
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExpanded graphite with boron-doping for cathode materials of high-capacity and stable aluminum ion batteries†
Ying Yang *ab,
Ruirui Zhaob and
Yong P. Chenacde
*ab,
Ruirui Zhaob and
Yong P. Chenacde
aDepartment of Engineering Science, Faculty of Innovation Engineering, Macau University of Science and Technology, Av. Wai Long, Macao SAR, 999078, China. E-mail: yangying632882@126.com
bInstitute of New Energy for Vehicles, School of Materials Science and Engineering, Tongji University, Shanghai, 201804, China
cAdvanced Institute for Materials Research (WPI-AIMR), Tohoku University, Sendai 980-8577, Japan
dDepartment of Physics and Astronomy, Elmore Family School of Electrical and Computer Engineering, Birck Nanotechnology Center, Purdue Quantum Science and Engineering Institute, Purdue University, West Lafayette, IN 47907, USA
eInstitute of Physics and Astronomy, Villum Center for Hybrid, Quantum Materials and Devices, Aarhus University, Aarhus-C 8000, Denmark
First published on 31st July 2024
Abstract
Recently, aluminum ion batteries (AIBs) have attracted more attention due to the reliable, cost-effective, and air-stable Al metal anode. Among various cathode materials of AIBs, graphite was paid more attention owing to its high-voltage plateau and stable properties in storing chloroaluminate anions (AlCl4−). However, its low capacity limits the real application and can not satisfy the requirements of modern society. To solve the above issue, herein, boron (B)-doping expanded graphite (B-EG) was prepared by thermal treatment of expanded graphite and boric acid together in a reduction atmosphere. Based on the structural and electrochemical characterization, the results show that B-doping amplifies the interlayer space of expanded graphite (EG), introduces more mesoporous structures, and induces electron deficiency, which is beneficial to accelerating the transfer and adsorption of active ions. The results indicate that the B-EG electrode exhibits excellent rate capability and a high specific capacity of 84.9 mA h g−1 at 500 mA g−1. Compared with the EG electrode, B-EG shows better cycle stability with the specific capacity of 87.7 mA h g−1 after 300 cycles, which could be attributed to lower pulverization and higher pseudo-capacitance contribution of B-EG after the introduction of B species.
1. Introduction
With the increasing demand for high-energy-density and safe secondary batteries driven by portable electronic devices, electric vehicles, and small-scale energy storage stations, alternative multivalent metal ion batteries, such as magnesium, zinc, and aluminium ion batteries, have garnered significant attention and research beyond lithium-ion batteries.1–4 Compared to other secondary batteries, aluminium ion batteries have been widely studied in recent years due to their abundant resources, low cost, environmental stability, and the capability to transfer three electrons of aluminium metal.5,6 Since 2015, H. J. Dai et al. utilized an ionic liquid as the electrolyte, graphite as the cathode material, and aluminium metal as the anode. The graphite electrode in this battery system demonstrated a reversible specific capacity of 70 mA h g−1 at a current density of 66 mA g−1.7 Subsequently, aluminium ion batteries with ionic liquid electrolytes have received widespread attention.To enhance the energy density of aluminium ion batteries in this system, researchers have primarily focused on searching cathode materials with high voltage platforms, high reversible capacities, and good cycling stability. In recent years, various materials including metal sulfides (MoS2,8 Co3S4 (ref. 9)), metal oxides (Co3O4,10 SnO2,11 TiO2 (ref. 12)), metal phosphides and phosphates (Cu3P,13 Co3PO4 (ref. 14)), conductive polymers (PANI),15 carbon materials (carbon paper),16 and graphite-based materials17,18 have been widely investigated as cathode materials for aluminium ion batteries. Among these materials, graphite-based materials have been extensively studied due to their highest voltage plateau around 2 V vs. Al/Al3+ and stable cycling performance. However, the relatively low specific capacity of graphite limits its commercial applications. To improve the specific capacity of graphite, researchers have mainly focused on constructing special morphologies with high surface area and introducing multiple defects and nanoscale voids. For instance, Zhang et al. synthesized polythiophene/graphite composites, which have a lamellar structure with a large surface to accommodate chloroaluminate anions (AlCl4−).19 At a current density of 1000 mA g−1, its specific capacity reached 113 mA h g−1. Additionally, Lee et al. prepared acid-treated expanded graphite (AEG) and base-etched graphite (BEG), which have turbostratically ordered and disordered structures, largely expanded d-spacing, and a high density of defective sites. At a current density of 4 A g−1, AEG exhibits a specific capacity of 88.6 mA h g−1 and BEG delivers a high specific capacity of 110 mA h g−1.20
Additionally, studies have shown that heteroatom doping (such as N,21 S,22 and P23) can expand the interlayer spacing of graphite-based materials, alter local electron distribution, and induce certain defects, thereby facilitating the storage of sodium ions, potassium ions, and others. In the current aluminium ion battery system, it has been demonstrated that the storage mechanism of Al/graphite cell during charging and discharging can be written as:7
| 4Al2Cl7− + 3e− → Al + 7AlCl4− | (1) |
| Cn + AlCl4− → Cn[AlCl4] + e− | (2) |
The AlCl4− ions are intercalated and deintercalated between the interlayer space of graphite during the charging and discharging process. As the transported ions are negatively charged chloroaluminate anions (AlCl4−), it is thus ideal to create some active sites with empty orbital to bind AlCl4− ions and accelerate their diffusion.
Herein, we demonstrated that two-dimensional (2D), boron-doping expanded graphite (B-EG), features as a novel cathode material that can promote intercalation of AlCl4−. B-EG was prepared by simple heat treatment of expanded graphite using boric acid as the boron source in a reductive inert atmosphere, aiming to modify the local electron configuration and enlarge the interlayer distance of expanded graphite. As boron is an important doping element that induces electron deficiency in EG, it could accelerate the diffusion of active ions and improve the adsorption of active ions, resulting in a lower pulverization, an improved specific capacity, and higher capacitance contribution.
