
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Boron-mediated one-pot access to salicylaldehydes via ortho-C–H hydroxylation of benzaldehydes†
Ruiyang Wang,
Xu Feng,
Boya Feng * and
Yu Chen*
* and
Yu Chen*
Jiangsu Key Laboratory for the Research and Utilization of Plant Resources, Jiangsu Province Engineering Research Center of Eco-cultivation and High-value Utilization of Chinese Medicinal Materials, Institute of Botany, Jiangsu Province and Chinese Academy of Sciences (Nanjing Botanical Garden Mem. Sun Yat-Sen), Nanjing, 210014, China. E-mail: boyafeng@gmail.com; ychen@jib.ac.cn
First published on 20th June 2024
Abstract
A novel protocol has been devised for the ortho-C–H hydroxylation of benzaldehydes. Directed by a transient imine group, the borylation of benzaldehydes, sequentially followed by the hydroxylation, furnishes diverse salicylaldehydes in a one-pot manner. The resultant salicylaldehydes could be readily applied in the downstream synthesis to produce bioactive molecules.
Introduction
Salicylaldehyde and its derivatives represent ubiquitous bioactive motifs found in cosmetics, natural products, and fragrances.1 They also function as valuable synthetic building blocks or even photocatalysts in the construction of various complex scaffolds.2 The conventional preparation of salicylaldehyde derivatives relies on the formylation of phenols through reactions such as the Reimer–Tiemann reaction, Vilsmeier–Haack reaction, and Duff reaction.3 Thanks to the rapid development of C–H activation, diverse opportunities of aromatic C–H hydroxylation have been unlocked, presenting alternative synthetic approaches to produce salicylaldehyde derivatives. Transition-metal catalyzed C–H hydroxylation, assisted by directing groups like pyridine and amide, enable the streamlined synthesis of various ortho-substituted phenols.4 However, the direct hydroxylation on the adjacent position of aldehyde has been less developed,5 likely hampered by the weak coordinating ability and the undesired oxidation tendency of –CHO group. The elegant catalyzed examples including a ruthenium system developed by Ackermann5a and a palladium system achieved by Sorensen (Scheme 1A).5b Despite their high regioselectivity, these protocols suffered from complicated operations, transition-metal loading, and costly reagents (hypervalent iodine or the F+ oxidant).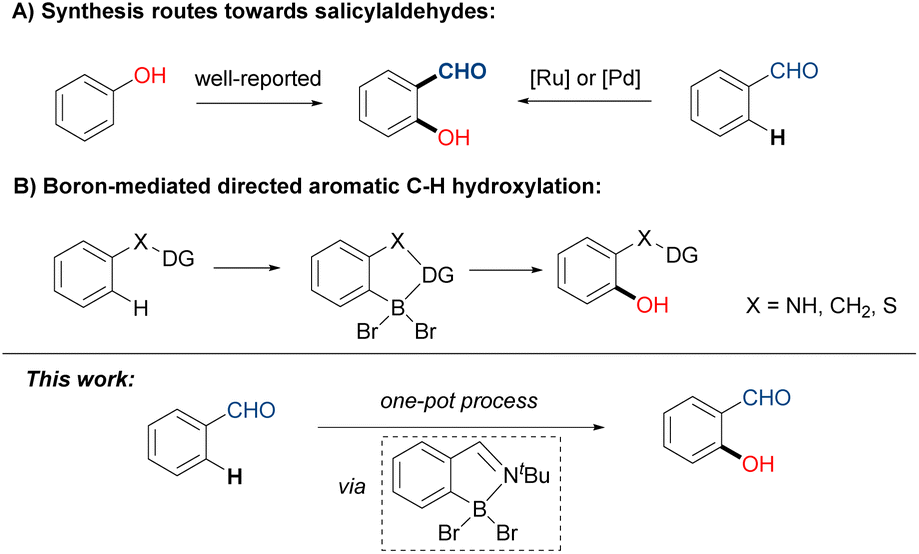 | ||
| Scheme 1 Synthetic strategies of ortho-substituted phenols: (A) generation of salicylaldehydes; (B) boron-mediated directed C–H hydroxylation. | ||
On the other hand, metal-free C–H borylation offers an alternative for regioselective C–H activation, as pioneering work by Maclean demonstrated.6 However, this method requires extremely high temperatures. In 2019, the BBr3-mediated borylation of indoles was developed by Shi and Ingleson, independently.7 Following the establishment of C–B bonds, Shi and co-workers further expanded their concept, realizing the hydroxylation of anilines and indoles.8 Ji's group also reported a boron-mediated hydroxylation with 3,4,5-tribromopyrazole as chelating group.9 Due to the nature of electrophilic aromatic substitution (SEAr), the BBr3-mediated transformations primarily depend on the electron-rich arenes (Scheme 1B), such as indoles,7 anilines,7a,10 phenols,11 thiophenols12 and pyrroles.13 Thus, the modification of electron-poor aromatic systems like benzaldehydes are challenging. To address the drawback, the adaption of an imine transient directing group (TDG) in boron-mediated borylation of benzaldehydes was elegantly proposed and illustrated by Rej and Chatani.14 Very recently, Ji and co-workers also demonstrated a borylation of benzophenones with hydrazone as traceless directing group.15 Inspired by Chatani's work, we anticipate the transient directing group strategy will be feasible for forging ortho-C–O bonds on ubiquitous benzaldehyde derivatives. We are thereby interested in combining sequential TDG installation/borylation/hydroxylation/TDG removal in one pot, targeting the straightforward synthesis of structurally significant salicylaldehyde motifs.
Results and discussion
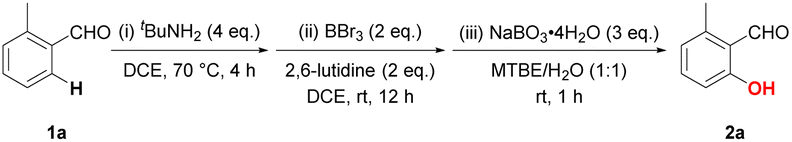
We initiated our investigation with the hydroxylation of 2-methylbenzaldehyde (1a) as the model substrate (Table 1). The reaction was carried out in three steps: (i) 2-methylbenzaldehyde was reacted with excess tBuNH2 (4.0 eq.) in DCE at 70 °C for 4 h for fully conversion to imine; (ii) the borylation was then carried out by the addition of BBr3 (2.0 eq.) and 2,6 lutidine (2.0 eq.) in DCE at room temperature for 12 h; (iii) cosolvent MTBE (methyl t-butyl ether) and H2O (v/v, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was injected followed by the addition of NaBO3·4H2O (3.0 eq.) to furnish the hydroxylation and removal of directing group. Pleasingly, the primary reaction conditions resulted in a total yield of 63% (entry 1). Either less amount of tBuNH2 or elevation of temperature (ii) led to a slightly diminished yield (entries 2 and 3). The overall yield remarkably decreased when the solvent for borylation process (ii) was altered to DCM (entry 4). The effect of base in borylation was also examined as product was hardly formed without base (entry 5). 2,6-Lutidine was recognized as an superior base balancing both borenium species generation and reversible acid/base interaction.16 On the contrary, triethylamine, pyridine and 2,3,5,6-tetramethylpyrazine were totally ineffective while the desired product 2a was detected in 31% yield with 2,4,6-trimethylpyridine (entries 6–9). Neat BBr3 appeared as same effective as its solution in dichloromethane while BCl3 was totally ineffective (entries 10 and 11). We further explored the reaction conditions with BBr3 (2N in DCM) as its more stable under laboratory-bench storage conditions and readily commercially available. A slight increase in BBr3 to 2.5 equivalent promoted the yield to 79% (entry 12). Further adjustments on the stoichiometry showed no benefits (entries 13 and 14). For the oxygenation process (iii), other common oxidants including oxone, hydrogen peroxide and sodium percarbonate failed to give effective yields (entries 15–17). The organic component of cosolvent utilized in step (iii) turned out to be an impressive factor for the transformation. THF was competitive while other ether solvents led to diminished yields (entries 18–20). The desired product 2a was merely obtained by the switch of imine directing group to rigid amantadine (entry 21).
