
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTuning the high-entropy perovskite as efficient and reliable electrocatalysts for oxygen evolution reaction†
Ruixue Wei a,
Gaoliang Fu*b,
Huafeng Qib and
Hewei Liub
a,
Gaoliang Fu*b,
Huafeng Qib and
Hewei Liub
aDepartment of Cerebrovascular Diseases, The Second Affiliated Hospital of Zhengzhou University, Zhengzhou 450052, Henan, China
bHenan Provincial Key Laboratory of Nanocomposites and Applications, Institute of Nanostructured Functional Materials, Huanghe Science and Technology College, Zhengzhou, Henan 450006, China. E-mail: fugl@hhstu.edu.cn
First published on 6th June 2024
Abstract
Due to their unique electronic structure, atomic arrangement and synergistic effect, high-entropy materials are being actively pursued as electrocatalysts for oxygen evolution reaction (OER) in water splitting. However, a relevant strategy to improve high-entropy materials is still lacking. Herein, substitutional doping on the La-site in high-entropy perovskite La1−xSrx(CrMnFeCoNi)0.2O3 is reported as an efficient OER catalyst. Sr doping is found to be crucial to enhancing the OER activity. The overpotential for the best catalyst La0.3Sr0.7(CrMnFeCoNi)0.2O3 is only 330 mV at 10 mA cm−2, achieving a reduction of 120 mV in overpotential compared to La(CrMnFeCoNi)0.2O3, which is attributed to the enhancement in intrinsic catalytic activity. Experimental evidences including in situ electrochemical impedance spectroscopy (EIS) and X-ray photoelectron spectroscopy (XPS) indicate Sr doping induces the formation of high-valence Cr6+, Mn4+, Fe4+, Co4+ and Ni3+ species, which can accelerate the faster charge transfer at the interface, thereby increasing the intrinsic catalytic activity. The assembled two-electrode overall water splitting system operates stably at 10 mA cm−2 for 200 h without attenuation. This work offers an important method for developing a high-performance, high-entropy perovskite OER catalyst for hydrogen production by electrochemical water splitting.
1. Introduction
Producing H2 via electrochemical water splitting is regarded as a promising route for replacing fossil fuels.1–3 The overall efficiency of the electrolytic process, however, is limited by the anodic oxygen evolution reaction (OER, 4OH− → 2H2O + O2 + 4e−) as a consequence of its sluggish four-electron transfer.4 Although noble metal-based catalysts such as IrO2 are state-of-the-art OER catalysts, their poor scarcity and high price restrict their enlarged application.5 The pursuit of inexpensive and efficient transition metal (TM)-based electrocatalysts is urgently needed to accelerate OER.4,6,7The ABO3 perovskite oxides have been intensively explored as active electrocatalysts because of their highly flexible structure and good oxygen catalytic activity.8 In the typical perovskite, the A-site represents rare-earth and alkaline cations and the B-site occupied by TM cations generally acts as the catalytic active site, where the changes in the B-site oxidation state or coordination can control the catalytic activity.8,9 Notably, Suntivich et al. proposed that the OER activity is related to the eg occupancy of TM at the B-site, with eg = 1 showing the highest OER activity.10 Various strategies have been subsequently employed to tune functionally active B-site TM cation to improve the OER performance,11 such as doping elements,12,13 introducing oxygen vacancy,14 and constructing heterogeneous grain boundaries.15–17 Although a certain enhancement in the OER activity is achieved, there is still much room for improvement.
High-entropy materials have recently received increasing attention in electrocatalysis owing to their unique bonding style and atomic arrangement in a lattice.18,19 In principle, high-entropy materials refer to single-phase solid solutions containing at least five main constituent metal elements uniformly mixed in an equal molar ratio.20 High-entropy materials have four specific effects, namely, the cocktail effect, high entropy effect, slow diffusion effect and lattice distortion effect, which greatly affect the catalytic performance through the reconstruction of electronic structures in the multi-element systems.18 Recently, Nguyen et al.21 reported a new type of B-site equimolar La(CrMnFeCoNi)0.2O3 and five non-equimolar high-entropy perovskites for the OER. All the high-entropy perovskites show superior performance to the single perovskite oxides, with the optimized La(CrMnFeCo2Ni)0.2O3 showing the highest OER activity.21 This shows that high-entropy perovskite exhibits great potential for OER, yet a relevant strategy to improve high-entropy materials is still lacking.
Perovskite La(CrMnFeCoNi)0.2O3 (La(5B0.2)O3) is a compositionally complex high-entropy perovskite oxide that demonstrates high catalytic activity for OER.21 Substitutional doping on the A-site can impart changes to the functionally active B-site TM cations.22–24 In this work, Sr doping is found to significantly enhance the OER activity of the La1−xSrx(CrMnFeCoNi)0.2O3 (0 ≤ x ≤ 0.7, La1−xSrx(5B0.2)O3). When x = 0.7 (i.e., 0.7Sr), the highest OER activity is observed with an overpotential of only 330 mV at a current density of 10 mA cm−2. In situ electrochemical impedance spectroscopy (EIS) shows that Sr doping facilitates faster electron transfer at the interface, which enhances the intrinsic catalytic activity of La1−xSrx(5B0.2)O3. The faster charge transfer is attributed to the formation of high-valence TM cations induced by Sr doping. A 200 h-stability test of an assembled electrolyzer at 10 mA cm−2 without obvious attenuation verifies the industrial prospects of the La0.3Sr0.7(5B0.2)O3 as an oxygen evolution electrocatalyst for hydrogen production from water splitting.
2. Experimental details
2.1 Synthesis and characterization of catalysts
High-entropy perovskite La(CrMnFeCoNi)0.2O3 nanoparticles (La(5B0.2)O3) were prepared using the sol–gel method described in detail elsewhere.12 Stoichiometric amounts of metal acetates or nitrates were dissolved in ultrapure water, followed by the addition of citric acid and acrylamide with vigorous stirring for 1 h to form a sol. The citric acid and acrylamide were in a 1.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4:1 molar ratio to the total metal cations. Sol was transferred to an oil bath at 80 °C to evaporate the water to form a gel. After that, the gel was successively heated at 150 °C for 6 h, calcined at 400 for 3 h, and further calcined at 750 for 3 h to form La(5B0.2)O3 nanoparticles. High-entropy La1−xSrx(CrMnFeCoNi)0.2O3 (x = 0.1, 0.3, 0.5, 0.7 and 0.9, La1−xSrx(5B0.2)O3) nanoparticles were synthesized following the same procedure, except for the addition of a certain amount of strontium nitrate.
:4:1 molar ratio to the total metal cations. Sol was transferred to an oil bath at 80 °C to evaporate the water to form a gel. After that, the gel was successively heated at 150 °C for 6 h, calcined at 400 for 3 h, and further calcined at 750 for 3 h to form La(5B0.2)O3 nanoparticles. High-entropy La1−xSrx(CrMnFeCoNi)0.2O3 (x = 0.1, 0.3, 0.5, 0.7 and 0.9, La1−xSrx(5B0.2)O3) nanoparticles were synthesized following the same procedure, except for the addition of a certain amount of strontium nitrate.
X-ray diffraction (XRD, BRUKER, D8 ADVANCE) using Cu Kα radiation was employed to characterize the crystal phase of the catalyst. The morphology, microstructure and size of the catalyst were identified through scanning electron microscopy (SEM, ZEISS, Sigma 300) and transmission electron microscopy (TEM, JEOL JEM-2100F). The Brunauer–Emmett–Teller (BET) surface areas were measured by utilizing N2 adsorption–desorption isotherms at 77 K using an ASAP 2020HD88 surface area analyzer. X-ray photoelectron spectroscopy (XPS) was collected using a Thermo Scientific K-Alpha with Al Kα radiation to investigate the oxidation states of TM cations. XPS data were fitted with CasaXPS software.
