
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical study on lithium storage performance of V-doped Ti2CO2 MXene†
Li Wanga,
Wenqi Liub,
Fan Baic,
Xichen Zhenga,
Chaofan Yina,
Jiawei Weia,
Juanjuan Maa,
Hongyu Baic and
Binbin Dong *a
*a
aHenan Key Laboratory of Green Building Materials Manufacturing and Intelligent Equipment, Luoyang Institute of Science and Technology, Luoyang, Henan 471023, PR China. E-mail: dongbb@lit.edu.cn
bJinan Central Business District Investment and Construction Group Co., Ltd., Jinan, Shandong 250013, PR China
cYanshi Zhongyue Refractory Co. LTD, Luoyang 471900, PR China
First published on 20th June 2024
Abstract
With the increasing application of lithium-ion batteries, the demand for high energy density, high-rate performance and high stability lithium-ion batteries is becoming more and more urgent. Ti2CO2 MXene, as a two-dimensional material with multilayer atomic structure and multiple active sites, has great advantages in lithium-ion battery electrode materials. However, the original Ti2CO2 MXene has been unable to meet the requirements of lithium-ion batteries due to its semiconductor properties. Doping is an effective means to regulate the conductivity and electrochemical properties of Ti2CO2 and improve the capacity of lithium-ion batteries and other energy storage devices. Hence, we use first-principles calculations to study the effect of V atom doping on the adsorption and diffusion of Li on the MXene surface. The density of states (DOS) and partial density of states (PDOS) of TiVCO2 and Ti2CO2 MXene indicated the transition of their conductive types from semiconductors to conductors. In addition, we observed that TiVCO2 has higher electrical conductivity and ion transport speed than the original Ti2CO2 MXene, and at the same time, Li atoms can be adsorbed well on the surface of MXene and show a lower diffusion energy barrier. Therefore, TiVCO2 is expected to become the anode material for the next generation of lithium-ion batteries and has good lithium storage performance.
1. Introduction
Due to the irreversible consumption of non-renewable energy sources and the advancements in the portable electronics and electric vehicle industries, the increasing demand for energy storage devices has spurred extensive research interest in the development of new storage systems.1 Lithium-ion batteries (LIBs) have garnered significant attention as the primary power source for portable electric vehicles and implantable medical systems, owing to their small size and high energy density.2–4 However, the safety and cost issues associated with LIBs have hindered their development. Consequently, the search for or development of new materials with high-rate performance, enhanced safety, improved cycling performance and stability, and higher capacity represents a major challenge for LIBs. In this context, the discovery of graphene5 and its exceptional properties has led to the exploration of numerous two-dimensional (2D) materials, such as germanene,6 silicene,7 hexagonal boron nitride (h-BN),8 black phosphorus,9 and transition metal dichalcogenides (TMDC).10 While most 2D materials have predominantly been applied in electronic devices due to their semiconductor characteristics, such as p–n junctions and field-effect transistors, there has also been research into their application in energy storage devices like supercapacitors, thermoelectric devices, and batteries. Notably, lithium-ion batteries based on 2D materials have garnered increasing attention due to their rapid ion diffusion properties. On the other hand, MXenes, as new members of the 2D materials family, have been demonstrated to be promising candidates for electrode materials in lithium-ion batteries and lithium-ion capacitors.11 Furthermore, to date, numerous different types of MXene materials have been synthesized through chemical exfoliation methods, and it is anticipated that many MXene materials are stable and experimentally obtainable.12MXene was a novel two-dimensional material that had recently been recognized as one of the best candidate anode materials for lithium-ion batteries. MXenes were derived from MAX phases with a closed-packed hexagonal structure and P63/mmc space group symmetry.13 MXene nanosheets were typically described by the general formula “Mn+1XnTx”, where “M” represents a transition metal, “X” denotes carbon and/or nitrogen, “Tx” stands for surface terminal groups such as –F, –O, and –OH, and n can take values from 1 to 3.14 Currently, extensive theoretical and experimental research has been conducted on MXene materials. Their electronic structure, electrical conductivity, surface chemistry, and electrochemical properties have been widely studied. The exceptional conductivity, unique layered structure, ultra-high specific surface area, and excellent ion transport capabilities of MXenes have been well-documented. However, most research to date has focused predominantly on single-component MXene materials. There was relatively little research on MXene electrode materials that incorporate transition metal impurity atoms.
Shunsuke et al.15 studied the formation energy of Na/Li adsorption on Ti2CTx (T = O, OH, F) sheets, showing that the Li–Ti2CO2 system was more stable than others. Michael Ashton et al.16 reported a reversible capacity of 192 mA h g−1 and a diffusion barrier of 0.63 eV for Ti2CO2 MXene through systematic density functional theory calculations. Yierpan Aierken et al.17 found, based on first-principles calculations, that the charge transfer of Li on mono-Ti2CO2 is 0.912e, which is higher than that of most MXene materials, indicating strong binding between Li atoms and the Ti2CO2 monolayer, resulting in stronger ion interactions in the Ti2CO2-based system. Additionally, theoretical studies show that Ti2CO2 has a low migration barrier (0.27 eV) and a high theoretical weight capacity (348.9 mA h g−1). However, according to previous studies and our own calculations, Ti2CO2 MXene behaves as a semiconductor, which makes it unfavorable for use as a negative electrode material for lithium-ion batteries.
