DOI:
10.1039/D4RA03352C
(Paper)
RSC Adv., 2024,
14, 24749-24755
Mesoporous NiMoO4 nanorod electrode materials for flexible and asymmetric energy storage devices
Received
7th May 2024
, Accepted 31st July 2024
First published on 9th August 2024
Abstract
Recently, ternary metal oxides as cathode materials have been the focus of research into supercapacitors owing to their high power density and cost-efficient features. The development of excellent electrode materials is the key to improving supercapacitor total performance. Herein, we report several kinds of NiMoO4 nanostructures grown on nickel foam using a simple hydrothermal strategy. The assembled hybrid devices show an energy density of 35.9 W h kg−1 at a power density of 2708 W kg−1. After repeated charging and discharging cycling and bending tests, they show excellent durability performance and mechanical stability performance.
1 Introduction
The creation of some sustainable energy sources is essential in light of the increasing popularity of flexible, portable electronics.1–5 Thereinto, supercapacitors have attracted wide attention as extremely efficient and highly powerful electrochemical energy storage systems.6–9 However, their energy density is far behind commercial demands, which limits their large-scale applications.10–13 Therefore, it is indispensable to explore desired electrode materials that combine high specific capacity and energy density simultaneously.14–17
Compared to binary transition metal oxides, the ternary NiMoO4 compound shows richer oxidation states because of the synergistic effect of bimetallic interactions.18–20 It results in highly reversible redox reactions and excellent electrochemical performance.21,22 In previous reports, Wang et al. prepared Pr2CrMnO6 electrode materials with a specific capacitance of 177.4 F g−1 at 2 A g−1.23 Co2S4@NiMo-LDH nanocomposites deliver a specific capacitance of 1346 F g−1 at 1 A g−1 and after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, the capacitance retention rate is 80%.24 He's group synthesized a NiMoO4–Ag/rGO product with 1132.8 F g−1 at 1 A g−1, which retains 95% of the initial capacitance after 1000 cycles.25 We also prepared NiMoCo-LDH materials on nickel foam using a hydrothermal route. It showed a specific capacitance of 1284.2 F g−1 at 1 A g−1 and a capacitance retention of 85.3% after 8000 cycles.26 Although much progress has been made in the past two decades, this has been made using excessively complex preparation routines to obtain similar specific capacities. We have reduced the complexity of the manufacturing process and production cost via a simple preparation method to increase the practical value of supercapacitors. NiMoO4 nanorods on NF for supercapacitors possess the advantages of low cost, excellent electrical conductivity, excellent chemical stability, light weight, flexibility and high porosity. The open space between the nanorods allows easy diffusion of electrolytes and avoids the use of polymer binders.27
000 cycles, the capacitance retention rate is 80%.24 He's group synthesized a NiMoO4–Ag/rGO product with 1132.8 F g−1 at 1 A g−1, which retains 95% of the initial capacitance after 1000 cycles.25 We also prepared NiMoCo-LDH materials on nickel foam using a hydrothermal route. It showed a specific capacitance of 1284.2 F g−1 at 1 A g−1 and a capacitance retention of 85.3% after 8000 cycles.26 Although much progress has been made in the past two decades, this has been made using excessively complex preparation routines to obtain similar specific capacities. We have reduced the complexity of the manufacturing process and production cost via a simple preparation method to increase the practical value of supercapacitors. NiMoO4 nanorods on NF for supercapacitors possess the advantages of low cost, excellent electrical conductivity, excellent chemical stability, light weight, flexibility and high porosity. The open space between the nanorods allows easy diffusion of electrolytes and avoids the use of polymer binders.27
In this work, we synthesize several NiMoO4 samples on nickel foam (NF) using a simple hydrothermal process. Many characterization studies are performed to assess the microstructural and capacitance performance of the as-obtained samples. By adjusting the Ni/Mo molar ratio, it is found that the NiMoO4-2 electrode material delivers a specific capacitance of 1194 F g−1 of 1 A g−1 when Ni/Mo = 1:1. Moreover, the as-prepared samples are used as cathodes in the assembly of asymmetric supercapacitors (ASC). They provide an energy density of 35.9 W h kg−1 at a power density of 2708 W kg−1. At the same time, the as-fabricated devices retain 89.2% of initial capacitance after 10000 cycles. Also, the bending experiments demonstrate their outstanding mechanical stability, indicating potential for applications in flexible electronics fields.