2.2 Electrochemical measurements
The electrochemical measurements were conducted using a three-electrode system with a reference electrode of Hg/HgO (1 M KOH) and a counter electrode of graphite rode. The working electrode was fabricated by dropping inks on an “L” type glassy carbon electrode (an area of 0.196 cm−2). The inks were made by adding catalysts (5 mg), acetylene black (1 mg) and Nafion solution (45 μL, 5 wt%) to a mixture (0.75 mL deionized water and 0.25 mL isopropanol) and sonicating for 30 min. The loading of the catalyst on the as-prepared electrode is 0.25 mg cm−2.Linear sweep voltammograms (LSVs) were performed in a 1 M KOH solution at a scan rate of 10 mV s−1. Tafel plots were derived from the LSV curves. EIS measurements proceeded under the conditions of a perturbation amplitude of 5 mV and a frequency ranging of 100 kHz–1 Hz. All the potentials relative to the Hg/HgO electrode were converted to potentials relative to the reversible hydrogen electrode (RHE) using the following equation: E(vs. RHE) = E(vs. Hg/HgO) + 0.098 V + pH × 0.0592 V. A two-electrode system for overall water splitting was carried out by dropping the catalyst ink on carbon paper. The catalyst drop-coated area was 0.5 cm2, and the loading of the catalyst was 0.5 mg cm−2.
3. Results and discussion
3.1 Structural characterization
The crystal phase for high-entropy La1−xSrx(5B0.2)O3 was analyzed by XRD. Fig. 1 shows the XRD patterns. The diffraction peaks for La(5B0.2)O3 centered at 22.9°, 32.7°, 40.2°, 46.8° and 58.2° correspond to the (100), (110), (111), (200) and (211) crystal planes of cubic structure (space group: Pm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m), respectively. From the extended XRD patterns in Fig. 1b, the main peak for 0.1Sr at 32.7° is cleaved, showing that a phase transition from cubic to rhombohedral structure (space group Rm) occurs. For x ≥ 0.3, the crystal phases return to cubic structure. A small amount of impurities attributed to SrCrO4 is observed for 0.7Sr. High concentrations of Sr9Ni7O21 impurities are observed in 0.9Sr (as shown in Fig. S1†), and thus only the OER performances of La1−xSrx(5B0.2)O3 (x = 0, 0.1, 0.3, 0.5, 0.7) are studied in the subsequent section, excluding 0.9Sr. Besides, the peaks for La1−xSrx(5B0.2)O3 shift toward high angles compared to those of La(5B0.2)O3, indicating a shortened lattice spacing after Sr doping. The change in lattice spacing with Sr doping implies the substitution of La3+ with Sr2+ at lattice sites, which agrees with those of Sr-doped LaCrO3,25 Sr-doped LaFeO3 (ref. 26) and Sr-doped LaNi0.8Fe0.2O3.23 As discussed in the subsequent sections, when an Sr2+ substituting La3+ site introduces a hole state, it leads to the oxidation of the TM cations at the B-site.27,28 Consequently, the lattice spacing is reduced for La1−xSrx(5B0.2)O3, which agrees with the reported La1−xSrx(5B0.2)O3 single-crystalline thin film.27
m), respectively. From the extended XRD patterns in Fig. 1b, the main peak for 0.1Sr at 32.7° is cleaved, showing that a phase transition from cubic to rhombohedral structure (space group Rm) occurs. For x ≥ 0.3, the crystal phases return to cubic structure. A small amount of impurities attributed to SrCrO4 is observed for 0.7Sr. High concentrations of Sr9Ni7O21 impurities are observed in 0.9Sr (as shown in Fig. S1†), and thus only the OER performances of La1−xSrx(5B0.2)O3 (x = 0, 0.1, 0.3, 0.5, 0.7) are studied in the subsequent section, excluding 0.9Sr. Besides, the peaks for La1−xSrx(5B0.2)O3 shift toward high angles compared to those of La(5B0.2)O3, indicating a shortened lattice spacing after Sr doping. The change in lattice spacing with Sr doping implies the substitution of La3+ with Sr2+ at lattice sites, which agrees with those of Sr-doped LaCrO3,25 Sr-doped LaFeO3 (ref. 26) and Sr-doped LaNi0.8Fe0.2O3.23 As discussed in the subsequent sections, when an Sr2+ substituting La3+ site introduces a hole state, it leads to the oxidation of the TM cations at the B-site.27,28 Consequently, the lattice spacing is reduced for La1−xSrx(5B0.2)O3, which agrees with the reported La1−xSrx(5B0.2)O3 single-crystalline thin film.27
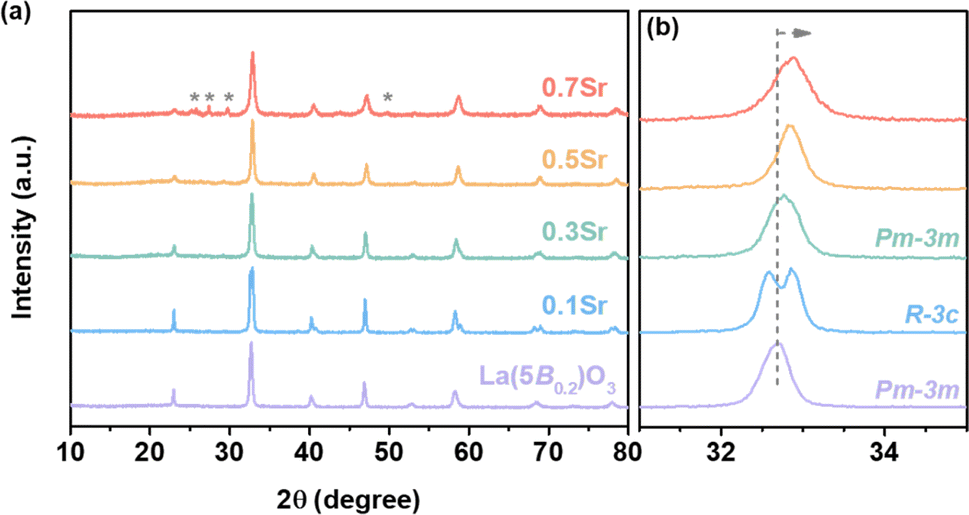 | ||
| Fig. 1 (a) XRD patterns for La1−xSrx(5B0.2)O3 and (b) expanded XRD patterns around the main diffraction peak. Diffraction peaks shift towards high angles as Sr doping. The sign * represents the impurity of CrSrO4. | ||
To clarify the morphology and particle size of the catalyst, high-entropy perovskite catalysts were examined using SEM and TEM images. Fig. 2a shows the SEM image for 0.7Sr, displaying the nanoparticles with a size of ∼50 nm. TEM images shown in Fig. 2b and c further showcase the particle size in the range of 30–60 nm, which coincides with the result of the SEM image. The selected area electron diffraction pattern (SAED, shown in Fig. 2d) presents diffraction rings/dots indexed to (100), (110), (111), (200) and (211) facets of the 0.7Sr catalyst phase, which coheres with the XRD patterns (as shown in Fig. 1). The high-resolution TEM image (HRTEM) shown in Fig. 2e exhibits a regular lattice fringe of the (110) crystalline planes with an interplanar spacing of 0.272 nm. The high-angle annular dark-field scanning TEM (HAADF-STEM) and elemental mapping (shown in Fig. 2f) confirm the homogenous distribution of all the elements in the 0.7Sr catalyst.
