
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unveiling photoinduced electron transfer in cobalt(III)-R-pyridine complexes anchored to anatase nanocrystals: photoluminescence and magnetic studies†
Ganeshraja Ayyakannu Sundaram *a,
Krishnamoorthy Anbalagan*b,
Mohammad Ahmad Wadaanc,
Jagan Rajamonid and
Vaithinathan Karthikeyan*e
*a,
Krishnamoorthy Anbalagan*b,
Mohammad Ahmad Wadaanc,
Jagan Rajamonid and
Vaithinathan Karthikeyan*e
aDepartment of Research Analytics, Saveetha Dental College and Hospitals, Saveetha Institute of Medical and Technical Sciences, Poonamallee High Road, Chennai, Tamil Nadu 600 077, India. E-mail: ganeshrajaas.sdc@saveetha.com; asgchem84@gmail.com
bDepartment of Chemistry, Pondicherry University, Kalapet, Pondicherry, 605 014, India. E-mail: kanuniv@gmail.com
cDepartment of Zoology, College of Science, King Saud University, P. O. Box. 2455, Riyadh, 11451, Saudi Arabia
dDepartment of Chemistry and Biochemistry, University of Missouri, St. Louis, MO 63121, USA
eSchool of Science and Technology, Hong Kong Metropolitan University, Ho Man Tin, Hong Kong. E-mail: vkarthik@hkmu.edu.hk
First published on 28th May 2024
Abstract
In this study, we synthesized mixed ligand complexes of the cis-[Co(tn)2(Rpy)Br]Br2 type using a novel mechanochemical approach. Characterization involved spectral measurements and single crystal X-ray diffraction analysis, confirming the structure of the cis-[Co(tn)2(4-Mepy)Br]Br2 complex. The single crystal refinement data revealed a monoclinic crystal system with a distorted octahedral geometry. The choice of the sixth ligand influenced the emission and magnetic properties, showing a ferromagnetic character in the Co(III)-complex environment. We investigated efficient electron transfer to the cobalt(III) center using TiO2 nanoparticles under UV-light irradiation. The adsorption characteristics of cis-[Co(tn)2(Rpy)Br]Br2 in aqueous 2-propanol varied, leading to surface compound formation. Under UV irradiation, the anatase surface exhibited remarkable adsorption capabilities, facilitating efficient electron transfer to the Co(III) center and resulting in a high photoefficiency for Co(II) formation. Our study has put forward a model for interfacial electron transfer (IET), taking into account the overlap between the TiO2 conduction band and the acceptor level of the Co center, as well as the electronic coupling between the donor level of the Ti center and the acceptor level of the Co center. This model sheds light on the accumulation of electrons for reducing the adhered complex ion. The IET process was corroborated by the conversion of 2-propanol into acetone, as verified by 1H NMR technique. Overall, our findings provide novel insights into the role of the Rpy moiety in modifying the structure of the TiO2-cobalt(III)-Rpy compound and propose a mechanism for IET reactions, thus advancing the field.
Introduction
Understanding the intricate photophysics of transition metal complexes tethered to semiconductor surfaces is crucial for designing artificial systems for solar energy conversion.1 Cobalt(III) compounds have emerged as promising candidates due to their ability to foster long-lived charge-separated states and exhibit rich photochemical properties.2,3 However, the electronic excitations and photoconversion mechanisms of these complexes are only partially understood.4,5 Beyond their application in photovoltaic cells for converting solar energy into electricity, Ru(II)-polypyridine complexes anchored to TiO2 surfaces via phosphonate linkers exhibit remarkable resistance to humidity and can emulate photosynthetic charge transfer events.6,7 Consequently, unraveling the intricacies of electron transfer mechanisms stands to impact the development of multicomponent molecular assemblies for photocatalysis and artificial photosynthesis. Previous theoretical endeavors have primarily focused on the comparative analysis of binding energies and vertical excitations, particularly when covalently attaching [Ru(tpy)2]2+ to TiO2 using carboxylates or phosphonate linkers.8,9 Similar investigations have extended to the attachment of dye-sensitized TiO2 nanoparticles.10,11Estimates of electron injection rates within the time-independent framework have been derived from the analysis of donor and acceptor orbital overlap in both adsorbate and host-nanoparticle. Meanwhile, explicit quantum dynamics simulations have probed the time scales and mechanisms of interfacial electron transfer (IET), building upon earlier studies of electron injection in sensitized TiO2 semiconductors.12 The reported results herein offer fundamental insights into the electronic and structural properties of molecular assemblies, crucial for achieving efficient IET in catalyst-chromophore/TiO2 interfaces.13,14
Recent research endeavors outlined in this discussion have been spurred by the pursuit of advancing photovoltaic cells built upon sensitized nanocrystalline TiO2.15–18 Notably, investigations in this domain have revealed that while the fundamental photophysical properties of molecular components persist upon immobilization on a semiconducting surface, the interaction with said surface can significantly modulate the rates of individual photophysical processes.19–21 A compelling illustration of this phenomenon is observed in ruthenium polypyridyl complexes, which, when bound to TiO2, undergo a remarkable transformation from intrinsic photolability in solution to pronounced photostability.22,23 This transformation is ascribed to the exceptionally rapid sub-picosecond charge injection from the excited state of the surface-bound compound into the conduction band of the semiconductor.24 Furthermore, the subsequent back electron transfer process, involving conduction band electrons and the oxidized form of the sensitizer, occurs at a significantly slower rate than the forward electron transfer reaction.25 Consequently, this pronounced disparity in rates facilitates effective charge separation, rendering the TiO2 surface instrumental in achieving long-lived charge separation—a pivotal factor driving investigations in this field.26
In this study, we utilized a novel mechanochemical approach to synthesize cis-[Co(tn)2(Rpy)Br]Br2 mixed ligand complexes. Advanced instrumental analyses were employed to explore the geometry and the impact of aryl ligands on the emission and magnetic properties within the coordination sphere. The primary focus lies on investigating the luminescent and magnetic characteristics of these complexes, which also provide access to stable cobalt(III) monomers. Specifically, we aim to comprehensively understand the redox activity of excited nanocrystalline TiO2 and structurally modified cobalt(III)-aryl amine complexes, particularly focusing on the influence of ligand electron donor strength on the cobalt(III) complex's M−H loop. Our objective is to unravel the intricate interplay between photoinduced electron transfer phenomena and cobalt(III)-R-pyridine complexes immobilized onto anatase nanocrystals. This comprehensive study includes detailed synthesis methodology, characterization techniques, and analysis of photoluminescence and magnetic properties. By exploring complex interfacial dynamics, our research sheds light on unique electron transfer processes within this system, offering valuable insights to photochemical and materials science. Ultimately, the custom-designed metal complexes developed here hold promise for boosting photocatalytic activity and photon harvesting by facilitating efficient interfacial electron transfer processes. Notably, the validity of the IET process was confirmed through the conversion of 2-propanol into acetone, as validated by 1H NMR technique.
Experimental section
Materials and instrumental methods
Reagent-grade cobaltous chloride, 1,3-propanediamine (tn), R-pyridine (analytical reagent grade), KBr (spectral grade), nanocrystalline titanium dioxide (with a BET surface area of 150–200 m2 g−1, particle size of approximately 50 nm, and micropore volume of ∼0.027 cm3 g−1), and DMSO-d6 (spectral grade) were procured from Sigma-Aldrich and utilized without further purification. Triply distilled water was obtained using an all-glass apparatus over alkaline potassium permanganate. Elemental analyses were performed using an Elementar Vario EL III-Germany instrument.FT-IR spectra of the complexes were acquired using a Thermo Nicollet-6700 FT-IR instrument in the range of 4000–400 cm−1 (KBr pellet technique). Electronic absorption spectral studies were conducted on a double-beam spectrophotometer (Shimadzu 2450, Japan) with an integrating sphere attachment (ISR-2200).
Single crystals of the complex were examined using an Oxford Diffraction Xcalibur diffractometer with an Eos (Nova) detector, utilizing ω and φ scan modes. Diffraction measurements were carried out at 293(2) K using graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). Structure solution and refinement were accomplished by direct methods and full-matrix least squares on F2, respectively, utilizing the 32 bit Olex 2-1.1 version program. Other computer programs involved in data collection, cell refinement, data reduction, and absorption correction included CrysAlis PRO (Oxford Diffraction, 2009).
Steady-state fluorescence emission was recorded on a Spex FluoroLog-3 spectrofluorometer (Jobin-Yvon Inc.) equipped with a 450 W Xenon lamp. Time-resolved fluorescence decay measurements were conducted using a Nano-LED (λex = 295 nm) source for excitation (repetition rate 10 kHz). Photon collection utilized a TBX-4-X single-photon-counting detector, and lifetimes were determined by fitting the data to exponential decay models. The fitting analysis was performed using DAS6 v6.2-Horiba Jobin Yvon software, and magnetic measurements were conducted using a vibrating sample magnetometer (VSM) in powder form on Lakeshore-7404. The VSM technique allowed the observation of hysteresis curves for the all prepared complexes in a DC magnetic field.
1H NMR spectra of the acetone formation were obtained on a Bruker instrument, model Avance-II, operating at 400 MHz (∼9.4 Tesla) for Fourier-transform nuclear magnetic resonance. The complex was dissolved in DMSO-d6 with added TMS as an internal standard, and the solution was placed in an NMR tube for 1H NMR. For 1H NMR spectra, measurements comprised 256 scans with a 3.52 s repetition time.
Synthesis of cis-[Co(tn)2(Rpy)Br]Br2
Trans-[Co(tn)2Br2]Br, precursor complex was synthesized following the established procedure in the literature.27–29 The cis-[Co(tn)2(Rpy)Br]Br2 complexes were synthesized utilizing an simple mechanochemical approach from the precursor complex. To this end, one gram of trans-[Co(tn)2Br2]Br was transformed into a paste using distilled water and subsequently ground with an equimolar amount of R-pyridyl (where R = 4-CN, H, 4-Bz, 4-Me, 4-Et, and 4-MeNH) for 3–5 hours. The resulting mixture was left undisturbed overnight in a dark environment, and the solid product obtained underwent washing with ethanol before being recrystallized using distilled water. The structural details of the cis-[Co(tn)2(Rpy)Br]Br2 complexes are illustrated in Fig. 1.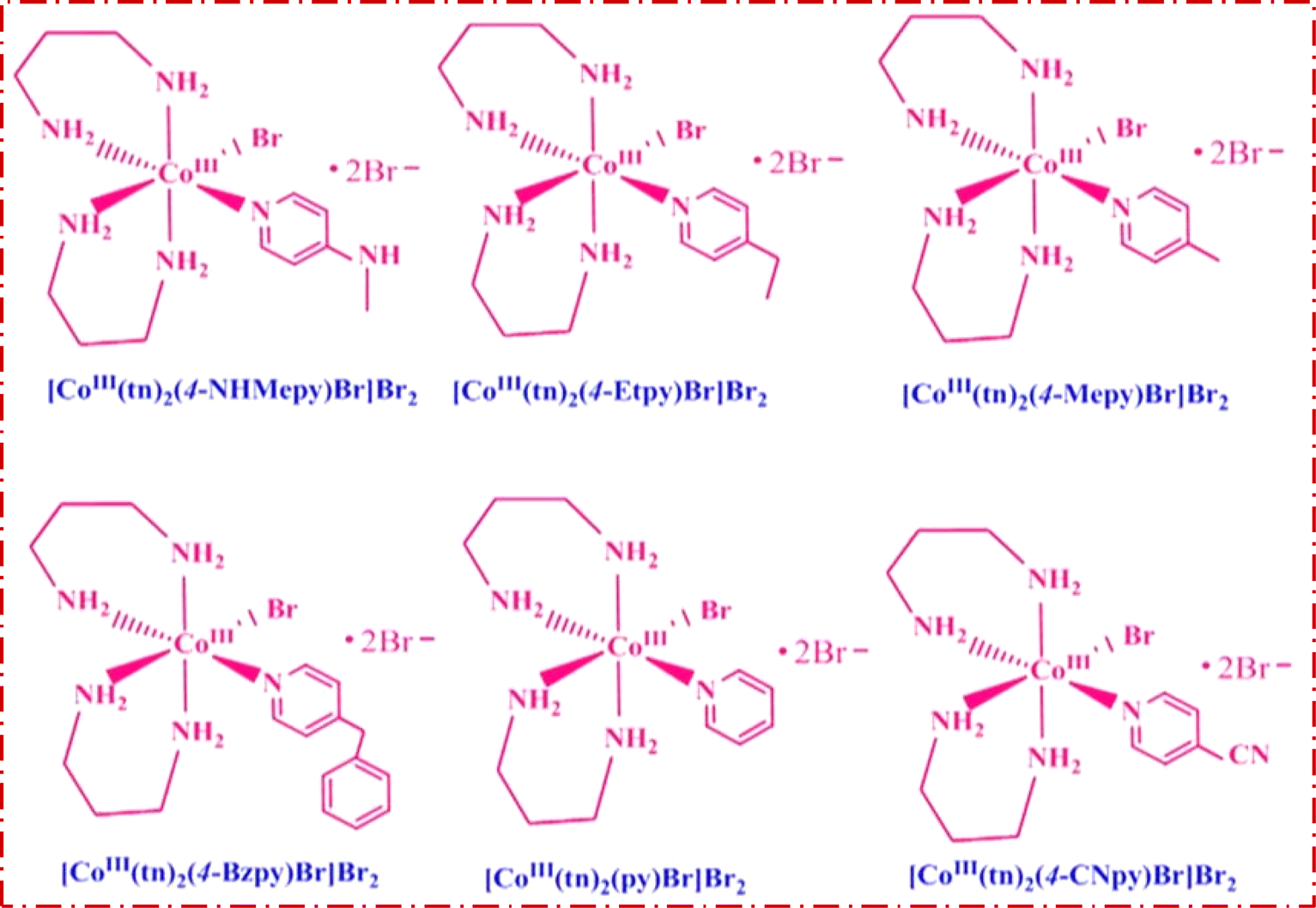 | ||
| Fig. 1 Structure of the cis-[Co(tn)2(Rpy)Br]Br2 complexes. | ||
cis-[Co(tn)2(4-MeNHpy)Br]Br2 (1)
Recrystallized from water to give dark pink colored powder, yield 0.813 g, 81%. Anal. calc. for C12H28CoN6Br3: C 25.97, H 5.09, N 15.14, Br 43.19%. Found: C 25.23, H 4.93, N 14.33, and Br 43.11%. FTIR (KBr disc) cm−1, 3504.06(b): O–H stretching vibrations of coordinated or adsorbed water molecules, 3207.16(w): stretching vibration of NH groups, 3070.63(w): stretching vibration of aromatic C–H bonds., 1613.25(s): the stretching vibration of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in the pyridine ring, 1416.68(s): the bending vibration of N–H bonds or CC vibrations, 1305.65(w): stretching vibration of C–N bonds, 1229.98(m): C–N stretching vibrations, 1156.79(w): C–H bending vibrations, 1100.04(w): C–H bending vibrations, 1072.89(m): to stretching vibrations of C–N bonds or C–H bending vibrations, 1012.03(m): stretching vibrations of C–N bonds, 928.97(w): C–H bending vibrations, 826.98(s): bending vibrations of C–H bonds, 757.89(w), 697.04(w), 651.80(w), 594.23(w) and 522.68(w): bending vibrations of C–H bonds or metal–ligand vibrations. UV-vis (water 1.80 × 10−3 M) λmax, nm (εmax M−1 cm−1), 359.9 (1.23 × 102), 518.3 (90.47).
