
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGlassy carbon electrode modified with a film of tetraruthenated nickel(II) porphyrin located in natural smectite clay's interlayer for the simultaneous sensing of dopamine, acetaminophen and tryptophan
Justin Claude Kemmegne-Mbouguen *a,
Guy Bertrand Tamnec,
Marcelline Carine Ngo-Ngwema,
Henrique Eisi Tomab,
Koiti Arakib,
Vera Regina Leopoldo Constantinob and
Lúcio Angnes*b
*a,
Guy Bertrand Tamnec,
Marcelline Carine Ngo-Ngwema,
Henrique Eisi Tomab,
Koiti Arakib,
Vera Regina Leopoldo Constantinob and
Lúcio Angnes*b
aLaboratory of Porous Materials for Sensors and Energy, Faculty of Science, University of Yaounde 1, P.O. Box 812, Yaounde, Cameroon. E-mail: justin.kemmegne@facsciences.uy1.cm
bUniversidade de São Paulo, Instituto de Química, Av. Professor Lineu Prestes, 748, CEP: 05508-000-São Paulo, Brazil. E-mail: luangnes@iq.usp.br
cDepartment of Chemistry, High Teacher Training College, University of Yaounde 1, P.O. Box 49, Yaounde, Cameroon
First published on 18th June 2024
Abstract
A supramolecular complex μ-meso-tetra(4-pyridyl) porphyrinate nickel(II)tetrakis[bis(bipyridine)(chloro)ruthenium(II)] ([NiTPyP{Ru(bipy)2Cl}4]4+) was intercalated into the interlayer space of natural smectite clay (shortened as Ba) collected in a Cameroonian deposit at Bagba hill. Physicochemical characterization of the resulting material using ultraviolet-visible spectroscopy (UV-vis), Fourier transform infrared (FTIR) spectroscopy, and X-ray diffraction (XRD) confirmed the intercalation of the porphyrin within the interlayer space of the clay. The intercalated clay was then used to form a stable thin film onto a glassy carbon electrode (GCE) by drop casting a suspension of the hybrid material. The GCE modified with the intercalated organoclay endowed the electrode with a larger electrochemically active surface area, good stability, high selectivity, and sensitivity toward dopamine (DA), acetaminophen (AC) and tryptophan (Trp). In addition, it was observed that the modified electrodes exhibited good and pH-dependent electrocatalytic properties toward these analytes. The simultaneous determination of DA, AC and Trp at [NiTPyP{Ru(bipy)2Cl}4]4+–Ba/GCE was thus possible without the interference of one analyte on the others, and the resulting calibration curve exhibits two segments for the three analytes. For DA, AC and Trp, the detection limits were found to be 0.8 μM, 0.3 μM and 0.3 μM, respectively. The [NiTPyP{Ru(bipy)2Cl}4]4+–Ba/GCE modified electrodes were successfully applied for the determination of AC in Paracetamol, a commercial product, and Trp in real pharmaceutical formulation samples.
1 Introduction
Chemically modified electrodes (CMEs) have increasingly attracted research attention over the past decades with the interest of better assessing the nature of chemical reactions at the electrodes. This has led to the continuous development of numerous devices for sensing applications that exhibit improved selectivity and sensitivity in different fields including clinical, industrial and environmental. One of the key ways to obtain sensors with enhanced analytical performance is the use of new electrode material with advantageous and improved properties compared to commonly used electrode substrates made of noble metals and graphite. Various materials have been used as electrode modifiers, including aluminosilicate, and in particular smectite clay and zeolite, which open up many opportunities in designing and implementing novel and improved sensors owing to their interesting properties including chemical and thermal stability, cationic exchange properties, swelling and porosity properties.Since its introduction in 1983 (by Gosh and Bard) in the field of electrochemistry,1 smectite clay has been largely used to modify electrode substrates and to build immunosensors, biosensors2,3 and various sensors4–7 despite its insulating character and barrier effect. This limitation was overcome using redox mediators situated in the interlayer of the clay and/or within the clay.4
Porphyrin and its derivatives have been investigated intensively owing to their rich redox and catalytic properties.8,9 Porphyrin and related compounds can undergo multiple electron and proton transfer reactions owing to the 18 extended π-conjugated system of the macrocycle complex. In addition, the π system is known to endow the porphyrin with the capability to undergo rapid redox processes with minimal energy although these processes are highly dependent on the central metal atom.10 When porphyrin macrocycles are coordinated to a metal, these electron (and proton) transfers can occur either in the central metal or in porphyrin's ligand.11 However, their applications in the field of CME are limited by the peeling of the porphyrin layer from the electrode surface when used as an electrode modifier, thus severely affecting their electrochemical activity.10 This limitation can be overcome by immobilizing porphyrin on host guest solids, which imposes shape selectivity and a favorable microenvironment that facilitates the approach of the substrate to the active catalytic center for an efficient and selective catalyst reaction. Their immobilization of solids has led to efficient and selective catalysts for oxidation reactions. Several solids have been used for porphyrin immobilization, including polymer, silica,12 natural13,14 and synthetic clay.15,16 Because of their flat surface, clay sheets can act as an ideal porphyrin host for building regular structures of guest porphyrins.17 The immobilized porphyrin/solid has been successfully exploited for the catalytic oxidation of alkane, cyclohexene and alcohol15 and as electrode modifiers for electrochemical sensing.4,14,18
In this work, we report the construction of a chemically modified glassy carbon electrode with a film of a natural clay/tetraruthenated nickel porphyrin hybrid material (denoted as NiTPyP{Ru(bipy)2Cl4}4+–Ba), obtained by intercalating a positively charged synthetic porphyrin between the natural smectite sheets. The stability of as-prepared glassy carbon film electrodes was studied using an anionic probe and then utilized to investigate (i) the electrochemical behavior of a mixture of dopamine (DA), acetaminophen (AC) and tryptophan (Trp) and (ii) the possible application as sensitive and selective electrochemical sensors for the simultaneous detection of these three important compounds in biological media. Owing to the electrocatalytic properties of nickel(II) porphyrin within the organoclay and interesting physicochemical properties of smectite, the organoclay film-modified electrodes exhibited a pH dependence on selectivity and remarkable sensitivity toward DA, AC and Trp. It is noteworthy that one of the major problems encountered in the electrochemical detection of DA is its interference with other biological compounds.10 Indeed, DA and derivatives, AC and Trp coexist in biological fluid and are electrochemically oxidizable at various electrodes.19 However, the electrochemical response at bare electrodes is not optimal because they show a sluggish electron transfer process, surface fouling and high overpotential.20 Numerous works have been devoted to enhancing electron transfer and reducing the overpotential for their electrochemical oxidation, thus favoring their simultaneous detection using chemically modified electrodes.21 Alongside the electrochemical method for the quantification of DA, AC and Trp, several other methods, including chromatography, spectroscopy and electrophoresis, have been successfully utilized.22–29 Generally, these methods require laborious sample preparation and organic solvents and demand expensive equipment; consequently, they are costly and not so suitable for routine analysis.
