
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis, in silico, and in vitro evaluation of pyrrol-2-yl-phenyl allylidene hydrazine carboximidamide derivatives as AChE/BACE 1 dual inhibitors†
Amit Sharma a,
Santosh Rudrawarbc,
Ankita Sharmad,
Sandip B. Bharated and
Hemant R. Jadhav*a
a,
Santosh Rudrawarbc,
Ankita Sharmad,
Sandip B. Bharated and
Hemant R. Jadhav*a
aPharmaceutical Chemistry Research Laboratory, Department of Pharmacy, Birla Institute of Technology and Science Pilani, Pilani Campus, Vidya Vihar, Pilani, RJ 333031, India. E-mail: hemantrj@pilani.bits-pilani.ac.in; Fax: +91-1596-244183; Tel: +91-1596-255 506
bInstitute for Glycomics, Griffith University, Gold Coast 4222, Australia
cSchool of Pharmacy and Medical Sciences, Griffith University, Gold Coast 4222, Australia
dNatural Products and Medicinal Chemistry Division, CSIR-Indian Institute of Integrative Medicine, Canal Road, Jammu – 181110, India
First published on 23rd August 2024
Abstract
Alzheimer's disease (AD) manifests as a progressive decline in cognitive function and mental behavior. Targeting two crucial enzymes associated with AD, acetylcholinesterase (AChE) and BACE 1 (Beta-site APP Cleaving Enzyme), in combination, holds promise for therapeutic breakthroughs. In this study, 40 derivatives of pyrrol-2-yl-phenyl allylidene hydrazine carboximidamide were designed based on prior research. These derivatives underwent synthesis and assessment for their inhibitory potential against AChE and BACE 1. ADME predictions indicated favorable physicochemical properties for these compounds. The findings offer novel avenues for exploring the dual inhibition of AChE and BACE 1 as a promising therapeutic strategy for AD.
1. Introduction
Alzheimer disease (AD) is a progressive neurodegenerative disorder causing cognitive decline and memory loss. AD is the most common cause of dementia that slowly degrades thinking and social interactions due to degradation of brain cells.1 Worldwide, as of 2023, around 55 million people have dementia, with over 60% living in low- and middle-income countries. As the proportion of older people in the population is increasing in nearly every country, this number is expected to rise to 78 million by 2030 and 139 million by 2050.2,3 Despite significant research, the underlying causes and precise molecular processes involved in AD remain unclear. At present, there are no therapeutic options that can prevent or permanently reduce the advancement of the disease. AD is presently treated using cholinesterase inhibitors (such as donepezil, galantamine, and rivastigmine) and N-methyl-D-aspartate (NMDA) receptor antagonist (memantine) (Fig. 1). As these treatments only provide relief from symptoms and do not modify the disease's progression, there is a pressing need for the development of disease-modifying agents for the treatment of AD.4 Various pathological factors have been identified as major contributors to the development and progression of AD.5 There is characteristic reduction in cholinergic neurotransmission caused by lower levels of acetylcholine (ACh). Other typical pathological factors of AD include the accumulation of senile plaques, which are made up of β-amyloid (Aβ) aggregates, and the formation of neurofibrillary tangles comprised of hyperphosphorylated tau protein 6.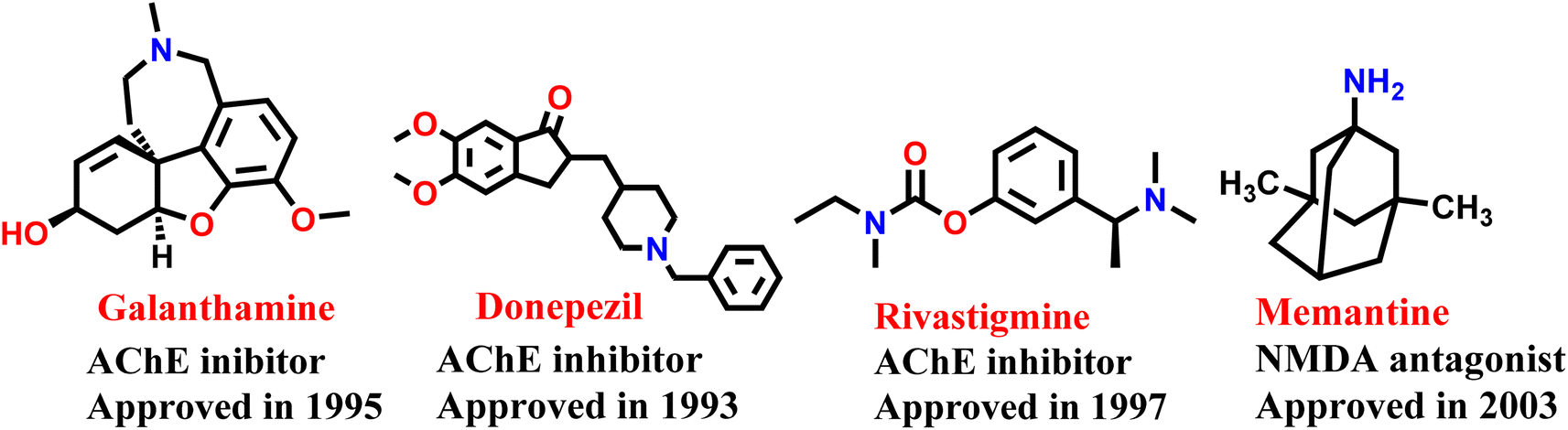 | ||
| Fig. 1 US FDA approved drugs for the treatment of Alzheimer's disease. | ||
The two most common hypotheses for AD are the amyloid pathway and the cholinergic pathway. The build-up of Aβ occurs when the amyloid precursor protein (APP) is broken down and leads to the formation of Aβ oligomers and plaques through the amyloidogenic pathway. In the nonamyloidogenic pathway, APP is broken down by α-secretase into fragments that are soluble in water, making it possible for them to be eliminated from the body. The formation of insoluble and waxy Aβ fragments (amyloid β 1–42 or Aβ 1–42), caused by the breakdown of APP through the activity of β-secretase (also called Aβ precursor cleaving enzyme or BACE1), is the hallmark of the amyloidogenic pathway.7 Targeting BACE 1 is crucial in the development of drugs for Alzheimer's disease leading to the inhibition of spontaneous aggregation of Aβ 1–42, a protein that tends to aggregate on its own.8 Aducanumab, an amyloid targetting monoclonal antibody, is the only recently approved therapy for modifying the course of Alzheimer's disease. While the effectiveness of aducanumab in slowing down the progression of Alzheimer's disease is still being debated, it has been shown to enhance memory and cognitive function impairments that are connected to Aβ plaques.9
Cholinesterase enzymes break Ach into choline and acetic acid. Found throughout the body, there are of two types: acetyl cholinesterase (AChE) and butyryl cholinesterase (BChE).10 The use of cholinesterase inhibitors is aimed at addressing cognitive dysfunction caused by the degradation of ACh.11 While AChE is primarily known for its symptom-relieving effects, studies have revealed that it also plays non-enzymatic roles in facilitating neurite growth, differentiation, adhesion, and synaptic maintenance.12 As Alzheimer's disease progresses, there is a notable reduction in the levels of acetyl choline. Moreover, it is recently reported that AChE triggers the formation of Aβ fibrils and plaques in vitro.13,14 The process is facilitated by the interaction between Aβ and AChE's peripheral anionic site (PAS). Consequently, compounds that can inhibit both the enzyme's catalytic active site (CAS) and peripheral binding site (PAS), referred to as dual-binding site AChE inhibitors, have been developed as potential anti-AD agents (Donepezil, Tacrine, Physostigmine, Decamethonium etc.).15–17
Illnesses with complex pathophysiology, such as AD, may necessitate treatment that concurrently affects multiple biological targets (polypharmacology). The inability to develop effective anti-AD medication has cast doubt on the conventional “one drug, one target, one illness” approach.18 Consequently, the exploration of dual- or multi-target directed ligands (MTDLs) has emerged as a potential paradigm for identifying novel small-molecular weight compounds, wherein a single molecule is used to target two or more enzymes simultaneously. It could be advantageous in effectively slowing down the progression of the disease, along with providing relief for its symptoms.19,20 Therefore, the present work was undertaken to identify dual AChE and BACE1 inhibitor.
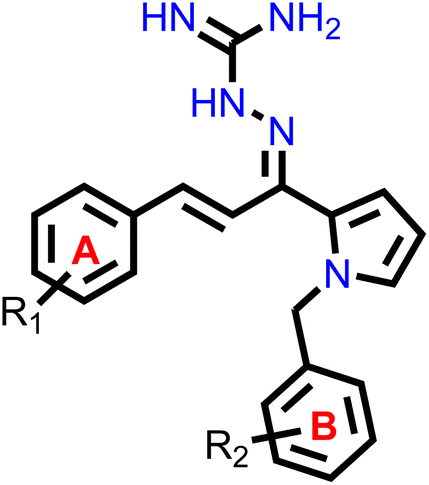
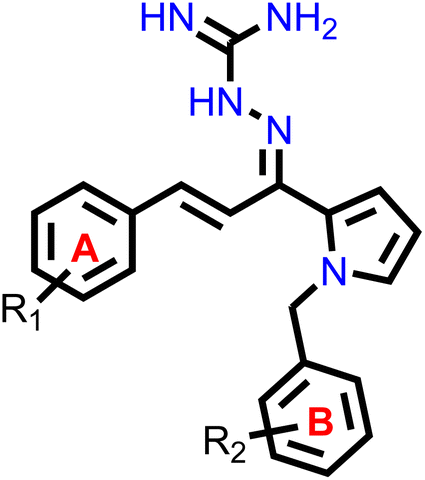
Our group had reported a novel class of allylidene hydrazine carboximidamide derivatives as potent BACE 1 inhibitors, having aminoguanidine substitution on 3 atom allyl linker with two aromatic groups on either side. The most potent compound had IC50 of 6.423 μM against BACE 1 (23). It was deduced that two aromatic rings separated by three carbon linker are needed to occupy the S1 and S3 cavity, while guanidine functionality binds with catalytic aspartate dyad of BACE 1. Recently, compounds based on N-benzyl triazole are reported to exhibit dual inhibition of AChE and BACE-1 with IC50 values of 1.43 and 10.9 μM, respectively.21 Donepezil also has a benzyl substitution and has two aromatic rings separated by a linker.22 Considering these similarities, it was inferred that incorporating additional ring in our BACE 1 inhibitor could impart AChE inhibitor property. It is reported that N-benzylated heterocyclic fragments can bind the CAS site of AChE.23 Therefore, N-benzyl pyrrole was considered on one side of 3 carbon linker having guanidine substitution as could help the structure to interact with the catalytic sites of AChE as well as BACE 1 (Fig. 2).24 It was hypothesized that –NH group of guanidine could form a hydrogen bond with ASP 32 and ASP 228 (key amino acid of BACE 1), ring A and B to occupy the S1, S3 cavities of BACE 1, and occupancy of PAS by N-benzyl pyrrole and PAS of AChE of other aromatic ring. Herein, this study of design, synthesis, and in vitro evaluation of pyrrol-2-yl-phenyl allylidene hydrazine carboximidamide derivatives as potential dual inhibitors of AChE and BACE 1 is presented.