In this context, this paper constructed a doped MXene material TiVCO2 based on the geometric lattice structure of Ti2CO2 using the Special Quasi-random Structure (SQS) method in the ATAT software. First-principles calculations were used to systematically study the electronic properties and electrochemical performance of V-doped Ti2CO2 MXene materials. To understand the electronic conductivity of TiVCO2 MXene materials, the total density of states (DOS) and partial density of states (PDOS) of Ti2CO2 and TiVCO2 MXene materials were calculated. At the same time, the adsorption energy and diffusion barriers of Li atoms on the surface of MXene materials were also calculated to study their adsorption and migration properties. Furthermore, this paper systematically studies the open-circuit voltage (OCV), binding energy, and maximum theoretical capacity of Li on the surface of MXene using density functional theory (DFT). By comparing the electrochemical performance data with the original Ti2CO2 MXene materials, this paper explores the feasibility of TiVCO2 MXene as an anode material for lithium-ion batteries. The theoretical calculation data indicate that TiVCO2 MXene materials are promising candidates for new lithium-ion battery anode materials.
2. Calculation method
Using density functional theory (DFT), all calculations were performed with the Vienna ab initio simulation package (VASP).18–20 The Perdew–Burke–Ernzerhof (PBE) functional within the Generalized Gradient Approximation (GGA) was used to describe the interactions between nuclei and electrons.21,22 The valence electronic states of 3s23p63d34s1 for Ti, 3s23p64s23d3 for V, 2s22p2 for C, 2s22p4 for O, and 1s22s1 for Li were taken into account. The mcsqs script in the Alloy Theoretic Automated Toolkit (ATAT) software was used to generate the Special Quasi-random Structure (SQS) of V-doped MXene materials. An energy cutoff of 520 eV was used for the plane wave expansion of the electronic wave functions. All calculations used a 3 × 3 × 1 Monkhorst–Pack k-point grid. The convergence criteria for atomic forces and energy were set to 0.02 e Å−1 and 10−5 eV, respectively. To avoid interactions due to the use of periodic boundary conditions, a real space gap of approximately 15 Å was set between adjacent slabs. Considering the weak interaction systems we are dealing with, van der Waals interactions were considered using the semi-empirical DFT-D2 method. Additionally, the Climbing-Image Nudged Elastic Band (CI-NEB) method implemented in VASP was used to determine the diffusion barriers of Li atoms on MXene surfaces.The adsorption energy (Ead) of Li atoms on the MXene surface is defined as follows:23
| Ead = E(MXene–Li) − E(MXene) − E(Li) |
The open-circuit voltage (OCV) can be calculated as:24
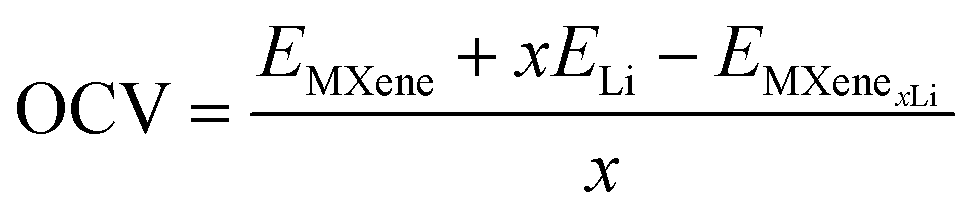
The binding energy per Li atom on the MXene surface is defined as:
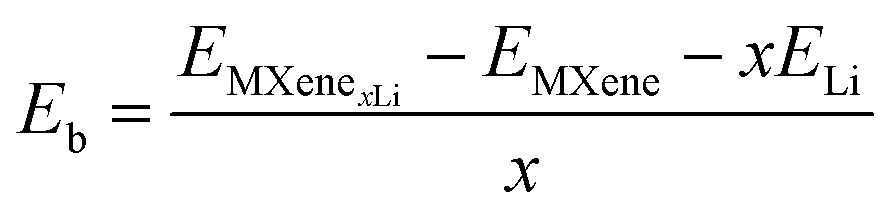
The theoretical capacity is defined as:25
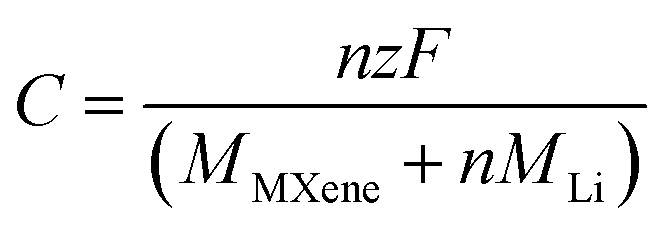
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 801 mA h mol−1), and MMXene and MLi are the molar masses of MXene and Li, respectively.
801 mA h mol−1), and MMXene and MLi are the molar masses of MXene and Li, respectively.
