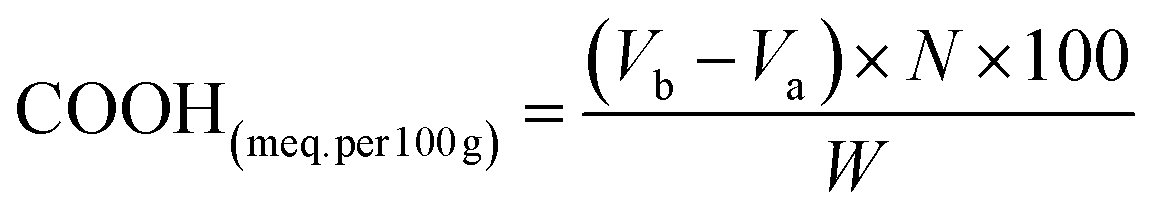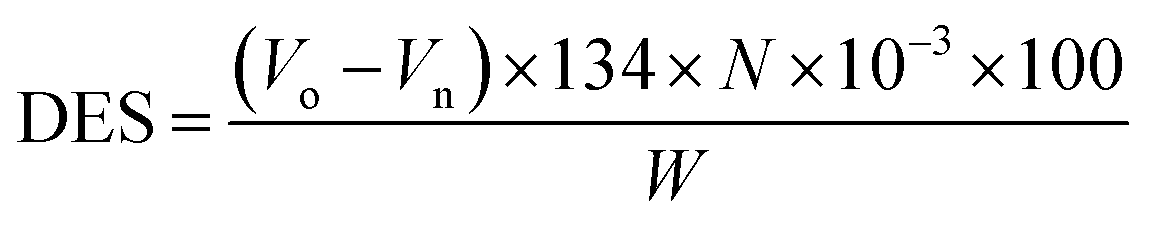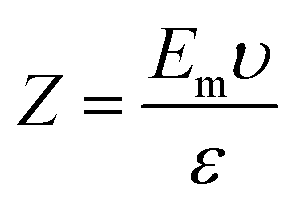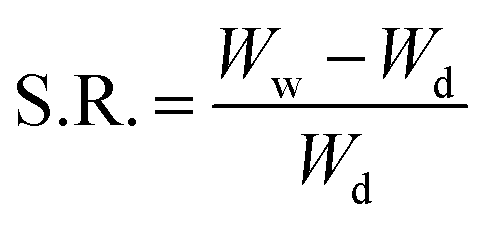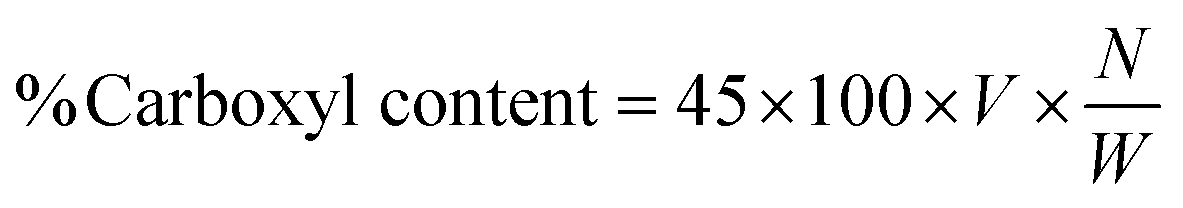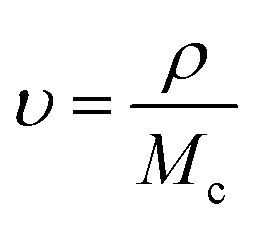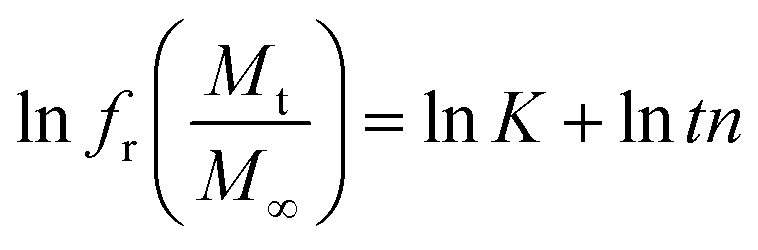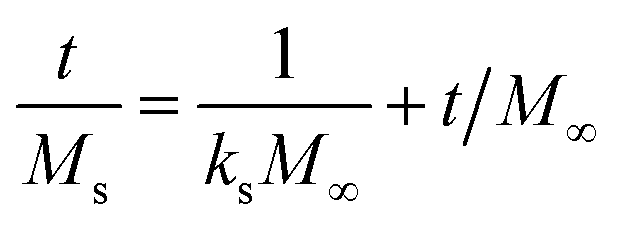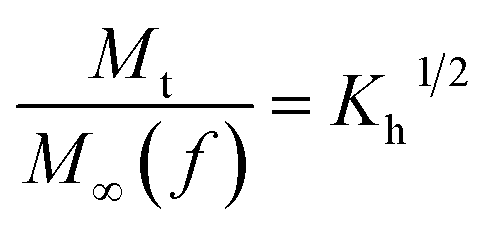DOI:
10.1039/D4RA02638A
(Paper)
RSC Adv., 2024,
14, 25174-25189
A yeast–malic acid crosslinker/polyacrylic acid hydrogel containing doxycycline for the treatment of periodontitis†
Received
8th April 2024
, Accepted 15th June 2024
First published on 12th August 2024
Abstract
Doxycycline (DX) is a drug of choice for the treatment of periodontitis, with the limitation of requiring a high dose, which may be overcome by the preparation of a targeted controlled-release hydrogel containing a newly synthesized yeast–malic acid crosslinker (YMC). YMC was synthesized via thermochemical modification of yeast with malic acid at 100–140 °C and compared with glutaraldehyde-saturated toluene (GST). Swelling capacity, acid and carboxyl content, scanning electron microscopy (SEM) imaging, Brunauer–Emmett–Teller (BET) analysis, viscosity, cross-linking density, DX loading and release behavior at pH 6.5, mucoadhesion, and antimicrobial and periodontal efficacy of the glutaraldehyde hydrogel (HGG) and YMC hydrogel (HGY) were compared. Changes from C–O (1421 cm−1) to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OOR (1702 cm−1) in the infrared spectroscopy, along with changes in the degree of substitution from 0 to 0.39, degree of esterification from 0 to 40 ± 1.5 and COOH content from 129 ± 0.5 to 290 ± 0.5 (meq. per 100 g), were found between yeast to YMC, respectively. The results revealed 1.5 times more dynamic swelling, 0.25-fold decrease in acid content, 2.3-fold increase in carboxyl content, and 1.2- and 2.1-fold increases in cross-linking density and viscosity of HGY as compared to HGG, respectively. The SEM and BET results revealed that HGY had a 2 times greater porous surface than HGG. HGY/DX was 35 ± 2% more effective in controlling periodontitis bacteria, decreased periodontal depth from 4 to 3.2 mm, and gingival index from 3 to 1 as compared to HGG/DX in patients suffering from periodontitis. HGY/DX not only serves as a tool for the controlled release of DX in periodontal pockets but also contributes to the treatment of gingival periodontitis.
OOR (1702 cm−1) in the infrared spectroscopy, along with changes in the degree of substitution from 0 to 0.39, degree of esterification from 0 to 40 ± 1.5 and COOH content from 129 ± 0.5 to 290 ± 0.5 (meq. per 100 g), were found between yeast to YMC, respectively. The results revealed 1.5 times more dynamic swelling, 0.25-fold decrease in acid content, 2.3-fold increase in carboxyl content, and 1.2- and 2.1-fold increases in cross-linking density and viscosity of HGY as compared to HGG, respectively. The SEM and BET results revealed that HGY had a 2 times greater porous surface than HGG. HGY/DX was 35 ± 2% more effective in controlling periodontitis bacteria, decreased periodontal depth from 4 to 3.2 mm, and gingival index from 3 to 1 as compared to HGG/DX in patients suffering from periodontitis. HGY/DX not only serves as a tool for the controlled release of DX in periodontal pockets but also contributes to the treatment of gingival periodontitis.
1 Introduction
Periodontal disease is the result of bacterial infections and inflammation of the gums and bones that support teeth, which ultimately results in tooth loss if it remains untreated.1 The most common causes of periodontitis are poor oral hygiene, dietary habits, and the presence of pathogenic bacteria at salivary pH. Actinomyces viscosus (A. viscosus) and Porphyromonas gingivalis (P. gingivalis) are the most common bacteria that cause gingival bleeding, tooth decay, and tooth mobility, which ultimately cause periodontitis.2 The prevalence of periodontitis is about 40% worldwide, among which approximately 70% of adults are approaching tooth loss.3 Periodontitis can be treated with high systemic doses of antibiotics, e.g., doxycycline (DX), which has severe side effects and low patient compliance. Previous literature showed limited treatment of periodontitis by DX due to its high dose and abrupt release from traditional dosage forms.4 Superficial loading of DX in hydrogels can be controlled by using a naturally occurring, simply modified crosslinker, which not only controls the release of DX but also prevents its abrupt release.
Yeast is a prime option for the preparation of superabsorbent composites, working as a cross-linker, an immunomodulatory agent with antimicrobial effects, and a biofilm disruptor due to the presence of its unique water-holding functional groups.5,6 The synthesis of yeast crosslinkers is possible with naturally occurring α-hydroxy acids such as lactic acid, glycolic acid, and citric acid, with the limitation of stability. The esterification of yeast with malic acid via thermochemical modification would be preferred due to its dicarboxylic-acid structure, which favors cross-linking of hydrogels, pH-dependent drug release, mucoadhesion and antimicrobial properties.7 Hydrogels obtained using yeast, with their maze-like nature, have become the best choice for the treatment of periodontitis due to their pore size, cross-linking density, dynamic and equilibrium swelling, acid and carboxyl contents, and viscosity parameters. By carefully selecting monomers and crosslinkers, hydrogels with the necessary characteristics, such as hydrophilicity and porosity, can be produced for antibacterial applications.8
The objectives of the current study were to synthesize semisynthetic yeast–malic acid crosslinker (YMC) with an additional property of chemical interaction with DX. Thermochemical modification of yeast with malic acid and its confirmation by FTIR spectroscopy, degree of substitution, and degree of esterification were successfully performed. The proposed newly synthesized YMC was used for the preparation of controlled-release, stable and non-toxic polyacrylic-acid-containing hydrogels. Glutaraldehyde-saturated toluene (GST)- and YMC-based hydrogels (HGG and HGY, respectively) were compared for their crosslinking density, DX loading and release capacity, antimicrobial potential, and radiographic analysis. An in-vivo clinical study was also conducted on periodontitis patients and the gingival index, tooth mobility index, and periodontal depth were examined before and after treatment with HGG/DX and HGY/DX using X-ray images of periodontal tissue with diseased teeth.
2 Materials and methods
2.1 Materials
Granulated yeast (M.W. = 274.31 g mol−1, Sigma-Aldrich, Germany) and DL-malic acid (M.W. = 134.09 g mol−1, Merck, Japan) were used for the synthesis of a novel superabsorbent yeast–malic acid crosslinker (YMC). Polyacrylic acid (density = 1.14 g mL−1 at 25 °C, Merck, Japan), glutaraldehyde (M.W. = 100.13 Da, Riedel-de Haën, Germany), and ammonium persulfate (APS, M.W. = 228.18 g mol−1, Riedel-de Haën, USA) were used for hydrogel synthesis. Toluene (M.W. = 92.14 g mol−1, MP Biomedicals, France) was purchased for the synthesis of glutaraldehyde saturated toluene (GST). NaCl, D-glucose, methanol (CH3OH, M.W. = 32.04) and hydrochloric acid (HCl, M.W. = 36.46) were purchased from Fluka (Germany) for the investigation of the biological activity of the hydrogel. Doxycycline hyclate (M.W. = 444.42 g mol−1, 99.98%) was generously gifted from the Drug Testing Laboratory, Punjab, Multan. All chemicals used were of analytical grade and water obtained from reverse osmosis (RO) was used for experimental analysis.