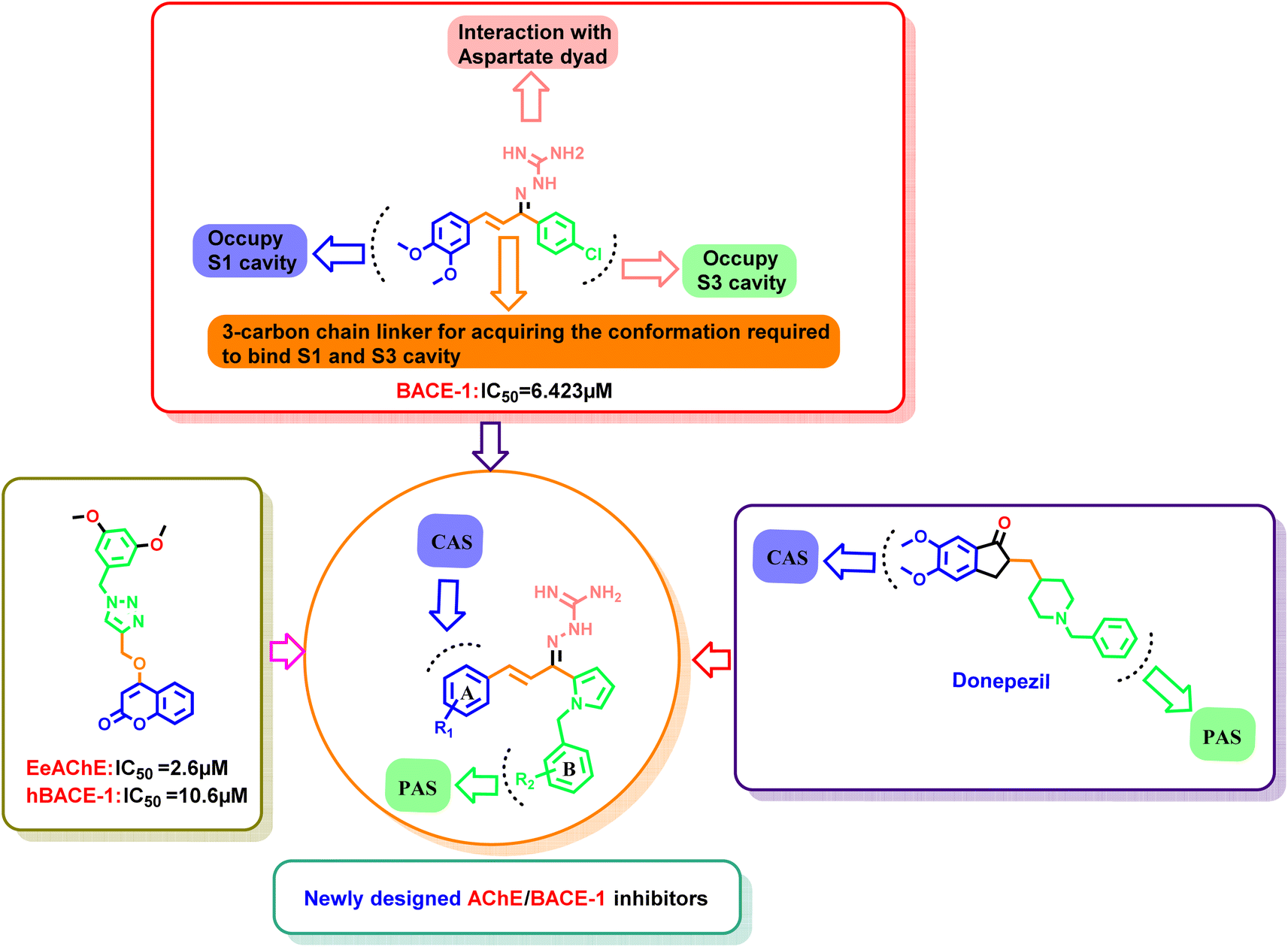 | ||
| Fig. 2 Design strategy using molecular hybridization approach. | ||
2. Results and discussion
2.1. Design of novel dual AChE/BACE 1 inhibitors
The X-ray crystal structure of AChE (4EY7) indicates two binding sub-sites-PAS and CAS, both of which are involved in binding acetylcholine. The PAS is noticeably separate from the choline binding pocket, while the CAS is believed to resemble the catalytic sub-sites found in other serine hydrolases. The AChE's active site features a catalytic triad consisting of Ser200, His440, and Glu327 located at the bottom of the pocket. Studies conducted through both kinetic and chemical analyses have suggested the involvement of a histidine residue in the active site. AChE's active site can be described as a long and narrow gorge, spanning approximately 20 Å in length. This pocket extends over halfway into the enzyme and gradually widens as it approaches the base. The gorge pocket contains only a few acidic amino acid residues; which includes Asp285, Glu273, Asp72 hydrogen bonded to Tyr334 and Glu199 respectively. Thus, AChE inhibitor must occupy both CAS (Trp86, Tyr337, Trp439 and Tyr341) and PAS (Trp286, Phe297, Phe295, Phe338, Tyr124 and Tyr72) (Fig. 3).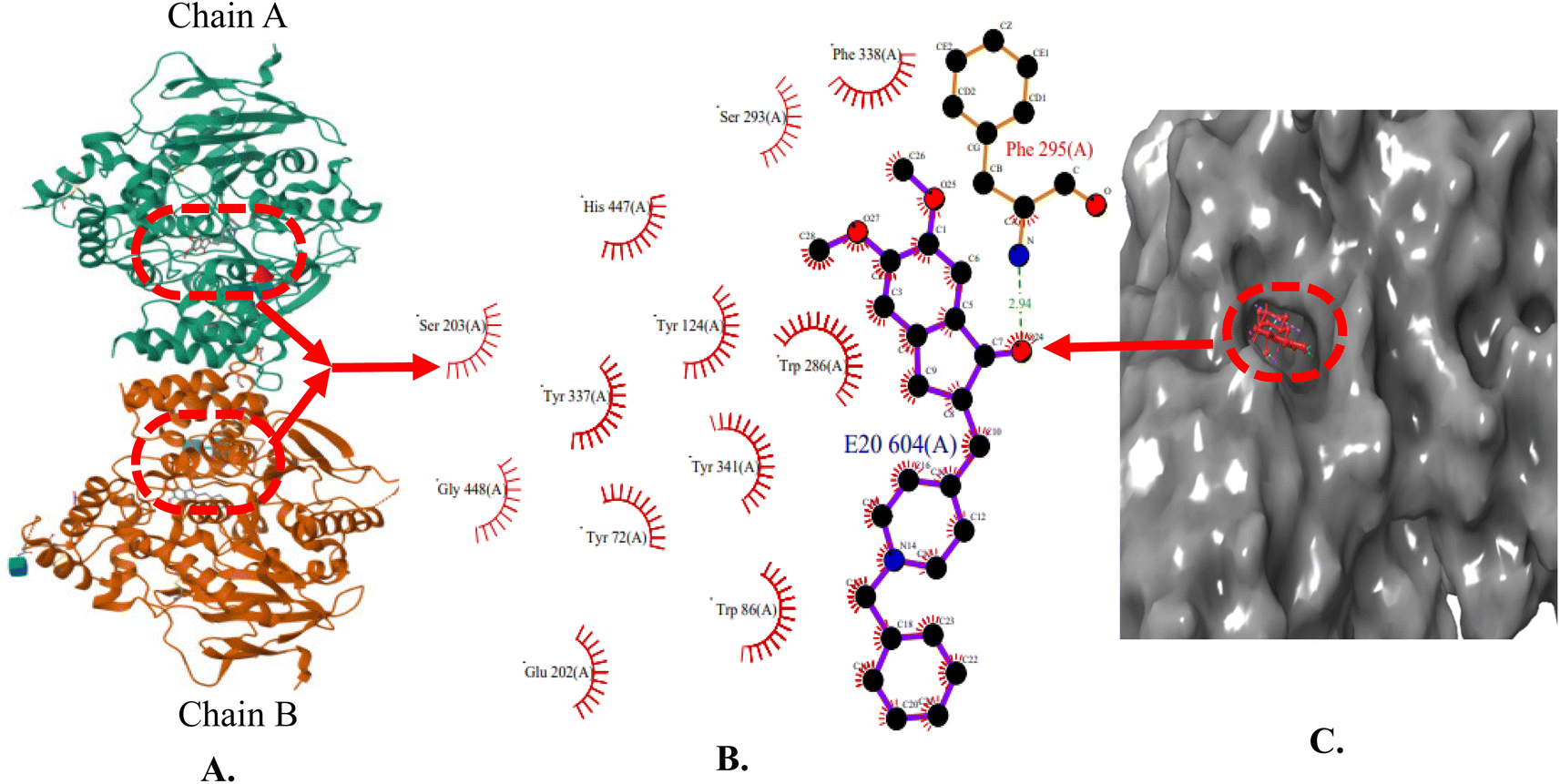 | ||
| Fig. 3 (A) X-ray crystallographic structure of AChE (4EY7, ribbon form); (B) prominent amino acid residues surrounded by co-crystallized ligand (E20) bound to chain A and B; (C) surface model of 4EY7 bounded co-crystallized ligand (red color). | ||
The X-ray crystal structure of beta-secretase (PDB ID: 6UWP) was used for gaining insight into the fundamental groups that are necessary for inhibiting BACE 1. In order to form H-bonding interactions with the catalytic residues (Asp-228 and Asp-32), a H-bond donor is needed. Further, large hydrophobic pockets (S1, S3, S2′) are present, wherein the side chains of Tyr71, Phe108, Trp115, Ile118, and Leu30 collectively shape the S1 hydrophobic cleft. Similarly, the S3 pocket has the side chains of Trp115 and Ile110, as well as the main chains of Gln12, Gly11, Gly230, Thr231, Thr232, and Ser35. S2′ consists of the amino acid residues Ile126, Trp76, Val69, Arg128, and Tyr198 (Fig. 4) [https://www.rcsb.org/].