 | ||
| Fig. 2 Structural characterization of representative 0.7Sr. (a) SEM image. (b and c) TEM image. (d) Corresponding SAED pattern. (e) High-resolution TEM image. (f) HAADF image and corresponding elemental mapping. | ||
3.2 OER activities
The OER activities of La1−xSrx(5B0.2)O3 catalysts were evaluated using a three-electrode system in 1 M KOH solution. Fig. 3a shows the LSV curves of the La1−xSrx(5B0.2)O3 with catalyst loading of 0.25 mg cm−2. It is clearly observed that the OER activities increase as the Sr doping increases. In particular, the 0.7Sr shows the best OER activity, which is comparable to the commercial IrO2. For the sake of comparison, the overpotentials reaching 10 mA cm−2, an important evaluation parameter for practical water splitting,29 were displayed in the histogram shown in Fig. 3b. The overpotential @ 10 mA cm−2 for La(5B0.2)O3 is 0.450 V, and it is reduced to 0.437, 0.400, 0.370 and 0.330 V for 0.1Sr, 0.3Sr, 0.5Sr and 0.7Sr, respectively. The overpotentials @20 mA cm−2 for La1−xSrx(5B0.2)O3 show a similar tendency, achieving a reduction of 122 mV for 0.7Sr in comparison with that of La(5B0.2)O3. The significant diminution in overpotential indicates that Sr incorporation has an extremely good effect on the OER activities of La1−xSrx(5B0.2)O3. Fig. 3c illustrates the plots of the corresponding Tafel curves. A lower Tafel slope typically implies faster OER kinetics.30 As observed, the Tafel slope gradually decreases with Sr doping, such as 64 mV dec−1 for La(5B0.2)O3 and 55 mV dec−1 for 0.7Sr. The reduction in the Tafel slope suggests better OER kinetics induced by Sr doping. The EIS at 1.60 V vs. RHE for La1−xSrx(5B0.2)O3 is shown in Fig. 3d. In the Nyquist plots, the observed semi arch is related to the interfacial charge transfer resistance for OER.31 Sr doping causes a shrinking semi arch, revealing faster charge transfer kinetics at the catalyst–electrolyte interface during the OER.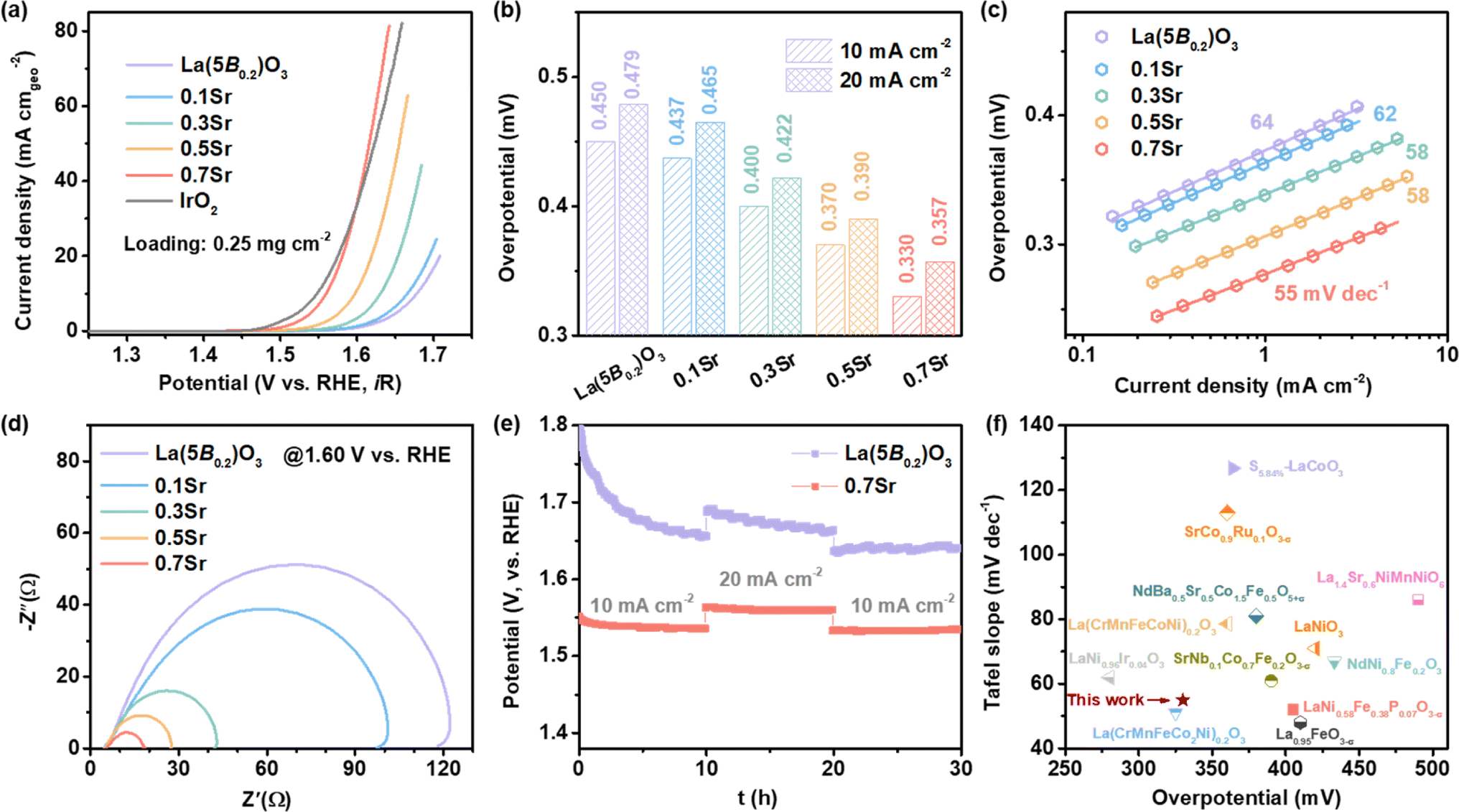 | ||
| Fig. 3 Electrochemical performances for La1−xSrx(5B0.2)O3. (a) LSV curves. (b) Overpotentials at 10 and 20 mA cm−2. (c) Tafel plots. (d) EIS at 1.60 V vs. RHE. (e) Chronopotentiometry curves for La(5B0.2)O3 and 0.7Sr. (f) Comparison of activity with the reported perovskite catalysts. Among them, the catalyst loading of La(CrMnFeCo2Ni)0.2O3 and La(CrMnFeCoNi)0.2O3 in ref. 21 is ∼2.4 mg cm−2. | ||
In addition, electrochemical stability is another important parameter for catalysts. Fig. 3e shows the potential–time curves of La(5B0.2)O3 and 0.7Sr at 10 mA cm−2 for 10 h, 20 mA cm−2 for 10 h and 10 mA cm−2 for 10 h. The potential @ 10 mA cm−2 for La(5B0.2)O3 rapidly decreases and then levels off to ∼1.66 V vs. RHE in the first ten hours. The rapid decrease in the potential is closely related to the catalyst surface reconstruction due to the strong oxidizing conditions during the OER process.6,32,33 Immediately thereafter, the OER was run at 20 mA cm−2 for 10 h and then returned to 10 mA cm−2 for 10 h. The final stabilization potential @10 mA cm−2 for La(5B0.2)O3 was 1.64 V vs. RHE. However, the same stability test procedure was performed on the 0.7Sr catalyst. The 0.7Sr catalyst displayed excellent electrochemical stability. Unlike La(5B0.2)O3, the potential for 0.7Sr reaching a current density of 10 mA cm−2 quickly stabilized in the first ten hours, preserving stability at subsequent tests at 20 mA cm−2 and 10 mA cm−2. We also contrasted the OER activity of 0.7Sr with other reported perovskite electrocatalysts (as shown in Fig. 3f).12,21,33–40 0.7Sr catalyst compared favorably with most other perovskite catalysts.
