
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBeyond transition block metals: exploring the reactivity of phosphine PTA and its oxide [PTA(O)] towards gallium(III)†
Antonella Guerriero *a,
Andrea Iencoa,
Thomas Hicksb and
Agostino Cilibrizzi*c
*a,
Andrea Iencoa,
Thomas Hicksb and
Agostino Cilibrizzi*c
aConsiglio Nazionale delle Ricerche, Istituto di Chimica dei Composti OrganoMetallici (ICCOM), Via Madonna del Piano 10, 50019 Sesto Fiorentino (Florence), Italy. E-mail: antonella.guerriero@iccom.cnr.it
bDepartment of Chemistry, King's College London, 7 Trinity Street, London SE1 1DB, UK
cInstitute of Pharmaceutical Science, King's College London, Franklin Wilkins Building, London SE1 9NH, UK. E-mail: agostino.cilibrizzi@kcl.ac.uk
First published on 4th July 2024
Abstract
The water-soluble cage-like phosphine PTA (1,3,5-triaza-7-phosphaadamantane) and its phosphine oxide derivative [PTA(O)] (1,3,5-triaza-7-phosphaadamantane-7-oxide) were used to explore their reactivity towards two gallium(III)-halide precursors, namely GaCl3 and GaI3, for the first time. By using various reaction conditions, a series of N-mono-protonated phosphine salts with [GaCl4]− or [I]− as counterions were obtained in all cases, while the formation of coordinated Ga-PTA and Ga-[PTA(O)] complexes was not observed. All compounds were characterized in solution using multinuclear NMR spectroscopy (1H, 13C{1H}, 31P{1H} and 71Ga) and in the solid state using FT-IR spectroscopy and X-ray crystal diffraction. The new Ga-phosphine salts resulted stable and highly soluble in aqueous solution at room temperature. Density functional theory (DFT) calculations were also performed to further rationalize the coordination features of PTA with Ga3+ metal ion, highlighting that the phosphorus–gallium bond is about twice weaker than the phosphorus–metal bond commonly established by PTA with transition metals such as gold. Furthermore, the mono-protonation of PTA (or [PTA(O)]) makes the formation of ionic gallium–PTA coordination complexes thermodynamically unstable, as confirmed experimentally by the formation of Ga–phosphine salts reported herein.
Introduction
The neutral adamantane-like phosphine 1,3,5-triaza-7-phosphaadamantane, also known as PTA (Fig. 1), is an established, versatile and attractive ancillary ligand used widely to access water-soluble metal coordination complexes in various fields such as chemical biology, catalysis and material chemistry.1,2 Besides the undemanding steric properties due to its small cone angle (103°), PTA shows high thermal and chemical stability compared to other alkyl and aryl-phosphines for example towards oxidation.3 The most relevant feature of this phosphine is its great solubility in water (S = ca. 235 g L−1).4 This has prompted extensive synthetic chemistry investigations, giving rise to the development of a wide number of organometallic transition metal compounds with photoluminescence properties and mostly active in catalysis under homogeneous aqueous or biphasic conditions. Typical catalytic reactions carried out in the presence of PTA (or PTA-based analogues) include hydrogenations and transfer hydrogenations,5–8 hydroformylations and hydrosilylations,9–12 allylic isomerization13,14 and organonitrile hydration.15,16 In parallel, the high stability and water solubility of PTA make it also an attractive scaffold to be incorporated into compounds and formulations for bio-medical use. In this context, the number of reports in the literature on the design and synthesis of biologically active PTA-based complexes has sharply grown in recent years.17–19 Among these, ruthenium complexes undoubtedly represent the largest group of PTA-based compounds with both catalytic and biological activity20,21 and, with regard to the latter, those belonging to the series of RAPTA-type metallodrugs with the general formula [RuCl2(η6-arene)(PTA)], showed strong antiangiogenic and antimetastatic activities.22–24 In a similar fashion, silver compounds containing PTA or some of its derivatives, such as the phosphine oxide [PTA(O)] analogue (Fig. 1), have demonstrated effective antiproliferative,25,26 antimicrobial and antifungal activities.27–29 Very recently, palladium allyl complexes bearing a combination of PTA and NHC ligands (NHC = N-heterocyclic carbenes) were tested in different tumor cell lines demonstrating high cytotoxicity, in many cases significantly higher than for cisplatin, while lower toxicity effects were observed in normal cells.30,31 In a similar fashion, PTA–rhenium–carbonyl compounds were investigated for their phototoxicity effects towards tumor cell lines,32,33 and water-soluble Re(V) complexes (i.e. [ReO2Cl(PTA)3] and [ReNCl2(PTA)3])34 were developed to access complexes for radiopharmaceutical applications.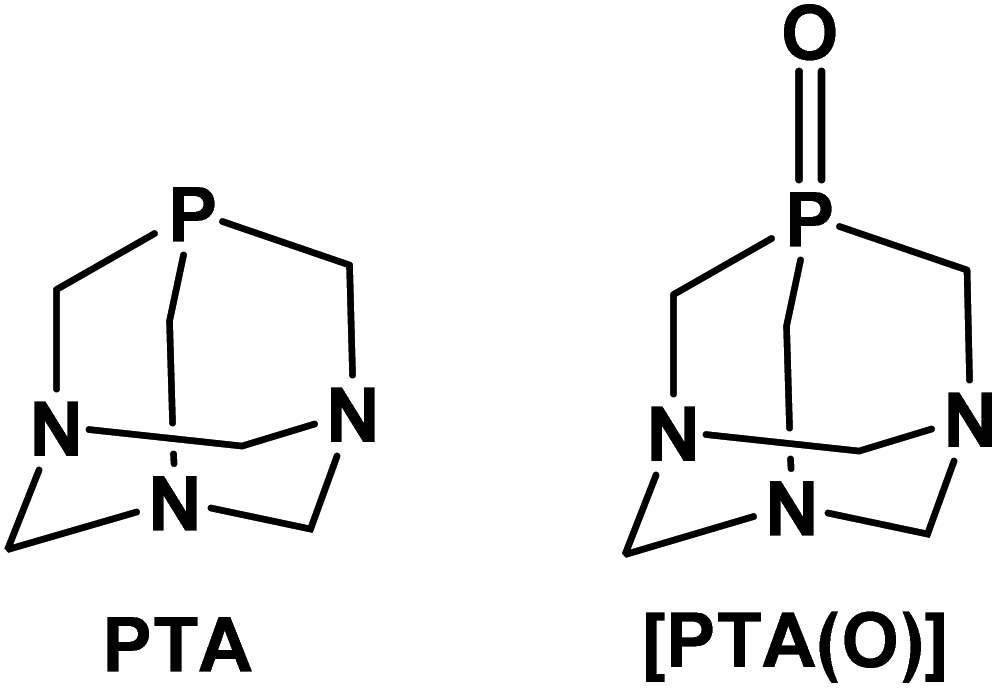 | ||
| Fig. 1 1,3,5-Triaza-7-phosphadamantane (PTA) and its oxide derivative [PTA(O)]. | ||
Although there is extensive literature reporting on the synthesis and application of PTA transition metal complexes, in particular with elements belonging to groups from 7 to 12, the coordination chemistry of this hydrophilic phosphine with other metals, such as those of group 13, is still largely unexplored. Among these elements, gallium represents an interesting metal for the development of modern semiconductor materials for the electronic industry35,36 as well as for the use in medicine to access new diagnostic and therapeutic agents.37–41 The idea behind the use of gallium in therapy originates from the high similarity between Ga3+ and Fe3+ ions, in terms of ionic radius, electronic configuration, coordination number and ligand-donor preferences.42 Moreover, Ga3+ and Fe3+ complexes with organic ligands are often isostructural.43,44 Nevertheless, unlike Fe3+, Ga3+ cannot be easily reduced under physiological conditions and, therefore, it cannot take part in redox reactions.45 By replacing iron in proteins, the most important being transferrin, gallium can be adsorbed in cells where iron is mostly required, such as proliferating cancer cells.46,47 Due to the iron-mimetic features, gallium-based compounds have also demonstrated efficacy as antimicrobials.48–50 In this regard, several Ga-containing agents, including gallium nitrate, gallium chloride, gallium maltolate and gallium citrate, are currently under investigation for the treatment of malignancies and microbial infections, both in preclinical studies and clinical trials.51–53 In addition to the therapeutic uses, radioactive gallium derivatives have been successfully applied in modern molecular imaging strategies and protocols adopted in the clinics.41 Of the three isotopes with nuclear properties (i.e. 66Ga, 67Ga, 68Ga), gallium-68 represents the radionuclide most widely investigated for PET (Positron Emission Tomography) imaging, most likely due to a combination of factors, including the suitable half-life (t1/2 = 68 min) and decay properties, as well as the easy access to 68Ge/68Ga generators in hospitals and clinically-oriented research centres.54,55 To enable suitable medical applications, the design of ligands able to stabilize Ga3+ formulations clearly becomes a crucial point to ensure sufficient stability of Ga-containing compounds (i.e. preventing the possible hydrolysis in vivo to [Ga(OH)4]−, a nephrotoxic species), as well as avoid trans-chelation by endogenous proteins, which can lead to a lower uptake in diseased tissues or also a decreased quality of PET images.40,56,57 A rational choice of the coordinating ligands may also offer the opportunity to finely tune bioavailability and biodistribution features, which are known to be strongly affected by the physico-chemical properties of the compounds, such as overall charge, size and hydrophilicity/lipophilicity balance. With regard to the potential applications of new gallium derivatives in the biological field, the final formulation is expected to possess appropriate hydrophilic character, along with high stability. In this context, PTA-based structures represent valid candidates to explore further, in order to access Ga-based biologically active molecules with enhanced drug-like properties.
Based on the overall lack of studies on the use of PTA as an effective ligand for metals other than those of the transition block, as well as the interesting role gained by gallium compounds in drug discovery, in the present work, we have investigated the chemistry and the reactivity of PTA with two selected gallium precursors. To date, numerous gallium(III) compounds with neutral alkyl and aryl phosphines are known, the majority being of the type [GaX3(PR3)] (where X = halide; PR3 = phosphine), in which the metal strongly favours the distorted tetrahedral quadrilateral coordination.58,59 Since Ga3+ is an oxophilic metal ion and complexes of gallium with phosphine oxides are also known,60,61 [PTA(O)] derivative (Fig. 1) was also used to test the reactivity in combination with GaX3. The characterization in solution and in the solid state of all the main reaction products is reported, along with supporting DFT (Density Functional Theory) calculations providing new and relevant insights on the mode of interaction of PTA with Ga3+ metal ion.