:1) was injected followed by the addition of NaBO3·4H2O (3.0 eq.) to furnish the hydroxylation and removal of directing group. Pleasingly, the primary reaction conditions resulted in a total yield of 63% (entry 1). Either less amount of tBuNH2 or elevation of temperature (ii) led to a slightly diminished yield (entries 2 and 3). The overall yield remarkably decreased when the solvent for borylation process (ii) was altered to DCM (entry 4). The effect of base in borylation was also examined as product was hardly formed without base (entry 5). 2,6-Lutidine was recognized as an superior base balancing both borenium species generation and reversible acid/base interaction.16 On the contrary, triethylamine, pyridine and 2,3,5,6-tetramethylpyrazine were totally ineffective while the desired product 2a was detected in 31% yield with 2,4,6-trimethylpyridine (entries 6–9). Neat BBr3 appeared as same effective as its solution in dichloromethane while BCl3 was totally ineffective (entries 10 and 11). We further explored the reaction conditions with BBr3 (2N in DCM) as its more stable under laboratory-bench storage conditions and readily commercially available. A slight increase in BBr3 to 2.5 equivalent promoted the yield to 79% (entry 12). Further adjustments on the stoichiometry showed no benefits (entries 13 and 14). For the oxygenation process (iii), other common oxidants including oxone, hydrogen peroxide and sodium percarbonate failed to give effective yields (entries 15–17). The organic component of cosolvent utilized in step (iii) turned out to be an impressive factor for the transformation. THF was competitive while other ether solvents led to diminished yields (entries 18–20). The desired product 2a was merely obtained by the switch of imine directing group to rigid amantadine (entry 21).
|
||
|---|---|---|
| Entry | Variation from the “primary conditions” | Yieldb |
| a Primary conditions: (i) 1a (0.4 mmol, 1 eq.) and tBuNH2 (1.6 mmol, 4 eq.) in 1,2-DCE (1.5 mL) at 70 °C for 4 h under argon, then evaporation under reduced pressure to remove excess tBuNH2, water and DCE; (ii) BBr3 (0.4 mL, 2N in DCM, 2.0 eq.) and 2,6-lutidine (0.8 mmol, 2 eq.) in 1,2-DCE (1 mL) at r.t. for 12 h under argon, then evaporation under reduced pressure; (iii) NaBO3·4H2O (1.2 mmol, 3 eq.) in MTBE/H2O (v/v = 1:1, 2 mL) at r.t. under argon for 1 h.b GC yield (internal standard: mesitylene). |
||
| 1 | None | 63 |
| 2 | tBuNH2 (2 eq.) | 46 |
| 3 | 40 °C(II) | 48 |
| 4 | DCM instead of DCE (II) | 25 |
| 5 | Without 2,6-lutidine | 1 |
| 6 | Triethylamine instead of 2,6-lutidine | 0 |