3.3 Investigation of reaction kinetics
To clarify the superior OER performance for the 0.7Sr catalyst, we first distinguished the contribution of catalytic intrinsic activity versus the surface area to OER performance. BET measurements were performed to determine the surface area of the catalysts (Fig. S3†), and the obtained values are listed in Table 1. As noticed, La(5B0.2)O3, 0.1Sr and 0.3Sr catalysts have similar BET surface areas. The BET surface area of 0.7Sr is 15.944 m2 g−1, which is ∼1.7 times higher than that of La(5B0.2)O3 (9.234 m2 g−1). To estimate the intrinsic catalytic activity, the current density for each catalyst is normalized to the BET surface area. Fig. 4a shows the BET-normalized LSV curves. It is clear that the normalized OER activities systematically increase as the Sr doping increases. To be more specific, the BET-normalized current density for La(5B0.2)O3 at 1.63 V vs. RHE is 0.111 mA cmBET−2 and increases to 1.477 mA cmBET−2 for 0.7Sr, which is ∼13.3 times higher than that of La(5B0.2)O3. Obviously, the enhanced OER performance for La1−xSrx(5B0.2)O3 is mainly ascribable to the significant increase in the intrinsic activity induced by Sr doping. As reported in other Sr-doped perovskite oxide systems, such as Sr-doped LaFeO3 (ref. 26) and Sr-doped LaNi0.8Fe0.2O3,23 Sr doping can effectively enhance TM d-O p hybridization. The increased p-d hybridization implies a reduced energy difference between TM d and O p orbitals, leading to a faster charge transfer between the active site of TM in contact with adsorbed reaction intermediates (i.e., *OH, *O, *OOH and *O2) for enhanced OER activity, which is confirmed by the subsequent EIS measurements.| La1−xSrx(5B0.2)O3 | BET surface area (m2 g−1) | ηj=10 (V) | J@1.63V vs. RHE (mA cmBET−2) | Tafel slope (mV dec−1) |
|---|---|---|---|---|
| La(5B0.2)O3 | 9.234 | 0.450 | 0.111 | 64 |
| 0.1Sr | 9.756 | 0.437 | 0.155 | 62 |
| 0.3Sr | 10.87 | 0.400 | 0.372 | 58 |
| 0.5Sr | 13.56 | 0.370 | 0.791 | 58 |
| 0.7Sr | 15.94 | 0.330 | 1.477 | 55 |
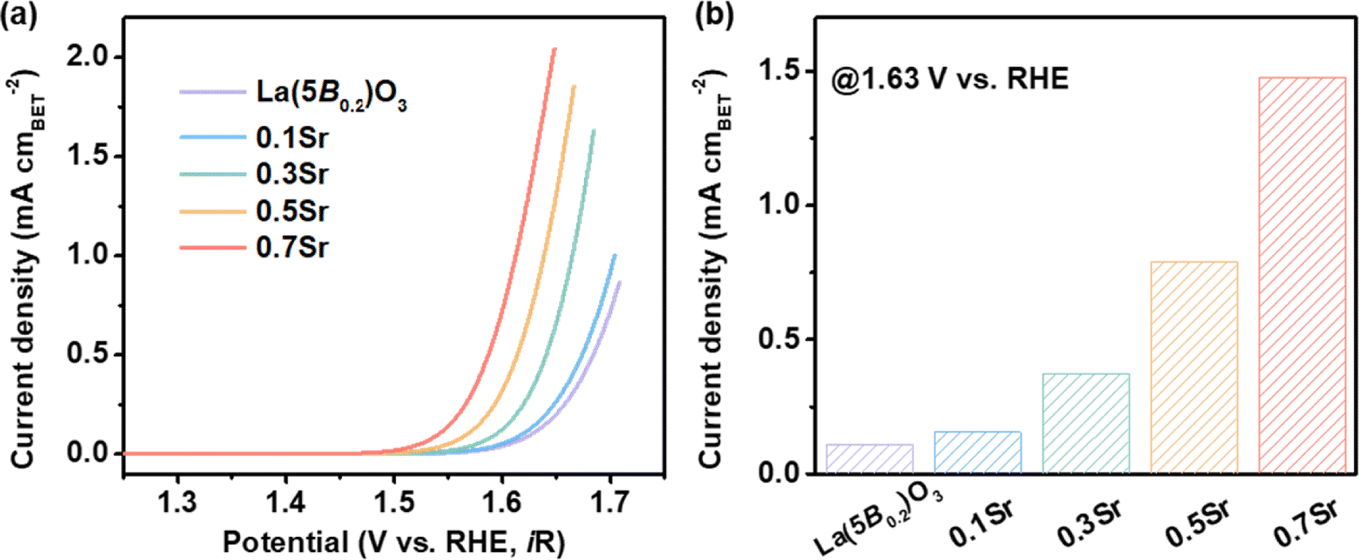 | ||
| Fig. 4 Intrinsic catalytic activity for La1−xSrx(5B0.2)O3. (a) LSV curves with the BET-normalized current density. (b) Comparison of the BET-normalized current densities at 1.63 V vs. RHE. | ||
In situ EIS measurements were conducted to investigate the kinetics of charge transfer at the electrode–electrolyte interface. Fig. 5b and c show the Nyquist plots of La(5B0.2)O3 and 0.7Sr in the range of 1.54–1.64 V vs. RHE, which can be fitted using the equivalent circuit (as shown in Fig. S4†) composed of electrolyte resistance, a constant phase element and a charge transfer resistance (Rct). The fitted Rct values are depicted in Fig. 5f. The Rct decreases as the potential increases. At the same potential, 0.7Sr displays a smaller Rct than that of La(5B0.2)O3, suggesting the faster charge transfer for 0.7Sr.
