin (Fig. 11).82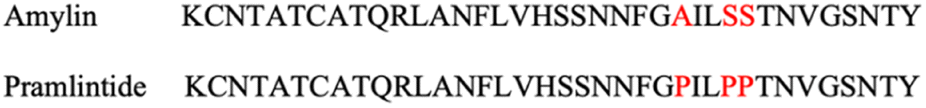 | ||
| Fig. 11 Human amylin and Pramlintide sequences. Highlighted in red are the proline substitutions.82 | ||
Human amylin is a hormone produced by β-cells when glucose levels rise; it regulates glucagon secretion and promotes satiety. In patients with type 1 diabetes, its production is absent because of the loss of β cells, whereas in patients with T2DM, its secretion is reduced.83 Amylin has an amidated C-terminus and a disulfide bridge between Cys2 and Cys7.84–86 Human amylin (hIAPP) is released into the bloodstream by the β-cells of the pancreas along with insulin after a meal.87–89 It binds to specific sites in pancreatic β-cells, inhibits insulin release, and may provide negative feedback inhibition of its own secretion.90 Previous preclinical studies91,92 indicate that amylin possesses the capacity to bind with high affinity to specific regions of the brain, including the area called Postrema, which has the greatest density of amylin receptors. Therefore, amylin is considered as a neuroendocrine hormone with an important role in regulating the rate of glucose influx into circulation after meals.93,94 However, hIAPP has a unique amino acid sequence that increases its tendency to misfold and form cytotoxic fibrils,95 which have been strongly associated with β-cell degeneration in T2DM.96 Moreover, His18 plays an important role in the binding of small oligomeric species of hIAPP to the cellular membrane.97 Its protonation state is very sensitive even on minor pH changes.98 This binding has been involved in disturbing cellular homeostasis, either by ion channel formation or by a non-specific general lipid bilayer disruption. Recent studies correlated the presence of amylin aggregates and their direct toxicity to β-cells.97,99 At higher concentrations, hIAPP rapidly aggregates by forming β-sheet structures. The formation of the aggregates leads to the loss of all the physiological hIAPP functions and to the appearance of cellular toxic effects. The formation of hIAPP amyloids is strongly dependent on the region 20–29 (known as the amyloidogenic region).100 Other studies101 have shown that hIAPP has the ability to generate hydrogen peroxide (H2O2) during amyloid fibril formation in vitro. On the contrary, the nontoxic and non-fibrillogenic analog rat amylin which is called (rIAPP), does not generate H2O2.101,102 Also, it is a well-known fact that rats do not develop T2DM. One of the reasons why rIAPP doesn't form aggregates unlike hIAPP is because their amino acid sequences differ from each other.
The native amylin is unstable for clinical use but scientists discovered that substituting 3 amino acids in the hIAPP sequence with 3 prolines at certain sites leads to the development of Pramlintide.103,104 Pramlintide collaborates with insulin to delay gastric emptying and inhibit glucagon release. It is recommended for patients with T1DM who require mealtime insulin and for those with T2DM who have uncontrolled blood sugar levels even though receiving optimal therapy with mealtime insulin, metformin, or sulfonylurea.105
Patients with T1DM and T2DM participated in numerous clinical trials to assess the safety and effectiveness of these medications. Studies that were double-blind and placebo-controlled were conducted to evaluate the effectiveness of pramlintide in patients with T2DM and T1DM. Patients treated with pramlintide showed a significant decrease in HbA(1c) in these investigations, which was followed by weight loss. Thus, pramlintide coupled to insulin therapy can enhance glycemic control without causing weight gain or hepatic, renal, or cardiovascular damage, according to these trials' findings.106
Gastrointestinal side effects are the main consequences of this drug; nausea is the most common symptom. In certain cases, when insulin doses are not reduced, pramlintide might lead to insulin-induced hypoglycemia.70
4 Discussion
Pharmacological interventions targeting reactive oxygen species (ROS) and protein aggregation have shown promise in mitigating diabetes-related complications. Antioxidant therapies, such as N-acetylcysteine (NAC), have demonstrated beneficial effects in reducing oxidative stress and improving glycemic control.107 Additionally, small molecules like resveratrol have been shown to inhibit protein aggregation and ameliorate diabetes-associated pathology.108 However, some pharmacological interventions have yielded negative findings. For instance, bardoxolone methyl did not show significant improvement in renal outcomes in diabetic nephropathy.109 These negative findings highlight the need to address limitations and challenges associated with specific drug candidates or intervention strategies.Future directions in pharmacological studies for diabetes include identifying and validating novel drug targets, exploring combination therapies, and personalizing treatment approaches. Advanced screening methods and computational approaches can aid in identifying potential drug candidates. Moreover, the development of targeted drug delivery systems and conducting long-term studies on safety and clinical benefits are important considerations for optimizing treatment regimens.
