
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing C–S and C–N bond formation with ultrasound assistance: lipase-catalyzed synthesis of 2,4-disubstituted thiazole derivatives from arylethanones and thioamides†
Priya Mahaur,
Khushbu Rajput,
Vishal Singh,
Vandana Srivastava * and
Sundaram Singh*
* and
Sundaram Singh*
Department of Chemistry, Indian Institute of Technology (BHU), Varanasi 221005, U.P., India. E-mail: vsrivastava.apc@iitbhu.ac.in; sundaram.apc@iitbhu.ac.in; Tel: +91-9453365168 Tel: 91-9451658650
First published on 5th July 2024
Abstract
The present study explores an innovative approach for the efficient synthesis of 2,4-disubstituted thiazole derivatives, a class of compounds with diverse biological and pharmaceutical significance. This research presents lipase as a highly effective and environmentally friendly catalyst for thiazole synthesis. Under mild circumstances, the condensation of aryl ethenone, KBrO3, and thioamide is aided by using ultrasonic energy. Moreover, we harness the power of ultrasound irradiation to accelerate the reaction, reducing reaction times and improving product yields. The lipase-catalyzed, ultrasound-assisted synthesis presented in this study represents a greener and more sustainable alternative to traditional synthetic pathways for these important compounds, offering promising potential for applications in medicinal chemistry and drug development. This approach holds the promise of advancing the field of thiazole synthesis, contributing to more sustainable and efficient chemical processes.
Introduction
Thiazole and its derivatives are extremely helpful substances in a variety of chemistry-related sectors, including medicine and agriculture. For example, the thiazolium ring in vitamin B1 acts as an electron sink and is crucial for the decarboxylation of a-keto acids in their coenzyme form.1 This heterocyclic system offers various applications in creating medications for managing inflammatory conditions,2 hypertension,3 bacterial infections,4 and HIV infections.5 Aminothiazoles are familiar as ligands of estrogen receptors6 and a novel class of adenosine receptor antagonists.72-Aminothiazole is a thiazole-containing heterocyclic amine. It is used as a precursor material in the synthesis of several compounds, including sulfur drugs, fungicides, biocides, dyes, thyroid inhibitors for hyperthyroidism treatment, and chemical reaction accelerators.8 In light of their potential biological characteristics and pharmacological significance, 2-amino thiazoles have attracted significant interest in pharmaceutical chemistry.9,10
Since thiazole is included in many medications and natural items, it is one of the most significant heteroaromatic units. The most common medications with a thiazole unit are as follows: the anti-inflammatory drug Fanetizole, antifungal agent Abafungin, histamine H2 receptor antagonist Famotidine, anti-inflammatory agent Fentiazac, and anti-cancer agent Pamicogrel as shown in Fig. 1.11,12
 | ||
| Fig. 1 Some of the medicinal compounds contain thiazole skeleton. | ||
The existence of these patterns in natural substances and their diverse biological functions has sparked interest in synthesizing them.13,14 Nowadays, there are numerous methods for the synthesis of thiazole moiety. Hantzsch thiazole synthesis is the most extreme and effective method, which involves the condensation of α-halo ketones with thioamide/thiourea.15,16