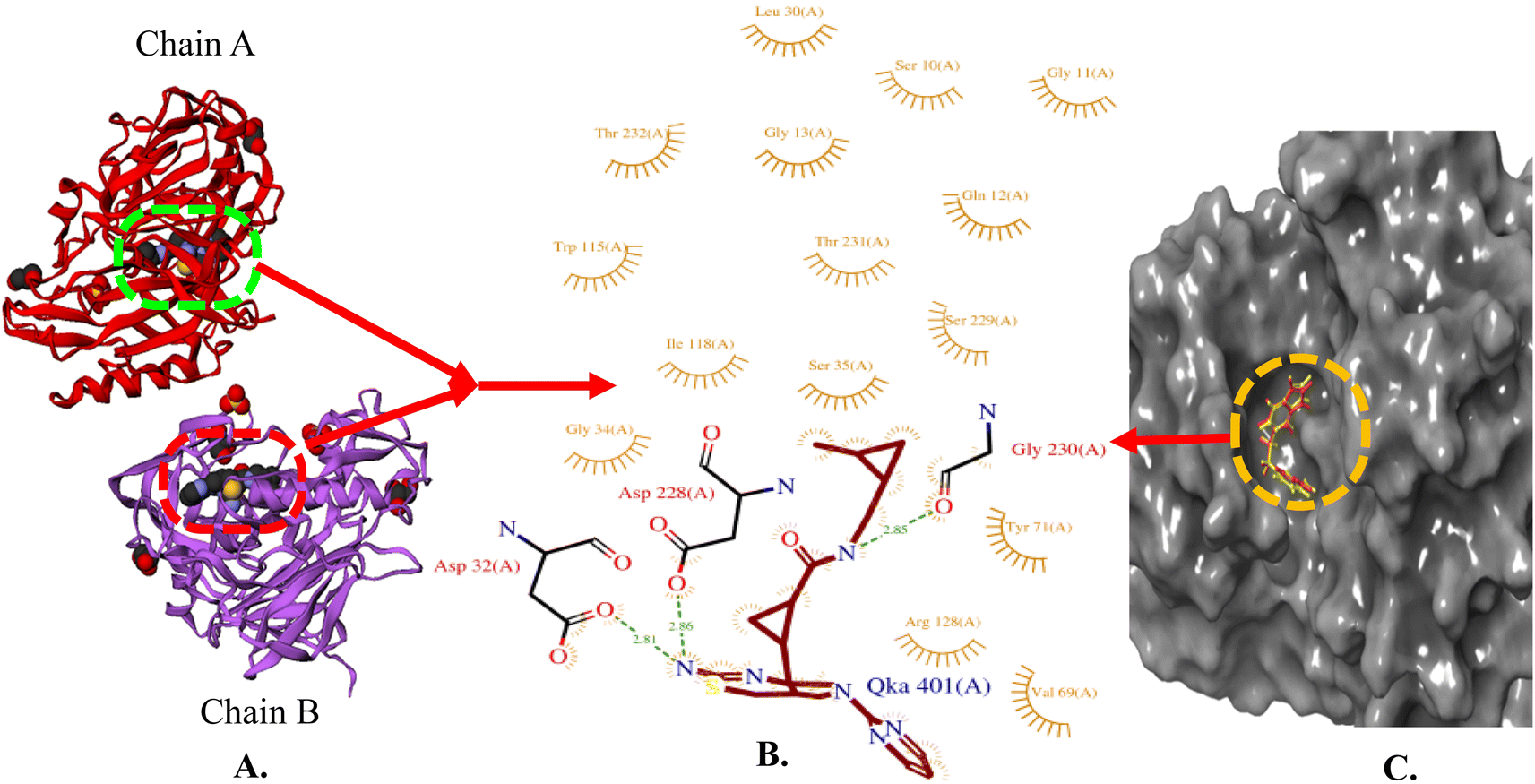 | ||
| Fig. 4 (A) X-ray crystallographic structure of BACE 1 (6UWP, ribbon form); (B) prominent amino acid residues surrounded by co-crystallized ligand (QKA) bound to chain A and B; (C) surface model of 6UWP bounded co-crystallized ligand (red color). | ||
Our previous results with allylidene hydrazinecarboximidamide derivatives showed that for optimal BACE 1 inhibition, a three-atom linker with aromatic groups on both ends to occupy the S1 and S3 cavities, as well as a hydrogen bond donor for binding to the aspartate dyad is essential.25 Previous studies have shown that N-benzylated heterocyclic fragments have the ability to target the CAS site in AChE,23 which aligned with objective of introducing AChE inhibition. Thus, instead of substituting phenyl ring on either sides of linker, N-benzylated pyrrole on one end (ring B), substituted phenyl ring (ring A) on other end, and allylidene hydrazine scaffold on the linker was hypothesized.
2.2. Chemistry
The synthesis of all the final compounds 1a–j, 2a–r, 3a–l were carried out as per the procedure detailed in Scheme 1. In the first step, N-benzyl pyrrole was benzylated with substituted benzyl bromides using base (potassium hydroxide) in the presence of dimethyl formamide (DMF). The Claisen–Schmidt condensation was followed by the addition of substituted benzaldehydes in alcoholic basic condition resulting in the formation of chalcone derivatives. Nucleophilic addition of aminoguanidine hydrochloride to the chalcones provided the final compounds. The progress of the reactions was monitored by Thin Layer Chromatography (TLC) analysis (Silica gel G60 F254, Merck). 1H and 13C NMR spectra were recorded on a Bruker Avance II 400 spectrometer (400 MHz) using DMSO-d6 as solvent. HRMS-ESI+ mode analysis was performed on Agilent Technologies 6545, Version-Q-TOF B.06.01(B6172 SP1).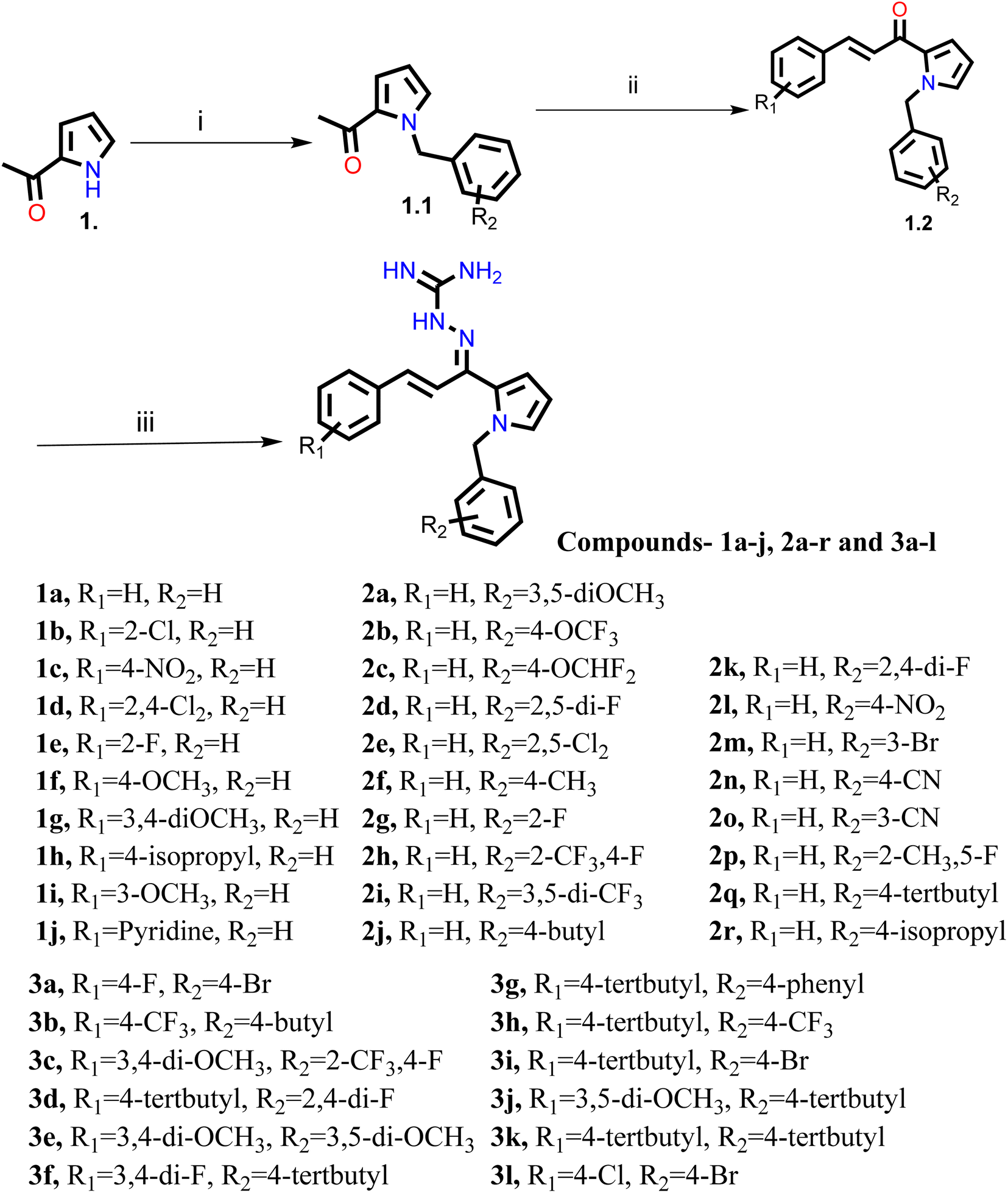 | ||
| Scheme 1 Reagents and conditions: (i) substituted benzyl bromide, KOH, DMF, ultra-sonication, 25–27 °C, 1 h (ii) substituted benzaldehyde, ethanolic NaOH, ultra-sonication, 20–25 °C, 30 min (iii) aminoguanidine·HCl, conc. HCl, EtOH, ultra-sonication, 30–35 °C, 2.0 h. | ||
2.3. AChE/BACE 1 inhibitory activity
The inhibitory activities of pyrrol-2-yl-phenyl allylidene hydrazine carboximidamides 1a–j, 2a–r, and 3a–l against AChE and BACE 1 were evaluated by the Ellman assay and fluorescence resonance energy transfer (FRET) assay, respectively using donepezil and β-secretase inhibitor IV as reference standards. The results are shown in Tables 1–3.
|
||||
|---|---|---|---|---|
| Compound | R1 | R2 | eeAChE | hBACE 1 |
| IC50 (μM) ± SD | IC50 (μM) ± SD | |||
| Results are presented as mean ± SD, n = 3, eeAChE indicates AChE from electric eel and hBACE 1 indicates human BACE 1. | ||||
| 1a | H | H | 215.30 ± 0.14 | 169.90 ± 1.27 |
| 1b | 2-Cl | H | 173.95 ± 0.92 | 183.75 ± 0.92 |
| 1c | 4-NO2 | H | 43.25 ± 0.41 | 194.30 ± 0.99 |
| 1d | 2,4-Di-Cl | H | 55.45 ± 0.91 | 207.95 ± 1.91 |
| 1e | 2-F | H | 172.30 ± 1.56 | 174.45 ± 1.48 |
| 1f | 4-OCH3 | H | 139.15 ± 0.35 | 173.05 ± 1.06 |
| 1g | 3,4-Di-OCH3 | H | 145.90 ± 2.40 | 114.20 ± 1.70 |
| 1h | 4-Isopropyl | H | 203.80 ± 1.70 | 107.55 ± 1.20 |
| 1i | 3-OCH3 | H | 206.65 ± 0.49 | 134.05 ± 0.21 |
| 1j | Pyridine | H | 95.60 ± 2.40 | 13.74 ± 0.77 |
|
||||
|---|---|---|---|---|
| Compound | R1 | R2 | EeAChE | hBACE 1 |
| IC50 (μM) ± SD | IC50 (μM) ± SD | |||
| Results are presented as mean ± SD, n = 3, eeAChE indicates AChE from electric eel and hBACE 1 indicates human BACE 1. | ||||
| 2a | H | 3,5-Di-OCH3 | 231.21 ± 2.26 | 95.75 ± 3.45 |
| 2b | H | 4-OCF3 | 231.66 ± 1.27 | 86.19 ± 0.38 |
| 2c | H | 4-OCH-di-F | 195.83 ± 0.99 | 15.35 ± 0.24 |
| 2d | H | 2,5-Di-F | 153.34 ± 1.98 | 107.45 ± 3.04 |
| 2e | H | 2,5-Di-Cl | 75.57 ± 3.09 | 178.25 ± 0.92 |
| 2f | H | 4-CH3 | 176.75 ± 0.64 | 85.74 ± 2.43 |
| 2g | H | 2-F | 163.35 ± 3.18 | 100.83 ± 0.57 |
| 2h | H | 2-CF3,4-F | 57.09 ± 3.91 | 74.24 ± 1.80 |
| 2i | H | 3,5-Di-CF3 | 77.56 ± 0.66 | 139.11 ± 1.13 |
| 2j | H | 4-Butyl | 55.36 ± 4.19 | 132.75 ± 1.22 |
| 2k | H | 2,4-Di-F | 61.12 ± 1.48 | 133.42 ± 1.84 |
| 2l | H | 4-NO2 | 43.57 ± 0.66 | 193.35 ± 0.07 |
| 2m | H | 3-Br | 51.14 ± 0.22 | 134.43 ± 3.11 |
| 2n | H | 4-CN | 56.05 ± 1.87 | 112.12 ± 0.71 |
| 2o | H | 3-CN | 75.42 ± 1.03 | 147.83 ± 0.28 |
| 2p | H | 2-CH3,5-F | 80.13 ± 0.16 | 145.03 ± 0.28 |
| 2q | H | 4-Tertbutyl | 48.52 ± 0.23 | 167.51 ± 0.71 |
| 2r | H | 4-Isopropyl | 50.37 ± 0.91 | 191.26 ± 1.41 |
|
||||
|---|---|---|---|---|
| Compound | R1 | R2 | EeAChE | hBACE 1 |
| IC50 (μM) ± SD | IC50 (μM) ± SD | |||
| Results are presented as mean ± SD, n = 3, eeAChE indicates AChE from electric eel and hBACE 1 indicates human BACE 1. | ||||
| 3a | 4-F | 4-Br | 86.09 ± 0.33 | 165.65 ± 1.34 |
| 3b | 4-CF3 | 4-Butyl | 57.35 ± 1.12 | 204.63 ± 0.42 |
| 3c | 3,4-Di-OCH3 | 2-CF3,4-F | 58.19 ± 0.44 | 203.75 ± 2.33 |
| 3d | 4-Tertbutyl | 2,4-Di-F | 104.31 ± 0.14 | 221.84 ± 1.27 |
| 3e | 3,4-Di-OCH3 | 3,5-Di-OCH3 | 57.23 ± 2.47 | 162.22 ± 2.43 |
| 3f | 3,4-Di-F | 4-Tertbutyl | 126.06 ± 0.71 | 135.92 ± 0.71 |
| 3g | 4-Tertbutyl | 4-Phenyl | 76.08 ± 0.79 | 185.41 ± 1.84 |
| 3h | 4-Tertbutyl | 4-CF3 | 71.17 ± 0.49 | 179.15 ± 2.26 |
| 3i | 4-Tertbutyl | 4-Br | 83.94 ± 0.74 | 193.43 ± 2.12 |
| 3j | 3,5-Di-OCH3 | 4-Tertbutyl | 69.63 ± 1.51 | 177.22 ± 2.83 |
| 3k | 4-Tertbutyl | 4-Tertbutyl | 48.06 ± 0.52 | 178.25 ± 0.92 |
| 3l | 4-Cl | 4-Br | 123.44 ± 3.11 | 192.35 ± 1.77 |
| Donepezil | 0.04 ± 0.01 | |||
| β-Secretase inhibitor IV | 0.02 ± 0.01 | |||
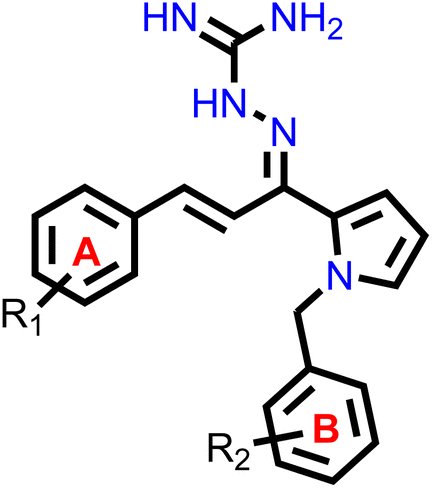
Table 1 indicates the results for compounds 1a–j of first series where ring A was substituted with electron withdrawing and donating substituents and ring B was unsubstituted. Overall, results indicate that the hypothesis that extension of our reported compound could result in dual inhibition of AChE and BACE 1 was correct. The compounds were active against both enzymes however, with introduction of AChE inhibition, the BACE 1 inhibitory potential was decreased.
