
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAir-through-precursor suction-augmented replica molding for fabrication of anisotropic microparticles in gas-impermeable molds†
Seok Joon Mun,
Wookyoung Jang ,
Jun Hee Choi,
Yong Jun Lim and
Ki Wan Bong*
,
Jun Hee Choi,
Yong Jun Lim and
Ki Wan Bong*
Department of Chemical and Biological Engineering, Korea University, Seoul 02841, Republic of Korea. E-mail: bong98@korea.ac.kr
First published on 12th August 2024
Abstract
Replica molding (REM) is a powerful technique for fabricating anisotropic microparticles. Current REM methods rely on the use of gas-permeable molds for defect-free castings and facile particle recovery. However, they often encounter limitations on either technical accessibility or producible particle diversity. While the use of gas-impermeable molds presents a promising solution to these challenges, particle production within such molds necessitates addressing two critical issues: precursor loading and particle recovery. This study introduces a REM methodology specifically tailored to enable the production of anisotropic microparticles within gas-impermeable molds. To address the issue of precursor loading, our approach incorporates the air-through-precursor suction method, employing a degassed polydimethylsiloxane block to effectively eliminate air bubbles trapped in microwells. Additionally, fluorosilane pretreatment of the mold surface, along with the polyvinyl alcohol film formation, significantly enhances particle recovery up to 249-fold while ensuring particle homogeneity. This methodology demonstrates high adaptability to various gas-impermeable molds and curing techniques. The practical feasibility is illustrated through the successful production of functional composite microparticles that can be effectively utilized for oxygen sensing and self-assembly, challenging in conventional REM.
Introduction
Anisotropic microparticles, in contrast to spherical counterparts, exhibit orientation-specific geometrical features, making them valuable in various academic and practical domains such as multiplex sensing,1–4 drug delivery,5–7 photonics8,9 and self-assembly.10–12 For example, in multiplex sensing, the unique shapes of anisotropic microparticles serve as graphical codes, allowing a particle to be easily distinguished from others without additional labelling. In self-assembly, alterations in the shape of magnetic microparticles enables the deliberate manipulation of assembled structures and electromagnetic properties of fluids. These distinctive attributes have naturally generated significant interest in developing fabrication methods for functionalized microparticles with diverse morphologies.Particle replication in nonwetting templates (PRINT) emerges as an appealing technique for the production of anisotropic particles.13–15 This method offers notable advantages, including the capacity to fabricate particles with varied geometries and achieve high productivity. In the PRINT process, a liquid precursor solution is confined within multiple arrays of negative features on a perfluoropolyether (PFPE) mold and subsequently cured through methods such as ultraviolet radiation, solvent evaporation, lyophilization, or heat. The utilization of PFPE for both mold and cover, characterized by low surface free energy, facilitates effective drainage of excess precursor material through the application of pressure. This, in turn, enables the production of isolated particles without the formation of interconnected films. While PRINT has successfully produced anisotropic particles smaller than 50 nm, limitations still exist. Wells of micrometer size or larger may lead to issues with air bubble entrapment during precursor loading, resulting in the production of non-uniform particles. Moreover, the costly and complex manufacturing process for PFPE molds diminishes their accessibility for widespread use in laboratory settings.
Recently, replica molding (REM) employing a polydimethylsiloxane (PDMS) mold has garnered considerable attention for anisotropic microparticle fabrication.16–18 This is primarily attributed to the commercial accessibility, cost-effectiveness, and gas-permeability of PDMS molds. Despite the potential formation of a scum precursor layer on the PDMS mold due to its wettability, the oxygen inhibition layer created between the mold and cover during free-radical polymerization ensures the production of isolated microparticles. The oxygen inhibition layer, formed near the walls of microwells, further facilitates particle recovery by impeding the adhesion of particles to the walls. For effective precursor loading, a specialized degassed micromolding lithography technique can be employed with PDMS molds.17 This technique involves a degassing procedure of the PDMS mold before precursor loading, thereby transforming the gas-permeable mold into an air suction pump. This transformation enables the rapid elimination of trapped air bubbles by absorbing them, enhancing the efficiency of precursor loading.
Notwithstanding the advantages of PDMS-based REM, certain limitations arise concerning the compatibility of precursor solutions. Specifically, the integration of nonpolar organic solvents into the precursor solution is highly hindered due to their tendency to induce swelling or deformation of the PDMS mold.19,20 This swelling phenomenon poses a threat to the uniformity and resolution of resulting particles, making the incorporation of nanomaterials such as quantum dots, carbon nanotubes, and magnetic nanoparticles challenging, as these materials are typically well-dispersed in nonpolar organic solvents.
Gas-impermeable micromolds exhibiting high resistance to organic solvents are promising candidates applicable in REM processes without the limitations of conventional REM techniques. However, the inherent gas impermeability of these molds introduces two primary challenges in their utilization for microparticle synthesis. First, achieving complete precursor loading becomes a task due to the challenge of removing trapped air bubbles within the microwells. Second, recovering particles is arduous due to particle adhesion caused by the absence of an oxygen inhibition layer near the walls of microwells.
In this study, we introduce a versatile REM method that can effectively produce anisotropic microparticles within gas-impermeable micromolds. The air-through-precursor suction-augmented process is harnessed to attain complete precursor loading within the gas-impermeable molds. To facilitate particle recovery, the molds undergo pretreatment with fluorosilane, followed by the formation of a polyvinyl alcohol film after the curing of the loaded precursor material. These combined methodologies enable the successful fabrication of anisotropic microparticles in a gas-impermeable mold, using precursor solutions containing organic solvents—an achievement that has proven challenging with conventional REM techniques. Notably, particle synthesis is achieved not only through photopolymerization but also thermal curing, further expanding the possibilities for versatile particle fabrication. Through the developed protocol, we successfully produce uniform composite hydrogel microparticles embedding nanoparticles that are well-dispersed in organic solvents, including ruthenium and magnetic nanoparticles. These microparticles exhibit significant potential for use in biosensing and self-assembly applications, demonstrating the practical utility of this novel approach.