Several techniques have been documented for thiazole synthesis, including the use of β-cyclodextrin in water,17 alkynyl(aryl)iodonium reagents,18 ammonium-12-molybdophosphate,19 oxodiphosphoniumsalt,20 and microwave irradiation,21 ionic liquid,22 and PEG.23 While numerous approaches exhibit various limitations, such as the employment of reactive or hazardous starting materials like α-halo ketones, the utilization of costly and corrosive catalysts, reliance on volatile organic solvents, tedious reaction workup, extended reaction durations, diminished yields, and stringent reaction conditions.15 Hence, a method for synthesizing thiazole that is affordable, safe for the environment, and useful, is needed. Aryl ethanones have versatile reactivity and various applications, making them vital to research in organic chemistry and pharmaceuticals. In synthesizing numerous medications, agrochemicals, and other fine heterocyclic compounds, aryl ethanones are widely used as a starting material.23 Because of their particular characteristics and reactivity, which are imparted by their aromatic nature and carbonyl functionality, they are valuable building blocks for designing and producing complex organic compounds. A sulfur atom bound to an amide functional group distinguishes thioamides as a family of chemical molecules. Thioamides have a wide range of uses in biological research and chemical synthesis.24 Potassium bromate is particularly useful as a brominating as well as a strong oxidizing agent. Lipase is recognized as an efficient and environmentally friendly bio-catalyst for organic synthesis because of its high selectivity and the requirement of mild reaction conditions.25 Many valuable processes, such as esterification,26 transesterification,27 regioselective acylation of glycols and menthols,28 and peptide synthesis,29 are catalyzed by lipases.30
In recent years, the application of ultrasound in organic synthesis has increased. Under ultrasonic irradiation, many chemical reactions can be carried out with a greater yield, a faster reaction time, and more comfortable settings.31,32 Ultrasonic activation, which is based on cavitation processes that lead to improved mass transfer, is a important green synthetic technique in organic chemistry that provides a versatile and simple pathway for a wide range of syntheses.33 Utilizing environmentally friendly solvents and energy efficiency stand out as the primary principles of green chemistry in organic synthesis.34 The main objective of green chemistry is to transition from traditional thermal methodologies to unconventional approaches with green solvents when synthesizing organic compounds with minimum waste.35
Results and discussion
Continuing our research efforts to create environmentally friendly reaction techniques, we present the synthesis of thiazoles from aryl ethanones and thioamides. This process involves the utilization of KBrO3 as a brominating agent and lipase as a bio-catalyst in aqueous medium (Scheme 1).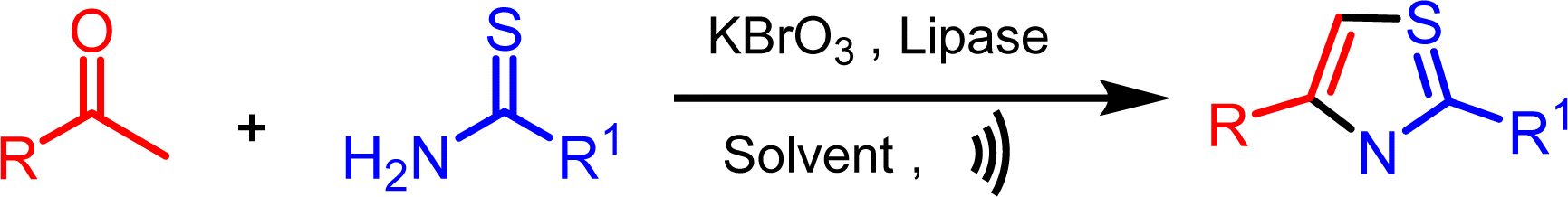 | ||
| Scheme 1 Lipase catalyzed synthesis of the thiazole. | ||
The optimization studies started with a reaction involving acetophenone (1a), thiobenzamide (2a) as substrates, KBrO3 as a brominating agent, and lipase as a biocatalyst, which was chosen as a model for synthesizing thiazole derivatives. The experiment was conducted at ambient temperature using 1.0 mmol of acetophenone (1a), 1.5 mmol of KBrO3, and 1.0 mmol of thiobenzamide (2a) in 10 mL of solvent with the addition of 5 mol% lipases. The experiment involved employing various solvents and both conventional and ultrasound irradiation techniques (Table 1). The model reaction was performed without a catalyst and solvent-free at room temperature (RT), but no reaction occurred even after a long time (Table 1, entry 1). Next, the model reaction was conducted at RT with catalyst and without solvent, but we got a low yield of the product under conventional (Table 1, entry 2).