2.2 Preparation of the yeast–malic acid crosslinker (YMC)
YMC was synthesized with slight modification of already reported method of Feng et al.,9 using different molar ratios of yeast and malic acid (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, and 2:1). Briefly, a homogenized solution of malic acid (33.33% w/v) in deionized water was gradually poured into a baker’s yeast extract solution (16.67% w/v) to synthesize YMC 1:1. The resulting mixture was placed into a Petri dish and heated in a hot-air oven (Memmert 9000 series), first for 4 days in a temperature range of 55–60 °C for complete removal of water, and then for 3 h at 100–140 °C. The resulting material was acidified with acetic acid (3.32% v/v) at pH 2.5, washed with water three times and dried under reduced pressure (97 psi). Similar procedures with different ratios of yeast and malic acid (1:2 and 2:1) were repeated for the synthesis of YMC 1:2 and YMC 2:1.
:1, 1:2, and 2:1). Briefly, a homogenized solution of malic acid (33.33% w/v) in deionized water was gradually poured into a baker’s yeast extract solution (16.67% w/v) to synthesize YMC 1:1. The resulting mixture was placed into a Petri dish and heated in a hot-air oven (Memmert 9000 series), first for 4 days in a temperature range of 55–60 °C for complete removal of water, and then for 3 h at 100–140 °C. The resulting material was acidified with acetic acid (3.32% v/v) at pH 2.5, washed with water three times and dried under reduced pressure (97 psi). Similar procedures with different ratios of yeast and malic acid (1:2 and 2:1) were repeated for the synthesis of YMC 1:2 and YMC 2:1.
2.3 Characterization of YMC
2.3.1 Physicochemical properties. FTIR spectroscopy was performed (400–4000 cm−1) for the detection of the functional groups of both yeast and YMC via ATR-FTIR (Bruker Alpha I, Germany), for the identification of –OH, COOH and COOR groups.10 Using an acid–base titration, the –COOH contents of yeast, YMC 1:2, YMC 1:1 and YMC 2:1 were calculated. Briefly, 1.01 g of yeast and 1.02 g of YMC were dissolved in 10 mL of 0.2 N NaOH solution. For calculation of the remaining moles of NaOH, phenolphthalein was used as an indicator and 0.1 N hydrochloric acid as a titrant, and the carboxyl content in terms of meq. per 100 g was calculated using following equation:| |
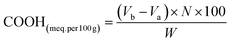 | (1) |
where Va and Vb (in mL) represent the volumes of HCl used for the titration in the absence and presence of yeast and YMC, respectively. N is the normality of HCl, and W (g) is the dry sample’s weight.
2.3.2 Degree of esterification and substitution. The ratio of reacted –OH groups to total initial –OH groups in the yeast (degree of esterification) and the average number of moles substituted per anhydrous glucose unit (degree of substitution) were calculated using potentiometric titration analysis (Metrohm, Titrino 877) and application of the Wurzburg method.11 Briefly, equal 1.021 g weights of ground of yeast and semisynthetic YMC were mixed separately in 50 mL of 75% v/v ethanol. The obtained slurries were continuously stirred at 500 rpm for 30 min, then kept in a water bath at 55 °C for 30 min. After cooling at room temperature, 0.51 M KOH solution (35 mL) was added for saponification of the esters and allowed to react for 72 h with continuous stirring. Using phenolphthalein as an indicator, the remaining moles of NaOH were back titrated with 0.5 M HCl. The degree of esterification and degree of substitution were calculated by using the following eqn (2) and (3):| |
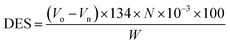 | (2) |
| |
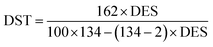 | (3) |
where V0 and Vn are the volumes of HCl used to titrate the blank sample and YMC, respectively, N is the normality of HCl used, and W is the dried weight of yeast or YMC. 134.09 (g mol−1) and 162.3 (g mol−1) are the molecular weights of malic acid and anhydrous glucose units, respectively, while 2 is the molar mass of two H atoms per reacted malic acid molecule.
2.3.3 Zeta potential and pH sensitivity. The value of pH necessary to effect net-zero charge on a solid surface in the absence of any biological indicator is called the point of zero charge (PZo). The PZo values of yeast and semisynthetic YMC were determined using already reported method of Dutta et al.12 All samples of semisynthetic YMC were equilibrated in distilled water and then poured into solutions with predetermined pH values ranging from 2–9, adjusted using 0.2 M HCl and NaOH solutions. The zeta potential was measured via electrophoretic mobility values using a Zetasizer Nano ZS (Malvern Panalytical 90 series, Germany). By using the Smoluchowski equation, the zeta potential was calculated:| |
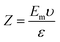 | (4) |
where Z is the zeta potential, Em is the electrophoretic mobility, υ is the viscosity of medium and ε is the dielectric constant, respectively.
2.4 Hydrogel preparation
Two types of hydrogels, glutaraldehyde hydrogel (HGG) and YMC hydrogel (HGY), were prepared via free-radical polymerization using the already reported but slightly modified method of Feng et al.13 Briefly, for the preparation of HGG, a previously homogenized aqueous solution of yeast was slowly poured into 15 mL polyacrylic acid, followed by the addition of 3 μL of previously prepared 2.5% glutaraldehyde-saturated toluene (GST), with magnetic stirring for 1 h.14 5 mL of APS solution (0.1%) as an initiator was added into the resulting mixture, which was placed in an ultrasonic water bath (Elmasonic, P300H). After 5 min, the mixture was transferred into a test tube for successive temperature 60 and 65 degree. 5 h at 55 °C and 24 h at 55 °C. In the case of HGY, the same procedure was repeated using semisynthetic YMC (5% w/v) instead of GST as the crosslinker. The resultant hydrogel was cut into round-shaped discs with the help of a mold (8 ± 0.2 mm) and stored in a tight container for further use.15
2.5 Physicochemical properties of hydrogels
2.5.1 Swelling kinetics study. To establish the application and compatibility of the hydrogels in saliva, the swelling ratio (g g−1) was determined in various physiological solutions prepared in deionized water, i.e., saline water (0.9%), D-glucose solution (5%), simulated urine (0.1% MgSO4, 0.06% CaCl2, 2% urea, and 0.8% NaCl), 16 and Hank’s solution (1% NaCl, 0.05% KCl, 0.0175% CaCl2, 0.0125% MgSO4, 0.0125% MgCl, 0.0125% Na2HPO4, 0.0075% KH2PO4, 0.0075% D-glucose, and 0.125% NaHCO3).17 These solutions were used for simulation of body fluids.18 Accurately weighed HGG and HGY discs were immersed in artificial saliva prepared using already reported method of Sarkar et al.19 (artificial saliva: 0.32 g L−1 NH4NO3, 0.15 g L−1 NaCl, 0.65 g L−1 K2HPO4, 0.25 g L−1 KCl, 0.02 g L−1 C5H3N4O3Na, 0.16 g L−1 C3H5O3Na, 0.25 g L−1 H2NCONH2, 0.32 g L−1 K3C6H5O7·H20 and 3.5 g L−1 goat mucin; pH 6.5 with 0.9 M NaOH)19 and in 0.05 M phosphate buffer medium (PBM: pH 7.4) to determine swelling behavior of HGG and HGY. After 60 and 120 min, the HGG and HGY were removed from the media, blotted on filter paper, and dried in an oven at 40 °C. The swelling ratio (S.R.) or dynamic swelling was measured using the following eqn (5):| |
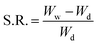 | (5) |
where Ww is the wet weight of the hydrogel after swelling, while Wd is the dry weight of the hydrogel.20
2.5.2 Acid value and % COOH. The acid values (number of free carboxylic acid groups) of HGG and HGY were determined using the already reported method of Mudassir et al.21 Briefly, accurately weighed 0.9 g portions of HGG and HGY were taken separately and dissolved in 20 mL acetone with the aid of ultrasonication. 1 mL of phenolphthalein (0.5%) solution was added and the mixture was titrated against 0.1 N KOH with the endpoint of a light pink color. The acid value (Av) and % carboxyl content were calculated using the following eqn (6) and (7), respectively:| |
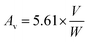 | (6) |
| |
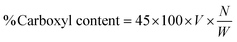 | (7) |
where V and W are the volume of KOH consumed and weight of HGG or HGY, respectively, 45 is the molecular weight of the carboxyl groups, and N is the normality of KOH.
2.5.3 SEM, BET and FTIR. HGG and HGY morphology was examined with scanning electron microscopy (SEM) (Hitachi High Tech S4900 FE-SEM). Gold was applied to HGG, HGY, HGG/DX and HGY/DX in a dry state and the samples placed on a sample holder with a 20 kV accelerating voltage and 10 μm resolution. The specific surface area (cm2 g−1), pore size (nm) and pore volume (cm3 g−1) of the prepared hydrogels, i.e., HGG and HGY, were evaluated by using a Brunauer–Emmett–Teller (BET) surface-area and pore-size analyzer (NOVA, Germany, 4200E: version 2.4) and Barret–Joyner–Halenda (BJH) principles.22 0.74 ± 0.09 g of HGG and 0.739 ± 0.09 g of HGY were accurately weighed, and first degassed under vacuum (15 × 10–3 torr) with heating at 120 °C to remove surface gases and moisture. The HGG and HGY were separately loaded into BET tubes and N2 adsorption and desorption data were evaluated by using Quantachrome (version 3.0).23 FTIR spectroscopy was performed (400–4000 cm−1) for determination of functional group interactions and the chemical compatibility of different reagents present in HGG, HGY and HGY/DX, via ATR-FTIR (Bruker Alpha I, Germany).10,24
2.5.4 Crosslinking and viscosity studies. The cross-linking ratio and cross-linking density were determined using the already reported method of Shokuhfar et al.25 The previously performed swelling studies were used to evaluate the cross-linking density of HGG and HGY according to Flory–Rehner’s theory. The equilibrium swelling ratio (υr) of both HGG and HGY was used to calculate the crosslinking density (υ) via the following eqn (8) and (9):| |
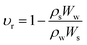 | (8) |
| |
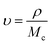 | (9) |
where ρs, ρw and ρ are the densities of the swelled HGG or HGY, water and crosslinker, respectively. Ww and Ws are the mass of water and swelled HGG or HGY, respectively. Mc is the molecular weight between cross-links calculated using the Flory–Rehner equation, and is referred to elsewhere as the crosslinking ratio.26After swabbing mucus from a goat oral cavity, it was subjected to centrifugation at 1500 rpm, a series of filtrations through 0.45 μm filters, dialysis and precipitation for removal of cellular debris. The resultant mucus was stored at −15 °C in an airtight container for further use.27 HGG and HGY were tested for their viscoelastic parameters using a plate-cone viscometer (Visco TM 158 RT20, Germany). Briefly, after being previously swelled in 40% methanolic solution, HGG and HGY were treated with 0.05 mL H2O2 and goat mucus and incubated for 35 min at 37.5 °C. Every 30 min, the dynamic viscosity at 1 Hz and shear stress in the range of 0.02 to 20 pa were calculated. The viscous modulus (G′′), elastic modulus (G′) and loss tangent θ (the ratio between the viscous properties and elastic properties of the polymer) were determined.
2.6 Ex vivo mucoadhesion strength
For evaluation of the mucoadhesion strength, chicken buccal membrane was taken after cleaning its contents and surface lipids, then frozen at −5 °C in PBM (pH 6.8) according to approved guidelines of the ethical committee of Bahauddin Zakariya University, Multan.28 A slightly modified calibrated physical balance was used; on the left side was a rubber cork with HGG or HGY connected to the bottom, linked to a piece of chicken membrane, which was connected to a weight. On the other side of the balance, the weight of water required to separate HGG or HGY from the mucosal surface was calculated. The average of triplicate data was reported for the evaluation of the bio-adhesive strength and adhesive force in Newton (N), using eqn (10).| |
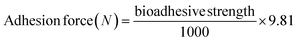 | (10) |
The ex vivo mucoadhesion duration was determined by modifying the already reported method of mudassir et al.21 The freshly removed chicken buccal mucosa was placed in a modified USP I (Erweka, GmBH, Germany) disintegration apparatus previously cleaned with normal saline. The mucosal membrane was adhered with cyanoacrylate adhesive tape and accurately weighed previously compacted 100 ± 2 mg HGG and HGY discs were adhered to the mucosal membrane with light pressure. The apparatus containing PBM (100 mM, pH 6.8) was operated at 0.5 dips per second (DPS) at 37 ± 0.5 °C and the detachment times of HGG and HGY were calculated.21
2.7 % DX loading and release
DX was loaded by immersing HGG and HGY into 5% aqueous solution of DX for 120 min (maximum swelling time) under a reduced pressure of −15 mmHg. Ascorbic acid (2.5%) was used to prevent the oxidation of DX. The % DX loading (%DL) was confirmed for 100 mg of HGG/DX and HGY/DX by using an already prepared standard curve for increasing concentrations of DX at λmax of 349 nm. The %DL was calculated using eqn (11).| |
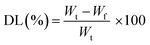 | (11) |
where Wt is the total weight of DX and Wf is that of the free drug.