| 7 | 2,4,6-Trimethylpyridine instead of 2,6-lutidine | 31 |
| 8 | Pyridine instead of 2,6-lutidine | 0 |
| 9 | 2,3,5,6-Tetramethylpyrazine instead of 2,6-lutidine | 1 |
| 10 | BBr3 (neat) instead of BBr3 (2N in DCM) | 63 |
| 11 | BCl3 instead of BBr3 | 0 |
| 12 | BBr3 (2.5 eq., 2N in DCM) | 79 |
| 13 | BBr3 (3.0 eq., 2N in DCM) | 74 |
| 14 | 2,6-Lutidine (1.2 eq.), BBr3 (1.5 eq., 2N in DCM) | 23 |
| 15 | Oxone instead of NaBO3·4H2O | 0 |
| 16 | H2O2 instead of NaBO3·4H2O | 2 |
| 17 | 2Na2CO3·3H2O2 instead of NaBO3·4H2O | 11 |
| 18 | 1,4-Dioxane instead of MTBE | 40 |
| 19 | THF instead of MTBE | 55 |
| 20 | Ether instead of MTBE | 31 |
| 21 | 1-AdNH2 instead of tBuNH2 | 1 |
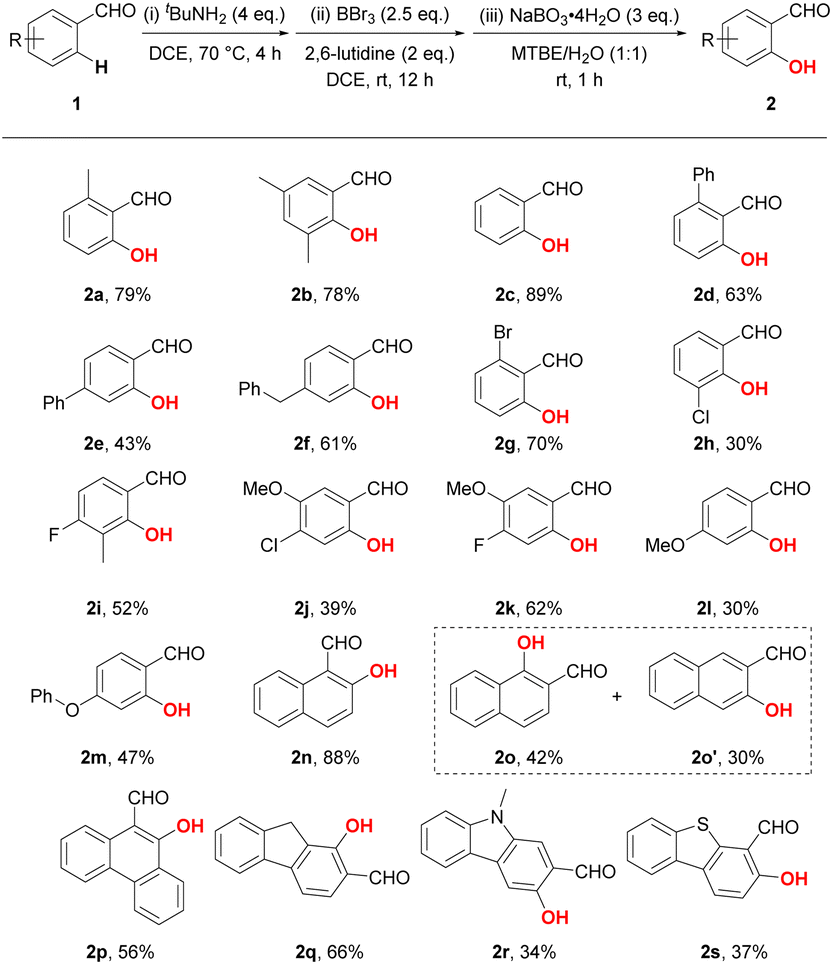
Based on the optimal reaction conditions, we subsequently tested the substrate scope with a number of benzaldehyde derivatives (Table 2). Similar to the standard product 2a, the introduction or deletion of a methyl group on the phenyl ring hardly affects the reaction. Treatment of aldehyde 1b and 1c under standard conditions led to good yields of 78% and 89%, respectively. However, diminished yields were detected when a bulkier phenyl substituent was introduced to either ortho- or para-position, reducing the yields to 63% and 43%, respectively (2d and 2e). The para-benzyl substituted benzaldehyde exhibited better reactivity, as product 2f was obtained in 61% yield. To our delight, electron-withdrawn halogen groups (F, Cl and Br) were compatible with our protocol, exemplified by the successful generation of products 2g–2k, which could also serve as useful synthetic handles for the further elaboration of salicylaldehydes. Due to the intrinsic electrophilicity of boron, the ortho-C–H hydroxylation preferentially react on the presumably electron-richer position (e.g. 1H NMR chemical for 1k: δH-6 = 7.45 ppm; δH-2 = 7.51 ppm).17 Despite the decomposition of substituents caused by BBr3, methoxy and phenyloxy group were also tolerated under the standard conditions with decreased yields (2l and 2m). Moreover, condensed (hetero)aromatic substrates could also undergo the hydroxylation reaction to furnish the desired products in moderate to good yields (2n–2s). Notably, the hydroxylation of 2-naphthaldehyde resulted in a mixture of 1-hydroxylated and 3-hydroxylated products which could be isolated with yields of 42% and 30%, respectively (2o and 2o′). On the other side of the coin, 9H-fluorene-2-carbaldehyde and 9-methyl-9H-carbazole-2-carbaldehyde delivered respectively sole products 2q and 2r, whereas their selectivity is opposite (determined by the HMBC NMR, Fig. S1 and S2†).