Interdisciplinary collaborations are crucial for advancing our understanding of diabetes and identifying new drug targets. By bringing together researchers from various fields such as molecular biology, biochemistry, pharmacology, genetics, and clinical medicine, valuable insights into disease mechanisms can be gained, leading to the development of targeted therapies.
Investing in basic research is essential for elucidating the molecular mechanisms underlying diabetes. By studying the interplay between reactive oxygen species (ROS), protein aggregation, and cellular damage, potential therapeutic targets can be identified. Basic research also helps uncover key signaling pathways, cellular components, and molecular targets that can be modulated to mitigate diabetes progression. Utilizing advanced technologies and methodologies is vital for enhancing knowledge in the field of diabetes research. Techniques such as high-throughput screening, genomics, proteomics, metabolomics, and bioinformatics provide valuable data for identifying new drug candidates and understanding their mechanisms of action. Integrating these cutting-edge technologies accelerates the discovery and development of effective treatments.
Moreover, openly sharing research findings, datasets, and methodologies, the scientific community can collectively build upon existing knowledge and promote collaborative research endeavors. Collaboration platforms, data repositories, and international consortia facilitate the exchange of information in this field.
5 Conclusions
Diabetes mellitus (DM) has emerged as a pressing concern in the realm of public health, demanding immediate attention. The increasing trajectory in the number of DM cases is anticipated to persist for several decades. At present, there is no permanent cure for DM. Although various treatment approaches have displayed encouraging outcomes in managing DM, the disease continues to pose a significant challenge. Consequently, it is crucial to address the issue in order to establish a robust, effective, and safe clinical management plan. Optimal regulation of metabolic factors such as glucose, blood pressure, and body weight is essential. Achieving these necessitates dietary choices, increased physical activity, and reduction in body weight. To manage this condition effectively and efficiently, there is a need for a focus on public policies that bolster healthcare access and resources.110 Furthermore, an increase in awareness will be essential for the prevention and management of this disease.3 As we can understand from above, many factors and variables influence the development of diabetes, and many of them are yet to be discovered, consequently, a proper and up-to-date diagnosis can be fundamental for the correct treatment of the disease to avoid long-term complications and reduce mortality. In essence, this review emphasizes the significance of understanding the intricate metabolic pathways and diabetes. By exploring and harnessing the potential of pharmacological interventions, there is a promising prospect of developing targeted and effective treatments to mitigate the impact of ROS and protein misfolding, further studies are needed to develop novel therapeutical approaches since science is still far from achieving a definite solution against this epidemic disease.Abbreviations
| AGEs | Advanced glycation end products |
| ROS | Reactive oxygen species |
| DM | Diabetes mellitus |
| T1DM | Type 1 diabetes mellitus |
| T2DM | Type 2 diabetes mellitus |
| GDM | Gestational diabetes mellitus |
| DAG | Diacilglycerol |
| PKC | Protein kinase C |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| RAGE | Receptors of advanced glycation endproducts |
| AD | Alzheimer's disease |
| Aβ | Amyloid-beta |
| IAPP | Islet amyloid polypeptide |
| PPARy | Peroxisome proliferator-activated receptor gamma |
| TNF α | Tumor necrosis factor alpha |
| IFN γ | Interferon gamma |
| IL-1α | Interleukin-1 alpha |
| NF-κB/ERK | Nuclear factor kappa B/extracellular signal-regulated kinase |
| MAPK | Mitogen-activated protein kinases |
| AG | Aminoguanidine |
| AOPP | Advanced oxidation protein products |
| PM | Pramlintide |
| MGO | Methylglyoxal |
| DACD | Diabetes associated cognitive decline |
| GLP-1 | Glucagon-like peptide-1 |
| DPP-4 | Dipeptidyl dipeptidase IV |
| BMI | Body mass index |
| AMPK | AMP-activated protein kinase |
Author contributions
All authors contributed equally to writing this review article. All authors read and approved the final version of this manuscript.Conflicts of interest
All authors declare that they have no conflicts of interest that could inappropriately influence this manuscript.Acknowledgements
This research has received no external funding.References
- K. L. Ong, et al., Global, regional, and national burden of diabetes from 1990 to 2021, with projections of prevalence to 2050: a systematic analysis for the Global Burden of Disease Study 2021, Lancet, 2023, 402(10397), 203–234, DOI:10.1016/S0140-6736(23)01301-6.
- Z. Tao, A. Shi and J. Zhao, Epidemiological Perspectives of Diabetes, Cell Biochem. Biophys., 2015, 73(1), 181–185, DOI:10.1007/s12013-015-0598-4.