2 Experimental section
2.1 Materials
The NF was purchased from Kunshan Guangjia New Material Co., Ltd. Ni(NO3)2·6H2O and urea were obtained from Shanghai Aladdin Biochemical Technology Co. Na2MoO4·2H2O and NH4F were sourced from Tianjin Damao Chemical Reagent Factory. N,N-Dimethylformamide (DMF), acetylene black, polyvinylidene fluoride (PVDF), N-methylpyrrolidone (NMP) were acquired from Taiyuan Lizhiyuan Technology Co., Ltd. All chemicals were directly used in the experiments without purification.
2.2 Preparation of nickel foam (NF)
A piece of NF (4 × 4 cm2) was immersed for 30 minutes in a solution of 1 M HCl to remove the surface grease and the oxide film layer. Subsequently, the nickel foam was washed repeatedly with anhydrous ethanol and deionized water under sonication and dried in an oven at 60 °C for 24 h.
2.3 Synthesis of NiMoO4 samples
NiMoO4 cathode materials were prepared by a simple one-step hydrothermal method. In detail, 0.5816 g of Ni(NO3)2·6H2O, 0.4839 g of Na2MoO4·2H2O, 0.037 g of NH4F and 0.06 g of urea were subsequently dissolved in varied molar ratios of 50 mL of deionized water (Table 1). After magnetic stirring for 0.5 h, a clean NF was placed in a reaction autoclave and heated at 150 °C for 6 h. Following room temperature cooling, the NF was washed three times with anhydrous ethanol and deionized water and calcined in the air at 350 °C for 2 h with a heating rate of 2 °C min−1. The light green materials were prepared. The corresponding samples were named as NiMoO4-1, NiMoO4-2, NiMoO4-3 and Ni(OH)2. The way to determine the mass loading is to take the difference between the mass per unit area of NF with active substance and the mass of NF per unit area. Their mass loadings were 2, 1.2, 1.3 and 1 mg cm−2, respectively.
Table 1 Reagent dosage ratio and load mass of electrode materials
| Labels |
Ni(NO3)2·6H2O |
Na2MoO4·2H2O |
Urea |
NH4F |
Molar ratios |
| Ni(OH)2 |
2 |
0 |
1 |
1 |
2:0 |
| NiMoO4-1 |
4 |
2 |
1 |
1 |
2:1 |
| NiMoO4-2 |
2 |
2 |
1 |
1 |
1:1 |
| NiMoO4-3 |
2 |
4 |
1 |
1 |
1:2 |
2.4 Preparation of activated carbon anode material
Activated carbon, acetylene black and PVDF were mixed in the ratio of 7:2:1 in an onyx mortar and ground by adding anhydrous ethanol. Each time, the ethanol in the agate mortar was completely ground and evaporated before the next grinding. After the third time, NMP solution was added dropwise to the agate mortar and ground well. The resulting slurry was uniformly coated on the pre-treated NF with a spatula and dried in a vacuum oven at 60 °C for 10 h.
2.5 Preparation of PVA–KOH gel electrolyte
To prevent the hydrogels from clumping and agglomerating, slow and uniform magnetic stirring was maintained throughout the preparation process. A beaker containing 20 mL of deionized water was first placed in a water bath. When the temperature of the beaker was heated to 60 °C, 2 g PVA was slowly poured into the beaker. When the temperature of the beaker was heated to 90 °C, 1.5 M KOH solution was slowly dropped into the PVA hydrogel in the water bath. The heating was stopped and the gel was cooled to room temperature. After the hydrogel was clarified, the PVA–KOH hydrogel was obtained.
2.6 Structure characterization
The as-synthesized materials' crystallographic structure was examined using an X-ray diffraction analyzer (XRD, 7000, Shimadzu). In the phase of testing, Cu target Kα radiation with a wavelength of 1.5405 Å was used with the tube voltage set at 40 kV. The range that was covered was 10 to 90°, and the speed of 10° min−1 was maintained. X-ray photoelectron spectra (XPS, ESCALAB250) measurement was performed to investigate the element composition. The X-ray photoelectron spectrometer from Thermo Fisher Scientific, USA, which was equipped by an Al-Kα target was used in this investigation. The experimental data was calibrated with the C 1s (284.8 eV) standard peak. Next, using a transmission electron microscope (TEM, JEM2100 Plus, 200 kV) and a microscope equipped with a scanning electron microscope (SEM, Hitachi-4800), respectively, that we examined their morphological aspects. Finally, a surface and aperture apparatus (JW-TB220A) was used to perform the specific surface area and aperture by the Barrett-Joyner-Hallenda (BJH, JW-TB220A) method.