2. Experimental sections
2.1 Preparation of B-doped expanded graphite
The expanded graphite was purchased from Jiangsu Xianfeng Nanomaterials Technology Co., Ltd, while boric acid was obtained from Shanghai Aladdin Biochemical Technology Co., Ltd. The reagents were used without any pre-treatment before use.0.1 g of expanded graphite (EG) and 1 g of boric acid (H3BO3) were separately weighed and ground evenly in a mortar following a mass ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10. Subsequently, 10 ml of deionized water was added, and the mixture was sonicated for 30 minutes until thoroughly mixed. The resulting mixture was then freeze-dried. After freeze-drying, as illustrated in Fig. 1, the mixture of EG and H3BO3 was placed in the center of a tube furnace and purged with a hydrogen–argon mixture. The temperature was ramped up to 800 °C at a rate of 2 °C min−1 and maintained for 3 hours at this temperature. Upon cooling to room temperature, the product was removed and immersed in 20 ml of deionized water, followed by sonication for 20 minutes. Subsequently, vacuum filtration was conducted, followed by washing with deionized water and ethanol. Finally, the filtered product was vacuum-dried at 60 °C for 12 hours to obtain boron-doped expanded graphite (B-EG).
:10. Subsequently, 10 ml of deionized water was added, and the mixture was sonicated for 30 minutes until thoroughly mixed. The resulting mixture was then freeze-dried. After freeze-drying, as illustrated in Fig. 1, the mixture of EG and H3BO3 was placed in the center of a tube furnace and purged with a hydrogen–argon mixture. The temperature was ramped up to 800 °C at a rate of 2 °C min−1 and maintained for 3 hours at this temperature. Upon cooling to room temperature, the product was removed and immersed in 20 ml of deionized water, followed by sonication for 20 minutes. Subsequently, vacuum filtration was conducted, followed by washing with deionized water and ethanol. Finally, the filtered product was vacuum-dried at 60 °C for 12 hours to obtain boron-doped expanded graphite (B-EG).
 | ||
| Fig. 1 The schematic illustration of the synthetic process for B-EG. | ||
2.2 Material characterization
The morphology of EG and B-EG was measured by Scanning electron microscopy (SEM) of Quanta 200 FEG. The crystal structure of prepared samples was characterized using the Panalytical X'Pert X-ray powder diffractometer from PANalytical B.V. in the range of 2θ = 20–60° with Cu Kα radiation (V = 40 kV, I = 40 mA, and λ = 1.5418 Å). The infrared spectral data was collected from the Bruker-Tenson 27 Fourier-transform infrared spectrometer from Bruker Optik GmbH. The microstructure evolution between EG and B-EG was measured by the Thermo Fisher Scientific-DXR Raman spectrometer with an excitation source of 532 nm. The Thermo Fisher X-ray photoelectron spectrometer was used to characterize the valence information of B and C. The pore structure and specific surface area of the samples were obtained using the JW-BK122W nitrogen physical adsorption instrument from Beijing Micromeritics Instrument Corporation. The thermal decomposition stability was investigated using the TA-Q50 thermogravimetric analyzer from TA Instruments Inc., USA from room temperature to 800 °C.2.3 Electrochemical measurements
The prepared B-EG and EG were separately mixed with the conductive additives of acetylene black and a binder of polytetrafluoroethylene (PTFE) in a mortar at a mass ratio of 70:20:10. A certain amount of ethanol was then added to form a slurry, which was coated onto the surface of molybdenum foil with a diameter of 8 mm. The coated electrodes were dried in a vacuum oven at 60 °C for 12 hours. The aluminum ions batteries were assembled in an argon-filled glove box (Mikrouna, H2O <0.1 ppm, O2 < 0.1 ppm), the prepared EG-related electrode was served as the cathode, aluminum foil was used as the anode, and glass fiber was used as the separator. An ionic liquid electrolyte, composed of 1-ethyl-3-methylimidazolium chloride ([EMIm]Cl) and aluminum chloride (AlCl3) salt with a molar ratio of 1:1.1, was used. Electrochemical tests including rate performance, charge–discharge cycling, and cycling stability were conducted using the Neware battery testing system within the voltage range of 0.5 V to 2.45 V vs. Al/Al3+ at room temperature. Before the electrochemical performance testing, the assembled batteries were rested for 12 hours at room temperature. Additionally, cyclic voltammetry (CV) measurements and Electrochemical Impedance Spectroscopy (EIS) were performed using an electrochemical workstation (EC-lab).
The AlCl4− ion diffusion coefficient, Dions, can be calculated from the Warburg region of EIS results using eqn (3):24
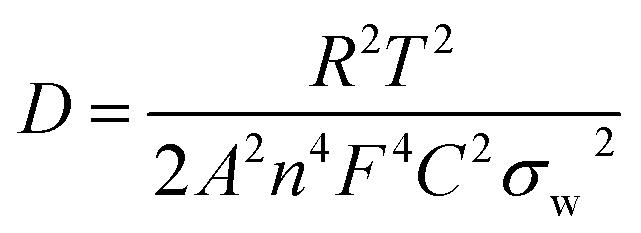 | (3) |
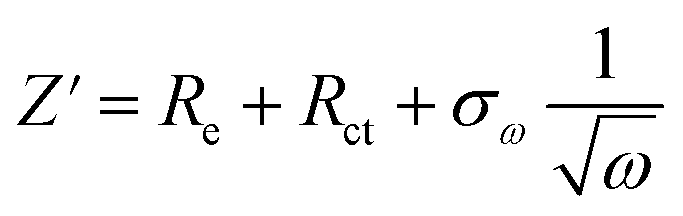 | (4) |
The ions diffusion coefficients (Dions, cm2 s−1) of all the cells are calculated using eqn (3) and (4).