The in vitro % release of DX from HGG/DX and HGY/DX was calculated using a USP II dissolution apparatus (Erweka) with 100 mM PBM (pH 6.8). Already immersed cellulose dialyzing membrane (12–14 kDa) was used as a diffusion barrier and the % release of DX from HGG/DX and HGY/DX (100 mg each) was calculated after rotating at 100 rpm at 37 ± 0.5°C. An aliquot of 5 mL was removed after predetermined time intervals at 0.55, 1.3, 2.5, 4.3, 6.6, 8.5, 10.5, 12.6, and 24 h and the concentration of DX was calculated using an already prepared standard curve for increasing concentrations of DX at 349 nm.29 Various kinetic models were applied, like power function (eqn (12)), Schott (eqn (13)), Higuchi kinetics (eqn (14)), Hixon–Crowell (eqn (15)) and Korsmeyer–Peppas (eqn (16)) models. The Akaike information criterion (AIC) for DX release was determined.30
| |
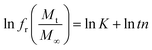 | (12) |
| |
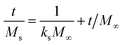 | (13) |
| |
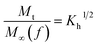 | (14) |
where
n,
K,
Kh,
α and
K3 are the HGG/DX or HGY/DX release exponent, characteristic constant, and Higuchi, Hixson–Crowell and Korsmeyer–Peppas rate constants, respectively.
t is the time,
fr is the fractional release,
Mt is the release at definite time interval, and
Ms is the the release after infinite time.
kS is the Schott constant,
M∞ is the release after infinite time,
Wi and
Wt are the initial and final amount of drug and
f is the fraction of drug release of HGG and HGY.
31
2.8 Antimicrobial assay
The antimicrobial assays of HGG/DX and HGY/DX were performed at the microbial laboratory of the Drugs Testing Laboratory, Punjab, Multan (ISO 17025) using P. gingivalis and A. viscosus. Briefly, 100 mg of swelled HGG/DX and HGY/DX (0.1 cm height, 2.8 and 1.0 cm diameter) were placed on top of agar plates already germinated with 100 μL of bacterial suspension. Metronidazole (MTZ) solution (0.6 mg mL−1) was used as a positive control. The Petri dishes were incubated for 24 h at 37 °C, the zone of inhibition was determined and the antibacterial assay was performed.32 For the time-kill assay, precisely weighed 100 mg portions of HGG/DX and HGY/DX were placed in sterile polystyrene tubes together with 1000 μL of nutritional broth and 20 μL (log 10 CFU mL−1) of bacterial solutions of P. gingivalis and A. viscosus. 5% MTZ solution in 1000 μL of culture media was used as a control. Anaerobic conditions were maintained and 30 μL of the solution were removed from each tube at each time point (0, 4, 8, 12 and 24 h), serially diluted to a concentration of 10−6. To count the number of colonies, 30 μL of each dilution were spread out on agar plates and incubated for 24 h. At each time point (0, 4, 8, 12 and 24 h) the ratio of colonies in the test tubes with HGG/DX or HGY/DX to those in the control tubes was calculated. Each experiment was repeated in triplicate.33
2.9 Effect of HGG/HGY on periodontitis
The experimental analysis was carried out in compliance with the current Declaration of Helsinki laws and the International Ethical Guidelines for Biomedical Research.34 The research ethical committee of the Faculty of Pharmacy, Bahauddin Zakariya University Multan, authorized all protocols for human subjects with provided informed consent.35 The study was conducted on patients infected with periodontitis according to inclusion criteria that participants had no periodontal maintenance therapy for six months prior. The exclusion criteria of this study were visible signs of disease as having comorbidity. Patients were divided into three groups: the first group (G1, n = 6) of infected patients received HGG/DX in each quadrant of the jaw. The second group (G2, n = 6) having periodontitis in molar areas received HGY/DX in each quadrant of the jaw. The third, control, group (G3, n = 6) with periodontitis at molar depth received oral Vibramycin (DX: 100 mg, Pfizer) capsules with Sip-C (vitamin C) tablets (Gluta One). All groups were used to identify changes in monitored parameters after induction of local anesthesia.
Clinical assessments were made at the beginning of the study (1st phase) and at the end of the study (2nd phase) after 2 months. The parameters assessed were body weight, probing depth, gingival index (GI), mobility index (MI), periodontal depth and bleeding index, evaluated using a periodontal probe (Dentaltix; CP 12); details of relevant indicators are shown in Table 1. Mobility was assessed with the help of two sterilized dental instrument handles. All parameters were recorded in each patient chart and tooth mobility was recorded using the scoring system shown in Table 1. Experimental locations were monitored, at 1, 3, 7, 14, 28 and 48 days before and following administration of HGG/DX and HGY/DX, for the following: retention of film in the pocket, verbal assessment of patient comfort and enhancement of periodontal tissue regeneration according to Handy Dentist software interpretation of the X-ray images of patients.36
Table 1 Different pathophysiological indicators for periodontitis patients
| Indicator |
Score |
Definition |
| Gingival margin and gingival papilla. |
| Periodontal depth |
Slight |
>3 mm but <5 mm |
| Moderate |
>5 mm but <7 mm |
| Advanced |
>7 mm |
| Gingival index (GI) |
0 |
GMPa are healthy |
| 1 |
GMP mildly inflamed, no bleeding |
| 2 |
GMP mildly inflamed, changes in color, absence of edema, punctate hemorrhage |
| 3 |
GMP moderately inflamed, changes in color, mild edema, and bleeding in gingival crevice |
| 4 |
GMP severely inflamed, changes in color, severe edema, with blood flowing out of gingival crevice |
| 5 |
GMP severely inflamed, changes in color, severe edema, ulceration, and spontaneous bleeding with blood observed flowing out of gingival crevice |
| Mobility index (MI) |
0 |
Physiological mobility |
| 1 |
Slight mobility (buccal palatal) |
| 2 |
Moderate mobility (buccal and mesial-distal) |
| 3 |
Severe mobility (tooth moves in and out of the socket) |
2.10 Statistical analysis
The data were analyzed by one-way analysis of variance (ANOVA) to compare the means of all values by calculating the level of significance, p < 0.05, mean and standard error of the mean (<1.5). ANOVA was carried out by using Microsoft Excel 365 and IBM SPSS Statistics 25.
3 Results and discussion
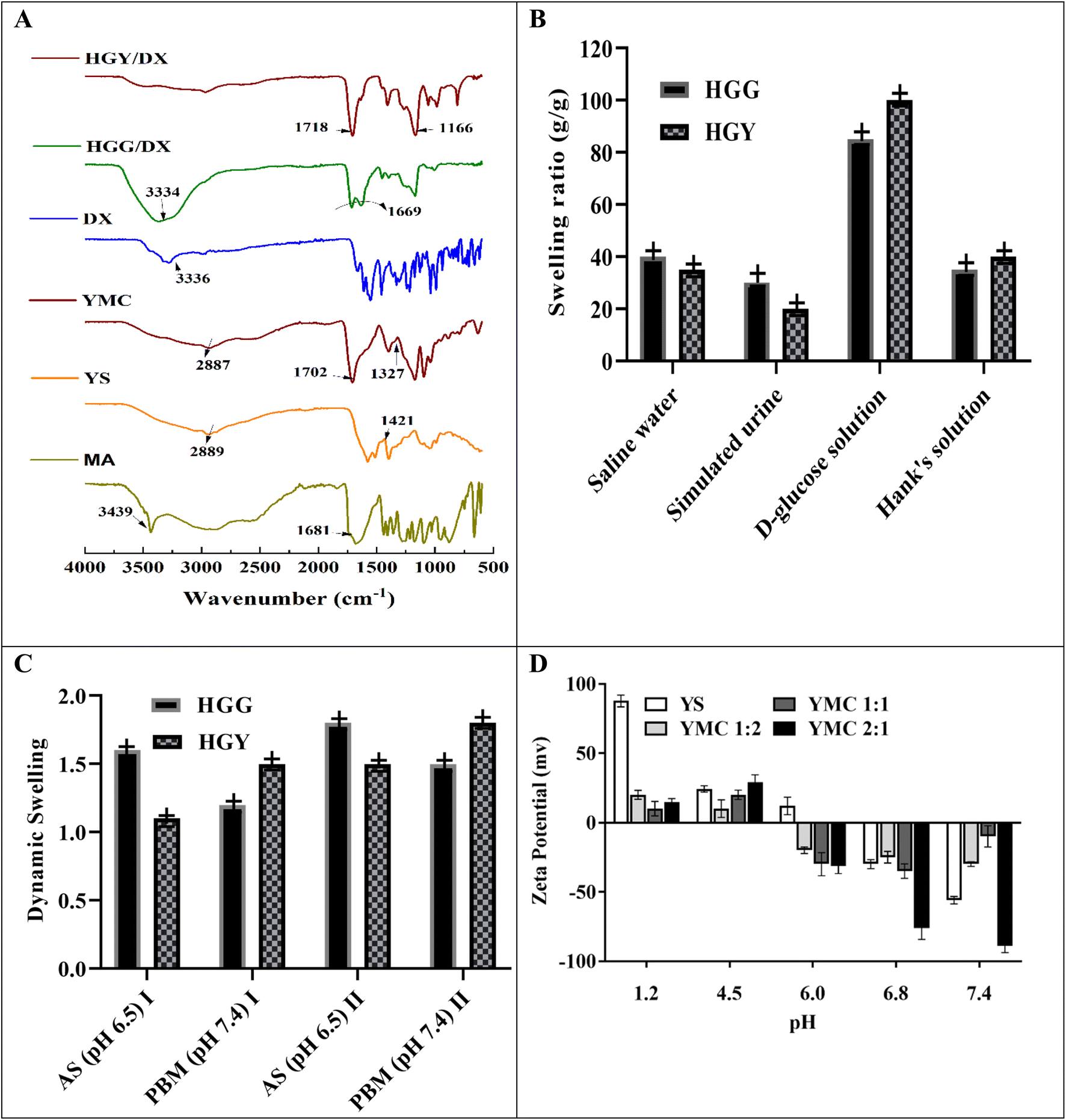
Periodontitis complications, like tooth mobility, gingivitis and tooth loss, can only be treated with a systemic high dose of DX, with various side effects like hypoplasia of the enamel, staining of teeth, gastrointestinal upset, and low patient compliance. The objective of the present study was to develop a controlled-release hydrogel containing DX for application in the periodontal pocket for the treatment of periodontitis. The semisynthetic crosslinker YMC was synthesized by thermochemical modification of yeast, and used to make HGY/DX. The rationale behind the use of YMC was less irritation, as evident from in vivo results. YMC not only improves the crosslinking density of HGY/DX, but also provides specific binding sites for DX to control its release, as clearly seen in Fig. 2(A). Similar types of interaction between DX and the carbonyl groups of YMC were also reported by Inan et al.37 DX bound to the carbonyl groups of malic acid in HGY/DX, which was responsible for a sustained-release effect up to 24 h, as shown in Fig. 4(D). The zeta potential of YMC was also negative at pH 6.8, which favors DX release from HGY/DX in the saliva of periodontitis patients. The negative charge over –COOH groups causes repulsion and loosens the hydrogel network, thus controlling DX release. DX loading was confirmed by FTIR spectroscopy, which showed that the 3344 cm−1 peak was suppressed in HGY/DX due to the chemical bonding of DX with COOH residues of malic acid. Similar studies of suppression of peaks were reported by Liu et al.38