N bond in the pyridine ring, 1416.68(s): the bending vibration of N–H bonds or CC vibrations, 1305.65(w): stretching vibration of C–N bonds, 1229.98(m): C–N stretching vibrations, 1156.79(w): C–H bending vibrations, 1100.04(w): C–H bending vibrations, 1072.89(m): to stretching vibrations of C–N bonds or C–H bending vibrations, 1012.03(m): stretching vibrations of C–N bonds, 928.97(w): C–H bending vibrations, 826.98(s): bending vibrations of C–H bonds, 757.89(w), 697.04(w), 651.80(w), 594.23(w) and 522.68(w): bending vibrations of C–H bonds or metal–ligand vibrations. UV-vis (water 1.80 × 10−3 M) λmax, nm (εmax M−1 cm−1), 359.9 (1.23 × 102), 518.3 (90.47).
cis-[Co(tn)2(4-Etpy)Br]Br2 (2)
Recrystallized from water to give dark pink colored powder, yield 0.724 g, 72%. Anal. calc. for C13H29CoN5Br3: C 28.18, H 5.28, N 12.64, Br 43.26%. Found: C 27.55, H 4.83, N 12.41, and Br 42.21%. FTIR (KBr disc) cm−1, 3222.63(b): O–H stretching vibrations of coordinated water molecules, 1680.51(w): the stretching vibration of the CN bond, 1433.07(s): the bending vibration of N–H bonds or CC vibrations, 1144.43(s): stretching vibrations of C–N bonds or C–H bending vibrations, 1088.11(s): stretching vibrations of C–N bonds, 980.16(w): stretching vibrations of C–N bonds, 625.31(m), 552.55(w) and 457.53(w): bending vibrations of C–H bonds or metal–ligand vibrations. UV-vis (water 1.84 × 10−3 M for UV region 1.84 × 10−4 M for visible region) λmax, nm (εmax M−1 cm−1), 265.06 (4.34 × 103), 358.64 (1.23 × 103), 516.30 (8.85 × 102).
cis-[Co(tn)2(4-Mepy)Br]Br2 (3)
Recrystallized from water to give dark pink colored powder, yield 0.867 g, 87%. Anal. calc. for C12H27CoN5Br3: C 26.69, H 5.04, N 12.97, Br 44.39%. Found: C 26.35, H 4.83, N 12.33, and Br 44.11%. FTIR (KBr disc) cm−1, 3437(b) and 3350.90(b): O–H stretching vibrations of coordinated water molecules, 1616.61(s): the stretching vibration of the CN bond in the pyridine ring, 1558.71(w): stretching vibrations of C–N bonds or C–H bending vibrations, 1503.62(m), 1443.28(m), 1424.97(m) and 1382.80(w): bending vibrations of N–H bonds or CC vibrations, 1230.69(m) and 1213.30(m): stretching vibrations of C–N bonds or C–H bending vibrations, 1072.04(w): stretching vibrations of C–N bonds or C–H bending vibrations, 1020.47(m) and 987.86(w): stretching vibrations of C–N bonds or C–H bending vibrations, 813.15(s): bending vibrations of C–H bonds, 722.99(m), 597.51(w) and 539.55(w): bending vibrations of C–H bonds, 493.71(s): bending vibrations of C–H bonds or metal–ligand vibrations. UV-vis (water 1.89 × 10−3 M for UV region 1.89 × 10−4 M for visible region) λmax, nm (εmax M−1 cm−1), 256.06 (6.86 × 104), 357.92 (1.22 × 103), 512.83 (8.45 × 102).
cis-[Co(tn)2(4-Bzpy)Br]Br2 (4)
Recrystallized from water to give dark pink colored powder, yield 0.793 g, 79%. Anal. calc. for C18H32CoN5Br3: C 35.03, H 5.23, N 11.35, Br 38.84%. Found: C 34.84, H 4.82, N 11.12, and Br 38.19%. FTIR (KBr disc) cm−1, 3732.52(w) and 3443.47(b): stretching vibrations of O–H bonds, likely from coordinated water molecules, 3212.84(w), 3119.87(w), 3060.38(w) and 3030.58(w): stretching vibrations of N–H bonds, 1662.80(w) and 1611.95(s): the stretching vibration of the CC and CN bonds in the benzene and pyridine rings, respectively, 1556.84(m), 1496.24(s), 1452.26(m) and 1423.59(s): bending vibrations of N–H bonds or CC vibrations, 1282.93(w) and 1223.04(s): stretching vibrations of C–N bonds, 1115.93(w), 1070.16(m), 1037.82(w), 1014.47(m), 925.34(w), 856.26(w) and 835.56(w): stretching vibrations of C–N bonds or C–H bending vibrations, 795.36(s), 743.79(s), 700.85(s), 670.81(w), 644.32(w) and 617.02(s): bending vibrations of C–H bonds, 559.30(m), 486.93(s) and 459.55(w): C–H bonds or metal–ligand vibrations. UV-vis (water, 1.77 × 10−3 M for UV region 1.77 × 10−4 M for visible region) λmax, nm (εmax M−1 cm−1), 257.21 (3.76 × 104), 350.26 (1.57 × 103), 516.30 (7.56 × 102).
cis-[Co(tn)2(py)Br]Br2 (5)
Recrystallized from water to give dark pink colored powder, yield 0.871 g, 87%. Anal. calc. for C11H25CoN5Br3: C 25.12, H 4.79, N 13.32, Br 45.57%. Found: C 24.55, H 4.33, N 13.23, and Br 44.91%. FTIR (KBr disc) cm−1, 3219.35(w), 3164.02(w), 3081.60(w) and 2925.59(w): stretching vibrations of N–H bonds, 1603.37(w) and 1562.07(w): stretching vibrations of the CN bond in the pyridine ring, 1435.35(s): bending vibrations of N–H bonds or CC vibrations, 1402.79(m), 1321.34(w), 1289.22(w) and 1250.70(w): bending vibrations of N–H bonds or C–H bending vibrations, 1143.89(s), 1095.21(s), 981.21(w), 926.11(w) and 886.70(w): stretching vibrations of C–N bonds or C–H bending vibrations, 738.33(w), 625.21(m), 553.07(w), 520.69(w) and 450.04(w): bending vibrations of C–H bonds or metal–ligand vibrations. UV-vis (water 3.17 × 10−3 for UV region 3.17 × 10−4 M for visible region) λmax, nm (εmax M−1 cm−1), 255.39 (1.02 × 104), 351.57 (8.42 × 102), 514.86 (4.15 × 102).
cis-[Co(tn)2(4-CNpy)Br]Br2 (6)
Recrystallized from water to give dark pink colored powder, yield 0.820 g, 82%. Anal. calc. for C12H25CoN6Br3: C 26.11, H 4.57, N 15.22, Br 43.42%. Found: C 25.55, H 4.13, N 15.03, and Br 42.82%. FTIR (KBr disc) cm−1, 3473.63(b): O–H stretching vibrations of coordinated water molecules, 3199.76(w), 3070.63(w) and 2941.51(w): stretching vibrations of N–H bonds, 1944.69(w): stretching vibrations of the C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N bond in the cyano group, 1716.87(s): stretching vibrations of the CN bond in a carbonyl group, 1670.81(m), 1602.55(s), 1549.10(m), 1465.21(m), 1423.26(s), 1373.91(w) and 1321.28(w): bending vibrations of N–H bonds or C–H bending vibrations, 1229.98(m), 1146.09(w), 1069.61(w), 1016.97(m), 933.07(w), 826.98(m), 757.89(w), 700.32(w) and 644.40(w): stretching vibrations of C–N bonds or C–H bending vibrations, 537.48(m): bending vibrations of C–H bonds or metal–ligand vibrations. UV-vis (water 1.81 × 10−3 M) λmax, nm (εmax M−1 cm−1), 357.9 (1.26 × 102), 512.1 (89.44).
N bond in the cyano group, 1716.87(s): stretching vibrations of the CN bond in a carbonyl group, 1670.81(m), 1602.55(s), 1549.10(m), 1465.21(m), 1423.26(s), 1373.91(w) and 1321.28(w): bending vibrations of N–H bonds or C–H bending vibrations, 1229.98(m), 1146.09(w), 1069.61(w), 1016.97(m), 933.07(w), 826.98(m), 757.89(w), 700.32(w) and 644.40(w): stretching vibrations of C–N bonds or C–H bending vibrations, 537.48(m): bending vibrations of C–H bonds or metal–ligand vibrations. UV-vis (water 1.81 × 10−3 M) λmax, nm (εmax M−1 cm−1), 357.9 (1.26 × 102), 512.1 (89.44).
Note: complex numbers from (1) to (6) have been assigned to each molecular formula provided in the above section. Hereafter, these complex numbers refer to the respective complexes.
Photocatalytic reduction of Co(III)-R-pyridine complexes
The IET process was investigated through the photoreduction of the cis-[Co(tn)2(Rpy)Br]Br2 complex employing TiO2 nanocrystals in water/aqueous 2-propanol solutions under 254 nm light irradiation. The photoefficiency of Co2+ ion formation from the above system was estimated using Kitson's method.30 Photoreduction experiments were conducted in a reactor vessel utilizing a 254 nm low-pressure mercury vapor lamp, housed in a Lab Guard fume hood and covered with a black polythene sheet to eliminate extraneous light. The photoreactor, a double-walled quartz vessel, contained a photolyte mixture of 100 mg of TiO2 nanocrystals in 80 mL of aqueous 2-proponal solution of cis-[Co(tn)2(Rpy)Br]Br2 (1.98 × 10−3 M), and 10 mL of 1 M NaNO3 (for stabilize pH) in the inner jacket, with cool water in the outer jacket. Before irradiation, the catalyst suspension was achieved through ultrasonic treatment and continuous magnetic stirring in the dark to attain equilibrium in cobalt(III) complex ion adsorption/desorption on the catalyst. The photolyte suspension was exposed to 254 nm light for specific irradiation periods (2–16 min). Subsequently, 4 mL aliquots were sampled, centrifuged to remove solid particles, and then subjected to spectral analysis. To minimize experimental error, each experiment was replicated at least three times for the same sample, and the mean value was calculated. The photoefficiency of Co2+ formation, expressed as a percentage (PE %), was determined using the formula: PE (%) = [(At − Ai)/At] × 100, where Ai and At are the absorbances of the photolysed solutions initially and at a defined time interval ‘t’, respectively.Results and discussion
Characterization of cobalt(III)-R-pyridyl complexes
The cobalt(III)-R-pyridine complexes, cis-[Co(tn)2(Rpy)Br]Br2, has been successfully synthesized, and its structural characteristics were comprehensively elucidated through a multi-faceted analytical approach. The determination of the complex's structure employed techniques such as FTIR, UV-vis spectroscopy, luminescence studies, and, notably, single crystal X-ray crystallography. The application of FTIR and UV-vis spectroscopy provided valuable insights into the molecular composition and electronic properties of the synthesized complex. Luminescence studies further probed the optical properties, adding a dimension to our understanding of the complex's behavior in excited states. However, the most definitive evidence supporting the cis structure of the aforementioned complexes was derived from the meticulous examination of their crystal structure using single crystal X-ray crystallography. This technique, known for its unparalleled precision in revealing molecular arrangements, unambiguously confirmed the cis configuration of the cobalt(III)-pyridine complex. The synthesis and structural elucidation of the cis-[Co(tn)2(Rpy)Br]Br2 complex were conducted through a synergistic application of diverse analytical methods, with single crystal X-ray crystallography serving as the definitive tool in confirming the cis configuration of the complex.The FT-IR spectra of complexes (1), (3), (5) and (6) are depicted in Fig. 2 and for remaining complexes (2) and (4) in Fig. S1.† Analysis of the IR data reveals distinctive peaks in the range of 3500 to 3400 cm−1, attributable to the antisymmetric and symmetric ν(O–H) stretching modes of lattice-bound water.31 The existence of stretching absorption bands within the range of 1650–1620 cm−1 affirms bending modes associated with ν(O–H), signifying the potential presence of the hydrated form of the molecule through hydrogen bonding in some of the complexes.26,32 However, it's important to note that this characteristic is not illustrated in the single crystal studies of the representative complex (4) discussed in the subsequent sections.