|
|||||||
|---|---|---|---|---|---|---|---|
| S. No. | Solvent | Lipase mol% | Temp. (°C) | Conventionala | Ultrasonicationb | ||
| Time (h) | Yieldc (%) | Time (min) | Yieldd (%) | ||||
| a Mixture containing 1.0 mmol each of 1a and 2a in 10 mL of solvent was stirred at room temperature (35 °C).b Mixture of 1.0 mmol each of 1a and 2a in 10 mL of solvent was subjected to irradiation at 750 W, 2000 J, 20% amplitude, at a temperature of 35 °C.c The pure isolated yield obtained under conventional conditions.d The pure isolated yield obtained under ultrasonication. | |||||||
| 1 | — | — | 35 | 5 | n.r | — | — |
| 2 | — | 5 | 35 | 5 | 20 | — | — |
| 3 | Ethanol | 5 | 35 | 5 | 38 | 25 | 49 |
| 4 | Methanol | 5 | 35 | 5 | 35 | 25 | 47 |
| 5 | Water | 5 | 35 | 5 | 40 | 25 | 52 |
| 6 | DCM | 5 | 35 | 5 | 28 | 25 | 48 |
| 7 | DMF | 5 | 35 | 5 | 25 | 25 | 47 |
| 8 | 1,4 Dioxane | 5 | 35 | 5 | 23 | 25 | 35 |
| 9 | Toluene | 5 | 35 | 5 | 20 | 25 | 33 |
| 10 | Xylene | 5 | 35 | 5 | 22 | 25 | 31 |
| 11 | Hexane | 5 | 35 | 5 | 25 | 25 | 33 |
| 12 | Benzene | 5 | 35 | 5 | 22 | 25 | 32 |
| 13 | Water | 10 | 35 | 3 | 58 | 10 | 97 |
| 14 | Water | 15 | 35 | 3 | 58 | 10 | 97 |
| 15 | Water | 20 | 35 | 3 | 55 | 10 | 95 |
| 16 | Water | 30 | 35 | 3 | 47 | 10 | 88 |
| 17 | Water | 10 | 40 | 3 | 43 | 10 | 85 |
| 18 | Water | 10 | 45 | 3 | 39 | 10 | 78 |
| 19 | Water | 10 | 50 | 3 | 25 | 10 | 67 |
| 20 | Water | 10 | 25 | 3 | 28 | 10 | 63 |
| 21 | Water | 10 | 20 | 3 | 23 | 10 | 61 |
| 22 | Water | — | 35 | 5 | n.r | 10 | n.r |

Next, the model reaction was conducted at RT with catalyst and in the presence of a different polar solvent like ethanol, methanol, water, dichloromethane (DCM), dimethylformamide (DMF), 1,4-dioxane resulting 2,4-diphenyl thiazole (3a) with 23–52% yield (Table 1, entries 3–8). In contrast, the product (3a) was obtained with a 20–33% yield when non-polar solvents such as toluene, xylene, hexane, and benzene were utilized (Table 1, entries 9–12). Water was the best solvent among all tested solvents, giving a 52% yield in 25 minutes under US conditions (Table 1, entry 5). Further, the load of lipase (10, 15, 20, and 30 mol%) was also varied for optimization of the reaction conditions, and the obtained results are shown in (Table 1, entries 13–16). We discovered that incorporating 10 mol% of lipase in the presence of water resulted in the highest product yield i.e. 97% within a short period (Table 1, entry 13). Following this, we examined how temperature influenced the yield of the product (3a). We observed a decrease in the product (3a) yield to 67% when the temperature was raised to 50 °C (Table 1, entry 19). Conversely, reducing the reaction temperature to 20 °C decreased the product (3a) yield to 61% (Table 1 entry 21). Next, the model reaction was conducted at RT without catalyst in water solvent, but we did not get our desired product (Table 1, entry 22).
After experiments, the optimal condition was established as follows: acetophenone (1a, 1.0 mmol), thiobenzamide (2a, 1.0 mmol), and potassium bromate (KBrO3, 1.5 mmol) with lipase (10 mol%) serving as the biocatalyst at 35 °C using water as a solvent under ultrasonication.
The utilization of ultrasound resulted in a quicker completion of the reaction with a high yield. The experiment involved conducting the model reaction at different energy levels, spanning from 500 to 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 J, to investigate the influence of ultrasound energy on both reaction time and yield. The findings are presented in Table 2.
000 J, to investigate the influence of ultrasound energy on both reaction time and yield. The findings are presented in Table 2.
| Entry | US energy (Joule) | Time (min) | Yield (%) |
|---|---|---|---|
| 1 | 500 | 20 | 80 |
| 2 | 1000 | 18 | 82 |
| 3 | 1500 | 16 | 85 |
| 4 | 2000 | 10 | 97 |
| 5 | 5000 | 10 | 85 |
| 6 | 7500 | 10 | 84 |
| 7 | 11000 |
10 | 82 |
The temperature at which enzymatic reactions occur is a crucial factor influencing both the catalytic properties and stability of enzymes. In the experiment, the model reaction was conducted at seven different temperatures ranging from 20 °C to 50 °C, revealing notable variations in yield at different temperatures (Fig. 2). As a result, 35 °C was identified as the optimum temperature for maximizing yield.