It was found that strong electron withdrawing substituents like 4-nitro or 2,4-dichloro on ring A, enhanced the AChE inhibition but decreased BACE 1 inhibitory activity. Conversely, substitution of electron donating groups on ring A enhanced the BACE 1 inhibitory potential but decreased the AChE inhibitory potency of compounds. It was noticed that substituting ring A alone with either electron donating or electron withdrawing moieties was not favorable for BACE 1 inhibition as compared to the earlier reported compound, the activity was decreased. However, if at ring A, phenyl is replaced with pyridine, BACE 1 inhibition is not impacted much as it exhibited an IC50 of 13.74 μM but affects AChE inhibition as the activity against AChE was poor.
In second series (compounds 2a–r), ring A was not substituted, and ring B was substituted with various electron withdrawing and donating substituents. It was observed that as compared to ortho, para substitution of electron withdrawing groups on ring B (compounds 2h, 2k, 2m, and 2n) was favorable for AChE inhibition. Also, for BACE 1 inhibition, substitution of electron donating substituents on ring B was favoured (compounds 2a, 2b, 2c, and 2f). Increasing the bulk at 4-position of ring B favoured AChE inhibition but compromised BACE 1 inhibition as can be seen in compounds 2j, 2q, and 2r. Overall, it can be noted that the electron withdrawing substitution on ring B favors AChE inhibition and electron donating groups promote BACE 1 inhibition.
In previous two series, electron withdrawing group substitution on any of the ring favored AChE inhibition and electron donating substituents on ring A or B were favorable to BACE 1 inhibition. Also, substitution of bulky groups had impacted the activity. Therefore, in the third series, both the rings (A and B) were substituted and in particular, the effect of bulky or electron withdrawing substituents was studied. It was expected that the substitution on both rings may potentiate the AChE inhibition. However, not much change in AChE inhibition was observed by substituting bulky or electron withdrawing substituents on both rings. Compound 3k which had tert-butyl substituent on both the rings was most potent AChE inhibitor of all these compounds showing IC50 value of 48.06 μM. But it did affect BACE 1 inhibition and the IC50 values increased indicating that bulky or electron withdrawing substituents on any ring, or both the rings was not favorable for BACE 1 inhibition.
2.4. Molecular modeling studies
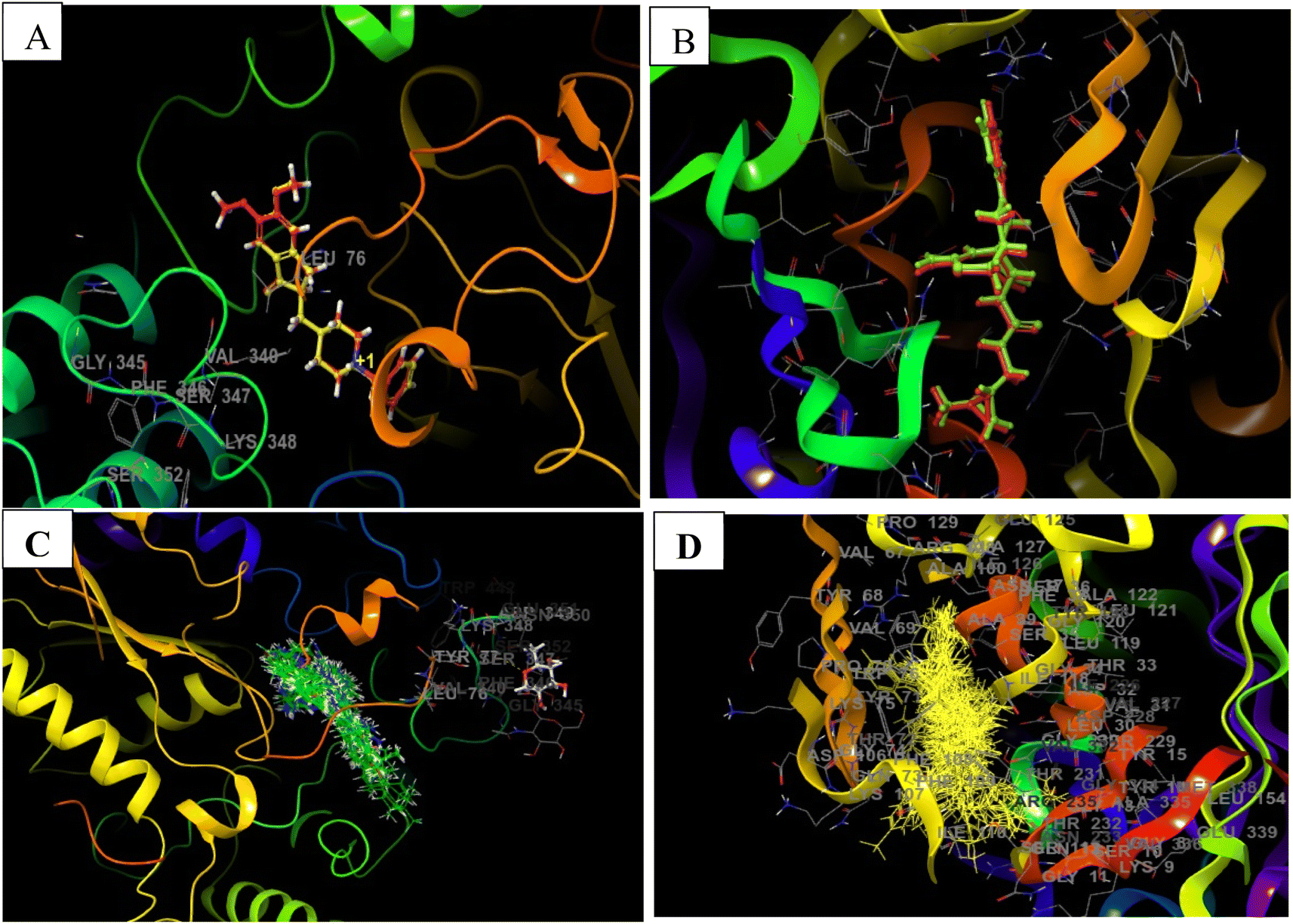 | ||
| Fig. 5 Docking validation of 4EY7 and 6UWP (A) redocked pose of Donepezil (red) in 4EY7 (AChE) superimposed on the co-crystallized ligand (yellow) (RMSD: 0.14 Å, Glide score: 14.56); (B) redocked pose of QKA (red) in 6UWP (BACE 1) superimposed on the co-crystallized ligand (green) (RMSD: 0.08 Å, Glide score: 9.64); (C) and (D) show overlapping of all of the docked compounds within the active site of 4EY7 and 6UWP, respectively. | ||
The analysis of series 1 compounds in the active site of both the enzymes (Fig. 6), as exemplified by compound 1j revealed the occupancy of substrate binding cavities and interactions with active site amino acids. It was seen that, along with occupancy of S1 cavity by ring A, ring B was accommodated in the S3 pocket of BACE 1. Its guanidinium moiety formed strong hydrogen bonding interactions with the key aspartate dyad Asp228 and Asp 32. In case of AChE, ring A was seen to accommodate in the CAS pocket with enhanced π–π stacked interactions with Leu130, Gly120, Tyr133 and ring B occupied PAS with π–π stacking with Glu202, Trp86, His447. Similar results were observed for series 2 compounds as indicated by compound 2h and are shown in Fig. 7.