Dopamine (DA) or 3,4-dihydroxyphenethylamine is a neurotransmitter of the family of catecholamines, which is fundamental in human metabolism. They are involved in many physiological processes, such as the functioning of the hormonal and nervous systems, and their abnormal concentration levels in humans lead to several neurological problems. N-(4-hydroxyphenyl) acetamide or AC is a non-opioid analgesic and antipyretic drug widely used to treat moderate pains and fevers. It is a suitable alternative to aspirin, and its overdose leads to the formation of some nephrotoxic metabolites.30. (2S)-2-amino-3-(1H-indol-3-yl) propanoic acid or Trp is an amino acid that is fundamental for the normal growth of humans. Due to its scarce presence in foods, Trp is supplemented in food products and added to certain pharmaceutical formulas. It is an important precursor for some biological compounds, including serotonin, melatonin and niacin. When it is not properly metabolized, Trp generates toxic metabolites in the brain, causing delusion and hallucination. Although it has been successfully detected and quantified using electrochemical techniques, its electrochemical response is sometimes unsatisfactory mainly because of the slow heterogeneous electron transfer rate at the sensor's surface.31
2 Experimental
2.1 Materials, chemicals, and reagents
The natural clay sample denoted Ba was sampled in a Baba Hill deposit in western Cameroon. It is formed mainly of smectite with a minor amount of feldspar. It was fully characterised by Tonle et al.32 and exhibited a BET surface area of 86 m2 g−1 and a cation exchange capacity of 0.89 meq g−1. The nickel supramolecular compound named μ-meso-tetra(4-pyridyl) porphyrinate nickel(II) tetrakis [bis(bipyridine) (chloro) ruthenium(II)] (TFMS)4, represented as [NiTPyP{Ru(bipy)2Cl4}4+], was synthesized as reported by Toma et al.33 The intercalation of the cationic complex onto the interlayer of the sodium fine fraction of clay (particle size below 2 μm) occurred spontaneously. All other chemicals and reagents used in the electrochemical studies were of analytical grade and were used as received. Dopamine (98%), acetaminophen (98%), tryptophan (≥98%), K2HPO4 (99%), KH2PO4 (99%), K3Fe(CN)6 (99%), NaCl (99.5%) and KCl (99.5%) were supplied by Sigma, while NaOH and CH3COOONa were obtained from Synth Chemical.2.2 Preparation of the organoclay composite NiTPyP{Ru(bipy)2Cl4}4+–Ba
The synthesis of the clay + nickel porphyrin (namely NiTPyP{Ru(bipy)2Cl4}4+–Ba) was achieved by intercalating the supramolecular complex into the clay interlayer. Experimentally, 120 mg of the sodium homoionic clay was suspended in 4 mL of dry ethanol containing NiTPyP{Ru(bipy)2Cl4}4+ (5 mg mL−1), which was dispersed by ultrasonication for 1 hour. Then, 20 mL of water was added to the mixture, which was sonicated for an additional 45 min and kept under continuous stirring for 96 hours. The suspension was allowed to rest overnight. The resulting supernatant was completely limpid, as shown by the photograph inserted in Fig. 1B. The supernatant was monitored spectrophotometrically and was found to be free from porphyrin, attesting to the effective association of NiTPyP{Ru(bipy)2Cl4}4+ with the clay.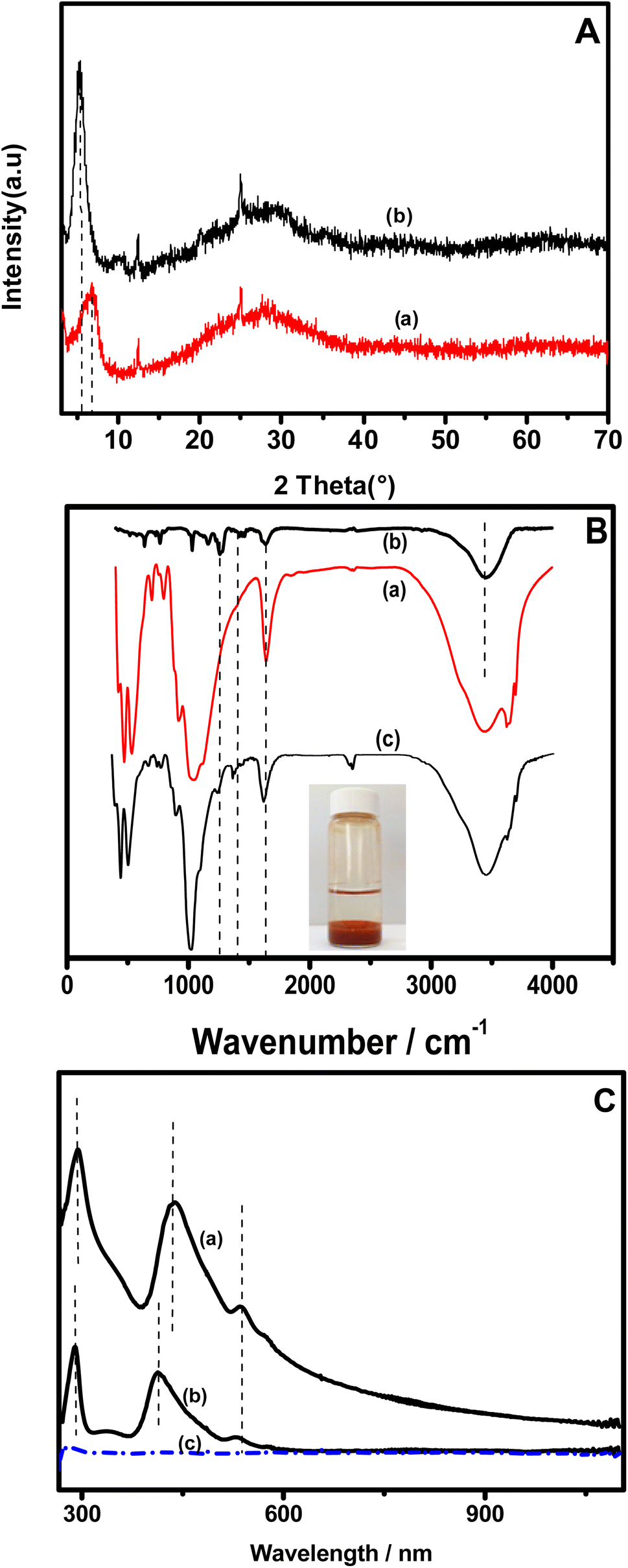 | ||
| Fig. 1 (A) XRD patterns of oriented film of pristine clay (a) and [NiTPyP{Ru(bipy)2Cl}4]4+–Ba (b); (B) FT-IR spectrum of pristine clay (a), NiTPyP{Ru(bipy)2Cl4}4+ (b) and NiTPyP{Ru(bipy)2Cl4}4+ (c); (C) UV-vis of NiTPyP{Ru(bipy)2Cl4}4+ in water/ethanol (a), NiTPyP{Ru(bipy)2Cl4}4+–Ba suspension (b), and supernatant after NiTPyP{Ru(bipy)2Cl4}4+ intercalation (c). Inset shows a photograph of a porphyrin solution after its treatment with Ba. | ||
2.3 Glassy carbon electrode film preparation
To prepare the glassy carbon electrode (Ø = 2 mm), its surface was first polished with alumina slurry (with three-grain sizes: 1, 0.3 and 0.05 μm), resulting in a mirror-like surface. On this surface, 5 μL of NiTPyP{Ru(bipy)2Cl4}4+–Ba suspension was drop casting onto the electrode surface and then was allowed to dry at room temperature for about 1 hour, and the resulting electrode was named NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE.2.4 Instrumentation
Cyclic voltammetry (CV) and square wave voltammetry (SWV) experiments were conducted using a PGSTAT-12 Autolab potentiostat/galvanostat associated with the GPES electrochemical analysis system (Eco Chemie, the Netherlands). Measurements were performed in a conventional three-electrode cell assembly consisting of an Ag/AgCl(KCl sat) reference electrode, a platinum wire counter electrode and the modified glassy carbon as the working electrode. Scanning electron microscopy (SEM) images of the clay and clay–porphyrin composite were obtained using a JSM-7401F field emission scanning electron microscope (FESEM, JEOL Ltd, Japan). The ultraviolet-visible (UV-vis) spectroscopy analysis was performed using Nicolet Evolution 100 (Thermo Electron Corporation). X-ray diffraction (XRD) patterns of oriented films of pristine clay and NiTPyP{Ru(bipy)2Cl4}4+–Ba were recorded using a Miniflex diffractometer (Rigaku, Japan) with Cu Kα (λ = 1.5406 Å) radiation, 30 kV, 15 mA, and Ni filter.3 Results and discussion
3.1 Physicochemical characterization of the organoclay NiTPyP{Ru(bipy)2Cl4}4+–Ba
The XRD patterns of the pristine clay before (curve a) and after the intercalation of NiTPyP{Ru(bipy)2Cl4}4+ into its interlayer space (curve b) are presented in Fig. 1A. One can observe that on pattern a, the d001 corresponding to the basal spacing at the lowest 2θ (6.42° for the intercalated clay) shifts to the lowest value (5.30° for the pristine clay), indicating an increase in the interlayer space. The basal spacing values were calculated using the Bragg equation and were found to be 12.6 Å and 17 Å, respectively, for the pristine clay (Ba) and organoclay (NiTPyP{Ru(bipy)2Cl4}4+–Ba). These results indicate that the basal spacing of the treated clay has expanded by ca. 4.4 Å as a result of the intercalation of NiTPyP{Ru(bipy)2Cl4}4+ within the interlayer space of Ba. The molecular diameter of tetraruthenated porphyrin is about 20 Å,34 which is 4 times the expended value of the organoclay, suggesting that the porphyrin's ring within the modified clay was likely laid flat parallel to the clay layers.35Fig. 1B presents the FT-IR spectra of the pristine clay (spectrum a), [NiTPyP{Ru(bipy)2Cl}4](TFMS)4 (spectrum b), and the porphyrin–intercalated clay (spectrum c). In plot (a), a broad band at 3640 is attributed to the stretching vibrational mode of the structural hydroxyl groups. The band at 1630 cm−1 was assigned to the O–H deformation of the interlayer water molecules. The broad intense band at ca. 1000 cm−1 is attributed to the asymmetric Si–O and Si–O–Si stretching modes. The [NiTPyP{Ru(bipy)2Cl}4]4+–Ba hybrid spectrum (plot c) shows the same characteristic bands of the clay, indicating that there is no significant change in clay structure. In addition to these bands, a good correlation can be observed between the vibrational bands of the hybrid material (plot b) and those of tetraruthenated porphyrin (plot c) in the 1000 to 1600 cm−1 range.
The band at 1647 cm−1 in plots b and c is attributed to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching of the pyridine substituent, and those at 1460 and 1400 cm−1 are assigned to CN and CC stretching modes of the porphyrin ring, respectively. In addition, a characteristic band of [NiTPyP{Ru(bipy)2Cl}4]4+ at 1669 cm−1, absent in the spectrum of pristine clay, was also observed in spectrum b. These changes denote the presence of tetraruthenated porphyrin in Ba.