- M. Z. Banday, A. S. Sameer and S. Nissar, Pathophysiology of diabetes: An overview, Avicenna J. Med., 2020, 10(04), 174–188, DOI:10.4103/ajm.ajm_53_20.
- M. H. Abutaleb, Diabetes Mellitus: An Overview, Pharm. Pharmacol. Int. J., 2016, 4(5), 406–411, DOI:10.15406/ppij.2016.04.00087.
- E. Chiefari, B. Arcidiacono, D. Foti and A. Brunetti, Gestational diabetes mellitus: an updated overview, J. Endocrinol. Invest., 2017, 40(9), 899–909, DOI:10.1007/s40618-016-0607-5.
- S. Grunbaum, Understanding Diabetes Mellitus Causes, Symptoms, and Complications, Biomed. J. Sci. Tech. Res., 2023, 50(3), 41622–41628, DOI:10.26717/bjstr.2023.50.007946.
- H.-H. Marcie, S. Mario and A. D. Deshpande, Epidemiology of diabetes and diabetes-related complications, Phys. Ther., 2008, 88, 1254–1264, DOI:10.2522/ptj.20080020.
- S. Asadi, et al., Assessing the possible association between MTHFR (rs1801133) and GPx-1 (rs1050450) polymorphisms with the risk of type 2 diabetes, diabetic neuropathy, and diabetic retinopathy, Mol. Biol. Rep., 2024, 51(1), 583, DOI:10.1007/s11033-024-09519-0.
- A. A. M. Ahmed, E. H. A. Salama, H. A. H. Shehata and A. A. Noreldin, Ischemia-modified albumin relation to glycemic state, neuropathy, and retinopathy in patients with type2 Diabetes mellitus, SVU Int. J. Med. Sci., 2024, 7(1), 17–25, DOI:10.21608/svuijm.2022.167939.1431.
- A. Kulkarni, A. R. Thool and S. Daigavane, Understanding the Clinical Relationship Between Diabetic Retinopathy, Nephropathy, and Neuropathy: A Comprehensive Review, Cureus, 2024, 16(3), e56674, DOI:10.7759/cureus.56674.
- B. Arif, et al., Attenuation of hyperglycemia and amadori products by aminoguanidine in alloxan-diabetic rabbits occurs via enhancement in antioxidant defenses and control of stress, PLoS One, 2022, 17(1), e0262233, DOI:10.1371/journal.pone.0262233.
- Y. Tan, M. S. Cheong and W. S. Cheang, Roles of Reactive Oxygen Species in Vascular Complications of Diabetes: Therapeutic Properties of Medicinal Plants and Food, Oxygen, 2022, 2(3), 246–268, DOI:10.3390/oxygen2030018.
- C. M. O. Volpe, P. H. Villar-Delfino, P. M. F. Dos Anjos and J. A. Nogueira-Machado, Cellular death, reactive oxygen species (ROS) and diabetic complications review-Article, Cell Death Dis., 2018, 9(2), 1745, DOI:10.1038/s41419-017-0135-z.
- M. Barbagallo, Type 2 diabetes mellitus and Alzheimer's disease, World J. Diabetes, 2014, 5(6), 889, DOI:10.4239/wjd.v5.i6.889.
- D. Cheng, J. Noble, M. X. Tang, N. Schupf, R. Mayeux and J. A. Luchsinger, Type 2 diabetes and late-onset Alzheimer's disease, Dement. Geriatr. Cogn. Disord, 2011, 31(6), 424–430, DOI:10.1159/000324134.
- C. C. Huang, et al., Diabetes mellitus and the risk of Alzheimer’s disease: A nationwide population-based study, PLoS One, 2014, 9(1), e87095, DOI:10.1371/journal.pone.0087095.
- C. Moran, R. Beare, T. G. Phan, D. G. Bruce, M. L. Callisaya and V. Srikanth, “Type 2 diabetes mellitus and biomarkers of neurodegeneration,” Alzheimer's Disease Neuroimaging Initiative (ADNI), Neurology, 2015, 85(13), 1123–1130, DOI:10.1212/WNL.0000000000001982.
- L. A. Beckett, M. C. Donohue, C. Wang, P. Aisen, D. J. Harvey and N. Saito, The Alzheimer's Disease Neuroimaging Initiative phase 2: Increasing the length, breadth, and depth of our understanding, Alzheimer's Dementia, 2015, 11(7), 823–831, DOI:10.1016/j.jalz.2015.05.004.
- A. M. Fjell, et al., The roots of Alzheimer's disease: Are high-expanding cortical areas preferentially targeted?, Cerebr. Cortex, 2015, 25(9), 2556–2565, DOI:10.1093/cercor/bhu055.