Experimental
The materials as well as detailed descriptions of mold and particle fabrication have been extensively documented in the ESI.†Results and discussion
Synthesis of anisotropic microparticles through APS REM
The process schematic of air-through-precursor suction-augmented replica molding (APS REM) for synthesizing microparticles in a gas-impermeable mold is illustrated in Fig. 1. To prevent adhesion of particles to microwells during precursor curing, a gas-impermeable mold was pretreated with fluorosilane, well-known for its low surface energy. The fluorosilane was deposited through vapor deposition in a vacuum desiccator subsequent to air plasma treatment of the mold (Fig. S1a†). Trichloro(1H,1H,2H,2H-perfluorooctyl)silane was specifically selected due to its documented ability to form a self-assembled monolayer on various substrates such as glass, PDMS, and silica. Although fluorosilane coating can be conducted without plasma treatment, we performed the plasma treatment to facilitate the coating process by generating diverse functional groups on the mold surfaces. While siloxane layers ranging from a few nanometers to hundreds of nanometers, rather than a monolayer, can be formed using this technique, this had little impact on particle geometry due to the relatively large size of the particles. The duration of the coating was determined based on contact angle measurements by dropping deionized water (DI water) on the flat surface of a gas-impermeable mold made of Norland Optical Adhesive 81 (NOA 81). Upon reaching a constant value of the contact angle after 30 minutes, the optimal deposition time for fluorosilane was established as 30 minutes (Fig. S1b†).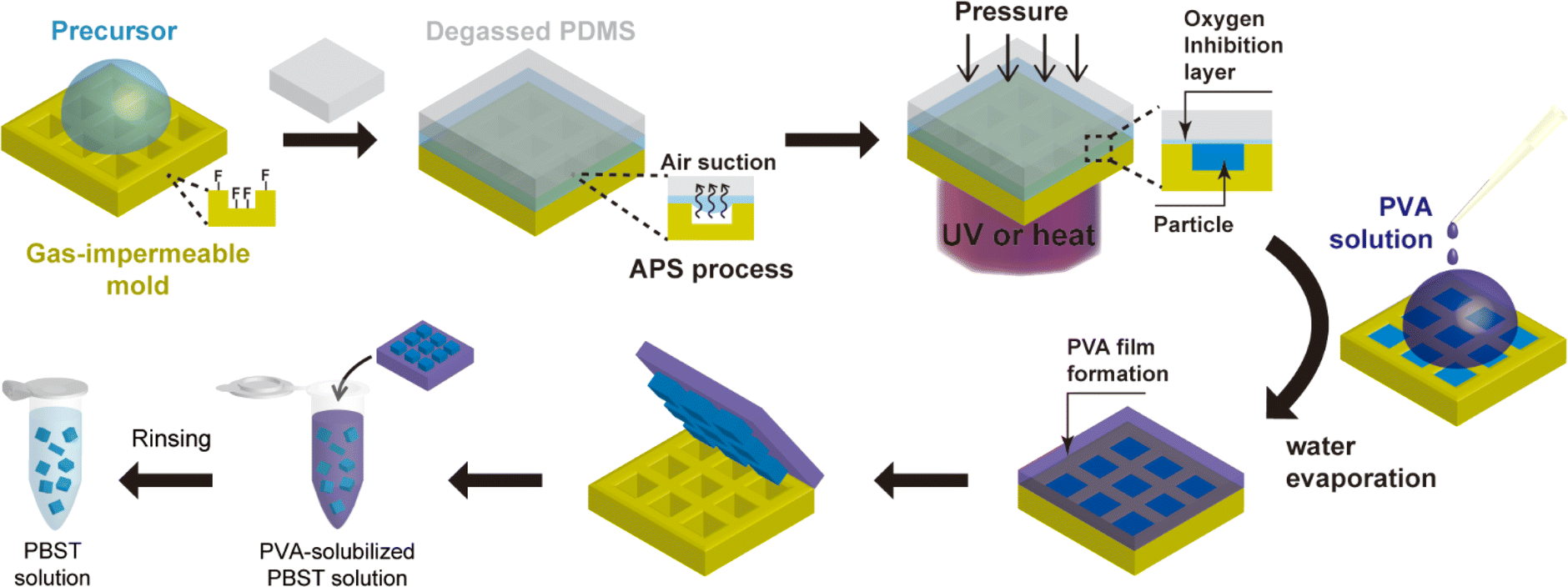 | ||
| Fig. 1 Schematic representation of APS REM for particle production in a gas-impermeable mold. The synthesis of anisotropic microparticles through air-through-precursor suction (APS)-augmented replica molding (REM) consists of three main steps: precursor loading by APS process, precursor curing, and particle recovery by poly(vinyl alcohol) (PVA) film formation. The gas-impermeable micromold is pretreated by fluorosilane before precursor loading. During the APS process, a degassed PDMS cover is utilized to absorb and eliminate air bubbles trapped in microwells, inducing the precursor solution to diffuse into the microwells. After precursor curing, polyvinyl siloxane (PVA) solution is dispersed onto the micromold and evaporated to create a PVA film for particle retrieval. | ||
The gas impermeable property of a mold poses a considerable obstacle to efficient removal of trapped air bubbles generated during the precursor loading process. While this is not a significant issue in sub-micron-sized wells, as seen in the PRINT method, it becomes a significant challenge in over-micron-sized wells. The trapped air bubbles hinder the synthesis of uniform microparticles in these larger wells. Furthermore, the low surface energy of the fluorosilane-coated mold can negatively impact the loading of the precursor solution by reducing the capillary forces that are typically involved in the precursor loading. To effectively address these challenges, we have adopted the APS method, which has recently been developed as an innovative technique for achieving rapid and complete precursor loading in REM.21 The APS method involves using a degassed PDMS block as a cover, strategically positioned to absorb and remove the air bubbles through the precursor layer instead of the mold. This innovative approach allows for swift and complete loading of diverse materials regardless of the mold's gas permeability. To the best of our knowledge, this is the first report that demonstrates the applicability of APS REM to particle synthesis. It is important to note that while an alternative method for precursor loading involves degassing the mold dispersed with a precursor solution in a vacuum desiccator, this approach may inadvertently result in excessive evaporation of the precursor. This is particularly pertinent when dealing with solutions containing organic solvents, potentially compromising the homogeneity of the synthesized microparticles.