3. Results and discussion
To confirm the morphology evolution after B species introduction, SEM images of EG and B-EG were collected (Fig. S1a and b†). The resulting data show that the EG presents a very thin and continuous folding nanosheet, however, after the introduction of B species into EG, the B-EG becomes small flat nanosheets and appears to accumulate. Furthermore, the related TEM and EDS results were collected and presented in Fig. 2. As shown in Fig. 2a and c, both EG and B-EG exhibit very thin and continuous folding nanosheets. However, after the introduction of B species into EG, the B-EG sample shows relatively fewer wrinkles, which is consistent with the SEM results.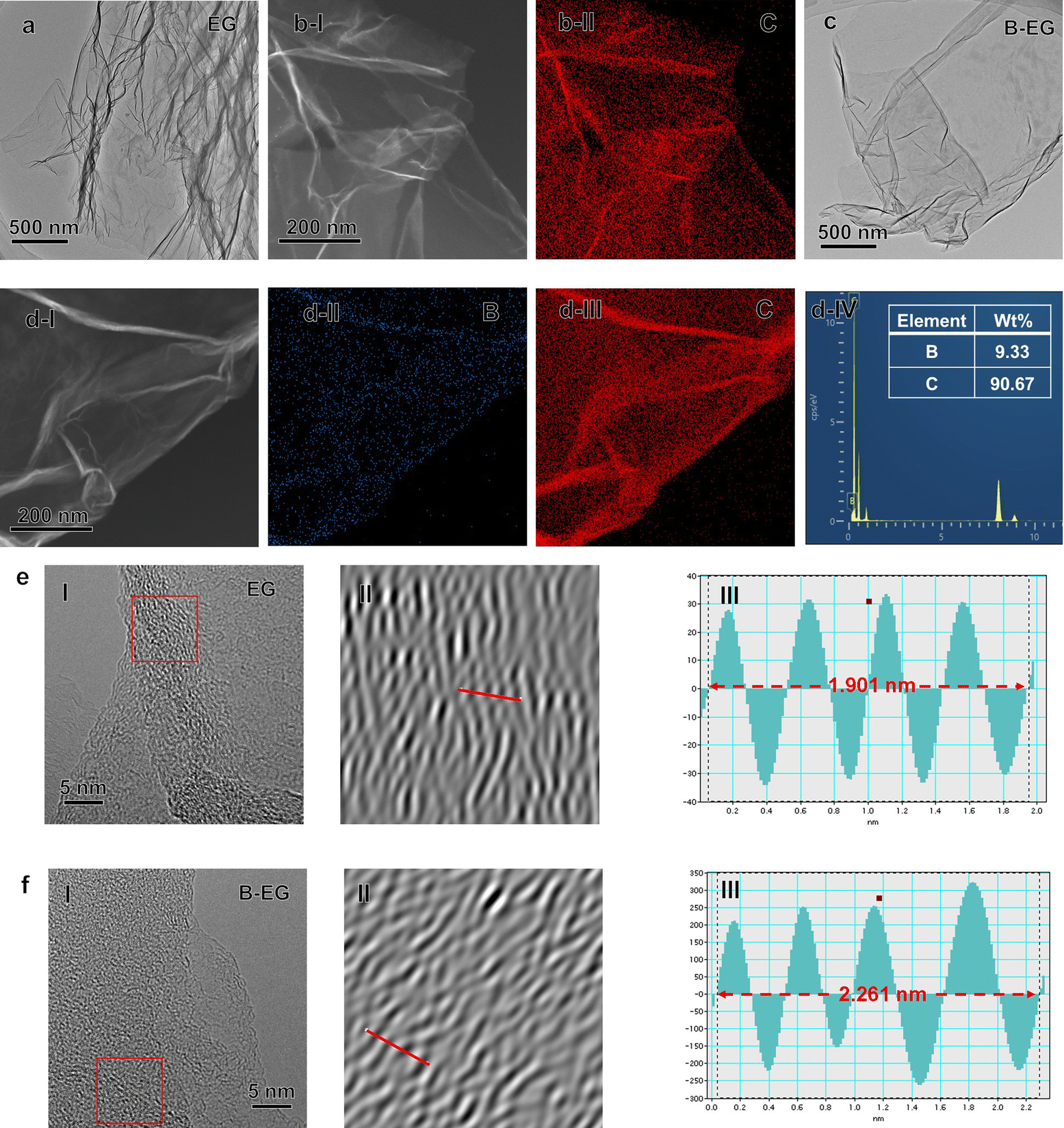 | ||
| Fig. 2 (a) TEM image of EG sample. (b) The EDS result of EG sample. (c) TEM image of B-EG sample. (d) The EDS result of B-EG sample. The related high-resolution TEM (HRTEM) results of (e) EG and (f) B-EG samples. (I) The HRTEM images, (II) the Inverse fast Fourier transform (IFFT) images of the red square areas in (I), and (III) the intensity profiles of the red lines scan across the lattice fringes. | ||
Additionally, the EDS results indicate that EG contains only C (Fig. 2b), while after B doping, B-EG shows the presence of B with content of approximately 9.33 wt% (Fig. 2d). The high-resolution TEM (HRTEM) images of the EG and B-EG samples (Fig. 2e and f) exhibit a turbulent structure with localized short-range ordered crystals. From the corresponding inverse fast Fourier transform (IFFT) profiles, it is observed that the interlayer distance of B-EG increases to 0.56 nm compared to 0.48 nm in EG, which is consistent with the following XRD results.
Fig. 3 depicts the X-ray diffraction (XRD) patterns and Fourier-transform infrared (FTIR) spectra of EG and B-EG. As shown in Fig. 3a, the boron doping process did not introduce boron oxide impurities and characteristic diffraction peaks of 2H-graphite (PDF No. 41-1487) were observed in both the XRD spectra of B-EG and EG, indicating no change in the crystal structure and composition after doping. Fig. 3b shows an enlarged view of the diffraction peaks within the green box in Fig. 3a. It can be observed that after boron doping, the diffraction peak of B-EG at 26.5° exhibited a slight shift towards the left compared to that of EG. According to the Bragg equation,26 this suggests an increase in the interlayer spacing after doping. Fig. 3c illustrates the FTIR spectra of EG and B-EG. It can be noted a highly intense band in the spectrum of EG and B-EG at 3447 cm−1 was due to the O–H.