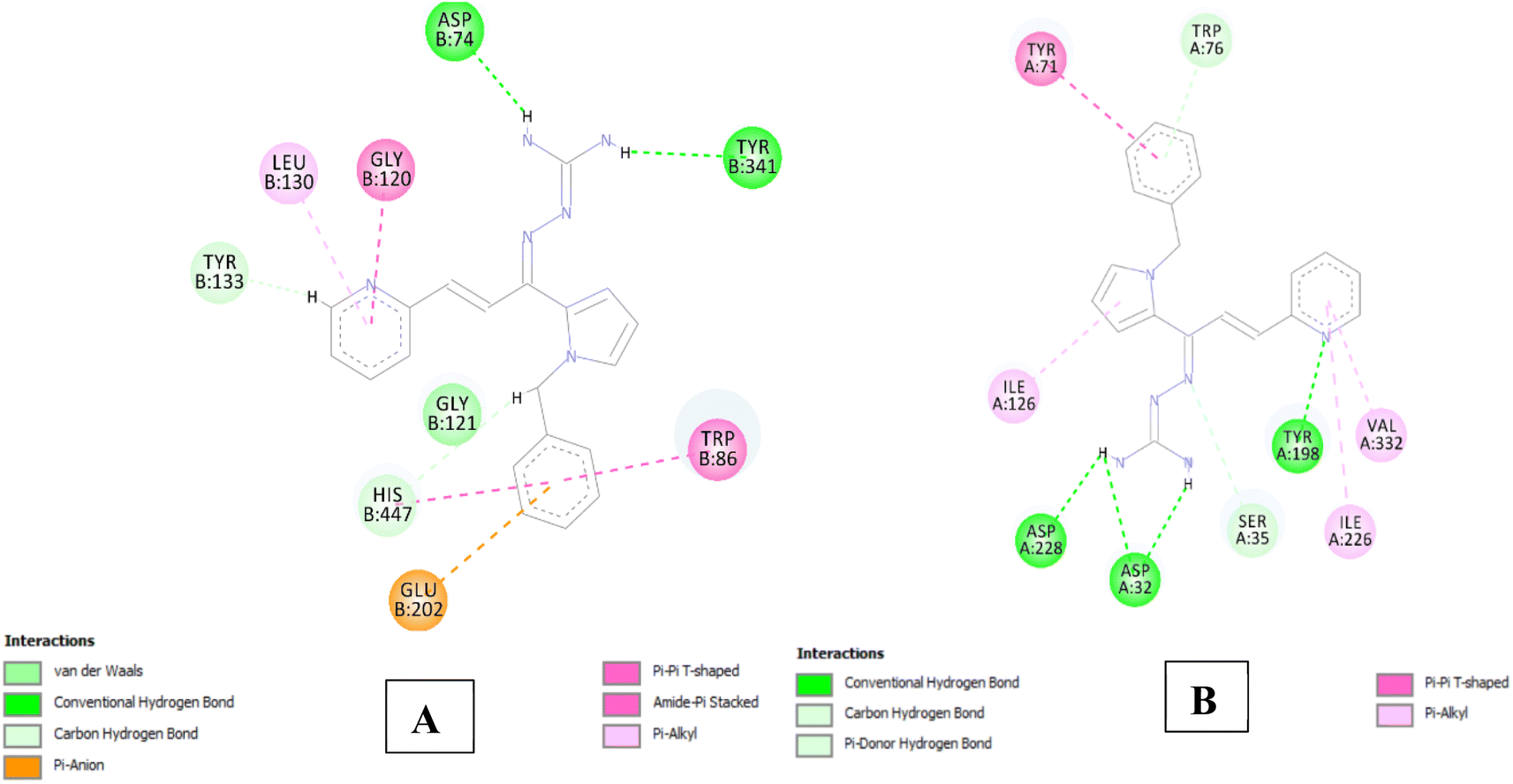 | ||
| Fig. 6 (A) 2D interaction plot of 1j in AChE (Glide score-12.26 kcal mol−1); (B) 2D interaction plot of 1j in BACE 1 (Glide score-8.03 kcal mol−1). | ||
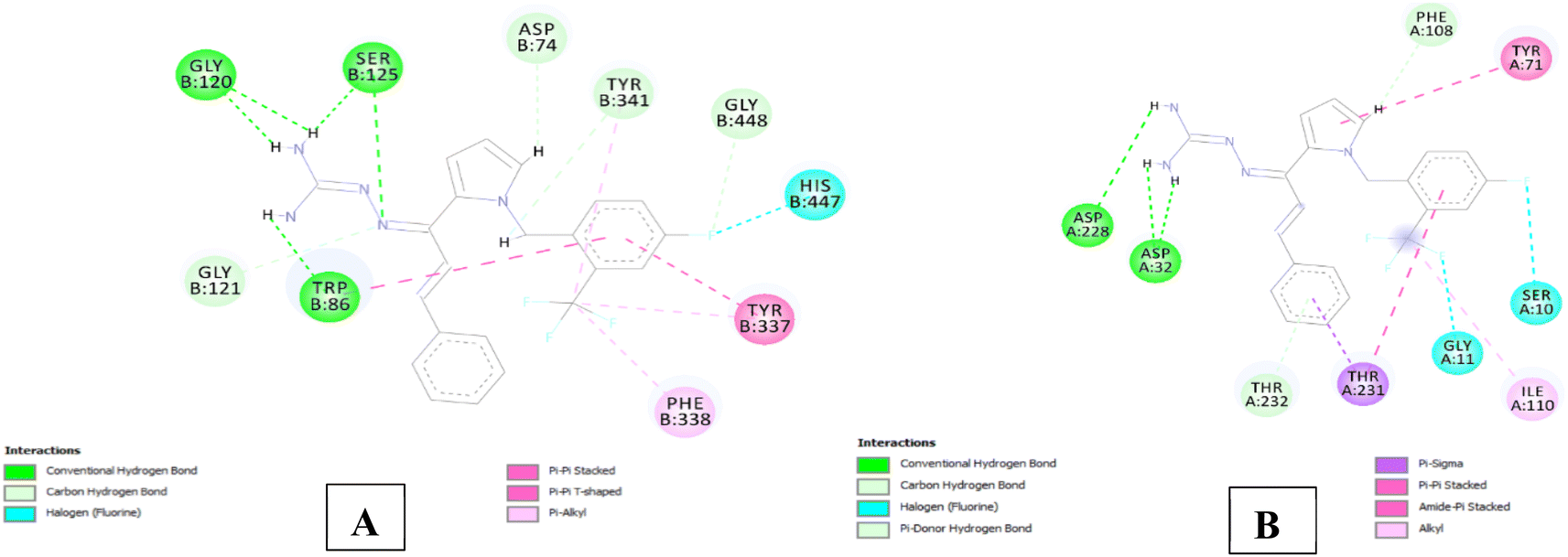 | ||
| Fig. 7 (A) 2D interaction plot of 2h in 4EY7 (Glide score-12.59 kcal mol−1); (B) 2D interaction plot of 2h in BACE 1 (Glide score-7.94 kcal mol−1). | ||
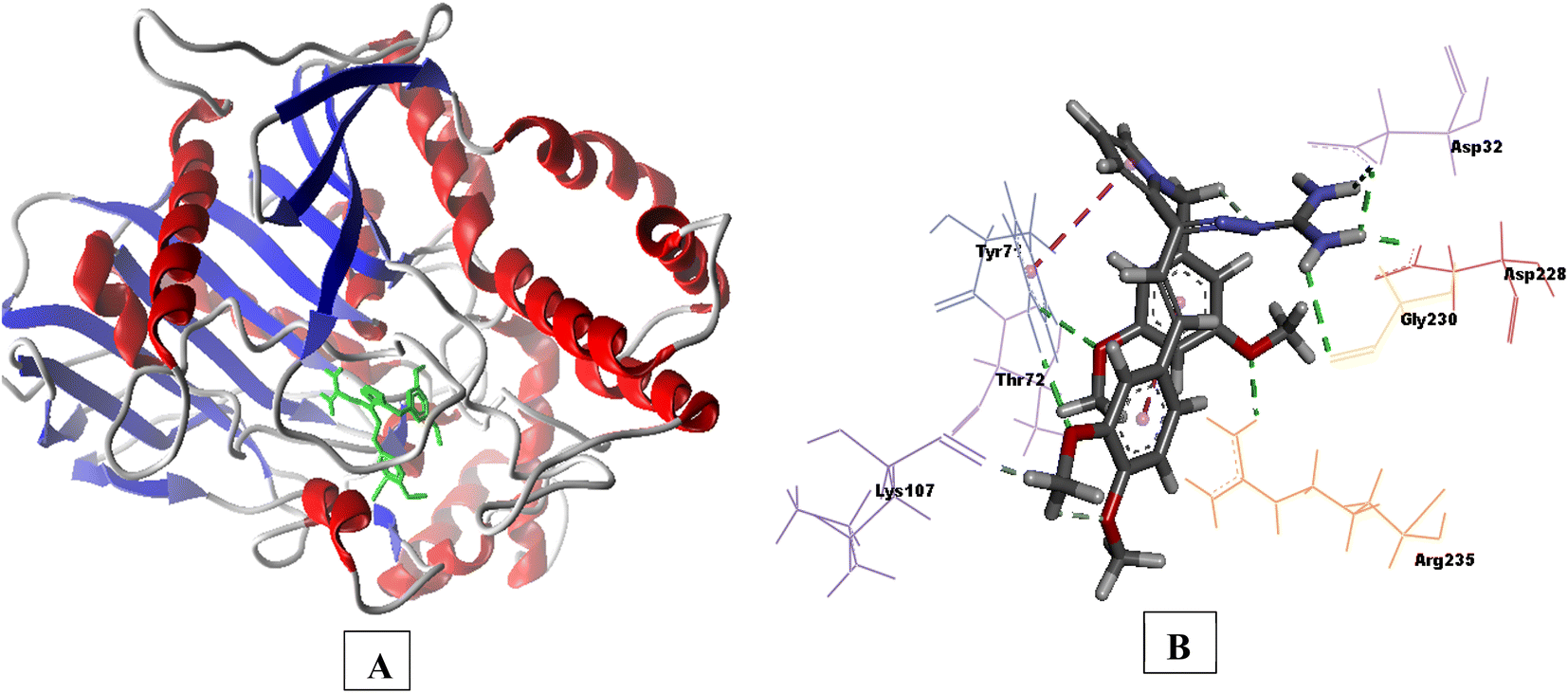
Compound 3e having substituted ring A and substituted ring B, showed moderate BACE 1 inhibition and displayed hydrogen bonding interactions with aspartate dyad Asp228, Asp 32. It was seen ring A occupies the S1 cavity (Tyr71, Lys107) and S3 cavity is occupied by ring B (Arg235, Thr72). In case of AChE, 3e was seen to accommodate the CAS pocket with enhanced π–π stacked interactions with Trp286, hydrogen bonded with Phe295, Tyr341 and occupy PAS with π–π stacked with Phe297, Trp86 were also observed and are shown in Fig. 8–10.