Results and discussion
Reactivity of PTA and [PTA(O)] with GaCl3 and GaI3
With the exception of thallium, the most stable oxidation state of Group 13 metals is 3+, giving to these ions a strong acidic character. Gallium(III) is a strong Lewis acid and typically coordinates electron-rich neutral or anionic donors (mainly O, N or S) forming stable complexes with coordination numbers from 3 to 6, depending on the nature of the surrounding ligands.62,63 Although phosphines are considered to be soft bases binding preferably soft acids, the number of complexes obtained from the reaction of alkyl and aryl monodentate phosphines with trihalides of gallium is significant.59 Di- and poly-phosphine adducts with gallium are also known,64,65 as well as complexes with amino-diphosphine pincer ligands (PNP), which can coordinate the metal in tridentate NP2-fashion or bidentate P2-fashion, depending on whether the pincer is in the anionic or neutral form.66 Noteworthy, a large number of complexes of formula [GaX3(PR3)] or [GaX3(POR3)] has been obtained through the reaction of GaX3 (X = Cl, Br or I) with phosphines or phosphine oxides in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio.61,67,68 For most of them, the molecular structure has been determined via X-ray crystallography, particularly in the case of compounds with monodentate tertiary phosphines. Indeed, besides the four-coordinate monomer [GaX3(PR3)], when reacted with PR3, the gallium(III) halides may also form four-coordinated ionic dimers of the type [GaX2(PR3)2][GaX4] and less frequently, five-coordinate dimers of formula [Ga2X6(PR3)2].69,70
:1 ratio.61,67,68 For most of them, the molecular structure has been determined via X-ray crystallography, particularly in the case of compounds with monodentate tertiary phosphines. Indeed, besides the four-coordinate monomer [GaX3(PR3)], when reacted with PR3, the gallium(III) halides may also form four-coordinated ionic dimers of the type [GaX2(PR3)2][GaX4] and less frequently, five-coordinate dimers of formula [Ga2X6(PR3)2].69,70
In the initial experiments with PTA phosphine, we used GaCl3 and GaI3 as metal precursors. When a solution of PTA in dichloromethane (DCM) was added under anhydrous conditions to the solution of GaCl3 in the same solvent, the fast formation of a white precipitate was observed (Scheme 1). As reported for other Ga–phosphine compounds, this solid was difficult to dissolve in most organic solvents but showed very high solubility in water. The 31P{1H} NMR spectrum of the DCM mother solution displayed a singlet at δ –93.39 ppm (Fig. S9†) while the white solid dissolved in D2O showed a singlet at δ –91.41 ppm (Fig. S7†), i.e. a value only slightly different from the 31P signal of free PTA in the same solvent (δP = –98.61 ppm). If this observation is atypical for PTA-transition metal compounds, where the coordination of the P atom to the metal usually determines a considerable shift of the 31P NMR peak (mostly to less negative values), it is not uncommon for certain [GaX3(PR3)] derivatives. Indeed, the 31P{1H} NMR signals of some gallium–phosphine–halides complexes are often detected at chemical shift unchanged or very similar to that of the free phosphine, especially when X = Cl. For example, compounds [GaCl3(PPh3)] and [GaBr3(PPh3)] exhibited low temperature (273 K) 31P{1H}NMR resonances as multiplets at δ –5.4 and at δ –10.7 ppm, respectively, i.e. values close to δP shift of uncoordinated PPh3 (δP = –5.0 ppm).61
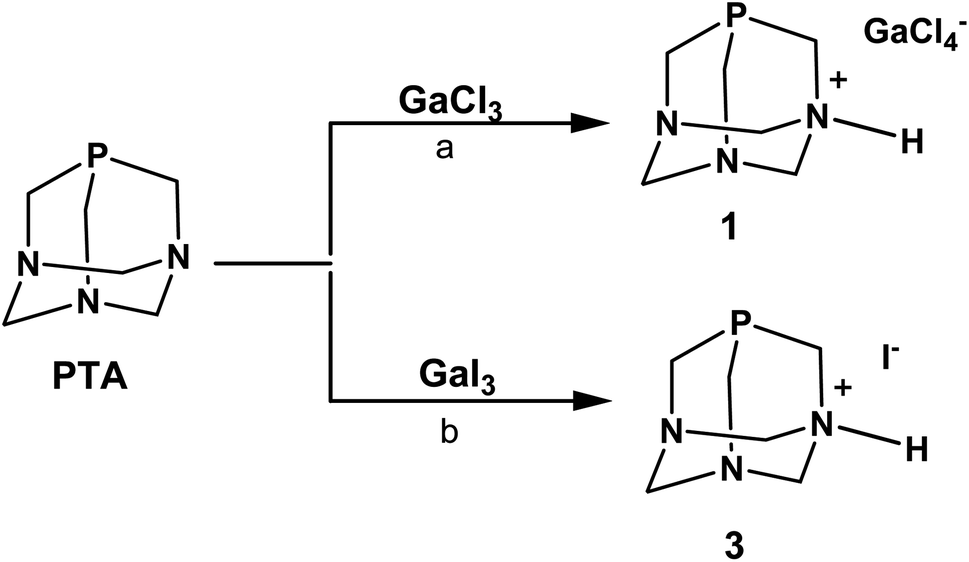 | ||
| Scheme 1 Products of reactions of PTA with GaCl3 and GaI3. Reaction conditions: (a) [Ga]:[P] = 1:1.2; DCM@rt; or DCM@0 °C; or DCM/Et3N@rt. (b) [Ga]:[P] = 1:1.2; EtOAc@rt; or Et2O@rt; or solid state reaction. | ||
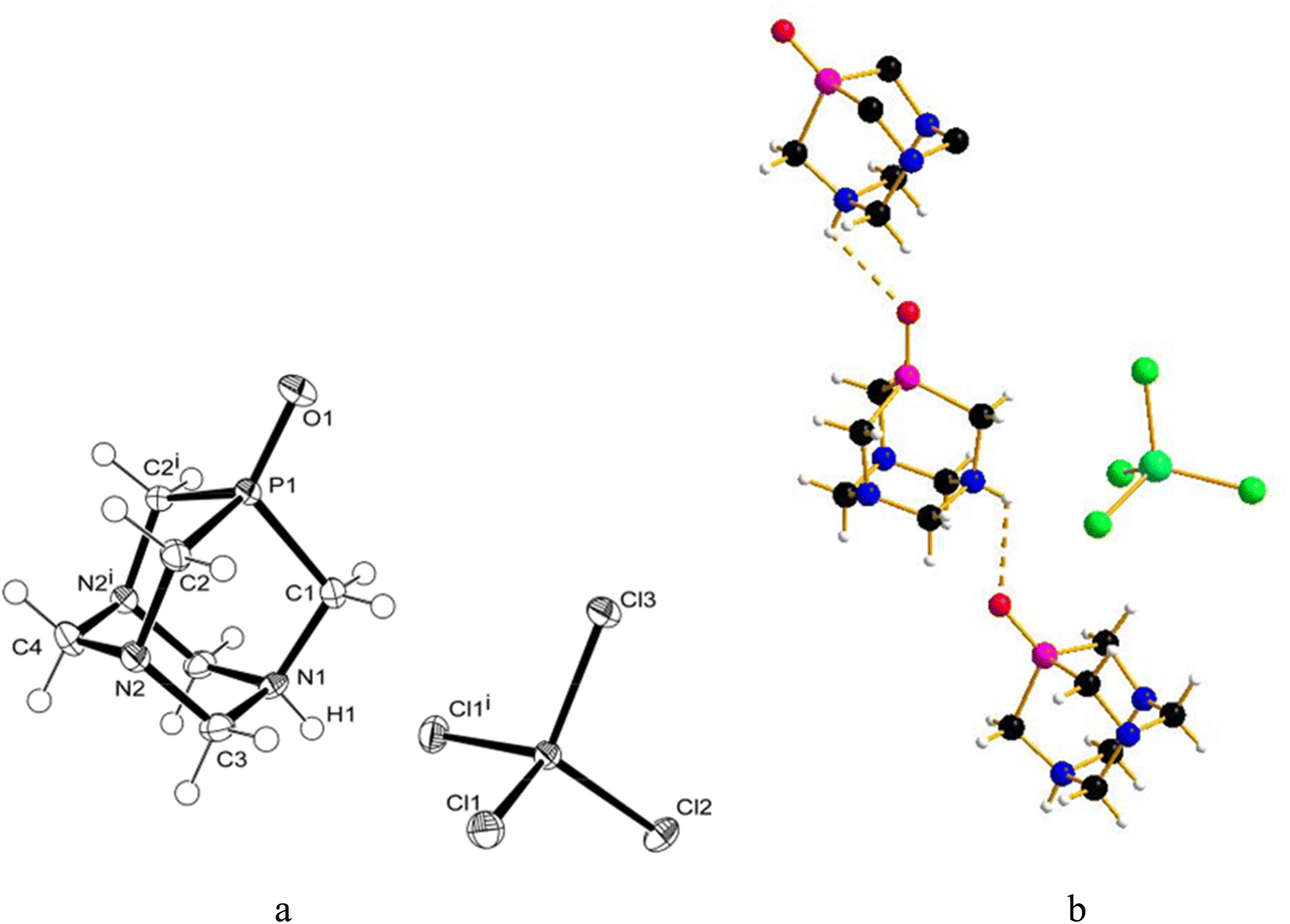
Similarly to 31P{1H} NMR, minimal differences in terms of multiplicity and chemical shift values in comparison to free PTA were also detected in 1H and 13C{1H} NMR spectra of the isolated product 1 (Fig. S5 and S6†). Therefore, all the NMR analyses were not conclusive in assigning the exact molecular structure, which was then determined by X-ray diffraction. Crystals suitable for this analysis were obtained by the slow evaporation of the mother DCM solution at room temperature. X-ray diffraction revealed that the structure of 1 consists of an N-monoprotonated [PTA-H]+ cation with [GaCl4]− as counteranion, as shown in Fig. 2. In [PTA-H]·[GaCl4] (1) P1, C1, N1, H1 and C4 atoms of the cation and Ga1, Cl2 and Cl3 of the anion are lying on the plane (see Fig. 2b). To date, only two non-coordinated protonated PTA have been reported in the literature with a metal complex as counteranion71,72 and 1 is the first structure that contains a main group atom as the counteranion. The P–C distances (average 1.856(3) Å) are higher than the range of coordinated and non-coordinated protonated PTA (1.838–1.851 Å obtained from a search in Cambridge Structural Database (CSD)73). In contrast, the N–C bonds of the protonated nitrogen are slightly longer than in the case of non-protonated N atoms (1.511(3) Å for N1–C1 and 1.531(2) Å N1–C3 and N1–C3i vs. 1.472(2) Å N2–C2, 1.436(2) Å N2–C3 and 1.467(2) Å N2–C4). Moreover, N1–C3 and N2–C3 distances are shorter than N1–C1 and N2–C2 distances, respectively. This general trend has also been confirmed by CSD search, highlighting that N–C distances of protonated PTA nitrogen atoms are on average 0.3 Å longer than distances of non-protonated N atoms. In the packing, [PTA-H]+ moieties are along the a axis (Fig. S32†). From the analysis of the fingerprint plot generated by the Hirshfeld surface around the [PTA-H]+ cation, 53.9% of contacts are with chlorine atoms followed by the hydrogen atoms of the neighbouring cation contacts (Fig. S34 and S35†).