- T. W. Schmitz and R. Nathan Spreng, Basal forebrain degeneration precedes and predicts the cortical spread of Alzheimer’s pathology, Nat. Commun., 2016, 7, 13249, DOI:10.1038/ncomms13249.
- H. Umegaki, Type 2 diabetes as a risk factor for cognitive impairment: Current insights, Clin. Interv. Aging, 2014, 9, 1011–1019, DOI:10.2147/CIA.S48926.
- G. F. Chen, et al., Amyloid beta: Structure, biology and structure-based therapeutic development, Acta Pharmacol. Sin., 2017, 38(9), 1205–1235, DOI:10.1038/aps.2017.28.
- S. Asthana, B. Mallick, A. T. Alexandrescu and S. Jha, IAPP in type II diabetes: Basic research on structure, molecular interactions, and disease mechanisms suggests potential intervention strategies, Biochim. Biophys. Acta, Biomembr., 2018, 1860(9), 1765–1782, DOI:10.1016/j.bbamem.2018.02.020.
- S. Schedin-Weiss, B. Winblad and L. O. Tjernberg, The role of protein glycosylation in Alzheimer disease, FEBS J., 2014, 281(1), 46–62, DOI:10.1111/febs.12590.
- K. Nowotny, T. Jung, A. Höhn, D. Weber and T. Grune, Advanced glycation end products and oxidative stress in type 2 diabetes mellitus, Biomolecules, 2015, 5(1), 194–222, DOI:10.3390/biom5010194.
- F. G. De Felice and S. T. Ferreira, Inflammation, defective insulin signaling, and mitochondrial dysfunction as common molecular denominators connecting type 2 diabetes to Alzheimer Disease, Diabetes, 2014, 63(7), 2262–2272, DOI:10.2337/db13-1954.
- C. Carvalho, M. S. Santos, C. R. Oliveira and P. I. Moreira, Alzheimer's disease and type 2 diabetes-related alterations in brain mitochondria, autophagy and synaptic markers, Biochim. Biophys. Acta, Mol. Basis Dis., 2015, 1852(8), 1665–1675, DOI:10.1016/j.bbadis.2015.05.001.
- A. H. Emwas, et al., Living with the enemy: from protein-misfolding pathologies we know, to those we want to know, Ageing Res. Rev., 2021, 70, 101391, DOI:10.1016/j.arr.2021.101391.
- C. M. O. Volpe, P. H. Villar-Delfino, P. M. F. Dos Anjos and J. A. Nogueira-Machado, Cellular death, reactive oxygen species (ROS) and diabetic complications review-Article, Cell Death Dis., 2018, 9(2), 119, DOI:10.1038/s41419-017-0135-z.
- A. Nenna, et al., Pharmacologic approaches against Advanced Glycation End Products (AGEs) in diabetic cardiovascular disease, Res. Cardiovasc. Med., 2015, 4(2), 5, DOI:10.5812/cardiovascmed.4(2)2015.26949.
- S. i. Yamagishi, Advanced Glycation End-Products, in Brenner's Encyclopedia of Genetics, Elsevier, 2nd edn, 2013, pp. 36–38, DOI:10.1016/B978-0-12-374984-0.00476-9.
- P. J. Thornalley, Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts, Arch. Biochem. Biophys., 2003, 419(1), 31–40, DOI:10.1016/j.abb.2003.08.013.
- W. K. Bolton and E. Abdel-Rahman, Pimagedine: a novel therapy for diabetic nephropathy, Expert Opin. Invest. Drugs, 2002, 11(4), 565–574, DOI:10.1517/13543784.11.4.565.
- W. Zheng, H. Li, Y. Go, X. H. Chan, Q. Huang and J. Wu, Research Advances on the Damage Mechanism of Skin Glycation and Related Inhibitors, Nutrients, 2022, 14(21), 4588, DOI:10.3390/nu14214588.
- A. König, H. Vicente Miranda and T. F. Outeiro, Alpha-Synuclein Glycation and the Action of Anti-Diabetic Agents in Parkinson's Disease, J. Parkinsons Dis., 2018, 8(1), 33–43, DOI:10.3233/JPD-171285.
- F. Magdaleno, et al., Aminoguanidine reduces diabetes-associated cardiac fibrosis, Exp. Ther. Med., 2019, 18(4), 3125–3138, DOI:10.3892/etm.2019.7921.
- D. Susic, J. Varagic, J. Ahn and E. Frohlich, Collagen Cross-link Breakers:A Beginning of a New Era in the Treatment of Cardiovascular Changes Associated with Aging,Diabetes,and Hypertension, Curr. Drug Targets: Cardiovasc. & Haematol. Disord., 2004, 4(1), 97–101, DOI:10.2174/1568006043481347.