N stretching of the pyridine substituent, and those at 1460 and 1400 cm−1 are assigned to CN and CC stretching modes of the porphyrin ring, respectively. In addition, a characteristic band of [NiTPyP{Ru(bipy)2Cl}4]4+ at 1669 cm−1, absent in the spectrum of pristine clay, was also observed in spectrum b. These changes denote the presence of tetraruthenated porphyrin in Ba.
Fig. 1C shows the UV-vis spectra of NiTPyP{Ru(bipy)2Cl4}4+ UV-vis in methanol/water solution (plot a) and that of suspension of the porphyrin–intercalated clay (plot b). On both plots, the band observed at 288 nm is attributed to the peripheral ruthenium complexes {MLCT RuII(dπ) → bpy(pπ*)}. In plot (a), the bands at 412 and 531 nm attributed to Soret and Q bands were found to have shifted for about 26.5 nm to higher wavelengths, respectively, suggesting that there is likely a significant interaction between the clay and the NiTPyP{Ru(bipy)2Cl4}4+ porphyrin moiety.36 Interestingly, these bands observable on plots a and b were absent on plot c of the supernatant obtained after the intercalation of the porphyrin into the clay (as depicted in the inset of Fig. 1C), demonstrating the absence of the porphyrin in this solution, and thus its complete intercalation of the porphyrin into the clay interlayer space.
3.2 Electrochemical behaviour of ionic probe at NiTPyP{Ru(bipy)2Cl4}4+–Ba film on GCE
The ability of the intercalated clay to form a stable thin film onto a glassy carbon substrate was studied using cyclic voltammetry (CV) by examining the electrochemical behaviour of a series of CVs recorded in 0.1 M KCl containing 0.5 mM [Fe(CN)6]3−. Fig. 2A presents multi-sweep CVs obtained upon repetitive sweeps at 50 mV s−1, exhibiting well-defined redox peaks with nearly constant peak currents. In addition, the CV recorded at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE exhibited a peak to peak potential ΔE (Epa − Epc) equal to 80 mV with a current peak ratio Ipc/Ipa equal to 1. These results are close to those of the theoretical values of peak-to-peak separation and the current ratio for an electrochemical reversible process.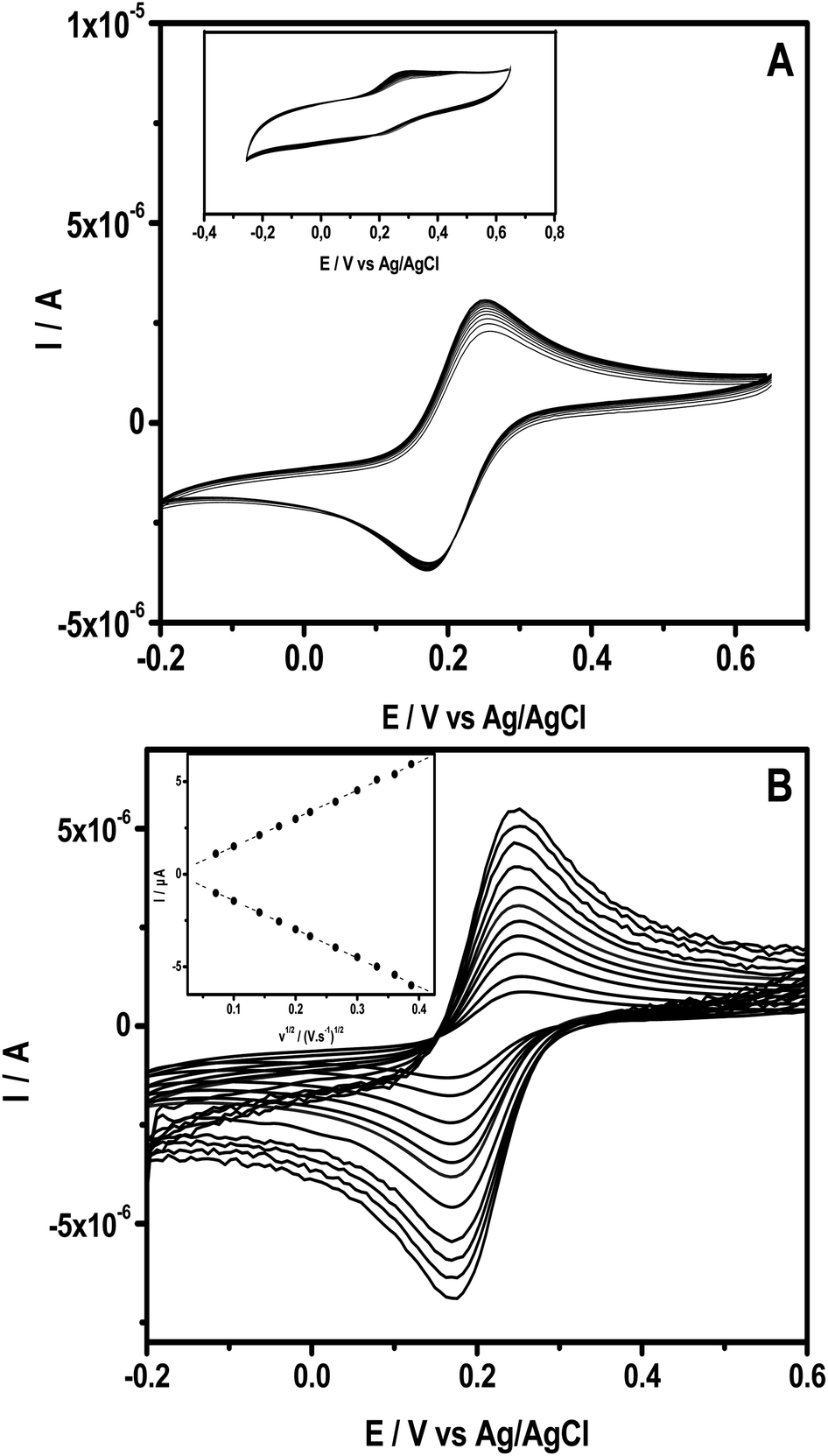 | ||
| Fig. 2 (A) Multisweep CVs recorded using NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE at scan rate 50 mV s−1 in 0.5 mM Fe(CN)63−(+0.1 M NaCl). Inset shows the same experiment using an electrode modified with Ba/GCE. (B) CVs at different scan rates. Inset shows the corresponding Ip versus v1/2. | ||
Interestingly, under the same conditions, as expected, the CVs recorded at pristine clay film GCE (Ba/GCE) exhibited a narrow peak, as shown in the inset of Fig. 1. This behavior is due to the repulsion of the negatively charged probe and the negatively charged clay sheets, while at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE, the electrostatic repulsion is overcome likely because of the presence of positively charged nickel-porphyrin within the clay's interspace and its catalytic activity. Fig. 2B shows several experiments in different scan rates, where the anodic and cathodic currents increased proportionally to the square root of the scan rate, as shown in the inset in Fig. 2B, which suggests that the process at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE is controlled by diffusion.
The surface area of the porphyrin clay-modified electrode NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE was estimated by applying the Randles–Sevcik equation for a reversible process:9,37,38
| Ip = 2.69 × 105 × DoAv1/2n3/2Co, |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 and A is the estimated surface of the modified electrode, which is found to be 0.04 cm2.
9 and A is the estimated surface of the modified electrode, which is found to be 0.04 cm2.
3.3 Electrochemical behavior of DA, AC and Trp at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE
The electrochemical behavior of DA, AC and Trp was studied using bare and modified GCE. This study began by utilizing cyclic voltammetry (CV). Fig. 3A presents the CV recorded within the potential window ranging from −0.3 V to +0.95 V at modified GCE (scan rate 50 mV s−1) in phosphate buffer (shortened PB) pH = 6, containing either no analyte (dashed line) or a mixture of analytes: DA (13 μM), AC (15.4 μM) and Trp (13.5 μM) (solid line). When the potential was scanned in the positive direction, no defined peak was observed in the absence of analyte in PB pH = 6, while in the presence of the three analytes, well-defined peaks appeared at +0.16 V, +0.45 V and +0.79 V. | ||
| Fig. 3 (A) CVs recorded in the absence (dashed line) and in the presence of a mixture of the analytes (DA = 13 μM, AC = 15.4 μM and Trp = 13.5 μM) on NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE. (B) CVs of NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE recorded in PB (pH = 6) in the presence of the same mixture of analytes at different scan rates. Insets show a plot of anodic peak currents versus the square root of scan rate (v1/2) (inset (a)), log of anodic peak currents versus the log of scan rate (inset (b)) and Ipa/v1/2 versus v1/2 (inset (c)). | ||
These peaks correspond to the oxidation of DA to dopamine O-quinine (scheme 1A), AC to acetyl-p-quinone imine (scheme 1B) and Trp to methylene imine (scheme 1C), respectively. On the reverse potential scan, two cathodic peak signals were observed at +0.06 V and +0.40 V, corresponding to a reduction in the product of the oxidation of AC and DA, respectively. Relative to Trp, it is well known that this compound presents an irreversible behavior.