2.7 Electrochemical characterization
The electrochemical performance of the samples was carried out at in an electrochemical workstation (Shanghai Chenhua CHI600E). The prepared material, Hg/HgO and Pt foils were utilized as working, reference and counter electrodes, respectively. The specific capacity (C g−1) of the electrodes was calculated by following equations.where I is named the discharge current (A), Δt is the discharge time (s) and m refers to the weight of active materials (g).
2.8 Assembly of the flexible supercapacitors
ASC were assembled with NiMoO4-2 samples as positive materials, active carbon as negative ones and PVA–KOH gel as an electrolyte. According to the conservation law of charge (Q+ = Q−), the loading of negative materials is governed by the following equations:| | |
m+/m− = Cm−(ΔV)−/Cm+(ΔV)+
| (3) |
where Q is the charge storage capacity, I is discharge current (A), t is discharge time (s), C is specific capacity (C g−1), m is the mass of the materials (g) and ΔV is the voltage window (V).
The energy density (E) and power density (P) of the NiMoO4-2 samples were calculated as follows:
where Δ
t refers to the discharge time, Δ
V represents the potential window, respectively.
3 Results and discussion
Firstly, we study the crystal structure and phase purity of the sample. From Fig. 1a, the diffraction peaks at 19.25°, 33.06°, 38.54°, 52.10°, 60.24° 62.72°, 70.47° and 73.12° belong to the (001), (100), (101), (102), (003), (111), (103) and (112) planes, respectively, of the Ni(OH)2 phase (JCPDS No. 14-0117). NiMoO4 phases locate the diffraction peaks at 27.3°, 29.85°, 33.40° and 53.54° (JCPDS No. 13-0128).28 It is found that the density of peaks at 2θ of 27.3° and 29.8° gradually weakens as Mo content increases. In addition, there is no appearance of other peaks, indicating the high crystallinity of the prepared samples.
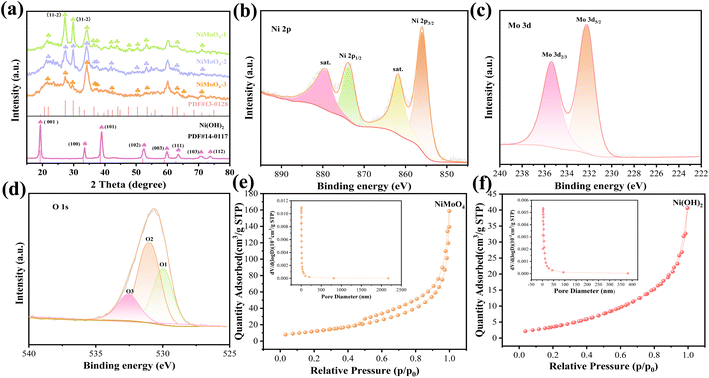 |
| | Fig. 1 Structural characterization. (a) XRD patterns. (b)–(d) XPS spectra of the NiMoO4-2 sample. (e) and (f) N2 adsorption–desorption isotherms (the insets: the pore size distribution curves). | |
XPS is then employed to exam the surface chemical composition of NiMoO4-2 electrode. From the Ni 2p spectra in Fig. 1b, two spin–orbit peaks at 856.1 and 873.8 eV correspond to Ni 2p3/2 and Ni 2p1/2, respectively. The peaks at 879.58 and 861.88 eV can be assigned to the satellites.29 The photoelectron spectra of Mo 3d (Fig. 1c) show the characteristic peaks of Mo 3d3/2 at 232.2 eV and Mo 3d5/2 at 235.4 eV. It proves that Mo6+ possesses a high oxidation state.30 Fig. 1d is divided into three peaks: O1 (530.1 eV), O2 (531.3 eV) and O3 (532.9 eV) peaks. They represent metal–oxygen bonds (M–O), –OH bonds and oxygen species in the adsorbed water/defect, respectively.31 Fig. 1e and f show the N2 isotherms of the two samples with a typical IV isotherm loop. It demonstrates that the samples possess a mesoporous structure and a broad pore-size distribution. The NiMoO4-2 products possess a larger specific area (48.589 m2 g−1) than Ni(OH)2 samples (18.054 m2 g−1). In addition, the total pore volumes of the NiMoO4-2 and Ni(OH)2 samples are 0.175 and 0.056 cm3 g−1, respectively. The results reveal that the introduction of Mo elements increase the specific surface area and active sites.