After achieving complete precursor loading, UV or heat was applied to initiate radical polymerization while exerting pressure on the mold. To prevent the formation of interconnective films, pressure close to 30 kPa was applied to the mold using a weight. Unlike PDMS mold, some gas-impermeable molds have relatively large elastic modulus and can resist deformation under pressure. For instance, the NOA 81 mold has an elastic modulus of 1.38 GPa, resulting in ignorable deformation of the mold features under the 30 kPa pressure (2.18 × 10−5 proportional deformation).
Following the curing of the precursor solution, any residual precursor material remaining on the mold surface was effectively removed through a thorough washing procedure employing ethanol. Although the issue of particle adhesion to the microwells had been mitigated by the deposition of fluorosilane, the retrieval of the cured microparticles in rigid microwells still remained a challenging task. As such, we adopted a strategy involving the formation of an aqueous polyvinyl alcohol (PVA) film to facilitate particle recovery. The PVA solution was uniformly applied to cover all the microwells on the mold. The concentration of the PVA solution was meticulously optimized at 5% (w/v). Lower concentrations were observed to be inadequate in forming a continuous PVA film, as they led to issues such as cracking, while higher concentrations necessitated prolonged periods and elevated temperatures for the dissolution of the PVA film to retrieve particles. Upon dispersion of the PVA solution, the micromold was placed within a vacuum to allow for the complete diffusion of the PVA solution into the voids of particles. Subsequently, the PVA solution underwent a thermal treatment, promoting the formation of a PVA film through water evaporation. Finally, the PVA film was delicately detached from the mold and subsequently dissolved in a buffer solution, followed by multiple washing steps to ensure the removal of any residual impurities.
Effect of fluorosilane deposition on particle recovery
To assess the impact of fluorosilane treatment on particle recovery in a gas-impermeable mold, a series of experiments were conducted to synthesize and retrieve microparticles using NOA 81 molds. The NOA 81 mold was created through sequential REM, utilizing a positively patterned SU-8 master template (visual representation can be found in Fig. S2†). Two variations of NOA 81 molds were prepared: one treated with fluorosilane, and the other left untreated. Additionally, two different precursor solutions were employed for the synthesis of microparticles, with one being hydrophilic and the other hydrophobic.Following the particle synthesis and recovery, the surfaces of both the bare and fluorosilane treated NOA 81 molds were examined (see Fig. 2a). As a result, the bare NOA 81 mold displayed inadequate particle recovery for both precursor solutions. In the hydrophilic precursor, the recovery rate was found to be 12.3% (number of replicates, N = 3), with some cured particles observed adhering to the microwell walls. In the case of the hydrophobic precursor, the recovery rate was 0.4% (N = 3), as most particles were either torn or adhered to the microwells. The variation in recovery rates between the two precursor solutions can be attributed to the stronger adhesion of particles produced from the hydrophobic precursor, which contained monomers of smaller molecular weight (250 Da) compared to those in the hydrophilic precursor (700 Da). As the monomer, poly(ethylene glycol) diacrylate was used. The smaller molecular weight of the monomer likely contributed to a higher degree of polymerization and increased interaction (i.e., adhesion) between the cured particles and the microwells. In contrast, the fluorosilane-treated NOA 81 mold exhibited almost complete particle recovery, achieving an average of 99.6% recovery rate (N = 3) with the hydrophilic precursor and 99.3% with the hydrophobic precursor. The fluorosilane treatment applied to the NOA 81 mold is assumed to facilitate particle recovery through three mechanisms. First, the interspacing layer formed by the fluorosilane acts as a physical barrier, effectively separating the walls of the microwells from the precursor solution. Additionally, the presence of fluorine molecules in the fluorosilane imparts water and oil-repellent properties (i.e., low wettability), thereby impeding the spreading of the precursor solution towards the microwell walls through the interspacing layer. These factors significantly reduce the interactions between the mold and the precursor, preventing particle adhesion during precursor curing. Once the precursor is cured, the only obstacle to particle recovery is the friction between the microwells and the particles. During the detachment step using a PVA film, the fluorine present on the walls reduces friction by virtue of its low surface energy, enabling particle recovery with minimal strain.
 | ||
| Fig. 2 Particle recovery in the bare and fluorosilane treated NOA 81 molds through a PVA film formation. (a) Images of the bare and fluorosilane treated NOA 81 molds after the recovery of particles produced from the hydrophilic and hydrophobic precursor solutions. The height of microwells was 25 μm. (b) Contact angles of hydrophilic and hydrophobic precursor solutions dropped on bare and fluorosilane treated NOA 81 films. (c) Schematic and images illustrating the diffusion of PVA solution into the cured microparticles, detached PVA film, and the recovered particles after washing processes. Brightfield and fluorescence images of the detached PVA film and the recovered microparticles after washing processes are presented. Scale bars indicate 100 μm. | ||
The wettable properties of the fluorinated and non-fluorinated NOA 81 molds, which are closely associated with the repelling properties of fluorine, were confirmed through contact angle measurements, as depicted in Fig. 2b. The bare NOA 81 mold exhibited favorable wettability for both hydrophilic and hydrophobic precursor solutions, as evidenced by contact angles close to 0°. In contrast, the NOA 81 mold treated with fluorosilane demonstrated relatively large contact angles of 88° and 70° for hydrophilic and hydrophobic precursors, respectively. The comparatively smaller increase in contact angle observed for the hydrophobic precursor can be attributed to the inherent hydrophobic nature of the treated silane. Given that repellent property increases with an increase in contact angle, the increased contact angles of the mold due to fluorosilane coating can be correlated with the observed enhancement in the recovery rate.