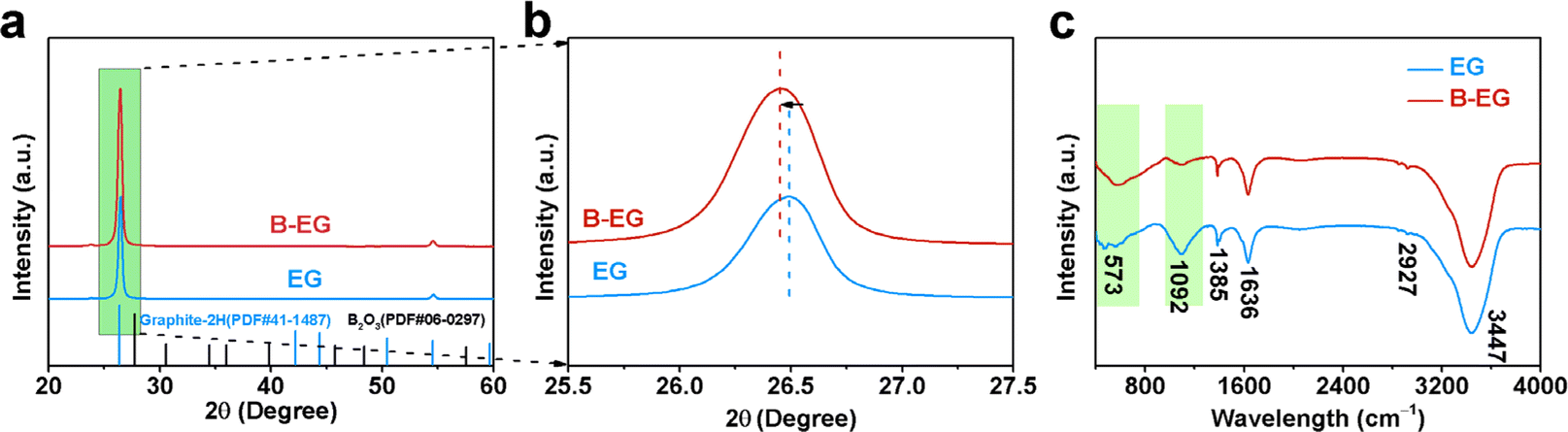 | ||
| Fig. 3 The microstructure evolution of EG and B-EG. (a) XRD patterns; (b) the enlarged XRD patterns at 26.5°; (c) FTIR spectra. | ||
Fig. 3 depicts the X-ray diffraction (XRD) patterns and Fourier-transform infrared (FTIR) spectra of EG and B-EG. As shown in Fig. 3a, the boron doping process did not introduce boron oxide impurities, and characteristic diffraction peaks of 2H-graphite (PDF No. 41-1487) were observed in both the XRD spectra of B-EG and EG, indicating no change in the crystal structure and composition after doping. Fig. 3b shows an enlarged view of the diffraction peaks within the green box in Fig. 3a. It can be observed that after boron doping, the diffraction peak of B-EG at 26.5° exhibited a slight shift towards the left compared to that of EG. According to the Bragg equation,26 this suggests an increase in the interlayer spacing after doping. Fig. 3c illustrates the FTIR spectra of EG and B-EG. It can be noted a highly intense band in the spectrum of EG and B-EG at 3447 cm−1 was due to the O–H stretching vibration indicating the presence of OH and/or COOH functional groups within the structure. The very weak band at 2927 cm−1 was due to the asymmetric stretching vibration of the C–H bond. C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibration of the graphitic domain (1636 cm−1), C–O stretching of epoxy groups (1385 cm−1), and C–O stretching vibration of the alkoxy group (1092 cm−1) were also observed.27 These spectral changes indicate the presence of carbonyl, ether, and hydroxyl groups in the chemical structure of EG and B-EG, implying that part of graphene oxide has been produced. After treating the EG with H3BO3, the peaks, at 1092 cm−1 in B-EG indexed to C–O stretching vibration, exhibited slight blue shifts and intensity reductions, which suggested the EG was reduced and doped by B.
C stretching vibration of the graphitic domain (1636 cm−1), C–O stretching of epoxy groups (1385 cm−1), and C–O stretching vibration of the alkoxy group (1092 cm−1) were also observed.27 These spectral changes indicate the presence of carbonyl, ether, and hydroxyl groups in the chemical structure of EG and B-EG, implying that part of graphene oxide has been produced. After treating the EG with H3BO3, the peaks, at 1092 cm−1 in B-EG indexed to C–O stretching vibration, exhibited slight blue shifts and intensity reductions, which suggested the EG was reduced and doped by B.
To further investigate the microstructure of prepared materials, Raman spectroscopy and X-ray photoelectron spectroscopy (XPS) were performed for characterization. Fig. 4a illustrates the Raman spectra of EG and B-EG. It could be seen that three peaks were observed, namely the D peak (1355.8 cm−1), G peak (1582.6 cm−1), and 2D peak (2716.8 cm−1), which can be attributed to defect-related peaks, graphite structure peaks, and a characteristic peak of multi-layered graphite structures.28 The intensity ratio (IG/ID) of the G and D peaks can be used to characterize the graphitization degree of the materials, the IG/ID ratio of EG and B-EG was 12.5 and 2.3, respectively, indicating the presence of more defects in B-EG after boron doping, resulting in a decrease in the graphitization degree. For further investigation into the surface composition and structural changes after boron doping, the XPS refine spectra of B1s and C1s, as shown in Fig. 4b and c respectively. After doping, the B1s peak appears in B-EG and is absent in EG (Fig. 4b). Additionally, compared to the refined XPS peak of C1s in EG, the B-EG sample shows a higher content of sp3-hybridized carbon, indicating successful doping of B into EG (Fig. 4c).
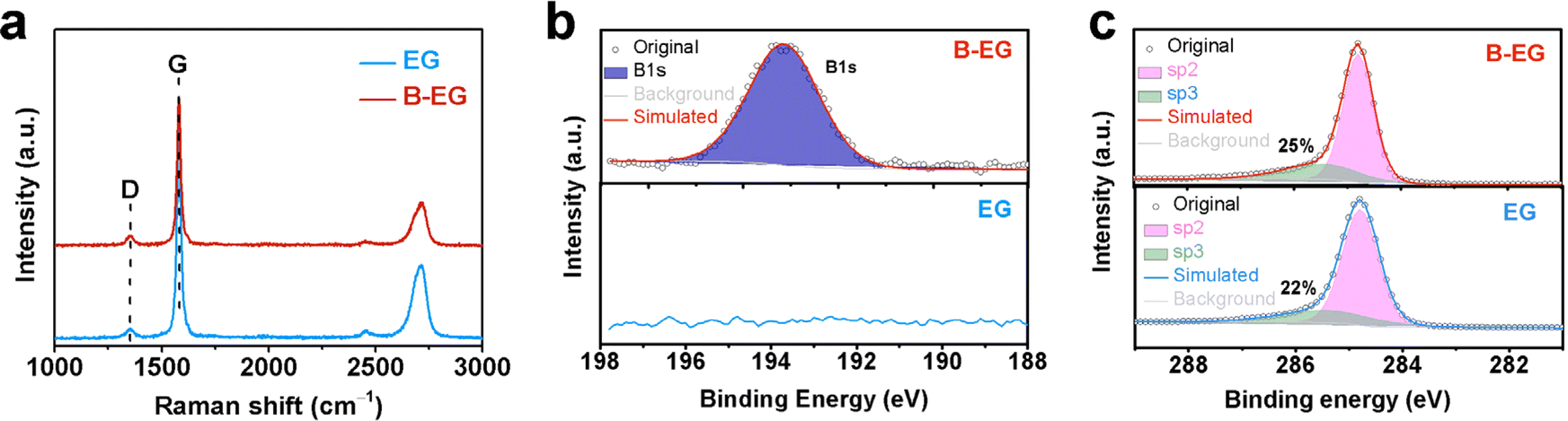 | ||
| Fig. 4 The microstructure of EG and B-EG: (a) Raman spectra; (b) the refined XPS of B1s; and (c) the refined XPS of C1s. | ||
To investigate the influence of boron doping on the pore structure, specific area, and thermal stability of the materials, nitrogen adsorption–desorption characterization and thermal analysis were conducted. Fig. S2a and b† show the nitrogen adsorption/desorption isotherms and corresponding Barrett–Joyner–Halenda (BJH) pore size distribution plots of EG and B-EG, respectively. As illustrated in Fig. S2a,† based on the curve shape and according to the IUPAC classification,29 the isotherm belongs to the typical type III adsorption/desorption isotherm with an H3 hysteresis loop, indicating the presence of abundant mesopores within the sample in the relative pressure range of 0.4 to 0.8. The specific BET surface areas of EG and B-EG were calculated from the isotherms as 41.4 and 19.8 m2 g−1, respectively, which may be attributed to boron doping.30 Fig. S2b† presents the corresponding BJH pore size distribution curves of the materials, indicating that the pore size distributions of EG and B-EG mainly reside in the mesoporous region, with an increase in mesopores observed after boron doping. Fig. S3† displays the thermal decomposition curves of EG and B-EG, revealing an enhancement in thermal stability after boron doping.