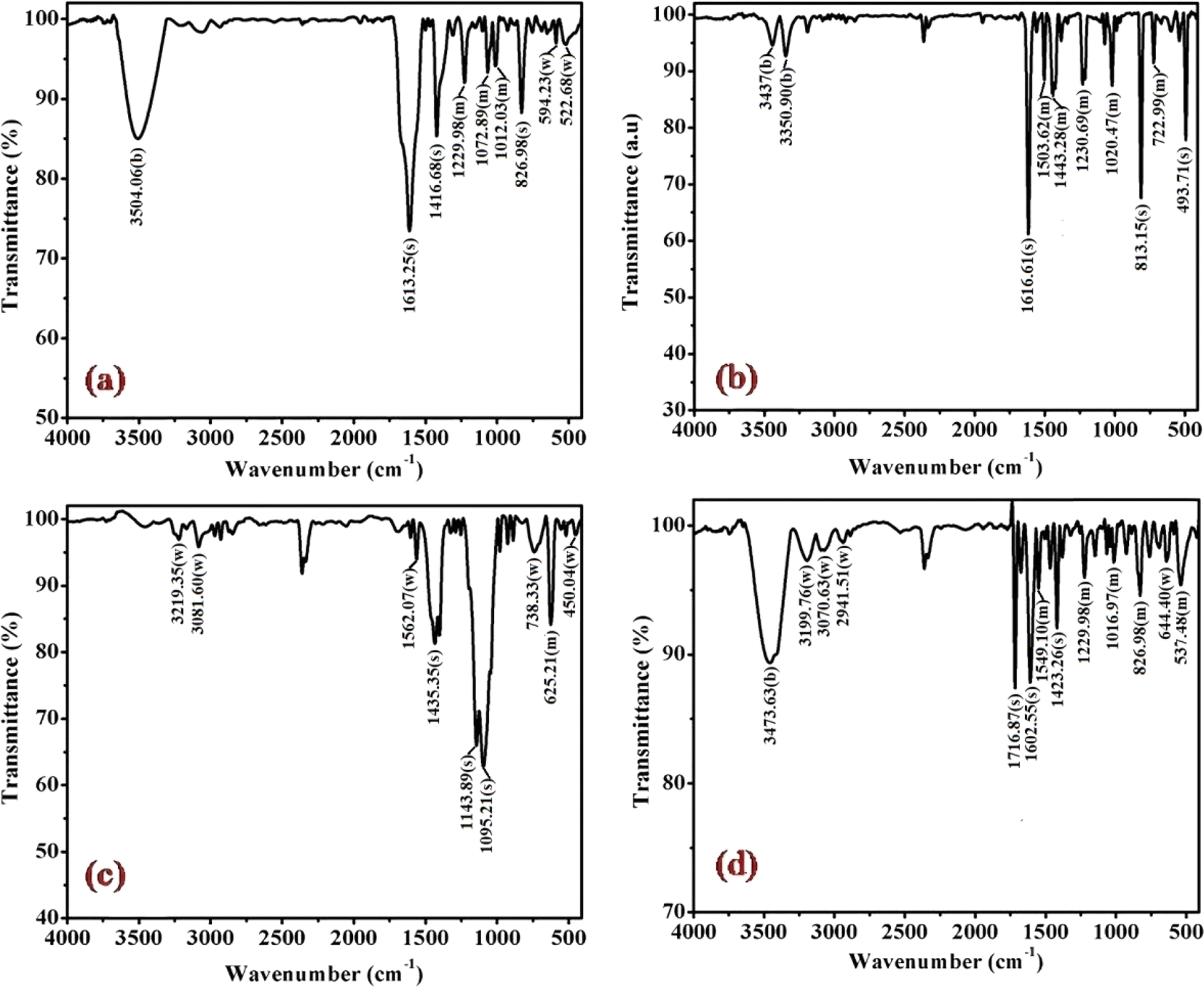 | ||
| Fig. 2 Displays the FTIR spectra of complexes (a) (1), (b) (3), (c) (5), and (d) (6), recorded in KBr at 298 K. | ||
Significant shifts in the antisymmetric ν(N–H) stretching modes at ∼3318–3005 cm−1 (metal-amine at ∼3300 cm−1) and deformation mode at ∼1632–1497 cm−1 (metal-amine ∼1600 cm−1) provide clear evidence of cobalt-amine coordination. Typically, [Co(N–N)2LX]2+ type molecules exhibit vibrations at 910–800 cm−1 in the –CH2 rocking region, with cis complexes manifesting more bands in this region than trans complexes.31,32 In this study, bands at ∼812, ∼868, and ∼910 cm−1 confirm the –CH2 rocking region, affirming the coordination of 1,3-propanediamine molecules to the metal ion. The band at 1617 cm−1 is assigned to the heterocyclic ring vibration, indicating coordination of the nitrogen atom of the 4-substituted pyridine ligand with the metal ion. Newly observed strong bands in the 492 cm−1 range may be attributed to M−N stretching. Bands in the 1320 cm−1 region, corresponding to NH2 deformation, further confirm the structural similarity of the Co(III) complexes to the earlier proposed [Co(en)2(RNH2)Cl]2+ ion.31 Distinctive bands at 1458 cm−1 and 1578–1590 cm−1 are observed, corresponding to CC and CN vibrations of the pyridine ring, respectively. Additionally, bands at ∼589 cm−1 and ∼590 cm−1 corresponding to Co–N(tn) and Co–N of NH2(tn) bending, respectively, are exhibited around 1650 cm−1.32 The FTIR spectral studies indicate that the remaining spectra align with identical peak patterns, as depicted in Fig. S1.† Confirmation from these studies suggests the presence of tn and R-py moieties within the coordination sphere, with a high likelihood of adopting a cis configuration, barring exceptions.
In practice, the identification of trans structures is often associated with a weak band in the range of approximately 625–700 nm, whereas the corresponding cis-CoL4X2 complexes typically exhibit absorption around 550 nm.32 The [Co(tn)2(Rpy)Br]Br2 complex manifests two distinct bands at 359 nm and approximately 516 nm, as illustrated in Fig. 3. Consistent with this pattern, the spectra of the remaining complexes exhibit analogous peaks, as depicted in Fig. S2.† The absorption bands at around 360 nm in these complexes are attributed to the ligand-to-metal charge transfer (LMCT) band, while those at approximately 515 nm correspond to the first absorption band, specifically the 1A1g → 1T1g transition. The observed positions and intensities strongly indicate a cis configuration rather than a trans structure for these complexes.33 Additionally, the electronic spectrum of these complexes bears resemblance to that of the cis-β-[Co(trien)Cl]2+ ion, suggesting a potential cis-β arrangement of ligands around the Co(III) ion.34
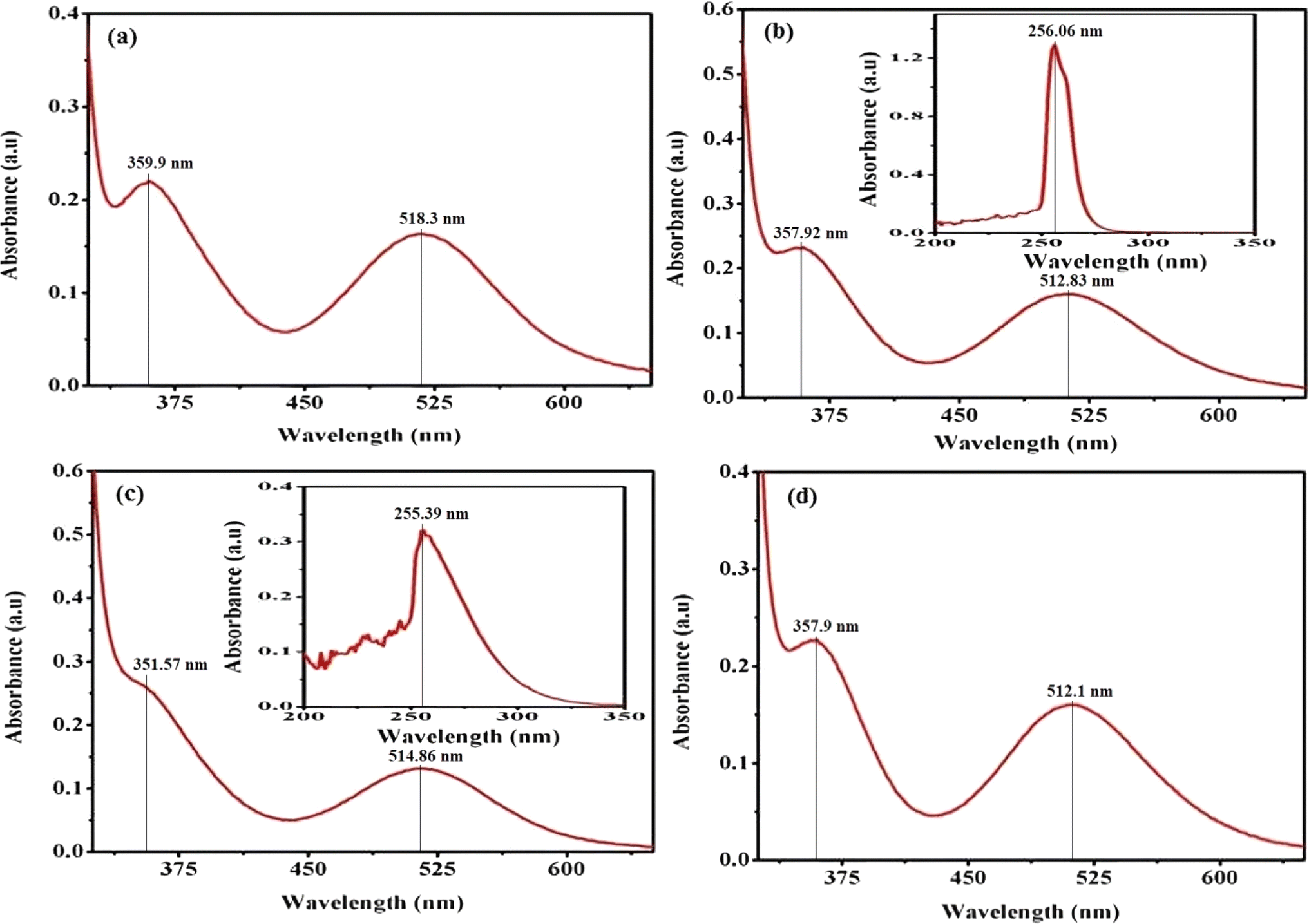 | ||
| Fig. 3 Presents the UV-vis electronic absorption spectra of complexes (a) (1), (b) (3), (c) (5), and (d) (6), recorded in water (300 μM) at 298 K. The insert focuses on the 200–350 nm range. | ||
Furthermore, the positions of these bands vary across the series of complexes. In cobalt(III) complexes, regular spin-allowed transitions are typically 1A1 → 1T1 and 1A1 → 1T2, which split into components for complexes with lower symmetry, such as [Co(tn)2(Rpy)Br]2+, resulting in 1A1 → 1A2 + 1Ea and 1A1 → 1B2 + 1Eb transitions. Notably, among these transitions, only the 1A1 → 1B2 is magnetically forbidden.35 The higher energy visible band is centered at 360 nm, while the low-energy band is expected around ∼515 nm, though the latter is obscured under the conditions of the present experimental setup.