Effect of PVA film-based recovery on homogeneity of particles
The homogeneity of composite particles plays a crucial role in ensuring their reliable utilization. To assess the preservation of particle homogeneity after recovery, we employed a polyvinyl alcohol (PVA) solution containing a fluorescent dye, Rhodamine B. Given its low molecular weight of 479.02 Da, Rhodamine B readily diffuses into the nanoporous structures of particles alongside the PVA solution. Following the curing of the precursor, we dispersed the fluorescent PVA solution onto the NOA 81 mold and subsequently evaporated it, resulting in the formation of a fluorescent PVA film (Fig. 2c). The PVA film was then detached and carefully cut along the vertical middle line. Brightfield and fluorescence images of the cross section were captured using a microscope. The acquired images clearly revealed the attachment of microparticles to the PVA film and the successful diffusion of the PVA solution into the internal structures of the particles, forming a PVA network. The entanglement of the PVA network within the particle structures would have provided sufficient tension to facilitate the lifting of the particles during the detachment of the PVA film. The PVA film was then dissolved in a buffer solution and rinsed multiple times to eliminate any remaining impurities within the particle structures. Subsequent imaging of the recovered particles using brightfield and fluorescence techniques revealed no detectable impurities remaining within the particles. This observation indicates that the homogeneity of the particles remains intact even after the PVA-based recovery processes.Synthesis of microparticles in an epoxy mold
To demonstrate the versatility of the developed procedures in this research, an epoxy micromold was chosen as another mold used for particle synthesis (Fig. 3a). This epoxy micromold offers excellent resistance to organic solvents and is impermeable to gases. The fabrication process employed for this micromold was identical to that used for the NOA 81 mold (Fig. S2†). The microparticles were synthesized using a hydrophobic precursor solution consisting of a polyethylene glycol (PEG)-based acrylate monomer, toluene, photoinitiator, and acrylate fluorescence dye (Rhodamine B). Given that the monomer and rhodamine B possess the same acrylate group, the fluorescence dye could be incorporated into the particles through covalent bonding during radical polymerization. As depicted in the images, particle recovery with significant recovery rate close to 100% and the synthesis of highly uniform microparticles were successfully achieved, with a coefficient of variation (CV) value of 4.2% observed among the particles (number of measured particles, n = 7).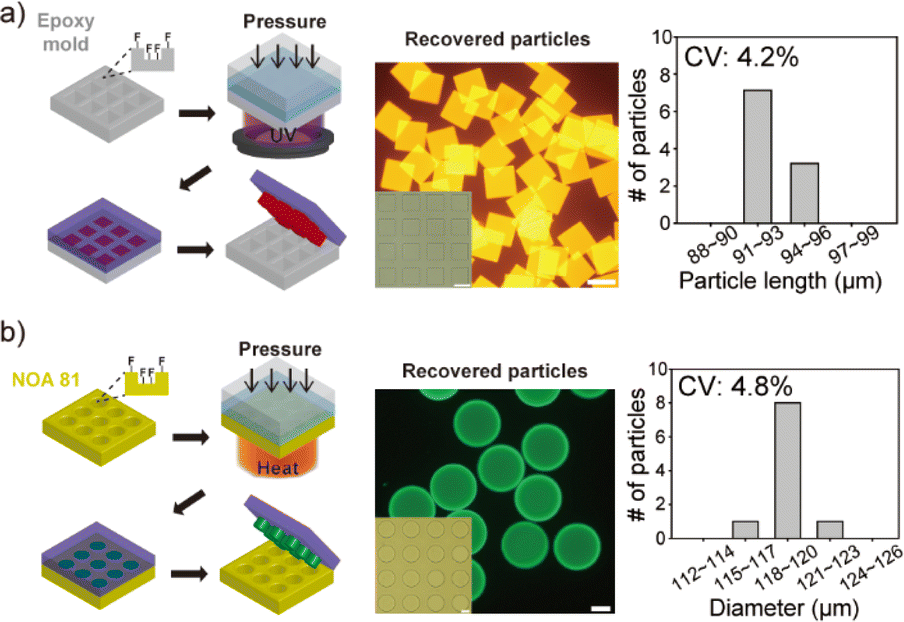 | ||
| Fig. 3 Synthesis of microparticles in an epoxy mold or through thermal curing. (a) Microparticle fabrication using an epoxy micromold, along with images showcasing the uniformity of the produced particles. Complete particle recovery and synthesis of homogeneous microparticles were achieved in the epoxy micromold. (b) Microparticle fabrication in a NOA 81 mold through thermal curing, accompanied by images demonstrating the uniformity of the resulting particles. Complete particle recovery and synthesis of uniform microparticles were achieved. Scale bars indicate 100 μm. | ||
Synthesis of microparticles through thermal curing
Photopolymerization is a commonly utilized method for curing precursors in particle synthesis. However, heat can serve as an efficient alternative, particularly in cases where achieving uniform exposure of UV light is challenging, such as in large-area molds where incident light tends to diverge.22 Unlike light, heat exhibits non-directional characteristics, allowing for its uniform application to a large and flat area. This characteristic makes heat highly advantageous for the mass production of particles. To investigate the viability of thermal curing, we conducted an experiment to synthesize microparticles using heat in a NOA 81 mold (Fig. 3b). A thermal initiator (azobisisobutyronitrile) in acetone and acrylate fluorescein were included in the precursor solution. The thermal initiator generates radicals upon heat absorption, thereby permitting the utilization of the oxygen inhibition layer for particle synthesis, akin to the process employed in photopolymerization. After loading the precursor into the NOA 81 mold, heat was applied for 1 minute using a hot plate set at 90 °C, while simultaneously applying pressure to the mold. Following the recovery process, the micromold and cured particles were observed and analysed. Consequently, complete particle recovery and uniform particle synthesis were successfully achieved, with a CV value of 4.8% observed among the particles (n = 7).Synthesis of ruthenium-embedding composite microparticles and their application to oxygen sensing