3. Results and discussion
3.1 Structure models
First, utilized the “mcsqs” script in the Alloy Theoretic Automated Toolkit (ATAT) software to generate special quasi-random structures (SQS) for V-doped MXene materials. In the ATAT software, stack five layers in the sequence of O–M (Ti, V)–C–M–O to establish a monolayer geometric model with TiVCO2 structure and electronic properties. In this work, a total of 26 different structures were predicted (Fig. S1†), the formation energy of different structures was calculated (Fig. S2†), and the most stable structure was selected. This model, based on the Ti2CO2 structure, replaced four Ti atoms in two layers of Ti atoms with V atoms, resulting in optimized lattice constants of a = b = 9.0127 Å for TiVCO2. To investigate Li adsorption on its surface, we constructed a 3 × 3 × 1 supercell consisting of 45 atoms, as depicted in Fig. 1.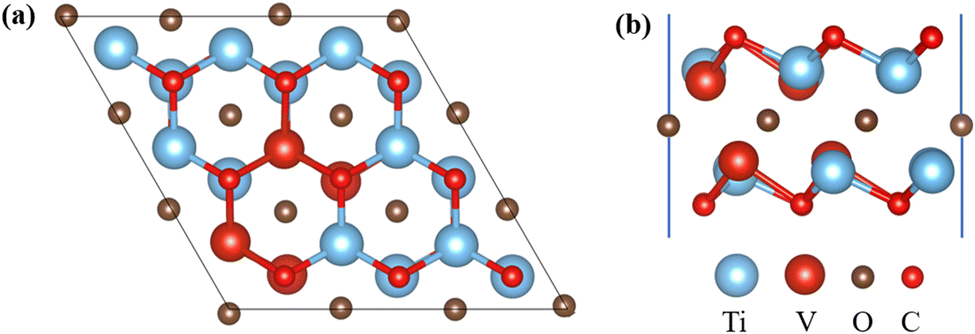 | ||
| Fig. 1 Geometric model of TiVCO2. Top (a) and side (b) view. | ||
3.2 Electronic structural properties
The electronic conductivity of a material is related to its electronic properties. In order to understand the effect of vanadium doping on electronic properties, the DOS and PDOS of Ti2CO2 and TiVCO2 were calculated, as shown in Fig. 2. Among them, the Fermi level was set to 0 eV, which is represented by a black dotted line in the figure. The total DOS is represented by gray lines, and the PDOS of Ti, V, C, and O are represented by green, light blue, dark blue, and brown respectively. As can be seen from the figure, the DOS at the Fermi level of Ti2CO2 is zero, while the DOS at the Fermi level of TiVCO2 after incorporating vanadium is not zero, which indicated that the material has metallic conductivity after incorporating vanadium. This is mainly due to the contribution of the d orbital of V to the density of states at the Fermi level. Transformed from a semiconductor into a conductor, the conductivity was significantly enhanced. In addition, the unpaired electrons of doping atoms will cause spin polarization of the material, which is manifested as spin-up and spin-down DOS diagram asymmetry. From Fig. 2b, it can be seen that the spin-up and spin-down DOS diagrams are asymmetric at energies of −4.6 eV, −3.1 eV, 0.6 eV, and 2.2 eV. What is more noteworthy was that in the energy range of −6 eV to −4 eV, the PDOS of the C 2p orbital and the V 3d orbital overlap significantly, which indicated that the C 2p orbital and the V 3d orbital are highly hybridized, forming a V–C bond. In the conduction band range, the peak at an energy of 0.7 eV was mainly contributed by the V 3d orbital, which showed that the incorporation of vanadium atoms makes a huge contribution to the improvement of the conductivity of Ti2CO2. In addition, it can be seen from the charge density difference diagram (Fig. S3†) of TiVCO2 after V doping that the introduction of V has an effect on the charge distribution of MXene, which may be the reason for the change of its electronic conductivity. According to the calculation results of DOS, TiVCO2 has metallic conductivity, which may make it suitable as an anode material for lithium-ion batteries.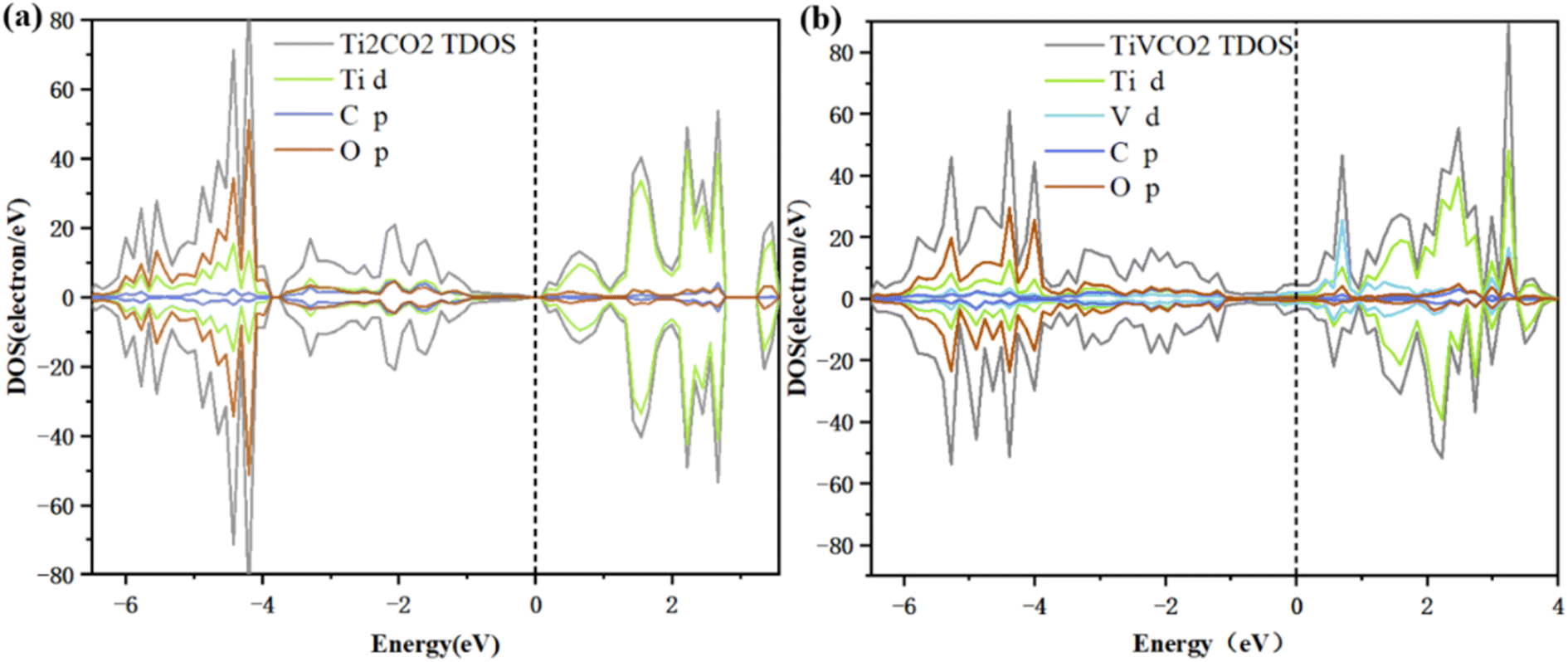 | ||