The complex ion, [Co(tn)2(Rpy)Br]Br2, exhibits a prominent band in the UV region. This phenomenon is interpreted through the lens of the linear combination of atomic orbitals - molecular orbital (LCAO-MO) theory, suggesting that these bands result from charge transfer events involving the p-orbitals of the halide ion and predominantly the dz2 orbital of the cobalt(III) ion. The π → π* transition attributed to the Rpy ligand occurs at a shorter wavelength, approximately 244 nm, as illustrated in the inset of Fig. 3(b) and (c). It's worth noting that at this concentration, the detection of the high-energy charge transfer band is either obscured in some cases or not observed.36
In pyridine systems, optical transitions from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO), π → π*, typically occur at 244 nm. However, the presence of acceptor/donor groups attached to the ring can alter the energies of LUMO/HOMO levels. The intensities of absorption due to d–d bands in [Co(tn)2(Rpy)Br]2+ parallel the magnitude of ε for [Co(en)(NH3)3CN]2+ ion.37 Notably, it has been reported that the molar extinction coefficient of the d–d band increases approximately tenfold when X is substituted by Cl → Br → I.32,38 Crucially, a weak band around 515 nm serves as a characteristic feature of cis structures.39 Moreover, cobalt(III) complexes with lower symmetry and aromatic amines are inclined to exist in the cis-form, making the cis configuration more probable in this context.
Upon comparing the various peaks observed in complexes (1) through (6), it becomes apparent that there is no discernible consistency in changes from complex (1) to complex (2). This lack of uniformity suggests that the sixth ligand of Rpy does not instigate systematic alterations in the optical characteristics of our synthesized complexes. However, a more in-depth examination reveals an intriguing trend: there is an approximate shortening of absorption wavelengths for both charge transfer bands and d–d bands from complex (1) with R = 4-MeNH ligand to complex (6) with R = 4-CN ligand in the sixth ligand of RPy within our prepared complexes. This trend is illustrated in Fig. 3.
This observed shift suggests that the presence of a releasing group in RPy leads to longer absorption wavelengths compared to a withdrawing group. Despite this influence, it's important to note that the sixth ligand's impact on the optical behavior of our complexes is modest and lacks a consistent pattern across the series. This indicates that while the ligand does contribute to variations in optical properties, there are likely other factors at play influencing the overall optical behavior. Further investigation into these factors could provide deeper insights into the underlying electronic structure governing the optical properties of these complexes.
Single crystal X-ray diffraction analysis
The single crystal refinement data for complex (3) reveals a symmetric structure with a twofold rotation axis, belonging to the monoclinic crystal system. The refined unit cell parameters are as follows: a = 9.058(2) Å, b = 16.135(4) Å, c = 13.128(3) Å, α = γ = 90°, β = 92.61(2)°, and a volume of 1916.68 Å3. The crystal structure adopts the space group P21/n, and the unit cell accommodates four formula units (z = 4). Additionally, further insights into the crystallographic details and intermolecular interactions within the complex are provided in the subsequent analysis.The crystal structure of the obtained cobalt(III) complex was elucidated through X-ray diffraction, revealing an octahedral coordination environment for the complex (3) complex. The single-crystal X-ray diffraction refinement affirmed that the molecule is classified under the monoclinic crystal system, featuring a distorted octahedral geometry, as illustrated in Fig. 4. Detailed insights into the molecular arrangement, bond angles, and interatomic distances were gleaned from this comprehensive structural analysis. Notably, the packing diagram derived from the X-ray diffraction data, as depicted in Fig. 5, highlights the absence of significant intermolecular interactions. This information is pivotal in understanding the spatial arrangement of individual molecules within the crystal lattice.
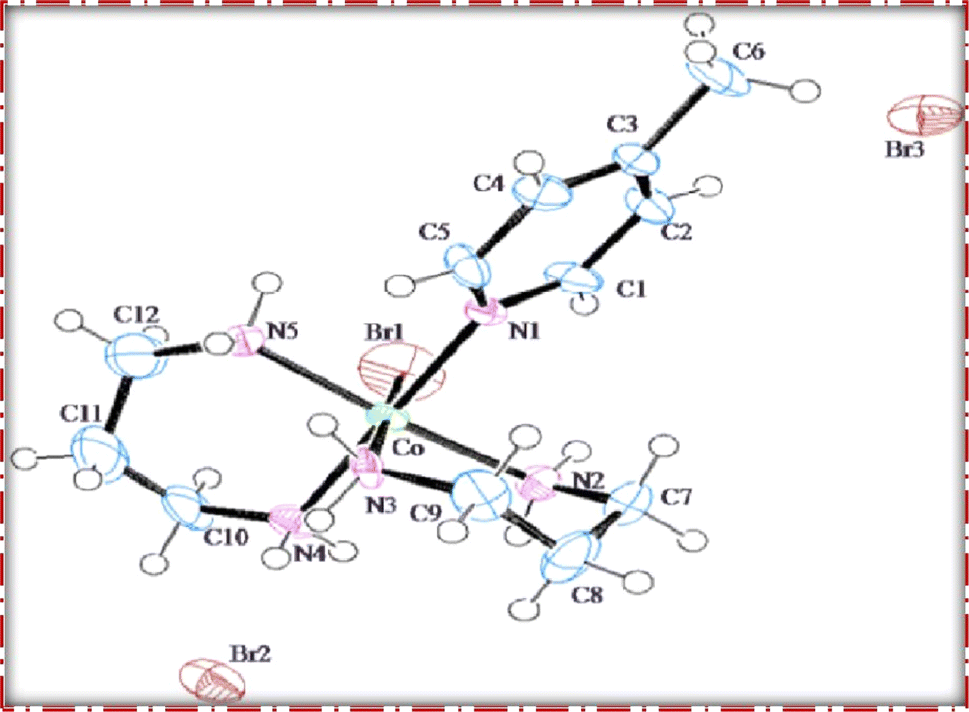 | ||
| Fig. 4 ORTEP diagram of [Co(tn)2(4-Mepy)Br]Br2 (3) complex. | ||
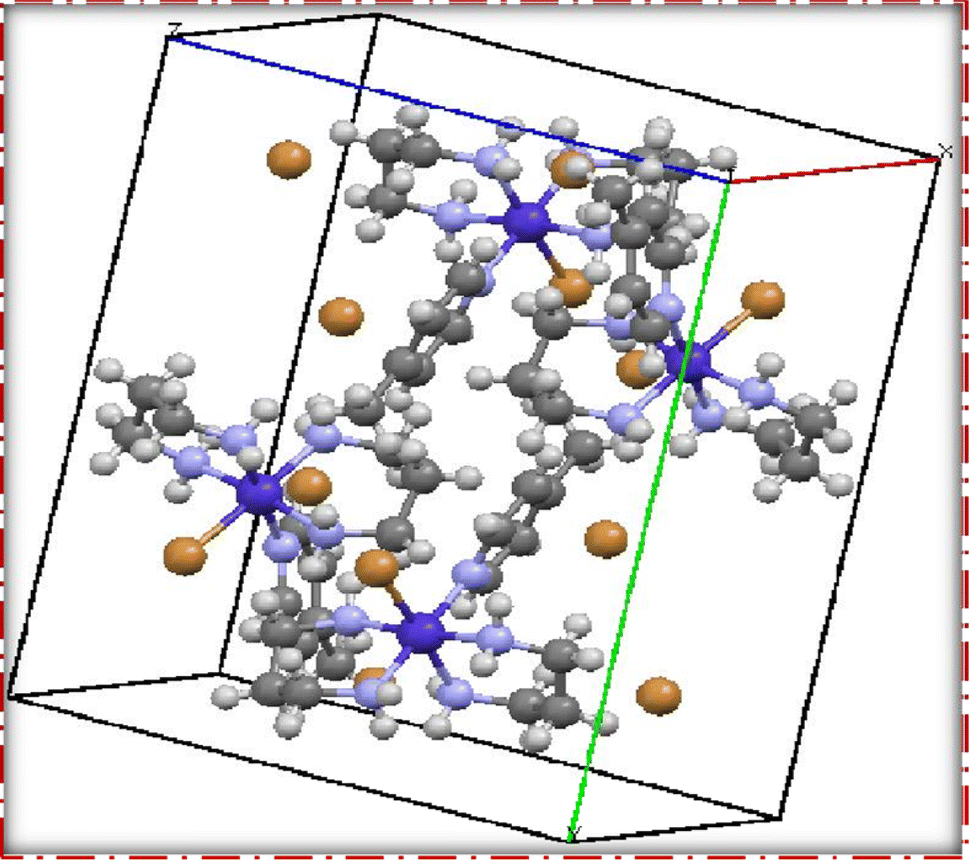 | ||
| Fig. 5 Packing diagram of [Co(tn)2(4-Mepy)Br]Br2 (3) complex. | ||
Moreover, the single crystal X-ray diffraction studies provided crucial validation for the structural integrity of our prepared representative complex. It is anticipated that a similar structural motif is maintained across the remaining complexes synthesized in this study, reinforcing the reliability and consistency of the synthetic methodology employed. The wealth of information obtained from these crystallographic analyses lays a robust foundation for the subsequent interpretation of the complex's properties and behaviors.
Fluorescence study of [Co(tn)2(Rpy)Br]Br2
Fig. 6, 7 and S3† illustrate the characteristic steady-state emission spectra of all prepared complexes (1) to (6) in water (∼180 μM). The emission spectra exhibit maxima at approximately λemi = 341 nm (50.12 × 103 cps), monitored in the wavelength range from 300 to 500 nm, under excitation at λexc = 315 nm (monitored wavelength ranging from 200 to 350 nm). Additionally, emission peaks at around λemi = 620 nm (8.69 × 103 cps) are observed, monitored in the wavelength range from 520 to 700 nm, upon excitation at λexc = 520 and 564 nm (monitored wavelength ranging from 400 to 600 nm). A comprehensive compilation of all emission data is provided in Table 1.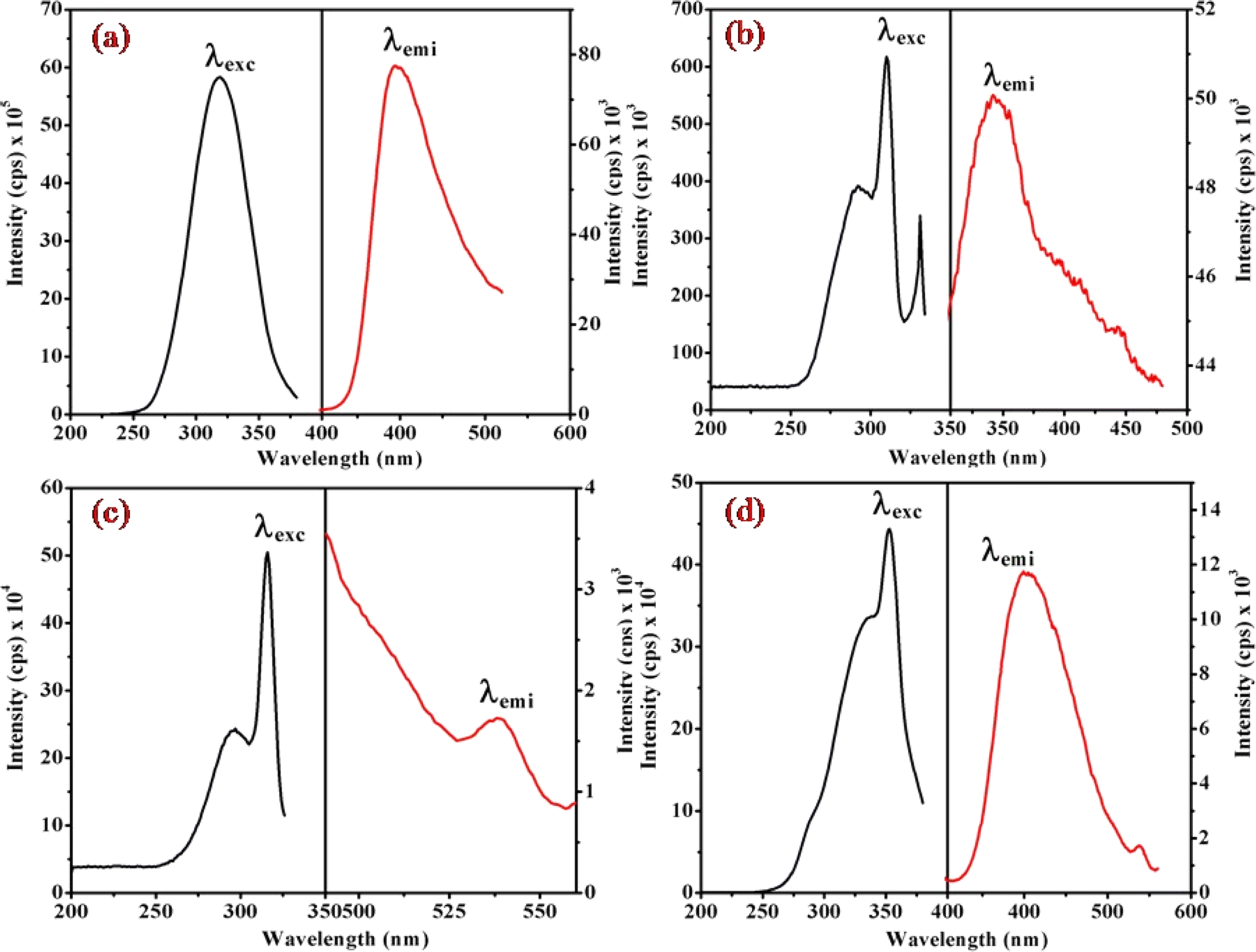 | ||
| Fig. 6 Presents the steady-state excitation and emission spectra of complexes (a) (1), (b) (3), (c) (5), and (d) (6), recorded at λmax = 256 nm in water (180 μM) at 298 K. | ||
 | ||
| Fig. 7 Displays the steady-state excitation and emission spectra of complexes (a) (1), (b) (3), (c) (5), and (d) (6), recorded at λmax = 520 nm in water (298 μM) at 298 K. | ||
| Complexes | PL recorded at λmax = 256 nm | PL recorded at λmax = 520 nm | ||
|---|---|---|---|---|
| λem (nm) | Intensity (cps) × 103 | λem (nm) | Intensity (cps) × 103 | |
| (1) | 396.60 | 77.99 | 544.60 | 58.21 |
| — | — | 607.60 | 31.08 | |
| (2) | 339.80 | 132.25 | 541.70 | 70.04 |
| 412.60 | 95.68 | 563.80 | 63.17 | |
| 432.40 | 95.91 | 618.40 | 62.08 | |
| (3) | 341.40 | 50.12 | 552.10 | 57.19 |
| — | — | 576.50 | 50.59 | |
| — | — | 635.70 | 55.56 | |
| (4) | 400.30 | 11.76 | 619.90 | 8.69 |
| (5) | 538.90 | 1.73 | 615.90 | 51.88 |
| (6) | 399.10 | 11.79 | 619.90 | 8.70 |
When examining the fluorescence behavior of complexes (1) through (6), we observed no precise systematic alterations in emission characteristics. However, a discernible trend emerged wherein there was an approximate lengthening of emission wavelengths from complex (1) with R = 4-MeNH ligand to complex (6) with R = 4-CN ligand in the sixth ligand of RPy within our prepared complexes. This suggests that the presence of a releasing group in RPy results in shorter emission wavelengths compared to a withdrawing group, as detailed in Table 1. While this indicates that the sixth ligand does modestly influence the emission behavior of our prepared complexes, it does not exhibit a consistent pattern of change across the series.