The practical utility of the developed process was demonstrated through the synthesis of functional composite microparticles incorporating a ruthenium complex, as depicted in Fig. 4. The choice of a ruthenium complex as the functional cargo was based on its susceptibility to quenching by oxygen, making it suitable for use as an oxygen biosensor.23,24 Additionally, ruthenium complexes are known for their photostability, high quantum yield, large Stokes shifts, and wide dynamic range in response to varying oxygen levels, further justifying their selection for this application. Notably, the immobilization of the ruthenium complex within a hydrogel matrix has gained significant attention as an efficient oxygen biosensor, particularly within biological disciplines such as cell culturing. This preference is attributed to the hydrogel's biocompatibility, which reduces the toxic effects of ruthenium nanoparticles on cells, as well as its anti-fouling properties, which minimize protein and cell adhesion.25,26 These characteristics make hydrogel-based ruthenium complex sensors valuable tools for oxygen sensing applications.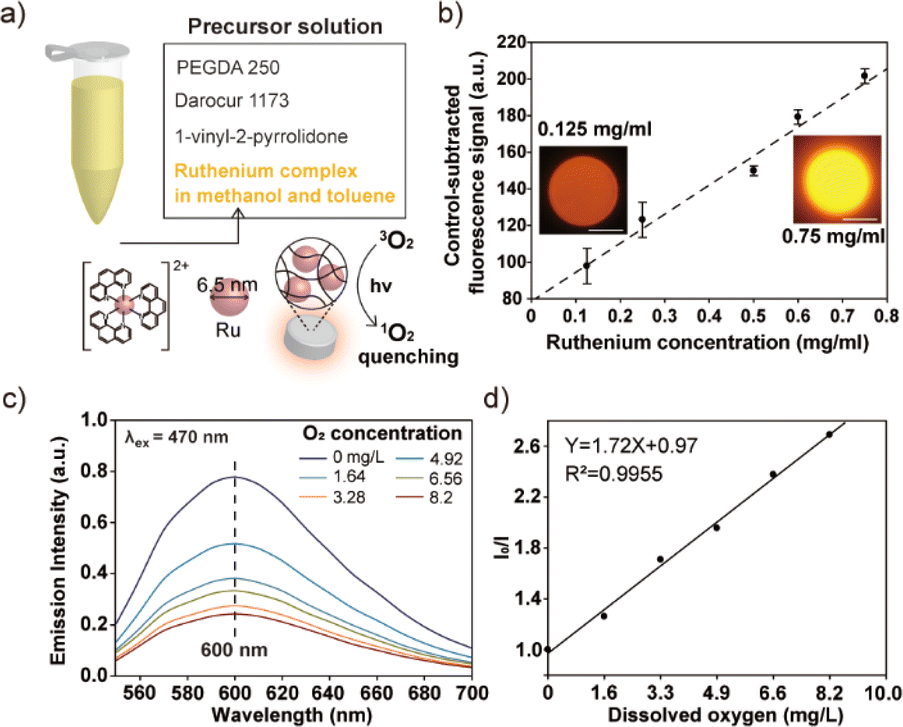 | ||
| Fig. 4 Synthesis of ruthenium-embedding composite hydrogel microparticles and their application to sensing of oxygen molecules. (a) Composition of the precursor solution and quenching effect of oxygen molecules on ruthenium nanoparticles. (b) Change in emission intensities with increasing ruthenium concentration in precursor solution (number of measured particles = 7). (c) Emission spectra of composite hydrogel microparticle at different oxygen concentrations in DI water. The excitation wavelength employed for these measurements was 470 nm. (d) Linear relationship between the reciprocal of relative phosphorescent intensity (I0/I) and dissolved oxygen. This relationship is represented by the Stern–Volmer equation. Scale bars indicate 100 μm. | ||
In order to solubilize the hydrophobic ruthenium nanoparticles, a solvent mixture comprised of methanol and toluene was employed. Additionally, a hydrophobic monomer, namely PEGDA 250, and a hydrophobic photoinitiator, specifically Darocur 1173, were utilized in the manufacturing of the precursor solution (Fig. 4a). This selection was made to prevent the agglomeration of the hydrophobic ruthenium nanoparticles when mixed. The particle synthesis process was carried out using photopolymerization. To assess and confirm the successful incorporation and stability of the ruthenium complex within the gel matrix, we conducted particle synthesis experiments in which we varied the concentration of the ruthenium complex in the precursor solution. We subsequently measured the fluorescent intensity of the resulting particles. As a result, we observed a linear increase in the fluorescent signals emanating from the composite microparticles, with a notably high coefficient of determination value (R2, 0.98), as presented in Fig. 4b. This linear relationship between the fluorescent intensity and the ruthenium concentration in a precursor solution serves as concrete evidence of the effective incorporation of the ruthenium complex within the gel matrix. Furthermore, to evaluate the long-term stability of the produced composite microparticles, we conducted extended stability tests. These tests verified that there was no leakage of nanoparticles from the gel matrix over time, thus affirming the robustness and reliability of the produced composite microparticles (refer to Fig. S3† for additional details). To evaluate the oxygen-sensing capabilities of the composite microparticles, we systematically controlled the oxygen level within a solution of deionized (DI) water containing the particles. This was accomplished by subjecting the solution to a degassing process within a vacuum desiccator, effectively reducing the concentration of dissolved oxygen. To achieve a near-zero air pressure condition, argon gas was introduced into the vacuum desiccator while simultaneously degassing the chamber. The emission spectrum emitted by the particles suspended in the solution was measured using a spectrophotometer upon exposure to 470 nm light (Fig. 4c). By applying the Stern–Volmer equation, which provides a mathematical description of the quenching of fluorescence by molecular oxygen, we analysed the emission intensities exhibited by the ruthenium-embedded particles under varying oxygen concentrations. The Stern–Volmer equation is represented as follows:
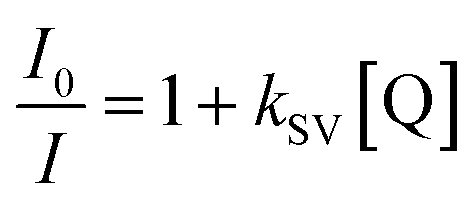 | (1) |
Synthesis of highly magnetized microparticles with significant uniformity
Anisotropic magnetic particles have emerged as prominent constituents in directed self-assembly processes, owing to their capacity to generate notable electrorheological and magnetorheological fluids when subjected to electric or magnetic fields.27–30 The manipulation of the macroscopic properties of these fluids is achievable through alterations in the geometrical characteristics of the particles, thereby conferring heightened efficiency to anisotropic microparticles in contrast to their spherical counterparts.10,31,32 The facilitation of self-assembly necessitates the conferment of substantial magnetic controllability upon the particles, a quality predicated on two pivotal factors: magnetization and the uniformity of magnetic particles. The magnitude of magnetization assumes significance in gauging the extent to which the magnetic particles can responsively interact with electromagnetic fields. Simultaneously, the uniformity of the magnetic particles plays a critical role in determining the precision with which they can be deliberately manipulated.Through conventional PDMS-based replica molding, the homogeneous incorporation of small-sized bare magnetic nanoparticles (MNPs) into anisotropic microparticles presented challenges owing to the inherent propensity of the nanoparticles to aggregate in non-organic solvents. Although the enhancement of nanoparticle stability can be achieved through surface functionalization with stable ligands such as PEG,33,34 this approach necessitates additional processing steps and entails compromises in the magnetization of MNPs.35 In the course of this research, we achieved a successful synthesis of magnetic anisotropic microparticles that incorporated a homogeneously dispersed population of 10 nm-sized bare MNPs. This accomplishment was realized through the APS REM technique within a NOA 81 mold. The precursor solution, depicted in Fig. 5a, comprised a toluene-based ferrofluid, PEGDA 250, toluene, Darocur 1173, and 1-vinyl-2-pyrrolidone. To prevent MNPs aggregation, a substantial proportion (80%, v/v) of the precursor solution was composed of toluene. However, this heightened toluene content led to rapid evaporation of the precursor during the loading step, paradoxically resulting in MNPs aggregation. To address this challenge, the PDMS substrate underwent degassing while immersed in toluene, thereby ensuring the retention of sufficient toluene within the PDMS structure prior to its utilization in particle synthesis. This procedural refinement significantly mitigated precursor evaporation and effectively averted MNPs agglomeration. The magnetic properties of the resulting magnetic particles were assessed through a hysteresis loop analysis (i.e., B–H curve) conducted using a vibrating sample magnetometer (VSM) (Fig. 5b). This measurement was progressed at room temperature, employing a magnetic field range of −0.8 T to 0.8 T. The obtained curve demonstrated the characteristic magnetization cycle indicative of a superparamagnetic material. This outcome underscored the retention of the superparamagnetic property of the bare MNPs even after their encapsulation within the polymer matrix. The value of saturation magnetization for the composite particles was derived from the following equation applicable in high-field conditions.
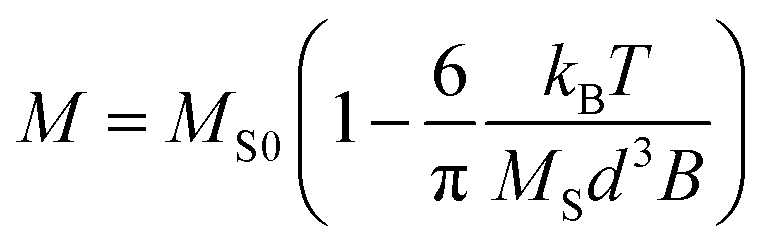 | (2) |
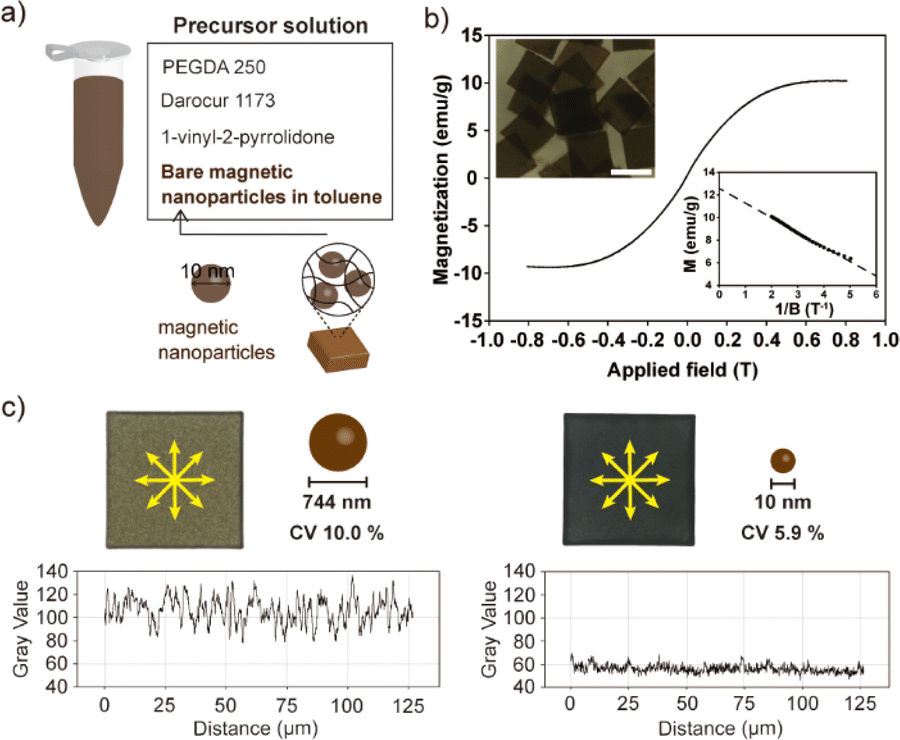 | ||
| Fig. 5 Synthesis of bare MNPs-embedding anisotropic magnetic microparticles and assessment of MNPs uniformity. (a) Composition of the precursor solution and size of the MNPs encapsulated within microparticles. (b) Vibrating sample magnetometer data of the produced magnetic composite hydrogel microparticles. The inset graph illustrates a linear relationship between the reciprocal of the applied magnetic field and the magnetization value of the composite microparticles. (c) Comparison of uniformity within magnetic particles of 744 nm MNPs and 10 nm MNPs. Scale bar indicates 100 μm. | ||