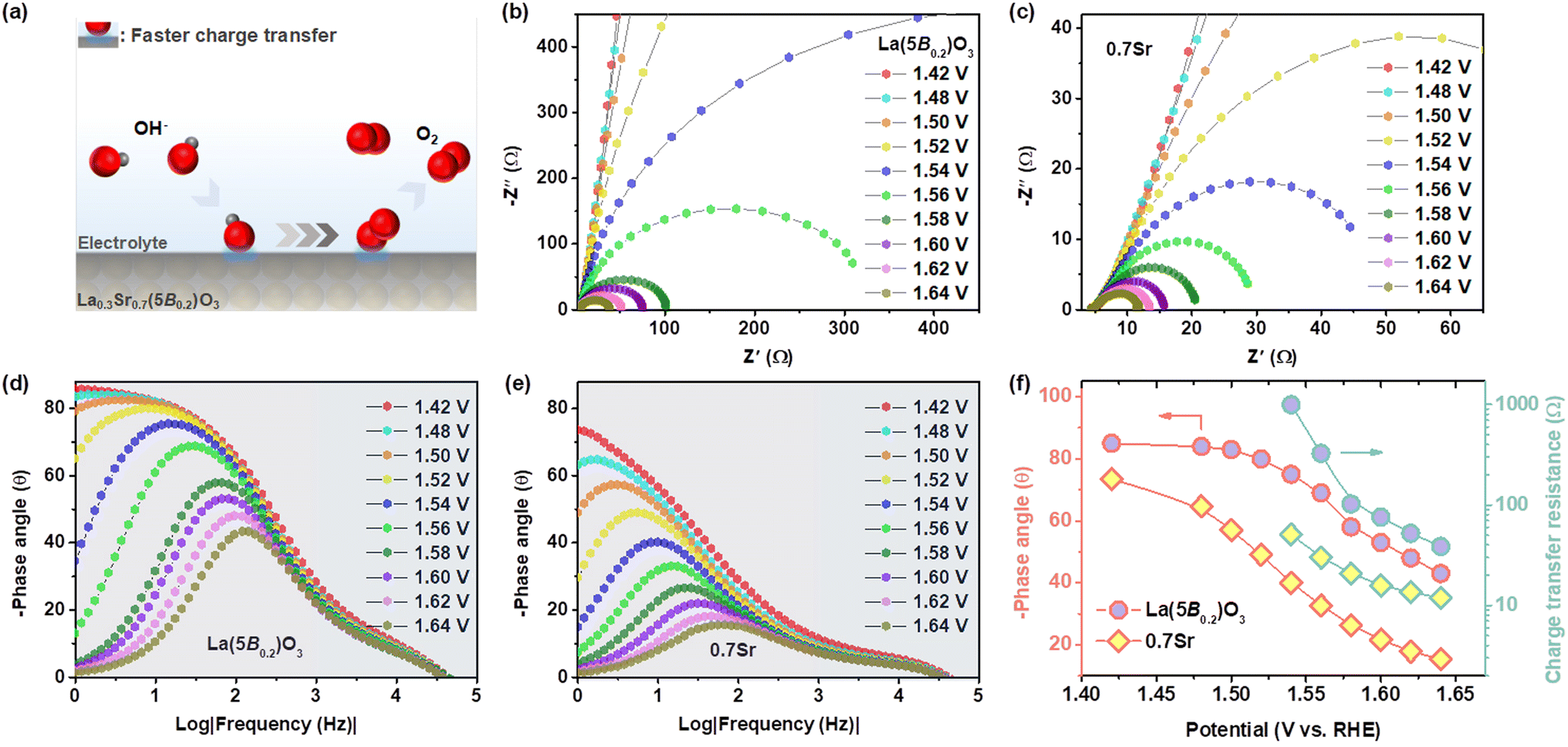 | ||
| Fig. 5 (a) Schematic illustration explaining the charge transfer of OER intermediates at the interfaces. EIS of (b) La(5B0.2)O3 and (c) 0.7Sr at different potentials. Bode phase plots of (d) La(5B0.2)O3 and (e) 0.7Sr at different potentials. (f) Phase peak angles and Rct of La(5B0.2)O3 and 0.7Sr. | ||
In addition, Fig. 5d and c exhibit the Bode phase plots for La(5B0.2)O3 and 0.7Sr. The smaller peak at the high-frequency region (1–100 kHz) and the main peak at the low low-frequency region (0.001–1 kHz) correspond to the conductivity of the catalyst inner layer and the OER charge transfer at the catalyst–electrolyte interface, respectively.41 The lower phase angle of 0.7Sr at the high-frequency region compared to that of La(5B0.2)O3 reveals good catalyst inter-layer electrical conduction for 0.7Sr. Within the low-frequency region, the phase angle of the main peak diminishes as the applied bias potential increases. The phase angle values of La(5B0.2)O3 and 0.7Sr at different potentials are depicted in Fig. 5f. In comparison with La(5B0.2)O3, 0.7Sr has a smaller phase peak angle at the same potential, confirming that the interaction between the catalyst with adsorbed reaction intermediates enhances the charge transfer and thus accelerates the OER kinetics. A schematic illustration of the 0.7Sr catalyst is shown in Fig. 5a.
3.4 Origin of the high OER performance for 0.7Sr
To gain some insights into the origin of the enhancement in OER activity for 0.7Sr, XPS spectra were conducted to check the chemical states of TM at the B-site in La(5B0.2)O3 and 0.7Sr, as shown in Fig. 6. For La(5B0.2)O3, the Cr 2p core level spectra (shown in Fig. 6a) display the spin–orbital doublets corresponding to Cr 2p3/2 and Cr 2p1/2. The Cr 2p3/2 peak is deconvoluted into two peaks, Cr3+ at 576.0 eV and Cr6+ at 579.0 eV, with a Cr6+/Cr3+ ratio of 1.10. Accordingly, the Mn4+ and Mn3+, Fe3+ and Fe2+, as well as Co3+ and Co2+ peaks are found in the Mn 2p, Fe 2p and Co 2p core level spectra, respectively (as shown in Fig. 6b–d and Tabel S1†). The ratios of Mn4+/Mn3+, Fe3+/Fe2+ and Co3+/Co2+ are 0.72, 3.32 and 1.35, respectively. In the Ni 2p spectra, the Ni 2p3/2 overlaps with La 3d3/2; thus, the Ni 2p1/2 is used to study the oxidation state of Ni. As depicted in Fig. 6e, only Ni2+ is found. The formation of Ni2+ in La(5B0.2)O3 is due to the super-exchange interaction, which is usually observed in perovskite oxides, such as the exchange of Ni2+–O–Mn4+ in perovskite LaNi0.5Mn0.5O3 (ref. 42) and exchange of Co2+–O–Mn4+ in LaCo0.5Mn0.5O3.43 The results of XPS analysis of La(5B0.2)O3 agree well with the previous study by Nguyen et al.21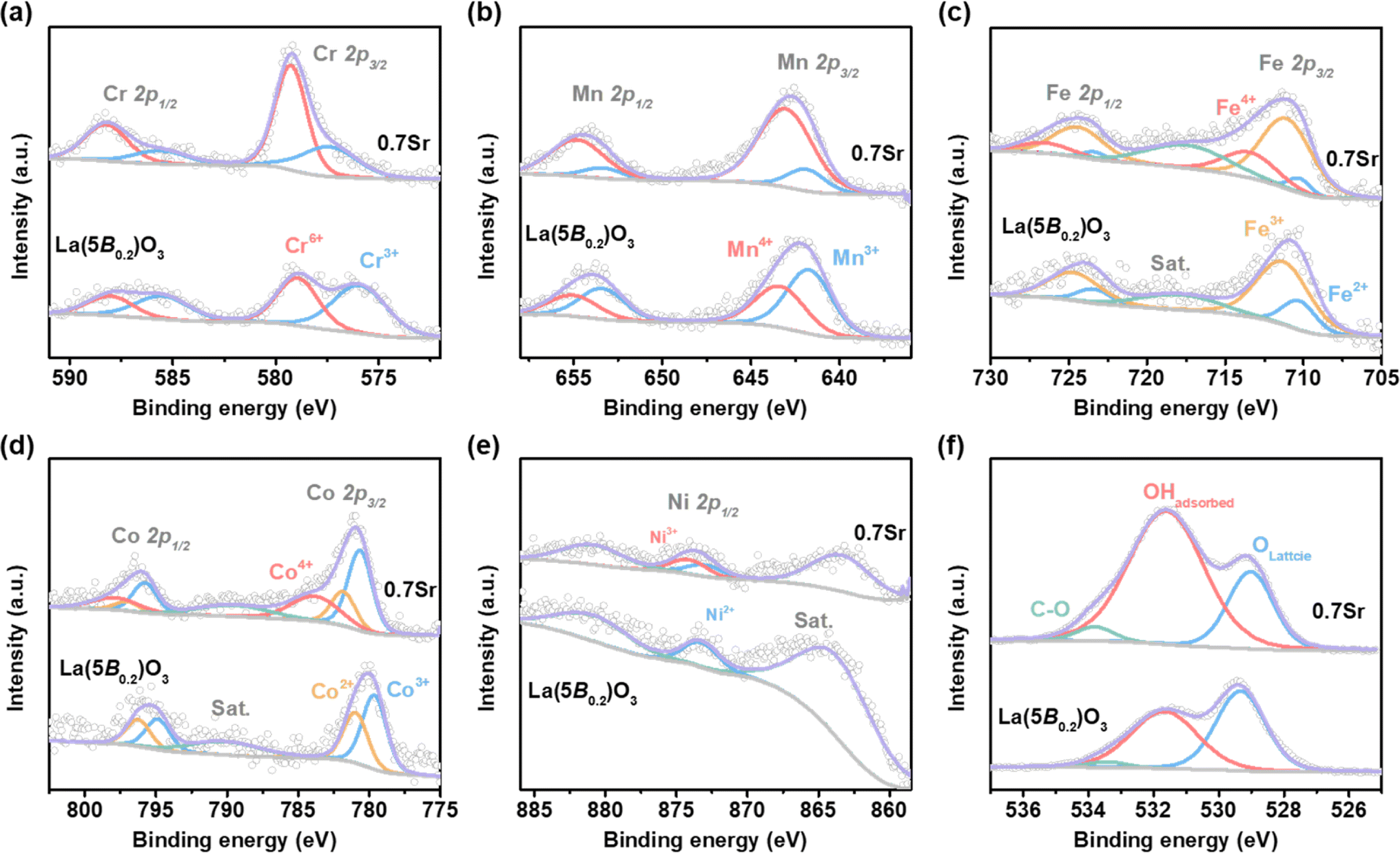 | ||
| Fig. 6 XPS spectra for La(5B0.2)O3 and 0.7Sr. (a) Cr 2p, (b) Mn 2p, (c) Fe 2p, (d) Co 2p, (e) Ni 2p and (f) O 1s. | ||
