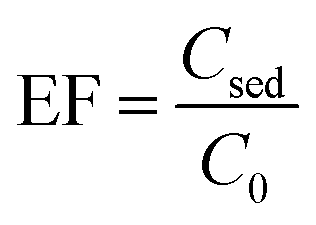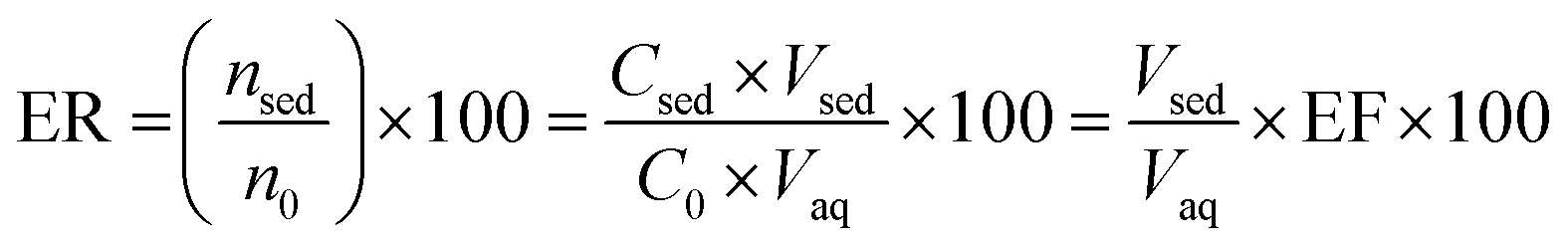DOI:
10.1039/D4RA02795G
(Paper)
RSC Adv., 2024,
14, 28035-28043
A fast and simple procedure for the synthesis of a zinc and 1,4-benzene dicarboxylic acid metal–organic framework and its evaluation as a sorbent for dispersive micro solid phase extraction of pesticide residues†
Received
15th April 2024
, Accepted 12th August 2024
First published on 3rd September 2024
Abstract
In this work, Zn-1,4-benzenedicarboxylate metal–organic framework was synthesized by a simple hydrothermal process and used in dispersive micro solid phase extraction of some pesticide from various fruit juice and water samples. Seven widely consumed pesticides in agriculture (chlorpyrifos, haloxyfop-R-methyl, oxadiazon, diniconazole, clodinafop-propargyl, fenpropathrin, and fenaxoprop-p-ethyl) were selected as target analytes. In this work, dispersive micro solid phase extraction was followed by a liquid phase microextraction method to achieve more enrichment of the analytes, and the enriched analytes were quantified using a gas chromatography-flame ionization detector. The sorbent was authenticated by Fourier transform infrared spectrophotometry, X-ray diffraction, energy dispersive X-ray analysis, and scanning electron microscope imaging. The factors affecting the extraction efficiency of the developed method were investigated, and the validation of the method under the optimized extraction conditions presented satisfactory results for precision and trueness, with limits of detection and quantification in the ranges of 0.50–0.90 and 1.5–2.7 μg L−1, respectively. Enrichment factors and extraction recoveries were in the ranges of 239–392% and 47–78%, respectively. One river water and some fruit and vegetable juice samples were analyzed by the recommended method, and the obtained recoveries were between 90% and 102%.
1. Introduction
The use of pesticides is a common and essential practice in different stages of cultivation to control pests, preserve crop quality, and enhance their yield. Although they have many advantages, their overuse can lead to detrimental effects on untargeted organisms, including humans, through contaminated water and leaving their residues in agricultural products.1 Hence, pesticide residue detection and quantification in water and agricultural products have become the subject of many analytical studies.2–4 Common analytical methods for the determination of pesticide residues utilize chromatography techniques such as gas chromatography (GC)5,6 and liquid chromatography.7,8 Generally, due to the complex matrices of food samples and the low concentration of pesticide residues in them, utilizing an efficient sample preparation process prior to instrumental analysis is needed. Solid phase extraction (SPE) is an efficient and commonly used method for the purification and enrichment of pesticide residues in food samples.9–11 Dispersive micro solid phase extraction (D-μSPE) is a variant of SPE and is used in food analysis frequently.12,13 In D-μSPE, sorbent particles are dispersed into a sample solution and a close contact is established between the solute molecules in the sample solution and the sorbent particles.14 Therefore, analytes are adsorbed onto the sorbent particles and most of the other components of the sample remain in the liquid phase.15–18 The prominent advantage of dispersive modes of SPE is the increasing contact area between the analytes and sorbent. Also, labors of performing SPE in the classic mode such as high and controlled vacuum or pressure for passing the sample solution through the cartridge and clogging the cartridge (when the samples having suspended particles are processed) are eliminated.19 Up to now, various materials have been utilized as the sorbent for the extraction of pesticide residues from fruit and vegetable juices such as molecularly imprinted polymers,20 carbon nanoparticles,21 graphene,22 functionalized silica,23 and metal–organic frameworks (MOFs).24 MOFs are a class of inorganic–organic hybrid crystals with porous structures. High adsorption ability, pore size and surface function tunability, synthetic versatility, and large specific surface area make them ideal for use in various fields such as gas storage,25 catalysis,26 drug delivery,27 chemical sensors,28 and luminescent materials.29 They also are capable of being applied as the chromatographic stationary phase30, and adsorbents for SPE31 and solid phase microextraction.32
In this study, MOF-5 consisting of zinc (as the metal ion) and 1,4-benzene dicarboxylic acid (terephthalic acid) (as the organic linker) was synthesized by a hydrothermal procedure and its efficiency as a sorbent for D-μSPE of some pesticide residues from water and some fruit and vegetable juices was evaluated. The successful synthesis of the MOF-5 was verified by using techniques such as Fourier transform infrared (FTIR), X-ray diffraction (XRD), energy dispersive X-ray (EDX), and scanning electron microscopy (SEM) imaging. A dispersive liquid–liquid phase extraction (DLLME) step was performed after the D-μSPE process and the extracted pesticides were quantified using a GC-flame ionization detector (FID).
2. Experimental
2.1. Solutions and reagents
Analytical-grade zinc nitrate hexahydrate, terephthalic acid, acetone, acetonitrile, isopropanol, methanol, potassium chloride, hydrochloric acid (37%, w/w), sodium sulfate, sodium hydroxide, ammonia solution (25%, w/v), and sodium chloride were obtained from Merck (Darmstadt, Germany). 1,1,2,2-Tetrachloroethane (1,1,2,2-TCE), 1,1,2-trichloroethane (1,1,2-TCE), carbon tetrachloride, and 1,2-dibromoethane (1,2-DBE) were from Janssen (Beerse, Belgium). All the studied pesticides including chlorpyrifos, fenpropathrin, oxadiazon, fenoxaprop-p-ethyl, haloxyfop-R-methyl, diniconazole, and clodinafop-propargyl (purity > 98%) were from Dr Ehrenstorfer company (Augsburg, Germany). A stock solution of the mentioned pesticides (100 mg L−1 of each pesticide) was prepared in methanol and diluted with deionized water daily for use in the optimization and validation steps.
2.2. Samples
Fresh apples, watermelon, and onions were bought from local stores (Tabriz, Iran). They were squeezed using a juice extractor. The river water sample was collected from the Meydan Chay River (Tabriz, Iran). Prior to the extraction process, all the juices were diluted at a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 with deionized water and used in the experiments. River water was subjected to the proposed method without any dilution or treatment.
:1 with deionized water and used in the experiments. River water was subjected to the proposed method without any dilution or treatment.