The encapsulation of small-sized MNPs imparts a heightened level of uniformity to the magnetic particles, thereby conferring a distinct advantage for precise and deliberate control when subjected to applied electromagnetic fields. In order to assess the uniformity of the synthesized magnetic particles, an additional set of magnetic particles containing larger MNPs (744 nm) was fabricated and subjected to comparative analysis. The concentration of MNPs in the precursor was adjusted to be the same when synthesized using 10 nm MNPs. The density variation of MNPs across different points within the two types of magnetic particles was quantitatively evaluated utilizing ImageJ software (version 1.52a) (Fig. 5c). This analysis revealed that magnetic particles containing 10 nm MNPs exhibited superior uniformity, with a CV of 5.9%, in contrast to those containing 744 nm MNPs, which exhibited a higher CV of 10.0%.
Conclusions
This study introduces an innovative and versatile REM methodology for the fabrication of anisotropic microparticles within gas-impermeable molds, with a particular emphasis on expanding the spectrum of available precursor solutions. The process, denoted as “air-through-precursor suction-augmented replica molding” (APS REM), is integrated with the surface treatment procedure involving fluorosilane and the application of a polyvinyl alcohol (PVA) film for efficient particle synthesis and recovery. This combined approach effectively facilitates the production of anisotropic microparticles from precursor solutions, even those incorporating organic solvents.In light of the fact that APS REM employs a polymerization method fundamentally similar to that of the PRINT technique, it is also estimated to be suitable for synthesizing sub-micron sized particles smaller than 50 nm. However, it is noteworthy that other parameters, including the concentration of PVA solution, need to be adjusted to maintain the homogeneity of the sub-micron particles. Additionally, this technique can be readily scaled up by incorporating roll-to-roll processing and adjusting the mold size from laboratory scale to commercial scale.13
The efficacy of the developed techniques is substantiated by their successful application in the synthesis of uniform composite hydrogel microparticles containing nanoparticles that are homogeneously dispersed within organic solvents. These microparticles exhibit significant potential for utilization in biosensing and self-assembly applications, thereby underscoring the practicality and utility of this pioneering methodology. Furthermore, the versatility of the APS REM process is demonstrated through its adaptability to diverse mold materials, such as NOA 81 and epoxy, and various curing modalities, including photopolymerization and thermal curing.
Notwithstanding the notable advantages of APS REM, it is imperative to address concerns associated with the non-uniform application of pressure to the mold during precursor curing, often resulting in film formation instead of isolated particles. The exploration of a purpose-designed apparatus for the uniform application of pressure presents a promising avenue for future research endeavors. For instance, previous study has devised a uniform pressure apparatus rooted in Pascal's law, specifically designed for application in micro/nanoimprint lithography. This approach holds the potential to enhance the reproducibility of particle synthesis based on the APS REM technique.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Seok Joon Mun: conceptualization and design of the study, performing the experiment, interpretation of data, writing – original draft, reviewing, and editing. Wookyoung Jang, Jun Hee Choi: performing the experimental and theoretical analysis. Yong Jun Lim: performing the experiment. Ki Wan Bong: conceptualization and supervision of the project, funding acquisition, writing – original draft, reviewing, and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Korea Research Institute of Defense Technology Planning and Advancement (KRIT) grant funded by Defense Acquisition Program Administration (DAPA) (no. KRIT-CT-21-034, 2023) and the Technology Innovation Program (20018111, Development of super-fast multiplex technology for the examination of diagnosis of infectious disease and in-body response test) funded by the Ministry of Trade, Industry and Energy (MOTIE, Korea).References
- N. W. Choi, J. Kim, S. C. Chapin, T. Duong, E. Donohue, P. Pandey, W. Broom, W. A. Hill and P. S. Doyle, Anal. Chem., 2012, 84, 9370–9378 CrossRef CAS PubMed.
- H. J. Lee, Y. H. Roh, H. U. Kim, S. M. Kim and K. W. Bong, Lab Chip, 2019, 19, 111–119 RSC.
- W. Choi, E. Park, S. Bae, K. H. Choi, S. Han, K. H. Son, D. Y. Lee, I. J. Cho, H. Seong and K. S. Hwang, Small, 2022, 18, 2105538 CrossRef CAS.
- S. J. Mun, W. Jang, H.-S. Park, Y. J. Lim, T.-J. Yang and K. W. Bong, Biosens. Bioelectron., 2023, 241, 115670 CrossRef CAS.
- B. Almería, W. Deng, T. M. Fahmy and A. Gomez, J. Colloid Interface Sci., 2010, 343, 125–133 CrossRef.
- G. Giani, S. Fedi and R. Barbucci, Polymers, 2012, 4, 1157–1169 CrossRef.