The origin of the emission bands can be attributed to the spin-allowed transition (1A1 → 1T2g) state emission, with a secondary weak emission band stemming from the second spin-allowed transition (1A1 → 1T1g). Additional insights into the emission decay kinetics were gleaned through single photon counting emission measurements on the cobalt complexes. Intriguingly, the kinetics observed in air-equilibrated water solutions exhibit a triphasic nature, suggesting a multifaceted emission process. These emission studies not only contribute to the understanding of the photophysical properties of the cobalt(III) complex but also offer valuable data for unraveling the intricate processes underlying its emission behavior in different excitation regimes.
In the luminescence lifetime experiments, the recorded data exhibit triple exponential decays, as illustrated by the example fit presented in Fig. 8. Notably, the sum of the squares of residuals indicates a random distribution over the regions used to determine lifetimes, suggesting a well-fitted model as the deviations are evenly distributed around zero. The obtained luminescence lifetime results provide insightful information about the excited-state dynamics of the [Co(tn)2(Rpy)Br]2+ ion. The triple-exponential decays observed in the data suggest the presence of at least three distinct excited-state species. This can be rationalized by considering different spatial locations or conformations of the [Co(tn)2(Rpy)Br]2+ ion within the studied environment.
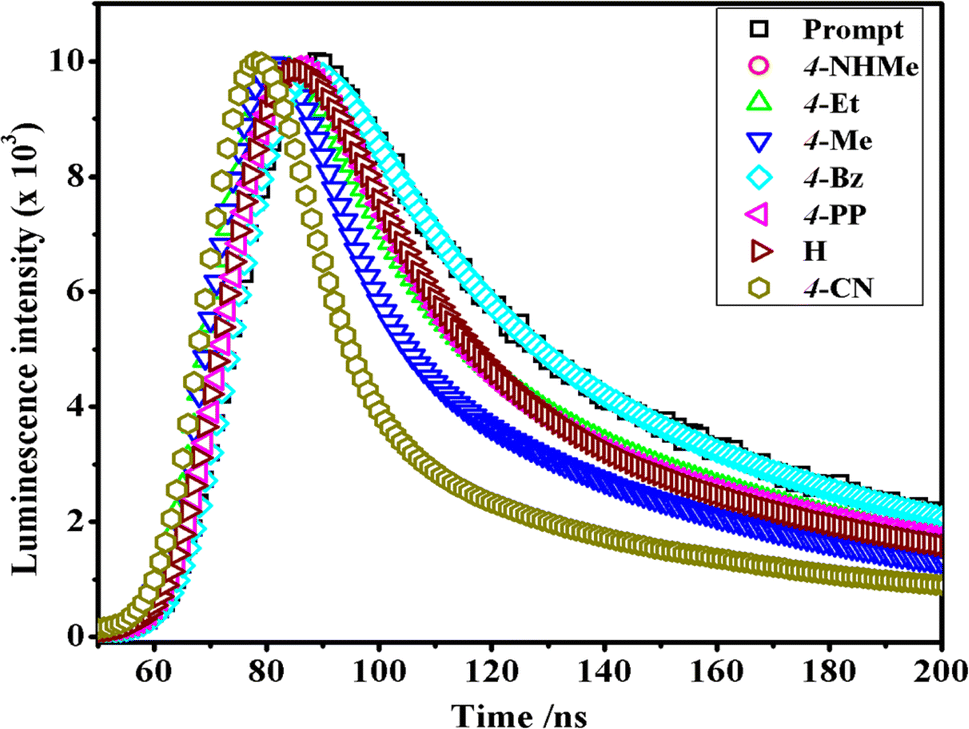 | ||
| Fig. 8 Comparison of time-resolved luminescence profiles for [Co(tn)2(Rpy)Br]2+ complexes in water solution (∼298 μM) at 298 K. | ||
Table 2 outlines the luminescence lifetime decay characteristics for the [Co(tn)2(Rpy)Br]2+ ion. The triple exponential decays indicate the coexistence of multiple excited-state species, shedding light on the complex nature of the luminescent behavior exhibited by the investigated cobalt(III) complex. This comprehensive analysis of luminescence lifetime data not only contributes to a nuanced understanding of the excited-state dynamics but also underscores the intricate nature of the photochemical processes governing the [Co(tn)2(Rpy)Br]2+ ion.
| Complexes | τ1 (ns) | τ2 (ns) | τ3 (ns) | χ2 |
|---|---|---|---|---|
| (1) | 0.701 | 4.348 | 16.172 | 1.03 |
| (2) | 0.767 | 4.382 | 16.040 | 1.05 |
| (3) | 3.280 | 0.359 | 13.933 | 1.12 |
| (4) | 1.028 | 4.639 | 8.596 | 1.03 |
| (5) | 0.917 | 4.588 | 1.201 | 1.11 |
| (6) | 0.827 | 4.448 | 1.251 | 1.18 |
Magnetic measurement of cis-[Co(tn)2(Rpy)Br]Br2 complexes
Magnetic hysteresis loops, representing a distinctive fingerprint of ferromagnetism, were experimentally observed as a crucial aspect of our investigation. The vibrating sample magnetometry (VSM) technique was employed to characterize the hysteresis curves of complexes (1)–(6) in a DC magnetic field. Fig. 9 and S4† illustrates the hysteresis loops for the cobalt(III) complexes, plotting moment/mass (emu g−1) against field (Oe). The presence of hysteresis loops in the magnetic measurements strongly suggests the manifestation of ferromagnetic behavior within the cobalt(III) complexes. Intriguingly, the intrinsic coercivity, remanent magnetization, and molar magnetic susceptibility values, derived from the VSM analysis and detailed in Table 3, reinforce this observation. The results collectively point towards the weak ferromagnetic nature of all [Co(tn)2(Rpy)Br]Br2 complexes, coupled with significant magnetization.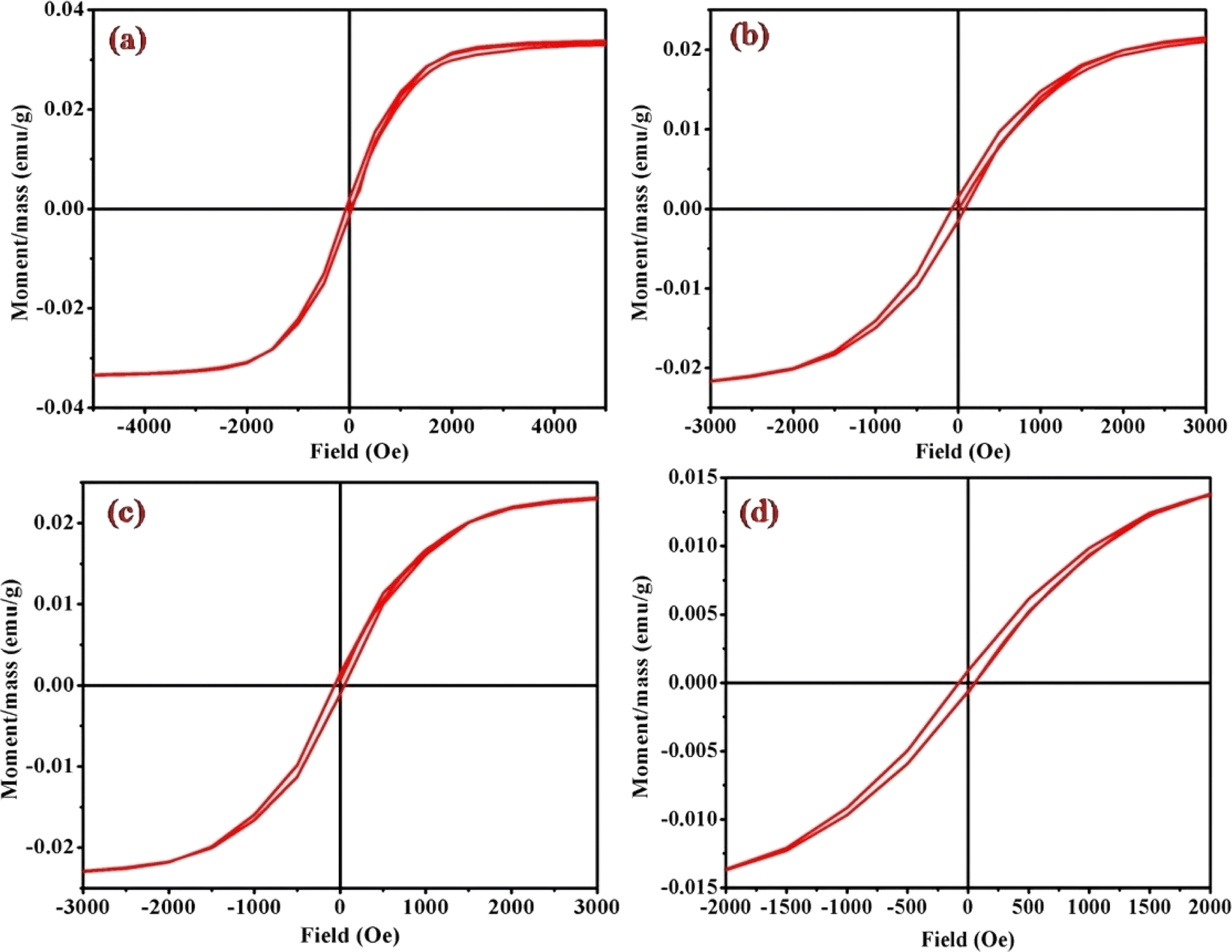 | ||
| Fig. 9 Magnetic hysteresis loops for complexes (a) (1), (b) (3), (c) (5), and (d) (6), measured at 298 K. Note the varied axis scales. All data have been corrected for diamagnetism. The correlation between the molar magnetic susceptibility (χmol) and the acidity constant (pKa) of Rpy provides valuable insights into the magnetic properties of the complexes (1)–(6) in Table 4. The observed linear relationship, expressed as χmol = 2.02 × 10−4 – 0.0551 cm3 mol−1 when pKa = 9.70–1.90, indicates that the magnetic susceptibility increases linearly with a decrease in acidity constant. This intriguing correlation suggests a direct influence of the electronic nature of the axial ligand (Rpy) on the magnetic behavior of the complexes. | ||
| Complexes | pKa (Rpy) | Ms × 10−3 (emu g−1) | χmass × 10−3 (cm3) | χmol × 10−4 (cm3) | Hc, Oe | Mr ×10−3 (emu g−1) | μr (emu g−1) |
|---|---|---|---|---|---|---|---|
| (1) | 9.70 | 33.82 | 2.09 | 2.02 | 55 | 1.65 | 1.002 |
| (2) | 6.02 | 15.59 | 3.41 | 3.38 | 82 | 1.18 | 1.003 |
| (3) | 6.02 | 23.19 | 4.10 | 3.96 | 78 | 1.50 | 1.004 |
| (4) | 5.59 | 13.20 | 32.80 | 36.24 | 65 | 0.91 | 1.033 |
| (5) | 5.25 | 23.94 | 37.00 | 34.85 | 57 | 1.28 | 1.024 |
| (6) | 1.90 | 16.06 | 558.00 | 551.50 | 65 | 0.76 | 1.016 |
A closer examination of the magnetic data unveils a consistent variation in molar magnetic susceptibility, intricately linked to the unique nature of the complex ion [Co(tn)2(Rpy)Br]Br2. Specifically, this variation correlates with the presence of the sixth ligand in the coordination environment, denoted as R in the Rpy ligand. The nature of R, influenced by the attachment of electron-withdrawing or donating groups, plays a crucial role in modulating the magnetic properties of the complexes. Remarkably, despite R being distantly positioned in relation to the metal center, the magnetic behavior of the complexes exhibits a discernible relationship with the acidity constant (pKa). This intriguing correlation further emphasizes the intricate interplay between the molecular structure and the magnetic properties observed in complexes (1)–(6).