2.3. Apparatus
Determination of the extracted pesticides was carried out on a Shimadzu 2014 gas chromatograph with a Zerbon™-1 capillary column (5% phenyl and 95% dimethylpolylsiloxane) (Phenomenex, Torrance, CA, USA). Column dimensions and film thickness were as follows: 30 m × 0.25 mm and 0.25 μm, respectively. The temperature programming of the column oven was: 60 °C for 1 min and ramped to 300 °C at 18 °C min−1 (kept for 6 min). Each run time was nearly 21 min. The gas chromatograph was equipped with a split/splitless injection port and an FID. The injection port was operated at 300 °C in a splitless/split mode (split ratio and sampling time were 1:10 and 1 min, respectively). Helium gas as the carrier gas (99.999%) was flown at a constant linear velocity of 30 cm s−1. The make-up gas was helium at 30 mL min−1. The temperature of FID was set at 300 °C and flow rates of hydrogen and air were 30 and 300 mL min−1, respectively. pH adjustment was performed using a Metrohm-654 pH meter (Herisau, Switzerland). A D-7200 Hettich centrifuge (Kirchlengern, Germany) was used for centrifugation. Vortex agitation was performed using an L46 vortex (Labinco, Breda, The Netherlands). EDX analysis and SEM (Tescan, Czech) imaging were conducted to study the surface composition and morphology of the prepared sorbent. The FTIR spectrum of the prepared sorbent was recorded using a Bruker FTIR spectrophotometer (Billerica, USA). The XRD pattern of the synthesized sorbent was recorded on a Siemens AG (Karlsruhe, Germany) XRD apparatus operated at 35 kV.
2.4. MOF-5 synthesis
For the synthesis of MOF-5, 500 mg of terephthalic acid was dissolved in 10 mL of concentrated ammonia solution (25%, v/v) and dropwise added to 100 mL 1 mol L−1 solution of Zn(NO3)2·6H2O prepared in degassed deionized water. The reaction was continued at 80 °C for 2 h under stirring. The reaction product was collected by vacuum filtration, washed with deionized water, and dried at room temperature.
2.5. Extraction procedure
2.5.1. D-μSPE. Five milligrams of MOF-5 were added to a 10 mL test tube poured with 5 mL of the sample solution (see Section 2.2) and/or model solution at a concentration of 100 μg L−1 of each pesticide. Then, sodium chloride was dissolved in it at a concentration of 5% (w/v). The resultant mixture was vortexed for 5 min to disperse the sorbent particles into the solution. Thereafter, it was centrifuged for 7 min at 7000 rpm and the supernatant was discarded. Afterward, 2 mL of isopropanol was added to the sorbent particles and the analytes were desorbed by vortexing for 5 min. Finally, centrifugation was performed (for 7 min at 7000 rpm) and the supernatant was utilized as a disperser in the subsequent DLLME step.
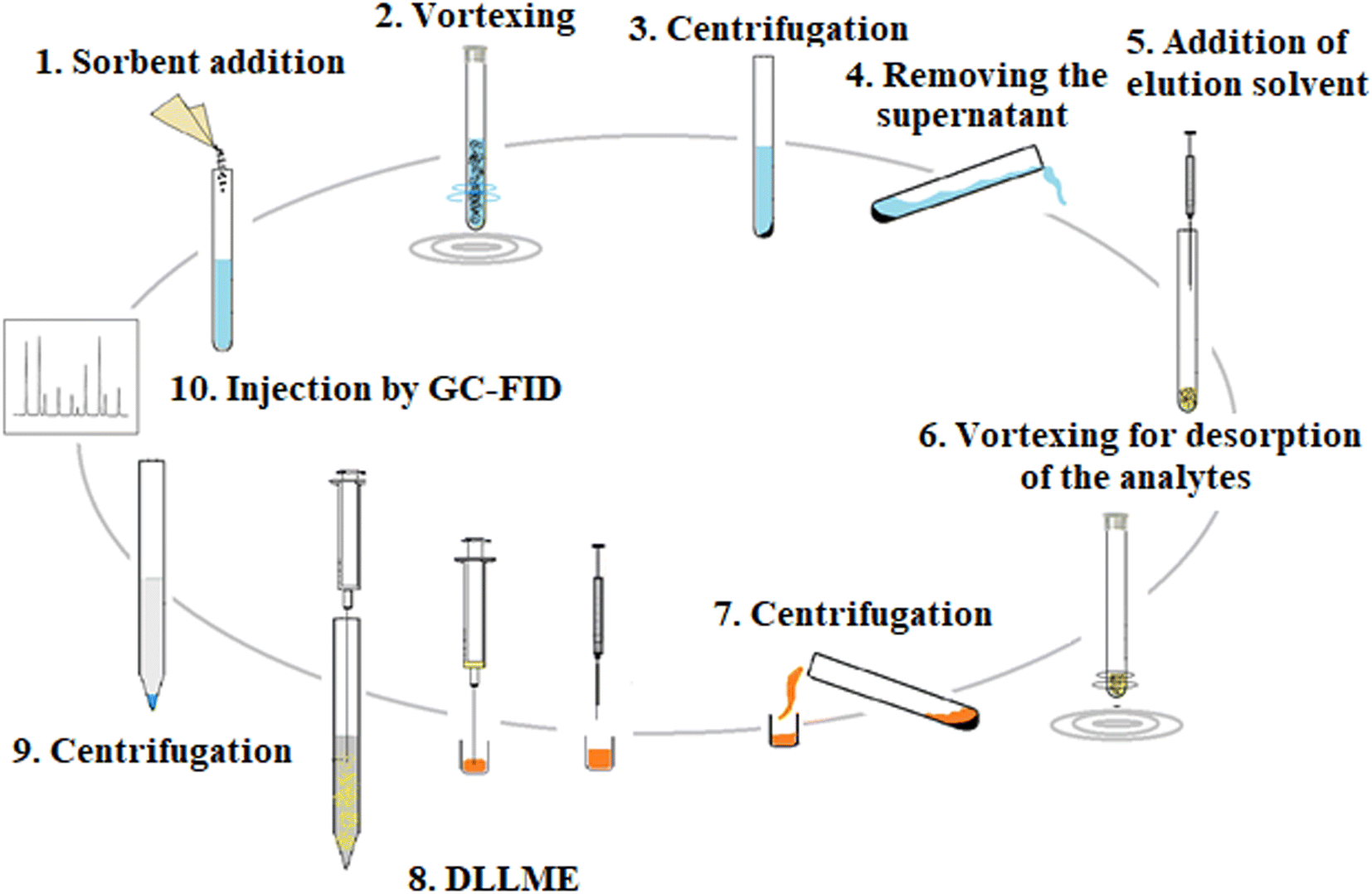
2.5.2. DLLME. To further enrich the extracted analytes from the previous step, the obtained eluate was mixed with 35 μL 1,2-DBE as an extractant. The obtained solution was speedily injected into 5 mL 5% (w/v) NaCl solution and placed into a 10 mL conical bottom glass test tube. A cloudy solution was formed. It was centrifuged at 7000 rpm for 7 min and a 10 ± 1 μL organic phase was sedimented. One microliter of the sedimented phase was injected into GC-FID to quantify the analytes. The overall extraction time was almost 30 min. The proposed D-μSPE-DLLME method is schematically shown in Fig. 1.