| Fig. 2 Total density of states (DOS) and partial density of states (PDOS) of (a) Ti2CO2 and (b) TiVCO2. The Fermi level was set to 0 eV. | ||
3.3 Adsorption sites and adsorption energy
As an electrode material, the electrochemical performance of TiVCO2 is closely related to the adsorption and diffusion behavior of surface Li atoms. First, it is necessary to confirm the optimal adsorption position of Li atoms on the surface of MXene material. For general Ti2CO2 materials with only one transition metal and one functional group, there are usually three non-equivalent adsorption positions, as shown in Fig. 3a, which are the top of the C atom (fcc) and the top of the Ti atom (hcp). and the top of the oxygen atoms.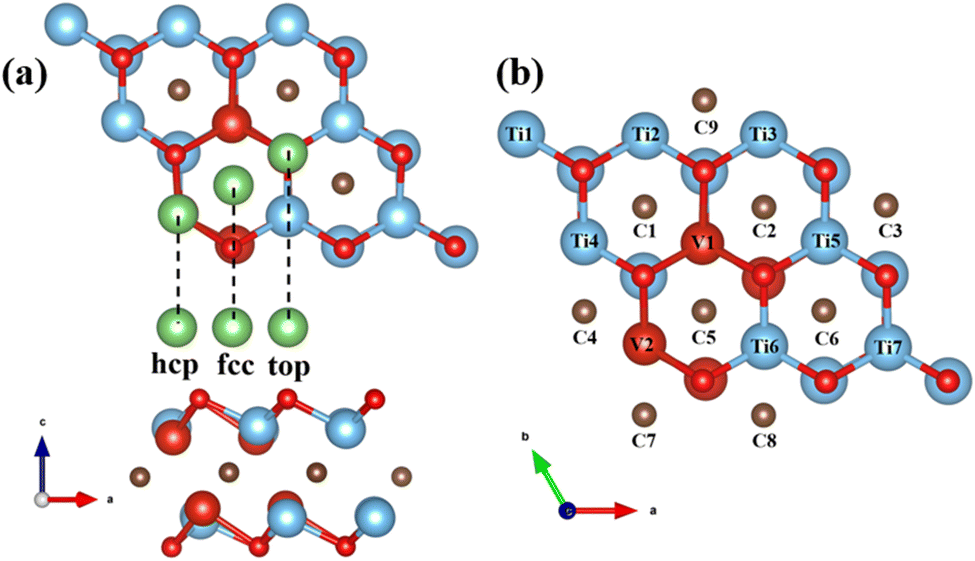 | ||
| Fig. 3 (a) Top view and side view of three possible lithium-ion adsorption sites on MXene, (b) possible lithium-ion adsorption sites on TiVCO2. | ||
Similarly, MXene materials with multiple transition metals can also be classified according to the above positions. However, since the incorporation of vanadium atoms destroys the symmetry of Ti2CO2, the surface environment of V-doped Ti2CO2 was more complex than that of the original Ti2CO2 MXene, and the adsorption positions directly above Ti, C, and O atoms were no longer equivalent. Therefore, for TiVCO2, it was necessary to consider the possibility of Li atom adsorption directly above each atom. However, due to the particularity of the position directly above the oxygen atom, when the potential around the oxygen atom is not symmetrical, it is usually unstable as an adsorption position. Therefore, the position directly above the oxygen atom was not taken into account in this article. Other adsorption positions are shown in the Fig. 3b.
The calculation results are shown in Table 1. The adsorption energy of Li atoms on the surface ranges from −1.92 to −2.57 eV, all of which are negative values. The more negative the adsorption energy, the more stable the system between the adsorbed atoms and the surface. Therefore, Li atoms can be stable adsorbed on the MXene surface. It should be emphasized that the adsorption energy of Li atoms above C atoms is greater than that of transition metal atoms, indicating that Li atoms will be preferentially adsorbed at the fcc position. This result is similar to the adsorption performance of pristine MXene–Li ions. The reason for this phenomenon may be that the pore size of the “fcc” site is larger, which is more suitable for adsorbing Li ions.