After that, we study the morphologies and structures of the samples. As shown in Fig. 2a and e, the Ni(OH)2 microplates overlap with each other. They present an average thickness of 100 nm. With the addition of Mo, the crystal structure and growth rate of the NiMoO4 material change compared to the Ni(OH)2 sample, resulting in a change in the morphology. From Fig. 2b and f, when the ratio of Ni/Mo is 2:1, the NiMoO4-1 sample evolves into a rod-like nanostructure with a high specific surface area relative to the nanoplate. When the Ni/Mo molar ratio decreased to 1:1 (Fig. 2c and g), the nanorod density increased. It provides more transmission channels for ion diffusion and storage.32 At this time, the nanorods have a diameter of 100 nm. As shown in Fig. 2d and h, the nanorods begin to aggregate and adhere when the ratio of Ni/Mo is further reduced to 1:2, which blocks the electron charge channels. A low magnification TEM image (Fig. 2i) shows that NiMoO4-2 sample has a diameter of 100 nm, which is consistent with the above SEM results. The corresponding HRTEM (Fig. 2j) reveals that the calculated d-spacing of 0.427 nm is well matched with the NiMoO4-2 crystal phase (JCPDS No. 13-0128). From Fig. 2k, the element mappings demonstrate that Ni, Mo and O elements are uniformly distributed on the surface of the nanorod.
 |
| | Fig. 2 Morphology characterization. (a) and (e) SEM image of the Ni(OH)2 nanosheets. (b) and (f) SEM image of the NiMoO4-1 nanorods. (c) and (g) SEM image of the NiMoO4-2 nanorods. (d) and (h) SEM image of the NiMoO4-3 nanorods. (i) TEM image of NiMoO4-2 samples. (j) HRTEM image. (k) EDS element mappings. | |
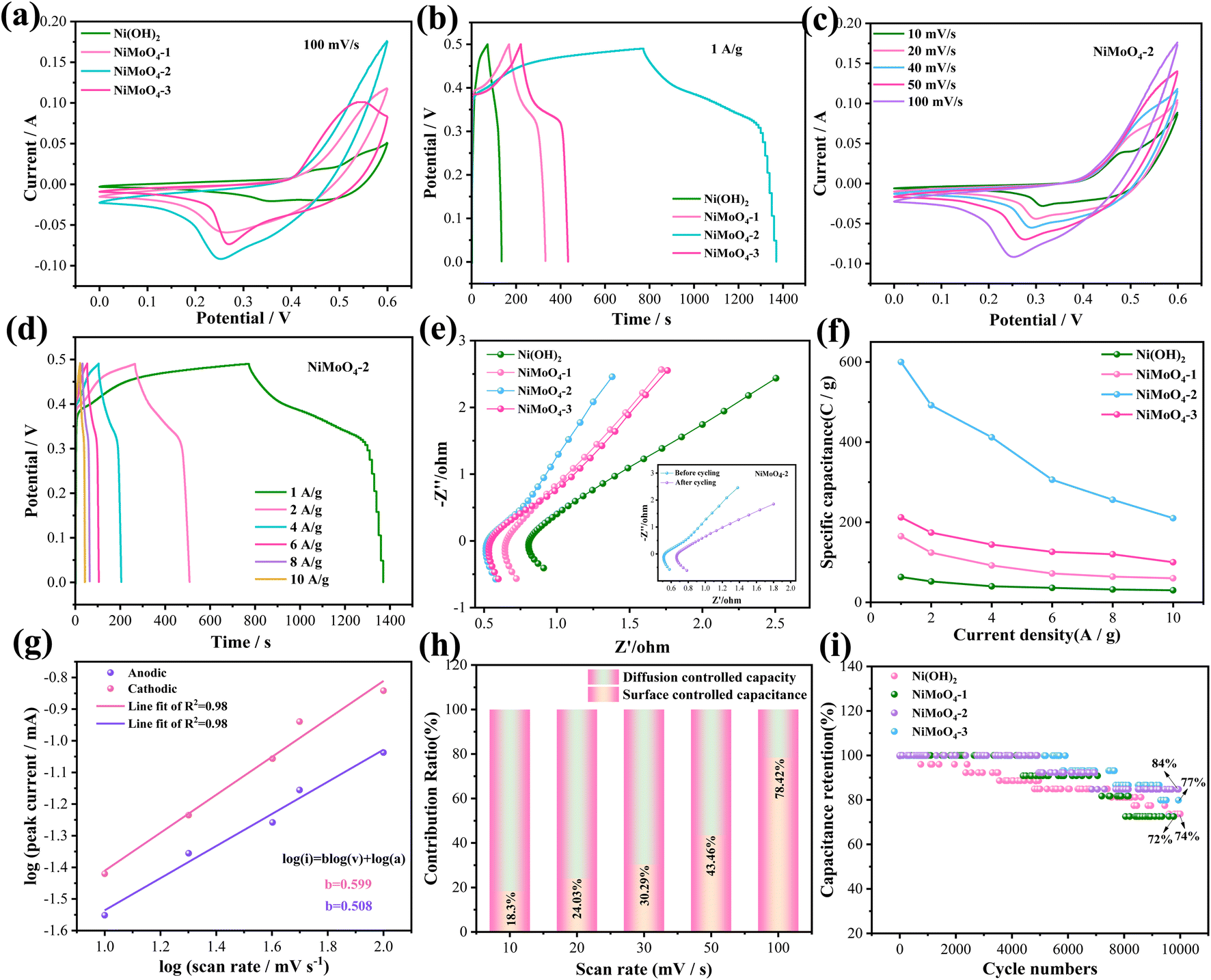
Then we examine the electrochemical performance of all the samples in a three-electrode system. Fig. 3a presents the CV curves of the several electrode materials. The curve area of as-prepared NiMoO4-2 product is larger than that of other products. The reaction dynamics during the electrochemical process can be described as follows:33
| | |
NiMoO4 ↔ Ni2+ + MoO42−
| (5) |
| | |
Ni2+ + 2OH− ↔ Ni(OH)2
| (6) |
| | |
Ni(OH)2 + OH− ↔ NiOOH + H2O + e−
| (7) |
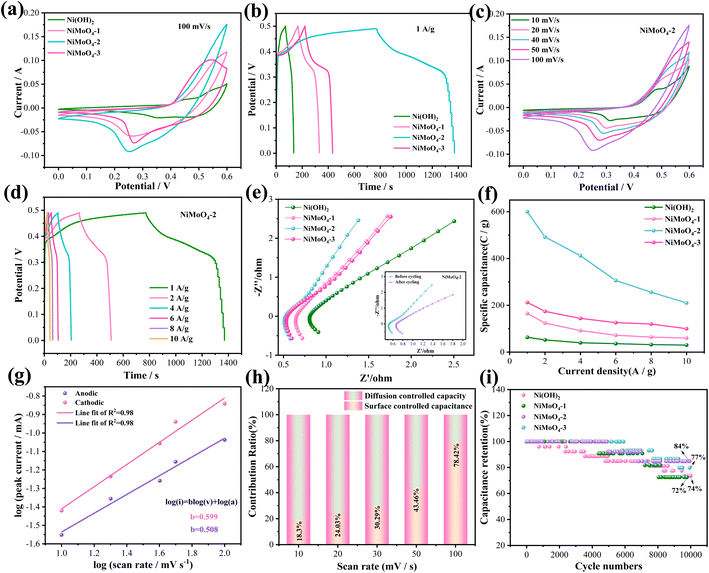 |
| | Fig. 3 Electrochemical performance of the electrode materials. (a) CV curves. (b) GCD curves. (c) CV curves of NiMoO4-2 sample at different scan rates. (d) GCD curves of NiMoO4-2 sample at different current densities. (e) Nyquist plots. (f) Specific capacitance. (g) b value of NiMoO4-2. (h) Contribution ratio between surface and diffusion-limited capacities. (i) Cycling performance. | |