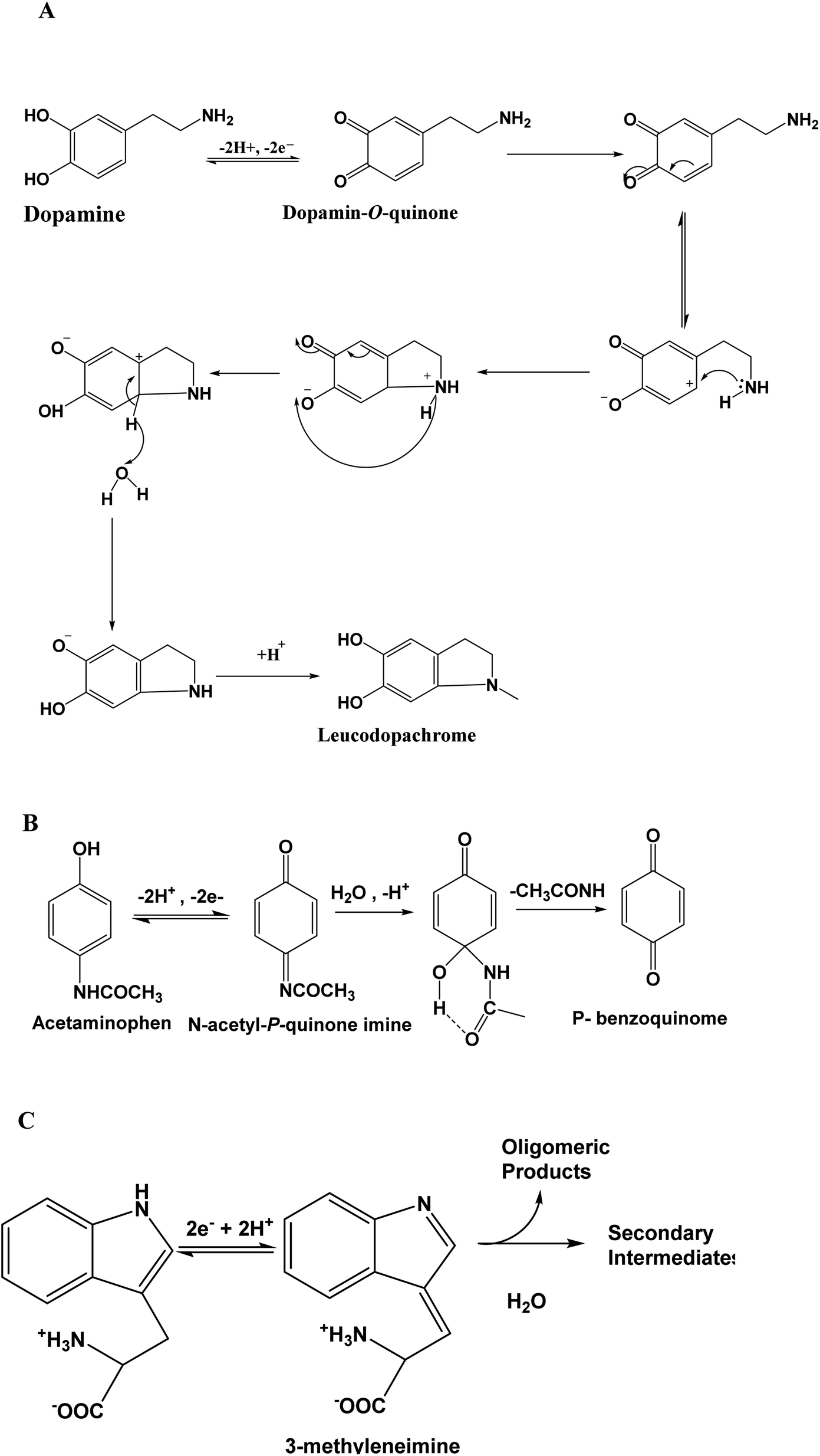
These results also indicate that the redox processes of DA and AC are quasi-reversible. For the three analytes, their electrochemical oxidation response depended on the scan rate, and the oxidation peak current (Ipa) increased with an increase in the scan rate (v). In addition, Ipa was proportional to the scan rate (10 to 40 mV s−1), as exhibited in the (inset (a)) in Fig. 3B, suggesting that the oxidation of DA, AC and Trp at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE are diffusion-controlled processes at such scan rates. For higher scan rates, a deviation from this behavior was observed. Interestingly, the plots of log Ipa vs. logv for the three analytes are linear (inset (b)) with a slope of 0.30 for DA and Trp, and 0.36 for AC close to the theoretical value 0.5 for a diffusion-controlled process. From the data collected in Fig. 3, the sweep rate normalized the oxidation current density, in which Ipa/v1/2 versus the square root of v (inset (c)) led to a characteristic shape of an EC mechanism. This suggests that after the electrooxidation step, the product of the electrooxidation of DA (dopamine O-quinine), AC (dopamine O-quinine) and Trp (methylene imine) undergo a subsequent chemical transformation, as shown in Scheme 1.
The electrochemical oxidation responses of DA, AC and Trp obtained using square wave voltammetry (SWV) at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE under the same experimental conditions used in Fig. 3A were compared with the oxidation responses recorded at pristine clay-modified GCE (Ba/GCE) and bare GCE, as shown in Fig. 4. This figure presents (a) the blank of the GCE (signal obtained just in the presence of electrolyte); (b) signal recorded for the bare electrode in the presence of 13 μM of DA, 15.4 μM of AC and 13.5 μM of Trp mixture; (c) signal recorded for the analytes with the Ba/GCE electrode; and (d) signal obtained with the NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE. This last curve displays three well-defined oxidation peaks situated at +0.24 V, +0.44 V and +0.77 V corresponding to the oxidation of DA, AC and Trp, respectively. The small elevation observed at +0.12 V is assigned to Ni(II)/Ni(III). Comparing these results with the ones obtained at the Ba/GCE (c), this exhibited a lower current intensity and a shift in the peak potential of ca. 132 mV, 131 mV and 91 mV compared with the oxidation potential peak of the DA, AC and Trp, respectively.
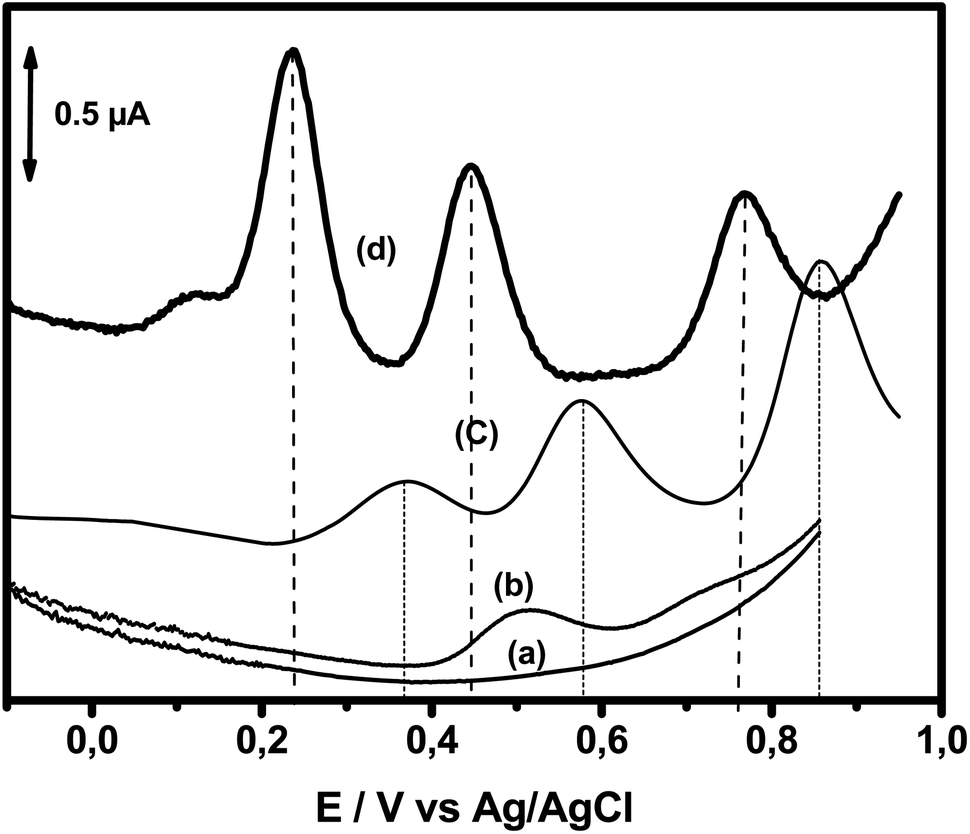 | ||
| Fig. 4 SWVs recorded in PB (pH = 6) in the absence of the analytes at bare (curve a) and at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE in the presence of 13 μM of DA, 15.4 μM of AC and 13.5 μM of Trp mixture at bare GCE (curve b), Ba/GCE (curve c) and at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE (curve d). | ||
Interestingly, in the SWV recorded at bare GCE, one oxidation peak was observed at +0.50 V corresponding to the oxidation of DA with a small signal at +0.70 V, probably corresponding to the oxidation of AC. The increase in selectivity and sensitivity observed at Ba/GCE compared with those of unmodified GCE can be explained by the favorable electrostatic attraction between negatively charged clay sheets and the three analytes in their cationic form (DA (pKa = 8.93), AC (pKa = 9.38) and Trp (pKa = 9.39)), thus likely favoring the presence of DA, AC and Try at the Ba/GCE interface, respectively. The enhancement of the oxidation peak current and the overpotential decrease observed in comparison with these results obtained at Ba/GCE highlight the electrocatalytic behavior of the Ni-porphyrin within the clay toward mainly DA and AC on the one hand and, on the other hand, demonstrate the synergic effect of the clay and Ni-porphyrin constituents of the organoclay.