| Adsorption site | Adsorption energy (eV) | Adsorption site | Adsorption energy (eV) |
|---|---|---|---|
| C1 | −2.44 | Ti1 | −1.93 |
| C2 | −2.45 | Ti2 | −2.03 |
| C3 | −2.16 | Ti3 | −2.04 |
| C4 | −2.44 | Ti4 | −2.00 |
| C5 | −2.57 | V1 | −2.45 |
| C6 | −2.24 | Ti5 | −1.93 |
| C7 | −2.45 | V2 | −2.45 |
| C8 | −2.25 | Ti6 | −1.95 |
| C9 | −2.30 | Ti7 | −2.03 |
In order to describe the adsorption mechanism in more detail, the charge transfer between them was analyzed using charge difference and Bader charge analysis. The charge difference calculation formula is:
| Δρ = ρ(MXene–Li) − ρMXene − ρLi |
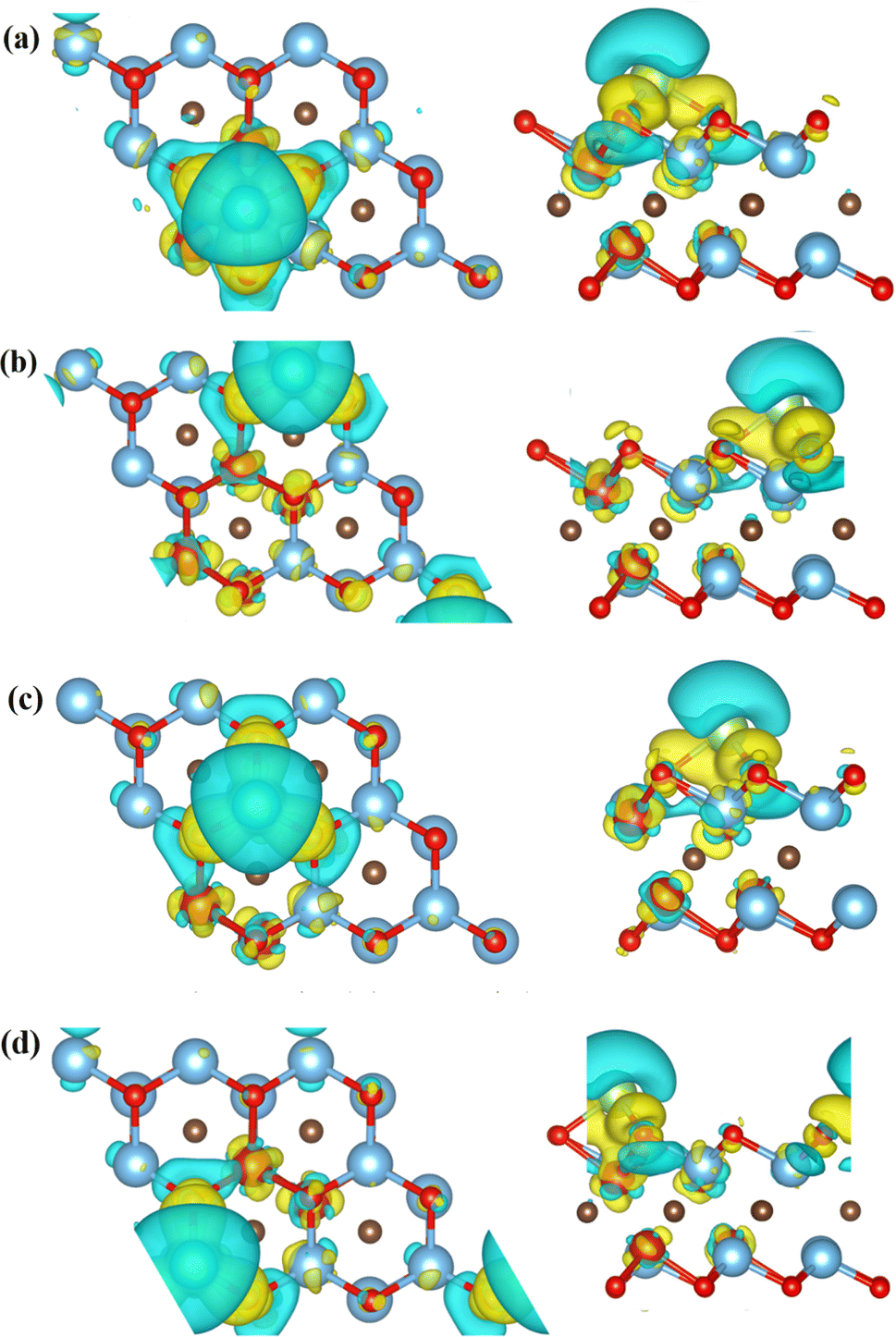 | ||
| Fig. 4 Differential charge diagrams of Li atoms adsorbed on (a) C5, (b) Ti3, (c) V1, (d) V2 respectively. | ||
3.4 Diffusion of lithium ions on MXene surface
The migration rate of Li atoms is an important criterion for evaluating the rate performance of electrode materials, and the migration rate of Li atoms on electrode materials depends on the diffusion energy barrier between the two most stable adsorption sites of Li atoms. Considering that the incorporation of vanadium atoms results in a complex surface potential environment of TiVCO2 MXene, the article considered the diffusion paths of multiple Li atoms on the TiVCO2 surface and obtained four representative diffusion paths through optimization calculations. As shown in Fig. 5c–f, according to the positions marked previously, the diffusion paths are C1 → C4, C5 → V1 → C2, C5 → C7 and Ti2 → V1 respectively. For comparison, the article also considers the diffusion path of Li atoms on the original Ti2CO2 MXene material. The diffusion path was selected along the high symmetry lines between energetically favorable adsorption sites on the surface, respectively path1 and path2, as shown in Fig. 5a and b. It can be found through a calculation that the diffusion energy barrier of Li in the migration path designed in this article was in the range of 0.20–0.45 eV. Among them, the C5 → V1 → C2 diffusion path has the lowest energy barrier (0.20 eV), and the C1 → C4 diffusion path has the highest energy barrier (0.45 eV). For original Ti2CO2 MXene, the diffusion barriers of Li atoms on its surface are 0.84 eV and 0.32 eV respectively, which are higher than TiVCO2 MXene. This result showed that vanadium doping reduced the diffusion barrier of lithium atoms on the surface of MXene, making it suitable as an anode material for lithium-ion batteries.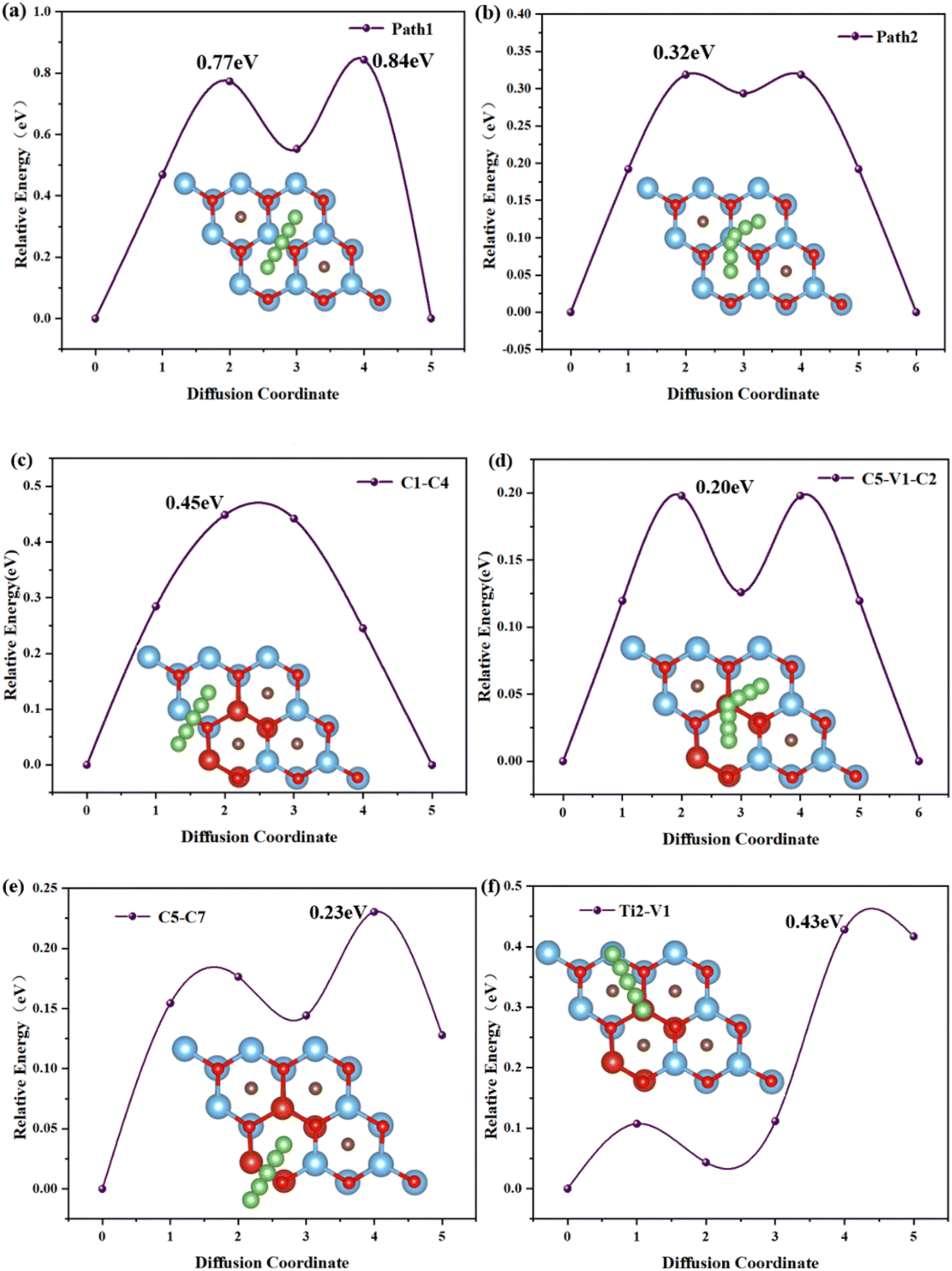 | ||