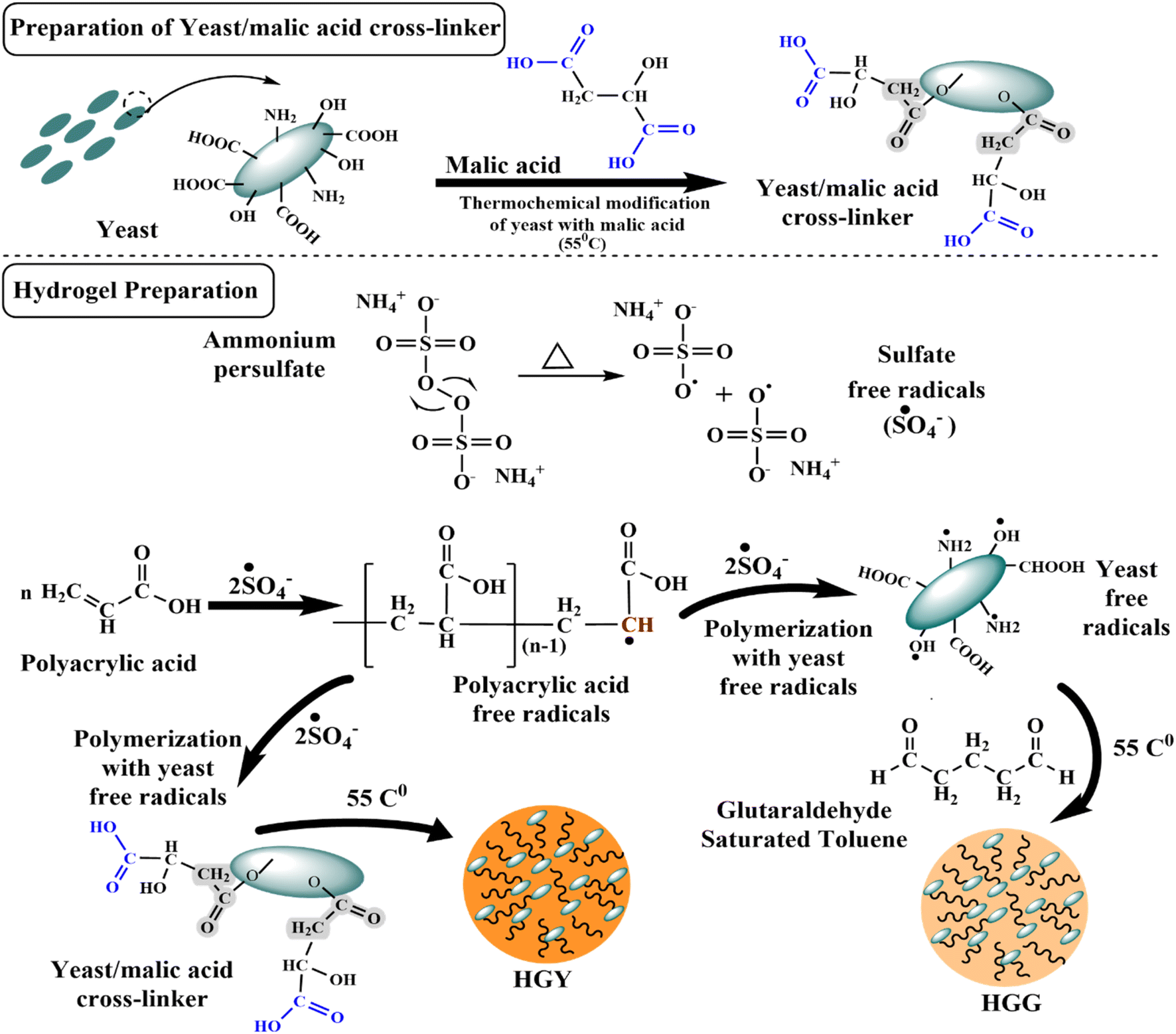
3.1 YMC and its confirmation
Fig. 1 illustrates the suggested mechanism for thermochemical modification-based preparation of the superabsorbent YMC composite, in which two –COOH groups of malic acid interacted with two free –OH groups of yeast to generate ester linkages via covalent bonding, with a third –OH group forming a hydrogen bond. During the second phase, several malic acid anhydrides were generated, which subsequently reacted with yeast –OH groups to build hollow pockets.39 The chemical transformation of yeast into YMC at room temperature (25 °C) was confirmed by FTIR spectroscopy in terms of the possible interaction between yeast and malic acid. The FTIR spectra shown in Fig. 2(A), ranging from 500–4000 cm−1, showed the characteristic transmission peaks of yeast as strong and broad bands at 2889 cm−1, 3306 cm−1 (OH and NH bending vibrations), 1647 cm−1 (amino bending vibration and carbonyl in amideI), 1548 cm−1 (NH2 in amideII), 1421 cm−1 (N in amideIII) and 1244 cm−1 (CO stretching vibration), and bands ranging from 1250 cm−1 to 850 cm−1 (cyano group, CN). As a result of the thermochemical process, the peak at 1421 cm−1 was moved to 1702 cm−1 (ester hydroxyl and carboxylic stretching vibrations), those at 2289 cm−1 and 1547 cm−1 were weakened (COOH), and one at 1722 cm−1 was strengthened (COOR).40
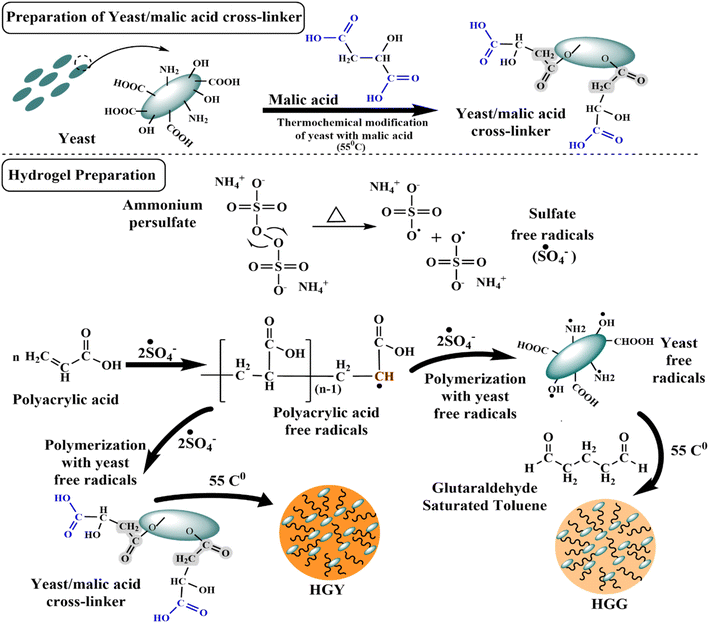 |
| | Fig. 1 Chemical scheme for the synthesis of the yeast–malic acid crosslinker (YMC) and hydrogels, i.e., HGG (glutaraldehyde hydrogel) and HGY (YMC hydrogel), using polyacrylic acid (PAA) and GST (glutaraldehyde saturated toluene) or YMC, respectively. | |
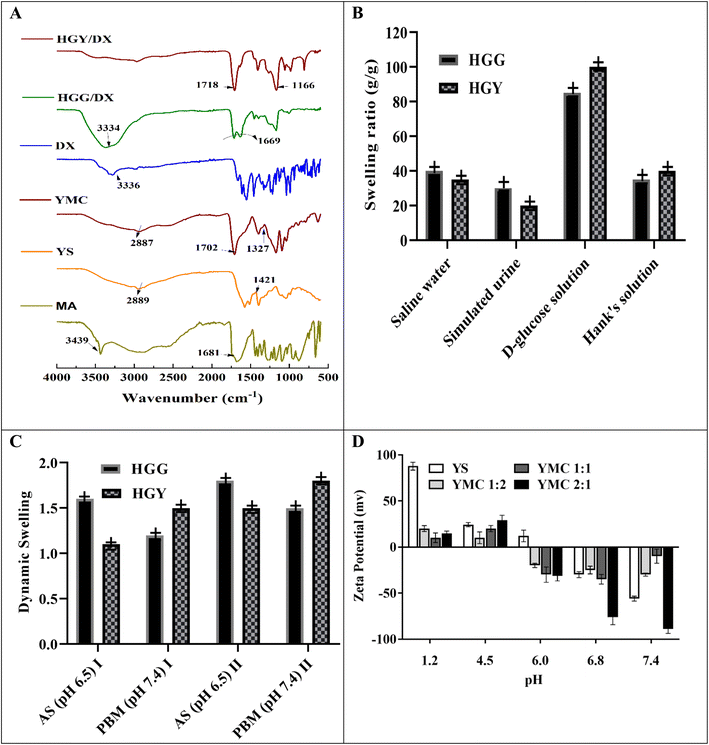 |
| | Fig. 2 (A) FTIR analysis of various components of the hydrogels (MA: malic acid, YS: yeast, YMC: yeast–malic acid crosslinker, DX: doxycycline); (B) swelling ratios of HGG and HGY in various physiological fluids; (C) dynamic swelling of HGG and HGY in AS (artificial saliva, pH 6.5) and PBM (phosphate buffer medium, pH 7.4), after (I) 60 min and (II) 120 min; (D) zeta potentials of YS (yeast), YMC 1:1, YMC 1:2 and YMC 2:1. | |
Table 2 illustrates that among all the crosslinkers YMC 1:1 had 0.75 times more carboxyl content due to the presence of a higher number of –COOH groups, with this increasing from 129 meq. per 100 g to 290 meq. per 100 g compared to yeast. However, as the temperature increased above 120 °C, the carboxyl content was reduced from 290 meq. per 100 g to 200 meq. per 100 g, which may be due to faster crosslinking that enabled dramatic additional crosslinking between two nearby free –COOH groups of YMC at higher temperature. Similar studies were reported by S.-D. Zhang et al.41 using yeast and citric acid, and showed that increased temperature effected a reduction in the carboxyl content. To further explore the reaction efficiency changes with temperature, a titration approach was used to measure the degree of substitution and degree of esterification between the –COOH groups of malic acid and –OH groups of yeast. YMC 1:1 had a 2 times greater degree of esterification and degree of substitution compared to YMC 1:2. With an increase in concentration of either yeast or malic acid, the degree of esterification and degree of substitution were decreased due to the steric hindrance of bulky –COOH functional groups. As the temperature rose up to 100–145 °C, this ultimately led to an increase in the degree of esterification and degree of substitution. Furthermore, the –OH groups of yeast were also significantly more reactive to the anhydride formed from malic acid.42
Table 2 Physicochemical evaluation of YMC, HGG and HGY, % DL and percentage of drug release at 24 h with various model-based approaches for in vitro drug-release parameters (K0 = rate constant, R2 = regression value, AIC = Akaike information criterion) of DX
| Characterization of YMCa |
| Yeast/YMC |
Carboxyl content (meq. per 100 g) |
Degree of esterification (%) |
Degree of substitution |
PZo (point of zero charge) |
| Yeast–malic acid composite. Ammonium persulfate. Drug loading. |
| Yeast |
129 ± 0.5 |
0 |
0 |
4.66 ± 0.2 |
| YMC 1:2 |
234 ± 0.5 |
20 ± 1.01 |
0.22 ± 0.01 |
3.95 ± 0.2 |
| YMC 1:1 |
290 ± 0.5 |
40 ± 1.01 |
0.39 ± 0.01 |
3.15 ± 0.2 |
| YMC 2:1 |
220 ± 0.5 |
25 ± 1.01 |
0.34 ± 0.01 |
3.85 ± 0.2 |
| Characterization of hydrogels |
HGG |
HGY |
| Cross linker |
GST (3.00 ± 0.02 μL) |
1.25 mL (5%) YMC |
| APSb (g) |
0.005 ± 0.001 |
0.005 ± 0.001 |
| Acid value (Av) |
0.45 ± 0.1 |
0.35 ± 0.1 |
| Carboxyl content (meq. per 100 g) |
139 ± 4 |
319 ± 5 |
| Cross-linking ratio (Mc, g mol−1) |
450 ± 10 |
360 ± 10 |
| Cross-linking density (mol m−3) |
3800 ± 20 |
4500 ± 20 |
| Viscous modulus (G′′) |
70 ± 2 |
150 ± 2 |
| Elastic modulus (G′) |
95 ± 5 |
160 ± 5 |
| Loss tangent (θ) |
0.35 ± 0.001 |
0.22 ± 0.001 |
| % DLc and in vitro drug release models |
HGG |
HGY |
| DL (%) |
45 ± 2 |
60 ± 2 |
| Power function model |
n |
0.543 ± 0.01 |
0.476 ± 0.01 |
| K × 103 |
76.8 ± 0.01 |
67.3 ± 0.01 |
| R2 |
0.994 ± 0.01 |
0.998 ± 0.01 |
| Schott kinetic model |
Ms |
25.223 ± 0.12 |
35.890 ± 0.12 |
| ks × 103 |
20.985 ± 1.5 |
29.002 ± 1.5 |
| R2 |
0.967 ± 0.01 |
0.984 ± 0.01 |
| Higuchi’s model |
Kh (h1) |
25.008 ± 0.15 |
27.799 ± 0.15 |
| R2 |
0.944 ± 0.01 |
0.929 ± 0.01 |
| AIC |
25.082 ± 1.5 |
29.986 ± 1.5 |
| Hixon–Crowell |
α |
0.002 ± 0.001 |
0.002 ± 0.001 |
| R2 |
0.999 ± 0.01 |
0.995 ± 0.01 |
| AIC |
12.854 ± 1.5 |
14.793 ± 1.5 |
| Korsmeyer–Peppas model |
K3 |
1.564 ± 0.01 |
1.124 ± 0.01 |
| R2 |
0.948 ± 0.01 |
0.997 ± 0.01 |
| AIC |
30.617 ± 1.5 |
26.432 ± 1.5 |
The zeta potentials of both yeast and semisynthetic YMC were used to evaluate the surface charge distribution and dependency of the surface characteristics. PZo is the pH value that effects net-zero charge on a solid surface in the absence of biosorption and is considered a surface index, i.e., how a surface charge changes as a function of pH. Similar findings published previously by Feng et al.9 were consistent with our results. The zeta potential of yeast was positive for acidic media, while it was negative for alkaline media, due to its negative surface charge and the zwitterion effect between COOH, OH and NH2. Meanwhile, the PZo values of YMC were found to be +3.95 ± 0.12, +3.15 ± 0.12 and +3.85 ± 0.12, respectively, in decreasing order: YMC 1:2 > YMC 2:1 > YMC 1:1.