To investigate the effect of boron doping on the electrochemical properties of expanded graphite, Fig. 5 compares the aluminium storage performance of EG and B-EG electrodes. Fig. 5a illustrates the rate performance of EG and B-EG. It could be seen that the specific capacity of B-EG reaches 81.6 mA h g−1 at a current density of 500 mA g−1, which is higher than that of EG (61.3 mA h g−1). With current densities increasing from 1000, 1200, 1500, 2000, to 4000 mA g−1, the specific capacities of B-EG are 65.4, 54.5, 50.3, 44.6, and 22.6 mA h g−1, respectively. Upon returning to a current density of 500 mA g−1, its specific capacity again rises to 84.9 mA h g−1, demonstrating excellent rate performance. To investigate the effect of boron doping on the cycling stability of expanded graphite, Fig. 5b depicts the cycling performance of EG and B-EG electrodes at a current density of 500 mA g−1. As shown in Fig. 5b, compared to that of EG electrode, the specific capacity of B-EG electrode remains stable during cycling without attenuation. After 300 cycles, its specific capacity remains at 87.7 mA h g−1, with an initial capacity increase in the first 30 cycles, possibly due to complete electrolyte infiltration leading to electrode activation. Moreover, after the first CV curve of EG and B-EG, the second and three CV curves are overlapped (Fig. S4†), also indicating the good electrochemical stability of EG-based materials. To further illustrate the effect of B doping on the aluminium storage process of expanded graphite, Fig. 5c compares the second cycle CV curves of EG and B-EG at a scan rate of 0.2 mV s−1 within the potential window of 0.5 to 2.45 V (vs. Al/Al3+). Generally, the intercalation of aluminum into graphite occurs through the insertion of AlCl4− ions between graphite layers to form graphite intercalation compounds.7 As shown in Fig. 5c, after B doping, there are no significant changes in the number and position of peaks in the CV of B-EG, which remain similar to graphite. Fig. 5d shows the charge–discharge curves of EG and B-EG electrodes during the second cycle at a current density of 500 mA g−1. The shapes of the charge–discharge curves for EG and B-EG are similar, indicating that B doping did not alter the reaction mechanism.7,31,32 However, after B doping, the electrode polarization of B-EG (0.26 V) is smaller than that of EG (0.33 V), and the reversible specific capacity of B-EG is larger than that of EG. Moreover, the EIS was collected and used to estimate the ions diffusion coefficient (Dions) as shown in Fig. S5.† The impedance plots shown in Fig. S5a† can be divided into a high-frequency component (partial semicircle) and a low-frequency component (straight slopping line along the imaginary axis). The semicircle in the high-frequency range represents charge-transfer resistance, which is directly related to the electroactive surface area and electrical conductivity of the electrode materials. It could be seen that the B-EG shows a smaller radius compared to that of EG sample. In addition, the low-frequency part is related to the ions diffusion as shown in Fig. S5b.† Thus, the estimated Dions of B-EG is about 2.46 × 10−18, which is higher than that of EG (1.42 × 10−18). These results suggest B doping can reduce the internal resistance of the electrode and promote the diffusion of ions and electrons.
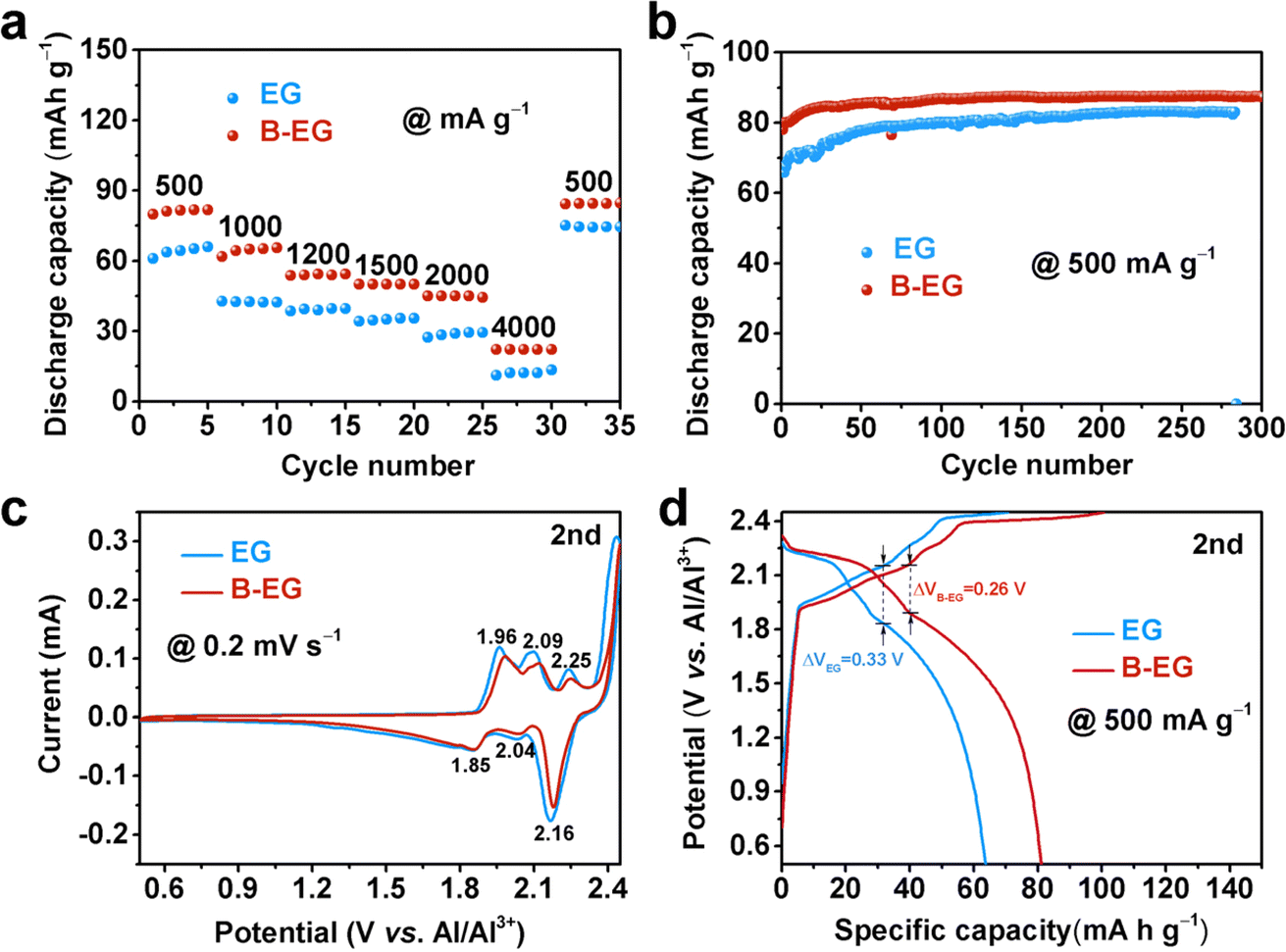 | ||