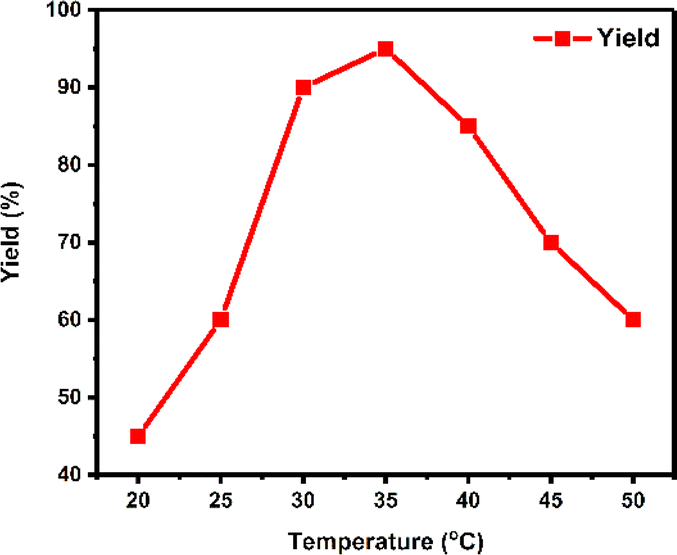 | ||
| Fig. 2 The effect of temperature on enzyme activity. | ||
The pH activity profile of a lipase enzyme was examined across a broad range, spanning from range of 4.1 to 5.5 (acetate buffer solutions), 5.8 to 8.0 (phosphate buffer solutions) and 8.0 to 10.0 (alkaline borate buffer solutions). It was noted that the enzyme demonstrates its highest activity at pH 8.0 (Fig. 3). We also evaluated the present catalyst's reusability by conducting a reaction involving acetophenone, KBrO3, and thiobenzamide. After each reaction, the organic layer containing our desired product was separated, and the aqueous layer containing the lipase catalyst was directly reused in subsequent runs without additional treatment. The lipase catalyst was successfully reused for 6 cycles and the yield was 95, 92, 91, 85, 80, and 65% in cycles 1, 2, 3, 4, 5, and 6 respectively. While the initial three runs exhibited no notable yield loss, the subsequent three cycles showed an immediate decrease in yield (Fig. 4). The relative enzyme activity was 0.97, 0.99, 0.93, 0.94, and 0.81 after cycles 1, 2, 3, 4, and 5 respectively.
 | ||
| Fig. 3 The effect of pH on enzyme activity. | ||
 | ||
| Fig. 4 Reusability of enzyme. | ||
After optimizing the reaction conditions, we proceeded to explore the range of substrates that could participate in the reaction. We focused on various aryl ethanones and documented the outcomes in Tables 3 and 4. A series of substituted aryl ethanones (1a–1m) with thiobenzamide were studied using ideal reaction conditions for the one-pot synthesis of 2,4-disubstituted thiazoles to evaluate the electronic influence of substituents on the aromatic ring. The results showed that electron-withdrawing groups (F, Cl, Br, NO2, CN) (1b–1g) on the benzene ring of aryl ethanone produced good to excellent yields (85–96%) of the relevant compounds. In contrast, benzene rings containing electron-donating groups (OMe, Me) (1h–1k) had excellent yields ranging from 85% to 96%. We focused on other aromatic heterocycles ethanones 2-acetylthiophene and 2-acetylfuran (1l–1m) in light of the previously described findings and it gave (78–85%) yield.
 |
 |
Aryl ethanones reacted with substituted thiobenzamides, including (F, Cl, NO2, CH3) (2b–2g), and heterocyclic (2h–2i), aliphatic thiobenzamides (2j–2k), resulting in the formation of the corresponding products with satisfactory yield. The structures of the newly synthesized compounds (3a–3za, 3aa–3ak) presented in this report were thoroughly validated through analytical and spectral analyses, including IR, 1H NMR, 13C NMR, and mass spectrometry.
To determine the feasibility of applying the proven method for synthesizing 2,4-disubstituted thiazole (3a) on a gram scale using ultrasonication, an experiment was conducted with acetophenone (1a) (5.0 mmol, 1.0 equiv.), thiobenzamide (2a) (5.0 mmol, 1.0 equiv.), and KBrO3 (7.5 mmol, 1.5 equiv.) with lipase (10 mol%) as the biocatalyst at 35 °C in water under standard conditions (Scheme 2). The reaction progress was monitored by thin-layer chromatography (ethyl acetate: n-hexane, 1:4). Upon completion, the mixture was diluted with dichloromethane and washed with water. The organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure, yielding our product (3a) and purified by recrystallization in ethanol.