Overall, these findings indicate that the thermochemical process used to synthesize YMC effectively increased the acidity and carboxyl content of the yeast, particularly at temperatures between 100–120 °C. This increase in acidity and carboxyl content is attributed to the formation of ester linkages between the malic acid and yeast surface groups, contributing to the unique properties of YMC as a crosslinker.
3.2 Hydrogel synthesis
Two types of hydrogel, i.e., HGG and HGY (with the optimal YMC 1:1), were synthesized by using free-radical polymerization of yeast with polyacrylic acid, as already reported by Das et al.43 Yeast was first activated and converted into its free-radical form in the presence of ammonium persulfate (APS), which generated free radicals for the initiation of radical formation in polyacrylic acid, as shown in Fig. 1. APS was used to initiate the polymerization reaction in an ultrasonic water bath to further boost the reaction kinetics.
In the first phase of hydrogel synthesis, polyacrylic acid and a crosslinker, either GST or YMC, were attached to the surface of yeast via hydrogen bonding. In the second phase, which involved heating in an ultrasonic water bath, APS was changed into free radicals of sulfate anions (˙SO4−). Next, the polyacrylic acid monomer reacted with the sulfate anion radicals to produce free radicals of polyacrylic acid. During the formation of polyacrylic acid free radicals, the sulfate anion radicals removed hydrogen species from the amino and carboxy groups to generate more reactive amino and alkyl hydroxy radicals. In the same way, yeast was also changed into free radicals by APS under ultrasonic conditions (temperature, 60 °C; frequency, 37 kHz; power, 100%). The polyacrylic acid free radicals could bind with the surface of yeast macroradicals to initiate chain growth on the surface of yeast macroradicals. During propagation of the reaction, the vinyl groups of both GST and YMC react simultaneously to make a spongy network hydrogel. The hollow cavities in HGG and HGY were due to the yeast, which retained two times more water than a normal hydrogel.44
The proposed polymerization network of yeast and polyacrylic acid was reinforced with GST and the semisynthetic YMC in HGG and HGY, respectively. HGG exhibited a light-yellow color, likely due to the chemical properties of glutaraldehyde.45 In contrast, HGY appeared golden, possibly due to the incorporation of malic acid-derived moieties. These color differences provide visual cues regarding the composition and crosslinking mechanisms, with potential implications for the treatment of periodontitis.
3.3 Characterization of hydrogels
As shown in Fig. 2(A), FTIR studies showed prominent ester peaks in the case HGY at 1718 cm−1. The spectra between 3500 to 3000 cm−1 confirmed that the –NH2, –OH and –COOH peaks of DX were suppressed in the case of HGY/DX, which indicated that DX was chemically bonded with the malic acid residues of HGY. Similar results were also explained by Inan et al.37 The SEM analysis results in Fig. 3A and B showed that HGG had a plainer surface as compared to HGY, which may be due to a more porous structure and high number of –COOH groups in HGY. The more porous and rougher surface of HGY/DX confirmed DX loading, which would thus cause controlled release of DX from inside the matrix of the hydrogel, as evidenced by Bertz et al.46 The SEM images of HGG/DX and HGY/DX shown in Fig. 3C and D confirmed the loading of DX not only on the surface of HGY/DX, but also incorporated into the matrix area.
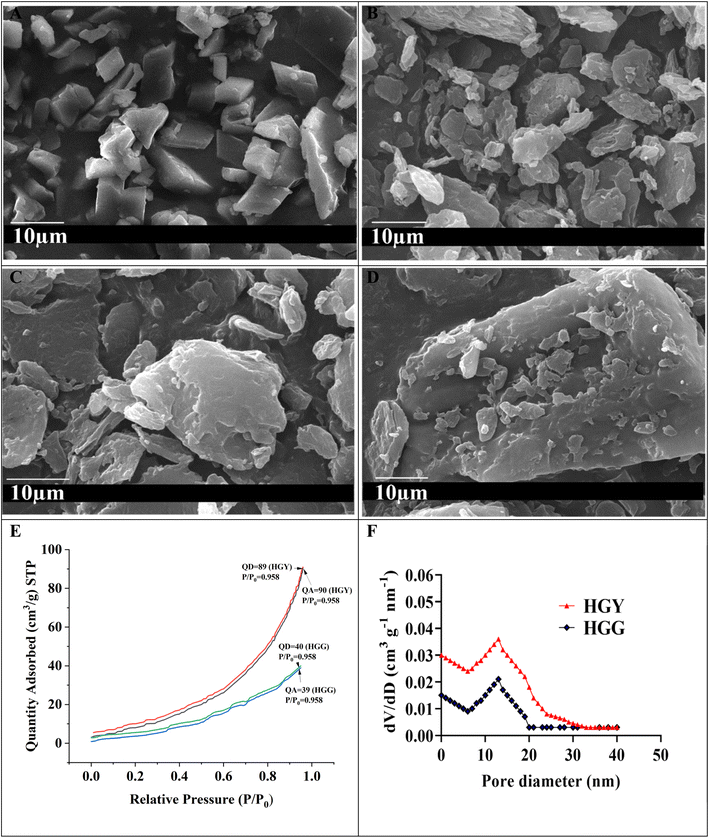 |
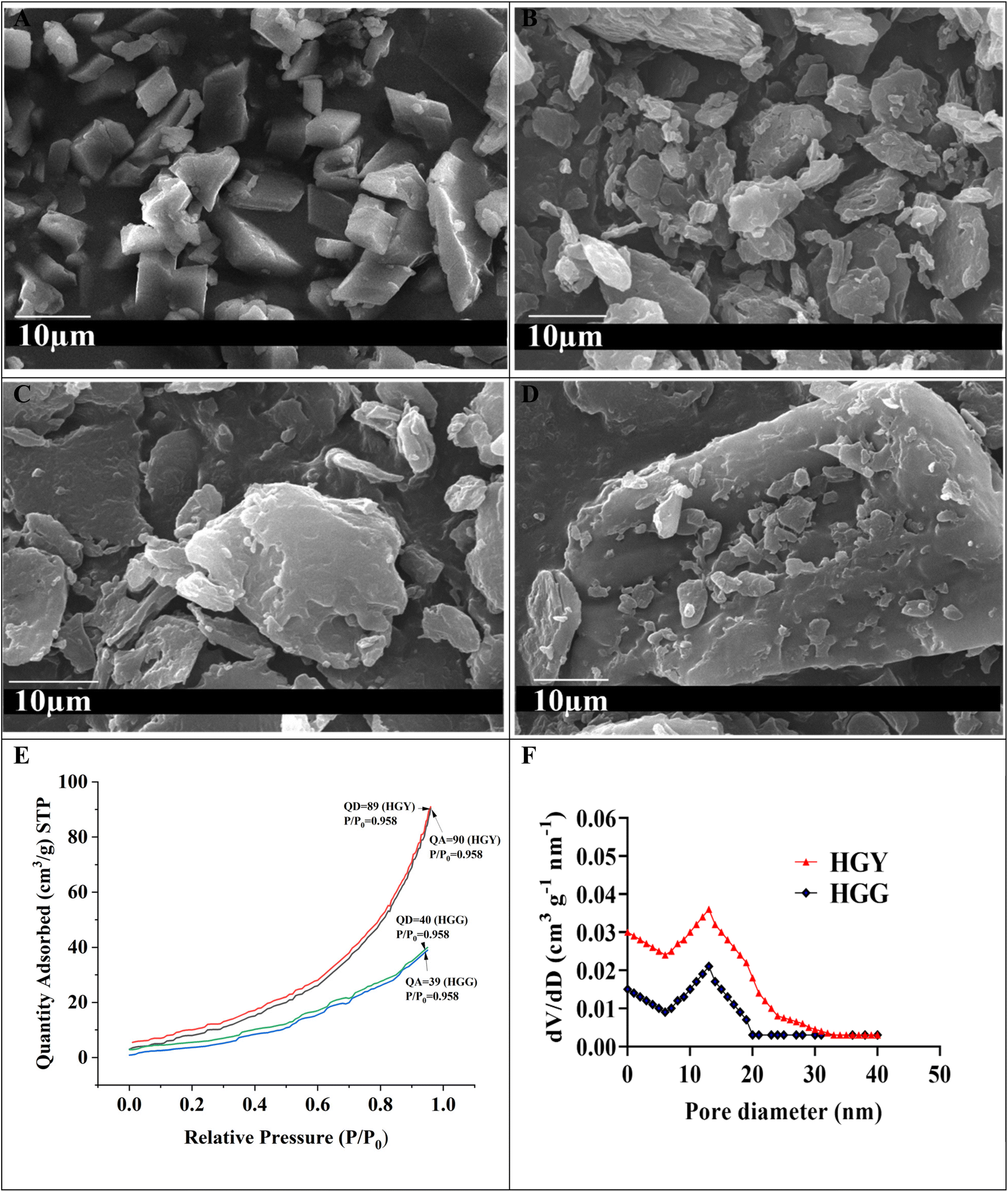
| | Fig. 3 (A and B) SEM results of HGG and HGY, respectively, showing that HGY had a more porous and rougher surface; (C and D) SEM images of HGG/DX and HGY/DX, respectively, showing that DX was loaded not only on the surface but also inside the matrix of HGY/DX; (E and F) BET analysis of HGG and HGY (QA: quantity adsorbed, QD: quantity desorbed). | |
The porous structure of HGY was also confirmed by BET, specific surface area (cm2 g−1), and pore volume (cm3 g−1) analysis. Fig. 3(E) shows type IV N2 desorption and N2 adsorption isotherms (relative pressure P/Po: 0 to 1) for HGG and HGY. The adsorption and desorption of N2 almost coincided due to the reversible adsorption of a monolayer of N2 in the case of HGY. The curve for HGY had significant hysteresis, which confirmed a more mesoporous structure of HGY as compared to HGG. The reason for this is that HGG had high GST loading and stronger linkages between the polyacrylic acid chains and yeast, allowing a more compact and plainer structure as compared to HGY. In the case of HGY, YMC had 2.5 times more crosslinking density as compared to GST, allowing a more porous structure of HGY as compared to HGG. HGY showed a broader distribution of pore sizes with a significant presence of both mesopores (2–50 nm) and possibly larger pores. The presence of larger pores can facilitate more significant drug loading, while the smaller mesopores can aid in sustained release of DX. HGG exhibits a narrower pore size distribution, suggesting fewer and more uniform pore sizes, primarily in the mesoporous range. HGG’s narrower distribution suggests a more uniform pore structure, which might limit its loading capacity and release dynamics compared to HGY. The pore size distributions (dV/dD: cm3 g−1 nm−1) of HGG and HGY were calculated using the BET data of adsorption and desorption of N2. According to Fig. 3(F), the pore size (nm) was mainly distributed around 39.544 ± 0.1 for HGY, as compared to 23.464 ± 0.1 for HGG, while the specific surface area was increased from 22.463 ± 2 m2 g−1 for HGG to 52.129 ± 2 m2 g−1 for HGY.