| Fig. 5 The electrochemical properties of EG and B-EG: (a) rate capability, (b) cycle stability, (c) CV curves of the second cycle at a scan rate of 0.2 mV s−1, and (d) charge–discharge curves of the second cycle at a current density of 500 mA g−1. | ||
Since the electrochemical performance of materials is related to the kinetics of the electrochemical energy storage process, the reasons for the enhancement of the electrochemical performance of EG electrodes due to boron doping were further investigated. Regularly, in cyclic voltammetry (CV) curves, the total current response at different scan rates is composed of the relevant current for the slow diffusion-controlled process (idiff) and the current required for rapid surface oxidation–reduction reactions and double-layer formation (icap), expressed by the following formula:33,34
| i(v) = icap + idiff = k1v + k2v1/2 = avb | (5) |
|
logi(v) = loga + blogv
| (6) |
i versus logν, i is the response current in the CV curve, and ν is the scan rate. By calculating the b value, the energy storage process can be estimated to be diffusion-controlled or capacitance-contributed. Fig. 6a and b show the CV curves of EG and B-EG electrodes at different scan rates, and by fitting them with eqn (6), the b values at different voltages in Fig. 6c and d are obtained. As shown in the inset, the b value is greater than 0.8, indicating the energy storage capability of EG and B-EG could be contributed by both capacitance and the slow semi-infinite diffusion-controlled faradaic process in the bulk phase. The k1 is calculated using eqn (5), which helps to obtain the value of capacitance contribution k1v at different scan rates and determine the capacitance contribution during aluminium storage. As shown in Fig. 6e, the capacitance contribution of B-EG is greater than that of EG at different scan rates. At a scan rate of 1.0 mV s−1, the capacitance contribution percentage of the B-EG electrode is 87%, whereas, under the same conditions, the capacitance contribution percentage of EG electrode is 84%. This indicates that boron doping enlarges the interlayer spacing, promotes ion diffusion, and thereby enhances the electrochemical performance.
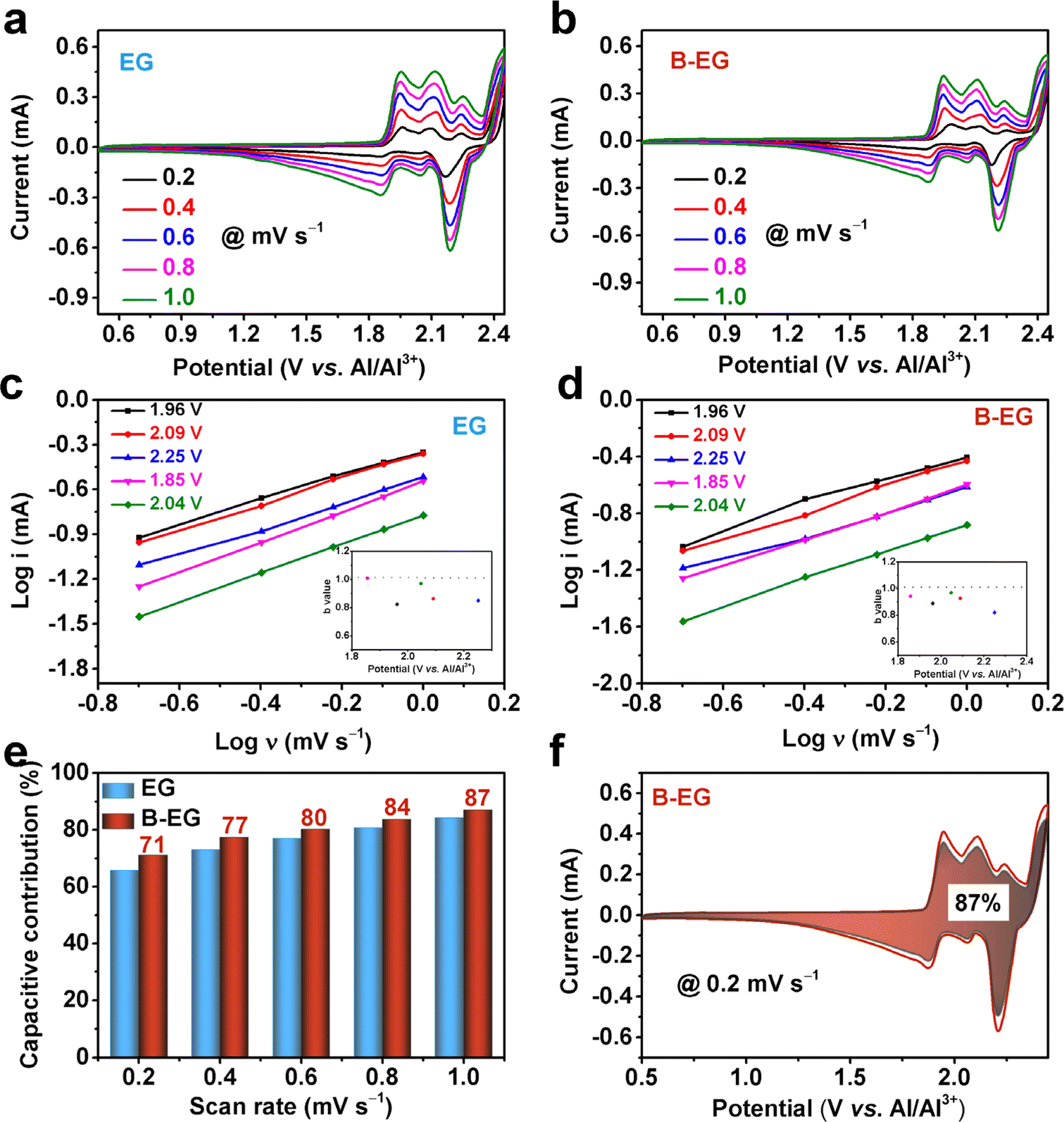 | ||
| Fig. 6 The Al3+ storage mechanism of EG and B-EG: CV curves at different scan rate of 0.2, 0.4, 0.6, 0.8, 1.0 mV s−1 of (a) EG and (b) B-EG; the relationship curves of logν and logi of (c) EG and (d) B-EG, the inset shows the b values at different potential; (e) pseudo-capacitance contribution at various scan rate of EG and B-EG; (f) the pseudo-capacitance contribution of B-EG at scan rate of 1.0 mV s−1. | ||
4. Conclusions
This study utilized boric acid as a B source to prepare B-doped expanded graphite by a thermal treatment method. The introduction of boron led to an expansion of the interlayer spacing, an increase in mesopores, and the generation of electron efficiency in the expanded graphite. These structural changes directly affected the aluminum ion storage performance of boron-doped expanded graphite as a cathode material for aluminum-ion batteries. The research results indicate that after boron doping, the mechanism of aluminum ion storage remained unchanged, but the rate performance and cycling stability are significantly improved, and electrode polarization is decreased during aluminum ion storage. Through kinetic analysis of the energy storage process, the results suggest that the enhancement of the electrochemical performance of boron-doping expanded graphite may be attributed to the increase in capacitance contribution. This design provides new insights for the development of graphite as a cathode material for aluminum-ion batteries.Data availability