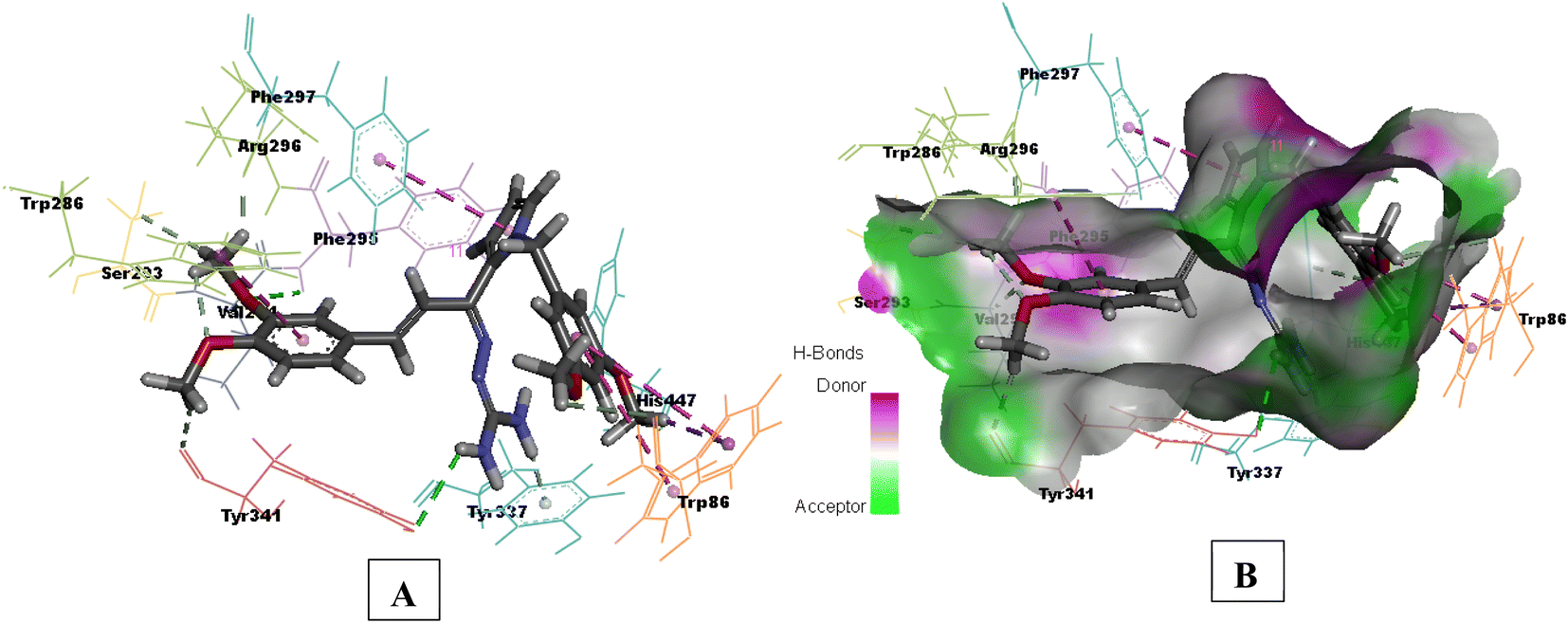 | ||
| Fig. 8 (A) 3D interaction plot of 3e in 4EY7 (Glide score-11.47 kcal mol−1); (B) H-bond surface model (donor/acceptor) of 4EY7 bound with 3e. | ||
 | ||
| Fig. 9 (A) Secondary structure view of 4EY7 with docked compound 3e within the active site (green color); (B) 3D interaction plot of 3e in 6UWP (Glide score-9.87 kcal mol−1). | ||
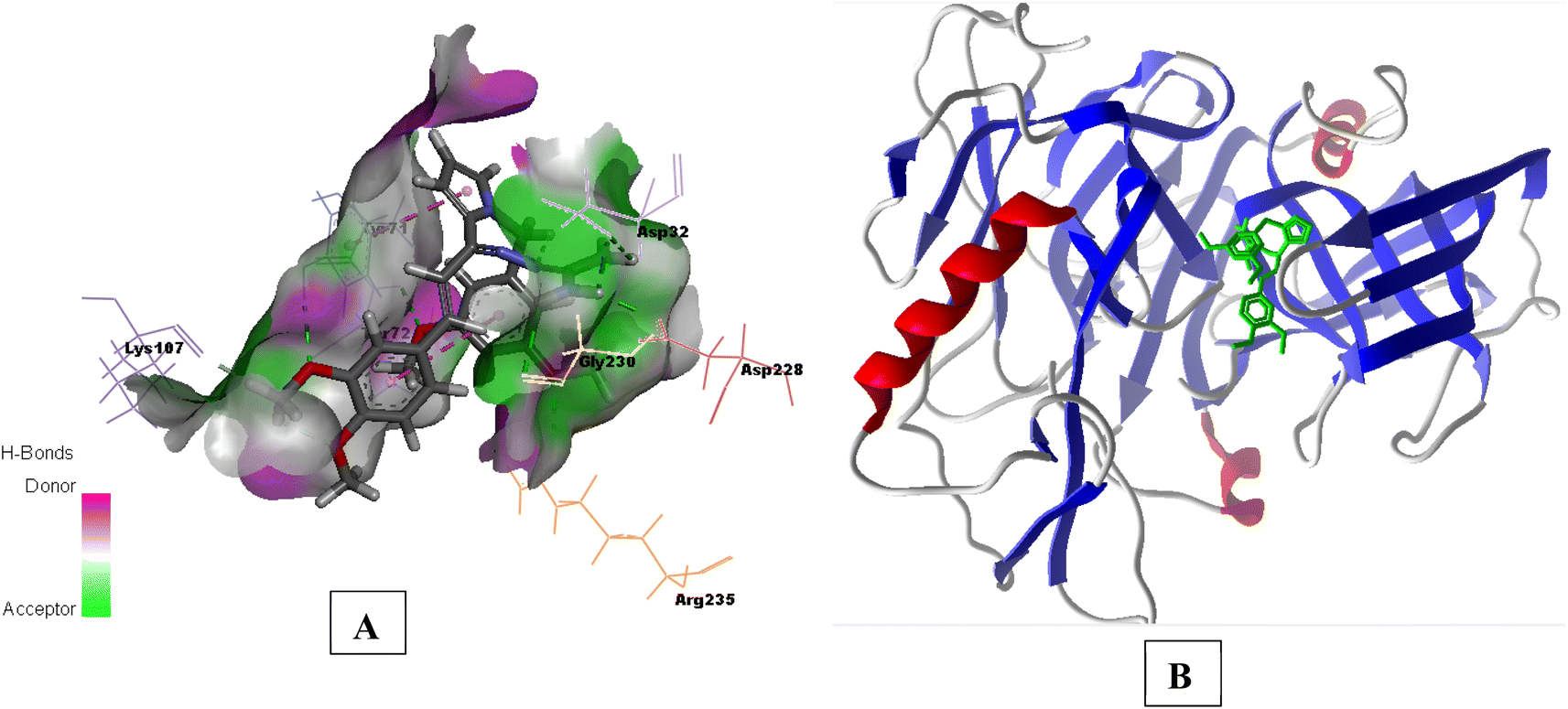 | ||
| Fig. 10 (A) H-bond surface model (donor/acceptor) of 6UWP bound with 3e; (B) secondary structure view of 6UWP with docked compound 3e within the active site (green color). | ||
The interactions of representative compounds with BACE 1 and AChE, along with their Glide score, are given in Tables 4 and 5, respectively (available in ESI).†
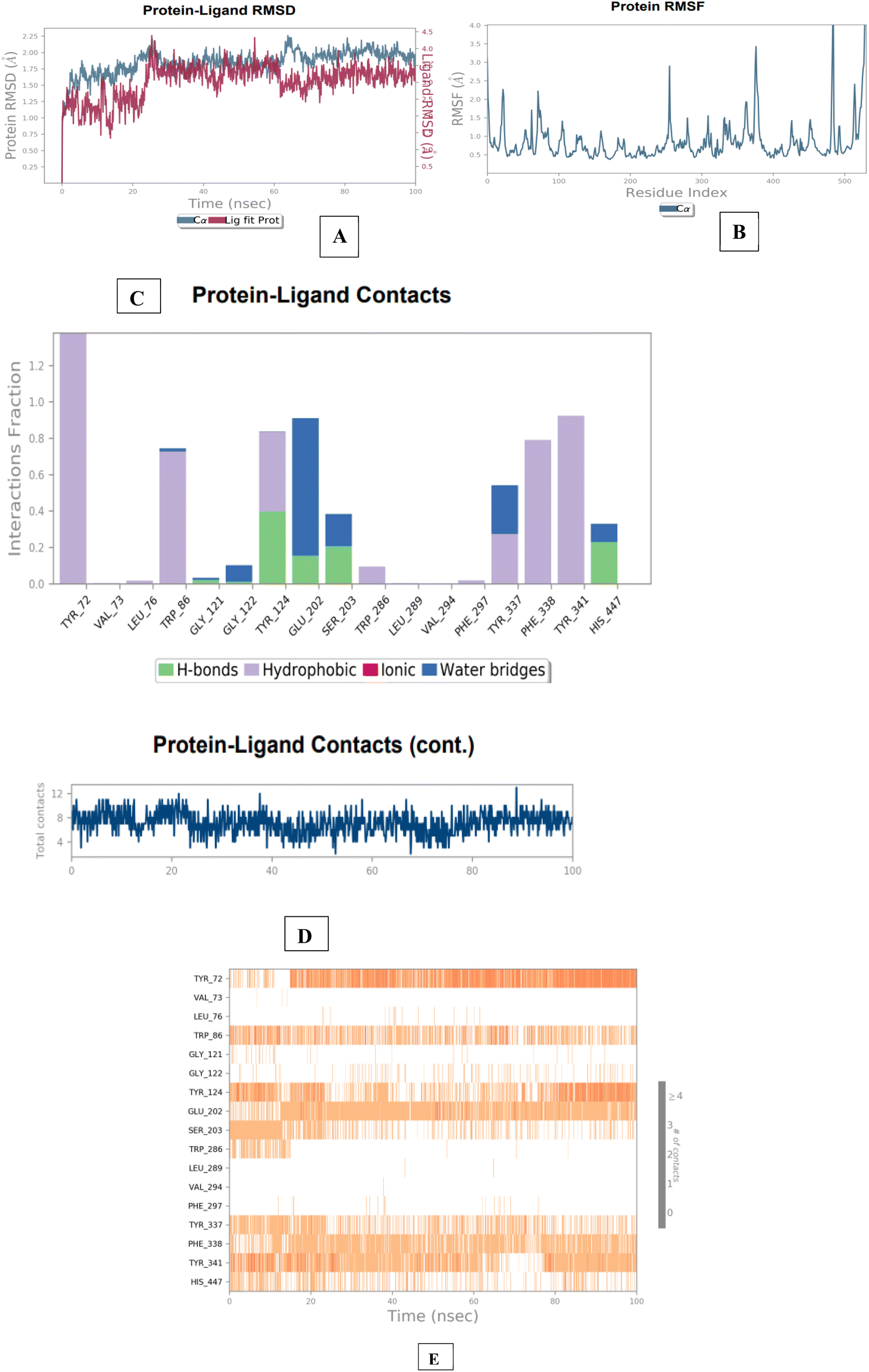 | ||
| Fig. 11 Molecular dynamics studies of 3e-AChE (4EY7) docked complex. [A] Comparative RMSD of AChE (protein backbone) and compound 1d in the protein–ligand complex throughout the simulation period of 100 ns. [B] Graphical representation showing protein RMSF throughout the simulation period of 100 ns. [C] Histogram showing interaction fractions with active amino acid residues. [D and E] Timeline representation showing interaction with all the amino acid residues at each time frame. | ||
The results of the BACE 1 molecular dynamics simulation showed that compound 3e established strong interactions with aspartate dyad residues (Asp32 and Asp228) with least fluctuations. The amino-guanidinium functionality involved in H-bonding interaction with Asp32 accounted throughout the trajectories. Asp228, a one of the catalytic aspartate dyad residues interacted with the N-atom of amino-guanidinium core of compound 3e through the creation of salt bridge. The MD findings suggested that compound 3e showed more stable and balanced interaction in AChE and BACE 1 active sites (Fig. 12).