| a Reaction conditions: (i) 1 (0.4 mmol, 1 eq.) and tBuNH2 (1.6 mmol, 4 eq.) in 1,2-DCE (1.5 mL) at 70 °C for 4 h under argon, then evaporation under reduced pressure; (ii) BBr3 (0.5 mL, 2N in DCM, 2.5 eq.) and 2,6-lutidine (0.8 mmol, 2 eq.) in 1,2-DCE (1 mL) at r.t. for 12 h under argon, then evaporation under reduced pressure; (iii) NaBO3·4H2O (1.2 mmol, 3 eq.) in MTBE/H2O (1:1, 2 mL) at r.t. under argon for 1 h. |
|---|
 |
To further illustrate the utility of our protocol, 8 mmol synthesis of 2c and 2g were conducted. As shown in Scheme 2, the salicylaldehydes 2c and 2g were isolated in yields of 80% and 70%, respectively, with no starting material left, proving the scale-up robustness of the protocol. The downstream derivation of formyl and phenol group has also been exploited. Valuable bioactive heterocycles including substituted coumarins and benzofuran could be easily prepared from the salicylaldehyde motifs.5a,18 Moreover, the compatible –Br group could be further functionalized, exemplified by alkynylation and arylation. Further applications of the resultant salicylaldehyde in antimicrobials are undergoing in our laboratory.
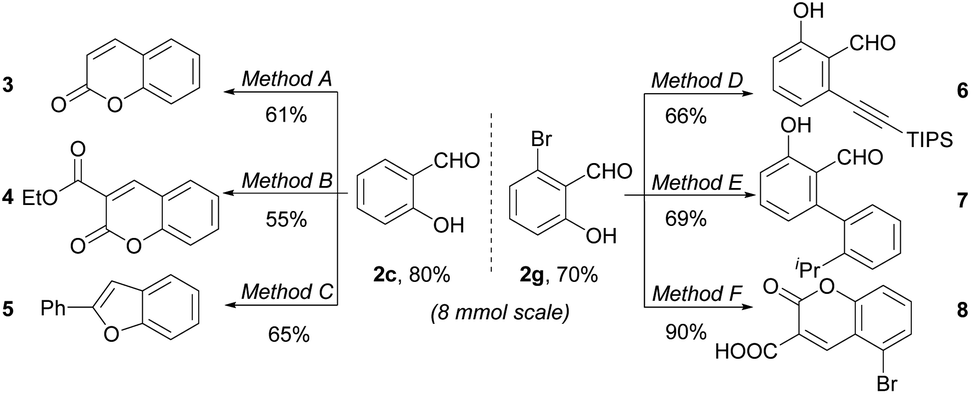 | ||
Scheme 2 Examples for downstream derivation. Reaction conditions: method A: (1) MeI, NaH, THF, DMF, 0 °C, 2 h, then r.t., 3 h; (2) Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCOOEt, DCM, 40 °C, 4 h; (3) BBr3, DCM, 50 °C, 4 h. Method B: ethyl cyanoacetate, FeCl3, EtOH, 80 °C, 24 h. Method C: (1) Ph3PCBr2, DCM, 0 °C, 0.5 h, then r.t., 2 h; (2) PhB(OH)2, Pd(OAc)2(5 mol%), CuI (5 mol%), PPh3, K3PO4, 1,4-dioxane, 100 °C, 24 h. Method D: Pd(dppf)Cl2 (10 mol%), (triisopropylsilyl)acetylene, CuI (20 mol%), Et3N, 90 °C, overnight. Method E: 2-isopropylphenylboronic acid, Pd(PPh3)4 (3 mol%), Na2CO3, H2O/MeOH/DME, 80 °C, overnight. Method F: (1) 2,2-dimethyl-1,3-dioxane-4,6-dione, ammonium acetate, H2O, r.t. 3 h; (2) HCl, pH = 4.5. CHCOOEt, DCM, 40 °C, 4 h; (3) BBr3, DCM, 50 °C, 4 h. Method B: ethyl cyanoacetate, FeCl3, EtOH, 80 °C, 24 h. Method C: (1) Ph3PCBr2, DCM, 0 °C, 0.5 h, then r.t., 2 h; (2) PhB(OH)2, Pd(OAc)2(5 mol%), CuI (5 mol%), PPh3, K3PO4, 1,4-dioxane, 100 °C, 24 h. Method D: Pd(dppf)Cl2 (10 mol%), (triisopropylsilyl)acetylene, CuI (20 mol%), Et3N, 90 °C, overnight. Method E: 2-isopropylphenylboronic acid, Pd(PPh3)4 (3 mol%), Na2CO3, H2O/MeOH/DME, 80 °C, overnight. Method F: (1) 2,2-dimethyl-1,3-dioxane-4,6-dione, ammonium acetate, H2O, r.t. 3 h; (2) HCl, pH = 4.5. | ||
Experimental
General procedure for the ortho-C–H hydroxylation
(i) An oven-dried Schlenk tube equipped with a stirring bar was charged with benzaldehyde 1 (0.4 mmol, 1.0 equiv.), tBuNH2 (168 μL, 4.0 equiv.) under argon. Then solvent DCE (1 mL) was added at room temperature. The reaction mixture was placed in a preheated oil bath at 70 °C, and stirred for 4 h.(ii) Next, tBuNH2 and solvent was removed under reduced pressure. 2,6-Lutidine (85.7 mg, 2.0 equiv.), DCE (1 mL) and BBr3 (0.5 mL, 2N in DCM, 2.5 equiv.) were added under argon. The reaction mixture was stirred at r.t. for 12 h.