The data supporting this article have been included within the manuscript and its ESI.†Author contributions
Y. Y. designed the research and carried out the main experiments. R. Z. helped with the electrochemical tests. Y. Y. wrote the manuscript and Y. C. revised the manuscript. All authors discussed the results and reviewed the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported by the Macao Young Scholars Program under Grant No. AM2021010, the National Natural Science Foundation of China under Grant No. 52002277, and the China Postdoctoral Science Foundation under Grant No. 2019M651574.References
- Y. Zhang, S. Q. Liu, Y. J. Ji, J. M. Ma and H. J Yu, Emerging Nonaqueous Aluminum-Ion Batteries: Challenges, Status, and Perspectives, Adv. Mater., 2018, 30, 1706310 CrossRef PubMed.
- G. A. Elia, K. Marquardt, K. Hoeppner, S. Fantini, R. Y. Lin, E. Knipping, W. Peters, J.-F. Drillet, S. Passerini and R. Hahn, An Overview and Future Perspectives of Aluminum Batteries, Adv. Mater., 2016, 28, 7564–7579 CrossRef CAS PubMed.
- S. K. Das, S. Mahapatrab and H. Lahana, Aluminium-ion batteries: developments and challenges, J. Mater. Chem. A, 2017, 5, 6347–6367 RSC.
- S. K. Das, Graphene: A Cathode Material of Choice for Aluminum-Ion Batteries, Angew. Chem., Int. Ed., 2018, 57, 2–14 CrossRef.
- Q. F. Li and N. J. Bjerrum, Aluminum as anode for energy storage and conversion: a review, J. Power Sources, 2002, 110, 1–10 CrossRef CAS.
- Y. Li, J. Yang and J. Song, Nano energy system model and nanoscale effect of graphene battery in renewable energy electric vehicle, Renewable Sustainable Energy Rev., 2017, 71, 645–651 CrossRef CAS.
- M. C. Lin, M. Gong, B. G. Lu, Y. P. Wu, D. Y. Wang, M. Y. Guan, M. Angel, C. X. Chen, J. Yang, B. J. Hwang and H. J. Dai, An ultrafast rechargeable aluminium-ion battery, Nature, 2015, 520, 325–336 CrossRef PubMed.
- Q. P. Zhou, D. W. Wang, Y. Wang, L. B. Ni, H. H. Zhang and J. Zhao, Ultrathin MoS1.68Se0.32 Alloy Nanoflakes: An Intercalation-Type Positive Electrode Material for Rechargeable Aluminum-Ion Battery, J. Phys. Chem. C, 2022, 126, 2679–2688 CrossRef CAS.
- H. C. Li, H. C. Yang, Z. H. Sun, Y. Shi, H. M. Cheng and F. Li, A highly reversible Co3S4 microsphere cathode material for aluminum-ion batteries, Nano Energy, 2019, 56, 100–108 CrossRef CAS.
- J. N. Zheng, T. Xu, G. L. Xia and X. B. Yu, Oxygen defect engineering endows Co3O4 nanosheets with advanced aluminum ion storage, J. Mater. Chem. A, 2022, 10, 18322 RSC.
- H. Y. Lu, Y. X. Wan, T. Y. Wang, R. Jin, P. T. Ding, R. Wang, Y. Wang, C. Teng, L. L. Li, X. L. Wang, D. S. Zhou and G. Xue, A high performance SnO2/C nanocomposite cathode for aluminum-ion batteries, J. Mater. Chem. A, 2019, 7, 7213–7220 RSC.
- S. T. Wang, K. V. Kravchyk, S. Pigeot-Rémy, W. Q. Tang, F. Krumeich, M. Wörle, M. I. Bodnarchuk, S. Cassaignon, O. Durupthy, S. L. Zhao, C. Sanchez and M. V. Kovalenko, Anatase TiO2 Nanorods as Cathode Materials for Aluminum-Ion Batteries, ACS Appl. Nano Mater., 2019, 2, 6428–6435 CrossRef CAS.
- G. Y. Li, J. G. Tu, M. Y. Wang and S. Q. Jiao, Cu3P as a novel cathode material for rechargeable aluminum-ion batteries, J. Mater. Chem. A, 2019, 7, 8368–8375 RSC.