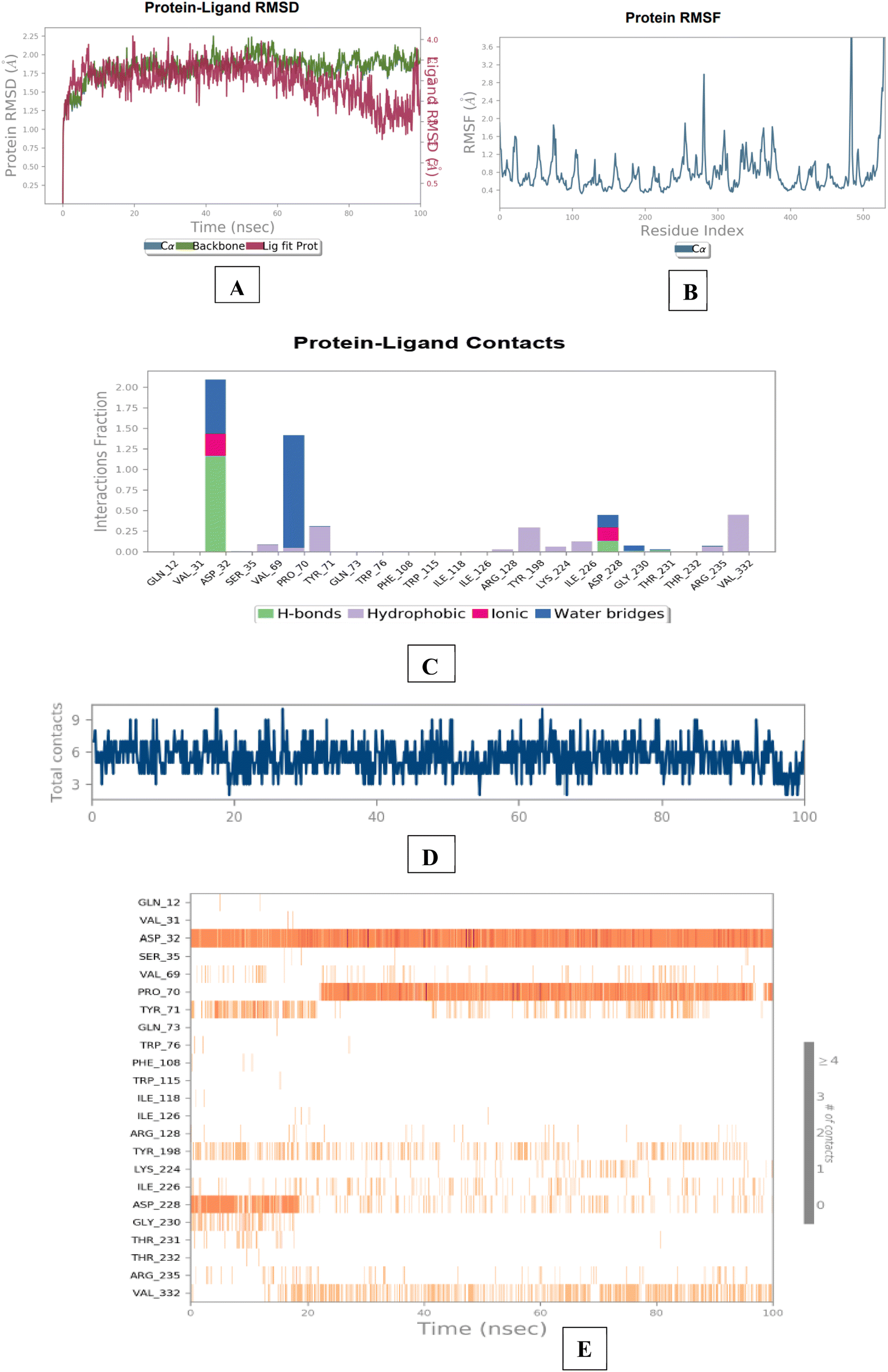 | ||
| Fig. 12 Molecular dynamics studies of 3e-BACE 1 (6UWP) docked complex [A] comparative RMSD of BACE 1 (protein backbone) and compound 3e in the protein–ligand complex throughout the simulation period of 100 ns. [B] Graphical representation showing protein RMSF throughout the simulation period of 100 ns. [C] Histogram showing interaction fractions with active amino acid residues; [D and E] timeline representation showing interaction with all the amino acid residues at each time frame. | ||
2.5. Structure–activity relationship
Based on the AChE and BACE 1 inhibitory activity of the compounds, a preliminary analysis of their structure–activity relationship was done. The potential of the compounds to inhibit AChE and BACE 1 varied with respect to the substitutions on the ring A and B respectively. In the case of molecular modeling studies of AChE, ring B occupy the CAS site of AChE, while the ring A is anchored to the PAS site. This same binding pattern is mainly driven by hydrogen bonding and π–π stacking interactions, in which the aromatic ring B of the N-benzyl interacts with the Trp86 of CAS, ring A interacts with Trp286 of PAS, pyrrole interacts with residue Tyr337 Tyr341, Phe338 and guanidine interacts with the catalytic amino acid residues of AChE (Ser203, Glu202 and His447). The presence of substitution (1c and 1d) on ring A resulted in better AChE inhibition (38.07% and 26.75% of inhibition) in comparison to their unsubstituted counterparts (1a). This might have provided an additional binding stability to the ligand through enhanced hydrophobic interactions with the CAS and PAS of AChE. When conducting modeling studies on BACE 1, it is observed that ring B occupies the S1 region, ring A is anchored to the S3 site and guanidine group can form H-bonding interactions with catalytic aspartate dyad that is, Asp32 and Asp228 respectively. When ring A alone was replaced with pyridine (compounds 1j), it was observed that compound 1j had high docking score and high (43.62%) inhibition. One possible explanation could be attributed to the robust interactions that were observed during the docking simulation: (i) ring B occupied S1 active site region with the pyrrole scaffold π–π stacked to Phe108; (ii) aminoguanidinium nitrogen formed hydrogen bonding interactions with catalytic aspartate dyad (Asp228 and Asp32). Introducing EDG and EWG on ring A alone gave less potent molecules probably due to weaker interactions with the enzymatic active site of BACE 1. Placing 4-butyl and 2-CF3,4-F on ring B (2j and 2h), although showed good interactions in docking, were more favorable for balanced AChE/BACE 1 dual inhibition, which may be attributed to strong hydrophobic interactions in S1 and S3 region of BACE 1, occupying CAS and PAS of AChE and interacts with the catalytic amino acid residues of BACE 1 and AChE (Asp32/Asp228 and Glu202).2.6. Theoretical prediction of the drug-likeness and ADME properties of representative target compounds by QikProp analysis
The net desired pharmacological activities of drugs/drug candidates depend on their in vitro potential and the pharmacokinetic fate (i.e., ADME). Generally, drug discovery and development is a complicated and lengthy process that deals with huge financial and time investments. Due to the unintended side/adverse effects and lack of site delivery, numerous drug candidates fail during the drug discovery program. Drug candidates' fate can be predicted using numerous in silico molecular and/or data modeling methods to reduce the attrition rate during the discovery process. These approaches offer an option to reduce the overall attrition rate during the drug discovery process.The drug-like characteristics of target compounds were evaluated using Schrödinger QikProp software, which indicated that the properties are comparable to that of donepezil (Table 4). It includes molecular weight of the molecule [mol MW (130–725)], estimated number of hydrogen bonds that the solute would donate to water molecules in an aqueous solution [donor HB (0–6.0)], estimated number of hydrogen bonds that would be accepted by the solute from water molecules in an aqueous solution [acceptor HB (2.0–20.0)] as well as the other important factors like total solvent accessible surface area [SASA (300–1000)], predicted brain/blood partition coefficient [QPlogBB (−3.0 to 1.2)], predicted octanol/water partition coefficient [QPlogo/w (−2.0 to 6.5)], predicted qualitative human oral absorption [% human oral absorption (0 to 100% scale)] and predicted IC50 value for blockage of HERG K+ channels [QplogHERG (concern below −5)]. The results indicated that the compounds from series I, II, and III possess “drug-like” properties.
| Compound | Mol_Wta | Donor HBb | Acceptor HBc | SASAd | QplogBBe | Qplogo/wf | % human oral absorptiong | QplogHERGh |
|---|---|---|---|---|---|---|---|---|
| a Molecular weight of the compound.b Hydrogen bond donor in a molecule.c Hydrogen bond acceptor with in a molecule.d Total solvent accessible surface area in square angstroms using a probe with a 1.4 Å radius.e Predicted brain/blood partition coefficient.f Predicted octanol/water partition coefficient.g Predicted human oral absorption on 0 to 100% scale.h Predicted IC50 value for blockage of HERG K+ channels. | ||||||||
| 1c | 388.42 | 4.0 | 4.5 | 659.17 | −2.115 | 3.417 | 82.95% | −6.192 |
| 1d | 412.32 | 4.0 | 3.5 | 667.12 | −0.792 | 5.029 | 96.03% | −6.114 |
| 1j | 344.41 | 4.0 | 4.5 | 620.42 | −1.215 | 3.557 | 100.00% | −6.197 |
| 2c | 409.43 | 4.0 | 3.5 | 657.33 | −0.831 | 4.961 | 100.00% | −6.122 |
| 2h | 429.41 | 4.0 | 3.5 | 690.37 | −1.029 | 5.032 | 93.99% | −6.629 |
| 2j | 399.53 | 4.0 | 3.5 | 795.21 | −1.684 | 5.607 | 96.49% | −7.389 |
| 2k | 379.41 | 4.0 | 3.5 | 628.50 | −0.906 | 4.423 | 100.00% | −5.867 |
| 2l | 388.42 | 4.0 | 4.5 | 655.03 | −1.936 | 3.511 | 86.51% | −6.225 |
| 2m | 422.32 | 4.0 | 3.5 | 633.64 | −0.883 | 4.549 | 100.00% | −5.969 |
| 2n | 368.44 | 4.0 | 5.0 | 651.49 | −1.780 | 3.413 | 88.63% | −6.268 |
| 2p | 375.44 | 4.0 | 3.5 | 622.41 | −1.246 | 4.038 | 100.00% | −5.426 |
| 2q | 399.53 | 4.0 | 3.5 | 679.57 | −0.935 | 5.252 | 100.00% | −5.725 |
| 3e | 463.53 | 4.0 | 6.5 | 748.31 | −1.307 | 4.603 | 100.00% | −5.683 |
| 3k | 455.64 | 4.0 | 3.5 | 787.74 | −1.034 | 6.554 | 100.00% | −5.912 |
| Donepezil | 379.49 | 0.0 | 5.5 | 707.05 | 0.057 | 4.293 | 100.00% | −6.588 |
3. Conclusion
Herein, pyrrol-2-yl-phenyl allylidene hydrazine carboximidamide derivatives have been identified as dual AChE/BACE 1 inhibitors. All the analogs showed dual inhibition and in silico binding with both the enzymes. The compounds also showed excellent drug-likeness and ADME characteristics in silico. These in vitro results collectively warrant further investigation in suitable animal models for in vivo efficacy.4. Experimental protocols
4.1. General procedures
All chemicals and reagents were purchased from commercial sources and used as such unless specifically mentioned. Thin-layer chromatography (TLC) was used to monitor the progress of the reactions and checked by pre-coated TLC plates (E. Merck Kieselgel 60 F254 with fluorescence indicator UV254). Components were visualized by irradiation with ultraviolet light (254 nm) or other visualizing agents such as iodine vapors, potassium permanganate, etc. Compounds were purified over silica gel (230–400 mesh) using a solvent mixture specified in individual experiments. All solvents used for chromatographic purification were distilled before use. All final compounds were characterized by 1H NMR, 13C NMR spectroscopy using CDCl3 or DMSO-d6. 1H NMR spectra were recorded on a Bruker Avance 400 MHz spectrometer Chemical shifts are given in parts per million (ppm), (δ relative to residual solvent peak for 1H). Data are reported as following: chemical shift, multiplicity (s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quartet, m = multiplet, br = broad signal), and integration. HRMS-ESI+ mode analysis was performed on Agilent Technologies 6545, Version-Q-TOF B.06.01(B6172 SP1) mass instrument.4.2. AChE inhibition assay
The inhibitory effect of compounds on EeAChE was assessed using a modified Ellman assay. Each compound was accurately weighed and dissolved in molecular biology grade DMSO to create a 10 mM stock solution. AChE was dissolved in 0.1 M phosphate buffer (pH 7.2) to prepare enzyme stock solution at 1 mg mL−1 concentration, which was then further diluted with PBS to yield working solutions with enzyme activity of 2.5 units per mL. Working solutions of DTNB and ATChI were also prepared in PBS at concentrations of 0.3 mM, 10 mM, and 10 mM respectively. Test compound concentrations ranging from 1 to 100 μM were prepared from the stock solutions by dilution with PBS. In the experimental procedure, 20 μL of enzyme solution (AChE), 20 μL of test compound solution (inhibitor), and 140 μL of 0.3 mM DTNB solution were added to each well of a 96-well plate. Control wells contained the same components as the test wells, with the addition of the proportion of DMSO present in the test compound solution to account for any interference from DMSO. The assay solutions, with and without inhibitor, were pre-incubated at 25 °C for 20 minutes before the addition of 20 μL of 10 mM ATChI solution. The reaction was monitored by measuring absorbance at 412 nm at 1-minute intervals for a total of 10 minutes using a 96-well microplate reader, as the absorbance is directly proportional to enzymatic activity. Each experiment was performed in triplicate, and donepezil was used as the reference standard. Blank solutions, containing all components except enzyme solution, were prepared alongside the test solutions to account for nonenzymatic substrate hydrolysis. Percent inhibition values were calculated from the slopes of absorbance readings over different time intervals for both control and test compounds. IC50 values were determined graphically from the observed percentage inhibition values at different inhibitor concentrations using GraphPad Prism 6 software and are reported as mean ± SD (an average of three experiments).4.3. BACE 1 inhibition assay