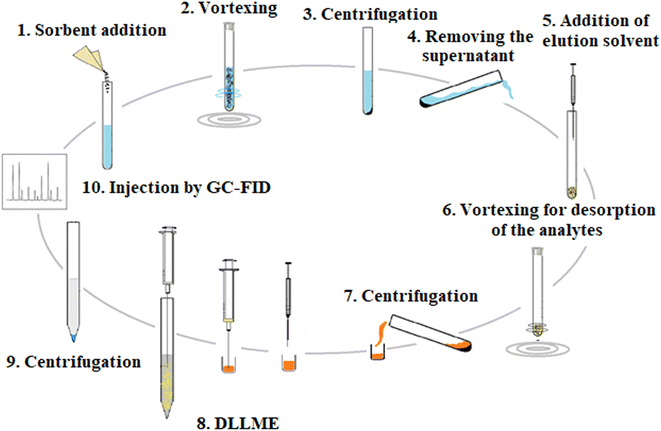 |
| | Fig. 1 Extraction procedure steps. | |
2.6. Calculation of the enrichment factor (EF) and extraction recovery (ER)
Two important factors of EF and ER indicating the efficiency of the proposed method were calculated from eqn (1) and (2), respectively:| |
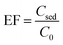 | (1) |
| |
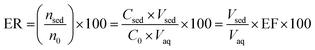 | (2) |
where C0 and Csed are the initial concentrations of analytes in the sample solution and the sedimented phase, respectively. n0 and nsed are the total and extracted amounts of the analyte, respectively. Vaq and Vsed are the volumes of the sample solution and sedimented extractant, respectively.
3. Results and discussion
3.1. Characterization of the synthesized MOF-5
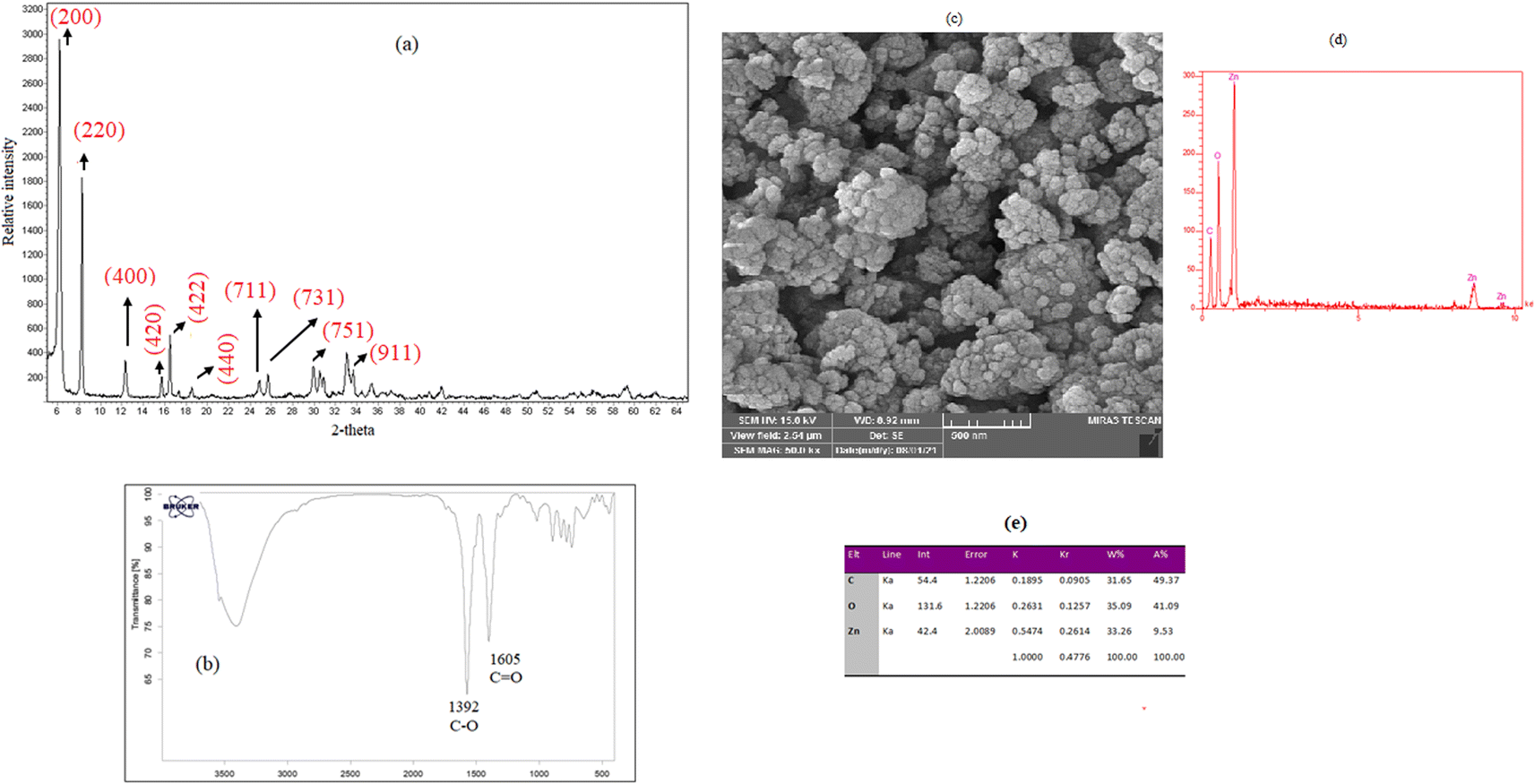
Characterization of the crystalline structure of the prepared MOF-5 was performed by XRD analysis. The obtained XRD pattern (Fig. 2a) showed that the diffractions at 2θ of 6.1°, 8.1°, 12.2°, 15.9°, 16.1°, 18.4°, 24.5°, 25.9°, 29.8°, and 33.8° are related to the crystal planes of (200), (220), (400), (420), (422), (440) (711), (731), (751), and (911), respectively, and they are matched with those from the previous reports in the literature.33,34 FTIR spectroscopy was used to confirm the functional groups in MOF-5 (Fig. 2b). Two peaks at 1392 and 1605 cm−1 are assigned to stretching vibrations of C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds of terephthalic acid, respectively. The morphology of MOF-5 particles was investigated by SEM. As shown in Fig. 2c, MOF-5 particles exhibit spherical and irregular shapes. The EDX analysis (Fig. 2d and e) indicates Zn, C, and O peaks with weight percentages of 33.26%, 31.65%, and 35.09%, respectively. The BET analysis was performed to investigate the surface properties of the prepared sorbent. The following values were obtained: average pores diameter; 24.365 nm; total pores volume: 0.111 cm3 g−1, and surface area; 18.227 m2 g−1.
O bonds of terephthalic acid, respectively. The morphology of MOF-5 particles was investigated by SEM. As shown in Fig. 2c, MOF-5 particles exhibit spherical and irregular shapes. The EDX analysis (Fig. 2d and e) indicates Zn, C, and O peaks with weight percentages of 33.26%, 31.65%, and 35.09%, respectively. The BET analysis was performed to investigate the surface properties of the prepared sorbent. The following values were obtained: average pores diameter; 24.365 nm; total pores volume: 0.111 cm3 g−1, and surface area; 18.227 m2 g−1.
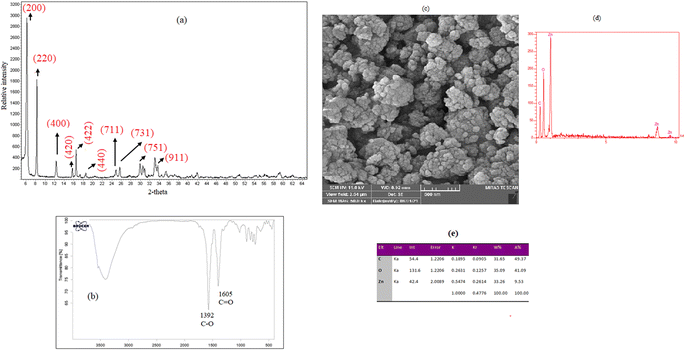 |
| | Fig. 2 (a) XRD pattern, (b) FTIR spectrum, (c) SEM images, (d) EDX spectrum (e), and EDX data of the synthesized sorbent. | |
3.2. Optimization of sorbent composition and weight
The amount of terephthalic acid was changed in the synthesis process in the range of 100–750 mg to reach an optimal composition for the prepared sorbent. The amount of Zn(NO3)2·6H2O was kept constant at 29.75 g. The reaction product in each case was collected, washed, and dried. Twenty milligrams of each product were subjected to the proposed procedure. Considering the results in Fig. S1,† the difference in ERs of the analytes by the use of each product as the sorbent is not significant (p values > 0.05). Therefore, MOF-5 was synthesized in each case. Given that the yield of the synthesis reaction was high using 500 mg of terephthalic acid, it was selected for the preparation of the sorbent.