| Fig. 5 Diffusion energy barrier and path of lithium ions on the surface of Ti2CO2 (a and b) and TiVCO2 (c–f) monolayers. | ||
3.5 Electrochemical performance
Based on the above analysis, the article believes that Li atoms can be stably adsorbed on the MXene surface, and the energy barrier for Li-ion diffusion is low. However, the above results only consider the adsorption and migration of one Li atom on the surface of TiVCO2 MXene, and the adsorption process of multiple Li atoms can better describe the situation of the material during use. Therefore, the article calculated the open circuit voltage (OCV), binding energy (Eb) and theoretical lithium storage capacity to evaluate the cumulative effect of multiple lithium atoms on the surface of TiVCO2 MXene. According to the adsorption energy results discussed above, lithium atoms were preferentially adsorbed at the C position with more negative adsorption energy (Fig. 6a), and the second layer was adsorbed above the transition metal atoms, as shown in Fig. 6b. The article provided images of the changes in OCV and Eb of Li atoms on the surface of Ti2CO2 and TiVCO2 MXene as a function of specific capacity, respectively, as shown in Fig. 7a and b. It can be easily seen from the figure that the initial value of the specific capacity of TiVCO2 was larger than that of Ti2CO2. However, as the specific capacity increases, the specific capacity of TiVCO2 gradually approaches that of Ti2CO2. From Fig. 7b, contrary to the open circuit voltage, the greater the number of adsorbed Li atoms, the smaller the negative value of the binding energy. The negative value of the binding energy illustrated the stability of Li atom adsorption.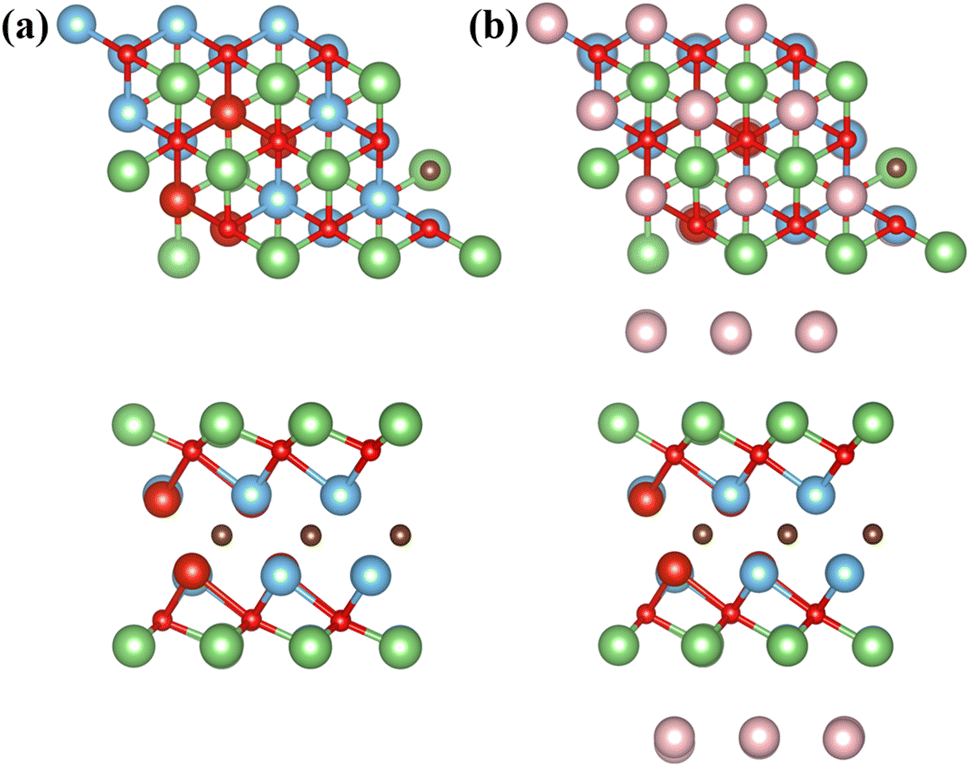 | ||
| Fig. 6 Top view and side view of optimized MXene, (a) single layer Li and (b) double layer Li on both sides. The green and pink balls represent the first and second layers of lithium atoms respectively. | ||
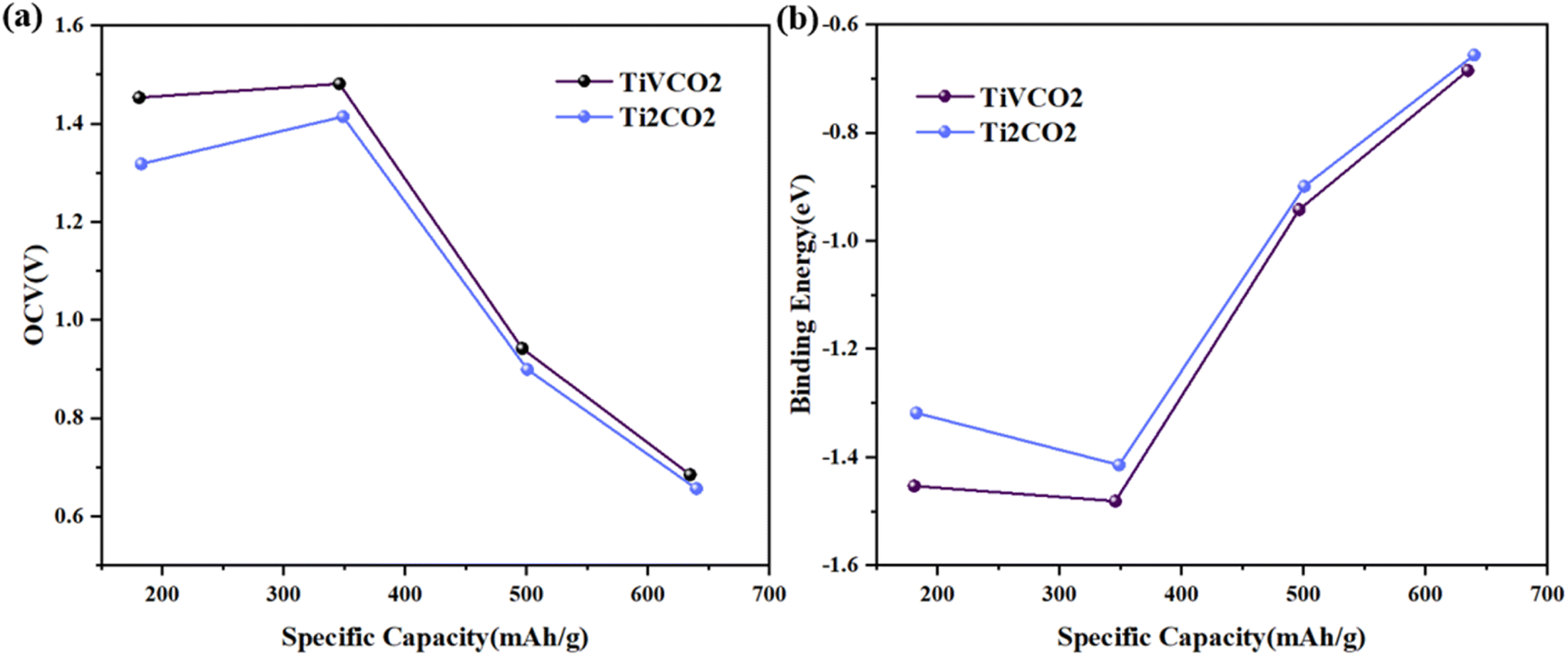 | ||