With regard to 0.7Sr, the partial substitution of La3+ with Sr2+ for La1−xSrx(5B0.2)O3 could introduce hole states, which pushes the valence state of TM at B-sites higher. As shown in Fig. 6 and Table S1,† more high-valence Cr6+ and Mn4+ were detected in 0.7Sr compared with La(5B0.2)O3, with Cr6+/Cr3+ ratio and Mn4+/Mn3+ ratio reaching 2.62 and 5.09, respectively. In addition, the peak around 713.7 eV for 0.7Sr shown in Fig. 6c corresponds to Fe4+ 2p3/2, suggestive of the forming high-valence Fe4+. In the Co 2p XPS spectra (Fig. 6d), an additional peak at 784.0 eV is observed for 0.7Sr in comparison with La(5B0.2)O3, indicating the partial oxidation of Co3+ to Co4+ for 0.7Sr.44 Moreover, a positive shift of 0.6 eV could be estimated for the Ni 2p1/2 peak (Fig. 6e and S5†), which is attributed to the formation of Ni3+. In a nutshell, Sr doping promotes the generation of more high-valence Cr6+, Mn4+, Fe4+, Co4+ and Ni3+ compared to La(5B0.2)O3. This is consistent with the scenario of the oxidation of B-site TM cations to compensate for the charge imbalance induced by Sr2+ doping.
The formation of high-valence TM cations induced by Sr doping is responsible for the higher OER activity of 0.7Sr. Based on molecular orbital theory, Suntivich et al. proposed the eg-filling activity descriptor for perovskite oxides in which the OER activities are closely related to the eg occupancy of the B-site cations, with eg = 1 showing the highest OER performance. In the 0.7Sr, the generating Fe4+ (high spin: t2g3eg1) and Ni3+ (low spin: t2g6eg1) with a nominal eg orbital occupancy of one electron is an important factor to enhance the OER activity. Moreover, Sr2+ substitution for La3+ introduces abundant hole states, thereby forming higher-valence cations (such as Mn4+, Fe4+, Co4+ and Ni3+), which can enhance the B-site cations TM 3d–O 2p hybridization.10,45 The greater hybridization not only accelerates the charge transfer between B-site cations and adsorbed reaction intermediates10 but also enhances the affinity of hydroxide,46 which is another factor that results in higher OER activity for 0.7Sr. The O 1s core level spectra (shown in Fig. 6f) and the deconvolution results (Table S1†) demonstrate the enhanced hydroxide affinity on 0.7Sr.26 In addition, although the Cr6+ (t02ge0g) in 0.7Sr is not conducive to OER activity based on the eg-filling activity descriptor, the soluble Cr6+ can promote surface reconstruction. As discussed subsequently, the surface reconstruction of the catalyst strongly correlates with the OER stability.
The surface structural reconstruction during OER in alkaline solution can determine the electrochemical OER durability.6,47 In general, the surface of perovskite catalysts undergoes structural reconstruction during OER to form a highly active oxyhydroxide layer, which is accompanied by the leaching of cations as well as dissolution-redeposition behavior.47,48 In the 0.7Sr, the soluble Sr2+ could easily leach out from the perovskite structure, leading to structural collapse, which can be further accelerated by the soluble Cr6+. The soluble Cr6+ leaching to promote surface reconstruction is discovered in the CoCr2O4 catalyst.49 Moreover, the dissolution of the Mn, Fe, Co and Ni cations, being rather insoluble species, can be redeposited on the perovskite surface to generate an amorphous reconstructed layer. Fig. S6† shows the TEM image of 0.7Sr after OER, in which an amorphous layer is observed at the catalyst surface. The yielding reconstructed layer is likely to be the Mn/Fe/Co/Ni oxyhydroxide species, which contributes to the good electrochemical stability shown in Fig. 3e.