Although Mo is not involved in the electrochemical process, it can improve the conductivity of the hybrid electrode. The GCD curve of four samples at 1 A g−1 are presented in Fig. 3b. It can be found that the specific capacity of the NiMoO4 material shows a growing and subsequently declining pattern as the addition ratio of Mo element increases. The reason for this is that the nanorod consistency increases with the increase of Mo ratio in the early stage, which in turn makes the material obtain more effective active sites and the electrochemical performance is enhanced. At Ni/Mo = 1:1, the nanorod density reaches the optimum and the specific capacity reaches the maximum. With the further increase of the addition ratio of Mo element, the density of the nanorod structure of NiMoO4 material increases again and adhesion occurs, which affects the adequate contact between the electrolyte and the active material as well as the smoothness of the electron charge inlet and outlet channels. Thus, the effective active area of the electrode material was reduced, leading to a decrease in the specific capacitance. Fig. 3c indicates their CV curves at various scan rates. Furthermore, the peak currents (anodic and cathodic) present a slight increase as the scan rate increases. It could be attributed to the reaction kinetics of the electrode materials. As shown on the NiMoO4-2 electrode GCD curves (Fig. 3d), there are obvious charging and discharging platforms at different current densities, indicating their battery-type characteristics. At the current density of 1, 2, 4, 6, 8 and 10 A g−1, the specific capacitance of the electrode materials is 1194, 984, 824, 612, 512 and 420 F g−1, respectively. Electrochemical impedance spectroscopy (EIS) of the devices are shown in Fig. 3e. For a high-frequency region, NiMoO4-2 electrode possesses an internal resistance (Rs) value of 0.53 Ω, which is superior to Ni(OH)2 (0.81 Ω), NiMoO4-1 (0.65 Ω) and NiMoO4-3 (0.55 Ω) electrode materials. The larger the linear slope of the low frequency region, the smaller the diffusion impedance (Zw). The inset shows the EIS of the device before and after the cycling. Fig. 3f indicates the specific capacitance of the samples at 1, 2, 4, 6, 8 and 10 A g−1. NiMoO4-2 electrode delivers three times larger specific capacitance than NiMoO4-1 and NiMoO4-3 electrodes at a current density of 1 A g−1. It also maintains a moderate specific capacitance at high current densities. Fig. 3g depicts