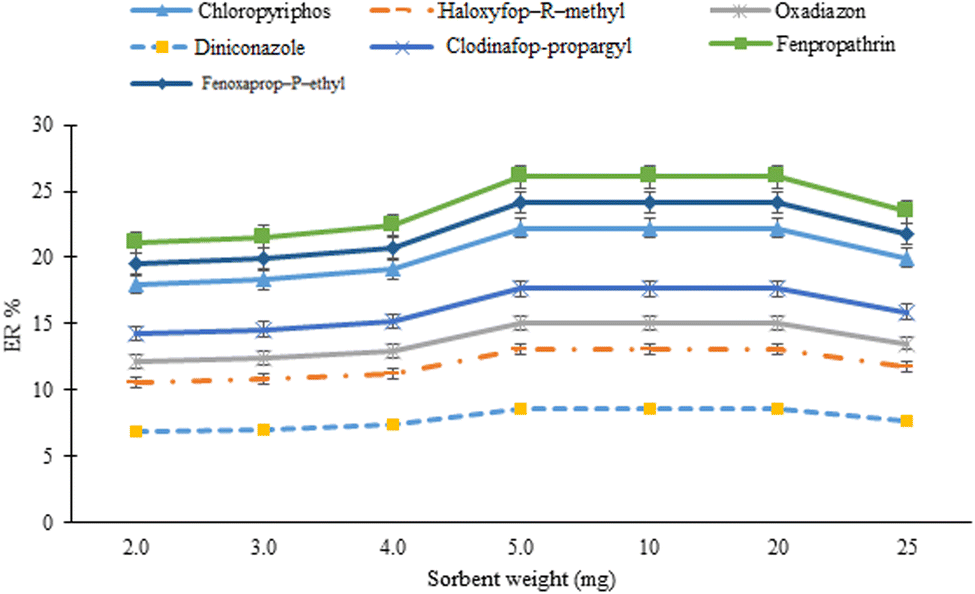
The weight of the sorbent was investigated in the range of 2–25 mg (2.0, 3.0, 4.0, 5.0, 10, 20, and 25 mg). The results from Fig. 3 show that after increasing the sorbent weight from 2 to 5 mg, the ERs of the analytes increased. Further increasing the sorbent amount has no significant effect on the extraction efficiency of the method. Increasing the method efficiency by increasing the sorbent amount is attributed to increasing the active sites for the adsorption of the analytes. However, by further increasing the sorbent amount, aggregation of the sorbent particles and incomplete elution of the analytes limit any additional improvement in the efficiency of the method. Therefore, 5 mg of the sorbent was used in the rest of the study.
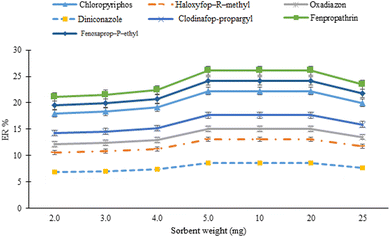 |
| | Fig. 3 Optimization of sorbent amount. Extraction conditions: sorbent amount, 20 mg; sample volume, 5 mL deionized water spiked with 100 μg L−1 of each analyte; terephthalic acid amount used for MOF synthesis, 500 mg; vortexing time in adsorption and desorption steps, 9 min; elution solvent (volume), acetonitrile (1 mL); DLLME aqueous phase, 5 mL deionized water; extraction solvent (volume), 1,2-DBE (45 μL); and centrifugation time and rate; 7 min and 7000 rpm, respectively. Error bars indicate minimum and maximum of three repeated determinations. | |
3.3. Optimization of D-μSPE conditions
3.3.1. Ionic strength of sample solution. Ionic strength is an important factor in microextraction techniques. Increasing ionic strength can represent two contrary effects: (I) a decrease in the solubility of the analyte in aqueous solution and favor their transferring into the extractive medium and (II) an increase in viscosity of the aqueous solution and a negative influence on the kinetic of the extraction. To investigate the effect of this factor, 5 mL of 2.5%, w/v, solutions of NaCl, KCl, and Na2SO4 spiked with 100 μg L−1 of each analyte were used as a sample solution, separately. The results in Fig. S2(a)† indicate that NaCl is a bit better than other salts and it can be related to better solvation of NaCl ions with water and release of the organic compounds.To optimize the concentration of NaCl, it was varied in the range of 1–10%, w/v. The results shown in Fig. S2(b)† indicate that the extraction efficiency is maximum at 5%, w/v, NaCl and decreases thereafter. Increasing the method efficiency by adding NaCl shows a salting-out effect. However, at higher concentrations, increasing the sample solution viscosity reduces the analytes migration rate and decreases the method efficiency. Attending to the results, 5%, w/v, NaCl was used for further studies.
3.3.2. Sample solution pH. The effect of pH on the extraction efficiency of the method was tested by adjusting the pH of the aqueous solution over the range of 3–9. The obtained results in Fig. S3† show that the maximum ER of each analyte was obtained in the pH range of 4–8, which matches the pH of the used samples in this study. Hydrolysis or dissociation of the analytes is the major reason for decreasing the method's efficiency.35 Therefore, the following experiments were performed without pH adjusting.
3.3.3. Optimization of adsorption and desorption time. In the dispersive mode of SPE, vortex mixing is usually used to form better interfacial contact between the analytes and sorbent particles in the adsorption step and effective elution of the analytes from the sorbent particle's surface in the desorption step. Therefore, it is an important parameter and affects the performance of the method. Additionally, the D-μSPE process needs optimum time durations to complete the adsorption and desorption of the analytes. Hence, vortex mixing times of adsorption and desorption steps were investigated in the range of 1–9 min. According to the results (Fig. S4†), the ERs increase with increasing vortex mixing time from 1–5 min and then remain almost constant in both adsorption and desorption steps. Increasing the analytical data by increasing adsorption and desorption time is related to the increasing contact area of the sorbent with the elution solvent. It must be noted that 5 min is sufficient time for adsorption or desorption of the analytes. Hence, 5 min time was opted for vortex mixing time for both of adsorption and elution steps in the subsequent experiments.