As depicted in Fig. 5, the pH of the supporting electrolytic solution influences the sensitivity and selectivity of the organoclay film electrodes toward the studied analytes. This figure presents the SWVs recorded at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE in buffer solution with different pH levels, ranging from 4 to 7 and containing a mixture of DA (13 μM), AC (15.4 μM) and Trp (13.5 μM). Interestingly, a close look at the oxidation process of DA shows the following behaviour: at pH = 7, a main peak is observed at ∼+0.18 V; when the pH decreases to 6, a small hump appears at about +0.12 V and the main peak still appears at ∼+0.24 V. At pH = 5, two peaks of similar magnitude were observed (+0.18 V and +0.36 V). At pH 4, a noticeable change occurred in the shape of the SWV response, and the oxidation peaks of AC and DA overlapped, producing a unique peak located at +0.57 V with two shoulders at +0.22 V and at +0.44 V. The peak at lower potential +0.22 V (pH 4), +0.18 V (pH 5) and +0.11 V (pH 6) was assigned to redox NiIII+/NiII+ of the porphyrin, as stated in the literature by some authors.42–45 This decrease in selectivity at pH 4 is likely because the nickel center of NiTPyP{Ru(bipy)2Cl4}4+ porphyrin is electrochemically less active under slightly acidic conditions (pH < 5), while the ruthenium centre activity is enhanced at more positive potential.46
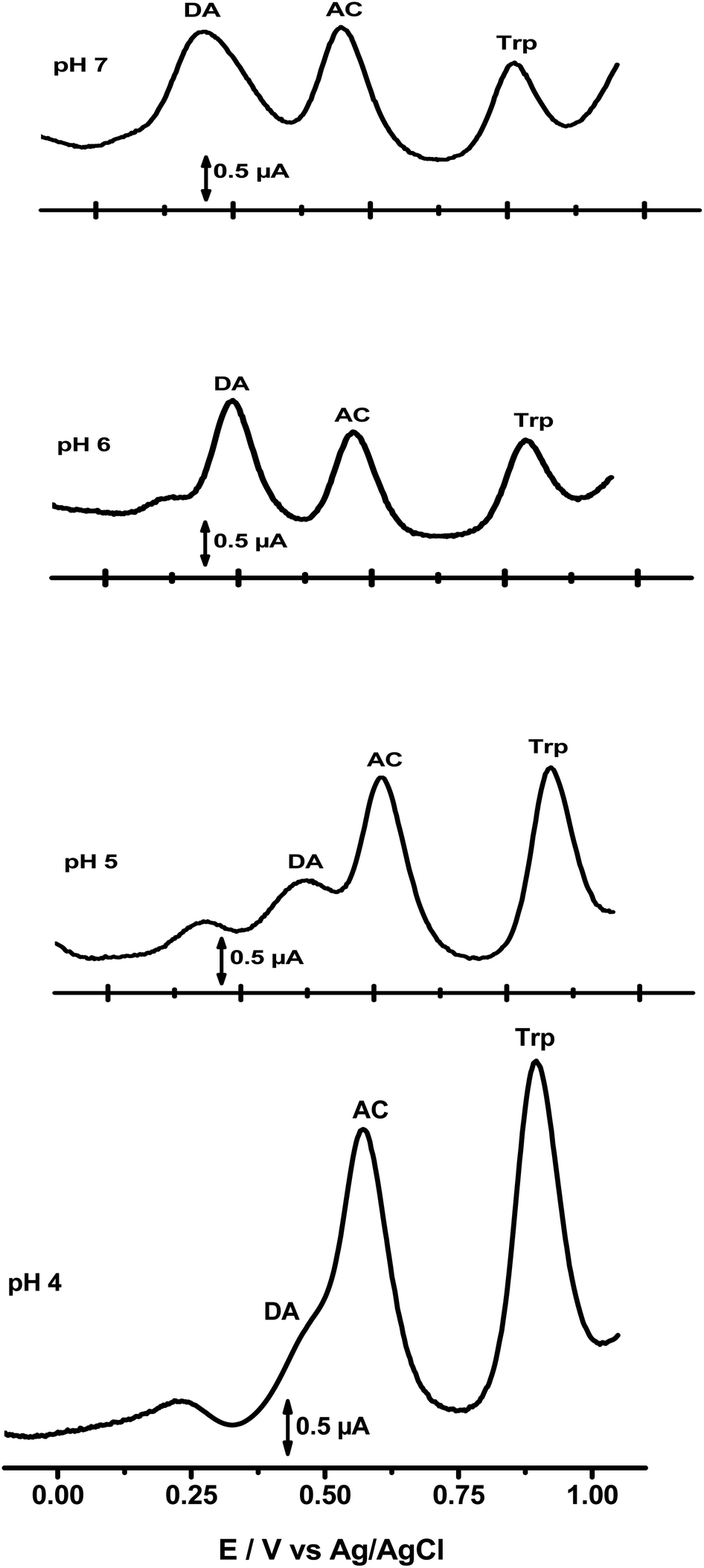 | ||
| Fig. 5 SWVs recorded at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE at different pH levels containing DA, AC and Trp mixture. | ||
In addition, the oxidation peak potential of DA, AC and Trp shifted with the pH of the electrolytic solutions. These oxidation peaks were also found to shift negatively with pH increase, linearly depending on the pH except for DA, and obey the following equations:
| For AC: Ep = −0.054 pH + 0.787 (R2 = 0.999); |
| For Trp: Ep = −0.053 pH + 1.075 (R2 = 0.993). |
These results indicate the involvement of protons in the electrochemical oxidation of the three analytes at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE. In addition, the slopes obtained by plotting Ep versus pH for AC and Trp were close to the theoretical value of 59 mV pH−1 at 25 °C, indicating that the number of electrons and protons involved in the oxidation process are equal. For DA, Ep vs. pH was not linear (R2 = 0.8764), suggesting that the process involving the oxidation of DA is more complex than those involving AC and Trp. This fact makes the evaluation and description of multiple processes that occur simultaneously (or in sequence) much more complicated. Thus, the electrochemical process of DA, for example, could be as follows at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE:10
| 2Ni(II) TPyP{Ru(bipy)2Cl4}4+–Ba → 2Ni(III) TPyP{Ru(bipy)2Cl4}4+–Ba + 2e− |
| 2Ni(III) TPyP{Ru(bipy)2Cl4}4+–Ba + DAred → 2Ni(II)TPyP{Ru(bipy)2Cl4}4+–Ba + DAox |
By considering the peak-to-peak separation between the oxidation peak of DA, AC and Trp and the highest and most favourable oxidation peak current of the three analytes, the PB (pH 6) was selected for their simultaneous sensing.
Another important parameter that can affect the electrochemical response of NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE is the film thickness of organoclay NiTPyP{Ru(bipy)2Cl4}4+–Ba on the GCE, which can be tuned by drop coating 5 μL of different suspensions obtained by varying the amount of organoclay (ranging from 5 to 40 mg) dispersed in 5 mL of distilled water. It was observed (figure not shown) that the SWV response recorded in PB (pH = 6) containing 10 μM of AC was found to increase when the film onto GCE was made by drop coating (1 mg mL−1) up to the one made with (5 mg mL−1) of NiTPyP{Ru(bipy)2Cl4}4+–Ba and then levelled off and decreased when more concentrated suspensions were used for the electrode modification. These results suggested that the increase until 5 mg mL−1 increased the number of active sites, but a higher amount of modifier produced a barrier effect of the NiTPyP{Ru(bipy)2Cl4}4+–Ba's film. Thus, in the next step, the film-modified electrode used as a working electrode was made by dropping 5 μL of NiTPyP{Ru(bipy)2Cl4}4+–Ba suspension (5 mg mL−1) onto GCE.