3.5 Alkaline water splitting performances
Owing to the excellent performance of the 0.7Sr catalyst in the three-electrode system at hand, a two-electrode system (0.7Sr‖20%Pt/C) was assembled to evaluate the performance of practical alkaline overall water splitting in 4 M KOH solution (inset in Fig. 7a). Each electrode area is 0.5 cm−2 with a catalyst loading of 0.5 mg cm−2. Fig. 7a shows the corresponding polarization curves at a scan rate of 5 mV s−1. The 0.7Sr‖20%Pt/C electrolyzer requires 1.627 V to reach the current density of 10 mA cm−2, far superior to that of La(5B0.2)O3‖20%Pt/C electrolyzer (1.762 V). Moreover, 0.7Sr‖20%Pt/C electrolyzer runs at a current density of 10 mA cm−2 for 200 h with a decay of 0.12 mV h−1 (shown in Fig. 7b), indicating excellent electrochemical stability. All the results prove that the 0.7Sr catalyst is an outstanding oxygen-evolution electrocatalyst for alkaline water splitting with great potential for application.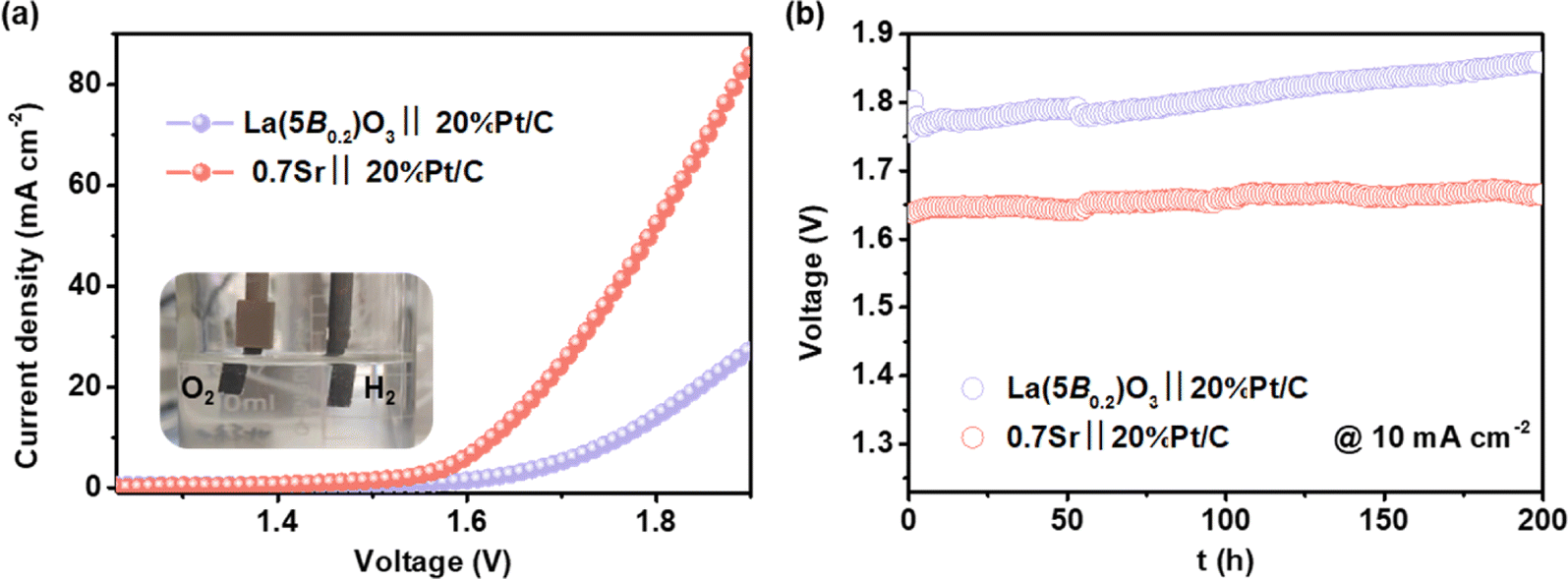 | ||
| Fig. 7 (a) Polarization curves of La(5B0.2)O3‖20%Pt/C and 0.7Sr‖20%Pt/C two-electrode overall water splitting systems. (b) Voltage-time tests for the two-electrode overall water splitting systems at 10 mA cm−2. | ||
4. Conclusions
In conclusion, high-entropy perovskite La1−xSrx(CrMnFeCoNi)0.2O3 nanoparticles are synthesized using the sol–gel method. Due to the acceleration of interfacial charge transfer, Sr doping can significantly enhance the OER activity of high-entropy La1−xSrx(CrMnFeCoNi)0.2O3. Among them, the best electrocatalyst La0.3Sr0.7(CrMnFeCoNi)0.2O3 can operate at 10 mA cm−2 in 1 M KOH only with an overpotential of 330 mV and deliver a 13.3-fold enhancement in the intrinsic catalytic activity compared to La(CrMnFeCoNi)0.2O3. XPS spectra confirmed that Sr2+ substitution for La3+ introduces abundant hole states, thereby forming higher-valence cations (such as Mn4+, Fe4+, Co4+ and Ni3+), which could accelerate the charge transfer and result in higher OER activity. The assembled electrolyzer with a two-electrode system runs at a current density of 10 mA cm−2 for 200 h without obvious voltage attenuation. This work offers an important method for developing a high-performance, high-entropy perovskite OER catalyst for hydrogen production by electrochemical water splitting.Author contributions
G. F. directed the project. H. Q. and H. L. prepared the samples and performed electrochemical measurements. R. W. conducted the XRD, SEM measurements. R. W. and G. F. discussed the results and wrote the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Key Scientific Research Projects of Higher Education Institutions in Henan Province (23A150050), Henan Medical Science and Technology Research Program Joint Construction Project (LHGJ20230326), Natural Science Foundation of Henan Province of China (222300420328, 242300420343), the Starting Research Fund from Huanghe Science and Technology College (02032388) and the Huanghe Science and Technology College Students’ Innovation and Entrepreneurship Training Program (2024xscxcy003). The authors thank the Shiyanjia Lab (http://www.shiyanjia.com) for the TEM test.References
- R. Zheng, Z. Liu, Y. Wang, Z. Xie and M. He, Joule, 2022, 6, 1148 CrossRef CAS.
- A. Raveendran, M. Chandran and R. Dhanusuraman, RSC Adv., 2023, 13, 3843 RSC.
- X. Zhao, Y. Li, C. Zhao and Z.-H. Liu, Small, 2020, 16, 2004973 CrossRef CAS PubMed.
- J. Corbin, M. Jones, C. Lyu, A. Loh, Z. Zhang, Y. Zhu and X. Li, RSC Adv., 2024, 14, 6416 RSC.
- Y. Gu, Y. Min, L. Li, Y. Lian, H. Sun, D. Wang, M. H. Rummeli, J. Guo, J. Zhong, L. Xu, Y. Peng and Z. Deng, Chem. Mater., 2021, 33, 4135 CrossRef CAS.
- L. Wu, Z. Guan, D. Guo, L. Yang, X. a. Chen and S. Wang, Small, 2023, 19, 2304007 CrossRef CAS PubMed.
- L. Gao, X. Cui, C. D. Sewell, J. Li and Z. Lin, Chem. Soc. Rev., 2021, 50, 8428 RSC.
- K. Wang, C. Han, Z. Shao, J. Qiu, S. Wang and S. Liu, Adv. Funct. Mater., 2021, 31, 2102089 CrossRef CAS.
- J. S. Yoo, X. Rong, Y. Liu and A. M. Kolpak, ACS Catal., 2018, 8, 4628 CrossRef CAS.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou and Z. Shao, Small, 2017, 13, 1603793 CrossRef PubMed.
- M. Qu, X. Ding, Z. Shen, M. Cui, F. E. Oropeza, G. Gorni, V. A. de la Peña O'Shea, W. Li, D.-C. Qi and K. H. L. Zhang, Chem. Mater., 2021, 33, 2062 CrossRef CAS.
- S. Ingavale, M. Gopalakrishnan, C. M. Enoch, C. Pornrungroj, M. Rittiruam, S. Praserthdam, A. Somwangthanaroj, K. Nootong, R. Pornprasertsuk and S. Kheawhom, Small, 2024, 20, 2308443 CrossRef CAS PubMed.
- C. Hu, X. Wang, T. Yao, T. Gao, J. Han, X. Zhang, Y. Zhang, P. Xu and B. Song, Adv. Funct. Mater., 2019, 29, 1902449 CrossRef.
- X. Xu, Y. Pan, L. Ge, Y. Chen, X. Mao, D. Guan, M. Li, Y. Zhong, Z. Hu, V. K. Peterson, M. Saunders, C.-T. Chen, H. Zhang, R. Ran, A. Du, H. Wang, S. P. Jiang, W. Zhou and Z. Shao, Small, 2021, 17, 2101573 CrossRef CAS PubMed.
- Y. Zhu, Q. Lin, Z. Hu, Y. Chen, Y. Yin, H. A. Tahini, H.-J. Lin, C.-T. Chen, X. Zhang, Z. Shao and H. Wang, Small, 2020, 16, 2001204 CrossRef CAS PubMed.
- T.-Q. Gao, Y.-Q. Zhou, X.-J. Zhao, Z.-H. Liu and Y. Chen, Adv. Funct. Mater., 2024, 2315949, DOI:10.1002/adfm.202315949.