- Y.-W. Yang, et al., Increased risk of Parkinson disease with diabetes mellitus in a population-based study, Medicine, 2017, 96(3), e5921, DOI:10.1097/MD.0000000000005921.
- X. Yue, H. Li, H. Yan, P. Zhang, L. Chang and T. Li, Risk of Parkinson Disease in Diabetes Mellitus: An Updated Meta-Analysis of Population-Based Cohort Studies, Medicine, 2016, 95(18), e3549, DOI:10.1097/MD.0000000000003549.
- G. Münch, et al., Crosslinking of α-synuclein by advanced glycation endproducts — an early pathophysiological step in Lewy body formation?, J. Chem. Neuroanat., 2000, 20(3–4), 253–257, DOI:10.1016/s0891-0618(00)00096-x.
- H. Vicente Miranda, et al., Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies, Brain, 2017, 140(5), 1399–1419, DOI:10.1093/brain/awx056.
- J. Ortega-Castro, M. Adrover, J. Frau, J. Donoso and F. Muñoz, Cu2+ complexes of some AGEs inhibitors, Chem. Phys. Lett., 2009, 475(4–6), 277–284, DOI:10.1016/j.cplett.2009.05.074.
- G. García-Díez, R. Ramis and N. Mora-Diez, Theoretical Study of the Copper Complexes with Aminoguanidine: Investigating Secondary Antioxidant Activity, ACS Omega, 2020, 5(24), 14502–14512, DOI:10.1021/acsomega.0c01175.
- G. García-Díez and N. Mora-Diez, Theoretical Study of the Iron Complexes with Aminoguanidine: Investigating Secondary Antioxidant Activity, Antioxidants, 2020, 9(8), 756, DOI:10.3390/antiox9080756.
- E. Mascolo and F. Vernì, Vitamin B6 and Diabetes: Relationship and Molecular Mechanisms, Int. J. Mol. Sci., 2020, 21(10), 3669, DOI:10.3390/ijms21103669.
- T. P. Degenhardt, et al., Pyridoxamine inhibits early renal disease and dyslipidemia in the streptozotocin-diabetic rat, Kidney Int., 2002, 61(3), 939–950, DOI:10.1046/j.1523-1755.2002.00207.x.
- R. G. Khalifah, J. W. Baynes and B. G. Hudson, Amadorins: Novel Post-Amadori Inhibitors of Advanced Glycation Reactions, Biochem. Biophys. Res. Commun., 1999, 257(2), 251–258, DOI:10.1006/bbrc.1999.0371.
- F. Chiazza, et al., Protective Effects of Pyridoxamine Supplementation in the Early Stages of Diet-Induced Kidney Dysfunction, BioMed Res. Int., 2017, 2017, 2682861, DOI:10.1155/2017/2682861.
- A. Stitt, et al., The AGE Inhibitor Pyridoxamine Inhibits Development of Retinopathy in Experimental Diabetes, Diabetes, 2002, 51(9), 2826–2832, DOI:10.2337/diabetes.51.9.2826.
- E. J. Lewis, et al., Pyridorin in type 2 diabetic nephropathy, J. Am. Soc. Nephrol., 2012, 23(1), 131–136, DOI:10.1681/ASN.2011030272.
- S. Ahmad, et al., Inhibitory effect of metformin and pyridoxamine in the formation of early, intermediate and advanced glycation end-products, PLoS One, 2013, 8(9), e72128, DOI:10.1371/journal.pone.0072128.
- R. Ramis, et al., How Does Pyridoxamine Inhibit the Formation of Advanced Glycation End Products? The Role of Its Primary Antioxidant Activity, Antioxidants, 2019, 8(9), 344, DOI:10.3390/antiox8090344.
- E.-T. Wu, J.-T. Liang, M.-S. Wu and K.-C. Chang, Pyridoxamine prevents age-related aortic stiffening and vascular resistance in association with reduced collagen glycation, Exp. Gerontol., 2011, 46(6), 482–488, DOI:10.1016/j.exger.2011.02.001.
- M. Murakoshi, et al., Pleiotropic effect of pyridoxamine on diabetic complications via CD36 expression in KK-Ay/Ta mice, Diabetes Res. Clin. Pract., 2009, 83(2), 183–189, DOI:10.1016/j.diabres.2008.11.008.
- P. A. Voziyan and B. G. Hudson, Pyridoxamine: The Many Virtues of a Maillard Reaction Inhibitor, Ann. N. Y. Acad. Sci., 2005, 1043(1), 807–816, DOI:10.1196/annals.1333.093.