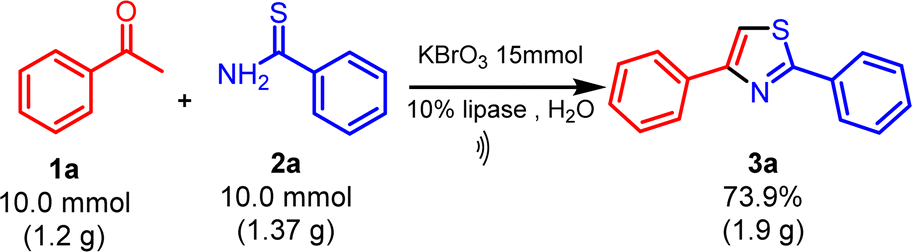 | ||
| Scheme 2 Gram-scale reaction for the synthesis of 2,4-disubstituted thiazole. | ||
Reaction mechanism
In order to clarify a conceivable reaction mechanism, a set of carefully conducted experiments were carried out using optimized reaction conditions, as illustrated in (Scheme 3). In an alternative experimental method, the reaction took place with the inclusion of radical scavengers, namely TEMPO ((2,2,6,6-tetramethylpiperidin-1-yl)oxy) at a concentration of 2 equivalents, as illustrated in (Scheme 3a). Remarkably, this setup yielded a 90% yield of the desired product. This outcome strongly suggests that the formation of the thiazole compound does not proceed through a radical mechanistic pathway. In a subsequent experiment, a control reaction was conducted involving the reaction of acetophenone, KBrO3, and thiobenzamide. Two separate trials were performed, one with the addition of lipase and the other without lipase. The reaction with lipase yielded the desired product with 90% yield in (Table 1, entry 13), whereas the reaction without lipase resulted in a lower yield of 20% in (Table 1, entry 22). Following that, the control reaction took place in an aqueous medium involving acetophenone, KBrO3, lipase, resulting in the desired product with a substantial yield (Scheme 3b). The findings as mentioned above unveiled how KBrO3 plays a part in the bromination process of acetophenone, leading to the formation of 2-bromo acetophenone. The lipase was found to affect all the steps to be catalyzed by lipase. By consolidating the aforementioned outcomes with previously documented literature,30,36 we anticipate a plausible reaction mechanism, illustrated in Fig. 5. Initially, acetophenone 1 undergoes enolization to form A under basic condition, and A converts to B in the presence of KBrO3 and aqueous media. Subsequently, B undergoes intermolecular nucleophilic substitution with enolization isomer of thioamide 2 to form C intermediate. C undergoes intramolecular nucleophilic addition to form D, which undergoes dehydration to form the desired product in the presence of lipase. | ||
| Scheme 3 Control experiments in support of the mechanism. | ||
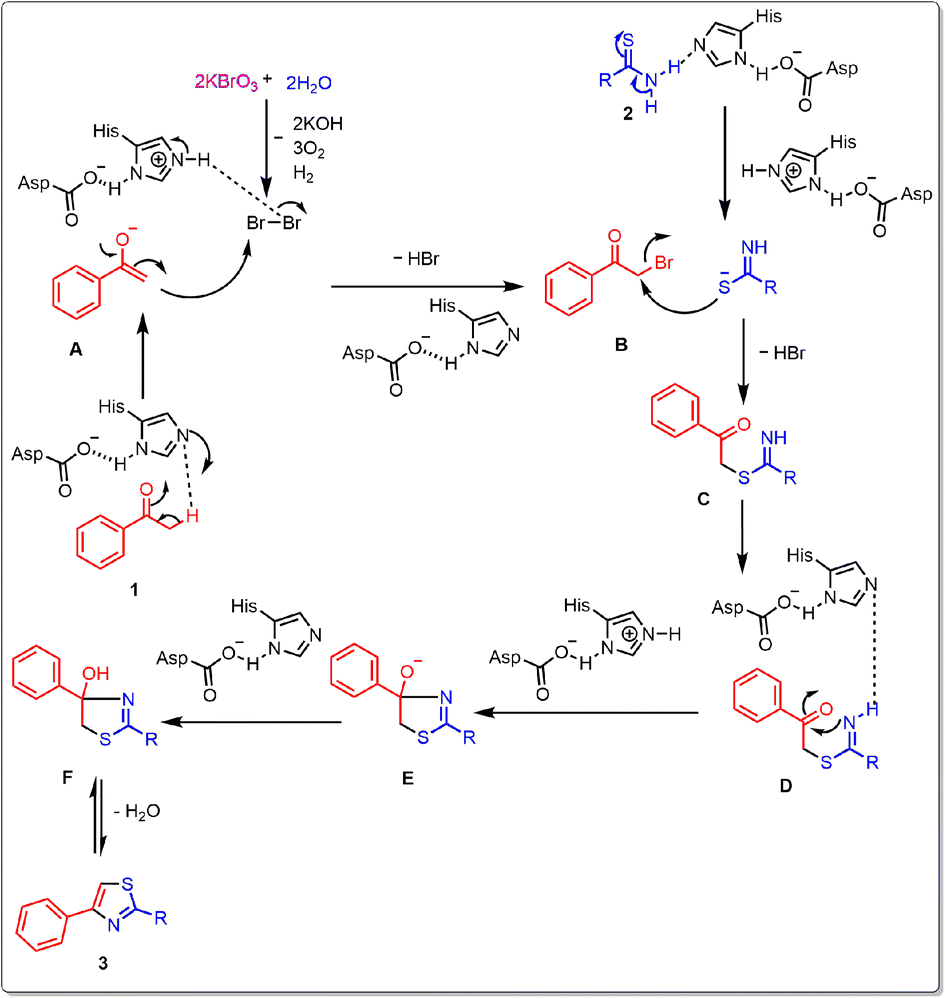 | ||