3.3.1 Swelling kinetics. The swelling capacities of HGG and HGY were correlated with the absorption effectiveness of polyacrylic acid and yeast, as shown in Fig. 2(B and C). Pseudo-first order and pseudo-second-order kinetic models were used with the assumption that the swelling rate is proportional to the number of pores in HGG and HGY. The previously reported yeast contained hollow pockets due to large hydrophilic groups, facilitating faster permeation of water into the hydrogel.47 As shown in Fig. 2(B), HGY was further evaluated for its swelling ratio in physiological solutions under simulated conditions and the results were in the order of D-glucose solution > saline water > Hank’s solution > simulated urine. The maximum swelling ratio (g g−1), i.e., 101 ± 2, was observed in D-glucose solution because it increases the charge intensity over the surface of the hydrogel, which thus takes more swelling medium as compared to in the other physiological solutions. The swelling ratio (g g−1) of HGY was 2.5 times greater than that of HGG due to the increase in the number of –COOH groups, which increased their ionization. 48 In periodontitis, the glucose concentration is slightly higher than normal. So, in diseased saliva, swelling would be significantly increased, thus promoting drug release in diseased saliva, as reported by Dabhi et al.49
3.3.2 Acid value and carboxyl content. The acid value and carboxyl content are critical parameters for the swelling and pH-dependant characteristics of hydrogels. The acid value was determined via a simple acid–base titration of –COOH groups. According to Table 2, the acid values of HGG and HGY were 0.45 and 0.35, while the carboxyl contents (meq. per 100 g) were 139 and 319, respectively, indicating an inverse correlation of free acids and carboxyl content, due to free –OH groups being linked with –COOH groups in the case of HGY.50
3.3.3 Viscosity and crosslinking properties. The order of viscosity parameters was as follows: for the viscous modulus (G′′) HGY > HGG, and for the ratio of the viscous modulus to elastic modulus (θ) HGG > HGY, as shown in Table 2. The effects of pH on the viscosity of HGG and HGY were also measured in normal saliva and diseased saliva, as shown in Fig. 4(A and B). Diseased saliva (pH 5–6) is more acidic due to the presence of bacterial toxins, bacterial strains and more mucins. The viscosity of HGY was increased 1.24 times as compared to that of HGG, as mucin forms strong interactions with YMC residues. The viscosity parameter was also critical for patient compliance and drug-release scenarios. The greater the dynamic viscosity (Pa s), the more controlled the released of DX. In diseased saliva, the dynamic viscosity (Pa s) was increased due to the increased cross-linking density of HGY/DX and low pH of diseased saliva, as shown in Fig. 4(A and B).51
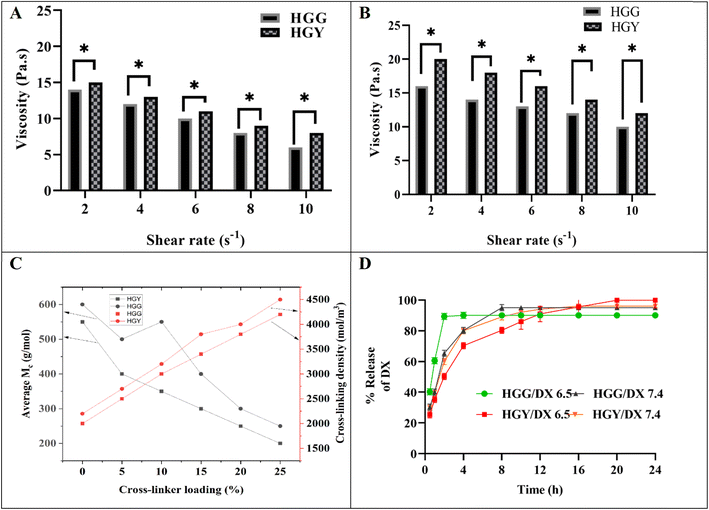 |
| | Fig. 4 (A and B) Viscosity (Pa s) of HGG and HGY in simulated saliva with pH 7 ± 0.22 and simulated diseased saliva with pH 6.24 ± 0.25, respectively. (C) Cross-linking ratio (g mol−1) with reference to the cross-linking density (mol m−3) of HGG or HGY, and (D) DX release from HGG/DX and HGY/DX at pH 6.5 and 7.4. | |
The determination of the crosslinking ratio and crosslinking density was essential for selecting the appropriate crosslinking agent, and was carried out using the Flory–Rehner equation. The previously calculated equilibrium swelling ratios (g g−1) of both HGG and HGY were used to determine the crosslinking density. The effect of % crosslinker loading on crosslinking ratio (Mc) is shown in Fig. 4(C); HGG has 600 g mol−1 while HGY has 500 g mol−1 respectively. An increase in crosslinker loading from 0 to 25% led to a decrease in the crosslinking ratio. This phenomenon was more evident in HGY as compared to HGG and can be attributed to the greater stability, swelling, and efficacy of the YMC crosslinker in HGY. YMC made the matrix of HGY more consistent, adhesive, and porous, resulting in an increased cross-linking density as compared to HGG.
3.4 Ex vivo mucoadhesion strength and time
The mucoadhesive strength of HGY containing semisynthetic YMC was 0.8 ± 0.05 N, as compared to 0.42 ± 0.05 N for HGG, due to presence and linking of –COOH groups with the cysteine residues of the buccal mucin. Furthermore, dipole–dipole interactions of mucin and –COOH, with the addition of hydrogen bonding, were also considered as a source of the of higher mucoadhesive strength of HGY. The results indicated that HGY demonstrated 2 times longer mucoadhesion time compared to HGG, even after 8 h, as depicted in Fig. 5(B). In HGG, mucoadhesion primarily relied on the inherent activity of –OH groups, which provided weaker van der Waals forces.52,53
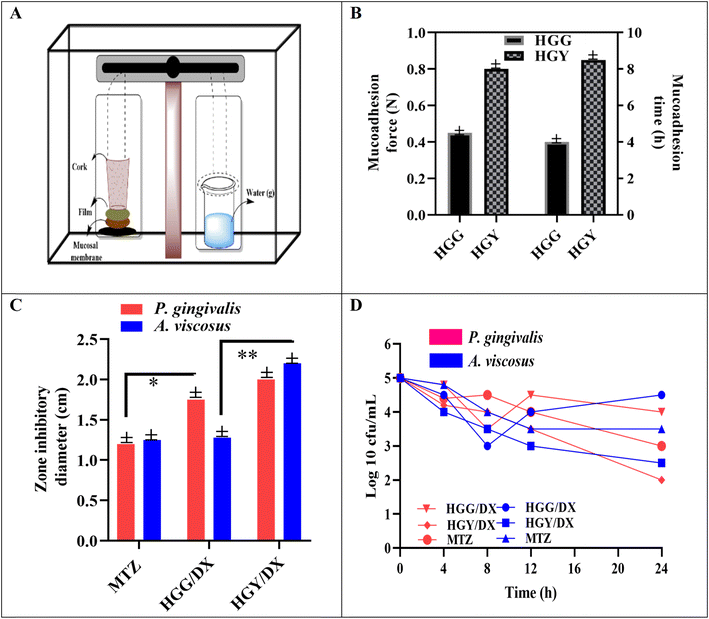 |
| | Fig. 5 (A) Modified physical balance and (B) mucoadhesion force (N) along with mucoadhesion time (h) of HGG and HGY. (C) Antimicrobial zone of inhibition and (D) time kill assay of HGG/DX and HGY/DX. | |
3.5 % DX loading and release
After dissolving DX in an aqueous solution with ascorbic acid as an antioxidant, the % DL of HGG and HGY was calculated. The % loading efficiencies of HGG and HGY at equilibrium were observed to be 45 ± 2 and 60 ± 2% for HGG and HGY, respectively, showing direct relation between the swelling ratio and drug loading, which may be due to the lower pKa value of polyacrylic acid at pH 7. The % release of DX from HGG/DX and HGY/DX in simulated artificial saliva (pH 7.4) and diseased saliva (pH 6.5) is shown in Fig. 4(D). More than 80% of the DX in HGG/DX was released in 4 h, while in the case of HGY/DX, controlled release of DX was observed, i.e., 30% in 3 h and more than 90% in 24 h. The phenomenon of abrupt release was more dominant in HGG/DX as compared to HGY/DX, which may be due to the chemical interaction of DX with newly prepared YMC even at the surface of HGY.
The obtained dynamic % release data was applied to various kinetic models. As shown in Table 2, value of “n” lies between 0.45–0.55 for the power function model, indicating a non-Fickian release pattern that depends upon the swelling and deswelling of HGG/DX and HGY/DX.27 The Schott kinetic model and Higuchi’s model suggest solvent permeation and diffusion-controlled release for HGY/DX as compared to HGG/DX, respectively. The Korsmeyer–Peppas model describes the release of the drug from the polymer matrix. The AIC value of less than 30 in the case of HGY/DX showed that the release is diffusion permeation dependent. The higher surface area and pore volume of HGY/DX than HGG/DX, as evident from BET analysis, promotes more permeation of solvent (at pH 6.5 and 7.4) and thus facilitates controlled DX release. In periodontitis, salivary pH is slightly acidic, which promotes DX release from HGY/DX due to its higher viscosity as compared to HGG/DX. For HGG/DX, the abrupt release of DX was better explained by a modified Korsmeyer–Peppas model with the addition of “β”, which is a burst effect that was negligible in the case of HGY/DX. The Hixson–Crowell model suggests that the release rate is influenced by the erosion of the hydrogel matrix, which changes the surface area for both HGG/DX and HGY/DX. HGY/DX exhibited superior swelling properties compared to HGG/DX, as evidenced by a higher swelling ratio (g g−1). This increased swelling leads to a greater change in surface area, which enhances the controlled release of DX for HGY/DX. Firstly, the burst release of DX within 3 h was due to DX present on the surface of HGY/DX. Secondly, DX release was controlled for 24 h due to diffusion of DX inside the matrix of HGY/DX through a solvent exchange mechanism. The regression values obtained from the release-data plots were 0.949 and 0.962 for HGG/DX and HGY/DX, respectively.54 The controlled release over 24 h with a reduced burst effect enhances patient compliance and improves therapeutic outcomes in the case of HGY/DX.
3.6 Antimicrobial assay
The antimicrobial activity of HGG/DX and HGY/DX was analyzed via the zone of inhibition and time kill assay under ISO 5 (particle count <1) in the Drug Testing Laboratory, Multan, Punjab. The activities of a standard (MTZ), HGG/DX and HGY/DX were studied in PBM (pH 6.0–8.0). The zone of inhibition against P. gingivalis calculated after 48 h for HGG/DX was 1.9 ± 0.08 mm and that for HGY/DX was 2.2 ± 0.08 mm, as compared to MTZ (1.25 ± 0.08 mm). Furthermore, in the case of A. viscosus, HGY/DX showed remarkably increased inhibition (2.25 ± 0.08 mm) as compared to HGG/DX (1.35 ± 0.08 mm). This significant decrease in growth of A. viscosus may be due to the effect of a high loading of vitamin C in HGY/DX, which not only prevented oxidation of DX but also gave a synergistic antibacterial effect with DX.55 De Francesco et al. also reported similar synergistic behavior of vitamin C and DX.55 This was in line with earlier studies that showed the benefits of combination therapy and how their synergistic effects can increase the effectiveness of antibiotics. The hypothesis that combining medications can lead to more effective bacterial growth prevention (e.g., high vitamin C loading in HGY/DX) is supported by this consistency.56 Findings from Tada et al. were also supported by the observed synergistic impact of ascorbic acid and dextran, indicating that ascorbic acid can augment the efficacy of antimicrobial drugs.57 The sustained release effect of DX in HGY/DX led to a long-term decline in bacterial colonies, which is consistent with earlier studies emphasizing the value of sustained release formulations in antimicrobial therapy.58 These long-lasting impacts may help prolong the duration of therapeutic concentrations in the periodontal pocket, improving the efficacy of therapy.