the slope values of anode and cathode peaks. The b values of NiMoO4-2 nanorod are 0.508 and 0.599, respectively. This implies that surface capacitance and diffusion control both contribute to the current response of the NiMoO4-2 electrode. Fig. 3h shows the surface capacitive contribution and diffusion controlled one of NiMoO4-2 at different scan rates. The diffusion controlled capacity reduces with the scan rate increasing. This is due to the limited diffusion time. The surface capacitive contributions reach 78.4% at 100 mV s−1, which indicates fast reaction kinetics. Fig. 3i presents the cycling curves of four electrode materials at the current density of 10 A g−1. It is found that NiMoO4-2 electrode material keeps a retention rate of 84% after 10000 cycles, which is better than other electrode materials. NiMoO4-2 electrode material during 1500 times, it is completely saturated by the electrolyte, thus increasing the specific capacity of the material. However, as the number of cycles increased, the internal microstructure of the electrode material collapsed, thus decreasing the specific capacity.
Also, we fabricate ASC with a sandwich structure to explore the practical applications. Fig. 4a indicates the CV curves of active carbon and NiMoO4-2//AC samples at 100 mV s−1. The device reaches a theoretical potential of 1.6 V. From Fig. 4b, the CV curves of the device retain similar shapes and prove to have good reversibility as scan rates increase. The corresponding GCD curves (Fig. 4c) reveal that the device presents a specific capacitance of 115 F g−1 at a current density of 1 A g−1. Fig. 4d indicates the EIS of the device with a Rs value of 1.28 Ω, which proves its excellent electrical conductivity and the inset above left shows the appropriate equivalent circuit. The lower right inset shows the increase in the Rs value to 1.99 after cycling, indicating the decay of the capacitance. As shown in Fig. 4e, the device achieves the energy density of 36.03 W h kg−1 when the power density is 2699 W kg−1, and it can retain 25.5 W h kg−1 at 16200 W kg−1. Table 2 lists the power and energy densities of several reported devices.34–37 It demonstrates that our assembled device is dominant compared with the others. Fig. 4f shows the capacitance contribution of NiMoO4-2//AC at different scan rates. The role of diffusion control is gradually substituted by surface-controlled behavior. The dominance of surface-controlled capacitance can be attributed to the limited ion diffusion time at high scan rates.