| Fig. 5 Plausible mechanisms for the synthesis 2,4 disubstituted thiazole derivatives. | ||
Conclusions
We have described a novel, environmentally friendly process for synthesizing thiazoles that employs water as a green solvent and lipase as a biocatalyst. This method's key characteristics include increased reaction rate, easy reaction conditions, ease of work-up procedure, atom economy, the lack of isolating α-bromoketones, high yields, and green chemistry follows such as avoiding organic solvents, toxic and wasteful catalysts.Data availability
All data, such as 1H, 13C NMR, mass, and trapping experiments for the manuscript to support the product formation, are available in the ESI.†Conflicts of interest
The authors declare no competing interests.Acknowledgements
The authors are thankful for the instrumentation facilities provided by CIFC and IIT (BHU) and the financial support provided by UGC (SRF).References
- R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719–3726 CrossRef CAS.
- F. Haviv, J. D. Ratajczyk, R. W. DeNet, F. A. Kerdesky, R. L. Walters, S. P. Schmidt, J. H. Holms, P. R. Young and G. W. Carter, J. Med. Chem., 1988, 31, 1719–1728 CrossRef CAS PubMed.
- W. C. Patt, H. W. Hamilton, M. D. Taylor, M. J. Ryan, D. G. Jr. Taylor, C. J. C. Connolly, A. M. Doherty, S. R. Klutchko and I. Sircar, J. Med. Chem., 1992, 35, 2562–2572 CrossRef CAS PubMed.
- K. Tsuji and H. Ishikawa, Bioorg. Med. Chem. Lett., 1994, 4, 1601–1606 CrossRef CAS.
- F. W. Bell, A. S. Cantrell, M. Hoegberg, S. R. Jaskunas, N. G. Johansson, C. L. Jordan, M. D. Kinnick, P. Lind and J. M. Jr. Morin, J. Med. Chem., 1995, 38, 4929–4936 CrossRef CAS PubMed.
- B. E. Fink, D. S. Mortensen, S. R. Stauffer, Z. D. Aron and J. A. Katzenellenbogen, Chem. Biol., 1999, 6, 205–219 CrossRef CAS PubMed.
- J. E. Van Muijlwijk-Koezen, H. Timmerman, R. C. Vollinga, J. Frijtag von Drabbe Künzel, M. De Groote, S. Visser and A. P. IJzerman, J. Med. Chem., 2001, 44, 749–762 CrossRef CAS PubMed.
- A. Gallardo-Godoy, J. Gever, K. L. Fife, B. M. Silber, S. B. Prusiner and A. R. Renslo, J. Med. Chem., 2011, 54, 1010–1021 CrossRef CAS PubMed.
- D. Das, P. Sikdar and M. Bairagi, Eur. J. Med. Chem., 2016, 109, 89–98 CrossRef CAS PubMed.