Examining the intrinsic coercivity (Hc) in the range of 55 to 82 Oe provides further evidence of the magnetic behavior within the complexes (1)–(6). The room temperature magnetic hysteresis loop measurements confirm the ferromagnetic nature of the cobalt(III) complexes, with variations in magnetic character correlating with the basicity of the axial ligand. This observation aligns with similar findings reported for certain low-spin cobalt(III) complexes.40
Interestingly, the anticipated non-linear relationship between the magnetic properties and the substituents in the pyridyl ligand is revealed to be more complex than initially expected. Comparable investigations have demonstrated correlations between the demagnetization properties of mixed-ligand nicotinate N-oxide metal(II) complexes, emphasizing the intricate nature of such relationships.40 Furthermore, existing literature highlights clear correlations between d–d band positions and the electronic effects of ligand substituents. Reports on metal complexes with pyridyl amine also reinforce the association of physicochemical properties with the electronic nature of substituents.41
The magnetic characteristics of the complexes (1)–(6) exhibit a nuanced interplay with the electronic properties of the axial ligand and its basicity. The non-linear correlation between molar magnetic susceptibility and pKa values adds a quantitative dimension to the understanding of these relationships. The observed ferromagnetic behavior and its variation underscore the complex nature of the magnetic properties in coordination complexes, providing valuable insights for further investigations in this intriguing field of study.
Cobalt(III)-R-pyridyl complexes: photoexcitation on nanoscale TiO2
Upon UV-light excitation (λ = 254 nm), the nano-TiO2//cobalt(III)-(Rpy) surface compound initiated IET reactions. The efficiency of IET was observed to be dependent on the coordination environment of the complex, with a notable influence from the nature of the Rpy ligand. Building upon our previous investigation of Co(III)-alkyl amine complexes with TiO2, where we proposed a mechanism for interfacial electron transfer involving the formation of Co2+ ions implanted in nanocrystalline TiO2, the same mechanism is applied in this study.42 Our investigation encompasses the characterization of electronic excitations, electron injection time scales, and interfacial electron transfer mechanisms, along with the photoefficiency of IET and the photo-degradation of 2-propanol. Specifically, we concentrate on TiO2 surfaces modified by Co(III)-pyridyl complexes tethered via nitrogen linkers. The focal point lies in detailing the electronic excitations and injection time scales, which are influenced by the nature of the molecular adsorbates and the modes of attachment.The cobalt(III)-Rpy complex serves as a proficient UV light absorber, yet it undergoes decomposition upon prolonged light exposure, primarily attributed to ligand-to-metal charge transfer (LMCT) bands at 356 nm. Introduction of nano-TiO2 (anatase) significantly enhances the complex degradation efficiency, resulting in a higher generation of cobalt(II). In Fig. 10(a), UV-vis absorption spectra for [Co(tn)2(4-Etpy)Br]2+ complex ion is presented at distinct irradiation time intervals. Notably, a blue shift in the absorption maxima occurs at λ = 357 → 351 nm, accompanied by a red shift at λ = 512 → 518 nm.
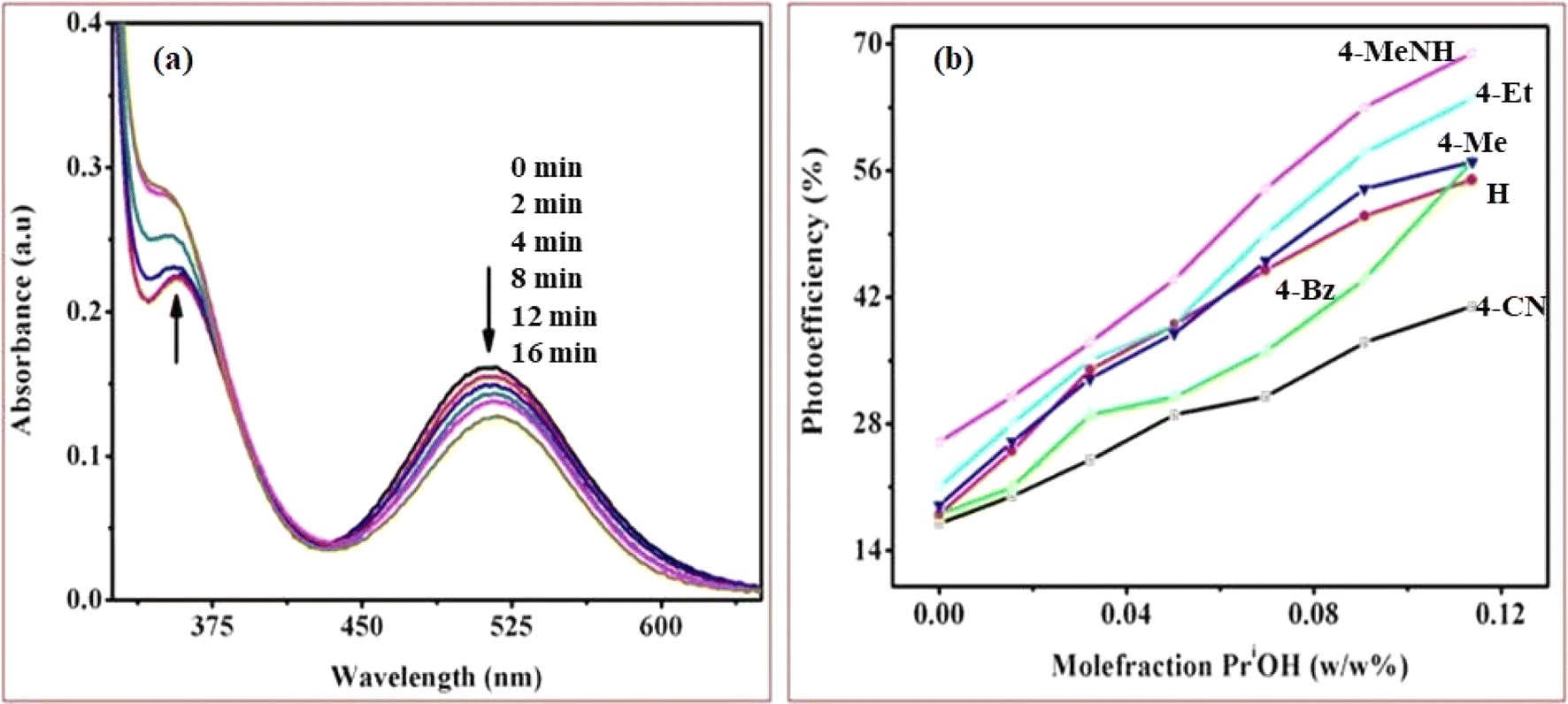 | ||
| Fig. 10 (a) UV-vis absorption spectra were obtained during the photocatalytic irradiation of the [Co(tn)2(4-Etpy)Br]2+ complex ion with nano-TiO2 suspension in pure water at different time points and (b) Graph illustrating the correlation between the mole fraction of water (w/w % PriOH + water) and photoefficiency, determined through spectrophotometer measurements during the photo-irradiation of a series of [Co(tn)2(Rpy)Br]2+ complex ions (R = 4-CN, H, 4-Bz, 4-Me, 4-Et, and 4-MeNH). The complex concentration was maintained at 1.68 × 10−3 M in water/2-propanol mixtures with nanocrystalline TiO2. Experimental conditions included an ionic strength of 1 M NaNO3, pH ∼7, and a temperature of 298 K. | ||
The absence of an isosbestic point in the UV-vis spectra indicates complex decomposition during the reduction process, suggesting perturbation of the Co(III) center due to IET. The photolytic solutions exhibit a photoefficiency of Co2+ formation by TiO2 (ecb−/hvb+)/scavenger of [Co(tn)2(4-Etpy)Br]2+, which increases with the concentration of 2-propanol, showing heightened activity at higher concentrations (Fig. 10(b)). This suggests that the observed photoefficiency (as detailed in Table 5) of Co2+ formation is a summation of individual electron transfer reactions, specifically involving (i) excited nano-TiO2:[Co(tn)2(Rpy)Br]2+ + nano-TiO2 + hν(λ = 254 nm) → Co2+ + products and (ii) ligand-to-metal charge transfer transition in the cobalt(III) complex: [Co(tn)2(Rpy)Br]2+ + hν(λ = 254 nm) → Co2+ + products. However, the former path is predominant and necessitates further in-depth investigation.
The nanocrystalline titania has a more pronounced impact on the photoreduction of the [Co(tn)2(Rpy)Br]2+ complex compared to its polycrystalline counterpart in Table 4. Nanocrystalline materials, characterized by a grain size typically less than 100 nm, exhibit unique properties due to the significant volume of their microstructure composed of interfaces, particularly grain boundaries. In contrast to conventional coarse-grained polycrystalline materials, nanocrystalline counterparts often demonstrate enhanced and distinct properties.43 In polycrystalline wide band gap oxide semiconductors like ZnO and TiO2, the photoconductivity (PC) decay is observed to be notably slow, persisting over extended periods ranging from hours to days.42,44 This phenomenon, often termed ‘persistent PC,’ has prompted various proposed explanation.45–47 In some instances, models have been employed to fit experimental data, aiding in the determination of electronic parameters such as the energy distribution of charge carrier traps. Previous studies on photocatalytic degradation of pollutants have explored different approaches. For instance, investigations using Co(II)-tetrasulfophthalocyanine grafted on TiO2 via a silane reagent have been documented, along with studies involving polycrystalline TiO2 samples impregnated with Cu(II)-phthalocyanine.48,49 These findings underscore the importance of nanocrystalline materials in influencing photoreduction processes and the distinct behavior exhibited by such materials, shedding light on potential advancements in photocatalytic applications.