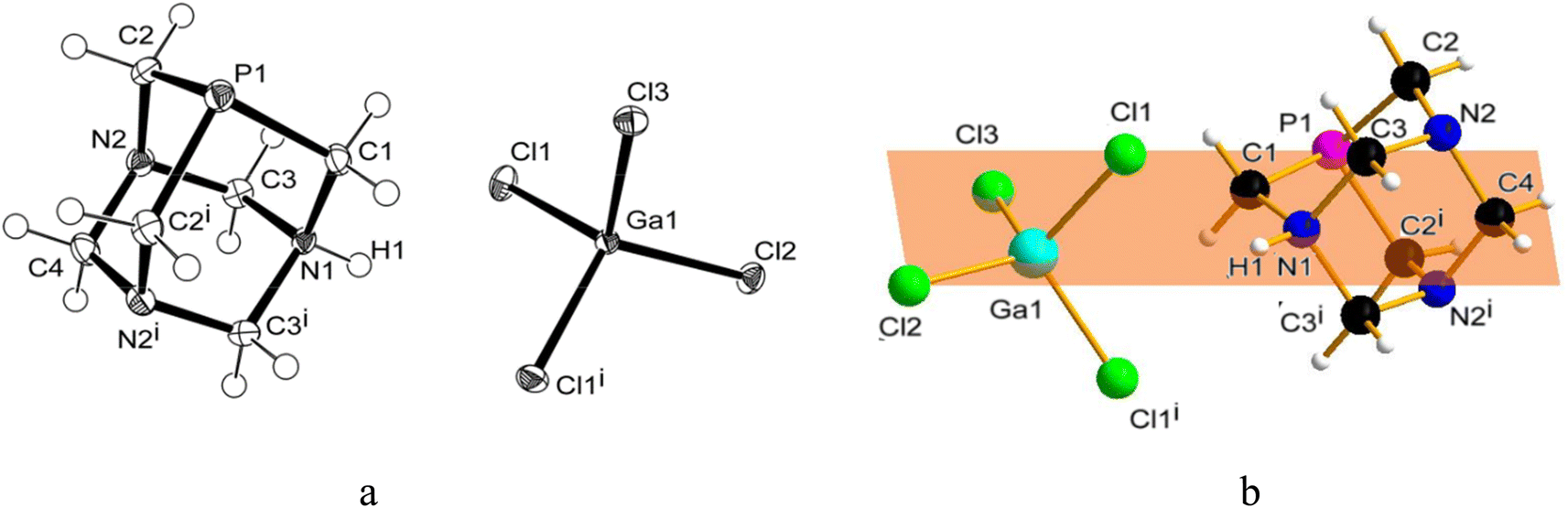 | ||
| Fig. 2 (a) Molecular structure (ORTEP diagram) of [PTA-H]·[GaCl4] (1) showing the atom labelling scheme. Thermal ellipsoids are shown at a 50% probability level. (b) Molecular structure highlighting the mirror plane. Color code: C, black; H, white; N, blue; P, purple; Cl, green; Ga, light blue selected bond lengths (Å) and angles (°): P1–C1 1.854(3); P1–C2 1.8566(19); N1–C1 1.511(3); N1–C3 1.531(2); N2–C2 1.472(2); N2–C3 1.436(2); N2–C4 1.467(2); Ga1–Cl1 2.1862(5); Ga1–Cl2 2.1582(6); Ga1–Cl3 2.1737(6); C1–P1–C2 96.58(8); C2–P1–C2#1 96.68(12); C1–N1–C3 111.81(12); C3–N1–C3#1 108.64(18); C3–N2–C4 109.88(15); C2–N2–C3 112.13(14); C2–N2–C4 111.57(16); N2–C2–P1 113.85(12). Symmetry transformations used to generate equivalent atoms: x, −y + 1/2, z. | ||
To further confirm the correct formulation of derivative 1 as [PTA-H]·[GaCl4], we performed inductively coupled plasma mass spectrometry (ICP-MS) experiments focusing at determining the concentrations of gallium and phosphorus in 1. The ICP-MS results indicate that the content of gallium found in 1 has a robust agreement with calculated values (i.e. 0.12 ± 0.004 vs. 0.12 mg mL−1, Table 1) and a complete match between found vs. calculated values was also recorded for [P] (Tables 1 and S2†).
| Compound | [Ga] found (mg mL−1) | [Ga] calculated (mg mL−1) | [P] found (mg mL−1) | [P] calculated (mg mL−1) |
|---|---|---|---|---|
| [PTAH][GaCl4] (1) | 0.12 ± 0.004 | 0.12 | 0.05 ± 0.001 | 0.05 |
| [PTA(O)H][GaCl4] (2) | 0.09 ± 0.003 | 0.09 | 0.04 ± 0.001 | 0.04 |
The N-protonation of PTA in 1 was also validated by the infrared analysis (KBr pellets, Fig. S1†) with bands at 3165 and 2969 cm−1 (i.e. N–H stretching) and at 1622 cm−1 (i.e. N–H bending). The presence of [GaCl4]− was further confirmed by 71Ga NMR spectra run on both DCM mother solution of 1 and on the isolated compound dissolved in D2O. In the first case, a large signal at δ 263.43 ppm (with C6D6 insert) was observed (Fig. S10†), while a broad signal at δ 0.00 ppm was detected in the 71Ga NMR spectrum of 1 in D2O (Fig. S8†). This last value corresponds to the gallium hexahydrate [Ga(H2O)6]3+ – i.e. a species commonly formed when gallium salts are dissolved in aqueous solution, as a consequence of the rapid halogen/water exchange on the NMR time scale.74,75 In the end, diffusion-ordered NMR spectroscopy (1H-DOSY) experiments were also performed to assess the diffusion properties of 1 in water solution compared to PTA. The experiments indicated that 1 and PTA ligand had very similar diffusion properties and close hydrodynamic radii (3.36 Å and 3.92 Å for 1 and PTA, respectively; Fig. S23, S24, S27 and S28†), thus confirming the presence in solution of a species of comparable size with PTA and ruling out the formation of a coordination complex with Ga3+ ions.
The formation of the phosphonium cation [R3PH]+ was observed in certain [GaX3(PR3)] or gallium–diphosphine complexes when their chlorocarbon solutions (and their deuterated analogues) were exposed to air and this tendency was particularly evident with alkyl phosphines, probably due to their strong base character.61,76 For instance, compound [GaCl3(PPh3)] is quantitatively converted into [Ph3PH]·[GaCl4] by treatment with HCl gas in CH2Cl2, as well as crystals of [Me2PhPH]·[GaCl4] have been obtained by vapour diffusion of n-hexane into a DCM solution of [GaCl3(Me2PhP)] under nitrogen.61 In the case of PTA, the pKa is known to be in the range of 5.7–6.0 and this basicity does not relate to the phosphorus but to one of the nitrogen atoms.77,78 Indeed, when PTA is reacted with 0.1 M HCl, the ammonium-phosphine salt [PTA-H]·[Cl]78 is obtained and the 31P{1H} NMR in D2O of this N-protonated derivative shows a singlet at δ –89.90 ppm, a value very close to that observed for 1.
In order to decrease the residual acidity of GaCl3, the reaction with PTA was repeated under the same conditions described above after a pre-treatment of GaCl3 with triethylamine in DCM, or additionally performing the reaction at low temperature (0 °C). In both cases, after the addition of PTA, a white precipitate quickly formed showing the same solubility and signals of 1 in the NMR spectra. From the spectroscopic data of all experiments described, we did not have any evidence for the formation of [GaCl3(PTA)] or [GaCl3(PTA-H)]+ complexes. Moreover, the latter is thermodynamically unstable as shown by DFT calculations (see below).
When GaI3 was used as metal precursor, the addition of 1.2 equivalents of PTA to the solution of gallium iodide in ethyl acetate maintained in the dark produced again the formation of a white precipitate soluble in water (Scheme 1). The 31P{1H} NMR run in D2O showed a singlet at δ –91.39 ppm, which is about the same value observed for 1 (Fig. S19†). The 71Ga NMR showed no discernible peaks indicating that no gallium species were present, and the FT-IR in KBr revealed the presence of bands in the range 2857–2532 cm−1 (stretching) and at 1625 cm−1 (bending) indicative of the presence of a charged N–H group (Fig. S3†). The X-ray diffraction analysis of the needle-like crystals of this compound produced from a warm ethanolic solution supported the formula of this derivative as [PTA-H]·[I] (3) in accordance with the elemental analysis and spectroscopic data. Unfortunately, the poor quality of the crystals did not allow a detailed analysis of the structure (see Fig. S33†). As for the chloride precursor, GaI3 induces the N-protonation of the phosphine but in this case with iodide as the counterion. The formation of derivative 3 was also observed when GaI3 and PTA (1:1.2 ratio) were reacted in dry diethyl ether and even when the reaction was performed in the solid state by grinding and mixing the two starting materials as powders.
Based on results obtained with PTA, we decided to explore the reactivity of the analogue phosphine oxide, [PTA(O)] (Fig. 1), considering that the oxidation of the phosphorus centre in PTA results in decreased basicity of the nitrogen atoms, with a calculated pKa of 2.52 for [PTA(O)].3 Furthermore, the phosphine oxide is a hard donor compared to the phosphine itself and, additionally, it is established that Ga3+ is an oxophilic metal ion.