3.4 Simultaneous determination of DA, AC and Trp
SWV was also used for the selective determination of DA, AC and Trp in their mixed liquor utilizing NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE because the large difference between the oxidation peak potentials, estimated to be 0.22 V for ΔEDA–AC and 0.30 for ΔEAC–Trp, favored their simultaneous determination when in mixture. Fig. 6 illustrates the SWVs recorded at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE with three oxidation peaks of DA, AC and Trp at +0.22 V, +0.44 V and +0.74 V, respectively, when increasing amounts of one analyte (Fig. 6A for DA, Fig. 6B for AC and Fig. 6C for Trp) were added to PB (pH = 6), while those of the other two species remained fixed. As shown in this figure, in all the three cases, the oxidation's peak current of the targeted analyte increased when an increasing amount of this analyte was added to the electrolytic solution containing a fixed amount of the two others, and the oxidation peak current due to the oxidation of the latter was kept constant upon addition of the target analyte. The insets exhibited in Fig. 6 clearly show that the plots of oxidation peak current versus analyte concentration are composed of two segments of straight lines with different slopes. Thus, when successive addition of DA was made in PB (pH = 6) containing AC (15.4 μM) and Trp (10.2 μM), the plots of oxidation current versus concentration were linear, ranging from 3 μM to 15.0 μM (R2 = 0.998) and from 15 μM to 23.0 μM (R2 = 0.995). The electrooxidation current of AC recorded in the same buffer in the presence of 15.4 μM of DA and 10.5 μM of Trp was linearly dependent on the AC's concentration from 2.2 μM to17.6 μM (R2 = 0.997) and from 17.6 μM to 55.2 μM (R2 = 0.997). Finally, for Trp, the oxidation current was recorded in PB (pH 6) containing DA (11.5 μM) and AC (11.4 μM), and the two linear segments were situated at intervals from 1.5 μM to 10.2 μM (R2 = 0.999) and from 10.2 μM to 23.4 μM (R2 = 0.997).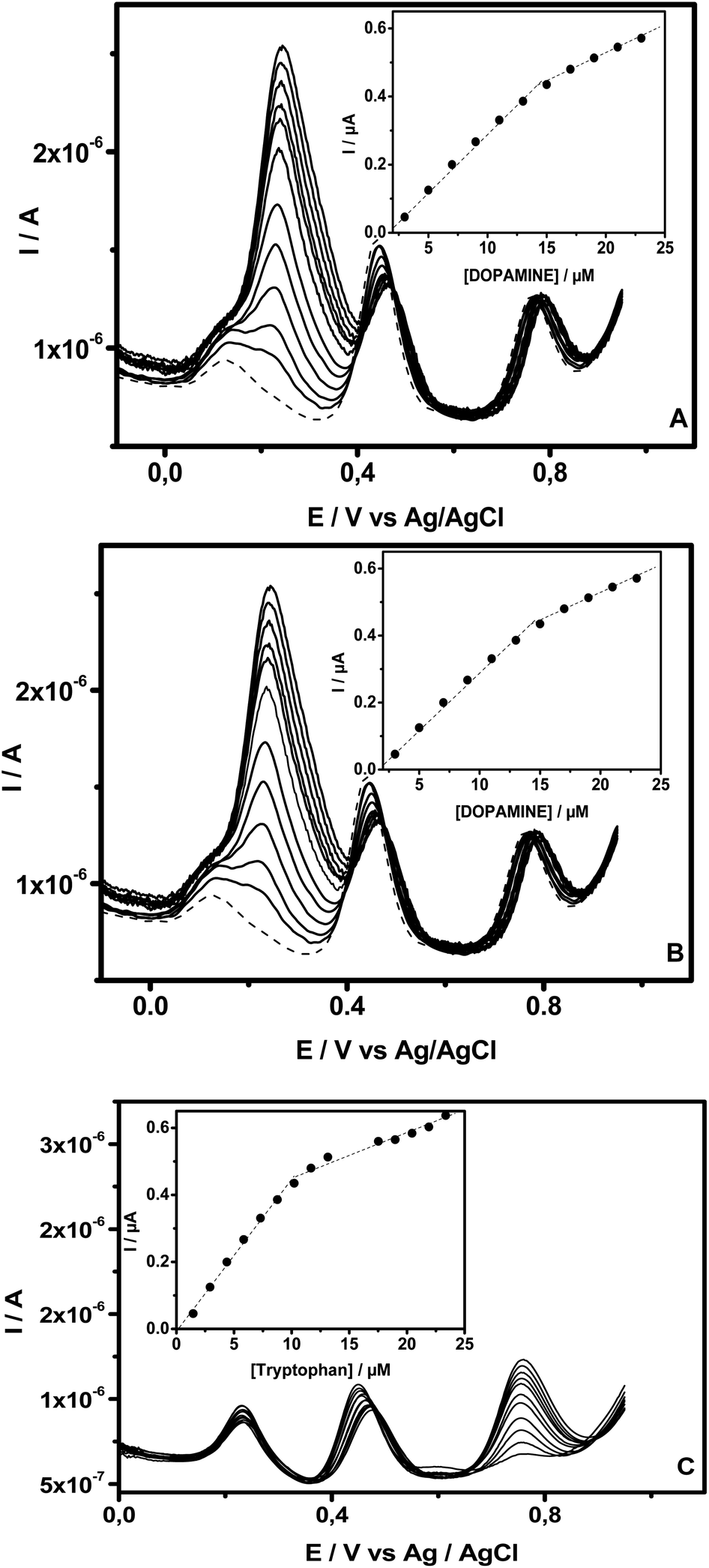 | ||
| Fig. 6 SWVs of NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE recorded in 0.1 M NaCl, and PB (pH 6) added increasing amounts of (A) DA in the presence of a constant concentration of AC (15.4 μM) and Trp (10.5 μM); (B) AC in the presence of a constant concentration of DA (15 μM) and Trp (10.5 μM); (C) Trp in the presence of fix amount of DA (11.5 μM) and AC (11.4 μM). The inset shows the corresponding calibration curve. | ||
These results indicate that the selective determination of these analytes in the presence of the two other compounds (within the concentration ranges explored here) exhibited no obvious interference. The sensibilities and limits of detection estimated from the first segment of the straight line were 0.03 μA μM−1 and 0.82 μM for DA; 0.06 μA μM−1 and 0.30 μM for AC and 0.05 μA μM−1 and 0.30 μM for Trp, respectively. These performances were compared with other detection limits reported in the literature, as displayed in Table 1. From the data gathered in Table 1, there is no doubt that the proposed sensors exhibit detection limits in the same order of magnitude as those reported by other authors.
| Electrodes | Linear range (μM) | Detection limit (μM) | Reference | |
|---|---|---|---|---|
| a Imidazole derivative multi-wall (IMWCNT); 8,9-dihydroxy-7-methyl-12H-benzothiazolo[2,3-b]quinazolin-12-one modified multiwall carbon nanotubes paste electrode (DMBQ-MCNTPE); nickel hydroxide nanoparticle (NHNP); gold nanoparticles (AuNP). | ||||
| Poly(new coccine)/GCE | DA | 0.7–200 | 0.2 | 47 |
| Pyrolytic carbon films | 18–270 | 2.3 | 48 | |
| MWCNT/GCE | 3–200 | 0.8 | 49 | |
| SWCNT/GCE | 0.4–150 | 0.22 | 50 | |
| AuNP/polyimidazole/GCE | 5.0–268 | 0.08 | 51 | |
| IMWCNT/GCE | 113.6–416.7 | 0.38 | 52 | |
| NiTPyPRu(bipy)2–Ba/GCE | 3–15 and 15–23 | 0.82 | This work | |
| Poly(new coccine)/GCE | AC | 1.5–120 | 0.3 | 47 |
| Pyrolytic carbon films | 15–225 | 1.4 | 48 | |
| MWCNT/GCE | 3–300 | 0.6 | 49 | |
| SWCNT/GCE | 0.2–100 | 0.12 | 50 | |
| DMBQ-MCNTPE/CPE | 5.0–500 | 1 | 53 | |
| MWCNTs–NHNPs–MCM-41/GCE | 0.05–2 and 4–30 | 0.01 | 54 | |
| NiTPyPRu(bipy)2–Ba/GCE | 1–17.6 and 17.6–55.2 | 0.2 | This work | |
| Poly(new coccine)/GCE | Trp | 0.6–50 | 0.2 | 47 |
| AuNP/polyimidazole/GCE | 3.0–34.0 and 84.0–464.0 | 0.7 | 51 | |
| IMWCNT/GCE | 227.3–833.3 | — | 52 | |
| DMBQ-MCNTPE | 10.0–800 | 4 | 53 | |
| MWCNTs–NHNPs–MCM-41/GCE | 0.5–5 and 5–50 | 0.11 | 54 | |
| NiTPyPRu(bipy)2–Ba/GCE | 1.4–10.3 and 10.3–23.4 | 0.3 | This work | |
3.5 Interference study
The influence of potential interfering species, including ascorbic acid, uric acid, glucose, saccharin, sodium carbonate, citric acid, tyrosine, and epinine compounds, on the simultaneous determination of DA, AC and Try (10 μM) was investigated in PB (pH = 6) using NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE. It was observed that uric acid, epinine, ascorbic acid, and tyrosine interfered significantly with the electrochemical oxidation response of DA, AC and Trp whereas glucose, saccharin, sodium carbonate, and citric acid did not.3.6 Reproducibility and stability of the modified electrode