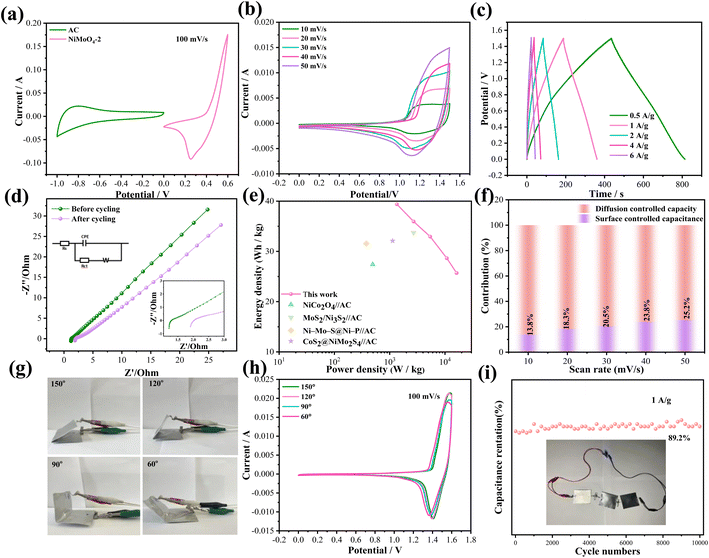 |
| | Fig. 4 Electrochemical performance of the asymmetric device. (a) CV curves at different scan rates. (b) CV curves with different scan rates and voltage windows. (c) GCD curves. (d) Nyquist plots. (e) Ragone plots. (f) Contribution ratio between surface and diffusion-limited capacities. (g) Digital photographs of the folded device. (h) CV curves with various bending angles. (i) Cycling performance. | |
Table 2 Comparison of the energy density and power density of several devices
| Electrode materials |
Energy density (W h kg−1) |
Power density (W kg−1) |
Ref. |
| NiCo2O4//AC |
27.4 |
493.2 |
34 |
| MoS2/Ni3S2//AC |
33.75 |
2700 |
35 |
| Ni–Mo–S@Ni–P//AC |
31.57 |
377.59 |
36 |
| CoS2@NiMo2S4//AC |
32.1 |
1130.3 |
37 |
| NiMoO4//AC |
36.03 |
2699 |
This work |
Finally, we explore the mechanical stability of the device. Fig. 4h shows the digital photos of the device bending at different angles. It is observed that the shapes of the CV curves maintain similar shapes with essentially the same area of integration when the device is twisted to 150°, 120°, 90° and 60°, respectively (Fig. 4g). From the cycling curves shown in Fig. 4i, the device retains 89.2% of its original capacitance at a current density of 1 A g−1 after 10000 cycles. A blue LED lamp may be lit by three ASC devices connected in series for almost 9 min, demonstrating their potential applications in future energy storage fields.
4 Conclusions
In summary, we have prepared several NiMoO4 cathode materials with different Ni/Mo molar ratios. The prepared NiMoO4-2 samples demonstrate superior electrochemical performance with a ratio of Ni and Mo of 1:1. The electrode materials show improved specific capacity and stability during cycles due to the synergistic effect of high conductivity Ni and Mo ions. In addition, the assembled ASC delivers high energy and power density and shows excellent mechanical stability. The reported synthetic protocol can be widened to prepare certain additional transition metals, such as sulfide and phosphide, for high-performance electrode materials for flexible capacitors.
Data availability
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
Author contributions
Qi He: conceptualization, software, data curation, writing-original draft preparation, methodology, Xingyu Liu: visualization, xiang wu: supervision, writing-reviewing and editing.
Conflicts of interest
The authors declare that they have no known competing financial interests.
References
- J. Jiang and J. Liu, Interdiscip. Mater., 2022, 1, 116–139 CrossRef.
- X. W. Wang, Y. C. Sun, W. C. Zhang and X. Wu, Chin. Chem. Lett., 2023, 34, 107593 CrossRef CAS.
- C. Costentin, Adv. Energy Sustainability Res., 2024, 5, 2300242 CrossRef CAS.
- Y. Liu and X. Wu, J. Energy Chem., 2023, 87, 334–341 CrossRef CAS.
- A. Andola, R. R. Pandey, Y. Kashyap, H. Pandey, H. Nakanishi and R. K. Pandey, Nanomaterials, 2023, 7, 1701–1712 Search PubMed.
- Y. B. Chen, J. J. You, Y. H. Chen, L. A. Ma, H. X. Chen, Z. H. Wei, X. Y. Ye and L. Zhang, CrystEngComm, 2022, 24, 5238–5250 RSC.
- K. Zhang, X. Gao, F. Yao, Y. Q. Xie, H. Bai, Y. J. Sun, R. Liu and H. Y. Yue, J. Colloid Interface Sci., 2023, 650, 105–111 CrossRef CAS PubMed.
- J. X. Liu, S. Q. Zhao and X. Wu, Chin. Chem. Lett., 2024, 35, 109059 CrossRef CAS.
- Y. T. Li, C. R. Zhao, A. Abdukader and X. Wu, RSC Adv., 2024, 14, 9594–9601 RSC.
- J. J. Zhang and X. Wu, Int. J. Miner., Metall. Mater., 2024, 31, 179–185 CrossRef CAS.
- M. Z. Dai, D. P. Zhao, H. Q. Liu, X. F. Zhu, X. Wu and B. Wang, ACS Appl. Energy Mater., 2021, 4, 2637–2643 CrossRef CAS.
- H. F. Huang, C. Li, F. X. Yan, F. L. Yuan, X. Q. Liang, W. Z. Zhou and J. Guo, Appl. Surf. Sci., 2023, 623, 157079 CrossRef CAS.
- J. Y. Liang, S. M. Qin, S. Luo, D. Pan, P. F. Xu and J. Li, J. Colloid Interface Sci., 2024, 653, 504–516 CrossRef CAS.