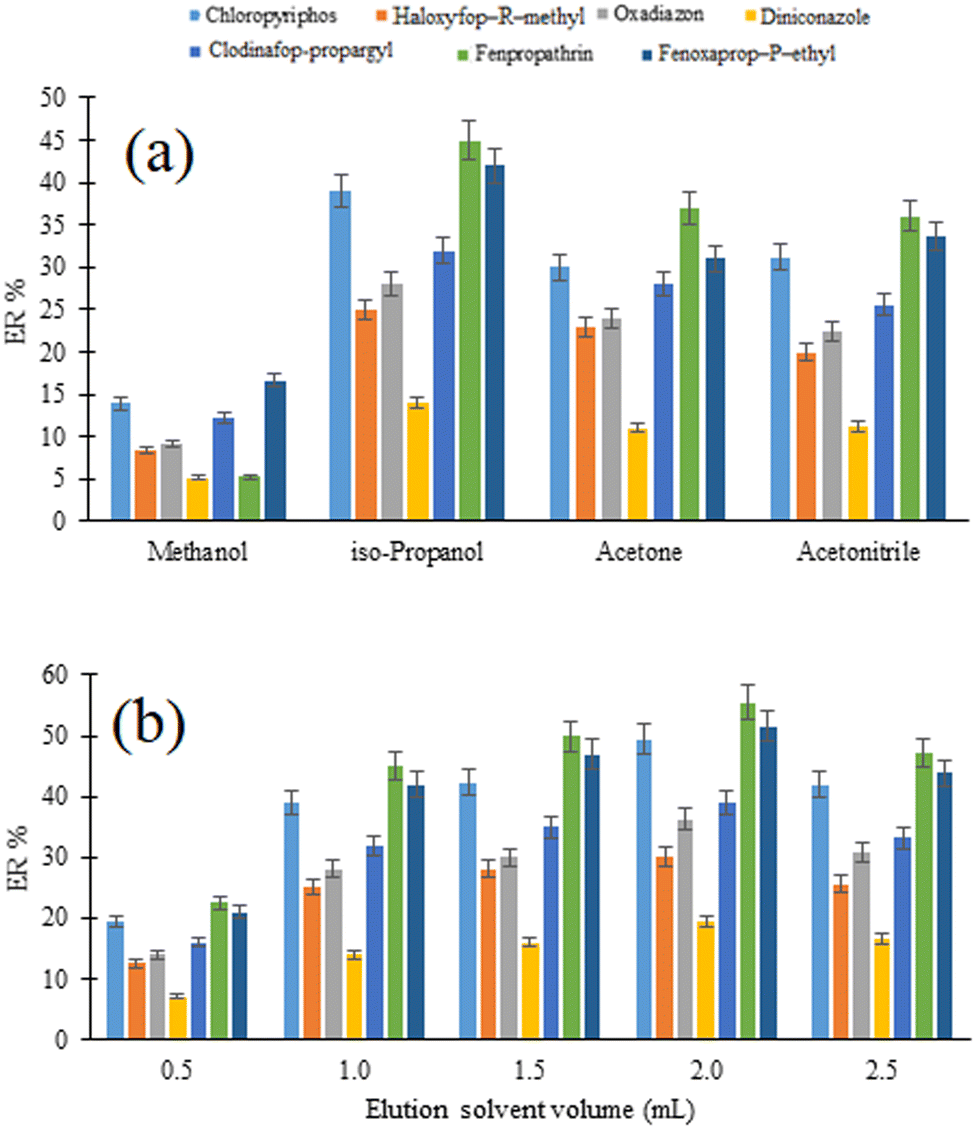
3.3.4. Selection of elution solvent type and volume. In the proposed D-μSPE-DLLME method, the selection of a viable elution solvent for quantitative desorption of the analytes from the surface of the sorbent particles is of great importance. Additionally, the dispersion efficiency of the extraction solvent used in the DLLME step depends on the type of the elution solvent. For this purpose, 1 mL of methanol, isopropanol, acetonitrile, and acetone were tested as the possible eluents and their desorption/dispersion capabilities are compared in Fig. 4a. Regarding the results, the highest ERs for the studied analytes are achieved using isopropanol as the elution solvent. It can be related to the higher solubility of the analytes in isopropanol or its better role as a disperser solvent in DLLME. Therefore, it was selected for use in the following experiments.
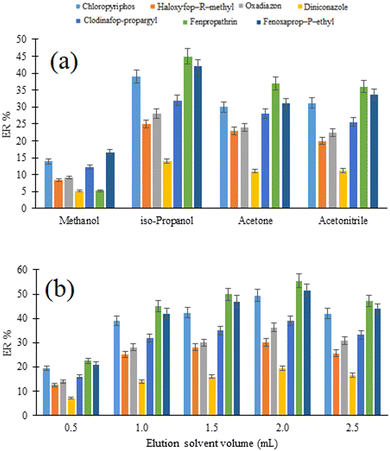 |
| | Fig. 4 Optimization of elution solvent type (a) and (b) volume. (a) Extraction conditions: are the same as those used in Fig. 3, except adsorption and desorption times were 5 min. (b) Extraction conditions: are the same as those used in (a), except iso-propanol was chosen as the elution solvent. | |
The effect of the elution solvent volume on the extraction efficiency of the method was evaluated using different volumes of isopropanol ranging from 0.5 to 2.5 mL and the outcomes are given in Fig. 4b. As can be seen, increasing the volume of isopropanol up to 2.0 mL results in increasing the extraction efficiency of the method. A decrease in the extraction efficiency in the volume of 2.5 mL can be assigned to the decreased polarity of the aqueous phase used in DLLME. This leads to increasing the analyte's solubility in the aqueous phase. Therefore, 2.0 mL was used in subsequent steps.
3.4. Optimization of DLLME conditions
3.4.1. Salt addition to the DLLME aqueous phase. Five milliliters of Na2SO4, NaCl, and KCl aqueous solutions at a concentration of 2.5% (w/v) were used as the DLLME aqueous phases to investigate the influence of salt addition in the DLLME step. The results shown in Fig. S5a† indicate that the addition of NaCl improves extraction efficiency for all of the analytes.Different concentrations of NaCl in the concentration range of 1–10%, w/v, were tested to investigate the influence of salt concentration. By increasing the salt concentration from 1 to 10%, w/v, the volume of the extractant was decreased from 45 to 25 μL and in all cases 10 ± 1 μL of the sedimented extractant was obtained. The results shown in Fig. S5b† indicate that the salt addition up to 5.0%, w/v, leads to an increase in ERs of the analytes and that increasing the NaCl concentration provides no further improvement in the extraction efficiency of the method. Increasing the method efficiency by the addition of salt can be related to decreasing the solubility of the analyte in the aqueous phase used in the DLLME step. Therefore, 5 mL NaCl solution at a concentration of 5.0%, w/v, was selected as the aqueous phase in the DLLME step.
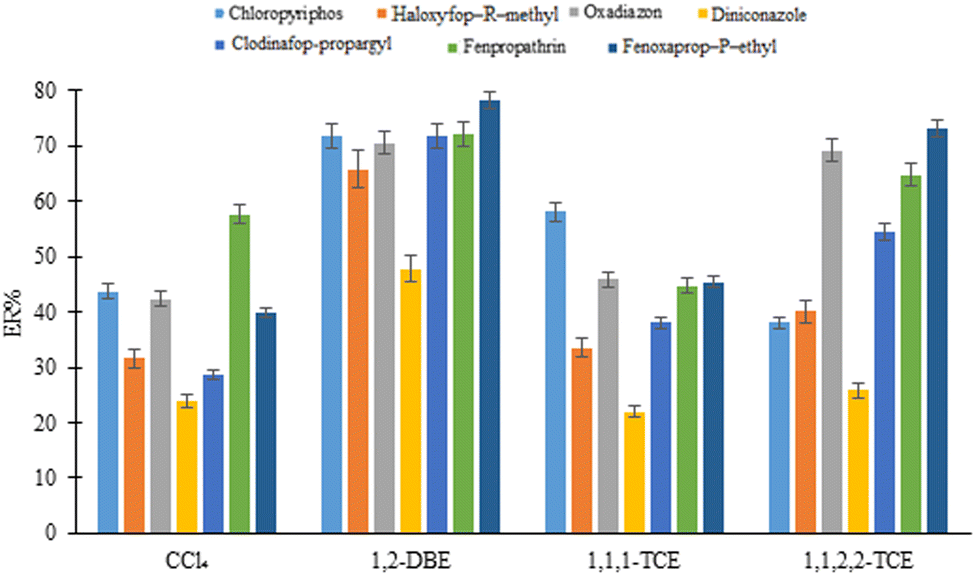
3.4.2. Selection of extraction solvent type and volume. Different density than water, low aqueous solubility, high extraction ability for the analytes, and good chromatographic behavior are the required conditions of a proper extraction solvent for the proposed D-μSPE-DLLME method. Considering the mentioned requirements, 1,1,2,2-TCE (50 μL), 1,2-DBE (35 μL), 1,1,1-TCE (40 μL), and carbon tetrachloride (30 μL) were examined and 10 ± 1 μL of the sedimented extractant was obtained in each case. The results (Fig. 5) illustrate that 1,2-DBE presents the highest efficiency among the tested solvents due to more solubility of the analytes in this solvent or its better dispersion by the disperser solvent and providing high contact area. Therefore, it was used in the further steps.