- S. Sha, R. Ge, Y. Li, J. M. Cairney, R. Zheng, S. Li, B. Liu, J. Zhang and W. Li, Front. Energy, 2023 DOI:10.1007/s11708-023-0892-6.
- K. Chu, J. Qin, H. Zhu, M. De Ras, C. Wang, L. Xiong, L. Zhang, N. Zhang, J. A. Martens, J. Hofkens, F. Lai and T. Liu, Sci. China Mater., 2022, 65, 2711 CrossRef CAS.
- A. Sarkar, R. Djenadic, D. Wang, C. Hein, R. Kautenburger, O. Clemens and H. Hahn, J. Eur. Ceram. Soc., 2018, 38, 2318 CrossRef CAS.
- T. X. Nguyen, Y.-C. Liao, C.-C. Lin, Y.-H. Su and J.-M. Ting, Adv. Funct. Mater., 2021, 31, 2101632 CrossRef CAS.
- J. T. Mefford, X. Rong, A. M. Abakumov, W. G. Hardin, S. Dai, A. M. Kolpak, K. P. Johnston and K. J. Stevenson, Nat. Commun., 2016, 7, 11053 CrossRef CAS PubMed.
- G. Fu, W. Li, J.-Y. Zhang, M. Li, C. Li, N. Li, Q. He, S. Xi, D. Qi, J. L. MacManus-Driscoll, J. Cheng and K. H. Zhang, Small, 2021, 17, 2006930 CrossRef CAS PubMed.
- Z. Shen, M. Qu, J. Shi, F. E. Oropeza, V. A. de la Peña O'Shea, G. Gorni, C. M. Tian, J. P. Hofmann, J. Cheng, J. Li and K. H. L. Zhang, J. Energy Chem., 2022, 65, 637 CrossRef CAS.
- K. H. L. Zhang, Y. Du, A. Papadogianni, O. Bierwagen, S. Sallis, L. F. J. Piper, M. E. Bowden, V. Shutthanandan, P. V. Sushko and S. A. Chambers, Adv. Mater., 2015, 27, 5191 CrossRef CAS PubMed.
- Z. Shen, Y. Zhuang, W. Li, X. Huang, F. E. Oropeza, E. J. M. Hensen, J. P. Hofmann, M. Cui, A. Tadich, D. Qi, J. Cheng, J. Li and K. H. L. Zhang, J. Mater. Chem. A, 2020, 8, 4407 RSC.
- A. R. Mazza, E. Skoropata, J. Lapano, J. Zhang, Y. Sharma, B. L. Musico, V. Keppens, Z. Gai, M. J. Brahlek, A. Moreo, D. A. Gilbert, E. Dagotto and T. Z. Ward, Phys. Rev. B, 2021, 104, 094204 CrossRef CAS.
- K. H. L. Zhang, Y. Du, P. V. Sushko, M. E. Bowden, V. Shutthanandan, S. Sallis, L. F. J. Piper and S. A. Chambers, Phys. Rev. B: Condens. Matter Mater. Phys., 2015, 91, 155129 CrossRef.
- C. Wei and Z. J. Xu, Small Methods, 2018, 2, 1800168 CrossRef.
- H. Wang, M. Cui, G. Fu, J. Zhang, X. Ding, I. Azaceta, M. Bugnet, D. M. Kepaptsoglou, V. K. Lazarov, V. A. de la Peña O’Shea, F. E. Oropeza and K. H. L. Zhang, Sci. China Chem., 2022, 65, 1885 CrossRef CAS.
- G. Fu, X. Wen, S. Xi, Z. Chen, W. Li, J.-Y. Zhang, A. Tadich, R. Wu, D.-C. Qi, Y. Du, J. Cheng and K. H. L. Zhang, Chem. Mater., 2019, 31, 419 CrossRef CAS.
- Y. Wu, X. Wang, T. She, T. Li, Y. Wang, Z. Xu, X. Jin, H. Song, S. Yang, S. Li, S. Yan, H. He, L. Zhang and Z. Zou, Small, 2024, 20, 2306464 CrossRef CAS PubMed.
- G. Fu, L. Zhang, R. Wei, H. Liu, R. Hou, Z. Zhang, K. Yang and S. Zhang, Small, 2024, 2309091, DOI:10.1002/smll.202309091.
- J.-W. Zhao, C.-F. Li, Z.-X. Shi, J.-L. Guan and G.-R. Li, Research, 2020, 2020, 6961578 CAS.
- J. Li, L. Zheng, B. Huang, Y. Hu, L. An, Y. Yao, M. Lu, J. Jin, N. Zhang, P. Xi and C.-H. Yan, Small, 2022, 18, 2204723 CrossRef CAS PubMed.
- Y. Zhu, W. Zhou, J. Yu, Y. Chen, M. Liu and Z. Shao, Chem. Mater., 2016, 28, 1691 CrossRef CAS.
- N.-I. Kim, Y. J. Sa, T. S. Yoo, S. R. Choi, R. A. Afzal, T. Choi, Y.-S. Seo, K.-S. Lee, J. Y. Hwang, W. S. Choi, S. H. Joo and J.-Y. Park, Sci. Adv., 2018, 4, eaap9360 CrossRef PubMed.
- Y. L. Gaoliang Fu and J. Wang, Journal of Huanghe S&T College, 2023, 25, 23 Search PubMed.
- Y. Zhu, W. Zhou, Y. Zhong, Y. Bu, X. Chen, Q. Zhong, M. Liu and Z. Shao, Adv. Energy Mater., 2017, 7, 1602122 CrossRef.
- J. Dai, Y. Zhu, Y. Yin, H. A. Tahini, D. Guan, F. Dong, Q. Lu, S. C. Smith, X. Zhang, H. Wang, W. Zhou and Z. Shao, Small, 2019, 15, 1903120 CrossRef PubMed.
- J. Fan, X. Zhang, M. Han, X. Xiang, C. Guo, Y. Lin, N. Shi, D. Xu, Y. Lai and J. Bao, Small, 2024, 20, 2303927 CrossRef CAS PubMed.
- Y. Tong, J. Wu, P. Chen, H. Liu, W. Chu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2018, 140, 11165 CrossRef CAS PubMed.
- R. I. Dass and J. B. Goodenough, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 014401 CrossRef.
- J. Qian, J. Li, B. Xia, J. Zhang, Z. Zhang, C. Guan, D. Gao and W. Huang, Energy Storage Mater., 2021, 42, 470 CrossRef.
- A. Grimaud, K. J. May, C. E. Carlton, Y.-L. Lee, M. Risch, W. T. Hong, J. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 1 Search PubMed.
- W. T. Hong, K. A. Stoerzinger, Y.-L. Lee, L. Giordano, A. Grimaud, A. M. Johnson, J. Hwang, E. J. Crumlin, W. Yang and Y. Shao-Horn, Energy Environ. Sci., 2017, 10, 2190 RSC.
- Y. Sun, R. Li, X. Chen, J. Wu, Y. Xie, X. Wang, K. Ma, L. Wang, Z. Zhang, Q. Liao, Z. Kang and Y. Zhang, Adv. Energy Mater., 2021, 11, 2003755 CrossRef CAS.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B.-J. Kim, J. Durst, F. Bozza, T. Graule, R. Schäublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925 CrossRef CAS PubMed.
- Y. Duan, J. Y. Lee, S. Xi, Y. Sun, J. Ge, S. J. H. Ong, Y. Chen, S. Dou, F. Meng, C. Diao, A. C. Fisher, X. Wang, G. G. Scherer, A. Grimaud and Z. J. Xu, Angew. Chem., Int. Ed., 2021, 60, 7418 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02680b |
| This journal is © The Royal Society of Chemistry 2024 |