- C. X. Li, S. H. Dong, P. Wang, C. X. Wang and L. W. Yin, Metal–Organic Frameworks-Derived Tunnel Structured Co3(PO4)2@C as Cathode for New Generation High-Performance Al-Ion Batteries, Adv. Energy Mater., 2019, 9, 1902352 CrossRef CAS.
- S. Wang, S. Huang, M. J. Yao, Y. Zhang and Z. Q. Niu, Engineering Active Sites of Polyaniline for AlCl2+ Storage in an Aluminum-Ion Battery, Angew. Chem., Int. Ed., 2020, 59, 11800–11807 CrossRef CAS PubMed.
- R. D. Mckerracher, A. Holland, A. Cruden and R. G. A. Wills, Comparison of carbon materials as cathodes for the aluminium-ion battery, Carbon, 2019, 144, 333–341 CrossRef CAS.
- J. Vatsala Rani, V. Kanakaiah, T. Dadmal, M. Srinivasa Rao and S. Bhavanarushi, Fluorinated Natural Graphite Cathode for Rechargeable Ionic Liquid Based Aluminum-Ion Battery, J. Electrochem. Soc., 2013, 160, A1781–A1784 CrossRef.
- J. Wei, W. Chen, D. M. Chen and K. Yang, An amorphous carbon-graphite composite cathode for long cycle life rechargeable aluminum ion batteries, J. Mater. Sci. Technol., 2018, 34, 983–989 CrossRef CAS.
- S. M. Zhao, H. X. Chen, J. L. Li and J. X. Zhang, Synthesis of polythiophene/graphite composites and their enhanced electrochemical performance for aluminum ion batteries, New J. Chem., 2019, 43, 15014 RSC.
- J. S. Kim, M. R. Raj and G. Lee, High-Defect-Density Graphite for Superior-Performance Aluminum-Ion Batteries with Ultra-Fast Charging and Stable Long Life, Nano-Micro Lett., 2021, 13, 171 CrossRef CAS PubMed.
- K. Share, A. P. Cohn, R. Carter, B. Rogers and C. L. Pint, Role of Nitrogen-Doped Graphene for Improved High-Capacity Potassium Ion Battery Anodes, ACS Nano, 2016, 10, 9738–9744 CrossRef CAS PubMed.
- M. Chen, W. Wang, X. Liang, S. Gong, J. Liu, Q. Wang, S. J. Guo and H. Yang, Sulfur/Oxygen Codoped Porous Hard Carbon Microspheres for High-Performance Potassium-Ion Batteries, Adv. Energy Mater., 2018, 8, 1800171 CrossRef.
- Y. Qian, S. Jiang, Y. Li, Z. Yi, J. Zhou, T. Q. Li, Y. Han, Y. S. Wang, J. Tian, N. Lin and Y. T. Qian, In Situ Revealing the Electroactivity of P-O and P-C Bonds in Hard Carbon for High-Capacity and Long-Life Li/K-Ion Batteries, Adv. Energy Mater., 2019, 9, 1901676 CrossRef.
- B. H. Li, C. P. Han, Y. B. He, C. Yang, H. D. Du, Q. H. Yang and F. Y. Kang, Facile synthesis of Li4Ti5O12/C composite with super rate performance, Energy Environ. Sci., 2012, 5, 9595 RSC.
- S. Y. Yang, X. Y. Wang, X. K. Yang, Y. S. Bai, Z. L. Liu, H. B. Shu and Q. L. Wei, Determination of the chemical diffusion coefficient of lithium ions in spherical Li[Ni0.5Mn0.3Co0.2]O2, Electrochim. Acta, 2012, 66, 88–93 CrossRef CAS.
- Y. Yang, S. Z. Ren, S. B. Ma, C. Hao and M. Ji, Hollow Tin Dioxide Microspheres With Multilayered Nanocrystalline Shells for Pseudocapacitor, Electrochim. Acta, 2015, 155, 437–446 CrossRef CAS.
- W. Liu, H. J. Li, J. L. Jin, Y. Z. Wang, Z. Zhang, Z. D. Chen, Q. Wang, Y. G. Chen, E. Paek and D. Mitlin, Synergy of Epoxy Chemical Tethers and Defect-Free Graphene in Enabling Stable Lithium Cycling of Silicon Nanoparticles, Angew. Chem., Int. Ed., 2019, 58, 16590–16600 CrossRef CAS PubMed.
- S. Roscher, R. Hoffmann and O. Ambacher, Determination of the graphene–graphite ratio of graphene powder by Raman 2D band symmetry analysis, Anal. Methods, 2019, 11, 1224–1228 RSC.
- C. Sangwichien, G. L. Aranovich and M. D. Donohue, Density functional theory predictions of adsorption isotherms with hysteresis loops, Colloids Surf., A, 2002, 206, 313–320 CrossRef CAS.
- E. Vunain, D. Kenneth and T. Biswick, Synthesis and characterization of low-cost activated carbon prepared from Malawian baobab fruit shells by H3PO4 activation for removal of Cu(II) ions: equilibrium and kinetics studies, Appl. Water Sci., 2017, 7, 4301–4319 CrossRef CAS.
- D. Y. Wang, C. Y. Wei, M. C. Lin, C. J. Pan, H. L. Chou, H. A. Chen, M. Gong, Y. P. Wu, C. Z. Yuan, M. Angell, Y. J. Hsieh, Y. H. Chen, C. Y. Wen, C. W. Chen, B. J. Hwang, C. C. Chen and H. J. Dai, Advanced rechargeable aluminium ion battery with a high-quality natural graphite cathode, Nat. Commun., 2017, 8, 14283 CrossRef CAS PubMed.
- C. J. Pana, C. Yuana, G. Z. Zhua, Q. Zhang, C. J. Huang, M. C. Lin, M. Angella, B. J. Hwang, P. Kaghazchic and H. J. Dai, An operando X-ray diffraction study of chloroaluminate anion-graphite intercalation in aluminum batteries, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 5670–5675 CrossRef PubMed.
- D. L. Chao, C. R. Zhu, P. H. Yang, X. H. Xia, J. L. Liu, J. Wang, X. F. Fan, S. V. Savilov, J. Y. Lin, H. J. Fan and Z. X. Shen, Array of nanosheets render ultrafast and high-capacity Na-ion storage by tunable pseudocapacitance, Nat. Commun., 2016, 7, 12122 CrossRef CAS PubMed.
- V. Augustyn, P. Simon and B. Dunn, Pseudocapacitive oxide materials for high-rate electrochemical energy storage, Energy Environ. Sci., 2014, 7, 1597–1614 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03161j |
| This journal is © The Royal Society of Chemistry 2024 |