- S. Kassab, et al., Cognitive dysfunction in diabetic rats is prevented by pyridoxamine treatment. A multidisciplinary investigation, Mol. Metabol., 2019, 28, 107–119, DOI:10.1016/j.molmet.2019.08.003.
- S. Ugaji, et al., Pyridoxamine improves diabetes-evoked delayed bone repair in mice, J. Transl. Sci., 2021, 7(3), 2–6, DOI:10.15761/jts.1000413.
- P. H. Roham, S. N. Save and S. Sharma, Human islet amyloid polypeptide: A therapeutic target for the management of type 2 diabetes mellitus, J. Pharmaceut. Biomed. Anal., 2022, 12(4), 556–569, DOI:10.1016/j.jpha.2022.04.001.
- Wavebreak pipeline, https://wavebreaktx.com/pipeline/, Accessed March 2024.
- D. C. Rodriguez Camargo, et al., Surface-Catalyzed Secondary Nucleation Dominates the Generation of Toxic IAPP Aggregates, Front. Mol. Biosci., 2021, 8, 757425, DOI:10.3389/fmolb.2021.757425.
- Neurimmune pipeline, https://www.neurimmune.com/research-development/pipeline, accessed March 2024.
- S. Chamberlain, H. Gabriel, W. Strittmatter and J. Didsbury, An Exploratory Phase IIa Study of the PPAR delta/gamma Agonist T3D-959 Assessing Metabolic and Cognitive Function in Subjects with Mild to Moderate Alzheimer's Disease, J. Alzheimer's Dis., 2020, 73(3), 1085–1103, DOI:10.3233/JAD-190864.
- J. L. Cummings, A. M. L. Osse and J. W. Kinney, Alzheimer's Disease: Novel Targets and Investigational Drugs for Disease Modification, Drugs, 2023, 83(15), 1387–1408, DOI:10.1007/s40265-023-01938-w.
- M. A. Mengstie, et al., Endogenous advanced glycation end products in the pathogenesis of chronic diabetic complications, Front. Mol. Biosci., 2022, 9, 1002710, DOI:10.3389/fmolb.2022.1002710.
- M. C. Jones, Therapies for Diabetes: Pramlintide and Exenatide, Am. Fam. Physician, 2007, 75(12), 1831–1835 Search PubMed.
- B. J. Hoogwerf, Exenatide and pramlintide: New glucose-lowering agents for treating diabetes mellitus, Cleve. Clin. J. Med., 2006, 73(5), 477–484, DOI:10.3949/ccjm.73.5.477.
- B. Gallwitz, Exenatide in type 2 diabetes: Treatment effects in clinical studies and animal study data, Int. J. Clin. Pract., 2006, 60(12), 1654–1661, DOI:10.1111/j.1742-1241.2006.01196.x.
- M. Wronka, J. Krzemińska, E. Młynarska, J. Rysz and B. Franczyk, The Influence of Lifestyle and Treatment on Oxidative Stress and Inflammation in Diabetes, Int. J. Mol. Sci., 2022, 23(24), 15743, DOI:10.3390/ijms232415743.
- J.-d Wu, et al., Effect of exenatide on inflammatory and oxidative stress markers in patients with type 2 diabetes mellitus, Diabetes Technol. Therapeut., 2011, 13(2), 143–148, DOI:10.1089/dia.2010.0048.
- L. L. Want and R. Ratner, Exenatide and pramlintide: New therapies for diabetes, Int. J. Clin. Pract., 2006, 60(12), 1522–1523, DOI:10.1111/j.1742-1241.2006.01219.x.
- H. Nasri and M. Rafieian-Kopaei, “Metformin: Current knowledge,” 2014, J. Res. Med. Sci., 2014, 19(7), 658–664 Search PubMed.
- K. Cho, et al., Antihyperglycemic mechanism of metformin occurs via the AMPK/LXRα/POMC pathway, Sci. Rep., 2015, 5, 8145, DOI:10.1038/srep08145.
- G. Zhou, et al., Role of AMP-activated protein kinase in mechanism of metformin action, J. Clin. Invest., 2001, 108(8), 1167–1174, DOI:10.1172/jci200113505.
- K. Cusi and R. A. DeFronzo, Metformin: a review of its metabolic effects, Diabetes Rev., 1998, 6, 89–131, DOI:10.1172/jci200113505.
- X. Hou, et al., Metformin reduces intracellular reactive oxygen species levels by upregulating expression of the antioxidant thioredoxin via the AMPK-FOXO3 pathway, Biochem. Biophys. Res. Commun., 2010, 396(2), 199–205, DOI:10.1016/j.bbrc.2010.04.017.