(iii) Then, solvent was removed under reduced pressure, followed by the addition of MTBE (1 mL), sodium perborate tetrahydrate (185 mg, 1.2 mmol, 3.0 equiv.) and water (1 mL) under argon. The reaction mixture was stirred at r.t. for another 1 h. Afterwards, the reaction mixture was extracted with ethyl acetate (10 mL × 3). The combined organic layer was further purified by column chromatography on silica gel (200–300 mesh) to afford the corresponding product 2.
Conclusions
In conclusion, we have developed a novel one-pot procedure for the rapid synthesis of salicylaldehydes. The BBr3-mediated ortho-C–H hydroxylation assisted by a transient imine directing group features high regioselectivity, mild reaction conditions and good compatibility. By patiently tuning the reaction conditions, TDG installation/borylation/hydroxylation/TDG removal, are combined in one pot, omitting isolation of intermediates. The strategy rapidly assembles structurally diverse salicylaldehydes, which are capable to serve as key intermediates in the synthetic and medicinal chemistry.Data availability
A data availability statement (DAS) is required to be submitted alongside all articles. Please read our full guidance on data availability statements (https://www.rsc.org/journals-books-databases/author-and-reviewer-hub/authors-information/prepare-and-format/data-sharing/) for more details and examples of suitable statements you can use.Author contributions
BF conceived the project; RW and BF performed the research; XF contributed to the chemical characterization/data analysis; RW and BF wrote the final manuscript; BF and YC revised the manuscript and supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (No. 22207047, 32070360), the Natural Science Foundation of Jiangsu Province (BK20220749) and the Open fund of Jiangsu Key Laboratory for the Research and Utilization of Plant Resources (JSPKLB202212).References
- R. E. Kirk, D. F. Othmer, M. Grayson and D. Eckroth, Kirk-Othmer Encyclopedia of Chemical Technology, Wiley, New York, 4th edn, 2004 Search PubMed.
- (a) M. M. Heravi, V. Zadsirjan, M. Mollaiye, M. Heydari and A. T. K. Koshvandi, Russ. Chem. Rev., 2018, 87, 553–585 CrossRef CAS; (b) Y. J. Zhuang, J. P. Qu and Y. B. Kang, J. Org. Chem., 2020, 85, 4386–4397 CrossRef CAS PubMed.
- (a) W. Kantlehner, Eur. J. Org Chem., 2003, 2003, 2530–2546 CrossRef; (b) M. E. Jung and T. I. Lazarova, J. Org. Chem., 1997, 62, 1553–1555 CrossRef CAS; (c) T. V. Hansen and L. Skattebøl, Org. Synth., 2012, 89, 220–229 CrossRef CAS; (d) X.-w. Fu, W.-c. Pu, G.-l. Zhang and C. Wang, Res. Chem. Intermed., 2014, 41, 8147–8158 CrossRef.
- Z. Iqbal, A. Joshi and S. Ranjan De, Adv. Synth. Catal., 2020, 362, 5301–5351 CrossRef CAS.