The electrode-to-electrode reproducibility of the developed sensors was studied by preparing 5 independent modified electrodes following the procedure reported in the experimental section under optimal conditions and used to record the electrochemical oxidation response of DA, AC and Trp in PB (pH = 6). The standard deviation calculated for the 5 electrodes using SWVs resulted in relative standard deviations of 1.86% for DA, 2.41% for AC and 2.32% Trp, confirming the good reproducibility of the fabrication method of these electrodes. Long-term stability was also evaluated by performing successive measurements over two weeks. Measurements using AC 15.4 μM were performed almost daily using the NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE film electrode. After the measurements, the electrode was stored at 4 °C in PB (pH 6). The electrode retained 95% of the signal after two weeks of use.3.7 Real sample analysis
To evaluate the potential of the new sensor developed in this study for applications in the analysis of real samples, drugstore samples containing acetaminophen and tryptophan were analysed. Paracetamol 750 (i.e. a tablet of this drug with the commercial name of paracetamol contains 750 mg of acetaminophen) and tryptophan syrup (40 mg in 10 mL) were selected for this purpose. Prior to their analysis using the standard addition method, the drugs were diluted with an adequate amount of PB (pH = 6). To carry out the analysis of AC, a tablet of paracetamol was first diluted in water (200 mL of volumetric flask). After the complete dilution of the tablet, 1.00 mL of this solution was diluted with PB (pH = 6) in a 100 mL volumetric and mixed well. The tryptophan solution was prepared by the direct transference of 1.00 mL of the syrup to a 100 mL volumetric flask, completed with PB buffer. From these diluted samples, 0.50 mL of either AC or Trp real samples were transferred to a voltammetric cell containing 9.50 mL of PB (pH = 6) under stirring. After that, the first scan was run, and an SWV (dashed line) recorded at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE is presented in Fig. 7. This was followed by 5 injections (100 μL) in a sequence of standard solutions into the voltammetric cell, with the respective SWVs (solid line) recorded after each standard addition. Fig. 7A and B present the recorded SWVs at NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE for AC and Trp, respectively. The linear regression of the points presented in the inset indicates an intersection in the x axis at −12.53 μM for AC and – 8.89 μM for Trp, which are very close to the calculated value (12.44 μM for AC and 9.79 μM), indicating that the experimental value corresponds to 100.7% and 90.8% of the calculated value, respectively.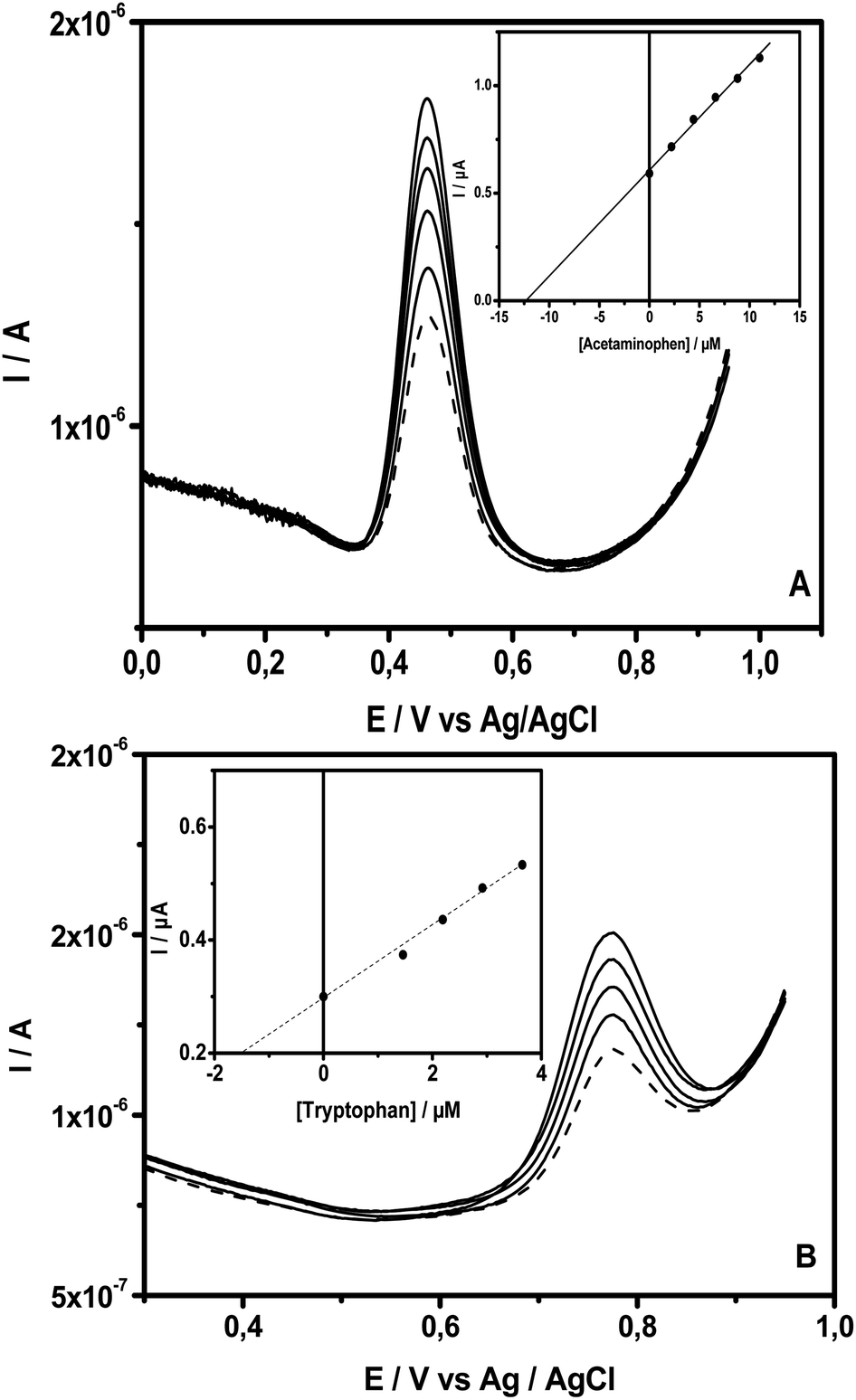 | ||
| Fig. 7 SWVs of NiTPyP{Ru(bipy)2Cl4}4+–Ba/GCE recorded in PB (pH = 6) with the addition of (A) AC and (B) Trp drug solution (dashed line), followed by the successive addition of their standard (solid line) under the same condition, as depicted in Fig. 6. Insets show AC and Trp concentrations versus the peak current obtained. | ||
4 Conclusion
Clay + tetraruthenated nickel porphyrin was prepared using the batch method by the intercalation of the positively charged porphyrin on the natural Cameroonian smectite clay interlayer (NiTPyP{Ru(bipy)2Cl4}4+–Ba). The organoclay composite was used to modify the surface of the glassy carbon electrode, and the thin film immobilized on the surface of the electrode exhibited high electrocatalytic activity towards DA, AC and Trp. The modified electrodes were then employed as sensors for selective and sensitive simultaneous detection and quantification of these analytes under optimal conditions in which no interference was observed between them. These sensors were found to favor the attainment of wide concentration ranges (3 to 23 μM for DA, 2.2 to 55.2 μM for AC and 1.5 to 23.4 μM for Trp) and low detection limits. Further, electrochemical sensors were applied in the quantification of AC and Trp in pharmaceutical preparation, thus indicating that the sensors have promising applications for routine analyses of these three analytes in biological real samples.Author contributions
J. C. Kemmegne-Mbouguen: conceptualisation, resource, funding acquisition, supervision, formal analysis, methodology, investigation, validation, writing original draft, writing – review and editing G. B. Tamne: methodology, investigation, writing original draft, writing – review and editing M. C. Ngo-Ngwem: investigation, writing original draft, writing – review and editing H. E. Toma: methodology, investigation, writing original draft, writing – review and editing K. Araki: methodology, investigation, writing original draft, writing – review and editing V. R. L. Constantino: methodology, investigation, writing original draft, writing – review and editing L. Angnes: conceptualisation, funding acquisition, resource, supervision, methodology, investigation, validation, writing original draft, writing – review and editing.Conflicts of interest
There is no conflict to declare.Acknowledgements
The authors gratefully acknowledge the Royal Society ACBI programme (grant no. AQ15002 allowed to Prof Kemmegne-Mbouguen) and the National Council for Research (CNPq) (grants no. 3240274276, 465389/2014-7, 308996-2023-2 and process no. 306504-2011-1 allowed to Prof. Kemmegne-Mbouguen and Prof. L. Angnes) for their financial support.References
- P. K. Ghosh and A. J. Bard, J. Phys. Chem., 1984, 88, 5519–5526 CrossRef CAS.
- J. Kemmegne Mbouguen, E. Ngameni and A. Walcarius, Anal. Chim. Acta, 2006, 578, 145–155 CrossRef CAS PubMed.
- J. K. Mbouguen, E. Ngameni and A. Walcarius, Biosens. Bioelectron., 2007, 23, 269–275 CrossRef CAS PubMed.