Despite these assumptions, the overall performances of [PTA(O)] towards reactions with GaCl3 and GaI3 were highly similar to those of PTA (Scheme 2). Namely, the formation of a white precipitate, highly soluble in water, was observed with both halide precursors. The products of reactions showed singlets at δ –3.11 (using GaCl3) and –3.04 ppm (using GaI3) in the 31P{1H} NMR spectra run in D2O (Fig. S13 and S22†), i.e. values only slightly different from that of the free ligand in the same solvent (δP = –2.92 ppm). In further experiments, [PTA(O)] was reacted with GaCl3 in a mixture of MeOH/DCM (1:1.4), and crystals were obtained by slow evaporation of the residual solution after filtration of the precipitate. The X-ray diffraction analysis revealed the formation of [PTA(O)–H]·[GaCl4] (2, Fig. 3a), where again the ligand is protonated at one nitrogen atom and the gallium tetrachloride is the counterion. Also, in this case, either a mirror plane cuts [PTA(O)–H]+ and [GaCl4]− species, and as for 1, P–C bonds were slightly influenced by the protonation. On the N-coordinated PTA chain, the P1–C1 distance is 1.830(4) Å, while in the non-coordinated one, the P1–C2 distance is 1.811(3) Å. The N–C distances around the protonated nitrogen atom (N1–C1 1.507(5) Å; N1–C3 1.531(4) Å) are longer than those in the non-protonated nitrogen atoms (N2–C2 1.478(4) Å; N2–C3 1.444(4) Å; N2–C4 1.466(4) Å). These trends are confirmed by a survey on CSD.73 Atom O1 forms a hydrogen bond with H1 of the protonated N1 of a neighbour [PTA(O)–H]+ molecule (O1⋯N1′ 2.872(5) Å symmetry operation to generate the atoms: −1/2 + x, 3/2 − y, 1/2 − z). The latter value is close to the upper limit for the O⋯N distance range for P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H–N hydrogen bonds (2.76–2.89 Å obtained from a search in Cambridge Structural Database (CSD)) indicating a weak interaction. In the packing diagram, a 1D hydrogen-bonded chain along the a axis can be recognized (Fig. 3b) and the fingerprint plot generated by the Hirshfeld surface shows that 7% of the contacts around the [PTA(O)–H] cation is with oxygen atoms of the neighbour molecule (Fig. S36 and S37†). As in the case of compound 1, the presence of [GaCl4]− was confirmed from the 71Ga NMR spectrum of the DCM/MeOH mother solution of 2, showing a large signal at δ 246.25 ppm (with C6D6 insert, Fig. S16†), and by 71Ga NMR spectrum of 2 in D2O, with a broad peak at δ 0.00 ppm (Fig. S14†). Furthermore, 1H-DOSY NMR experiments revealed very similar diffusion properties and hydrodynamic radii between 2 and [PTA(O)] (3.52 Å and 3.72 Å for 2 and [PTA(O)], respectively; Fig. S25, S26, S29 and S30†) and data obtained through ICP-MS analysis support the formulation of 2, with a complete match between found vs. calculated for the content of both gallium and phosphorus (Tables 1 and S2†).
O⋯H–N hydrogen bonds (2.76–2.89 Å obtained from a search in Cambridge Structural Database (CSD)) indicating a weak interaction. In the packing diagram, a 1D hydrogen-bonded chain along the a axis can be recognized (Fig. 3b) and the fingerprint plot generated by the Hirshfeld surface shows that 7% of the contacts around the [PTA(O)–H] cation is with oxygen atoms of the neighbour molecule (Fig. S36 and S37†). As in the case of compound 1, the presence of [GaCl4]− was confirmed from the 71Ga NMR spectrum of the DCM/MeOH mother solution of 2, showing a large signal at δ 246.25 ppm (with C6D6 insert, Fig. S16†), and by 71Ga NMR spectrum of 2 in D2O, with a broad peak at δ 0.00 ppm (Fig. S14†). Furthermore, 1H-DOSY NMR experiments revealed very similar diffusion properties and hydrodynamic radii between 2 and [PTA(O)] (3.52 Å and 3.72 Å for 2 and [PTA(O)], respectively; Fig. S25, S26, S29 and S30†) and data obtained through ICP-MS analysis support the formulation of 2, with a complete match between found vs. calculated for the content of both gallium and phosphorus (Tables 1 and S2†).
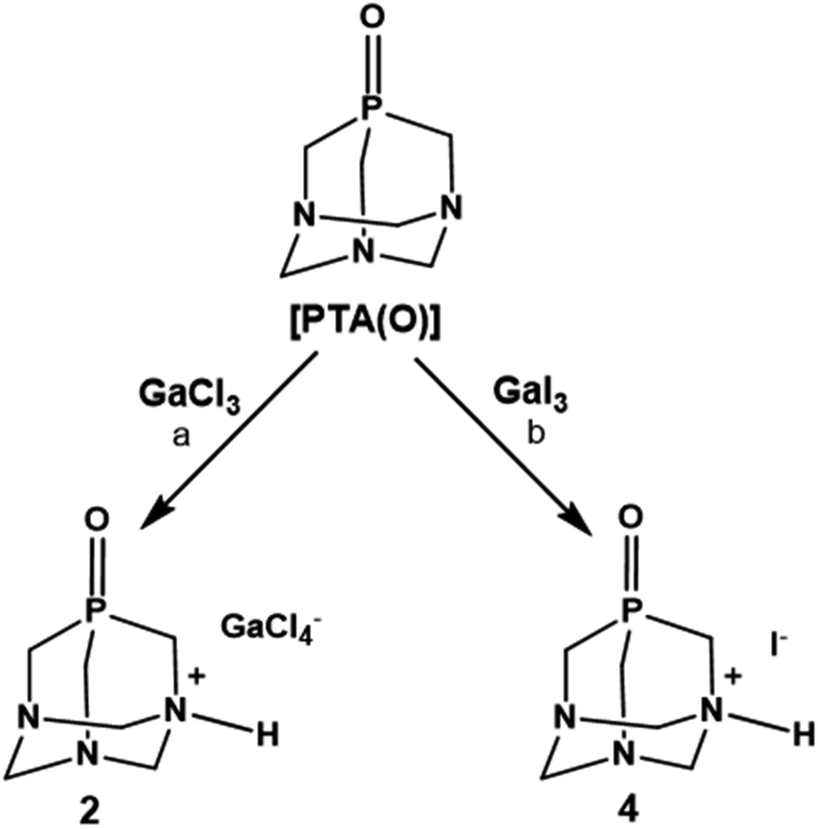 | ||
| Scheme 2 Products of reactions of [PTA(O)] with GaCl3 and GaI3. Reaction conditions: (a) [Ga]:[P(O)] = 1:1.2; DCM@rt; or MeOH/DCM@rt. (b) [Ga]:[P(O)] = 1:1.2; H2O@rt; or DCM@rt. | ||
 | ||
| Fig. 3 (a) Molecular structure (ORTEP diagram) of [PTA(O)-H]·[GaCl4] (2) showing the atom labelling scheme. Thermal ellipsoids are shown at a 50% probability level. (b) Propagation of the 1D hydrogen bond of the [PTA(O)-H]+ chain. Hydrogen bonding interactions are highlighted as dashed lines. Color code: C, black; H, white; O, red; N, blue; P, purple; Cl, green; Ga, light blue. Selected bond lengths (Å) and angles (°): P1–O1 1.479(3); P1–C2 1.811(3); P1–C1 1.830(4); N1–C1 1.507(5); N1–C3 1.531(4); N2–C2 1.478(4); N2–C3 1.444(4); N2–C4 1.466(4); Ga1–Cl2 2.1642(11); Ga1–Cl1 2.1801(9); Ga1–Cl3 2.1816(11); O1–P1–C2 118.02(12); C2–P1–C2′ 101.4(2); O1–P1–C1 115.3(2); C1–P1–C2 100.57(13); C1–N1–C3 111.4(2); C3–N1–C3′ 108.9(3); C3–N2–C4 109.7(3); C3–N2–C2 113.0(2); C4–N2–C2 112.0(3). Symmetry transformations used to generate equivalent atoms: x, −y + 3/2, z. | ||