| Fig. 7 (a) Change curve of open circuit voltage with specific capacity of Ti2CO2 and TiVCO2. (b) The relationship between the binding energy of each Li atom and the specific capacity of MXene. | ||
4. Conclusion
This paper explores the possibility of MXene material (TiVCO2) as an anode material for lithium-ion batteries through computational simulation methods using VASP, ATAT and other software as carriers, based on generalized first principles, and using methods such as charge difference and Bader charge analysis. Through horizontal comparison with original MXene (Ti2CO2), the lithium storage performance of high-entropy MXene (TiVCO2) as an anode material for lithium-ion batteries was studied. By calculating the DOS and PDOS of Ti2CO2 and TiVCO2, this paper systematically studied their electronic structure and bonding mechanism. The non-zero DOS at the Fermi level and the increase in the DOS above the Fermi level indicated that after the incorporation of V atoms. The electronic structure has changed, from the original semiconductor to a conductor, and the conductivity has been significantly improved. In addition, this article studies the adsorption performance by calculating and analyzing the adsorption sites and adsorption energies of Li atoms on the surface of MXene materials. The negative adsorption energies indicated the stability of adsorption. At the same time, this paper uses charge difference and Bader charge analysis methods to further study the adsorption mechanism of Li on MXene. On average, each Li atom transfers 0.917|e| electrons to the surface of the MXene material, further indicating the adsorption of Li on its surface. By designing paths and using the climbing transition state calculation method (CI-NEB) to find the minimum diffusion energy barrier of Li on the MXene surface, and studying its surface diffusion characteristics, TiVCO2 has a lower diffusion barrier (0.20 eV) than Ti2CO2, indicating that The TiVCO2 MXene material has better ion mobility and is the guarantee of high-speed electrode materials.In summary, this article further proves that TiVCO2 MXene has better conductivity, adsorption and diffusion properties than the original MXene (Ti2CO2). High conductivity, high ion transport rate and low diffusion barrier make it a good alternative anode material for lithium-ion batteries with very impressive lithium storage performance.