- R. Aggarwal, M. Hooda, N. Jain, D. Sanz, R. M. Claramunt, B. Twamley and I. Rozas, J. Sulfur Chem., 2022, 43, 12–21 CrossRef CAS.
- K. Shibasaki and H. Togo, Eur. J. Org Chem., 2019, 2019, 2520–2527 CrossRef CAS.
- G. Yin, X. Wang, Y. Wang, T. Shi, Y. Zeng, Y. Wang, X. Peng and Z. Wang, Org. Biomol. Chem., 2022, 20, 9589–9592 RSC.
- V. S. C. Yeh, Tetrahedron, 2004, 60, 11995–12042 CrossRef CAS.
- P. Wipf, Chem. Rev., 1995, 95, 2115–2134 CrossRef CAS.
- M. K. Muthyala and A. Kumar, J. Heterocycl. Chem., 2012, 49, 959–964 CrossRef CAS.
- X. Wang, Y. Zhu, T. Zhou, W. Yang, H. Fu, H. Chen and M. Ma, ChemistrySelect, 2022, 7, e202201316 CrossRef CAS.
- M. Narender, M. S. Reddy, R. Sridhar, Y. V. D. Nageswar and K. R. Rao, Tetrahedron Lett., 2005, 46, 5953–5955 CrossRef CAS.
- P. Wipf and S. Venkatraman, J. Org. Chem., 1996, 61, 8004–8005 CrossRef CAS PubMed.
- B. Das, V. Saidi Reddy and R. Ramu, J. Mol. Catal. Chem., 2006, 252, 235–237 CrossRef CAS.
- S.-L. You and J. W. Kelly, J. Org. Chem., 2003, 68, 9506–9509 CrossRef CAS PubMed.
- G. W. Kabalka and A. R. Mereddy, Tetrahedron Lett., 2006, 47, 5171–5172 CrossRef CAS.
- T. M. Potewar, S. A. Ingale and K. V. Srinivasan, Tetrahedron, 2007, 63, 11066–11069 CrossRef CAS.
- D. V. Jawale, D. L. Lingampalle, U. R. Pratap and R. A. Mane, Chin. Chem. Lett., 2010, 21, 412–416 CrossRef CAS.
- P. W. Tan and J. Seayad, Tetrahedron Lett., 2019, 60, 151338 CrossRef.
- Z.-B. Xie, N. Wang, L.-H. Zhou, F. Wan, T. He, Z.-G. Le and X.-Q. Yu, ChemCatChem, 2013, 5, 1935–1940 CrossRef CAS.
- K. R. Kiran and S. Divakar, J. Biotechnol., 2001, 87, 109–121 CrossRef CAS PubMed.
- G. D. Yadav and K. M. Devi, Chem. Eng. Sci., 2004, 59, 373–383 CrossRef CAS.
- M. Therisod and A. M. Klibanov, J. Am. Chem. Soc., 1987, 109, 3977–3981 CrossRef CAS.
- L.-Q. Zhang, Y.-D. Zhang, L. Xu, X.-L. Li, X. Yang, G.-L. Xu, X.-X. Wu, H.-Y. Gao, W.-B. Du, X.-T. Zhang and X.-Z. Zhang, Enzyme Microb. Technol., 2001, 29, 129–135 CrossRef CAS.
- B. P. Dwivedee, S. Soni, M. Sharma, J. Bhaumik, J. K. Laha and U. C. Banerjee, ChemistrySelect, 2018, 3, 2441–2466 CrossRef CAS.
- M. Mamaghani and S. Dastmard, Ultrason. Sonochem., 2009, 16, 445–447 CrossRef CAS PubMed.
- A. Mishra, S. Singh, M. A. Quraishi and V. Srivastava, Org. Prep. Proced. Int., 2019, 51, 345–356 CrossRef CAS.
- J.-T. Li, Y. Yin and M.-X. Sun, Ultrason. Sonochem., 2010, 17, 363–366 CrossRef CAS PubMed.
- V. Singh, K. Rajput, A. Mishra, S. Singh and V. Srivastava, Chem. Commun., 2023, 59, 14009–14012 RSC.
- K. Rajput, V. Singh, A. Kamal, H. Kumar Singh, S. Singh and V. Srivastava, New J. Chem., 2023, 47, 22276–22280 RSC.
- H. R. Lobo, B. S. Singh and G. S. Shankarling, Catal. Lett., 2012, 142, 1369–1375 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03290j |
| This journal is © The Royal Society of Chemistry 2024 |