The slow diffusion of ethanol into a deuterated aqueous solution containing the product of the reaction between GaI3 and [PTA(O)], again performed in the dark, produced crystals, which after analysis with X-ray diffraction, proved to be the ammonium-phosphine oxide of formula [PTA(O)-H]·[I] (4, Fig. 4a). In this case, the salt crystallizes in a less symmetric space group (P21/c) and both species are found in the asymmetric unit. The geometrical trends in the adamantane cage are confirmed and in agreement with those already discussed for 1 and 2. Also for 4, atom O1 forms a hydrogen bond with H1 resulting in a 1D hydrogen-bonded chain along the b axis (Fig. 4b) (O1⋯N1′ 2.692(2) Å symmetry operation to generate the atoms: −x, −1/2 + y, 3/2 − z) being the O⋯N distance more than 0.05 Å shorter than the expected range (2.76–2.89 Å) for PO⋯H–N hydrogen bonds. The different shapes of the anions in 2 and 4 play a role in the resulting hydrogen bond interaction. In the fingerprint plot, for the [PTA(O)-H] cation in 4, the contacts are mostly with hydrogen and oxygen atoms of the cations (70% and 10%, respectively) and with the iodine anion (15%) (Fig. S38 and S39†). In addition to the signals due to the protonated N–H groups, the FT-IR spectra (KBr pellets, Fig. S2 and S4†) of both 2 and 4 showed strong bands due to the stretching of PO groups at 1174 and 1155 cm−1 respectively, values slightly shifted compared to that of [PTA(O)] (ν(PO) = 1167 cm−1).4,79
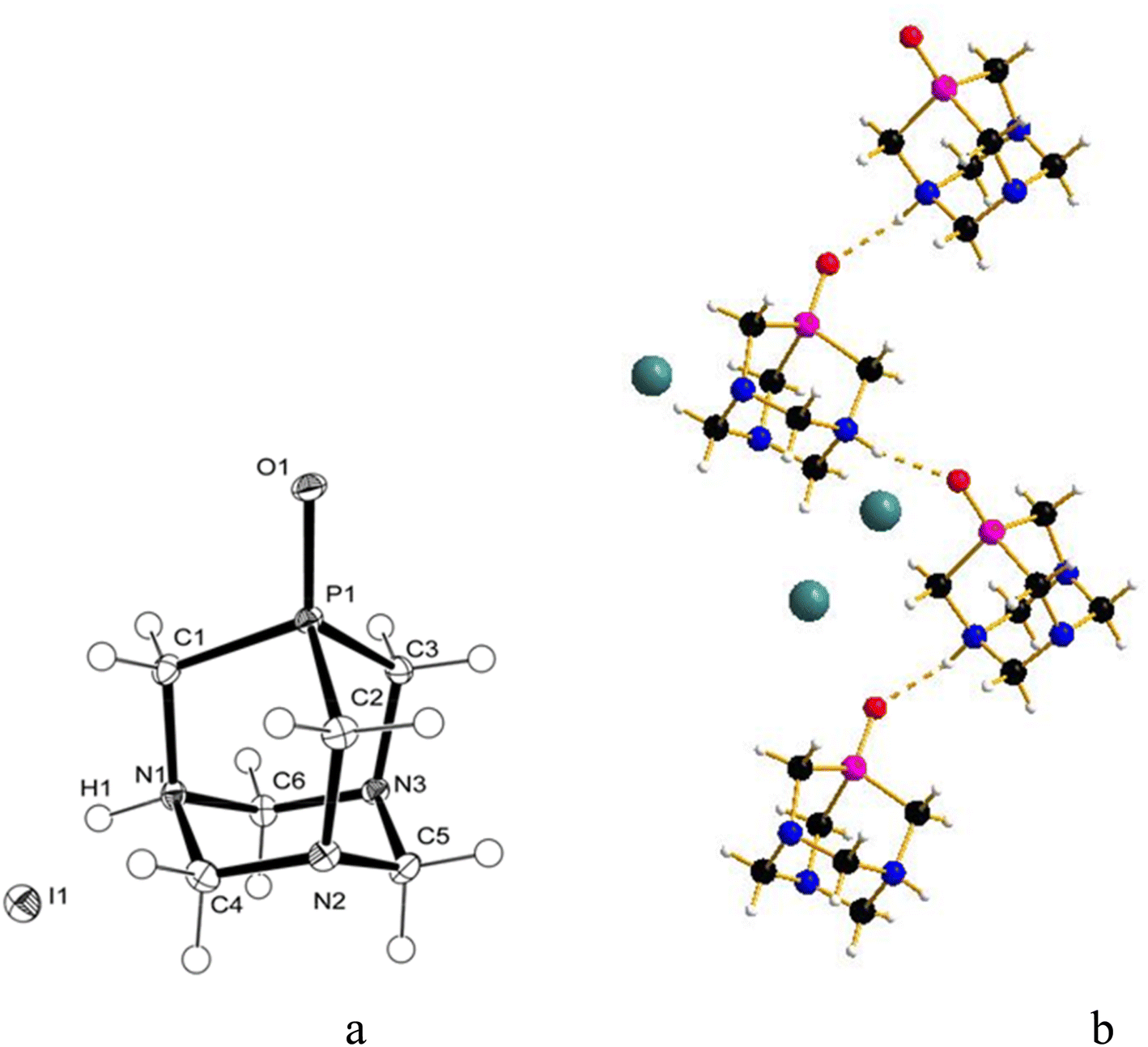 | ||
| Fig. 4 (a) Molecular structure (ORTEP diagram) of [PTA(O)-H]·[I] (4) showing the atom labelling scheme. (b) 1D hydrogen bond of [PTA(O)-H]+ chain. Hydrogen bonding interactions are highlighted as dashed lines. Color code: C, black; H, white; O, red; N, blue; P, purple; I, dark green. Thermal ellipsoids are shown at a 50% probability level. Selected bond lengths (Å) and angles (°): P1–C1 1.8175(13); P1–C2 1.8184(14); P1–C3 1.8160(14); P1–O1 1.4931(10); N1–C1 1.5005(17); N1–C4 1.5233(17); N1–C6 1.5288(17); N1–H1 0.897(18); N2–C4 1.4393(17); N2–C5 1.4719(17); N2–C2 1.4841(18); N3–C6 1.4390(17); N3–C5 1.4710(17); N3–C3 1.4864(17); O1–P1–C1 112.40(6); O1–P1–C3 119.29(6); C1–P1–C3 100.60(6); O1–P1–C2 118.37(6); C1–P1–C2 100.61(6); C3–P1–C2 102.62(6); C1–N1–C4 110.92(10); C1–N1–C6 110.72(10); C4–N1–C6 108.66(10); C4–N2–C5 109.69(10); C4–N2–C2 112.59(11); C5–N2–C2 112.47(11); C6–N3–C5 110.14(11); C6–N3–C3 113.43(10); C5–N3–C3 111.81(10); N1–C1–P1 109.83(8); N2–C2–P1 108.35(9); N3–C3–P1 108.33(9). | ||
As already mentioned, all compounds described herein showed high solubility in water, in particular compounds 1 and 2 with S20°C = 50 g L−1 and 63 g L−1, respectively. Compounds 1–4 resulted to be stable as solids although 4 showed the tendency to turn yellow after ca. one week. The stability of gallium–PTA salts 1 and 2 in aqueous solution was assessed by performing NMR experiments in D2O over time. Limited changes in the 31P{1H} NMR (Fig. 5) and 1H signals (Fig. S31b and S31d†) were detected for both compounds after one week in solution at room temperature and only a minimal decomposition was noted after one month (ca. 4.4% and 3.8% for 1 and 2 respectively, based on 31P). Also, no peaks other than the sharp singlets at δ 0.00 ppm were observed in the 71Ga NMR spectra of both samples over the same range of time (Fig. S31a and S31c†). The good stability shown by the gallium salts, in addition to the lack of formation of the toxic hydrolysis derivative [Ga(OH)4]− (δGa = 222 ppm)80 in the experimental conditions tested, are very promising results in view of the potential application of these derivatives in the biological field.
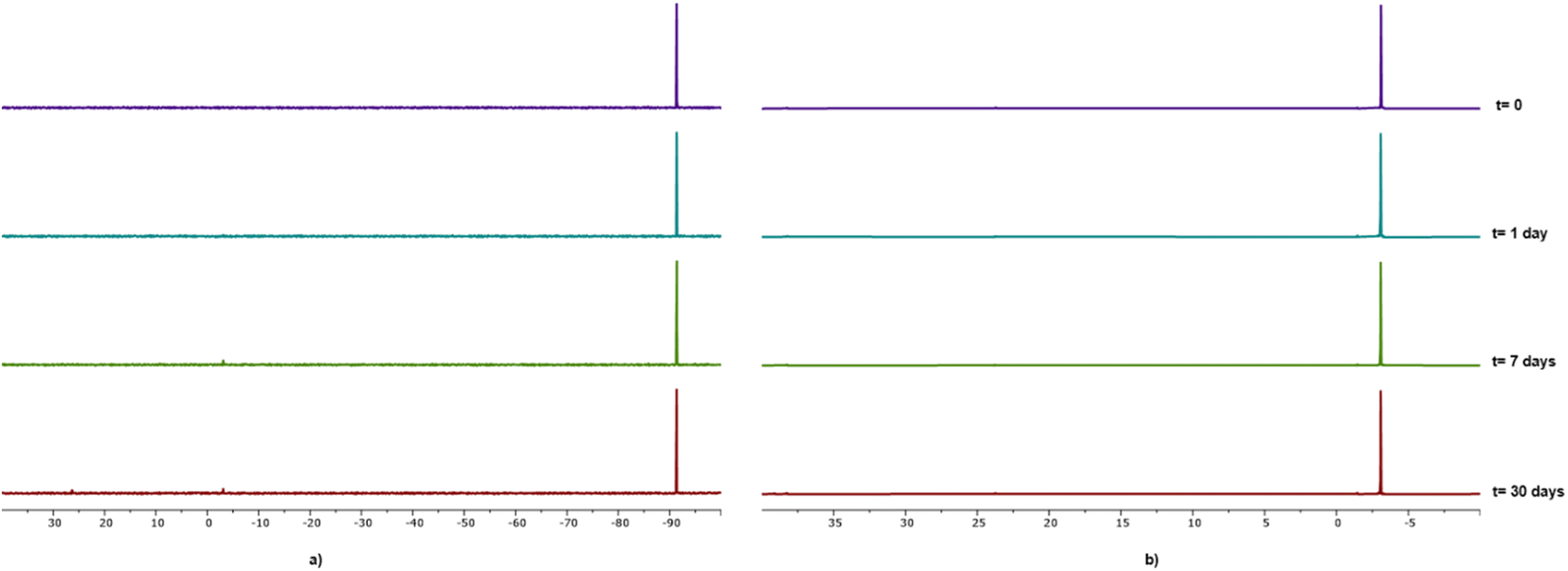 | ||
| Fig. 5 31P{1H} NMR spectra of D2O solutions (0.025 M) of [PTA-H]·[GaCl4] 1 (a) and [PTA(O)-H]·[GaCl4] 2 (b) recorded at several times. The spectra were recorded with a transmitter frequency offset of −40 ppm and a spectral width of 200 ppm. Spectral widths shown in the figure were expanded in the region where signals were detected. Each spectrum was normalised to the tallest peak intensity. | ||
DFT studies on the coordination of PTA with Ga3+ metal ion
On the basis of the results obtained by reacting PTA and [PTA(O)] with gallium-halides, we performed a DFT analysis to rationalize the lack of formation of any gallium-phosphine (or gallium-phosphine oxide) coordination species. Crystal structures of transition metal complexes containing protonated PTA molecules are well known, although less common than those of complexes with neutral PTA (i.e. 50 vs. 600 hits for protonated vs. unprotonated transition metal complexes based on CSD search). In some cases, the N-protonation of PTA takes place simultaneously with the formation of the coordination complex, but there are also examples of protonation occurring on the isolated complex through subsequent acid treatment.79,81Thus, the two models of [(PTA)GaCl3] (5) and [(PTA-H)GaCl3] (6) complexes (Fig. 6) were considered and optimized in the gas phase.