- W. Chen, L. C. Yan, Z. H. Song and L. Xiao, Electrochim. Acta, 2024, 144329 CrossRef CAS.
- T. F. Yi, L. Y. Qiu, J. Mei, S. Y. Qi, P. Cui, S. Luo, Y. R. Zhu, Y. Xie and Y. B. He, Sci. Bull., 2020, 65, 546–556 CrossRef CAS.
- N. Bu, J. Xiang, S. Loy, W. D. Yang, Y. F. Di, R. D. Zhao, F. Wu, D. Ma, M. T. Li and J. Li, Mater. Chem. Phys., 2022, 288, 126419 CrossRef CAS.
- B. Xing, J. Zhao, S. Jiang, M. Pang, Q. L. Pan and Y. Geng, Synth. Met., 2021, 271, 116638 CrossRef CAS.
- X. Y. Huai, J. X. Liu and X. Wu, Crystengcomm, 2023, 25, 5310–5315 RSC.
- A. S. Keshari and P. Dubey, Dalton Trans., 2022, 51, 3992–4009 RSC.
- X. Y. Zhang, Z. Li, Z. Y. Yu, L. Wei and X. Guo, Appl. Surf. Sci., 2020, 505, 144513 CrossRef CAS.
- P. Bandyopadhyay, G. Saeed, N. H. Kim and J. H. Lee, Chem. Eng. J., 2020, 384, 123357 CrossRef CAS.
- Y. Zeng, J. Z. Liao, B. B. Wei, Z. Huang, W. J. Zhu, J. X. Zheng, H. F. Liang, Y. Z. Zhang and Z. C. Wang, Chem. Eng. J., 2021, 409, 128297 CrossRef CAS.
- R. K. Muddelwar, J. Pani, A. B. Lad, K. U. Kumar, V. M. Gaikwad and H. Borkar, Mater. Chem. Phys., 2023, 302, 127726 CrossRef CAS.
- C. Cheng, Y. J. Zou, F. Xu, C. L. Xiang, Q. L. Sui, J. Zhang, L. X. Sun and Z. M. Chen, J. Energy Storage, 2022, 52, 105049 CrossRef.
- B. J. Huang, D. C. Yao, J. J. Yuan, Y. R. Tao, Y. X. Yin, G. Y. He and H. Q. Chen, J. Colloid Interface Sci., 2022, 606, 1652–1661 CrossRef CAS PubMed.
- H. Q. Liu, D. P. Zhao, Y. Liu, Y. L. Tong and X. Wu, Sci. China Mater., 2020, 64, 581–591 CrossRef.
- C. C. Wang, D. T. Wu, Y. Qin and Y. Kong, Chem. Commun., 2021, 57, 4019–4022 RSC.
- M. M. Wang, Q. Zhang, Y. L. Wang and X. Liu, Inorg. Chem., 2023, 62, 17555–17564 CrossRef CAS PubMed.
- G. G. Wang, Q. Y. Chen, J. Zhang, X. G. An, Q. Liu, L. Xie, W. T. Yao, X. P. Sun and Q. Q. Kong, J. Colloid Interface Sci., 2023, 647, 73–80 CrossRef CAS.
- Y. C. Sun, X. W. Wang, A. Umar and X. Wu, RSC Adv., 2022, 12, 14858–14864 RSC.
- X. Y. Liu, M. D. Wang, A. Umar and X. Wu, Dalton Trans., 2023, 52, 10457–10464 RSC.
- M. P. Dabir, S. M. Masoudpanah and M. Mamizadeh, J. Energy Storage, 2023, 70, 107951 CrossRef.
- Y. B. Chen, J. J. You, Y. H. Chen, L. A. Ma, H. X. Chen, Z. H. Wei, X. Y. Ye and L. Zhang, CrystEngComm, 2022, 24, 5238–5250 RSC.
- D. P. Zhao, F. Hu, A. Umar and X. Wu, New J. Chem., 2018, 42, 7399–7406 RSC.
- J. X. Liu and X. Wu, J. Solid State Electrochem., 2023, 27, 2571–2577 CrossRef CAS.
- X. Y. Lei, S. C. Ge, T. Y. Yang, Y. L. Lu, Y. L. Chueh and B. Xiang, J. Power Sources, 2020, 477, 229022 CrossRef CAS.
- K. Zhang, X. Gao, F. Yao, Y. Q. Xie, H. Bai, Y. J. Sun, R. R. Liu and H. Y. Yue, J. Colloid Interface Sci., 2023, 650, 105–111 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2024 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*