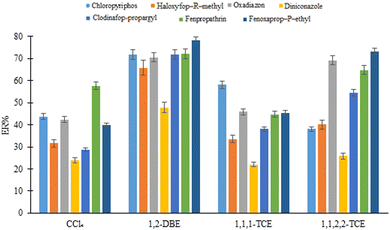 |
| | Fig. 5 Selection of extraction solvent in DLLME. Extraction conditions: are the same as those used in Fig. 4(b), except 5.0%, w/v, NaCl was dissolved in the DLLME aqueous phase. | |
To study the effect of extraction solvent volume on the method efficiency, the volume of 1,2-DBE was altered in the range of 35–55 μL. According to the results (data not shown here), the analytical signals of the analytes decrease with increasing volume of the extractant due to the dilution effect. On the other hand, using volumes lower than 35 μL led to obtaining an insufficient sedimented organic phase, which reduced the repeatability of results. Considering a proper balance between the analytes enrichment and repeatability, 35 μL 1,2-DBE was selected for the subsequent experiments.
3.5. Method validation
The calibration curves plotting was performed by preparing various solutions at concentrations of 3, 10, 25, 50, 100, 250, 500, and 1000 μg L−1 and subjecting them to the method. It is noted that the maximum concentration of the calibration curve for haloxyfop-p-ethyl and diniconazole was 500 μg L−1. Satisfactory coefficients of determination in the range of 0.994–0.997 were obtained for all analytes. The precision of the method was expressed using relative standard deviations (RSDs). Intra-and inter-day precisions were in the ranges of 5–6 and 6–8% (n = 5), respectively. The limit of detection (LOD) and quantification (LOQ) values, determined at the signal-to-noise ratios of 3 and 10, were in the ranges of 0.50–0.90 and 1.5–2.7 μg L−1, respectively. The ER and EF values were between 47 and 78% and 239 and 392, respectively. Table 1 summarizes the values.
Table 1 Analytical features of the developed D-μSPE-DLLME-GC-FID method for pesticides analysis
| Analytes |
LODa |
LOQb |
LRc |
r2d |
eRSD% |
EF ± SDf |
ER ± SDg |
| Intra-day |
Inter-day |
| Limit of detection (S/N = 3) (μg L−1). Limit of quantification (S/N = 10) (μg L−1). Linear range (μg L−1). Coefficient of determination. Relative standard deviation for intra- (n = 5) and inter-day (n = 5) precisions at a concentration of 25 μg L−1 of each analyte. Enrichment factor ± standard deviation (n = 3). Extraction recovery ± standard deviation (n = 3). |
| Chlorpyrifos |
0.90 |
2.7 |
2.7–1000 |
0.995 |
6 |
7 |
359 ± 24 |
71 ± 5 |
| Haloxyfop-p-ethyl |
0.50 |
1.5 |
1.5–500 |
0.994 |
5 |
7 |
329 ± 25 |
65 ± 5 |
| Oxadiazon |
0.50 |
1.5 |
1.5–1000 |
0.994 |
6 |
8 |
353 ± 16 |
70 ± 3 |
| Diniconazole |
0.90 |
2.7 |
2.7–500 |
0.994 |
5 |
6 |
239 ± 14 |
47 ± 3 |
| Clodinafob-propargyl |
0.90 |
2.7 |
2.7–1000 |
0.995 |
5 |
7 |
359 ± 35 |
71 ± 5 |
| Fenpropathrin |
0.62 |
1.8 |
1.8–1000 |
0.997 |
5 |
6 |
360 ± 15 |
72 ± 3 |
| Fenoxaprop-p-ethyl |
0.51 |
1.5 |
1.5–1000 |
0.996 |
6 |
8 |
392 ± 16 |
78 ± 3 |
3.6. Real samples analysis
It is a known fact that the efficiency of microextraction techniques can be influenced by the composition of the sample matrix. Therefore, river water, and fresh apple, watermelon, and onion juices were prepared as mentioned in Section 2.2, spiked with 50 and 100 μg L−1 for each analyte, and analyzed under the optimized experimental conditions. The found relative recoveries (RR) (expressed as the ratio of the analyte peak areas in real samples and deionized water spiked with the same concentration of analyte multiplied by 100) are listed in Table 2. The obtained RR values of the studied analytes are between 90 and 102%. The results indicate that the sample matrices have no significant effect on the extraction efficiency of the method. It should be mentioned that the obtained RR values in undiluted juice samples were relatively low. Therefore, they were diluted with deionized water at a ratio of 1:1 (v/v) before extraction. It is noteworthy that, none of the studied analytes was detected in the analyzed juice and water samples. Therefore, the studied pesticides were not present in the studied samples or their concentrations were below the method LODs.
Table 2 Results of relative recovery tests performed on the studied samples
| Analyte |
Mean relative recovery (%) ± standard deviation (n = 3) |
| Apple |
Watermelon |
Orange |
River water |
| All samples were spiked with each analyte at a concentration of 50 μg L−1 |
| Chlorpyrifos |
90 ± 5 |
92 ± 5 |
95 ± 5 |
99 ± 6 |
| Haloxyfob-p-ethyl |
90 ± 5 |
93 ± 5 |
95 ± 6 |
98 ± 5 |
| Oxadiazon |
91 ± 5 |
92 ± 6 |
94 ± 6 |
97 ± 5 |
| Diniconazole |
90 ± 5 |
93 ± 5 |
95 ± 5 |
98 ± 5 |
| Clodinafob-propargyl |
92 ± 6 |
93 ± 6 |
96 ± 5 |
97 ± 6 |
| Fenpropathrin |
91 ± 5 |
92 ± 5 |
96 ± 5 |
98 ± 5 |
| Fenoxaprop-p-ethyl |
90 ± 5 |
94 ± 5 |
95 ± 5 |
96 ± 5 |
|
| All samples were spiked with each analyte at a concentration of 100 μg L−1 |
| Chlorpyrifos |
95 ± 5 |
100 ± 5 |
96 ± 5 |
100 ± 6 |
| Haloxyfob-p-ethyl |
96 ± 5 |
102 ± 6 |
96 ± 5 |
99 ± 6 |
| Oxadiazon |
96 ± 6 |
98 ± 6 |
98 ± 6 |
95 ± 5 |
| Diniconazole |
97 ± 5 |
96 ± 5 |
98 ± 5 |
101 ± 6 |
| Clodinafob-propargyl |
95 ± 6 |
96 ± 6 |
98 ± 6 |
99 ± 6 |
| Fenpropathrin |
98 ± 5 |
98 ± 6 |
100 ± 5 |
100 ± 5 |
| Fenoxaprop-p-ethyl |
95 ± 5 |
95 ± 5 |
96 ± 6 |
95 ± 6 |
3.7. Comparison of the developed method and some other reported methods
The efficiency of the proposed method was compared with that of the other techniques applied for pesticide residue determination by comparing their figures of merit (Table 3). As can be seen, the developed method possesses lower LODs compared to other methods except the used for the analysis of oxadiazon in which an inherently sensitive instrument (ultra-performance liquid chromatography-tandem mass spectrometry) was used as the determination system.36 Wide linear ranges regarding the applied analysis system were obtained in comparison to other methods. Also, the EF and RSD values of the recommended method are higher or comparable with those of the mentioned methods. Generally, the developed D-μSPE-DLLME-GC-FID method can be considered a fast, low-cost, efficient, and reliable sample pretreatment method in the sample clean-up, extraction, and enrichment of the pesticide residues from various aqueous samples.