| pKa value of Rpy | Complexes | Catalyst used | PE (%) in water/PriOH (w/w)% | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 100/0 | 95/5 | 90/10 | 85/15 | 80/20 | 75/25 | 70/30 | |||
| a Photo-oxidation of 2-propanol into acetone. | |||||||||
| 1.90 (R = 4-CN) | (6) | — | 14 ± 0.5 | 18 ± 1.0 | 19 ± 1.0 | 21 ± 0.7 | 26 ± 0.8 | 28 ± 0.9 | 32 ± 0.6 |
| Poly-TiO2 | 16 ± 1.0 | 17 ± 0.5 | 21 ± 1.0 | 24 ± 0.8 | 29 ± 0.7 | 31 ± 0.6 | 37 ± 0.5 | ||
| Nano-TiO2 | 17 ± 0.7 | 20 ± 0.9 | 24 ± 0.5 | 29 ± 0.8 | 31 ± 0.6 | 37 ± 1.0 | 41 ± 1.4 | ||
| 5.25 (R = H) | (5) | — | 15 ± 0.8 | 21 ± 0.7 | 29 ± 1.0 | 34 ± 0.5 | 38 ± 1.4 | 40 ± 0.9 | 42 ± 1.2 |
| Poly-TiO2 | 14 ± 0.6 | 24 ± 1.0 | 32 ± 0.8 | 35 ± 0.8 | 41 ± 0.7 | 44 ± 0.9 | 48 ± 1.2 | ||
| Nano-TiO2 | 18 ± 1.0 | 25 ± 0.7 | 34 ± 0.6 | 39 ± 1.0 | 45 ± 0.5 | 51 ± 0.9 | 55 ± 0.5 | ||
| 5.59 (R = 4-Bz) | (4) | — | 12 ± 0.8 | 19 ± 1.0 | 24 ± 0.6 | 28 ± 0.7 | 32 ± 0.8 | 38 ± 0.5 | 41 ± 1.2 |
| Poly-TiO2 | 16 ± 0.6 | 23 ± 0.8 | 28 ± 1.0 | 30 ± 1.4 | 34 ± 0.5 | 38 ± 0.9 | 49 ± 0.8 | ||
| Nano-TiO2 | 18 ± 0.6 | 21 ± 0.8 | 29 ± 0.7 | 31 ± 0.5 | 36 ± 1.0 | 44 ± 0.9 | 57 ± 1.4 | ||
| 6.02 (R = 4-Me) | (3) | — | 12 ± 1.0 | 19 ± 1.0 | 26 ± 0.5 | 29 ± 0.8 | 34 ± 0.9 | 38 ± 0.8 | 49 ± 1.4 |
| Poly-TiO2 | 17 ± 1.0 | 23 ± 0.5 | 29 ± 0.6 | 34 ± 0.8 | 37 ± 0.9 | 45 ± 0.9 | 52 ± 0.7 | ||
| Nano-TiO2 | 19 ± 0.7 | 26 ± 0.5 | 33 ± 1.0 | 38 ± 0.6 | 46 ± 0.8 | 54 ± 1.4 | 57 ± 0.9 | ||
| 6.02 (R = 4-Et) | (2) | — | 18 ± 1.2 | 24 ± 0.6 | 28 ± 1.0 | 31 ± 0.5 | 36 ± 1.4 | 41 ± 0.8 | 49 ± 0.5 |
| Poly-TiO2 | 19 ± 1.0 | 27 ± 1.0 | 32 ± 0.9 | 37 ± 0.8 | 45 ± 0.5 | 48 ± 0.8 | 53 ± 1.4 | ||
| Nano-TiO2 | 21 ± 0.6 | 28 ± 0.5 | 35 ± 1.0 | 39 ± 0.9 | 49 ± 0.8 | 58 ± 0.5 | 64 ± 0.7 | ||
| 9.70 (R = 4-MeNH) | (1) | — | 19 ± 0.8 | 24 ± 0.5 | 29 ± 0.5 | 32 ± 1.0 | 46 ± 1.0 | 52 ± 0.8 | 53 ± 0.9 |
| Poly-TiO2 | 23 ± 0.7 | 28 ± 0.8 | 32 ± 0.5 | 38 ± 0.6 | 49 ± 1.0 | 58 ± 0.9 | 61 ± 0.5 | ||
| Nano-TiO2 | 26 ± 0.8 | 31 ± 0.6 | 37 ± 0.7 | 44 ± 0.5 | 54 ± 1.0 | 63 ± 0.9 | 69 ± 0.8 | ||
Photo-oxidation of 2-propanol leading to acetone production has been extensively studied. Previous literature reports highlight the remarkable catalytic activities of cobalt-doped TiO2 in the degradation of acetaldehyde, 2-chlorophenol, and 2,4-dichlorophenol in aqueous solutions.50–53 In our investigation, we systematically monitored the photo-oxidation process using NMR measurements, revealing a progressive increase in the signal intensity corresponding to the growth of the acetone peak as a function of light dosage.54 Table 5 presents 1H NMR signals associated with 2-propanol, identified at δ = 1.0 to 1.1 ppm (–CH3) and δ = 3.8 to 3.93 ppm (–CH), both before the initiation of photo-oxidation and at specific intervals during irradiation periods (refer to Fig. 11). Notably, a distinct 1H NMR signal at δ = 2.04 ppm emerges, signifying the formation of acetone, and its intensity (refer to Table 5) considerably increases over extended irradiation times.55
| Irradiating time (min) | 2× –CH3(d) P δ (ppm) | –CH(m) P δ (ppm) | 2× –CH3(s) A δ (ppm) | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| a Mechanism of photoinduced electron transfer reaction. | ||||||||||
| 0 | 1.014 | 1.002 | 3.723 | 3.738 | 3.753 | 3.769 | 3.784 | 3.799 | 3.814 | — |
| 8 | 1.001 | 1.017 | 3.739 | 3.755 | 3.770 | 3.785 | 3.801 | 3.816 | 3.831 | 2.050 |
| 16 | 1.006 | 1.022 | 3.717 | 3.732 | 3.747 | 3.763 | 3.778 | 3.793 | 3.808 | 2.040 |
| 45 | 1.007 | 1.023 | 3.724 | 3.740 | 3.755 | 3.770 | 3.786 | 3.801 | 3.816 | 2.045 |
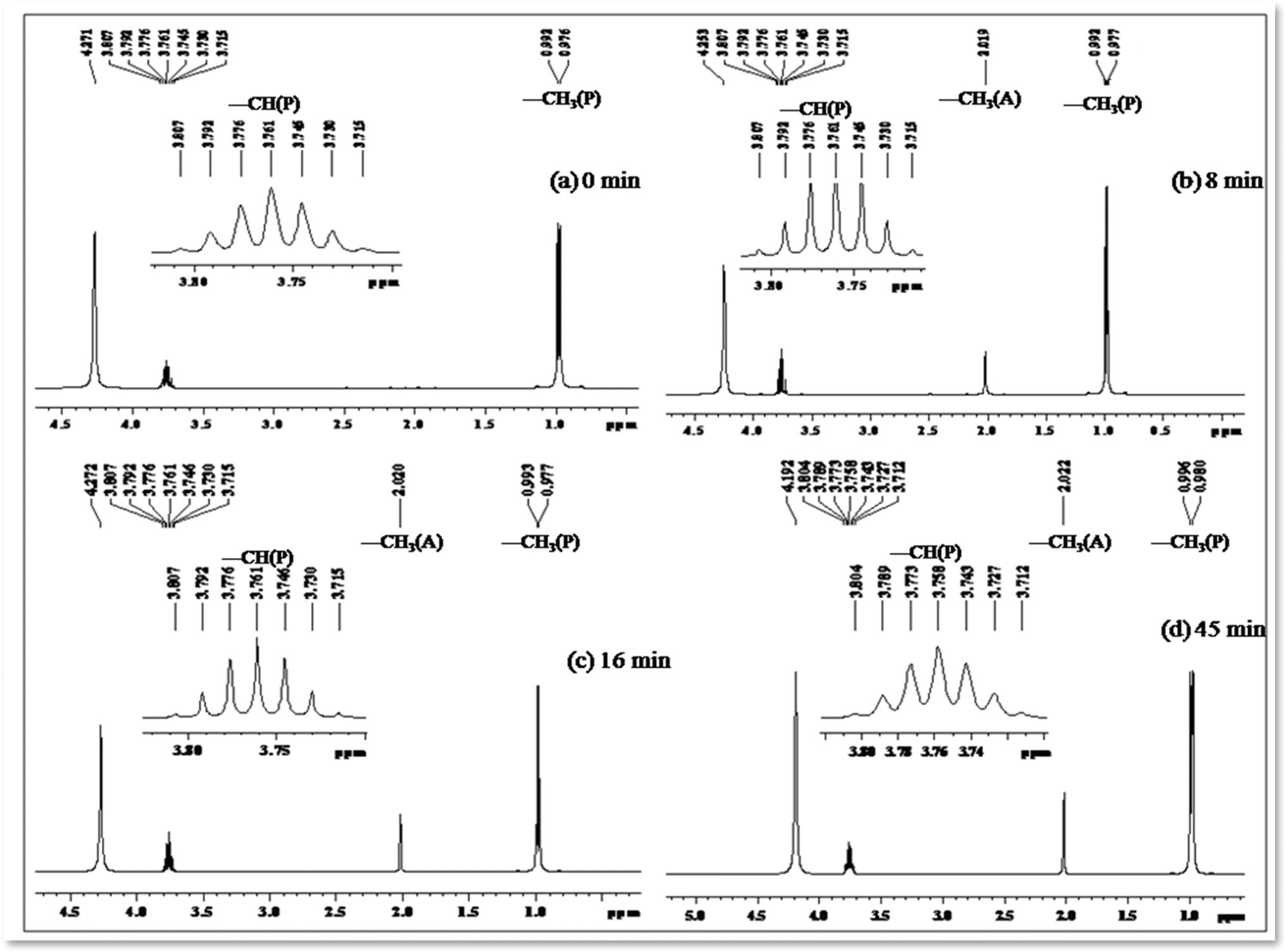 | ||
| Fig. 11 1H NMR spectrum (in DMSO-d6) depicting the decomposition profile of 2-propanol in the presence of nano-TiO2/[Co(tn)2(4-Etpy)Br]2+/UV-light (254 nm) at different time intervals: (a) 0, (b) 8, (c) 16, and (d) 45 minutes, respectively. ‘A’ and ‘P’ represent acetone and 2-propanol, respectively. The inset highlights the –CH signal of 2-propanol in the region 3.7–3.83 ppm. | ||
The integrated intensity of the acetone signals demonstrates a steady growth, accompanied by a gradual decrease in the signals corresponding to –CH and –CH3 (2-propanol). This observation implies that acetone originates from the oxidation of 2-propanol, a process catalyzed by the scavenging of valence band holes by alcohol, expressed as Co–TiO2(h+) + (CH3)2CHOH → CH3COCH3. These findings underscore the dynamic nature of the photo-oxidation process and provide valuable insights into the mechanistic aspects of acetone generation from 2-propanol oxidation.
Tables 4 and 5 clearly reveal that excited nanocrystalline TiO2 exhibits superior catalytic activity, a phenomenon intricately linked to the surface–substrate interaction. The surface affinity of molecules/ions presents a competitive aspect, necessitating the inevitable formation of a polycrystalline (or) nanocrystalline TiO2//cobalt(III)-(Rpy) surface compound. The augmentation in photocatalytic reduction stems from two primary factors: (i) Rpy's ability to modify the surface affinity of [Co(tn)2(Rpy)Br]2+ ions with the nano-TiO2 surface, and (ii) photoexcitation inducing the formation of microdomains in nano-TiO2 with distinctive hydrophobic/hydrophilic behavior.56,57
The incorporation of the sixth ligand (Rpy) in [Co(tn)2(Rpy)Br]2+, characterized by a hydrophobic tail, introduces variations in the surface adherence of the complex ion to the nano-TiO2 surface. However, it is important to note that the adsorption process is constrained by thermodynamic considerations. To provide a comprehensive understanding of these observations, we propose the following mechanistic equations (eqn (1) and (3)):
| nano-TiO2 + [Co(tn)2(Rpy)Br]2+ → nano-TiO2//[Co(tn)2(Rpy)Br]2 + | (1) |
| nano-TiO2(h+,VB)/(h+,tr) + nano-TiO2(e−,CB)/(e−,tr) + (Co3+)+(CH3)2CHOH → CH3COCH3 + Cosurf2+ + Co2+ | (2) |
| nano-TiO2(h+,VB)/(h+,tr) + nano-TiO2(e−,CB)/(e−,tr) + (Co3+) + (CH3)2CHOH → CH3COCH3 + Cosurf2+ + Coaq.2+ | (3) |
These equations elucidate the intricate processes involved in the interaction between nanocrystalline TiO2 and [Co(tn)2(Rpy)Br]2+ complex ion, and the subsequent photocatalytic reduction leading to the production of acetone and the involvement of surface-bound CoII species.
pKa dependent photoreduction of Co(III)-Rpy complexes
The photoreduction of Co(III)-Rpy complexes exhibits a notable pKa dependency, as evidenced in this investigation. A linear relationship between the photoefficiency (%) of [Co(tn)2(Rpy)Br]2+/TiO2 suspension and the acidity constant (pKa) of the Rpy ligand is observed, as illustrated in Fig. 12. The linear regression analysis of photoefficiency (%) against pKa reveals a slope that signifies the influence of the electron-donating or electron-withdrawing nature of the Rpy ligand on the photoefficiency. Specifically, the electron-donating group in Rpy, characterized by a higher pKa (>5.25), is found to enhance photoefficiency (%). Conversely, the electron-withdrawing group in Rpy, with a lower pKa value (pKa = 1.90), diminishes the photocatalytic behavior. This empirical observation underscores the significant impact of the coordination environment of the transition metal ion's metal center on the photochemical characteristics of the complex.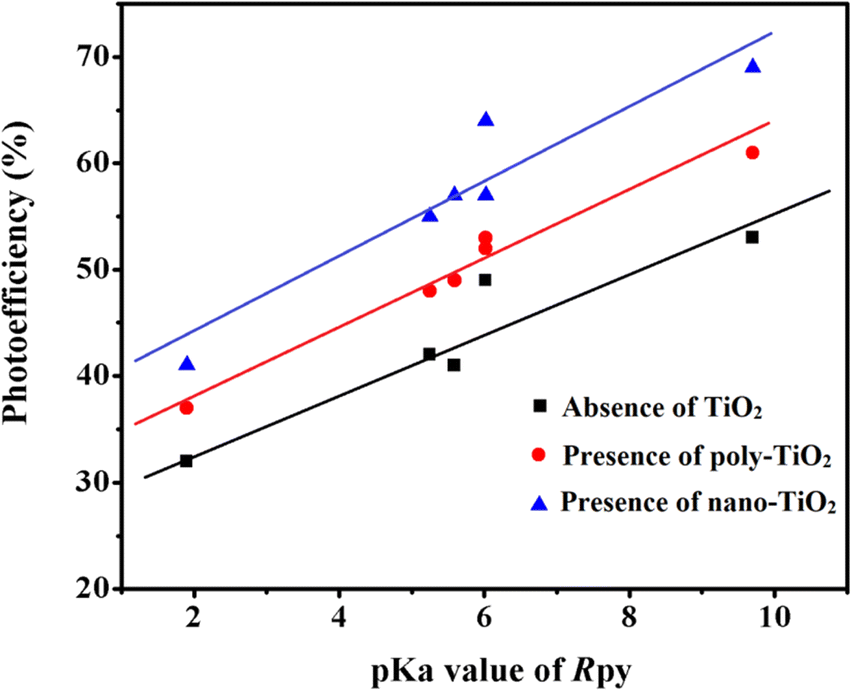 | ||
| Fig. 12 Linear relationship between photoefficiency and pKa value of aryl amine (R) in [Co(tn)2(Rpy)Br]2+ under varied conditions: absence of TiO2, presence of poly-TiO2, and nano-TiO2 in 70/30 (w/w)% water/PriOH. | ||
In addition to the pKa-dependent trends, it is noteworthy to explore the mechanistic underpinnings of how the electron-donating or withdrawing nature of Rpy influences the photoefficiency, providing a comprehensive understanding of the observed phenomena. Further investigations into the molecular–level interactions and electronic properties could contribute valuable insights to the design and optimization of photoactive complexes for enhanced catalytic performance.