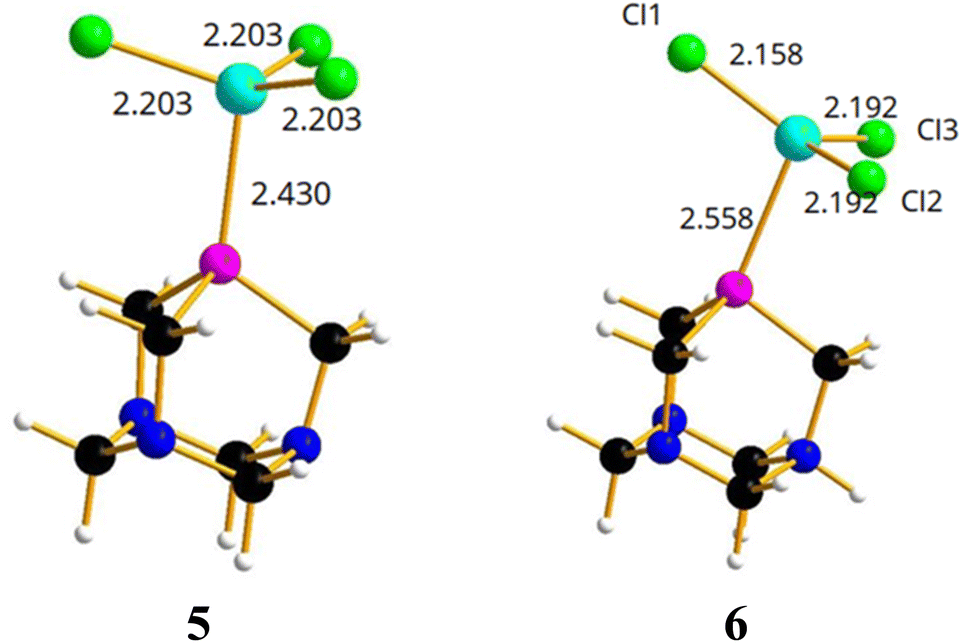 | ||
| Fig. 6 Optimized structures of [(PTA)GaCl3] (5) and [(PTA-H)GaCl3] (6) complexes. Color code: N, blue; P, pink; Cl, green; Ga, light blue; C, black; H, white. | ||
In [(PTA)GaCl3] (5), the gallium atom has a tetrahedral coordination and the Ga–P distance is calculated at 2.430 Å, a value comparable with 2.37 Å that is the average distance of Ga–P bond in the complexes of the type [GaCl3(PR3)] (based on CSD search). In contrast, the Cl–Ga–P angle (102.8°) is smaller than the average angle (107°) found in the CSD for the same complexes. Instead, the optimized model of [(PTA-H)GaCl3]+ (6, Fig. 6) shows a long Ga–P distance (2.558 Å), GaCl3 moiety is almost planar (i.e. the sum of the three Cl–Ga–Cl angles is 355°) and the gallium atom lacks the expected quasi tetrahedral coordination. This suggests that the complex could not exist in solution, and it is consistent with the lack of experimental evidence of its presence in solution.
From an energetic point of view (Table 2), the free energy calculated for the formation of 5 is –16.0 kcal mol−1, which is comparable to the free energy calculated for the [(PMe)3GaCl3] adduct (–19.8 kcal mol−1). For comparison, the free energies for PTA–gold complexes [(PTA)AuCl] have been calculated resulting in –43.3 kcal mol−1.82,83 Therefore, the coordination bond is twice as strong for the transition metal compared to the gallium atom. The presence of a very weak Ga–P bond in 6 has been further confirmed by the positive calculated free energy (+2.2 kcal mol−1; Table 2) for the formation of the protonated complex from PTA-H+ and GaCl3. With regard to the latter, there was indeed no experimental observation of coordination products even when the protonated derivative of the formula [PTA-H]·[Cl] was reacted with GaCl3 in DCM, or with GaI3 in diethyl ether for 24 h. Again, for comparison, the free energy calculated for the corresponding PTA-H–gold complex [(PTA-H)AuCl]+82,83 was –26.9 kcal mol−1 and from a geometrical point of view the P–Au calculated bond distance was 2.257 Å, slightly shorter than the corresponding distance (2.278 Å) in [(PTA-H)AuCl]. Similar results in terms of geometry and energy differences were obtained for the corresponding structures 5 and 6 using GaBr3 and GaI3 instead of GaCl3 (Table S3, Fig. S40 and S41†).
| Metal–phosphine–halide adduct formation | Calculated free energy (kcal mol−1) |
|---|---|
| PTA + GaCl3 → [(PTA)GaCl3] | ΔG = –16.0 |
| (PTA-H) + GaCl3 → [(PTA-H)GaCl3] | ΔG = +2.2 |
| P(Me)3 + GaCl3 → [(PMe)3GaCl3] | ΔG = –19.8 |
| PTA + AuCl → [(PTA)AuCl] | ΔG = –43.3 |
| (PTA-H) + AuCl → [(PTA-H)AuCl] | ΔG = –26.9 |
To summarize, the calculated Ga–P bond, where P is PTA or other phosphines, is always weaker than a common transition metal–phosphorus bond as demonstrated by the values reported in Table 2. The N-protonation of PTA carries an energy penalty of around 18 kcal mol−1 in the formation of both gallium and transition metal coordination complexes. As a consequence, the Ga–PTA complex cannot survive the perturbation induced by the N-protonation of PTA.
Conclusions
The reactivity of the cage-like phosphine PTA and its corresponding oxide [PTA(O)] towards the gallium(III) precursors GaCl3 and GaI3 has been investigated for the first time. The molecular structures of all products of reactions were unambiguously confirmed by a range of characterization techniques, including single-crystal X-ray diffraction, validating the formation of ionic compounds where the phosphine (or phosphine oxide) is protonated at one nitrogen atom and the anions are [GaCl4]− or [I]− depending on the gallium precursor. DFT calculations were performed to further rationalize our findings showing a favourable free energy for the formation of Ga–P bond, although a positive ΔG value was observed in the case of protonated phosphines. This result is in line with the lack of formation of any gallium-PTA coordination complexes from the metal-halide precursors GaCl3 and GaI3, being both expected to produce the fast protonation of PTA and [PTA(O)]. Based on our experimental results and theoretical calculations, the ionic derivatives 1–4 are the only species accessible in these conditions, thus alternative approaches need to be investigated in order to achieve coordination products of Ga3+ with PTA-based ligands.Experimental section
General procedures
All manipulations were carried out under a purified N2 atmosphere using standard Schlenk techniques unless otherwise noted. Deuterated solvents and all the gallium(III) precursors were bought from commercial suppliers and used without further purification. Doubly distilled water was used; dichloromethane (DCM), methanol (MeOH) and diethyl ether (Et2O) were distilled on calcium hydride, magnesium and sodium, respectively, and degassed prior to use. Ethyl acetate (EtOAc) was dried over molecular sieves (4 Å). The phosphine ligands PTA and [PTA(O)] were synthesized as reported in the literature.4 1H, 13C{1H}, 31P{1H} NMR spectra were recorded on a Bruker Avance II spectrometer (operating at 400.13, 100.61 and 161.98 MHz, respectively). The 13C and 31P NMR spectra were normally run with proton decoupling. 31P NMR spectra are reported in ppm relative to an external H3PO4 standard (0.0 ppm), with downfield positive shifts. 13C{1H} NMR spectra are reported in ppm relative to residual solvent resonances with downfield positive shifts. 71Ga NMR spectra of mother solutions were recorded with a C6D6 insert on a Bruker Avance II spectrometer (operating at 122.03 MHz). 71Ga NMR spectra of the isolated compounds and 1H-DOSY experiments were run on an Avance III HD spectrometer (operating at 122.03 MHz and 400.13 MHz, respectively). 1H-DOSY NMR (ledbpgp2s) experiments were performed at 298 K, with 16 increments varying gradient strength from 5% to 95% using a linear ramp with a diffusion time (d20) of 50 ms and a gradient pulse width (p30) of 1.3 ms. The diffusion constants for compounds 1 and 2 and for ligands PTA and [PTA(O)] were determined by fitting the integral of the isolated phosphor-coupled doublet of each compound using the T1/T2 function in the dynamics module of Topspin 3.6.5. The hydrodynamic radii of compounds 1 and 2 and of free ligands PTA and [PTA(O)] were then calculated using the Stokes–Einstein equation (for further details see ESI†). Infrared spectra were measured using KBr pellets on a PerkinElmer FT-IR Spectrum BX II instrument in the range 400–4000 cm−1. ICP-MS measurements were performed at the Metallomics Facility (King's College London, KCL), under GMP settings on a ThermoFisher iCAP TQ ICP-MS with Qtegra operating software (for further details see ESI†). Elemental analyses of compounds 1 and 2 were carried out at MEDAC Ltd (Chobham, Surrey, UK) using a Thermo Fisher Scientific CHNS-O Analyzer FlashEA 1112 Series (results are within ±0.4% of the theoretical values). Elemental analyses of compounds 3 and 4 were performed at the University of Florence, Italy, using a Thermo Fisher_FlashSmart elemental analyzer. The solubility of 1–4 in water was assessed by adding bidistilled H2O with a 100 μL Hamilton micro syringe to a 5 mg sample of each compound placed in a Schlenk flask, under slow stirring in a thermostated bath kept at 20 °C, until complete dissolution of the solid.O) 1174. Anal. Found (calcd) for C6H13Cl4GaN3OP·H2O: C, 18.03 (17.85); H, 3.42 (3.75); N, 10.20 (10.41).O) 1155. Anal. Found (calcd) for C6H13IN3OP·0.2CH2Cl2: C, 23.71 (23.41); H, 4.62 (4.25); N, 12.94 (13.21).| [PTA-H]·[GaCl4] | [PTA(O)-H]·[GaCl4] | [PTA(O)-H]·[I] | |
|---|---|---|---|
| Empirical formula | C6H13Cl4GaN3P | C6H13Cl4GaN3OP | C6H13IN3OP |