Table 3 Comparison of the developed method with other reported methods for pesticides determination
| Analyte |
Method |
Sample |
LRa |
LODb |
EFc |
dRSD% |
Ref. |
| Linear range (μg L−1). Limit of detection (μg L−1). Enrichment factor. Relative standard deviation. Magnetic dispersive solid phase extraction-high performance liquid chromatography-ultraviolet detection. Magnetic dispersive solid phase extraction-ultra high performance liquid chromatography-tandem mass spectrometry. Microwave-assisted extraction-dispersive solid phase extraction-gas chromatography-mass spectrometry. Dispersive micro solid phase extraction-dispersive liquid–liquid microextraction-gas chromatography-flame ionization detection. |
| Chlorpyrifos |
MDSPE-HPLC-UV e |
Water and rice |
10–100 |
1.0 |
401 |
4.8 |
36 |
| Diniconazole |
MDSPE-UHPLC-MS/MSf |
Water |
1.5–500 |
0.5 |
— |
3.7 |
37 |
| Oxadiazon |
MDSPE-HPLC-UVe |
Water |
0.5–10 |
0.07 |
180 |
7.2 |
38 |
| Fenpropathrin |
MWAE-DSPE-GC-MSg |
Radix astragali |
0.2 |
0.2–10 |
— |
— |
39 |
| Chlorpyrifos |
D-μSPE-DLLME-GC-FIDh |
Fruit and vegetable juices and river water |
2.7–1000 |
0.90 |
359 |
6 |
This study |
| Haloxyfob-p-ethyl |
1.5–500 |
0.50 |
329 |
5 |
| Oxadiazon |
1.5–1000 |
0.50 |
353 |
6 |
| Diniconazole |
2.7–500 |
0.90 |
239 |
5 |
| Clodinafob-propargyl |
2.7–1000 |
0.90 |
359 |
5 |
| Fenpropathrin |
1.8–1000 |
0.62 |
360 |
5 |
| Fenoxaprop-p-ethyl |
1.5–1000 |
0.51 |
392 |
6 |
4. Concluding remarks
In the present study, MOF-5 was synthesized by a simple and fast hydrothermal procedure and successfully applied for the sample cleanup and extraction of some pesticides from water and fruit juice samples. Appropriate LODs, EFs, and precision were achieved and the obtained experimental results proved that the D-μSPE method using the MOF-5 combined with DLLME has a good potential for the extraction of the selected pesticides. The use of only 5 mg of the sorbent, consumption of relatively low volumes of organic solvents, and simplicity, qualifies the method as an economically and environmentally benign technique for the pesticide analysis.
Abbreviations
| D-μSPE | Dispersive liquid–liquid microextraction |
| ER | Extraction recovery |
| EF | Enrichment factor |
| GC | Gas chromatography |
| LOD | Limit of detection |
| LOQ | Limit of quantification |
| LR | Linear range |
| SPE | Solid phase extraction |
| FTIR | Fourier transform infrared |
| XRD | X-ray diffraction |
| EDX | Energy dispersive X-ray |
| SEM | Scanning electron microscopy |
| DLLME | Dispersive liquid–liquid phase extraction |
| MOF | Metal–organic framework |
| FID | Flame ionization detector |
| 1,1,2,2-TCE | 1,1,2,2-Tetrachloroethane |
| 1,1,2-TCE | 1,1,2-Trichloroethane |
| 1,2-DBE | 1,2-Dibromoethane |
| RSD | Relative standard deviation |
| RR | Relative recovery |
Data availability
The data are confidential.
Conflicts of interest
The authors have declared no conflict of interest.
Acknowledgements
The authors gratefully acknowledge financial support from the University of Tabriz.
References
- J. R. Richardson, V. Fitsanakis, R. H. Westerink and A. G. Kanthasamy, Neurotoxicity of pesticides, Acta Neuropathol., 2019, 138, 343–362 CrossRef CAS PubMed.
- M. A. Farajzadeh, B. Feriduni and M. R. Afshar Mogaddam, Extraction and enrichment of triazole and triazine pesticides from honey using air-assisted liquid-liquid microextraction, J. Food Sci., 2014, 79, H2140–H2148 CrossRef PubMed.
- B. Gilbert-López, J. F. García-Reyes and A. Molina-Díaz, Sample treatment and determination of pesticide residues in fatty vegetable matrices: A review, Talanta, 2009, 79, 109–128 CrossRef PubMed.
- C. Torres, Y. Picó and J. Manes, Determination of pesticide residues in fruit and vegetables, J. Chromatogr. A, 1996, 754, 301–331 CrossRef CAS PubMed.
- M. A. Farajzadeh, B. Feriduni and M. R. Afshar Mogaddam, Determination of triazole pesticide residues in edible oils using air-assisted liquid-liquid microextraction followed by gas chromatography with flame ionization detection, J. Sep. Sci., 2015, 38, 1002–1009 CrossRef CAS PubMed.
- M. Nemati, M. A. Farajzadeh, M. R. Afshar Mogaddam, A. Mohebbi, A. R. Azimi, N. Fattahi and M. Tuzen, Development of a gas–controlled deep eutectic solvent–based evaporation–assisted dispersive liquid–liquid microextraction approach for the extraction of pyrethroid pesticides from fruit juices, Microchem. J., 2022, 175, 107196 CrossRef CAS.
- X. Liu, Z. Liu, L. Bian, Y. Ping, S. Li, J. Zhang, J. Wang, A. Van Schepdael and X. Wang, Determination of pesticide residues in Chilli and Sichuan pepper by high performance liquid chromatography quadrupole time-of-flight mass spectrometry, Food Chem., 2022, 387, 132915 CrossRef CAS.
- G. R. Algharibeh and M. S. AlFararjeh, Pesticide residues in fruits and vegetables in Jordan using liquid chromatography/tandem mass spectrometry, Food Addit. Contam.: Part B, 2019, 12, 65–73 CrossRef CAS PubMed.
- T. Albanis, D. Hela, T. Sakellarides and I. Konstantinou, Monitoring of pesticide residues and their metabolites in surface and underground waters of Imathia (N. Greece) by means of solid-phase extraction disks and gas chromatography, J. Chromatogr. A, 1998, 823, 59–71 CrossRef CAS.
- A. Di Muccio, P. Fidente, D. A. Barbini, R. Dommarco, S. Seccia and P. Morrica, Application of solid-phase extraction and liquid chromatography–mass spectrometry to the determination of neonicotinoid pesticide residues in fruit and vegetables, J. Chromatogr. A, 2006, 1108, 1–6 CrossRef CAS PubMed.
- M. Faraji, Y. Yamini and M. Gholami, Recent advances and trends in applications of solid-phase extraction techniques in food and environmental analysis, Chromatographia, 2019, 82, 1207–1249 CrossRef CAS.
- K. Moyakao, Y. Santaladchaiyakit, S. Srijaranai and J. Vichapong, Preconcentration of trace neonicotinoid insecticide residues using vortex-assisted dispersive micro solid-phase extraction with montmorillonite as an efficient sorbent, Molecules, 2018, 23, 883 CrossRef.