- Q. Truong Hoang, D. Y. Kim, H. S. Park, W. Jang, T. G. Nguyen Cao, J. H. Kang, Y. T. Ko, S. J. Mun, S. H. Bhang and M. S. Shim, Adv. Funct. Mater., 2024, 34, 2306078 CrossRef CAS.
- D. S. Bykov, S. Xie, R. Zeltner, A. Machnev, G. K. Wong, T. G. Euser and P. S. J. Russell, Light: Sci. Appl., 2018, 7, 22 CrossRef.
- J. Y. Sim, J. H. Choi, J. M. Lim, S. Cho, S. H. Kim and S. M. Yang, Small, 2014, 10, 3979–3985 CrossRef CAS PubMed.
- P. Panda, K. W. Bong, T. A. Hatton and P. S. Doyle, Langmuir, 2011, 27, 13428–13435 CrossRef CAS.
- C. K. Wong, X. Qiang, A. H. Müller and A. H. Gröschel, Prog. Polym. Sci., 2020, 102, 101211 CrossRef CAS.
- W. B. Rogers, W. M. Shih and V. N. Manoharan, Nat. Rev. Mater., 2016, 1, 1–14 Search PubMed.
- J. Xu, D. H. Wong, J. D. Byrne, K. Chen, C. Bowerman and J. M. DeSimone, Angew. Chem., Int. Ed., 2013, 52, 6580–6589 CrossRef CAS.
- S. S. Dunn, S. Tian, S. Blake, J. Wang, A. L. Galloway, A. Murphy, P. D. Pohlhaus, J. P. Rolland, M. E. Napier and J. M. DeSimone, J. Am. Chem. Soc., 2012, 134, 7423–7430 CrossRef CAS.
- J. P. Rolland, B. W. Maynor, L. E. Euliss, A. E. Exner, G. M. Denison and J. M. DeSimone, J. Am. Chem. Soc., 2005, 127, 10096–10100 CrossRef CAS PubMed.
- H. U. Kim, Y. H. Roh, S. J. Mun and K. W. Bong, ACS Appl. Mater. Interfaces, 2020, 12, 53318–53327 CrossRef CAS.
- H. U. Kim, Y. J. Lim, H. J. Lee, N. J. Lee and K. W. Bong, Lab Chip, 2020, 20, 74–83 RSC.
- C. L. Lewis, C.-H. Choi, Y. Lin, C.-S. Lee and H. Yi, Anal. Chem., 2010, 82, 5851–5858 CrossRef CAS PubMed.
- C.-H. Choi, B. Lee, J. Kim, J.-O. Nam, H. Yi and C.-S. Lee, ACS Appl. Mater. Interfaces, 2015, 7, 11393–11401 CrossRef CAS.
- R. Ganguly, Y. Choi, C.-S. Lee and C.-H. Choi, J. Colloid Interface Sci., 2021, 600, 373–381 CrossRef CAS PubMed.
- S. J. Mun, W. Jang, J. Y. Eom, H. U. Kim and K. W. Bong, Small, 2022, 18, 2204754 CrossRef CAS.
- D.-Z. Lin, P.-H. Yu, Z.-J. Zhang, Y.-C. Chen, S.-C. Lin and J.-Y. Jeng, Addit. Manuf., 2023, 103665 CAS.
- Y. Xu, D. Yang, S. Huo, J. Ren, N. Gao, Z. Chen, Y. Liu, Z. Xie, S. Zhou and X. Qu, Polym. Test., 2021, 93, 106957 CrossRef CAS.
- M. Verrucchi, G. E. Giacomazzo, P. S. Sfragano, S. Laschi, L. Conti, M. Pagliai, C. Gellini, M. Ricci, E. Ravera and B. Valtancoli, Langmuir, 2022, 39, 679–689 CrossRef.
- C. Shen, L. Bian, P. Zhang, B. An, Z. Cui, H. Wang and J. Li, J. Membr. Sci., 2020, 601, 117949 CrossRef.
- T. Wang, J. Deng, R. Ran, W. Shi, Y. Gao, X. Ren, J. Cao and M. Zhang, Chem. Eng. J., 2022, 449, 137486 CrossRef CAS.
- K. Deng, Z. Luo, L. Tan and Z. Quan, Chem. Soc. Rev., 2020, 49, 6002–6038 RSC.
- M. A. Kamal, A. V. Petukhov and A. Pal, J. Phys. Chem. B, 2020, 124, 5754–5760 CrossRef CAS PubMed.
- A. Saini, K. Theis-Bröhl, A. Koutsioubas, K. L. Krycka, J. A. Borchers and M. Wolff, Langmuir, 2021, 37, 4064–4071 CrossRef CAS PubMed.
- Q. Fan, Z. Li, C. Wu and Y. Yin, Precis. Chem., 2023, 1, 272–298 CrossRef CAS.
- J. Kim, S. E. Chung, S.-E. Choi, H. Lee, J. Kim and S. Kwon, Nat. Mater., 2011, 10, 747–752 CrossRef CAS.
- S. Y. Lee and S. Yang, Angew. Chem., Int. Ed., 2013, 52, 8160 CrossRef CAS.
- B. I. Kharisov, H. R. Dias, O. V. Kharissova, A. Vázquez, Y. Pena and I. Gomez, RSC Adv., 2014, 4, 45354–45381 RSC.
- K. Bhattacharjee and B. L. Prasad, Chem. Soc. Rev., 2023, 52, 2573–2595 RSC.
- I.-V. Ganea, A. Nan, C. Baciu and R. Turcu, Nanomaterials, 2021, 11, 679 CrossRef CAS.
- S. J. Mun, D. Ko, H. U. Kim, Y. Han, Y. H. Roh, B.-G. Kim, H. B. Na and K. W. Bong, Materials, 2020, 13, 4401 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra04719b |
| This journal is © The Royal Society of Chemistry 2024 |