- (a) F. Yang, K. Rauch, K. Kettelhoit and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 11285–11288 CrossRef CAS PubMed; (b) X. Y. Chen, S. Ozturk and E. J. Sorensen, Org. Lett., 2017, 19, 6280–6283 CrossRef CAS PubMed; (c) R. Trammell, L. D'Amore, A. Cordova, P. Polunin, N. Xie, M. A. Siegler, P. Belanzoni, M. Swart and I. Garcia-Bosch, Inorg. Chem., 2019, 58, 7584–7592 CrossRef CAS PubMed.
- R. L. Letsinger and D. B. MacLean, J. Am. Chem. Soc., 1963, 85, 2230–2236 CrossRef CAS.
- (a) J. Lv, X. Chen, X. S. Xue, B. Zhao, Y. Liang, M. Wang, L. Jin, Y. Yuan, Y. Han, Y. Zhao, Y. Lu, J. Zhao, W. Y. Sun, K. N. Houk and Z. Shi, Nature, 2019, 575, 336–340 CrossRef CAS PubMed; (b) S. A. Iqbal, J. Cid, R. J. Procter, M. Uzelac, K. Yuan and M. J. Ingleson, Angew. Chem., Int. Ed., 2019, 58, 15381–15385 CrossRef CAS PubMed.
- J. Lv, B. Zhao, Y. Yuan, Y. Han and Z. Shi, Nat. Commun., 2020, 11, 1316 CrossRef CAS PubMed.
- G. Wu, Z. Yang, X. Xu, L. Hao, L. Chen, Y. Wang and Y. Ji, Org. Lett., 2022, 24, 3570–3575 CrossRef CAS PubMed.
- (a) G. Wu, X. Fu, Y. Wang, K. Deng, L. Zhang, T. Ma and Y. Ji, Org. Lett., 2020, 22, 7003–7007 CrossRef CAS PubMed; (b) S. Rej, A. Das and N. Chatani, Chem. Sci., 2021, 12, 11447–11454 RSC; (c) G. Wu, X. Xu, S. Wang, L. Chen, B. Pang, T. Ma and Y. Ji, Chin. Chem. Lett., 2022, 33, 2005–2008 CrossRef CAS.
- (a) L. Niu, H. Yang, R. Wang and H. Fu, Org. Lett., 2012, 14, 2618–2621 CrossRef CAS PubMed; (b) O. Sadek, A. Le Gac, N. Hidalgo, S. Mallet-Ladeira, K. Miqueu, G. Bouhadir and D. Bourissou, Angew. Chem., Int. Ed., 2022, 61, e202110102 CrossRef CAS PubMed.
- (a) G. Wu, B. Pang, Y. Wang, L. Yan, L. Chen, T. Ma and Y. Ji, J. Org. Chem., 2021, 86, 5933–5942 CrossRef CAS PubMed; (b) X. Xu, G. Wu, Z. Yang, X. Liu, L. Hao, Y. Wang, Z. Ma and Y. Ji, Org. Lett., 2022, 24, 7163–7167 CrossRef CAS PubMed.
- Z. J. Wang, X. Chen, L. Wu, J. J. Wong, Y. Liang, Y. Zhao, K. N. Houk and Z. Shi, Angew. Chem., Int. Ed., 2021, 60, 8500–8504 CrossRef CAS PubMed.
- S. Rej and N. Chatani, J. Am. Chem. Soc., 2021, 143, 2920–2929 CrossRef CAS PubMed.
- Z. Yang, L. Hao, X. Xu, Y. Wang, G. Wu and Y. Ji, Org. Lett., 2023, 25, 5875–5879 CrossRef CAS PubMed.
- K. Yamazaki, S. Rej, Y. Ano and N. Chatani, Org. Lett., 2021, 24, 213–217 CrossRef PubMed.
- F. Claudi, M. Cardellini, G. M. Cingolani, A. Piergentili, G. Peruzzi and W. Balduini, J. Med. Chem., 1990, 33, 2408–2412 CrossRef CAS PubMed.
- X. He, Z. Yan, X. Hu, Y. Zuo, C. Jiang, L. Jin and Y. Shang, Synth. Commun., 2014, 44, 1507–1514 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra02994a |
| This journal is © The Royal Society of Chemistry 2024 |