The BACE 1 fluorescence resonance energy transfer (FRET) assay kit, obtained from Sigma-Aldrich (Product No. CS0010, Saint Louis), was used following the manufacturer's protocol. A stock solution of the substrate (500 μM) was prepared using DMSO, and aliquots were stored at −20 °C. Test compounds were dissolved in DMSO to create a 10 mM stock solution, and desired concentrations were achieved through further dilutions with fluorescent assay buffer (FAB, 50 mM sodium acetate buffer, pH 4.5) provided in the kit. Just before starting the assay, a BACE 1 enzyme solution (0.3 unit per μL) and a 50 μM substrate solution were prepared by dilution with FAB. The fluorometer was configured in well plate reader mode with excitation set at 320 nm and emission at 405 nm. For the assay, 10 μL of the test compound, 20 μL of substrate, and 68 μL of FAB were added to each well of a 96-well black polystyrene microplate. Two microliters of BACE 1 were then added, and fluorescence was immediately measured after enzyme addition. Following the initial reading (“time zero”), the plate was covered and incubated for 2 hours at 37 °C. After the incubation period, fluorescence intensities were measured again. The difference between the fluorescence readings at “time zero” and after 2 hours provided Δ fluorescence, which was used for subsequent calculations. Each concentration was assayed in triplicate. The background signal was determined in control wells containing all reagents except BACE 1 and was subsequently subtracted. Fluorescence intensities of assay solutions without inhibitor were also measured. The percentage of inhibition due to the presence of test compound was calculated by the following expression: 100 − (IFi/IFo × 100) where IFi and IFo are the respective fluorescence intensities obtained in the presence and in the absence of inhibitor. Percentage inhibition of enzyme by the compounds are reported as mean ± SD (an average of three experiments). BACE 1 inhibitor IV (Calbiochem IV; CAS no. 797035-11-1) (IC50 = 18 nM; literature value. 15 nM) were used as reference compounds.4.4. Computational studies
4.5. Statistical analysis
The experimental results were provided as mean standard deviation (±S.D.) GraphPad Prism 8.0 was used to analyze the data.Data availability
The authors confirm that the data supporting the findings of this study are available within the article and/or its ESI.†Author contributions
Amit Sharma collated the literature, carried out design and synthesis, and wrote the manuscript. Ankita Sharma and Sandip B. Bharate planned and carried out the screening and statistical analysis, Santosh Rudrawar and Hemant R. Jadhav conceptualized, monitored, reviewed, and supervised the entire work.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
For conducting NMR and MASS experiments, the authors gratefully acknowledge the Central Instrument Facility (CIF), Birla Institute of Technology and Sciences BITS, Pilani.References
- J. V. Shanmugam, B. Duraisamy, B. Chittattukarakkaran Simon and P. Bhaskaran, Alzheimer's disease classification using pre-trained deep Networks, Biomed. Signal Process. Control., 2022, 71, 103217, DOI:10.1016/j.bspc.2021.103217.
- https://www.who.int/news-room/fact-sheets/detail/dementia, accessed on 03-07-2024.
- J. Xie, R. Liang, Y. Wang, J. Huang, X. Cao and B. Niu, Progress in target drug molecules for Alzheimer's disease, Curr. Top. Med. Chem., 2020, 20, 4–36, DOI:10.2174/1568026619666191203113745.
- J. L. Cummings, G. Tong and C. Ballard, Treatment combinations for Alzheimer’s disease: current and future pharmacotherapy options, J. Alzheimer’s Dis., 2019, 67, 779–794, DOI:10.3233/JAD-180766.
- P. Scheltens, B. De Strooper, M. Kivipelto, H. Holstege, G. Chételat, C. E. Teunissen, J. Cummings and W. M. van der Flier, Alzheimer's Disease, Lancet, 2021, 397, 1577–1590, DOI:10.1016/S0140-6736(20)32205-4.
- K. Blennow and H. Zetterberg, Biomarkers for Alzheimer's disease: current status and prospects for the future, J. Intern. Med., 2018, 284, 643–663, DOI:10.1111/joim.12816.
- B.-M. Swahn, K. Kolmodin, S. Karlström, S. von Berg, P. Söderman, J. Holenz and S. Berg, Design and synthesis of β-site amyloid precursor protein cleaving enzyme (BACE 1) inhibitors with in vivo brain reduction of β-amyloid peptides, J. Med. Chem., 2012, 55, 9346–9361, DOI:10.1021/jm3009025.
- P. N. Nirmalraj, J. List, S. Battacharya, G. Howe, L. Xu, D. Thompson and M. Mayer, Complete aggregation pathway of amyloid β (1- 40) and (1-42) resolved on an atomically clean interface, Sci. Adv., 2020, 6, eaaz6014, DOI:10.1126/sciadv.aaz6014.
- M. Vaz, V. Silva, C. Monteiro and S. Silvestre, Role of aducanumab in the treatment of Alzheimer's disease: challenges and opportunities, Clin. Interventions Aging, 2022, 797–810, DOI:10.2147/CIA.S325026.
- P. N. Tripathi, P. Srivastava, P. Sharma, M. K. Tripathi, A. Seth, A. Tripathi, S. N. Rai, S. P. Singh and S. K. Shrivastava, Biphenyl-3-oxo-1, 2, 4-triazine linked piperazine derivatives as potential cholinesterase inhibitors with anti-oxidant property to improve the learning and memory, Bioorg. Chem., 2019, 85, 82–96, DOI:10.1016/j.bioorg.2018.12.017.
- S. K. Shrivastava, S. K. Sinha, P. Srivastava, P. N. Tripathi, P. Sharma, M. K. Tripathi and A. Tripathi, Design and development of novel p-aminobenzoic acid derivatives as potential cholinesterase inhibitors for the treatment of Alzheimer's disease, Bioorg. Chem., 2019, 82, 211–223, DOI:10.1016/j.bioorg.2018.10.009.
- M. Bartolini, C. Bertucci, V. Cavrini and V. Andrisano, β-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies, Biochem. Pharmacol., 2003, 65, 407–416, DOI:10.1016/S0006-2952(02)01514-9.
- R. M. Lane and S. G. Potkin, Albert Enz, Targeting acetylcholinesterase and butyrylcholinesterase in dementia, Int. J. Neuropsychopharmacol., 2006, 9, 101–124, DOI:10.1017/S1461145705005833.
- A. Alvarez, R. Alarcon, C. Opazo, E. O. Campos, F. Jose Munoz, F. H. Calderon and F. Dajas, Stable complexes involving acetylcholinesterase and amyloid-β peptide change the biochemical properties of the enzyme and increase the neurotoxicity of Alzheimer's fibrils, J. Neurosci., 1998, 18, 3213–3223, DOI:10.1523/JNEUROSCI.18-09-03213.1998.
- N. C. Inestrosa, A. Alvarez, C. A. Perez, R. D. Moreno, M. Vicente, C. Linker, O. I. Casanueva, C. Soto and J. Garrido, Acetylcholinesterase accelerates assembly of amyloid-β-peptides into Alzheimer's fibrils: possible role of the peripheral site of the enzyme, Neuron, 1996, 16, 881–891, DOI:10.1016/S0896-6273(00)80108-7.
- M. Bartolini, C. Bertucci, V. Cavrini and V. Andrisano, β-Amyloid aggregation induced by human acetylcholinesterase: inhibition studies, Biochem. Pharmacol., 2003, 65, 407–416, DOI:10.1016/S0006-2952(02)01514-9.
- G. Pepeu and M. G. Giovannini, Cholinesterase inhibitors and beyond, Curr. Alzheimer Res., 2009, 6, 86–96, DOI:10.2174/156720509787602861.
- C. Albertini, A. Salerno, P. de S. M. Pinheiro and M. L. Bolognesi, From combinations to multitarget-directed ligands: A continuum in Alzheimer's disease polypharmacology, Med. Res. Rev., 2021, 41, 2606–2633, DOI:10.1002/med.21699.
- R. Morphy and Z. Rankovic, Designed multiple ligands. An emerging drug discovery paradigm, J. Med. Chem., 2005, 48, 6523–6543, DOI:10.1021/jm058225d.
- J. Zhou, X. Jiang, S. He, H. Jiang, F. Feng, W. Liu, W. Qu and H. Sun, Rational design of multitarget-directed ligands: strategies and emerging paradigms, J. Med. Chem., 2019, 62, 8881–8914, DOI:10.1021/acs.jmedchem.9b00017.
- A. Sharma and S. B. Bharate, Synthesis and biological evaluation of coumarin triazoles as dual Inhibitors of cholinesterases and β-secretase, ACS Omega, 2023, 8, 11161–11176, DOI:10.1021/acsomega.2c07993.
- S. Ali, M. H. H. B. Asad, F. Khan, G. Murtaza, A. A. Rizvanov, J. Iqbal, B. Babak and I. Hussain, Biological evaluation of newly synthesized biaryl guanidine derivatives to arrest β-secretase enzymatic activity involved in Alzheimer’s disease, BioMed Res. Int., 2020, 2020(1), 8934289, DOI:10.1155/2020/8934289.
- P. Sharma, A. Tripathi, P. N. Tripathi, S. Kumar Prajapati, A. Seth, M. K. Tripathi, P. Srivastava, V. Tiwari, S. Krishnamurthy and S. K. Shrivastava, Design and development of multitarget- directed N-Benzylpiperidine analogs as potential candidates for the treatment of Alzheimer's disease, Eur. J. Med. Chem., 2019, 167, 510–524, DOI:10.1016/j.ejmech.2019.02.030.
- E. A. Dutysheva, I. A. Utepova, M. A. Trestsova, A. S. Anisimov, V. N. Charushin, O. N. Chupakhin, B. A. Margulis, I. V. Guzhova and V. F. Lazarev, Synthesis and approbation of new neuroprotective chemicals of pyrrolyl-and indolylazine classes in a cell model of Alzheimer's disease, Eur. J. Med. Chem., 2021, 222, 113577, DOI:10.1016/j.ejmech.2021.113577.
- P. Jain, P. K. Wadhwa, S. Rohilla and H. R. Jadhav, Rational design, synthesis and in vitro evaluation of allylidene hydrazinecarboximidamide derivatives as BACE 1 inhibitors, Bioorg. Med. Chem. Lett., 2016, 26, 33–37, DOI:10.1016/j.bmcl.2015.11.044.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03589e |
| This journal is © The Royal Society of Chemistry 2024 |