Fig. 5(D) shows the time kill assay of HGG/DX and HGY/DX, where colonies were counted over specific time intervals (4, 8, 12, 16, 20 and 24 h). HGG/DX showed a reduction in colonies from 5(log10) CFU mL−1 to 3.2(log10) CFU mL−1, while HGY/DX showed a remarkable reduction in colonies over the entire period, from 5(log10) CFU mL−1 to 2(log10) CFU mL−1 as compared to MTZ, i.e., 3.2(log10) CFU mL−1. In the case of HGG/DX, the maximum killing of 45% was observed in the first 8 h for both P. gingivalis and A. viscosus, with a slightly greater susceptibility shown by the A. viscosus species. However, in the case of HGY/DX, 40% killing was observed in the first 8 h and the remainder were killed in the next 24 h due to the sustained released effect of HGY/DX as compared to the control.
3.7 In vivo clinical study
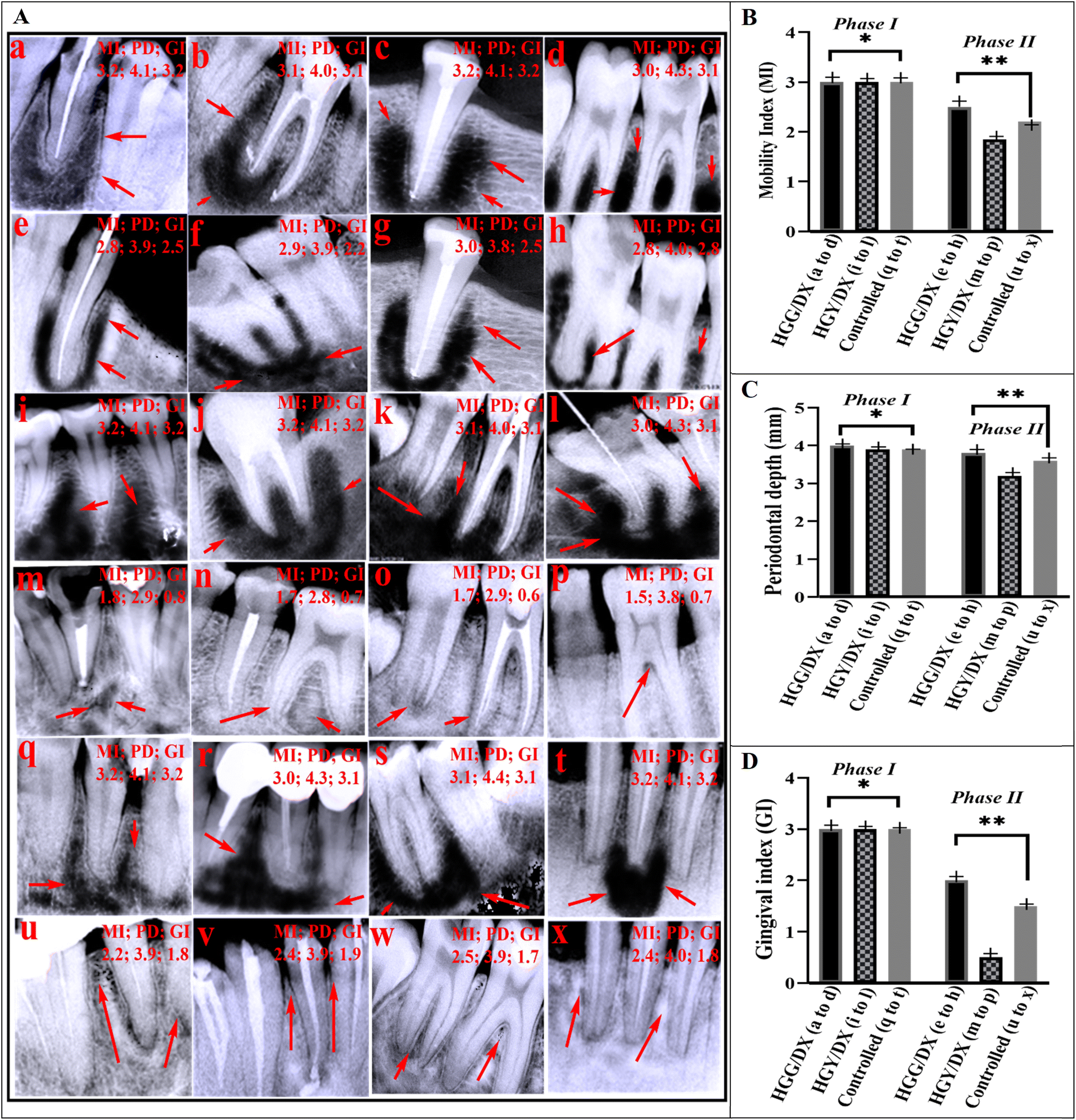
A total of 18 patients (8 females and 10 males; mean age, 30.38 ± 10.12 years) accounting for 232 teeth were included in our analysis. Excluded patients, which were four in number, had missing information (mostly systemic conditions) at the baseline, and had comorbidity. All included patients had follow-up treatment every month up to 6 months to determine the efficacy of the HGG/DX and HGY hydrogels. Fig. 6A(a–d) display the phase I X-ray images of patients treated with HGG/DX, and Fig. 6A(e–h) display the phase II X-ray images following therapy with HGG/DX. In Fig. 6A(i–l), the HGY/DX-treated group’s X-ray images (phase I) are shown, and in (m–p), the X-ray images after treatment (phase II) of the HGY/DX group are shown. The demineralization of tooth bone, which eventually leads to an increase in the MI and GI, was evident in the dark regions of the X-ray pictures. When these dark patches were replaced with white ones, the dental bone had begun to mineralize and repair, which eventually decreased the MI and GI.59 The phenomenon was more prominent in the HGY/DX group. Following the indicators in Table 1, the clinical findings for HGG/DX-treated groups showed a decrease in periodontal depth to 3.9 ± 0.1 mm, with no promising improvement, as compared to HGY/DX, which showed a decrease to 3.2 ± 0.1 mm. GI was improved from 3 (bleeding) to 1 (mildly inflamed), and MI was improved from 3 (severe mobility) to 2 (moderate mobility) in the case of HGY/DX, as compared to the control. The MI was not significantly improved in the case of HGG/DX due to the presence of GST, which was used as a cross-linker and caused irritation to the gingiva.
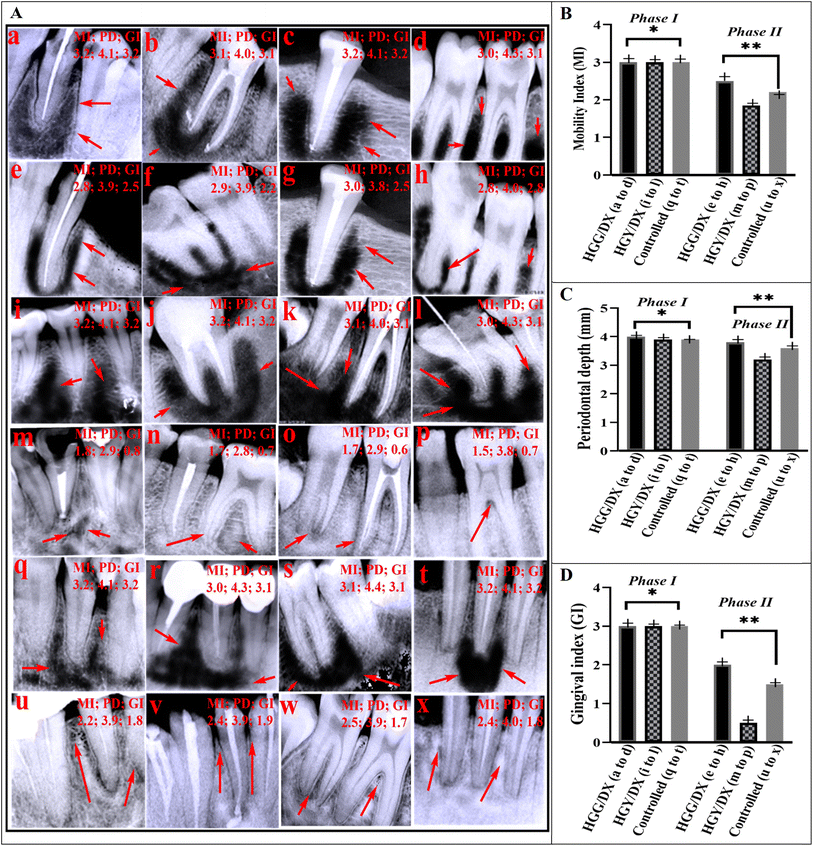 |
| | Fig. 6 (A) X-ray images of periodontitis patients. Images a–d, i–l, and q–t show phase I (before treatment) X-rays for HGG/DX, HGY/DX, and control groups, respectively. Images e–h, m–p, and u–x show phase II (after treatment) X-rays for HGG/DX, HGY/DX, and control groups, respectively (Handy Dentist; 3.1). (B–D) Data on the mobility index (MI), periodontal depth (mm), and gingival index (GI), respectively (*SDI (standard deviation index) < 1.25, **p < 0.05). | |
The current study used a cohort of 18 patients with a total of 232 teeth over a 6-month period to evaluate the in vivo clinical efficacy of HGG/DX and HGY/DX hydrogels in periodontal treatment. By comparing outcomes with a control group, statistical procedures such as analysis of variance (ANOVA) and p-values were used to interpret the results. The clinical results of the three groups (HGY/DX, HGG/DX, and control group) were compared using ANOVA. This statistical method allows for an evaluation of the variability both inside and across groups. ANOVA has been used in periodontal research in prior studies to determine important variations in treatment effects between different therapies.49 To assess the statistical significance of the observed results, it is essential to evaluate p-values. A low p-value (usually < 0.05) for both HGG/DX and HGY/DX suggested that it was improbable that the differences observed were the result of pure chance. Similar significance criteria have frequently been used in previous periodontal research to assess treatment outcomes.60 There were statistically significant improvements in GI and decreases in periodontal depth that were seen in the HGY/DX group.51 The utilization of ANOVA and p-values in this investigation enhances the statistical evaluation of the clinical effectiveness of HGY/DX and HGG/DX hydrogels in treating periodontal disease.61
4 Conclusion
In this study, thermochemically modified YMC resulted in a highly mucoadhesive conjugate of yeast that allowed controlled DX release. The optimized YMC 1:1 was used for the synthesis of HGY. The hydrogels HGG and HGY were cut into films and loaded with DX and the release profiles were investigated. Ex-vivo models were developed for the determination of the mucoadhesive strength and mucoadhesion time of HGG and HGY. For establishment of their antibacterial effect, zones of inhibition were determined and time kill assays carried out. After optimizing the formulation of HGG/DX and HGY/DX, they were used on human volunteers with periodontitis and the clinical outputs were observed with the help of X-ray radiographic techniques. Controlled release and less irritation were noted with HGY/DX in clinical outputs. Conclusively, by using the HGY/DX film in periodontitis, the dose of DX will be reduced, improving patient compliance and regeneration of dental tissue. Further optimization of the hydrogel formulation, more exploration of the potential of YMC, extensive biocompatibility studies to ensure safety in humans, and studies of combination therapies should be explored. Clinical trials of periodontitis could be conducted to evaluate the advantages and disadvantages of the proposed HGY/DX.