- J. C. Kemmegne-Mbouguen and L. Angnes, Sens. Actuators, B, 2015, 212, 464–471 CrossRef CAS.
- J. C. Kemmegne-Mbouguen, L. Angnes, E. Mouafo-Tchinda and E. Ngameni, Electroanalysis, 2015, 27, 2387–2398 CrossRef CAS.
- J. C. Kemmegne-Mbouguen, I. T. Kenfack, A. Walcarius and E. Ngameni, Talanta, 2011, 85, 754–762 CrossRef PubMed.
- M. C. N. Ngwem, J. C. Kemmegne-Mbouguen, H. W. Langmi, N. M. Musyoka and R. Mokaya, ChemistrySelect, 2022, 7, e202202308 CrossRef CAS.
- J. Li, W. Tu, J. Lei, S. Tang and H. Ju, Electrochim. Acta, 2011, 56, 3159–3163 CrossRef CAS.
- C. Wang, R. Yuan, Y. Chai, S. Chen, Y. Zhang, F. Hu and M. Zhang, Electrochim. Acta, 2012, 62, 109–115 CrossRef CAS.
- H. Xu, J. Xiao, L. Yan, L. Zhu and B. Liu, J. Electroanal. Chem., 2016, 779, 92–98 CrossRef CAS.
- H. M. Castro-Cruz and N. A. Macías-Ruvalcaba, Coord. Chem. Rev., 2022, 458, 214430 CrossRef CAS.
- M. Halma, A. Bail, F. Wypych and S. Nakagaki, J. Mol. Catal. A: Chem., 2006, 243, 44–51 CrossRef CAS.
- G. S. Machado, K. A. D. de Freitas Castro, F. Wypych and S. Nakagaki, J. Mol. Catal. A: Chem., 2008, 283, 99–107 CrossRef CAS.
- J. C. Kemmegne-Mbouguen, H. E. Toma, K. Araki, V. R. L. Constantino, E. Ngameni and L. Angnes, Microchim. Acta, 2016, 183, 3243–3253 CrossRef CAS.
- H. Kameyama, F. Narumi, T. Hattori and H. Kameyama, J. Mol. Catal. A: Chem., 2006, 258, 172–177 CrossRef CAS.
- M. Halma, K. Aparecida Dias de Freitas Castro, C. Taviot-Gueho, V. Prévot, C. Forano, F. Wypych and S. Nakagaki, J. Catal., 2008, 257, 233–243 CrossRef CAS.
- S. Takagi, M. Eguchi, D. A. Tryk and H. Inoue, J. Photochem. Photobiol., C, 2006, 7, 104–126 CrossRef CAS.
- J. C. Kemmegne-Mbouguen and E. Ngameni, Anal. Methods, 2017, 9, 4157–4166 RSC.
- D. Klein, J. Lichtmannegger, U. Heinzmann and K. H. Summer, J. Hepatol., 2000, 32, 193 CrossRef CAS PubMed.
- S. M. Macdonald and S. G. Roscoe, Electrochim. Acta, 1997, 42, 1189–1200 CrossRef CAS.
- E. Mouafo-Tchinda, J. C. Kemmegne-Mbouguen, C. P. Nanseu-Njiki, H. W. Langmi, C. Kowenje, N. M. Musyoka and R. Mokaya, RSC Adv., 2023, 13, 20816–20829 RSC.
- M. M. Yust, J. Pedroche, J. G. Calle, J. Vioque, F. Millán and M. Alaiz, Food Chem., 2004, 85, 317–320 CrossRef CAS.
- T. Bo and J. Pawliszyn, J. Chromatogr. A, 2006, 1105, 25–32 CrossRef CAS.
- C. Delgado-Andrade, J. A. Rufián-Hanares, S. Jiménez-Pérez and F. J. Morales, Food Chem., 2006, 98, 580–585 CrossRef CAS.
- T. Yoshitake, J. Kehr, S. Yoshitake, K. Fujino, H. Nohta and M. Yamaguchi, J. Chromatogr. B, 2004, 807, 177–183 CrossRef CAS PubMed.
- T. Németh, P. Jankovics, J. Németh-Palotás and H. Kőszegi-Szalai, J. Pharm. Biomed. Anal., 2008, 47, 746–749 CrossRef PubMed.
- N. Erk, Y. Özkan, E. Banoğlu, S. A. Özkan and Z. Şentürk, J. Pharm. Biomed. Anal., 2001, 24, 469–475 CrossRef CAS PubMed.
- A. Eustaquio, M. Blanco, R. D. Jee and A. C. Moffat, Anal. Chim. Acta, 1999, 383, 283–290 CrossRef CAS.
- M. L. Ramos, J. F. Tyson and D. J. Curran, Anal. Chim. Acta, 1998, 364, 107–116 CrossRef CAS.
- F. Patel, Med. Sci. Law, 1992, 32(4), 303–310 CrossRef CAS PubMed.
- V. I. Paz Zanini, R. E. Giménez, O. E. Linarez Pérez, B. A. López de Mishima and C. D. Borsarelli, Sens. Actuators, B, 2015, 209, 391–398 CrossRef CAS.
- I. K. Tonle, E. Ngameni and A. Walcarius, Electrochim. Acta, 2004, 49, 3435–3443 CrossRef CAS.
- K. Araki and H. E. Toma, J. Coord. Chem., 1993, 30, 9–17 CrossRef CAS.
- R. A. Timm, M. P. H. Falla, M. F. G. Huila, H. E. M. Peres, F. J. Ramirez-Fernandez, K. Araki and H. E. Toma, Sens. Actuators, B, 2010, 146, 61–68 CrossRef CAS.
- P. M. Dias, D. L. A. de Faria and V. R. L. Constantino, Clays Clay Miner., 2005, 53, 361–371 CrossRef CAS.
- X. Zhou and H. Ji, Chin. J. Catal., 2012, 33, 1906–1912 CrossRef CAS.
- S. Luo, H. Xiao, S. Yang, C. Liu, J. Liang and Y. Tang, Sens. Actuators, B, 2014, 194, 325–331 CrossRef CAS.
- F. Xu, F. Wang, D. Yang, Y. Gao and H. Li, Mater. Sci. Eng. C, 2014, 38, 292–298 CrossRef CAS PubMed.
- U. E. Majewska, K. Chmurski, K. Biesiada, A. R. Olszyna and R. Bilewicz, Electroanalysis, 2006, 18, 1463–1470 CrossRef CAS.
- D. Nematollahi, H. Shayani-Jam, M. Alimoradi and S. Niroomand, Electrochim. Acta, 2009, 54, 7407–7415 CrossRef CAS.
- V. I. P. Zanini, R. E. Gimenez, O. E. L. Pérez, B. A. L. de Mishima and C. D. Borsarelli, Sens. Actuators, B, 2015, 209, 391–398 CrossRef.
- K. Araki, M. J. Wagner and M. S. Wrighton, Langmuir, 1996, 12, 5393–5398 CrossRef CAS.
- T.-F. Kang, G.-L. Shen and R.-Q. Yu, Anal. Chim. Acta, 1997, 354, 343–349 CrossRef CAS.
- K. Araki and H. E. Toma, J. Chem. Res., 1994, 1501 Search PubMed.
- L. M. C. Ferreira, P. R. Martins, K. Araki, H. H. Toma and L. Angnes, Electroanalysis, 2015, 27, 2322–2328 CrossRef CAS.
- M. d. S. M. Quintino, H. Winnischofer, M. Nakamura, K. Araki, H. E. Toma and L. Angnes, Anal. Chim. Acta, 2005, 539, 215–222 CrossRef CAS.
- Y. Sun, P. Jia, Y. Bai, S. Liao, S. Wang and X. Zheng, Anal. Methods, 2013, 5, 5737–5745 RSC.
- G. P. Keeley, N. McEvoy, H. Nolan, S. Kumar, E. Rezvani, M. Holzinger, S. Cosnier and G. S. Duesberg, Anal. Methods, 2012, 4, 2048–2053 RSC.
- Z. A. Alothman, N. Bukhari, S. M. Wabaidur and S. Haider, Sens. Actuators, B, 2010, 146, 314–320 CrossRef CAS.
- B. Habibi, M. Jahanbakhshi and M. H. Pournaghi-Azar, Electrochim. Acta, 2011, 56, 2888–2894 CrossRef CAS.
- C. Wang, R. Yuan, Y. Chai, S. Chen, F. Hu and M. Zhang, Anal. Chim. Acta, 2012, 741, 15–20 CrossRef CAS PubMed.
- N. Nasirizadeh, Z. Shekari, H. R. Zare, M. Reza Shishehbore, A. R. Fakhari and H. Ahmar, Biosens. Bioelectron., 2013, 41, 608–614 CrossRef CAS PubMed.
- H. Karimi-Maleh, M. Moazampour, H. Ahmar, H. Beitollahi and A. A. Ensafi, Measurement, 2014, 51, 91–99 CrossRef.
- A. Babaei, E. Ansari and M. Afrasiabi, Anal. Methods, 2014, 6, 8729–8737 RSC.
| This journal is © The Royal Society of Chemistry 2024 |