The photogeneration efficiency (PE %) of Co2+ in our study exhibits intriguing dynamics, displaying a dual dependence: (i) on the 2-propanol content and (ii) a correlation with variations in the Rpy ligand of the complex. This non-consistency is attributed to the intricate interplay between nanoparticle surface characteristics and cobalt(III)-Rpy affinity, influencing subsequent interfacial electron transfer processes. The accumulation of the cobalt(III) complex onto the surface of nano-TiO2, forming a compact layer, is significantly influenced by the blocking effect of Rpy, dictated by its electron-withdrawing and electron-donating properties. Notably, Pellizzetti and colleagues have previously demonstrated a substantial alteration in the distribution of aromatic intermediates on the surface of TiO2, underscoring the impact of surface interactions on catalytic processes.58
In Fig. 12, the relationship between photoefficiency and the pKa value of the aryl amine of the Rpy ligand for six [Co(tn)2(Rpy)Br]2+ complexes is elucidated. Within this limited set of complexes, a discernible correlation emerges between PE and the pKa value of the ligand. Interestingly, there is a noticeable enhancement in PE with the aromatic ring's electronic nature of the Rpy ligand. This enhancement seems to approach a limiting value asymptotically, suggesting a saturation point in the influence of the aromatic nature on the photogeneration efficiency of Co2+. This intricate interplay between ligand properties, surface interactions, and electron transfer processes underscores the complexity of the system. Further exploration of a broader range of complexes and in-depth mechanistic investigations could unveil additional nuances, paving the way for fine-tuning and optimizing the photoreduction processes for enhanced catalytic performance.
Conclusion
This investigation presents a novel mechanochemical synthesis of mixed ligand complexes, specifically the cis-[Co(tn)2(Rpy)Br]Br2 type. The comprehensive characterization, involving spectral measurements and single crystal X-ray diffraction analysis, confirmed the structural integrity of these complexes. Intriguingly, the emission and magnetic properties exhibited a ferromagnetic character influenced by the choice of the sixth ligand in the Co(III)-complex environment. Exploring the electron transfer processes, the study demonstrated efficient electron transfer to the cobalt(III) center utilizing TiO2 nanoparticles under UV-light irradiation. The adsorption characteristics of cis-[Co(tn)2(Rpy)Br]Br2 in aqueous 2-propanol led to the formation of surface compounds. UV excitation of the nano-TiO2//cobalt(III)-(Rpy) surface compound initiated an IET reaction, dependent on the coordination environment of the complex, particularly the Rpy ligand. Spectral analysis revealed the photoefficiency of Coaq.2+ formation, with UV irradiation of the anatase surface demonstrating potent adsorption capabilities, facilitating efficient electron transfer to the Co(III) center and resulting in high photoefficiency of Co(II) formation. A proposed model for electron transfer considered the overlap of the TiO2 conduction band with the acceptor level (Co center) and the electronic coupling of the donor level (localized on Ti center) with the acceptor level (Co center). These pathways imply the accumulation of electrons, available for the reduction of the adhered complex ion. This study not only sheds light on the role of the Rpy moiety in modifying the TiO2-cobalt(III)-Rpy compound's compact structure and the redox power of the semiconductor surface but also proposes a mechanism for interfacial electron transfer reactions. The IET process was verified by the conversion of 2-propanol into acetone, as verified by 1H NMR technique. These findings contribute novel insights to the field, emphasizing the intricate interplay of ligand effects in coordination chemistry and the potential for advancing photocatalytic applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors express their sincere appreciation to the Researchers Supporting Project Number (RSP2024R466) King Saud University, Riyadh, Saudi Arabia. The authors express gratitude to the UGC-DAE CSR Consortium for Scientific Research for their support under Project Number CRS/2023-24/1378.References
- K. Maeda, Adv. Mater., 2019, 31, 1808205 CrossRef PubMed.
- R. Bakker, A. Bairagi, M. Rodríguez, G. L. Tripodi, A. Y. Pereverzev and J. Roithová, Inorg. Chem., 2023, 62, 1728 CrossRef CAS PubMed.
- A. M. May and J. L. Dempsey, Chem. Sci., 2024, 15, 6661 RSC.
- E. R. Batista and R. L. Martin, J. Am. Chem. Soc., 2007, 129, 7224 CrossRef CAS PubMed.
- M. Falkenstrom, O. Johansson and L. Hammarstrom, Inorg. Chim. Acta, 2007, 360, 741 CrossRef.
- X. Yang and M. H. Baik, J. Am. Chem. Soc., 2006, 28, 7476 CrossRef PubMed.
- G. Ayyakannu Sundaram, R. Kanniah, K. Anbalagan, K. Kulandaivelu and H. Valdés, Catalysts, 2024, 14, 169 CrossRef CAS.
- M. J. Lundqvist, M. Nilsing, P. Persson and S. Lunell, Int. J. Quantum Chem., 2006, 106, 3214 CrossRef CAS.
- M. Nilsing, P. Persson and L. Ojamae, Chem. Phys. Lett., 2005, 415, 375 CrossRef CAS.
- E. Marchini, S. Caramori and S. Carli, Molecules, 2024, 29, 293 CrossRef CAS PubMed.
- F. Aslan, Opt. Quantum Electron., 2024, 56, 645 CrossRef CAS.
- A. V. Müller, W. M. Wierzba, M. N. Pastorelli and A. S. Polo, J. Braz. Chem. Soc., 2021, 32, 1711 Search PubMed.
- C. Yang, T. Liu, P. Song, F. Ma and Y. Li, J. Mol. Liq., 2022, 368, 120797 CrossRef CAS.
- J. Yao, Y. Jiang, X. Gu, X. Guo, Y. Ying, Y. Wen, X. Liu, H. Yang and Y. Wu, J. Phys. Chem. C, 2023, 127, 248 CrossRef CAS.
- S. Rahman, A. Haleem, M. Siddiq, M. K. Hussain, S. Qamar, S. Hameed and M. Waris, RSC Adv., 2023, 13, 19508 RSC.
- Y. Kusumawati, A. S. Hutama, D. V. Wellia and R. Subagyo, Heliyon, 2021, 7, e08436 CrossRef CAS PubMed.
- H. Michaels and M. Freitag, ACS Appl. Energy Mater., 2022, 5, 1933 CrossRef CAS PubMed.
- R. Avilés-Betanzos, G. Oskam and D. Pourjafari, Energies, 2023, 16, 5617 CrossRef.
- P. O. Oladoye, M. Kadhom, I. Khan, K. H. Hama Aziz and Y. A. Alli, Green Chem. Eng., 2023 DOI:10.1016/j.gce.2023.12.004.
- H. Targhan, A. Rezaei, A. Aliabadi, A. Ramazani, Z. Zhao, X. Shen and H. Zheng, Sci. Rep., 2024, 14, 530 CrossRef CAS PubMed.
- D. Mathur, S. A. Díaz, N. Hildebrandt, R. D. Pensack, B. Yurke, A. Biaggne, L. Li, J. S. Melinger, M. G. Ancona, W. B. Knowlton and I. L. Medintz, Chem. Soc. Rev., 2023, 52, 7848 RSC.
- X. Y. Ng, K. W. Fong, L. V. Kiew, P. Y. Chung, Y. K. Liew, N. Delsuc, M. Zulkefeli and M. L. Low, J. Inorg. Biochem., 2024, 250, 112425 CrossRef CAS PubMed.
- S. Hennessey, R. González-Gómez, K. McCarthy, C. S. Burke, C. Le Houérou, N. K. Sarangi, P. McArdle, T. E. Keyes, F. Cucinotta and P. Farràs, ACS Omega, 2024, 9, 13872 CrossRef CAS PubMed.
- T. Gobbato, G. A. Volpato, A. Sartorel and M. Bonchio, Chem. Sci., 2023, 14, 12402 RSC.
- C. Ye, H. Cheng, S. Wrede, S. Diring, H. Tian, F. Odobel and L. Hammarström, J. Am. Chem. Soc., 2023, 145, 11067 CrossRef CAS PubMed.
- K. Pournemati, A. Habibi-Yangjeh and A. Khataee, ACS Appl. Nano Mater., 2024, 7, 2200 CrossRef CAS.
- K. Anbalagan, M. Tamilselvan, S. Nirmala and L. Sudha, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, E65, m836 CrossRef PubMed.
- K. Ravichandran, P. Ramesh, M. Tamilselvan, K. Anbalagan and M. N. Ponnuswamy, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, E65, m1174 CrossRef PubMed.
- K. Ravichandran, P. Ramesh, C. Maharaja Mahalakshmi, K. Anbalagan and M. N. Ponnuswamy, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, E65, m1458 CrossRef PubMed.
- R. E. Kitson, Anal. Chem., 1950, 22, 664 CrossRef CAS.
- K. Anbalagan and A. S. Ganeshraja, Inorg. Chem. Commun., 2013, 37, 59 CrossRef CAS.
- K. Nagamoto, Infra Red and Raman Spectra of Inorganic and Coordination Compounds, Wiley, New York, 1978 Search PubMed.
- S. K. Chawla, J. H. Aupers and D. C. Poey, Polyhedron, 1996, 15, 683 CrossRef CAS.
- S. P. R. Poonkodi and K. Anbalagan, Transition Met. Chem., 2001, 26, 212 CrossRef CAS.
- B. Bosnich and W. R. Kneen, Inorg. Chem., 1970, 9, 2191 CrossRef CAS.
- M. E. Vaschetto and B. A. Retamal, J. Phys. Chem., 1997, 101, 6945 CrossRef CAS.
- J. Cai, I. Bernal, F. Fronczek and S. F. Watkins, Struct. Chem., 1996, 7, 233 CrossRef CAS.
- M. Shahbaz, B. Dar, S. Sharif, M. A. Khurshid, S. Hussain, B. Riaz, M. Musaffa, H. Khalid, A. R. Ch and A. Mahboob, RSC Adv., 2024, 14, 9819 RSC.
- W. Jiang, Y. Gao, Y. Sun, F. Ding, Y. Xu, Z. Bian, F. Li, J. Bian and C. Huang, Inorg. Chem., 2010, 49, 3252 CrossRef CAS PubMed.
- Z. He, Z. M. Wang, S. Gao and C. H. Yan, Inorg. Chem., 2006, 45, 6694 CrossRef CAS PubMed.
- C. Shimokawa, S. Yokota, Y. Tachi, Y. Nishiwaki, N. Ariga and S. Itoh, Inorg. Chem., 2003, 42, 8395 CrossRef CAS PubMed.
- K. Anbalagan, A. S. Ganeshraja and C. Maharaja Mahalakshmi, Mat. Chem. Phys., 2012, 134, 747 CrossRef CAS.
- C. Suryanarayana, Int. Mater. Rev., 1995, 40, 41 CrossRef CAS.
- G. A. Sundaram, K. Namasivayam, L. Ramesh and S. P. D. Sadhasivam, J. Alloys Compd., 2023, 968, 172068 CrossRef.
- K. Anbalagan and I. S. Lydia, J. Phys. Org. Chem., 2011, 24, 45 CrossRef CAS.
- K. Shimakawa, K. Murata, S. Matsunomo and H. Naito, J. Non Cryst. Solids, 2006, 352, 1671 CrossRef CAS.
- G. A. Sundaram, S. Kumaravelu, W.-L. Tseng, P. V. Pham, A. S. K. Kumar and V. Parimelazhagan, Materials, 2024, 17, 892 CrossRef CAS PubMed.
- G. Mele, G. Ciccarella, G. Vasapollo, E. Garcia-Lopez, L. Palmisano and M. Schiavello, Appl. Catal., B, 2002, 38, 309 CrossRef CAS.
- A. S. Ganeshraja and K. Anbalagan, Int. J. Nanotechnol. Appl., 2012, 6, 127 Search PubMed.
- Q. J. Yang, H. Choi, Y. J. Chen and D. D. Dionysiou, Appl. Catal., B, 2008, 77, 300 CrossRef CAS.
- Y. Liu, J. H. Wei, R. Xiong, C. X. Pan and J. Shi, Appl. Surf. Sci., 2011, 257, 8121 CrossRef CAS.
- C. Han, M. Pelaez, V. Likodimos, A. G. Kontos, P. Falaras, K. O'Shea and D. D. Dionysiou, Appl. Catal., B, 2011, 107, 77 CrossRef CAS.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 269 CrossRef CAS PubMed.
- S. Matsuo, N. Sakaguchi, K. Yamada, T. Matsuo and H. Wakita, Appl. Surf. Sci., 2004, 228, 233 CrossRef CAS.
- S. Ardo and G. J. Meyer, J. Am. Chem. Soc., 2010, 132, 9283 CrossRef CAS PubMed.
- B. Lu, J. Zhang, G. Zhu, T. Liu, J. Chen and X. Liang, J. Nanobiotechnol., 2023, 21, 491 CrossRef CAS PubMed.
- M. V. Maevskaya, A. V. Rudakova, A. V. Emeline and D. W. Bahnemann, Nanomaterials, 2021, 11, 1526 CrossRef CAS PubMed.
- C. Minero, G. Mariella, V. Maurino and E. Pellizzetti, Langmuir, 2000, 16, 2632 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02648a |
| This journal is © The Royal Society of Chemistry 2024 |