- T. Matsui, M. Takeuchi, S. Yamagishi and Y. Ishibashi, Metformin inhibits advanced glycation end products (AGEs)-induced renal tubular cell injury by suppressing reactive oxygen species generation via reducing receptor for AGEs (RAGE) expression, Horm. Metab. Res., 2012, 44(12), 891–895, DOI:10.1055/s-0032-1321878.
- K. Harris, C. Boland, L. Meade and D. Battise, Adjunctive therapy for glucose control in patients with type 1 diabetes, Diabetes, Metab. Syndr. Obes., 2018, 11, 159–173, DOI:10.2147/DMSO.S141700.
- N. T. Shurrab and E. S. A. Arafa, Metformin: A review of its therapeutic efficacy and adverse effects, Obes. Med., 2020, 17, 100186, DOI:10.1016/j.obmed.2020.100186.
- T. Congiu, et al., Undercover toxic ménage à trois of amylin, copper (II) and metformin in human embryonic kidney cells, Pharmaceutics, 2021, 13(6), 830, DOI:10.3390/pharmaceutics13060830.
- S. VanDeKoppel, Managed Care Perspective on Three New Agents for Type 2 Diabetes, J. Manag. Care Pharm., 2008, 14(4), 363–380, DOI:10.18553/jmcp.2008.14.4.363.
- L. T. Meade, Practical use of exenatide and pramlintide for the treatment of type 2 diabetes, J. Pharm. Pract., 2009, 22(6), 540–545, DOI:10.1177/0897190009333164.
- K. R. Feingold, Oral and Injectable (Non-Insulin) Pharmacological Agents for the Treatment of Type 2 Diabetes, [Updated 2022 Aug 26], ed. K. R Feingold, B. Anawalt, M. R. Blackman and et al., South Dartmouth (MA), MDText.com, Inc., 2000, Available from: https://www.ncbi.nlm.nih.gov/books/NBK279141/ Search PubMed.
- S. J. Moon, I. Jung and C. Y. Park, Current advances of artificial pancreas systems: A comprehensive review of the clinical evidence, Diabetes Obes Metab, 2021, 45(6), 813–839, DOI:10.4093/dmj.2021.0177.
- M. Alghrably, D. Dudek, A. H. Emwas, Ł. Jaremko, M. Jaremko and M. Rowińska-Żyrek, Copper(II) and Amylin Analogues: A Complicated Relationship, Inorg. Chem., 2020, 59(4), 2527–2535, DOI:10.1021/acs.inorgchem.9b03498.
- M. Alghrably, I. Czaban, Ł. Jaremko and M. Jaremko, Interaction of amylin species with transition metals and membranes, J. Inorg. Biochem., 2019, 191, 69–76, DOI:10.1016/j.jinorgbio.2018.11.004.
- M. Samsom, et al., Pramlintide, an amylin analog, selectively delays gastric emptying: potential role of vagal inhibition, Am. J. Physiol. Gastrointest. Liver Physiol., 2000, 278(6), G946–G951, DOI:10.1152/ajpgi.2000.278.6.G946.
- T. A. Lutz and T. A. Lutz, The role of amylin in the control of energy homeostasis, Am. J. Physiol. Regul. Integr. Comp. Physiol., 2010, 298, 1475–1484, DOI:10.1152/ajpregu.00703.2009.-Amylin.
- J. E. Morley, et al., Amylin and Food Intake in Mice: Effects on Motivation to Eat and Mechanism of Action, Pharmacol. Biochem. Behav., 1997, 56(1), 123–129, DOI:10.1016/S0091-3057(96)00168-2.
- T. Tamura, C. Miyaura, I. Owan and T. Suda, Mechanism of action of amylin in bone, J. Cell. Physiol., 1992, 153(1), 6–14, DOI:10.1002/jcp.1041530103.
- A. Martínez, S. Kapas, M. Miller, Y. Ward and F. Cuttitta, Coexpression of Receptors for Adrenomedullin, Calcitonin Gene-Related Peptide, and Amylin in Pancreatic Cells, Endocrinology, 2000, 141, 406–411, DOI:10.1210/en.141.1.406.
- C. Weyer, D. Maggs, A. Young and O. Kolterman, Amylin Replacement With Pramlintide as an Adjunct to Insulin Therapy in Type 1 and Type 2 Diabetes Mellitus: A Physiological Approach Toward Improved Metabolic Control, Curr. Pharm. Des., 2005, 7(14), 1353–1373, DOI:10.2174/1381612013397357.
- M. Fineman, The human amylin analog, pramlintide, reduces postprandial hyperglucagonemia in patients with type 2 diabetes mellitus, Horm. Metab. Res., 2002, 34, 504–508, DOI:10.1055/s-2002-34790.