Ethical statement
Animal guidelines were implemented according to ARRIVE animal guidelines and Animal Ethical Committee of Bahauddin Zakariya University, Multan. The experimental analysis was carried out in compliance with the current Declaration of Helsinki laws and the International Ethical Guidelines for Biomedical Research. The ethical committee of Bahauddin Zakariya University in Multan authorized all protocols, and all human subjects provided informed consent.
Data availability
The data that support the findings of this study are available on request from the corresponding author, i.e., Muhammad Hanif.
Author contributions
Muhammad Hanif was the supervisor of all this research. Khalid Mahmood and Muhammad Asmatullah were the suppliers of chemicals and reagents. The writer, reviewer, and critical analyzer was Muhammad Qaiser. Muhammad Qaiser and Hafiz Muhammad Usman Abid were the analysts that performed the analysis of the formulation and in vivo activities. The patient clinical study was conducted by Muhammad Aqeel and Muhammad Qaiser in the Nishtar Institute of Dentistry. Dure Shahwar was the scientist who developed the chemical reaction. Syed Waqas Bukhari and Nabeela Ameer helped with manuscript writing. Fazal Rahman helped in manuscript writing.
Conflicts of interest
No potential conflict of interest was reported by the authors.
Acknowledgements
The researchers fully acknowledge the Higher Education Commission of Pakistan for this research project. The authors also aacknowledge the Drugs Testing Laboratory, Multan, for their support.
References
- R. G. Fischer, R. Lira Junior, B. Retamal-Valdes, L. C. D. Figueiredo, Z. Malheiros, B. Stewart and M. Feres, Braz. Oral Res., 2020, 34 DOI:10.1590/1807-3107bor-2020.vol34.0026
 .
. - P. M. Preshaw and S. M. Bissett, Endocrinol. Metab. Clin. North Am., 2013, 42, 849–867 CrossRef PubMed .
- E. F. Ruiz and A. B. Martínez, Avances en Periodoncia e Implantología Oral, 2005, 17, 111–118 Search PubMed .
- M. Madi, V. Pavlic, W. Samy and A. Alagl, Acta Odontol. Scand., 2018, 76, 71–76 CrossRef CAS PubMed .
- S. A. Gai and K. D. Wittrup, Curr. Opin. Struct. Biol., 2007, 17, 467–473 CrossRef CAS PubMed .
- E. T. Boder and K. D. Wittrup, Nat. Biotechnol., 1997, 15, 553–557 CrossRef CAS PubMed .
- S. Yucel, Z. O. Ozdemir, C. Kesgin, P. Terzioglu, S. Unlu, Y. Erdogan and K. Pusat, Int. J. Med. Health Sci., 2013, 7, 232–235 Search PubMed .
- S. Li, S. Dong, W. Xu, S. Tu, L. Yan, C. Zhao, J. Ding and X. Chen, Adv. Sci., 2018, 5, 1700527 CrossRef PubMed .
- D. Feng, B. Bai, H. Wang and Y. Suo, RSC Adv., 2015, 5, 104756–104768 RSC .
- K. Cheewatanakornkool, S. Niratisai and P. Sriamornsak, Asian J. Pharm. Sci., 2016, 11, 124–125 CrossRef .
- M. Elomaa, T. Asplund, P. Soininen, R. Laatikainen, S. Peltonen, S. Hyvärinen and A. Urtti, Carbohydr. Polym., 2004, 57, 261–267 CrossRef CAS .
- B. Dutta, S. Checker, K. Barick, H. Salunke, V. Gota and P. Hassan, J. Alloys Compd., 2021, 883, 160950 CrossRef CAS .
- D. Feng, B. Bai, C. Ding, H. Wang and Y. Suo, Ind. Eng. Chem. Res., 2014, 53, 12760–12769 CrossRef CAS .
- S. Majeed, M. Qaiser, D. Shahwar, K. Mahmood, N. Ahmed, M. Hanif, G. Abbas, M. H. Shoaib, N. Ameer and M. Khalid, RSC Adv., 2023, 13, 21521–21536 RSC .
- K. Wang, S. Z. Fu, Y. C. Gu, X. Xu, P. W. Dong, G. Guo, X. Zhao, Y. Q. Wei and Z. Y. Qian, Polym. Degrad. Stab., 2009, 94, 730–737 CrossRef CAS .
- B. R. Shmaefsky, Am. Biol. Teach., 1990, 52, 170–172 CrossRef .
- M. A. Abureesh, A. A. Oladipo and M. Gazi, Int. J. Biol. Macromol., 2016, 90, 75–80 CrossRef CAS PubMed .
- Y. Zhao, J. Kang and T. Tan, Polymer, 2006, 47, 7702–7710 CrossRef CAS .
- E. M. Krop, M. M. Hetherington, M. Holmes, S. Miquel and A. Sarkar, Food Hydrocolloids, 2019, 88, 101–113 CrossRef CAS .
- H. Park, X. Guo, J. S. Temenoff, Y. Tabata, A. I. Caplan, F. K. Kasper and A. G. Mikos, Biomacromolecules, 2009, 10, 541–546 CrossRef CAS PubMed .
- J. Mudassir and N. M. Ranjha, J. Polym. Res., 2008, 15, 195–203 CrossRef CAS .
- T. S. Rani, M. Subha, G. Venkata Reddy, Y. H. Kim and Y. S. Ahn, J. Appl. Polym. Sci., 2010, 115, 1675–1679 CrossRef CAS .
- R. Wang, X. Zhang, J. Zhu, J. Bai, L. Gao, S. Liu and T. Jiao, Colloids Surf., A, 2020, 598, 124860 CrossRef CAS .
- M. Hanif, N. Ameer, H. Akram, K. Mahmood, S. Bano, M. Qaiser, G. Abbas and H. M. A. Rahman, Polym. Bull., 2023, 80, 9833–9851 CrossRef CAS .
- A. Shokuhfar and B. Arab, J. Mol. Model., 2013, 19, 3719–3731 CrossRef CAS PubMed .
- G. Hoti, F. Caldera, C. Cecone, A. Rubin Pedrazzo, A. Anceschi, S. L. Appleton, Y. Khazaei Monfared and F. Trotta, Materials, 2021, 14, 478 CrossRef CAS PubMed .
- K. Anbu, T. More and A. Kumar, Indian J. Anim. Sci., 2003, 73, 1307–1311 CAS .
- N. P. Du Sert, A. Ahluwalia, S. Alam, M. T. Avey, M. Baker, W. J. Browne, A. Clark, I. C. Cuthill, U. Dirnagl and M. Emerson, PLoS Biol., 2020, 18, e3000411 CrossRef PubMed .
- P. Venkatesan, R. Manavalan and K. Valliappan, J. Basic Clin. Pharm., 2011, 2, 159 CAS .
- K. Ramteke, P. Dighe, A. Kharat and S. V. Patil, Scholars Acad. J. Pharm., 2014, 3, 388–396 Search PubMed .
- R. H. Moghaddam, S. Dadfarnia, A. M. H. Shabani, R. Amraei and Z. H. Moghaddam, Int. J. Biol. Macromol., 2020, 154, 962–973 CrossRef PubMed .
- D. Dey, S. Ghosh, R. Ray and B. Hazra, Phytother. Res., 2016, 30, 272–282 CrossRef CAS PubMed .
- T. A. V. Pham and T. T. P. Tran, J. Oral Biol. Craniofac. Res., 2023, 13, 332–336 CrossRef PubMed .
- M. D. Goodyear, K. Krleza-Jeric and T. Lemmens, BMJ, 2007, 335, 624–625 CrossRef .
- L. P. Nijhawan, M. D. Janodia, B. Muddukrishna, K. M. Bhat, K. L. Bairy, N. Udupa and P. B. Musmade, J. Adv. Pharm. Technol. Res., 2013, 4, 134–140 CrossRef PubMed .
- R. K. Agarwal, D. H. Robinson, G. I. Maze and R. A. Reinhardt, J. Controlled Release, 1993, 23, 137–146 CrossRef CAS .
- T. Inan, D. Dalgakiran, O. Kurkcuoglu and F. S. Güner, J. Polym. Res., 2021, 28, 408 CrossRef CAS .
- M. Liu, Z. Zhao and W. Yu, Chem. Eng. J., 2020, 393, 124748 CrossRef CAS .
- L. Rivera-Tarazona, V. Bhat, H. Kim, Z. Campbell and T. Ware, Sci. Adv., 2020, 6, eaax8582 CrossRef CAS PubMed .
- E. Burattini, M. Cavagna, R. Dell’Anna, F. M. Campeggi, F. Monti, F. Rossi and S. Torriani, Vib. Spectrosc., 2008, 47, 139–147 CrossRef CAS .
- S.-D. Zhang, Y.-R. Zhang, H.-X. Huang, B.-Y. Yan, X. Zhang and Y. Tang, J. Polym. Res., 2010, 17, 43–51 CrossRef CAS .
- A. Bendahou, A. Hajlane, A. Dufresne, S. Boufi and H. Kaddami, Res. Chem. Intermed., 2015, 41, 4293–4310 CrossRef CAS .
- D. Das, T. T. H. Pham and I. Noh, Colloids Surf., B, 2018, 170, 64–75 CrossRef CAS PubMed .
- E. Jabari and S. Nouzari, Iran. Polym. J., 1999, 8, 263–270 Search PubMed .
- M. S. Arshad, M. Qaiser, K. Mahmood, M. H. Shoaib, N. Ameer, N. Ramzan, M. Hanif and G. Abbas, Int. J. Biol. Macromol., 2022, 212, 314–323 CrossRef CAS .
- A. Bertz, S. Wöhl-Bruhn, S. Miethe, B. Tiersch, J. Koetz, M. Hust, H. Bunjes and H. Menzel, J. Biotechnol., 2013, 163, 243–249 CrossRef CAS .
- D. Feng, B. Bai, H. Wang and Y. Suo, New J. Chem., 2016, 40, 3350–3362 RSC .
- L. Li, J. Guo and R. Xiong, Polym. Test., 2021, 94, 106982 CrossRef CAS .
- M. R. Dabhi, S. A. Nagori, M. C. Gohel, R. K. Parikh and N. R. Sheth, Drug Delivery, 2010, 17, 520–531 CrossRef CAS PubMed .
- N. Nagata, T. Nakahara and T. Tabuchi, Biosci., Biotechnol., Biochem., 1993, 57, 638–642 CrossRef CAS .
- L. W. S. Cheong, P. W. S. Heng and L. F. Wong, Pharm. Res., 1992, 9, 1510–1514 CrossRef CAS .
- E. Cevher, D. Sensoy, M. A. Taha and A. Araman, AAPS PharmSciTech, 2008, 9, 953–965 CrossRef CAS PubMed .
- D. Rahmat, D. Sakloetsakun, G. Shahnaz, G. Perera, R. Kaindl and A. Bernkop-Schnürch, Int. J. Pharm., 2011, 411, 10–17 CrossRef CAS PubMed .
- J. R. Weiser and W. M. Saltzman, J. Controlled Release, 2014, 190, 664–673 CrossRef CAS PubMed .
- E. M. De Francesco, G. Bonuccelli, M. Maggiolini, F. Sotgia and M. P. Lisanti, Oncotarget, 2017, 8, 67269 CrossRef PubMed .
- A. Goc, A. Niedzwiecki and M. Rath, Int. J. Biol. Sci., 2016, 12, 1093 CrossRef CAS PubMed .
- A. Tada and H. Miura, Int. J. Environ. Res. Public Health, 2019, 16, 2472 CrossRef CAS .
- Q. Dai, R. Jia, H. Li, J. Yang and Z. Qin, ACS Sustain. Chem. Eng., 2024, 12, 1388–1404 CrossRef CAS .
- T. S. Kim, C. Obst, S. Zehaczek and C. Geenen, J. Periodontol., 2008, 79, 1141–1149 CrossRef PubMed .
- A. Dentino, S. Lee, J. Mailhot and A. F. Hefti, Periodontology, 2000, 2013(61), 16–53 Search PubMed .
- F. O. Correa, D. Gonçalves, C. M. Figueredo, A. S. Bastos, A. Gustafsson and S. R. Orrico, J. Clin. Periodontol., 2010, 37, 53–58 CrossRef CAS .
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *c,
Muhammad Hanif
*c,
Muhammad Hanif