- V. C. Fernandes, M. Freitas, J. P. Pacheco, J. M. Oliveira, V. F. Domingues and C. Delerue-Matos, Magnetic dispersive micro solid-phase extraction and gas chromatography determination of organophosphorus pesticides in strawberries, J. Chromatogr. A, 2018, 1566, 1–12 CrossRef CAS PubMed.
- A. Chisvert, S. Cárdenas and R. Lucena, Dispersive micro-solid phase extraction, Trends Anal. Chem., 2019, 112, 226–233 CrossRef CAS.
- B. Buszewski and M. Szultka, Past, present, and future of solid phase extraction: a review, Crit. Rev. Anal. Chem., 2012, 42, 198–213 CrossRef CAS.
- S. Bolat, S. Demir, H. Erer, F. Pelit, R. Dzingelevičienė, T. Ligor, B. Buszewski and L. Pelit, MOF-801 based solid phase microextraction fiber for the monitoring of indoor BTEX pollution, J. Hazard. Mater., 2024, 466, 133607 CrossRef CAS PubMed.
- G. Wu, J. Ma, S. Wang, H. Chai, L. Guo, J. Li, A. Ostovan, Y. Guan and L. Chen, Cationic metal-organic framework based mixed-matrix membrane for extraction of phenoxy carboxylic acid (PCA) herbicides from water samples followed by UHPLC-MS/MS determination, J. Hazard. Mater., 2020, 394, 122556 CrossRef CAS PubMed.
- J. Ma, G. Wu, S. Li, W. Tan, X. Wang, J. Li and L. Chen, Magnetic solid-phase extraction of heterocyclic pesticides in environmental water samples using metal-organic frameworks coupled to high performance liquid chromatography determination, J. Chromatogr. A, 2018, 1553, 57–66 CrossRef CAS PubMed.
- P. Tomai, A. Gentili, S. Fanali and Y. Picó, Multi-residue determination of organic micro-pollutants in river sediment by stir-disc solid phase extraction based on oxidized buckypaper, J. Chromatogr. A, 2020, 1621, 461080 CrossRef CAS PubMed.
- Y. Zhao, D. Du, Q. Li, W. Chen, Q. Li, Q. Zhang and N. Liang, Dummy-surface molecularly imprinted polymers based on magnetic graphene oxide for selective extraction and quantification of pyrethroids pesticides in fruit juices, Microchem. J., 2020, 159, 105411 CrossRef CAS.
- L. M. Ravelo-Pérez, J. Hernández-Borges and M. Á. Rodríguez-Delgado, Multi-walled carbon nanotubes as efficient solid-phase extraction materials of organophosphorus pesticides from apple, grape, orange and pineapple fruit juices, J. Chromatogr. A, 2008, 1211, 33–42 CrossRef PubMed.
- S. Mahpishanian and H. Sereshti, Three-dimensional graphene aerogel-supported iron oxide nanoparticles as an efficient adsorbent for magnetic solid phase extraction of organophosphorus pesticide residues in fruit juices followed by gas chromatographic determination, J. Chromatogr. A, 2016, 1443, 43–53 CrossRef CAS PubMed.
- L. Dinali, H. L. de Oliveira, L. S. Teixeira, B. W de Souza and K. B. Borges, Mesoporous molecularly imprinted polymer core@ shell hybrid silica nanoparticles as adsorbent in microextraction by packed sorbent for multi residue determination of pesticides in apple juice, Food Chem., 2021, 45, 128745 CrossRef.
- A. Amiri, R. Tayebee, A. Abdar and F. N. Sani, Synthesis of a zinc-based metal-organic framework with histamine as an organic linker for the dispersive solid-phase extraction of organophosphorus pesticides in water and fruit juice samples, J. Chromatogr. A, 2019, 1597, 39–45 CrossRef CAS.
- S. Ma and H. C. Zhou, Gas storage in porous metal–organic frameworks for clean energy applications, Chem. Comm., 2010, 246, 44–53 RSC.
- A. Dhakshinamoorthy, Z. Li and Z. H. Garcia, Catalysis and photocatalysis by metal organic frameworks, Chem. Soc. Rev., 2018, 47, 8134–8172 RSC.
- J. Della Rocca, D. Liu and W. Lin, Nanoscale metal–organic frameworks for biomedical imaging and drug delivery, Acc. Chem. Res., 2011, 44, 957–968 CrossRef CAS PubMed.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Metal–organic framework materials as chemical sensors, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS.
- M. D. Allendorf, C. A. Bauer, R. Bhakta and R. Houk, Luminescent metal–organic frameworks, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- J. Zhang and Z. Chen, Metal-organic frameworks as stationary phase for application in chromatographic separation, J. Chromatogr. A, 2017, 1530, 1–18 CrossRef CAS.
- X. Dai, X. Jia, P. Zhao, T. Wang, J. Wang, P. Huang, L. He and X. Hou, A combined experimental/computational study on metal-organic framework MIL-101 (Cr) as a SPE sorbent for the determination of sulphonamides in environmental water samples coupling with UPLC-MS/MS, Talanta, 2016, 154, 581–588 CrossRef CAS.
- A. Gutiérrez-Serpa, I. Pacheco-Fernández, J. Pasán and V. Pino, Metal–organic frameworks as key materials for solid-phase microextraction devices—a review, Separations, 2019, 6, 47 CrossRef.
- J. Hafizovic, M. Bjørgen, U. Olsbye, P. D. Dietzel, S. Bordiga, C. Prestipino, C. Lamberti and K. P. Lillerud, The inconsistency in adsorption properties and powder XRD data of MOF-5 is rationalized by framework interpenetration and the presence of organic
and inorganic species in the nanocavities, J. Am. Chem. Soc., 2007, 129, 3612–3620 CrossRef CAS PubMed.
- L. Huang, H. Wang, J. Chen, Z. Wang, J. Sun, D. Zhao and Y. Yan, Synthesis, morphology control, and properties of porous metal–organic coordination polymers, Microporous Mesoporous Mater., 2003, 58, 105–114 CrossRef CAS.
- A. Bokulic Petric, S. Stipicevic and A. Mesic, Stability of malathion, diazinon and chlorpyrifos in different water types – a review, J. Cent. Eur. Agric., 2023, 24, 3 Search PubMed.
- M. Shakourian, Y. Yamini and M. Safari, Facile magnetization of metal–organic framework TMU-6 for magnetic solid-phase extraction of organophosphorus pesticides in water and rice samples, Talanta, 2020, 218, 121139 CrossRef CAS.
- J. Dong, Z. Feng, S. Kang, M. An and G. Wu, Magnetic solid-phase extraction based on magnetic amino modified multiwalled carbon nanotubes for the fast determination of seven pesticide residues in water samples, Anal. Methods, 2020, 12, 2747–2756 RSC.
- B. Maddah, A. Sabouri and M. Hasanzadeh, Magnetic solid-phase extraction of oxadiazon and profenofos from environmental water using magnetite Fe3O4@ SiO2–C18 nanoparticles, J. Polym. Environ., 2017, 25, 770–780 CrossRef CAS.
- X. Mao, Y. Wan, A. Yan, M. Shen and Y. Wei, Simultaneous determination of organophosphorus, organochlorine, pyrethriod and carbamate pesticides in Radix astragali by microwave-assisted extraction/dispersive-solid phase extraction coupled with GC–MS, Talanta, 2012, 97, 131–141 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Sakineh Rahimzadeha,
Mohammad Reza Afshar Mogaddam
*ab,
Sakineh Rahimzadeha,
Mohammad Reza Afshar Mogaddam