| Formula weight | 369.68 | 385.68 | 301.06 |
| Temp (K) | 100(2) | 100(2) | 100(2) |
| Wavelength (Å) | 1.54178 | 1.54178 | 0.71073 |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic |
| Space group | Pnma | Pnma | P21/c |
| a (Å) | 13.3420(9) | 13.7699(10) | 7.8486(3) |
| b (Å) | 8.2734(6) | 8.1328(6) | 9.6385(3) |
| c (Å) | 11.9648(8) | 12.1428(9) | 12.7790(4) |
| a (deg) | 90 | 90 | 90 |
| b (deg) | 90 | 90 | 91.6520(10) |
| g (deg) | 90 | 90 | 90 |
| V (Å3) | 1320.72(16) | 1359.85(17) | 966.31(6) |
| Z | 4 | 4 | 4 |
| ρcalc (g cm−3) | 1.859 | 1.884 | 2.069 |
| μ (mm−1) | 11.235 | 11.002 | 3.439 |
| F(000) | 736 | 768 | 584 |
| θ range for data collection (deg) | 4.965 to 72.470° | 4.856 to 72.232° | 2.596 to 33.216° |
| Index ranges | −16 ≤ h ≤ 16, −10 ≤ k ≤ 9, −14 ≤ l ≤ 14 | −16 ≤ h ≤ 16, −10 ≤ k ≤ 9, −14 ≤ l ≤ 14 | −12 ≤ h ≤ 12, −14 ≤ k ≤ 14, −19 ≤ l ≤ 19 |
| Reflections collected | 16021 |
12108 |
53622 |
| Independent reflections | 1408 [Rint = 0.0563] | 1431 [Rint = 0.0665] | 3699 [Rint = 0.0427] |
| Completeness to θ = 25.0° | 99.8% | 99.6% | 100.0% |
| Data/restraints/parameters | 1408/0/84 | 1431/0/90 | 3699/0/116 |
| Goodness-of-fit on F2 | 1.102 | 1.077 | 1.039 |
| Final R indices [I > 2σ(I)] | R1 = 0.0229, wR2 = 0.0600 | R1 = 0.0409, wR2 = 0.1108 | R1 = 0.0159, wR2 = 0.0319 |
| R indices (all data) | R1 = 0.0251, wR2 = 0.0603 | R1 = 0.0424, wR2 = 0.1113 | R1 = 0.0205, wR2 = 0.0334 |
| Largest diff. peak and hole (e Å−3) | 0.911 and −0.378 | 0.753 and −0.812 | 0.677 and −0.419 |
Data availability
The data supporting this article (FT-IR spectra, 1H, 13C{1H}, 31P{1H} and 71Ga NMR spectra, 1H-DOSY NMR experiments, ICP-MS measurements, fingerprint plots for compounds 1, 2 and 4, details on DFT calculations) have been included as part of the ESI.†Author contributions
A. Guerriero: conceptualization, methodology, experimental work, supervision, funding acquisition. A. Cilibrizzi: conceptualization, methodology, supervision, funding acquisition. Thomas Hicks: 1H-DOSY and 71Ga NMR experiments. A. Ienco: single-crystal X-ray diffraction and DFT calculations. All the authors contributed to writing the manuscript and approved the final version.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This research work was financially supported by the National Council of Research of Italy (CNR) (Biennial Program 2022–2023) and by the Royal Society (RS) (International Exchanges 2021, IEC\R2\212003) through the Scientific Cooperation Agreement CNR/RS. The National Council of Research of Italy (CNR) and the Royal Society (RS) are kindly acknowledged. Dr Massimo Calamante (CNR-ICCOM) is acknowledged for elemental analyses measurements and Dr Graeme J. Stasiuk (KCL) for useful inputs on this research. The authors would like to thank the London Metallomics Facility (KCL) for access and support with ICP-MS experiments, as well as the KCL Chemistry Department Facilities for technical support and access to their NMR spectrometer. A. I. would like to thank the Centro di Cristallografia Strutturale (CRIST) of the University of Florence for the X-ray diffraction facilities and Dr Laura Chelazzi and Dr Samuele Ciattini for their valuable technical assistance.References
- A. Guerriero, M. Peruzzini and L. Gonsalvi, Coord. Chem. Rev., 2018, 355, 328–361 CrossRef CAS.
- J. Bravo, S. Bolaño, L. Gonsalvi and M. Peruzzini, Coord. Chem. Rev., 2010, 254, 555–607 CrossRef CAS.
- A. D. Phillips, L. Gonsalvi, A. Romerosa, F. Vizza and M. Peruzzini, Coord. Chem. Rev., 2004, 248, 955–993 CrossRef CAS.
- D. J. Daigle, T. J. Decuir, J. B. Robertson and D. J. Darensbourg, Inorg. Synth., 1998, 32, 40–45 CrossRef CAS.
- A. Guerriero, M. Erlandsson, A. Ienco, D. A. Krogstad, M. Peruzzini, G. Reginato and L. Gonsalvi, Organometallics, 2011, 30, 1874–1884 CrossRef CAS.
- D. A. Krogstad, A. Guerriero, A. Ienco, G. Manca, M. Peruzzini, G. Reginato and L. Gonsalvi, Organometallics, 2011, 30, 6292–6302 CrossRef CAS.
- S. Moret, P. J. Dyson and G. Laurenczy, Nat. Commun., 2014, 5, 4017 CrossRef CAS PubMed.
- M. Trivedi, P. Sharma, I. K. Pandey, A. Kumar, S. Kumar and N. P. Rath, Chem. Commun., 2021, 57, 8941–8944 RSC.
- N. Six, A. Guerriero, D. Landy, M. Peruzzini, L. Gonsalvi, F. Hapiot and E. Monflier, Catal. Sci. Technol., 2011, 1, 1347–1353 RSC.
- D. S. Ramarou, B. C. E. Makhubela and G. S. Smith, J. Organomet. Chem., 2018, 870, 23–31 CrossRef CAS.
- K. Hua, X. Liu, B. Wei, Z. Shao, Y. Deng, L. Zhong, H. Wang and Y. Sun, Green Chem., 2021, 23, 8040–8046 RSC.
- D. Anselmo, R. Gramage-Doria, T. Besset, M. V Escárcega-Bobadilla, G. Salassa, E. C. Escudero-Adán, M. Martínez Belmonte, E. Martin, J. N. H. Reek and A. W. Kleij, Dalton Trans., 2013, 42, 7595–7603 RSC.
- N. Ahlsten, H. Lundberg and B. Martín-Matute, Green Chem., 2010, 12, 1628–1633 RSC.
- E. Bolyog-Nagy, A. Udvardy, Á. Barczáné-Bertók, F. Joó and Á. Kathó, Inorg. Chim. Acta, 2017, 455, 514–520 CrossRef CAS.
- E. Bolyog-Nagy, A. Udvardy, F. Joó and Á. Kathó, Tetrahedron Lett., 2014, 55, 3615–3617 CrossRef CAS.
- W. L. Ounkham, J. A. Weeden and B. J. Frost, Chem.–Eur. J., 2019, 2, 10013–10020 CrossRef PubMed.
- F. Scalambra, P. Lorenzo-Luis, I. de los Ríos and A. Romerosa, Eur. J. Inorg. Chem., 2019, 2019, 1529–1538 CrossRef CAS.
- G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed.
- S. W. Jaros, M. Haukka, M. Florek, M. F. C. G. da Silva, A. J. L. Pombeiro, A. M. Kirillov and P. Smoleński, Materials, 2019, 12, 5–13 CrossRef PubMed.
- L. Hajji, C. Saraiba-Bello, F. Scalambra, G. Segovia-Torrente and A. Romerosa, J. Inorg. Biochem., 2021, 218, 111404 CrossRef CAS PubMed.
- F. Battistin, F. Scaletti, G. Balducci, S. Pillozzi, a. Arcangeli, L. Messori and E. Alessio, J. Inorg. Biochem., 2016, 160, 180–188 CrossRef CAS PubMed.
- A. Weiss, R. H. Berndsen, M. Dubois, C. Müller, R. Schibli, A. W. Griffioen, P. J. Dyson and P. Nowak-Sliwinska, Chem. Sci., 2014, 5, 4742–4748 RSC.
- P. Nowak-Sliwinska, J. R. van Beijnum, A. Casini, A. A. Nazarov, G. Wagnières, H. van den Bergh, P. J. Dyson and A. W. Griffioen, J. Med. Chem., 2011, 54, 3895–3902 CrossRef CAS PubMed.
- B. S. Murray, M. V. Babak, C. G. Hartinger and P. J. Dyson, Coord. Chem. Rev., 2016, 306, 86–114 CrossRef CAS.
- S. W. Jaros, U. Śliwińska-Hill, A. Białońska, D. S. Nesterov, P. Kuropka, J. Sokolnicki, B. Bazanów and P. Smoleński, Dalton Trans., 2019, 48, 11235–11249 RSC.
- S. W. Jaros, U. K. Komarnicka, A. Kyzioł, B. Pucelik, D. S. Nesterov, A. M. Kirillov and P. Smoleński, J. Med. Chem., 2022, 65, 11100–11110 CrossRef CAS PubMed.
- C. Pettinari, F. Marchetti, G. Lupidi, L. Quassinti, M. Bramucci, D. Petrelli, L. a Vitali, M. F. C. Guedes da Silva, L. M. D. R. S. Martins, P. Smoleński and A. J. L. Pombeiro, Inorg. Chem., 2011, 50, 11173–11183 CrossRef CAS PubMed.
- J. Kuchar, J. Rust, C. W. Lehmann and F. Mohr, Inorg. Chem., 2020, 59, 10557–10568 CrossRef CAS PubMed.
- A. M. Kirillov, S. W. Wieczorek, A. Lis, M. F. C. Guedes da Silva, M. Florek, J. Król, Z. Staroniewicz, P. Smoleński and A. J. L. Pombeiro, Cryst. Growth Des., 2011, 11, 2711–2716 CrossRef CAS.
- T. Scattolin, E. Bortolamiol, F. Visentin, S. Palazzolo, I. Caligiuri, T. Perin, V. Canzonieri, N. Demitri, F. Rizzolio and A. Togni, Chem.–Eur. J., 2020, 26, 11868–11876 CrossRef CAS PubMed.
- T. Scattolin, A. Piccin, M. Mauceri, F. Rizzolio, N. Demitri, V. Canzonieri and F. Visentin, Polyhedron, 2021, 207, 115381 CrossRef CAS.
- I. Chakraborty, S. J. Carrington, G. Roseman and P. K. Mascharak, Inorg. Chem., 2017, 56, 1534–1545 CrossRef CAS PubMed.
- S. C. Marker, S. N. MacMillan, W. R. Zipfel, Z. Li, P. C. Ford and J. J. Wilson, Inorg. Chem., 2018, 57, 1311–1331 CrossRef CAS PubMed.