Data availability
Data can be obtained from the authors. Data supporting the findings of this study are available from the corresponding authors upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The research was financially supported by National Natural Science Foundation of China (52202064), the Key Research and Development Project of Henan Province (No. 241111231600, 232300420329), Luoyang Major Science and Technology Innovation Project (2301009A), Henan Province Education Department of Key Scientific Research Project in Colleges and Universities (21B430012, 23B430012).References
- B. Dunn, H. Kamath and J.-M. Tarascon, Science, 2011, 334, 928–935 CrossRef CAS PubMed.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS PubMed.
- F. Du, H. Tang, L. Pan, T. Zhang, H. Lu, J. Xiong, J. Yang and C. J. Zhang, Electrochim. Acta, 2017, 235, 690–699 CrossRef CAS.
- J. O. Besenhard and G. Eichinger, J. Electroanal. Chem. Interfacial Electrochem., 1976, 68, 1–18 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- P. Vogt, P. De Padova, C. Quaresima, J. Avila, E. Frantzeskakis, M. C. Asensio, A. Resta, B. Ealet and G. Le Lay, Phys. Rev. Lett., 2012, 108, 155501 CrossRef PubMed.
- E. Bianco, S. Butler, S. Jiang, O. D. Restrepo, W. Windl and J. E. Goldberger, ACS Nano, 2013, 7, 4414–4421 CrossRef CAS PubMed.
- L. Song, L. Ci, H. Lu, P. B. Sorokin, C. Jin, J. Ni, A. G. Kvashnin, D. G. Kvashnin, J. Lou, B. I. Yakobson and P. M. Ajayan, Nano Lett., 2010, 10, 3209–3215 CrossRef CAS PubMed.
- H. Liu, A. T. Neal, Z. Zhu, Z. Luo, X. Xu, D. Tománek and P. D. Ye, ACS Nano, 2014, 8, 4033–4041 CrossRef CAS PubMed.
- Q. H. Wang, K. Kalantar-Zadeh, A. Kis, J. N. Coleman and M. S. Strano, Nat. Nanotechnol., 2012, 7, 699–712 CrossRef CAS PubMed.
- M. Naguib, J. Come, B. Dyatkin, V. Presser, P.-L. Taberna, P. Simon, M. W. Barsoum and Y. Gogotsi, Electrochem. Commun., 2012, 16, 61–64 CrossRef CAS.
- B. Anasori, M. R. Lukatskaya and Y. Gogotsi, Nat. Rev. Mater., 2017, 2, 16098 CrossRef CAS.
- Q. Zhong, Y. Li and G. Zhang, Chem. Eng. J., 2021, 409, 128099 CrossRef CAS.
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed.
- S. Kurahashi, S. Arabnejad, H. Ushiyama and K. Yamashita, J. Comput. Chem., Jpn., 2019, 18, 84–94 CrossRef CAS.
- M. Ashton, R. G. Hennig and S. B. Sinnott, Appl. Phys. Lett., 2016, 108, 023901 CrossRef.
- Y. Aierken, C. Sevik, O. Gülseren, F. M. Peeters and D. Çakır, J. Mater. Chem. A, 2018, 6, 2337–2345 RSC.
- W. Yi, G. Tang, X. Chen, B. Yang and X. Liu, Comput. Phys. Commun., 2020, 257, 107535 CrossRef CAS.
- V. Wang, N. Xu, J.-C. Liu, G. Tang and W.-T. Geng, Comput. Phys. Commun., 2021, 267, 108033 CrossRef CAS.
- K. Yuan, P. Hao, X. Li, Y. Zhou, J. Zhang and S. Zhong, New J. Chem., 2021, 45, 15234–15239 RSC.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Das, S. U. D. Shamim, M. K. Hossain, F. Ahmed, M. A. Hossain and M. O. Rahman, Appl. Surf. Sci., 2022, 600, 154173 CrossRef CAS.
- Q. Tang, Z. Zhou and P. Shen, J. Am. Chem. Soc., 2012, 134, 16909–16916 CrossRef CAS PubMed.
- Y. Xie, Y. Dall'Agnese, M. Naguib, Y. Gogotsi, M. W. Barsoum, H. L. Zhuang and P. R. C. Kent, ACS Nano, 2014, 8, 9606–9615 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra03618b |
| This journal is © The Royal Society of Chemistry 2024 |