- X.-X. Zhang, Y.-H. Pan, Y.-M. Huang and H.-L. Zhao, Neuroendocrine hormone amylin in diabetes, World J. Diabetes, 2016, 7(9), 189, DOI:10.4239/wjd.v7.i9.189.
- A. Young, Amylin and the integrated control of nutrient influx, Adv. Pharmacol., 2005, 52, 67–77, DOI:10.1016/S1054-3589(05)52004-0.
- B. Konarkowska, J. F. Aitken, J. Kistler, S. Zhang and G. J. S. Cooper, The aggregation potential of human amylin determines its cytotoxicity towards islet β-cells, FEBS J., 2006, 273(15), 3614–3624, DOI:10.1111/j.1742-4658.2006.05367.x.
- E. T. Beui, Hyalinization of the islets of Langerhans in nondiabetic individuals, Am. J. Pathol., 1959, 35(4), 801–805 Search PubMed.
- J. R. Brender, E. L. Lee, M. A. Cavitt, A. Gafni, D. G. Steel and A. Ramamoorthy, Amyloid fiber formation and membrane disruption are separate processes localized in two distinct regions of IAPP, the type-2-diabetes-related peptide, J. Am. Chem. Soc., 2008, 130(20), 6424–6429, DOI:10.1021/ja710484d.
- S. Jha, et al., PH dependence of amylin fibrillization, Biochemistry, 2014, 53(2), 300–310, DOI:10.1021/bi401164k.
- S. T. Ferreira, M. N. N. Vieira and F. G. De Felice, Soluble protein oligomers as emerging toxins in Alzheimer's and other amyloid diseases, IUBMB Life, 2007, 59(4–5), 332–345, DOI:10.1080/15216540701283882.
- T. A. Lutz and U. Meyer, Amylin at the interface between metabolic and neurodegenerative disorders, Front. Neurosci., 2015, 9, 216, DOI:10.3389/fnins.2015.00216.
- M. R. Hayden and S. C. Tyagi, Neural redox stress and remodeling in metabolic syndrome, type 2 diabetes mellitus, and diabetic neuropathy Word count, Med. Sci. Monit., 2004, 10(12), RA291 CAS.
- E. W. Jr, J. L. Scism-Bacon and L. C. Glass, Oxidative stress in type 2 diabetes: the role of fasting and postprandial glycaemia, Int. J. Clin. Pract., 2006, 60(3), 308–314, DOI:10.1111/j.1368-5031.2006.00825.x.
- B. J. Hoogwerf, Exenatide and pramlintide: New glucose-lowering agents for treating diabetes mellitus, Cleve. Clin. J. Med., 2006, 73(5), 477–484, DOI:10.3949/ccjm.73.5.477.
- B. J. Hoogwerf, K. B. Doshi and D. Diab, Pramlintide, the synthetic analogue of amylin: physiology, pathophysiology, and effects on glycemic control, body weight, and selected biomarkers of vascular risk, Vasc. Health Risk Manag., 2008, 4(2), 355–362, DOI:10.2147/vhrm.s1978.
- S. Padhi, A. K. Nayak and A. Behera, Type II diabetes mellitus: a review on recent drug based therapeutics, Biomed. Pharmacother., 2020, 131, 110708, DOI:10.1016/j.biopha.2020.110708.
- P. A. Hollander, et al., Pramlintide as an Adjunct to Insulin Therapy Improves Long-Term Glycemic and Weight Control in Patients With Type 2 Diabetes A 1-year randomized controlled trial, Diabetes Care, 2003, 26(3), 784–790, DOI:10.2337/diacare.26.3.784.
- R. V. Sekhar, GlyNAC (Glycine and N-Acetylcysteine) Supplementation Improves Impaired Mitochondrial Fuel Oxidation and Lowers Insulin Resistance in Patients with Type 2 Diabetes: Results of a Pilot Study, Antioxidants, 2022, 11(1), 154, DOI:10.3390/antiox11010154.
- P. K. Bagul and S. K. Banerjee, Application of resveratrol in diabetes: rationale, strategies and challenges, Curr. Mol. Med., 2015, 15(4), 312–330, DOI:10.2174/1566524015666150505155702.
- D. de Zeeuw, et al., Bardoxolone Methyl in Type 2 Diabetes and Stage 4 Chronic Kidney Disease, N. Engl. J. Med., 2013, 369(26), 2492–2503, DOI:10.1056/nejmoa1306033.
- C. Aloke, et al., Current Advances in the Management of Diabetes Mellitus, Biomedicines, 2022, 10(10), 2436, DOI:10.3390/biomedicines10102436.
| This journal is © The Royal Society of Chemistry 2024 |