- A. Marchi, E. Marchesi, L. Marvelli, P. Bergamini, V. Bertolasi and V. Ferretti, Eur. J. Inorg. Chem., 2008, 2, 2670–2679 CrossRef.
- R. R. Moskalyk, Miner. Eng., 2003, 16, 921–929 CrossRef CAS.
- L. Mochalov, A. Logunov and V. Vorotyntsev, Sep. Purif. Technol., 2021, 258, 118001 CrossRef CAS.
- L. R. Bernstein, Pharmacol. Rev., 1998, 50, 665–682 CAS.
- J. A. Lessa, G. L. Parrilha and H. Beraldo, Inorg. Chim. Acta, 2012, 393, 53–63 CrossRef CAS.
- C. R. Chitambar, Int. J. Environ. Res. Public Health, 2010, 7, 2337–2361 CrossRef CAS PubMed.
- G. Floresta, S. Memdouh, T. Pham, M. T. Ma, P. J. Blower, R. C. Hider, V. Abbate and A. Cilibrizzi, Dalton Trans., 2022, 12796–12803 RSC.
- M. Vorster, J. Buscombe, Z. Saad and M. Sathekge, Curr. Pharm. Des., 2018, 24, 787–794 CrossRef CAS PubMed.
- A. Cilibrizzi, V. Abbate, Y.-L. Chen, Y. Ma, T. Zhou and R. C. Hider, Chem. Rev., 2018, 118, 7657–7701 CrossRef CAS PubMed.
- L. Pilia, E. Sessini, F. Artizzu, M. Yamashita, A. Serpe, K. Kubo, H. Ito, H. Tanaka, S. Kuroda, J. Yamada, P. Deplano, C. J. Gómez-García and M. Mercuriet, Inorg. Chem., 2013, 52, 423–430 CrossRef CAS PubMed.
- U. Beckmann, E. Bill, T. Weyhermüller and K. Wieghardt, Eur. J. Inorg. Chem., 2003, 1768–1777 CrossRef CAS.
- C. R. Chitambar, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2044–2053 CrossRef CAS PubMed.
- P. Collery, B. Keppler, C. Madoulet and B. Desoize, Crit. Rev. Oncol. Hematol., 2002, 42, 283–296 CrossRef PubMed.
- C. R. Chitambar, in Metallo-Drugs: Development and Action of Anticancer Agents, De Gruyter, 2018, pp. 281–302 Search PubMed.
- E. Mitidieri, D. Visaggio, E. Frangipani, C. Turnaturi, D. Vanacore, R. Provenzano, G. Costabile, R. Sorrentino, F. Ungaro, P. Visca and R. d'Emmanuele di Villa Bianca, Pharmacol. Res., 2021, 170, 105698 CrossRef CAS PubMed.
- Y. Kaneko, M. Thoendel, O. Olakanmi, B. E. Britigan and P. K. Singh, J. Clin. Invest., 2007, 117, 877–888 CrossRef CAS PubMed.
- C. Bonchi, F. Imperi, F. Minandri, P. Visca and E. Frangipani, BioFactors, 2014, 40, 303–312 CrossRef CAS PubMed.
- C. H. Goss, Y. Kaneko, L. Khuu, G. D. Anderson, S. Ravishankar, M. L. Aitken, N. Lechtzin, G. Zhou, D. M. Czyz, K. McLean, O. Olakanmi, H. A. Shuman, M. Teresi, E. Wilhelm, E. Caldwell, S. J. Salipante, D. B. Hornick, R. J. Siehnel, L. Becker, B. E. Britigan and P. K. Singh, Sci. Transl. Med., 2018, 10, 1–12 Search PubMed.
- L. R. Bernstein, T. Tanner, C. Godfrey and B. Noll, Met.-Based Drugs, 2000, 7, 33–47 CrossRef CAS PubMed.
- S. R. Choi, B. Switzer, B. E. Britigan and P. Narayanasamy, ACS Infect. Dis., 2020, 6, 2582–2591 CrossRef CAS PubMed.
- M. D. Bartholomä, A. S. Louie, J. F. Valliant and J. Zubieta, Chem. Rev., 2010, 110, 2903–2920 CrossRef PubMed.
- I. Velikyan, Theranostics, 2014, 4, 47–80 CrossRef CAS PubMed.
- G. Bandoli, A. Dolmella, F. Tisato, M. Porchia and F. Refosco, Coord. Chem. Rev., 2009, 253, 56–77 CrossRef CAS.
- R. Cusnir, C. Imberti, R. C. Hider, P. J. Blower and M. T. Ma, Int. J. Mol. Sci., 2017, 18, 4–7 Search PubMed.
- N. C. Norman and N. L. Pickett, Coord. Chem. Rev., 1995, 145, 27–54 CrossRef CAS.
- J. Burt, W. Levason and G. Reid, Coord. Chem. Rev., 2014, 260, 65–115 CrossRef CAS.
- N. Burford, B. W. Royan, R. E. v. H. Spence, T. S. Cameron, A. Linden and R. D. Rogers, J. Chem. Soc., Dalton Trans., 1990, 1, 1521–1528 RSC.
- F. Cheng, H. L. Codgbrook, A. L. Hector, W. Levason, G. Reid, M. Webster and W. Zhang, Polyhedron, 2007, 26, 4147–4155 CrossRef CAS.
- A. J. Dawson, Chemistry of Aluminium, Gallium, Indium, and Thallium, Blackie academic and professional, 1993 Search PubMed.
- S. Aldridge, A. J. Downs and D. L. Kays, in The Group 13 Metals Aluminium, Gallium, Indium and Thallium: Chemical Patterns and Peculiarities, Wiley, 2011, pp. 148–245 Search PubMed.
- V. V. Shatunov, A. A. Korlyukov, A. V. Lebedev, V. D. Sheludyakov, B. I. Kozyrkin and V. Y. Orlov, J. Organomet. Chem., 2011, 696, 2238–2251 CrossRef CAS.
- D. C. Bradley, H. Chudzynska, M. M. Faktor, D. M. Frigo, M. B. Hursthouse, B. Hussain and L. M. Smith, Polyhedron, 1988, 7, 1289–1298 CrossRef CAS.
- K. Yurkerwich and G. Parkin, Inorg. Chim. Acta, 2010, 364, 157–161 CrossRef CAS.
- J. C. Carter, G. Jugie, R. Enjalbert and J. Galy, Inorg. Chem., 1978, 17, 1248–1254 CrossRef CAS.
- L. J. Baker, L. A. Kloo, C. E. F. Rickard and M. J. Taylor, J. Organomet. Chem., 1997, 545–546, 249–255 CrossRef CAS.
- A. J. Carty, Can. J. Chem., 1967, 45, 345–351 CrossRef.
- A. J. Carty, Can. J. Chem., 1967, 45, 3187–3192 CrossRef CAS.
- A. F. Alshamrani, T. J. Prior, B. P. Burke, D. P. Roberts, S. J. Archibald, L. J. Higham, G. Stasiuk and C. Redshaw, Inorg. Chem., 2020, 59, 2367–2378 CrossRef CAS PubMed.
- B. J. Frost, J. L. Harkreader and C. M. Bautista, Inorg. Chem. Commun., 2008, 11, 580–583 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171–179 CrossRef CAS PubMed.
- M. J. Taylor, Polyhedron, 1990, 9, 207–214 CrossRef CAS.
- J. W. Akitt, Aluminum, Gallium, Indium, and Thallium, in Multinuclear NMR, The Open University, Milton Keynes, Buckinghamshire, Springer US, Boston, MA, 1987, pp. 259–292 Search PubMed.
- M. Sigl, A. Schier and H. Schmidbaur, Z. Naturforsch., 1998, 53, 1313–1315 CrossRef CAS.
- D. J. Darensbourg, J. B. Robertson, D. L. Larkins and J. H. Reibenspies, Inorg. Chem., 1999, 38, 2473–2481 CrossRef CAS.
- K. J. Fisher, E. C. Alyea and N. Shahnazarian, Phosphorus, Sulfur Silicon Relat. Elem., 1990, 48, 37–40 CrossRef CAS.
- D. J. Darensbourg, N. W. Stafford, F. Joó and J. H. Reibenspies, J. Organomet. Chem., 1995, 488, 99–108 CrossRef CAS.
- Q. Zhou, C. Henoumont, L. Vander Elst, S. Laurent and R. N. Muller, Contrast Media Mol. Imaging, 2011, 6, 165–167 CrossRef CAS PubMed.
- D. N. Akbayeva, S. Moneti, M. Peruzzini, L. Gonsalvi, A. Ienco and F. Vizza, C. R. Chim., 2005, 8, 1491–1496 CrossRef CAS.
- Z. Assefa, B. G. McBurnett, R. J. Staples, J. P. Fackler, B. Assmann, K. Angermaier and H. Schmidbaur, Inorg. Chem., 1995, 34, 75–83 CrossRef CAS.
- Z. Assefa, M. A. Omary, B. G. McBurnett, A. A. Mohamed, H. H. Patterson, R. J. Staples and J. P. Fackler, Inorg. Chem., 2002, 41, 6274–6280 CrossRef CAS PubMed.
- Bruker, Bruker APEX2, Bruker AXS Inc., Madison, Wisconsin, 2012 Search PubMed.
- Bruker, Bruker SAINT, Bruker AXS Inc., Madison, Wisconsin, USA, 2012 Search PubMed.
- L. Krause, R. Herbst-Irmer, G. M. Sheldrick and D. Stalke, J. Appl. Crystallogr., 2015, 48, 3–10 CrossRef CAS PubMed.
- M. C. Burla, R. Caliandro, M. Camalli, B. Carrozzini, G. L. Cascarano, L. De Caro, C. Giacovazzo, G. Polidori and R. Spagna, J. Appl. Crystallogr., 2005, 38, 381–388 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- M. A. Spackman and J. J. McKinnon, CrystEngComm, 2002, 4, 378–392 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Jr Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- M. Dolg, H. Stoll, H. Preuss and R. M. Pitzer, J. Phys. Chem., 1993, 97, 5852–5859 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: FT-IR, 1H, 13C{1H}, 31P{1H} and 71Ga NMR spectra of compounds, 1H-DOSY NMR experiments, ICP-MS measurements, tests of stability in solution, additional details of crystallographic data and DFT calculations (PDF). CCDC 2341483, 2341485 and 2341486. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra02877e |
| This journal is © The Royal Society of Chemistry 2024 |